

Abstract
A key goal of whole genome sequencing (WGS) for human genetics studies is to interrogate all forms of variation, including single nucleotide variants (SNV), small insertion/deletion (indel) variants and structural variants (SV). However, tools and resources for the study of SV have lagged behind those for smaller variants. Here, we used a scalable pipeline22 to map and characterize SV in 17,795 deeply sequenced human genomes. We publicly release site-frequency data to create the largest WGS-based SV resource to date. On average, individuals carry 2.9 rare SVs that alter coding regions, affecting the dosage or structure of 4.2 genes and accounting for 4.0-11.2% of rare high-impact coding alleles. Based on a computational model, we estimate that SVs account for 17.2% of rare alleles genome-wide with predicted deleterious effects equivalent to loss-of-function coding alleles; ~90% of such SVs are non-coding deletions (mean 19.1 per genome). We report 158,991 ultra-rare SVs and show that ~2% of individuals carry ultra-rare megabase-scale SVs, nearly half of which are balanced or complex rearrangements. Finally, we infer the dosage sensitivity of genes and non-coding elements, revealing trends related to element class and conservation. This work will help guide SV analysis and interpretation in the era of WGS.
INTRODUCTION
Human genetics studies employ WGS to enable comprehensive trait mapping analyses across the full diversity of genome variation, including SVs (≥50 bp) such as deletions, duplications, insertions, inversions and other rearrangements. Prior work suggests a disproportionately large role for SVs (relative to their abundance) in rare disease biology1, and in shaping heritable gene expression differences in the human population2–4. Rare and de novo SV have been implicated in the genetics of autism5–9 and schizophrenia10–13, but few other complex trait association studies have directly assessed SV14,15.
One challenge for SV interpretation in WGS-based studies is the lack of high-quality publicly available variant maps from large populations. Our current knowledge is based primarily on three sources: (1) a large and disparate collection of array-based studies16–18, with limited allele frequency data and low resolution; (2) the 1000 Genomes Project callset4, which has been invaluable but is limited by the modest sample size and low coverage design; and (3) an assortment of smaller WGS-based studies with varied coverage, technologies, analysis methods, and levels of data accessibility7,8,19–21.
There is an opportunity to improve knowledge of SV in human populations via systematic analysis of large-scale WGS data resources generated by programs such as the NHGRI Centers for Common Disease Genomics (CCDG). A key barrier to the creation of larger and more informative SV catalogs is the lack of computational tools that can scale to the size of ever-growing datasets. To this aim, we have developed a highly scalable open source SV analysis pipeline22, and used it to map and characterize SV in 17,795 deeply sequenced human genomes.
RESULTS
A population-scale SV map
The samples analyzed here are derived from common disease case/control and quantitative trait mapping collections sequenced under the CCDG program, supplemented with ancestrally diverse samples from the PAGE consortium and Simons Genome Diversity Panel. The final ancestry composition includes 24% African, 16% Latino, 11% Finnish, 39% non-Finnish European, and 9% other diverse samples from around the world (Extended Data Table 1).
The tools and pipelines used for this work are described elsewhere22. Briefly, we developed a highly scalable software toolkit (svtools) and workflow for large-scale SV callset generation that combines per-sample variant discovery23, resolution-aware cross-sample merging, breakpoint genotyping24, copy number annotation, and variant classification (Extended Data Fig. 1). We created two distinct SV callsets using different reference genome and pipeline versions. The “B37” callset includes 118,973 high-confidence SV from 8,426 samples sequenced at the McDonnell Genome Institute and aligned to the GRCh37 reference genome. The “B38” callset includes 241,031 high-confidence SV from 23,175 samples sequenced at four CCDG sites and aligned to GRCh38 using the “functional equivalence” pipeline25 (see Methods). Of the 26,347 distinct samples in the union of the two callsets, aggregate-level sharing is permitted for 17,795; these comprise the official public release (Supplementary Files 1 and 2). For simplicity of presentation, most analyses below focus on the larger B38 callset (Supplementary Table 1).
We observed a mean of 4,442 high-confidence SV per genome, predominantly deletions (35%), mobile element insertions (27%), and tandem duplications (11%) (Fig. 1b, Extended Data Figs. 2 and 3). Variant counts and linkage disequilibrium patterns are consistent with prior studies using similar methods4,26, and most SVs are mapped to base-pair resolution (Extended Data Figs. 2 and 3). As expected, the site-frequency spectrum approximates that of SNV and indels, the size distribution shows increasing length with decreasing frequency, and principal components analysis reveals population structure consistent with self-reported ancestry (Fig. 1, Extended Data Figs. 2–4). Per-genome SV counts are broadly consistent and vary as expected based on ancestry, with more genetic variation in African-ancestry individuals and fewer singletons in Finns (Extended Data Figs. 2 and 3). Although we observe some technical variability due to cohort and sequencing center, these effects are mainly limited to small (<1 kb) CNVs detected solely by read-pair signals, which are sensitive to library preparation and alignment filtering methods (see Methods, Extended Data Fig. 3).
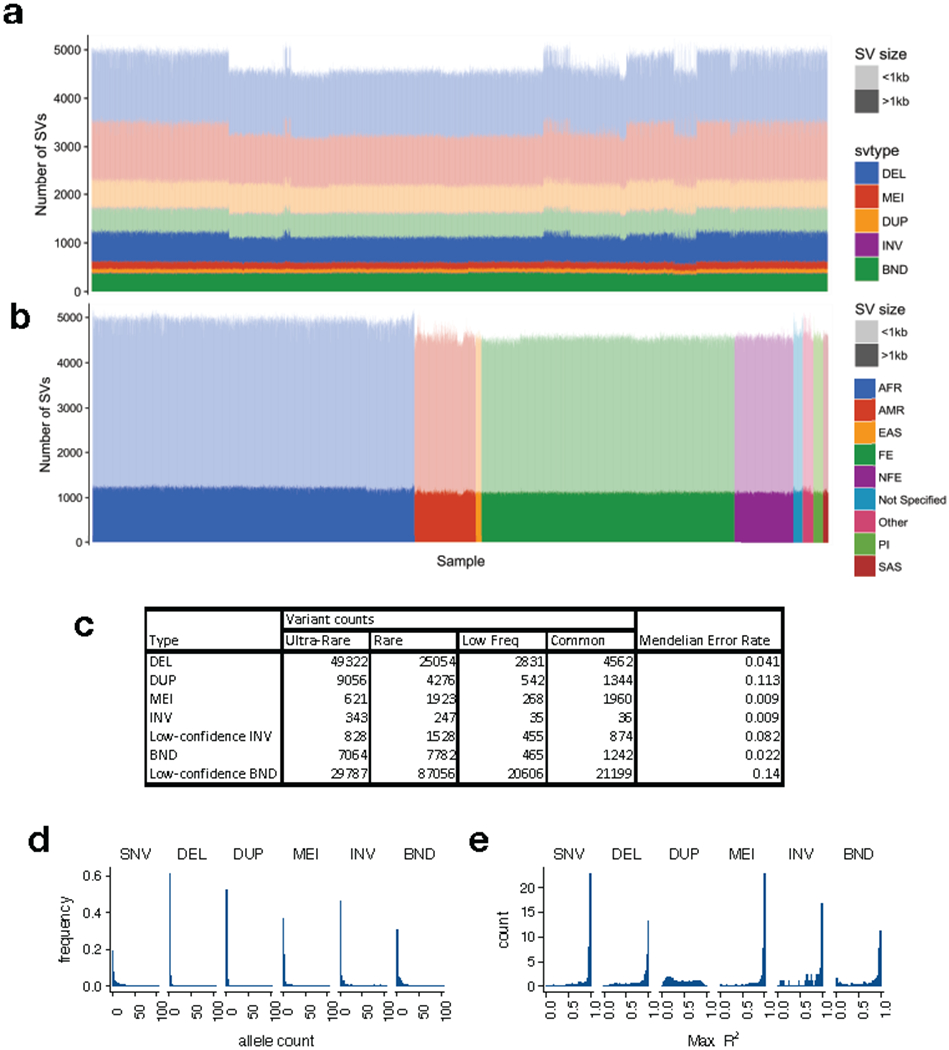
Figure 1.
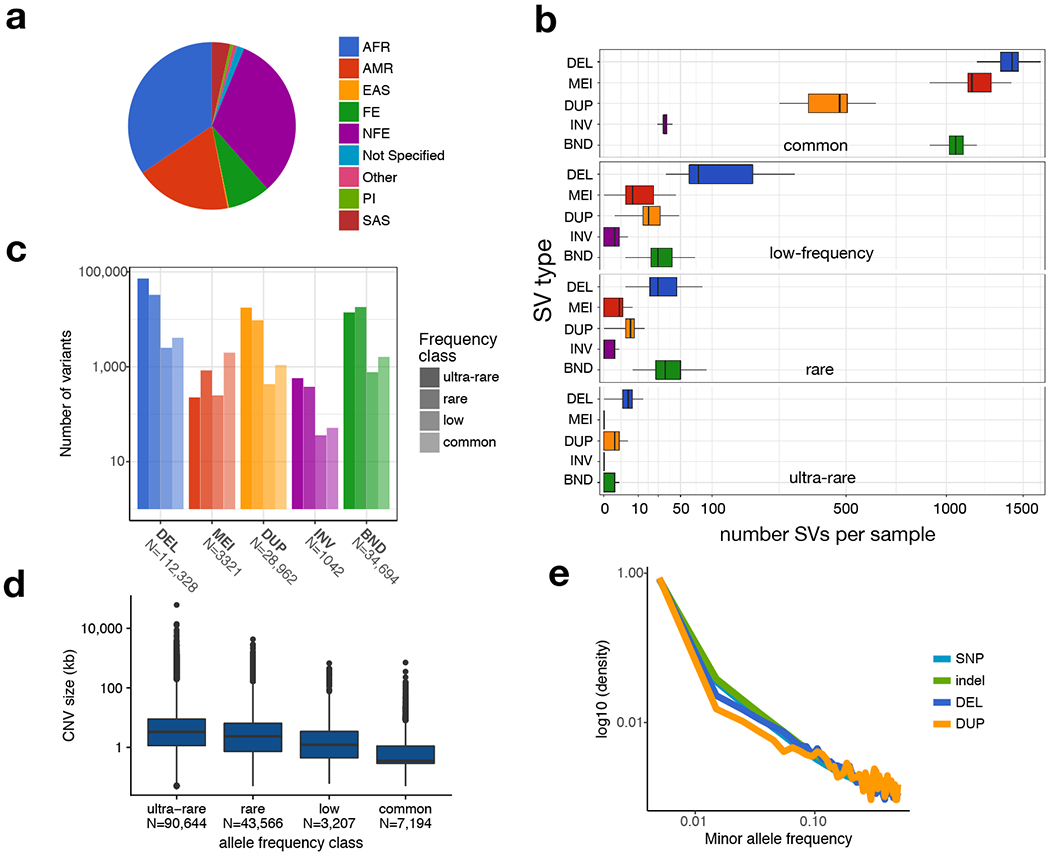
The public version of the B38 callset derived from 14,623 samples. (a) Self-reported ancestry. Abbreviations are as follows: AFR, African; AMR, admixed American; EAS, east Asian; FE, Finnish European; NFE, non-Finnish European; PI, Pacific Islander; SAS, South Asian. (b) Number of SVs per sample (x-axis, square-root scaled) by SV type (y-axis) and frequency class (panels labelled at top). SV classes are defined as: DEL, deletion; MEI, mobile element insertion; DUP, duplication; INV, inversion; BND, “break-end”, which is a generic term in the VCF specification for SV breakpoints that cannot be unequivocally classified. Minor allele frequency (MAF) bins are defined as: “ultra-rare” is private to an individual or family; “rare” is MAF<1%; “low-frequency” is 1%<MAF<5%; “common” is MAF>5%. (c) Number of high-confidence SVs by class and frequency bin. (d) CNV length distributions for each frequency class, defined as in part (b). (e) MAF distribution for SNV (N=85,687,916), indel (N=9,477,540), deletion (DEL, N=43,872) and duplication (DUP, N=10,805) variants for a subset of 4,298 samples for which GATK-based SNV/indel calls were also available. All boxplots in this figure indicate the median and the first and third quartiles.
We further characterized callset quality using independent data and analyses (see Supplementary Note) including (i) validation by deep coverage (>52-85x) long-read data from nine genomes, (ii) sensitivity relative to a comprehensive long-read callset27, (iii) inheritance patterns within a set of 3-generation pedigrees, and (iv) comparison to well-characterized short-read callsets4,27 (Supplementary Tables 2–4 and Extended Data Figs. 5–7). We achieve a validation rate of 84% by long-read data, with higher validation rates for the variant classes most relevant to the findings below: deletions (87%), rare SV (90%) and singleton SV (95%). Based on the validation rates of SV frequency classes and their relative abundance in the full dataset, we estimate a false discovery rate of 7.0%. Although overall sensitivity is low (49%) compared to long-read SV maps due to the inherent difficulty of detecting repetitive variants from short reads, it is comparable to published short-read callsets4,26,27, and substantially higher for functionally relevant subtypes such as SV larger than 1 kb (63%) and predicted high impact variants (82%).
Burden of deleterious rare SV
The contribution of rare SV to human disease remains unclear. Well-powered WGS-based trait mapping studies will ultimately be required to address this; however, the overall burden of predicted pathogenic mutations in the human population is informative and can be estimated from our data. Our analysis of 14,623 individuals identified 42,765 rare SV alleles (MAF<1%) predicted to decrease gene dosage (n=9,416), alter gene function (e.g., single exon deletion; n=26,337), or increase gene dosage (n=7,012). The majority of rare gene-altering SVs are deletions (54.5%), with fewer duplications (42.2%), and a small fraction of other variant types, primarily inversions and complex rearrangements that interrupt or rearrange exons. Of these, 23.4% affect multiple genes and 10.4% affect 3 or more genes, resulting in a mean of 4.2 SV-altered genes per individual. Based on a strict definition of loss-of-function (LoF) SV – gene disruptions and gene deletions affecting >20% of exons – we identified a mean of 1.39 rare SV-based gene LoF alleles per person. Analysis of 4,298 samples with SV and SNV/indel calls reveals that individuals carry a mean of 33.6 rare high-confidence LoF SNV and small indels (Fig. 2), consistent with prior studies28. Thus, SV accounts for 4-11.2% of rare, predicted high impact gene alterations in a population sample, depending on whether we consider all coding SV, or a strictly defined set of LoF variants (Fig. 2c). These are likely to be underestimates considering that the false negative rate of SV detection is typically higher than that of SNV and small indels24,27.
Figure 2.
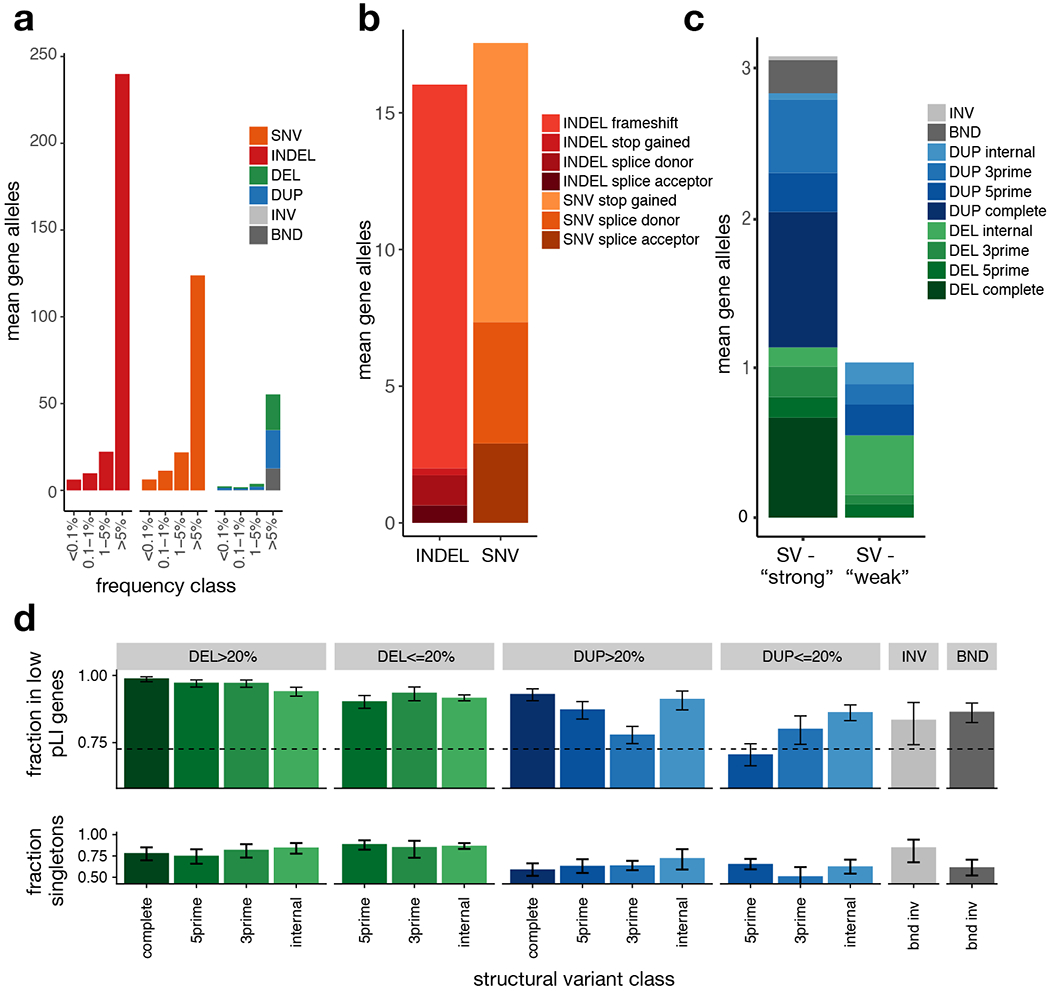
Burden of rare gene-altering SV. (a) Per-sample mean number of gene alterations by type and frequency class, in 4,298 samples. (b) Per-sample mean number of rare (<1% MAF) high-confidence PTV by type and VEP consequence. (c) Per-sample mean number of rare (<1% MAF) SV-derived gene alterations by type. DEL and DUP are classified into ‘strong’ (affecting >20% of exons of principal transcript) and ‘weak’ (affecting <20% of exons of principal transcript) and subclassified as ‘internal’ (variant overlaps at least one coding exon, but neither the 3’ nor 5’ end of the principal transcript), 3prime (variants overlaps the 3’ end of the transcript), 5prime (variant overlap the 5’ end of the transcript), and complete (variant overlaps all coding exons in principal transcript), (d) (top) Fraction of rare (<1% MAF), gene-altering variants occurring in low pLI (pLI<0.9) vs. high pLI (pLI>=0.9) genes, by type, size class, and gene region, in the B38 callset (N=14,623). Error bars indicate 95% confidence intervals (Wilson score interval). The dotted line indicates the expected fraction, assuming a uniform distribution of SV in coding exons. (bottom) Singleton rates for gene-altering variants by type in the B38 callset (N=14,623), restricted to genes with pLI>0.1. Error bars indicate 95% Wilson score confidence intervals. See Supplementary Table 5 for the number of variants in each category.
To characterize the relative impact of different coding SV classes we calculated two measures of purifying selection (Fig. 2d): (1) the fraction of variants that affect dosage tolerant genes with an LoF intolerance (pLI)28,29 score <0.9 (2) the fraction of variants present as “singletons” found in only one individual or family. By these measures, deletions are more deleterious than duplications, and complete gene deletions are the most deleterious class. Notably, based on the fraction of variants in dosage intolerant genes, complete gene duplications and sub-genic deletions affecting <20% of exons are relatively depleted, suggesting that many gene-altering SV are strongly deleterious, even if not predicted to completely obliterate gene function.
The above calculations ignore missense and non-coding variants that are expected to comprise a large fraction of rare functional variation. Predicting the impact of these variant types is challenging, but we can approximate their relative contribution to the deleterious variant burden under two simplifying assumptions: (1) impact prediction algorithms such as CADD30 and LINSIGHT31 are capable of ranking variants within a given class (SNV, indel, SV) by their degree of deleteriousness, and (2) the mean deleterious impact of a given set of variants is reflected by its singleton rate. The first assumption is somewhat tenuous, but should be valid here given that impact prediction inaccuracies are likely to affect all variant classes similarly; the second should hold under an infinite sites model of mutation, which is reasonable for the (N=4,298) samples used in this analysis. We note that other evolutionary forces such as positive selection, background selection, and biased gene conversion can also shape the site frequency spectrum; however, we expect that these forces would act similarly on the variant classes examined here, in a genome-wide analysis of a very large number of sites.
We used CADD and LINSIGHT to generate impact scores for SNV, indels, deletions and duplications (see Methods). As expected, these are highly correlated with singleton rate and variant effect predictions from VEP32 and LOFTEE28 (Fig. 3). We sought to identify “strongly deleterious” variants from each class by choosing impact score thresholds to match the singleton rate of the entire set of high-confidence LoF mutations. Individuals carried a mean of 121.9 such “strongly deleterious” rare variants, comprising 63% SNV, 19.8% indels and 17.2% SV (Fig. 3d). Given the relative numerical abundance of different rare variant classes, this suggests that a given rare SV is 841-fold more likely to be strongly deleterious than a rare SNV, and 341-fold more likely than a rare indel. Predicted deleterious SV are slightly larger than rare SV on the whole (median 4.5 vs. 2.8 kb). Whereas only a minority (13.1%) of predicted strongly deleterious SNV and indels are non-coding, 90.1% of predicted strongly deleterious rare SV are non-coding. In particular, the top 50% of non-coding deletions show similar levels of purifying selection (as measured by singleton rate) as high-confidence LoFs caused by SNV/indels (see Fig. 3c), implying that a typical individual carries 19.1 strongly deleterious rare non-coding deletion alleles. This suggests that non-coding deletions may have strong deleterious effects and play a larger than expected role in human disease.
Figure 3.
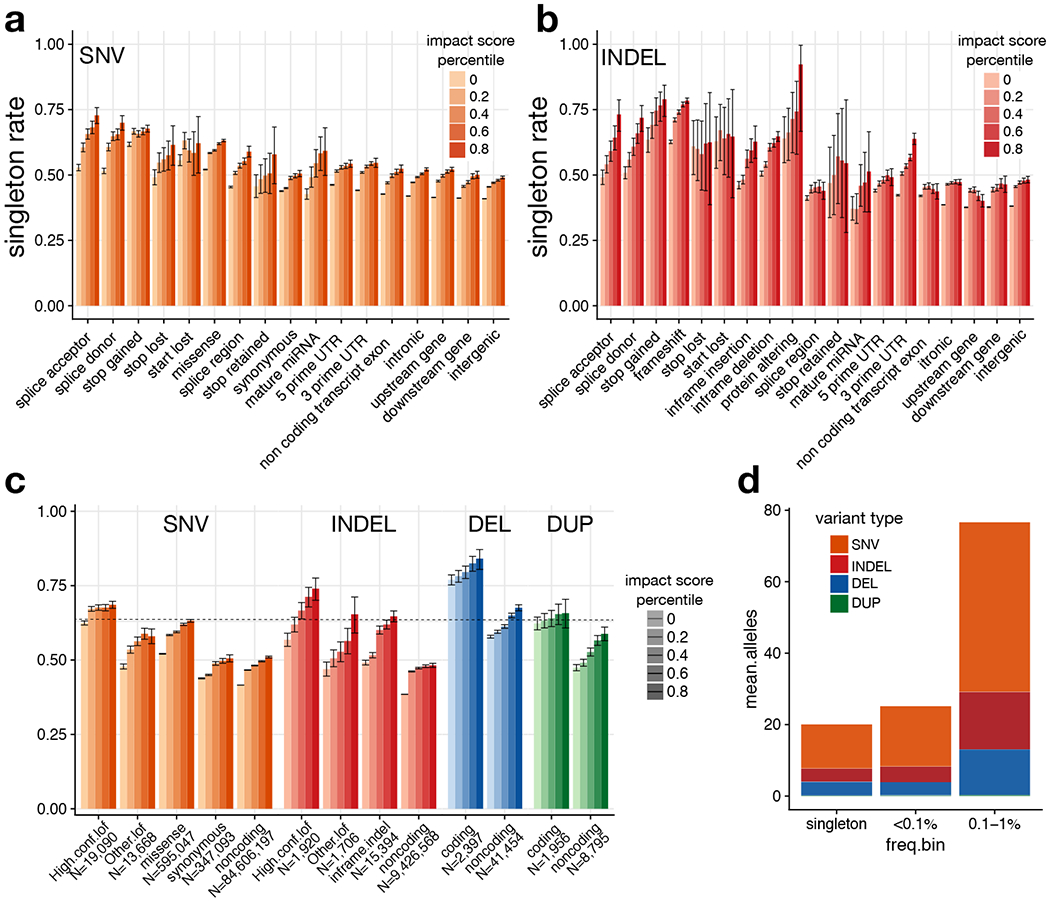
Estimation of genome-wide burden of high-impact functional alleles. (a) Singleton rates for SNV, by VEP consequence and percentile of combined VEP/CADD impact score. (b) Singleton rates for indels. (c) Singleton rates by variant type and percentile of combined VEP/CADD impact score. Here, “other LoF” indicates VEP-annotated protein-truncating variants (PTVs) that are not classified as high-confidence by LOFTEE. DELs and DUPs that intersect any coding exon of the principal transcript are classified as “coding”; otherwise they are “noncoding”. The horizontal line shows the singleton rate for all high confidence SNV/indel LoFs. (d) Per-sample mean number of “strongly deleterious” alleles genome-wide, by type and frequency class. In panels (a)-(c), error bars indicate the 95% confidence interval (Wilson score method). See Supplementary Table 6 for counts of variants in each category.
Landscape of ultra-rare SV
Most ultra-rare SV represent recent or de novo structural mutations, and thus the relative abundance of different ultra-rare SV classes sheds light on the underlying mutational processes at work. We identified 158,991 ultra-rare SV (105,175 high-confidence) present in only one of 14,623 individuals or private to a family. This corresponds to a mean of ~11.4 per individual (Extended Data Fig. 8a). Ultra-rare SV are mainly composed of deletions (5.2 per person) and duplications (1.3), with a smaller number of inversions (0.17).
It is of interest that ~40% of ultra-rare SV breakpoints in our dataset cannot be readily classified into the canonical forms of SV. This is a known limitation of short-read WGS, and such variants are often ignored. Formally, these SVs are of the generic “breakend” (BND) class33. We examined the 63,559 ultra-rare BNDs for insights into their composition and origin. Many (17.0%) appear to be deletions too small (<100 bp) to exhibit convincing read-depth support, and that our pipeline conservatively classifies as BNDs (e.g., complex SV can masquerade as deletions). 2.4% of the ultra-rare BND stem from 1,542 “retrogene insertions” caused by retroelement machinery acting on mRNAs. This set of retrogene insertions is ~10-fold larger than prior maps34–36 and will be valuable for future studies. 5.5% of ultra-rare BNDs are complex genomic rearrangements with multiple breakpoints in close proximity (<100 kb). The remainder are difficult-to-classify variants involving either local (49.9% ≤1 Mb), distant intra-chromosomal (5.7% >1 Mb), or inter-chromosomal alterations (27.2%), many (78.0%) of which are classified as low-confidence SV calls. This final class is likely caused primarily by repetitive element variation, but is also expected to be enriched for false positives.
A variety of sporadic disorders are caused by extremely large and/or complex SV, but knowledge of the frequency and architecture of these dramatic alterations in the general population is incomplete due to the limitations of array-based methods used in prior large-scale studies37, which fail to detect balanced events or resolve complex variant architectures. We observed 138 megabase-scale CNVs corresponding to a frequency of ~0.01 per individual; these include 47 deletions and 91 duplications, and affect a mean of 12.1 genes (Extended Data Fig. 8). Three individuals carried 2 megabase-scale CNVs, apparently due to independent mutations. We observed 19 reciprocal translocations (0.001 per individual), consistent with prior cytogenetic-based estimates38,39. Of these translocations, 14 affect one gene and 2 affect two genes, producing 1 predicted in-frame gene fusion (PI4KA:MGLL). We applied breakpoint clustering (as in 40) to identify ultra-rare complex rearrangements and discovered 33 complex SVs spanning >1 Mb (0.003 per individual). Most of these (20/33, 60.6%) involve three breakpoints; however, we observed 5 large-scale rearrangements with 5 or more breakpoints. Notably, when the entire SV size distribution is considered, 3.3% of ultra-rare SVs are complex variants, consistent with previous smaller-scale studies41–45.
Dosage sensitivity
A motivation for creating population-scale SV maps is to annotate genomic regions based on their tolerance to dosage changes and structural rearrangements, thus revealing the genes and non-coding elements most important (or dispensable) for human development and viability. The pLI score from ExAC/gnomAD28,29 has proven invaluable for this purpose but does not predict the effects of increased dosage or include non-coding elements.
We first generated DEL and DUP sensitivity scores for each gene based on the observed frequency of CNV in the combined dataset of 17,795 samples (as in 46, see Methods). The resulting scores correlate with the CNV scores from ExAC46, and with the DECIPHER haploinsufficiency score47 (Extended Data Fig. 9). Despite their relatively modest correlations with each other, all three measures are informative based on comparison to pLI, which was generated using an independent set of variants (SNV and indels). A combined score from multiple datasets performs better than any single score, and may be useful for interpreting rare SVs (Supplementary File 4).
We next performed a genome-wide analysis based on the frequency of dosage alterations in 1 kb genomic windows (see Methods). Our current dataset is not large enough to predict dosage sensitive non-coding elements based on the absence of variation; however, we can investigate the relative sensitivity of genomic features in aggregate. As expected, we observe a strong depletion of CNV near coding exons that varies by proximity to the nearest exon as well as pLI of the corresponding gene (Fig. 4a). We therefore estimated odds ratios for depletion of CNV in each functionally annotated region, stratified by distance and pLI of the nearest exon. The resulting dosage sensitivity scores mirror independent measures of selective constraint including LINSIGHT and PHASTCONS (Fig. 4b).
Figure 4.
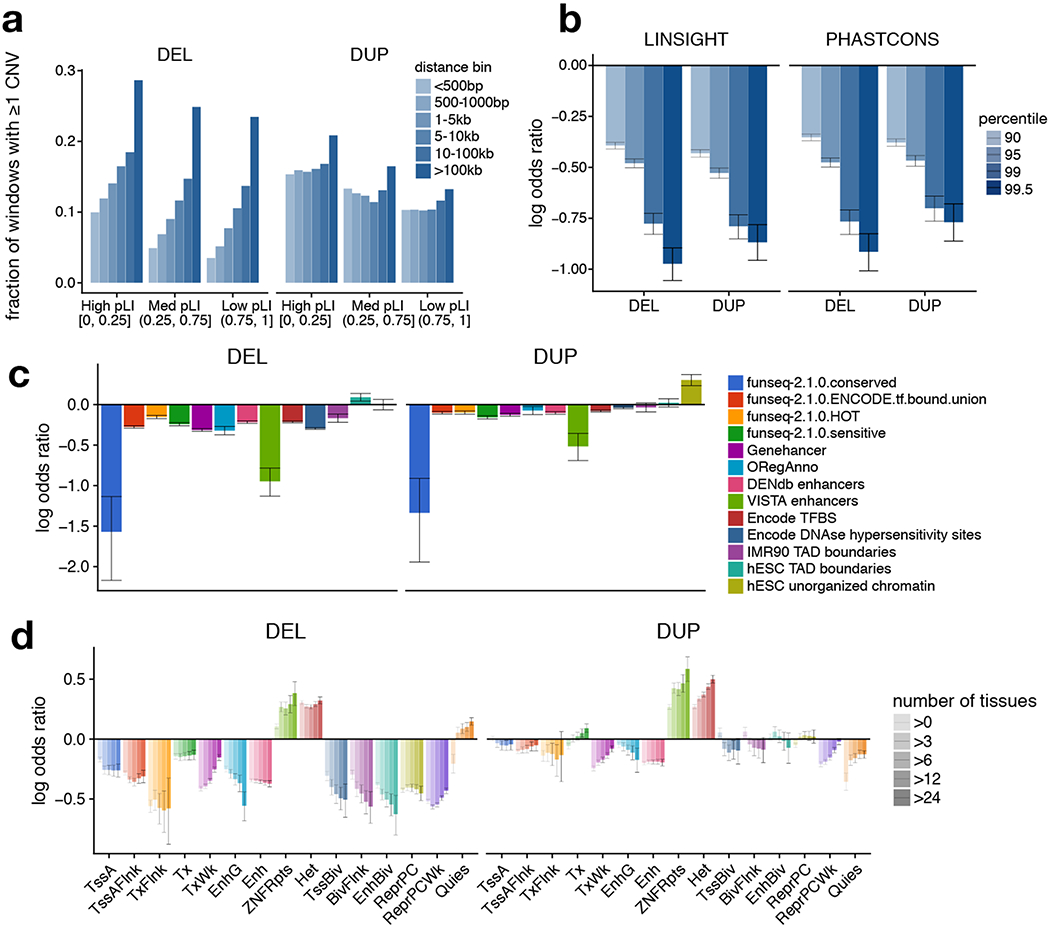
Dosage-sensitivity of functional annotations. (a) Fraction of 1 kb genomic windows containing at least one CNV, as a function of distance to the nearest coding exon and the pLI of that gene. (b) Depletion of CNV in conserved genomic regions. Log-odds ratios for the occurrence of CNV in highly conserved (based of LINSIGHT or PHASTCONS percentile) vs. less-conserved regions. Odds ratios are Cochran-Mantel-Haenszel estimates, stratified by distance to and pLI of nearest coding exon. (c) Log-odds ratios (estimated as in (b)) for the occurrence of CNV in 1 kb windows intersecting various functional annotation tracks. (d) Log-odds ratios (estimated as in (b)) for the occurrence of CNV in 1 kb windows overlapping roadmap segmentations, stratified by the number of roadmap tissues in which the region is observed. All error bars indicate 95% confidence intervals estimated by block bootstrap.
We also examined the relative dosage sensitivity of regulatory and epigenomic annotations from various projects48–53 (Fig. 4). Regulatory elements such as enhancers, polycomb repressors, DNase hypersensitivity sites, and transcription factor binding sites show strong sensitivity to dosage loss via deletion, whereas regions of inert non-coding annotations do not. The patterns of sensitivity to dosage gains via duplication are broadly similar, albeit weaker, with no obviously distinct patterns at (for example) enhancers, repressors or insulators. Dosage sensitivity of regulatory elements at “bivalent” genomic regions from ROADMAP is greater than their counterparts (e.g., enhancers vs. bivalent enhancers), suggesting that such elements may be under especially strong selection. Further, dosage sensitivity increases with the number of cell-types sharing a given annotation, suggesting a higher sensitivity for constitutive regulatory elements compared to those that act in a more cell-type specific manner.
DISCUSSION
Here, we have conducted the largest WGS-based study of SV in the human population to date. The sample size and use of deep (>20x) WGS allowed us to map rare SVs at high genomic resolution and estimate the relative burden of deleterious SV. Our data suggest that rare SV account for 4-11.2% of deleterious coding alleles and 17.2% of deleterious alleles genome-wide, an outsized contribution considering that SVs comprise merely ~0.1% of variants. Noteworthy is the burden of rare, strongly deleterious non-coding deletions apparent in our dataset: we estimate that a typical individual carries ~19 rare non-coding deletions that exhibit levels of purifying selection similar to LoF SNV and indels (of which there are ~34 per individual). These results indicate that comprehensive assessment of SV will improve power in rare variant association studies.
The public site-frequency maps reported here will also aid variant interpretation in smaller scale WGS-based studies (e.g., via allele frequency look-ups), in particular as they were generated via systematic joint analysis of large datasets from diverse populations (similar to ExAC/gnomAD28). One limitation is the high false negative rate for repetitive SV including mobile element insertions (MEI), short tandem repeats (STR) and multi-allelic CNVs (mCNV) due to the limitations of algorithms that rely on unique short-read alignments. Whereas we have reported a mean of 4,442 SV per genome, recent long-read analyses predict up to ~27,662 SV per genome, including STRs and other highly repetitive elements27. Although the inherent limitations of short-read WGS cannot be overcome, this resource could be made more comprehensive in future work with specialized algorithms tailored to MEI, STR and mCNV.
Finally, we have mined this resource to assess the dosage sensitivity of genes and non-coding elements. At genes, our results complement existing estimates from exome sequencing and microarray data. At non-coding elements, we observe strong correlations with measures of nucleotide conservation, purifying selection, regulatory element activity, and cell-type specificity. Although our current sample size is insufficient to assess dosage-sensitivity of individual non-coding elements, this will become feasible as large-scale WGS resources from ongoing international programs become available.
METHODS
Generation of the “Build 38” (B38) callset
Per-sample processing.
This callset is derived from 23,559 individuals that were part of the CCDG program as well as 950 Latino samples from the PAGE consortium. All data was produced at one of the four CCDG-funded sequencing centers and aligned to genome build GRCh38 using each individual center’s functionally equivalent pipeline implementation25. Per-sample calling was performed on 23,547 samples using LUMPY23 (v0.2.13), CNVnator54 (v0.3.3) and svtyper24 (v0.1.4). We excluded HLA, decoy or alternate contigs and regions of much higher than the expected copy number (>12 mean copies per genome across 409 samples) from SV calling with LUMPY (https://github.com/hall-lab/speedseq/blob/master/annotations/exclude.cnvnator_100bp.GRCh38.20170403.bed).
Per-sample QC.
We observed an excess of small (400 - 1000 bp) singleton deletions (i.e., present in only a single sample), suggesting a large number of false positives. On further investigation, this excess arose from differences between centers in library insert-size distribution. To reduce the number of false positive small deletions, deletions of ≤1000 bp were eliminated unless they had split read support in at least one sample. Subsequently, per-sample quality control was performed to eliminate outlier samples. We removed 213 samples where variant counts (for any SV type) were >6 median absolute deviations from the median count for that type.
Merging and cohort-level re-genotyping.
The remaining samples were processed into a single, joint callset using svtools22 (https://github.com/hall-lab/svtools) (v0.3.2), modified to allow for multi-stage merging. The code for this merging is available in a container via DockerHub (https://hub.docker.com/) (ernfrid/svtools_merge_beta@sha256:126ad19ad1aae53d05127df93105d83d236ddfb11a8aa65344f0d0aee936f919). Samples were merged using svtools lsort followed by svtools lmerge in batches of 1000 samples (or fewer) within each cohort. The resulting per-cohort batches were then merged again using svtools lsort and svtools lmerge to create a single set of variants for the entire set of 23,331 remaining samples. This site list was then used to genotype each candidate site in each sample across the entire cohort using svtyper (v0.1.4). Genotypes for all samples were annotated with copy number information from CNVnator. Subsequently, the per-sample VCFs were combined together using svtools vcfpaste. The resulting VCF was annotated with allele frequencies using svtools afreq, duplicate SVs pruned using svtools prune, variants reclassified using svtools classify (large sample mode), and any identical lines removed. For reclassification of chromosomes X and Y, we used a container hosted on DockerHub (ernfrid/svtools_classifier_fix:v1). All other steps to assemble the cohort above used the same container used for merging.
Callset tuning.
Using the variant calling control trios, we chose a Mean Sample Quality (MSQ) cutoff for inversions (INV) and breakends (BNDs) that yielded approximately a 5% Mendelian error rate (ME). Inversions passed if: MSQ ≥ 150; neither split-read nor paired-end lumpy evidence made up <10% of total evidence; each strand provided at least >10% of read support . BNDs passed if MSQ ≥ 250.
Genotype refinement.
Mobile element insertion (MEI) and deletion (DEL) genotypes were set to missing on a per-sample basis (https://github.com/hall-lab/svtools/blob/develop/scripts/filter_del.py, commit 5c32862) if the site was poorly captured by split-reads. Genotypes were set to missing if the size of the DEL or MEI was smaller than the minimum size discriminated at 95% confidence by svtyper (https://github.com/hall-lab/svtools/blob/develop/scripts/del_pe_resolution.py, commit 3fc7275). DEL and MEI genotypes for sites with allele frequency ≥0.01 were refined based on clustering of allele balance and copy number values within the datasets produced by each sequencing center (https://github.com/hall-lab/svtools/blob/develop/scripts/geno_refine_12.py, commit 41fdd60). In addition, duplications were re-genotyped with more sensitive parameters to better reflect expected allele balance for simple tandem duplications (https://github.com/ernfrid/regenotype/blob/master/resvtyper.py, commit 4fadcc4).
Filtering for size.
The remaining variants were filtered to meet the size definition of an SV (≥50 bp). The length of intra-chromosomal generic breakends (BNDs) was calculated using vawk (https://github.com/cc2qe/vawk) as the difference between the reported positions of each breakpoint.
Large callset sample QC.
Of the remaining samples, we evaluated per-sample counts of deletions, duplications, and generic breakends within the low allele frequency (0.1% - 1%) class. Samples with variant counts exceeding 10 median absolute deviations from the mean for any of the 3 separate variant classes were removed. In addition, we removed samples with genotype missingness >2% . These QC filters removed a total of 120 additional samples. Finally, we removed 64 samples that were identified as duplicates or twins in a larger set of data.
Breakpoint resolution
Breakpoint resolution was calculated using bcftools (v1.3.1) query to create a table of confidence intervals for each variant in the callset, but excluding secondary BNDs. Each breakpoint contains two 95% confidence intervals, one each around the start location and end location. Summary statistics were calculated in RStudio (v1.0.143; R version 3.3.3).
Self-reported ethnicity
Self-reported ethnicity was provided for each sample via the sequencing center and aggregated by the NHGRI Genome Sequencing Program (GSP) coordinating center. For each combination of reported ethnicity and ancestry, we assigned a super-population, continent (based on the cohort), and ethnicity. Samples where ancestry was unknown, but the sample was Hispanic, were assigned to the Americas (AMR) superpopulation. Summarized data are presented in Extended Data Table 1.
Sample relatedness
As SNV calls were not yet available for all samples at the time of the analysis, relatedness was estimated using large (>1 kb), high-quality autosomal deletions and mobile element insertions with allele frequency >1%. These were converted to plink format using plink (v1.90b3.38) and then subjected to kinship calculation using KING55 (v2.0). The resulting output was parsed to build groups of samples connected through first degree relationships (kinship coefficient > 0.177). Correctness was verified by the successful recapitulation of the 36 complete Coriell trios included as variant calling controls.
Callset summary metrics
Callset summary metrics were calculated by parsing the VCF files with bcftools (v1.3.1) query to create tables containing information for each variant/sample pairing or variant alone, depending on the metric. Breakdowns of the BND class of variation were performed using vawk to calculate orientation classes and sizes. These were summarized using Perl and then transformed and plotted using RStudio (v1.0.143; R version 3.3.3).
Ultra-rare variant analysis
We defined an ultra-rare variant as any variant unique to one individual or one family of first degree relatives. We expect the false positive rate of ultra-rare variants to be low because systematic false positives due to alignment issues are likely to be observed in multiple unrelated individuals. Therefore, we considered both high and low confidence variants in all ultra-rare analyses.
Constructing variant chains.
Complex variants were identified as in Chiang et al.3 by converting each ultra-rare SV to BED format and, within a given family, clustering breakpoints occurring within 100,000 bp of each other using bedtools56 (v2.23.0) cluster. Any clusters linked together by BND variants were merged together. The subsequent collection of variant clusters and linked variant clusters (hereafter referred to as chains) were used for both retrogene and complex variant analyses.
Manual review.
Manual review of variants was performed using IGV (v2.4.0). Variants were converted to BED12 using svtools (v0.3.2) for display within IGV. For each sample, we generated copy number profiles using CNVnator (v0.3.3) in 100 bp windows across all regions contained in the variant chains.
Retrogene insertions.
Retrogene insertions were identified by examining the ultra-rare variant chains constructed as described above. For each chain, we identified any constituent SV with a reciprocal overlap of 90% to an intron using bedtools (v2.23.0). For each variant chain, the chain was deemed a retrogene insertion if it contained one or more BND variants with +/− strand orientation that overlapped an intron. Additionally, we flagged any chains that contained non-BND SV calls, as their presence was indicative of a potential misclassification, and manually inspected them to determine if they represented a true retrogene insertion.
Complex variants.
We retained any cluster(s) incorporating 3 or more SV breakpoint calls, but removed SVs identified as retrogene insertions either during manual review or algorithmically. In addition, we excluded one call deemed to be a large, simple variant after manual review.
Large variants.
Ultra-rare variants >1 Mb in length were selected and any overlap with identified complex variants identified and manually reviewed. Of 5 potential complex variants, one was judged to be a simple variant and included as a simple variant while the rest were clearly complex variants and excluded. Gene overlap was determined as an overlap ≥1 bp with any exon occurring within protein-coding transcripts from Gencode v27 marked as a principal isoform according to APPRIS57.
Balanced Translocations.
Ultra-rare generic “breakend” (BND) variants, of any confidence class, connecting two chromosomes and with support (>10%) from both strand orientations were initially considered as candidate translocations. We further filtered these candidates to require exactly two reported strand orientations indicating reciprocal breakpoints (i.e. +−/−+, −+/+−, −−/++, ++/−−), no read support from any sample with a homozygous reference genotype, at least one split-read supporting the translocation from samples containing the variant, and <25% overlap of either breakpoint with any simple repeat (downloaded from ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/database/simpleRepeat.txt.gz).
Comprehensive annotations from the Gencode v27 GTF (ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_27/gencode.v27.annotation.gtf.gz) were used to determine the number of affected genes. A BED file of all introns was created by converting transcripts and exons to BED entries and subtracting all exons from their respective transcripts using bedtools (v2.23.0). To identify translocations affecting genes, the translocations were converted to BEDPE using svtools (v0.3.1), padded by 1 bp and intersected with introns using bedtools (v2.23.0). The number of unique chromosome/gene name pairs for each translocation was used to determine the number of affected genes affected by each breakpoint.
To determine if a translocation resulted in an in-frame fusion, we converted to BEDPE, padded by 1 bp and intersected the breakpoints with all introns using bedtools (v2.23.0). Each intron entry was then padded by 1 bp and intersected with the Gencode GTF file using bedtools (v2.23.0) and restricting to coding exons of the same transcript as the intron. Then, for each set of exons intersected by a given translocation, all combinations of transcripts were compared, taking into account their orientation and the orientation of the breakpoint, to determine if frame was maintained across the potentially fused exons. The resulting two candidate translocations were manually reviewed by reconstructing the transcript sequence of the fusion and translating the resulting DNA sequence using https://web.expasy.org/translate/ to confirm a single open reading frame was maintained.
Generation of the “Build 37” (B37) callset
Per-sample processing.
This callset was constructed starting from a set of 8,455 individuals: 8,181 samples from 8 cohorts sequenced at the McDonnell Genome Institute, as well as 274 samples from the Simons Genome Diversity Project downloaded from EMBL-EBI (https://www.ebi.ac.uk/ena/data/view/PRJEB9586). All samples passed standard production QC metrics and had a mean depth of coverage > 20X. Data were aligned to GRCh37 using the speedseq (v0.1.2) realignment pipeline. Per-sample SV calling was performed with speedseq sv (v0.1.2) using LUMPY (v0.2.11), cnvnator-multi, and svtyper (v0.1.4) on our local compute cluster. For LUMPY SV calling, we excluded high copy number outlier regions derived from >3,000 Finnish samples as described previously22 (https://github.com/hall-lab/speedseq/blob/master/annotations/exclude.cnvnator_100bp.112015.bed).
Per-sample QC.
Following a summary of per-sample counts, samples with counts of any variant class (DEL, DUP, INV, or BND) exceeding the median plus 10 times the median absolute deviation for that class were excluded from further analysis; 17 such samples were removed.
Merging.
The remaining samples were processed into a single, joint callset using svtools (v0.3.2) and the two-stage merging workflow (as described above): each of the 9 cohorts was sorted and merged separately in the first stage, and the merged calls from each cohort sorted and merged together in the second stage.
Cohort-level re-genotyping.
The resulting SV loci were then re-genotyped with svtyper (v0.1.4) and copy-number annotated using svtools (v0.3.2) in parallel, followed by combination of single-sample VCFs, frequency annotation, and pruning using the standard workflow for svtools (v0.3.2). A second round of re-genotyping with more sensitive parameters to better reflect expected allele balance for simple tandem duplications (https://github.com/ernfrid/regenotype/blob/master/resvtyper.py, commit 4fadcc4) was then performed, followed by another round of frequency annotation, pruning, and finally reclassification using svtools (v0.3.2) and the standard workflow.
Callset tuning and site-level filtering.
Genotype calls for samples in 452 self-reported trios were extracted, and Mendelian error rates calculated using a custom R script; we counted as a Mendelian error any child genotype inconsistent with inheritance of exactly one allele from the mother and exactly one allele from the father. Filtering was performed as described for the B38 callset: Inversions passed if: MSQ ≥ 150; neither split-read nor paired-end lumpy evidence made up <10% of total evidence; each strand provided at least >10% of read support. Generic breakends passed if MSQ ≥ 250. SV of length <50 bp were removed, according to our working definition of ‘structural variation’.
Final sample-level filtering.
Nine samples with retracted consents, and two hydatidiform mole samples were removed from the callset. Subsequently, the numbers of qc-passing, very rare (< 0.1% MAF) DEL, DUP, and BND per sample were determined. Excluding the samples in the Simons Genome Diversity cohort (which were expected, in general, to have unusually high counts of rare variants), we determined the median and median absolute deviation (MAD) of the per-sample counts of each type, and excluded outlier samples with a count exceeding the median+10*MAD of any type. Nine samples were removed in this way. Finally, kinship was estimated using KING (v2.0) based on high-quality, autosomal deletion and MEI calls with population allele frequency>1%. Each SV was annotated in the VCF according to the number of distinct, first-degree family clusters in which it was observed, as for the Build38 callset.
Principal components analysis.
A set of unrelated individuals (containing no first or second degree relatives) was extracted using KING (v2.0). PCA was performed using smartpca (version 13050) on a VCF of all high-quality DEL and MEI variant calls with population allele frequency > 1%. Eigenvectors were estimated based on the set of unrelated samples, and then all samples projected onto the eigenvectors.
Build38 (B38) SNV/indel callset generation and QC
Per-sample calling was performed at the Broad Institute as part of CCDG joint-calling of 22,609 samples using GATK58 (https://www.biorxiv.org/content/early/2018/07/24/201178) HaplotypeCaller v3.5-0-g36282e4. All samples were joint called at the Broad using GATK v4.beta.6, filtered for sites with an excess heterozygosity value of more than 54.69, and recalibrated using VariantRecalibrator with the following features: QD, MQRankSum, ReadPosRankSum, FS, MQ, SOR, and DP. Individual cohorts were subset out of the whole-CCDG callset using Hail v0.2 (https://github.com/hail-is/hail). Following SNV and indel variant recalibration, multiallelic variants were decomposed, and normalized with vt (v0.5)59. Duplicate variants and variants with symbolic alleles were then removed. Afterwards, variants were annotated with custom computed allele balance statistics, 1000 Genomes allele frequencies29, gnomAD based population data28, VEP (v88)60, CADD30 (v1.2), and LINSIGHT31. Variants having greater than 2% missingness were soft filtered. Samples with high rates of missingness (>2%) or with mismatches between reported and genetically-estimated sex (determined using plink v1.90b3.45 sex-check) were excluded. The LOFTEE plugin (v0.2.2-beta; https://github.com/konradjk/loftee) was used to classify putative LoF SNV and indels as high or low confidence.
Annotation of gene-altering SV calls
The VCF was converted to BEDPE format using svtools vcftobedpe The resulting BEDPE file was intersected (using bedtools (v2.23.0) intersect and pairtobed) with a BED file of coding exons from Gencode v27 with principal transcripts marked according to APPRIS57. The following classes of SV were considered putative gene-altering events: (1) DEL, DUP, or MEI intersecting any coding exon; (2) INV intersecting any coding exon and with either breakpoint located within the gene body; and (3) BND with either breakpoint occurring within a coding exon.
Gene-based estimation of dosage sensitivity
We followed the method of Ruderfer et al.46, to estimate genic dosage sensitivity scores using counts of exon-altering deletions and duplications in a combined callset comprising the 14,623 sample pan-CCDG callset plus 3,172 non-redundant samples from the B37 callset. Build37 CNV calls were lifted over to build38 as BED intervals using crossmap (v0.2.1)61. We determined the counts of deletions and duplications intersecting coding exons of principal transcripts of any autosomal gene. In Ruderfer et al.46, the expected number of CNVs per gene was modeled as a function of several genomic features (GC content, mean read depth, etc.), some of which were relevant to their exome read-depth CNV callset but not to our WGS-based breakpoint mapping lumpy/svtools callset. In order to select the relevant features for prediction, using the same set of gene-level annotations as in Ruderfer et al.46, we restricted to the set of genes in which fewer than 1% of samples carried an exon-altering CNV, and used l1-regularized logistic regression (from the R glmnet package62, v2.0-13), with the penalty chosen by 10-fold cross-validation. The selected parameters (gene length, number of targets, and segmental duplications) were then used as covariates in a logistic regression-based calculation of per-gene intolerance to DEL and DUP, similar to that described in Ruderfer et al.46. For deletions (or duplications, respectively), we restricted to the set of genes with <1% of samples carrying a DEL, to estimate the parameters of the logistic model. We then applied the fitted model to the full set of genes to calculate genic CNV intolerance scores as the residuals of the logistic regression of CNV frequency on the genomic features, standardized as z-scores and with winsorization of the lower 5th percentile.
Genome-wide estimation of deleterious variants
In order to estimate the relative numbers of deleterious SNV, indels, DELs and DUPs genome-wide in the normal population, we relied on a subset of 4,298 samples from the B38 callset for which we had joint variant callsets for both SNVs/indels (GATK) and SVs (lumpy/svtools). Each SNV and indel was annotated with CADD30 and LINSIGHT31 scores as described above. CADD and LINSIGHT scores were converted to percentiles and singleton rates calculated for variants above each score threshold. CADD and LINSIGHT scores were then calibrated to a standard scale by matching singleton rates. Each DEL and DUP was annotated with CADD and LINSIGHT scores, calculated as the mean of the top 10 single-base CADD or LINSIGHT scores, respectively, for the span of the CNV (similar to SVSCORE63). The CNV-level CADD and LINSIGHT scores were then standardized using the above calibration curves. Finally, each variant (SNV, indel, or CNV) was assigned a combined CADD-LINSIGHT score, calculated as the maximum of the 2 distinct scores.
The combined scores provided a means to rank, within each variant class, variants in order of deleteriousness. We calculated the singleton rate for the set of all LOFTEE high confidence protein-truncating SNV and indels in autosomal genes. We then estimated the number of deleterious variants of each type genome-wide by choosing the combined CADD-LINSIGHT score threshold as the minimum value such that the singleton rate for the set of higher-scoring variants was greater than or equal to the singleton-rate for LOFTEE high-confidence PTVs.
Annotation of non-coding elements
We divided the genome into 1 kb non-overlapping windows to investigate the rates of CNV occurrence relative to various classes of coding and non-coding elements, genome-wide. Windows intersecting assembly gaps or high-copy number outlier regions (as described above) and windows with fewer than 50% of bases uniquely mappable as determined using GEM-mappability (build 1.315)64 were excluded from analysis. Bed tracks of genomic annotations for the non-coding dosage sensitivity analysis were created as described below.
The phastcons-20way65 conservation track was downloaded from the UCSC genome browser (rsync://hgdownload.cse.ucsc.edu/goldenPath/hg38/phastCons20way/hg38.phastCons20way.wigFix.gz) and converted to bed format. The mean phastcons score for each 1 kb window was calculated using bedtools map. Quantiles of mean window-level phastcons scores were calculated and used as thresholds for the sensitivity analysis.
The LINSIGHT31 score track was downloaded from CSHL (http://compgen.cshl.edu/~yihuang/tracks/LINSIGHT.bw). The 1kb genomic windows were lifted over to hg19 using crossmap (v0.2.1), annotated with mean per-window LINSIGHT scores using bedtools map and lifted back to GRChb38. Quantiles of mean window-level LINSIGHT scores were calculated and used as thresholds for the sensitivity analysis.
Genehancer52 enhancers were downloaded from GeneCards (https://genecards.weizmann.ac.il/geneloc/index.shtml) and converted to bed format.
Vista51 enhancers were downloaded from LBL (https://enhancer.lbl.gov/cgi-bin/imagedb3.pl?page_size=20000;show=1;search.result=yes;page=1;form=search;search.form=no;action=search;search.sequence=1), restricted to human enhancers, converted to bed format and lifted over to GRChb38 using crossmap.
Encode48 DNAse hypersensitivity sites and transcription factor binding sites were downloaded from UCSC (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegDnaseClustered/wgEncodeRegDnaseClusteredV3.bed.gz, http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegTfbsClustered/wgEncodeRegTfbsClusteredV3.bed.gz) and lifted over to GRCHb38 using crossmap.
Oreganno66 literature-curated enhancers were downloaded from UCSC (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/oreganno.txt.gz) converted to bed format, and lifted over to GRChb38 using crossmap.
Sensitive50, transcription factor bound, ultra-conserved67, and HOT68 regions were downloaded from the funseq269 resources (http://archive.gersteinlab.org/funseq2.1.0_data).
Dragon enhancers were downloaded from DENdb70 (http://www.cbrc.kaust.edu.sa/dendb/src/enhancers.csv.zip), converted to bed format, lifted over to GRChb37, and filtered for score>2.
Chromatin interaction domains derived from Hi-C on hESC and IMR90 cells71 were downloaded from http://compbio.med.harvard.edu/modencode/webpage/hic/, and distances between adjacent topological domains calculated with bedtools. When the physical distance between adjacent topological domains was <400 kb, these were classified as TAD boundaries; otherwise, they were classified as unorganized chromatin. The TAD boundaries and unorganized chromatin data were converted to bed format and lifted over to GRCh38 using crossmap.
Roadmap chromatin state segmentations for 127 epigenomes were downloaded from Roadmap49 (https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/coreMarks/jointModel/final/) and lifted over to GRCh38. Bedtools multiinter was used to determine the number of epigenomes in which each segment was present.
Dosage sensitivity of non-coding elements
To maximize power, DEL and DUP calls from the non-redundant combination of the B37 and B38 callsets (as described above) were used for this analysis. Each window was further characterized by its distance to the nearest exon (the minimum distance between any point in the window and any point in the exon) and the pLI score of the gene corresponding to the nearest exon. The pLI score was set to zero for genes with pLI undefined. In the event that exons of 2 genes were equidistant to the window, the max of the two pLI scores was selected.
For a given SV type (DUP or DEL) and a given functional annotation (e.g., VISTA enhancers), each window was characterized by the presence or absence of one or more SV and the presence or absence of one of more genomic features. We observed a depletion of CNV in windows near exons, and in particular near exons of LoF-intolerant genes (see Fig. 5a). As such, we used a Cochran-Mantel-Haenszel estimate of the odds ratios for each SV type/functional annotation, while stratifying for the proximity to the nearest exon as well as that exon’s LOF-intolerance (pLI). Because adjacent windows are not strictly independent observations – i.e., CNV or features may overlap adjacent windows inducing some spatial correlations – we used a block bootstrap method (resampling was performed on blocks of 10 windows) to estimate robust confidence intervals.
Long-read validation
PacBio long-read sequences from nine 1000 Genomes Project (1KG) samples sequenced to deep coverage (>68-87x) at the McDonnell Genome Institute were used as an orthogonal means of validating SV calls. These PacBio data are available in SRA (see accessions in Supplementary Table 2) and were generated independently from the long-read data used by HGSVC to create the long-read SV callset used for sensitivity analyses described below27. The long-read sequences were aligned to GRCh38 using minimap272 (v2.16-r922; parameters -ax map-pb). Split-read alignments indicating putative SV were converted to BEDPE format56 as described previously23,24,73. Similarly, deletions or insertions longer than 50 bp contained within PacBio reads (as determined based on the cigar strings) were converted to BEDPE format. We used bedtools to judge the overlap between short-read SV calls and the long-read alignments. We judged an SV call to be validated when ≥2 long-reads exhibited split-read mappings in support of the SV call. For a long-read mapping to support an SV call, we required that it must predict a consistent SV type (e.g., deletion) and exhibit substantial physical overlap with the SV call, where overlap can be met by either of the following criteria: (i) the two breakpoint intervals predicted by the SV call and the two breakpoint intervals predicted by the long-read split-read mapping overlap with each other on both sides, as determined by bedtools pairtopair using 100 bp of “slop” (-type -is both -slop 100); or (ii) the SV call and the long-read split-read (or cigar-derived indel variant) exhibit 90% reciprocal overlap with one another (bedtools intersect -r -f 0.9). The above criteria for SV validation based on long-read support were selected based on extensive manual review of SV calls in the context of supporting data including read-depth profiles and long-read mappings from all 9 samples, and are the basis for the validation rates reported in the main text and in Supplementary Table 3. However, we also show the range of validation rates that are obtained when using more lenient or strict measures of physical overlap, and when requiring a varying number of supporting PacBio reads (Extended Data Fig. 5), in both carriers and non-carriers of SVs from various classes. We also note that 3 of the 6 singleton SV calls that are not validated by long-reads appear to be true variants based on manual review of read-level evidence, where it appears that long-reads failed to validate true short-read SV calls due to subtle differences in how coordinates were reported at local repeats. Our FDR estimates may be conservative due to these effects.
To conduct a comparison to HGSVC using the three samples shared between our datasets (NA19240, HG00514, HG00733), all non-reference, autosomal SV calls for each of the three samples were extracted from the CCDG B38 and HGSVC27 Illumina short-read callsets. For HGSVC variants detected solely by read-depth analysis, for which genotype information was not available, a variant was defined to be non-reference if its predicted copy-number differed from the mode for that site across the 9 samples in that callset (which includes the parents of NA19240, HG00514, and HG00733). The short-read calls from our study and HGSVC for the three relevant samples were converted to BEDPE format using svtools vcftobedpe. The 3 single-sample VCFs from the HGSVC PacBio long-read SV callset were converted to BEDPE format in similar fashion. For HGSVC Illumina calls (which had been taken from a callset comprising 3 only trios, rather than a large cohort) variants were classified as rare if seen in only 1 of the 6 trio founders and either absent from or observed at frequency <1% in the 1KG Phase3 SV callset.
Long-read SV truth set construction
In order to evaluate the sensitivity of our callset, we constructed a high-confidence truth set from the comprehensive HGSVC long-read SV callset created using reference-guided de novo assembly27. The assembly-based long-read truth set includes all autosomal SV reported by HGSVC27 that were also validated by split-read alignments from the PacBio data generated independently at our center. Here, an HGSVC call was judged to be validated by long-read data when 2 or more long reads exhibited split-read mappings or cigar-derived SV calls that match the HGSVC call in terms of the predicted SV type and breakpoint intervals, allowing 100 bp of “slop” to account for positional uncertainty (bedtools pairtopair -type -is both -slop 100). To account for the variant classification scheme of the HGSVC callset – which only has two variant categories, INS and DEL – we allowed INS variants to be validated by long-reads suggesting either insertion or tandem duplication variants. Variants were classified as STRs if either >50% of sequence from both reported breakpoint intervals or >50% of sequence contained in the outer span of the variant overlapped a GRCh38 track of simple repeats downloaded from the UCSC Table Browser. The interval spanned by each variant was converted to bed format and lifted over to hg19 using crossmap. A combined CADD/LINSIGHT score was calculated for each variant based on the mean of the top 10 CADD-scoring and the mean of the top 10 LINSIGHT-scoring positions, as described in the section “Genome-wide estimation of deleterious variants” above.
Liftover of the 1000 Genomes Phase3 SV Callset
The 1KG Phase3 SV callset was lifted over from GRCh37 to GRCh38 by first converting to BEDPE format using svtools vcftobedpe. The outer span of each variant was then converted to bed format and lifted over using crossmap61. For SVs that were not lifted over as contiguous intervals, discontiguous regions within 1 kb were merged using bedtools merge, and the largest of the merged variants were selected. The lifted-over bed interval was then converted back to BEDPE by padding each endpoint with 100 bp.
Assessment of sensitivity using the HGSVC long-read truth set
Sensitivity of the CCDG B38 and HGSVC Illumina short-read callsets to detect variants in the HGSVC long-read truth set was determined by converting each single-sample VCF to BEDPE format using svtools vcftobedpe and calculating overlaps using bedtools pairtopair, allowing for 100 bp of “slop”. For DEL calls, a variant was considered to be detected only if both breakpoints overlapped, and the type of the overlapping call was consistent with a deletion (i.e., DEL, MEI, CNV, or BND). For INS calls in the long-read callset, variants were considered detected if either breakpoint overlapped and the overlapping call was consistent with an insertion (i.e., DUP, INS, CNV, MEI, or BND).
Comparison with the 1KG Phase3 SV callset4 necessitated the use of a slightly different sensitivity metric, as 1KG analyzed the parents of HG00733 and NA19249, but not the trio offspring themselves. Since, with rare exception, germline variants present in the child should also be present in one of the two parents, the rate at which HGSVC long-read calls in the truth set were detected in at least one parent in each of the CCDG B38, HGSVC, and 1KG callsets serves as an informative alternate measure of “sensitivity”.
Genotype comparison to 1KG
Genotype comparisons were performed for the 5 parental samples (NA19238, NA19239, HG00513, HG00731, and HG00732) present in both the CCDG B38 and the 1KG Phase3 SV callsets. Each callset was subset (using bcftools) to the set of autosomal SVs with a non-reference call in at least one of the 5 parental samples and converted to BEDPE format. Variants in the 1KG callset detected using read-depth methods only were excluded. Bedtools pairtopair (100 bp slop, overlap at both breakpoints) was used to determine the set of variants called in both the 5-sample CCDG callset and 5-sample 1KG callset, requiring consistent SV type. For each variant site in each sample, genotypes from the 2 callsets were compared. Results were tallied, and concordance rates and kappa statistics (‘irr’ package) were calculated in R.
CEPH pedigree analysis
Analyses of the three-generation CEPH pedigrees were performed on the set of 576 samples contained in the B37 callset that remained after excluding 21 samples that had been deemed low-quality and/or possibly contaminated based on analysis of a SNV/indel callset (A. Quinlan, Pers. Comm.). The remaining samples comprise 409 trios, which were used in the estimation of transmission rates. The counts of all high-quality SVs called heterozygous in one parent, homozygous reference in the other, and non-missing in the offspring were used to estimate transmission rates by frequency class, with Wilson score confidence intervals calculated using R binconf.
Mendelian errors for all high-quality (filter=PASS) SV were calculated using plink (v1.90b3.45), with the output restricted to variant-trio observations in which all 3 genotypes (father, mother, and offspring) were non-missing. For each sample in the third generation (the “F2”; see Supplementary Fig. 6a) of any of the CEPH kindreds, Mendelian errors were counted by frequency class. The Mendelian error rate was calculated as the total number of Mendelian errors divided by the total number of non-reference, non-missing genotypes in F2 generation samples for variants of that frequency class. “De novo” variants were defined as variants private to a single family where both parental genotypes are 0/0 and the offspring genotype either 0/1 or 1/1, and were obtained by parsing the plink output. (Note that these variant counts are used as callset quality metrics and do not necessarily represent true de novo mutations.)
Transmission rates for putative de novo variants were calculated by restricting to all high-quality autosomal variants heterozygous in a second generation (“F1”) sample and homozygous reference in both of his/her parents (“P0” generation) and his/her F1 spouse. Each such variant was classified as transmitted if carried by any F2 offspring, with transmission rates calculated as the number of transmitted variants out of the total. “Missed heterozygous calls” were counted as the set of all family-private variants non-reference in at least two F2 offspring siblings, but homozygous reference in both of the F1 parents. The rate of missed heterozygous calls was calculated by dividing this count by the total count of family private variants carried by at least two F2 offspring siblings.
Extended Data
Extended Data Table 1.
(a) Ancestry, (b) ethnicity, and (c) continental origin of the samples analyzed in this study. For each table, the number of samples in the B37 and B38 callsets are shown separately, including the non-redundant combined set at right. Abbreviations are as follows: AFR, African; AMR, admixed American; EAS, east Asian; FE, Finnish European; NFE, non-Finnish European; PI, Pacific Islander; SAS, South Asian.
| a | Ancestry | Build 37 | Build 38 | Combined |
|---|---|---|---|---|
| AFR | 3683 | 5501 | 6170 | |
| AMR | 698 | 4165 | 4136 | |
| EAS | 65 | 929 | 972 | |
| FE | 2898 | 1207 | 2884 | |
| NFE | 682 | 9588 | 10254 | |
| Not Specified | 105 | 428 | 436 | |
| Other | 123 | 751 | 777 | |
| PI | 110 | 87 | 110 | |
| SAS | 62 | 519 | 558 | |
| b | Ethnicity | Build 37 | Build 38 | Combined |
| Hispanic | 586 | 2829 | 2829 | |
| Non-Hispanic | 3758 | 8022 | 10559 | |
| Not Specified | 4082 | 12324 | 12959 | |
| c | Continent | Build 37 | Build 38 | Combined |
| African | 66 | 24 | 66 | |
| Asian | 32 | 1272 | 1272 | |
| Caribbean | 279 | 1815 | 1815 | |
| East Asian | 43 | 0 | 43 | |
| European | 2985 | 1219 | 2971 | |
| North American | 4641 | 18563 | 19800 | |
| Oceanic | 41 | 18 | 41 | |
| Central Asian/Siberian | 26 | 0 | 26 | |
| South American | 274 | 264 | 274 | |
| South Asian | 39 | 0 | 39 | |
Extended Data Figure 1.
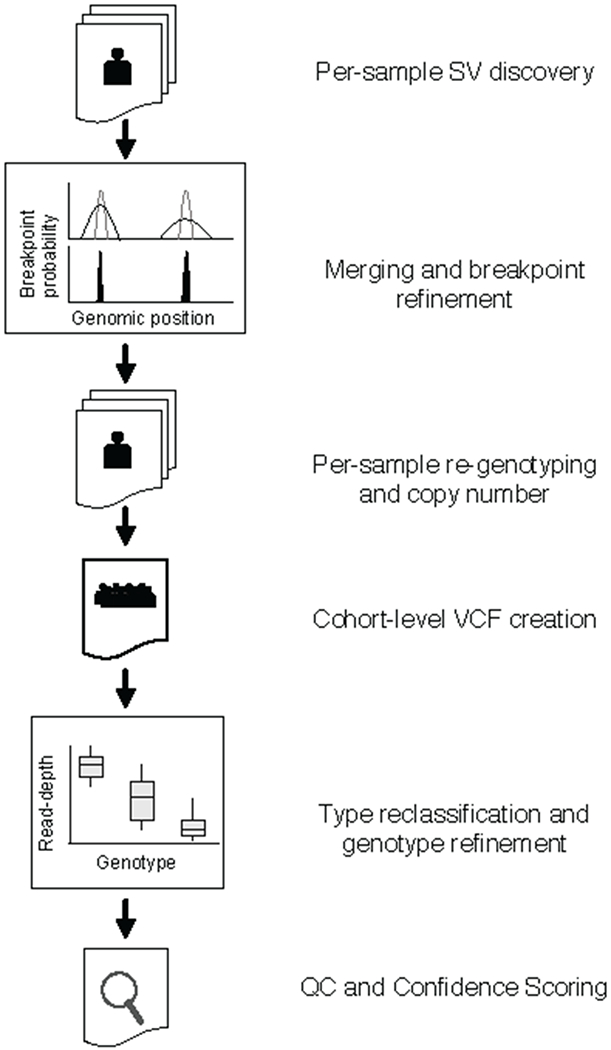
SV mapping pipeline. SV are detected within each sample using LUMPY. Breakpoint probability distributions are used to merge and refine the position of detected SV within a cohort, followed by parallelized re-genotyping, and copy number annotation. Samples are merged into a single cohort-level VCF file, variant types reclassified, and genotypes refined with svtools using the combined breakpoint genotype and read-depth information. Finally, sample-level QC and variant confidence scoring is conducted to produce the final callset.
Extended Data Figure 2.
The B37 callset. (a) Variant counts (y-axis) for each sample (x-axis) in the callset, ordered by cohort, where large (>1 kb) variants are shown in dark shades and smaller variants in light shades. (b) Variant counts per sample, where samples are ordered by self-reported ancestry according to the color scheme at right, using the abbreviations described in Supplementary Table 1. Note that African-ancestry samples show more variant calls, as expected. (c) Table showing the number of variant calls by variant and frequency class, and Mendelian error rate by variant type. (d) Histogram of allele count for each variant class, showing alleles with counts ≤100. (e) Linkage disequilibrium of each variant class as represented by max R2 value to nearby SNVs, for N=1581 samples. Note that these distributions mirror those from our prior SV callset for GTEx3, which was characterized extensively in the context of eQTLs.
Extended Data Figure 3.
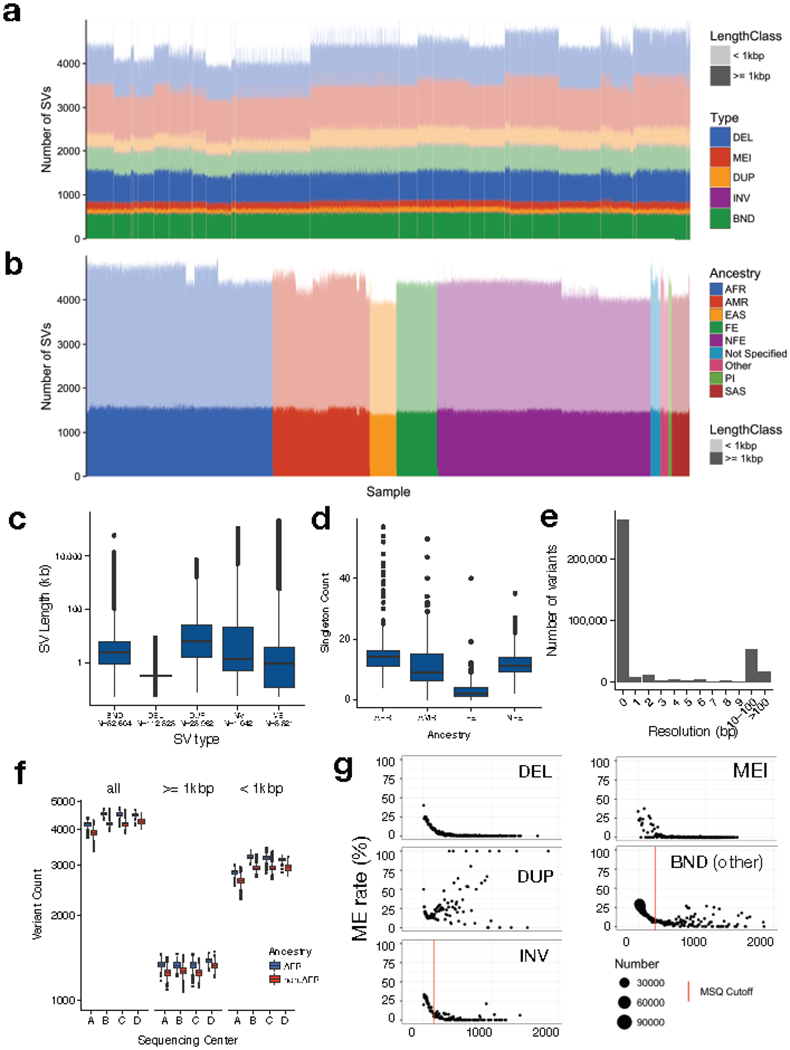
. Note that African-ancestry samples show more variant calls, as expected. Note also that there is some residual variability in variant counts due to differences in data from each sequencing center, but that this is mainly limited to small tandem duplications (see part a), primarily at STRs. (c) SV length distribution by variant class (d) Distribution of the number of singleton SVs detected in samples from different ancestry groups according to the abbreviations in Supplementary Table 1. Only groups with ≥1,000 samples in the B38 callset are shown, and each group was subsampled down to 1,000 individuals prior to allele frequency re-calculation. (e) Histogram showing the resolution of SV breakpoint calls, as defined by the length of the 95% confidence interval of the breakpoint-containing region defined by LUMPY, after cross-sample merging and refinement using svtools. Data are from N=360,614 breakpoints, 2 per variant. (f) Distribution of the number of SVs detected per sample in WGS data from each sequencing center (x-axis) for African and non-African samples, showing all variants (left), and those larger (middle) and smaller (right) than 1 kb in size. Per-center counts are as follows: Center A (1527 AFR, 2080 Non-AFR), Center B (408 AFR, 2745 Non-AFR), Center C (2953 AFR, 2226 Non-AFR), Center D (150 AFR, 2534 Non-AFR). (g) Plots of Mendelian error (ME) rate (y-axis) by mean sample quality (MSQ) for each variant class, where dot size is determined by point density (see right) and the threshold used to determine high and low confidence SVs is shown by the vertical lines. All boxplots indicate the medians and first and third quartiles; whiskers extend 1.5 times the interquartile distance.
Extended Data Figure 4.
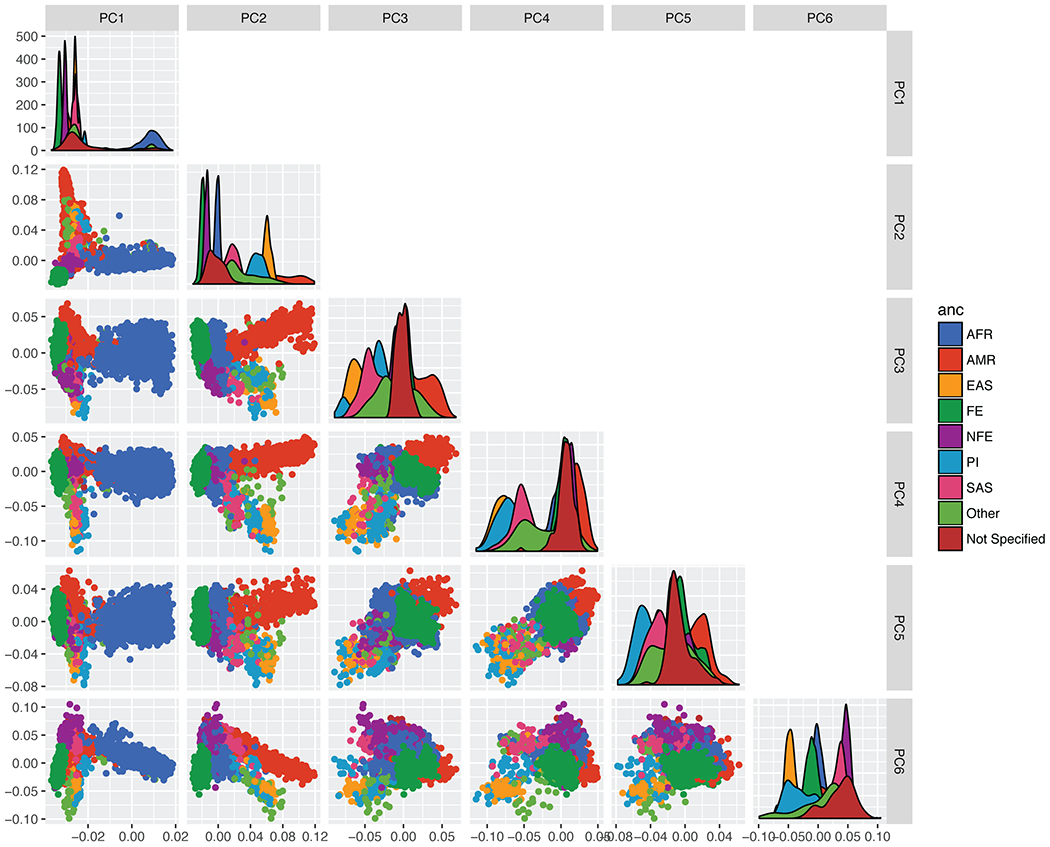
Principal components analysis for the B37 callset. PCA were calculated using an LD-pruned subset of high-confidence DEL and MEI variants, with MAF>1%. Ancestry is based on self-report, using the color scheme at right, using the ancestry abbreviations described in Extended Data Table 1.
Extended Data Figure 5.
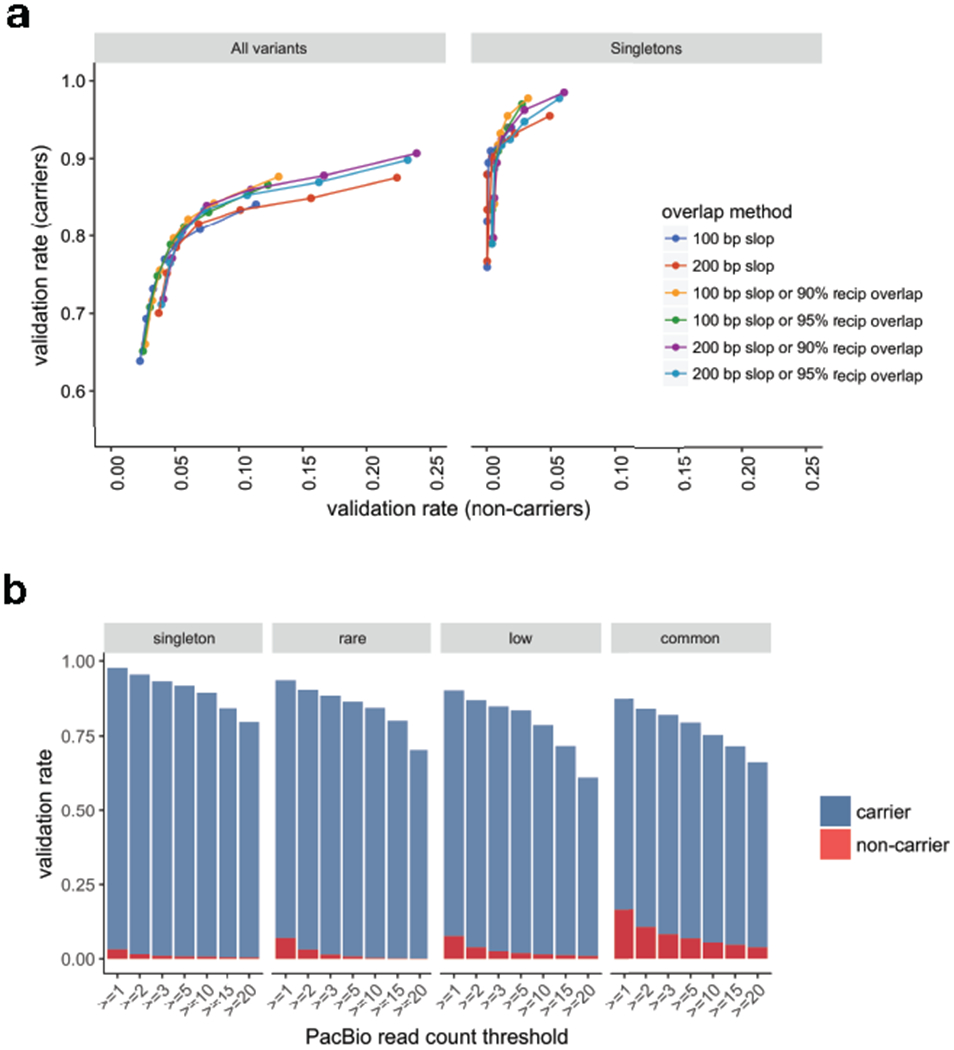
is based on the “100 bp slop or 90% reciprocal overlap” method, requiring ≥2 PacBio reads. (b) Validation rates by frequency class for variant carriers and non-carriers with increasing PacBio supporting read thresholds are shown using the same overlap method as in Supplementary Table 3. Variant counts per frequency class are as follows: singleton (N=133), rare (N=734), low frequency (N=1,361), and common (N=7,677).
Extended Data Figure 6.
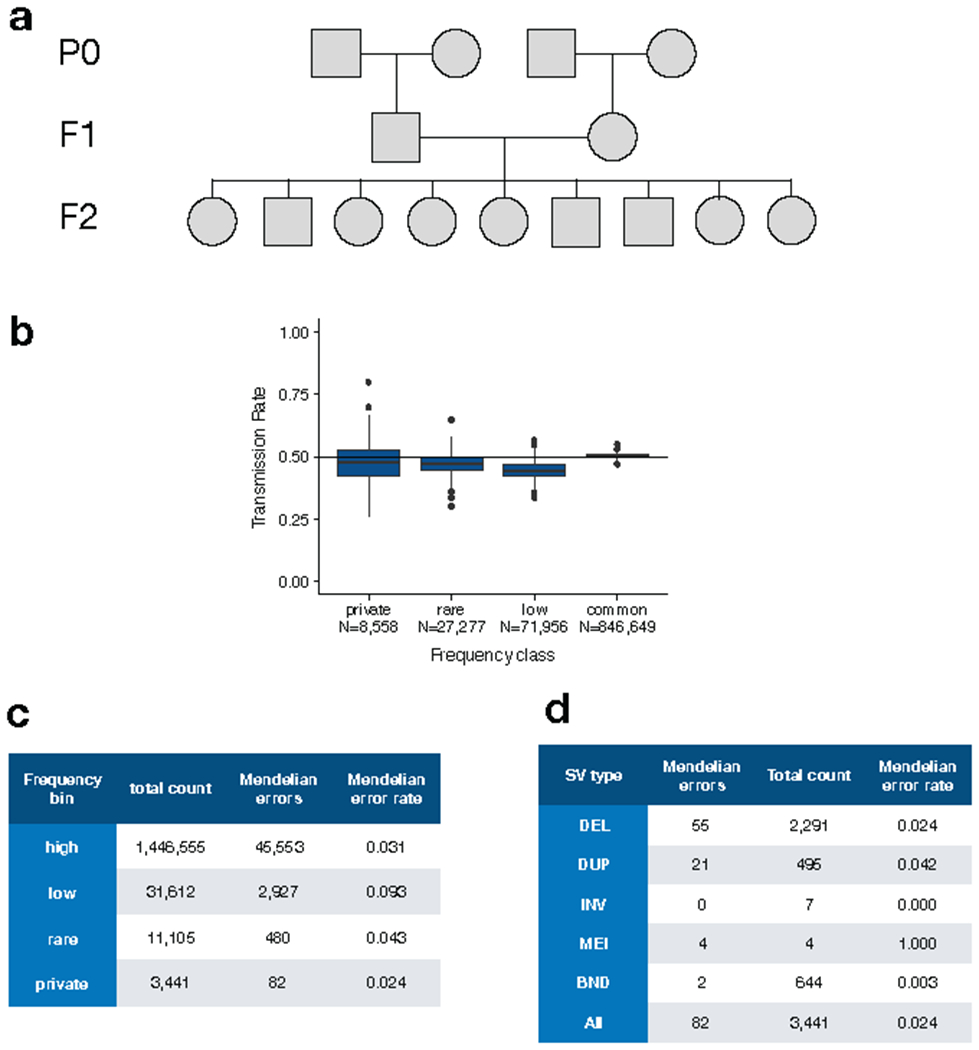
Mendelian inheritance analysis in a set of 3-generation CEPH pedigrees comprising 409 parent-offspring trios. (a) Example structure of a single CEPH pedigree indicating nomenclature of the parental (P0), first (F1) and second generation (F2). (b) Transmission rate of SVs from different allele frequency classes including SVs that are private to a single family (private), rare (<1%), low-frequency (“low”; 1-5%) and common (>5%). (c) Table showing the number and rate of Mendelian errors by allele frequency class. (d) Table showing the number and rate of Mendelian errors for SVs private to a single family, for each SV type.
Extended Data Figure 7.
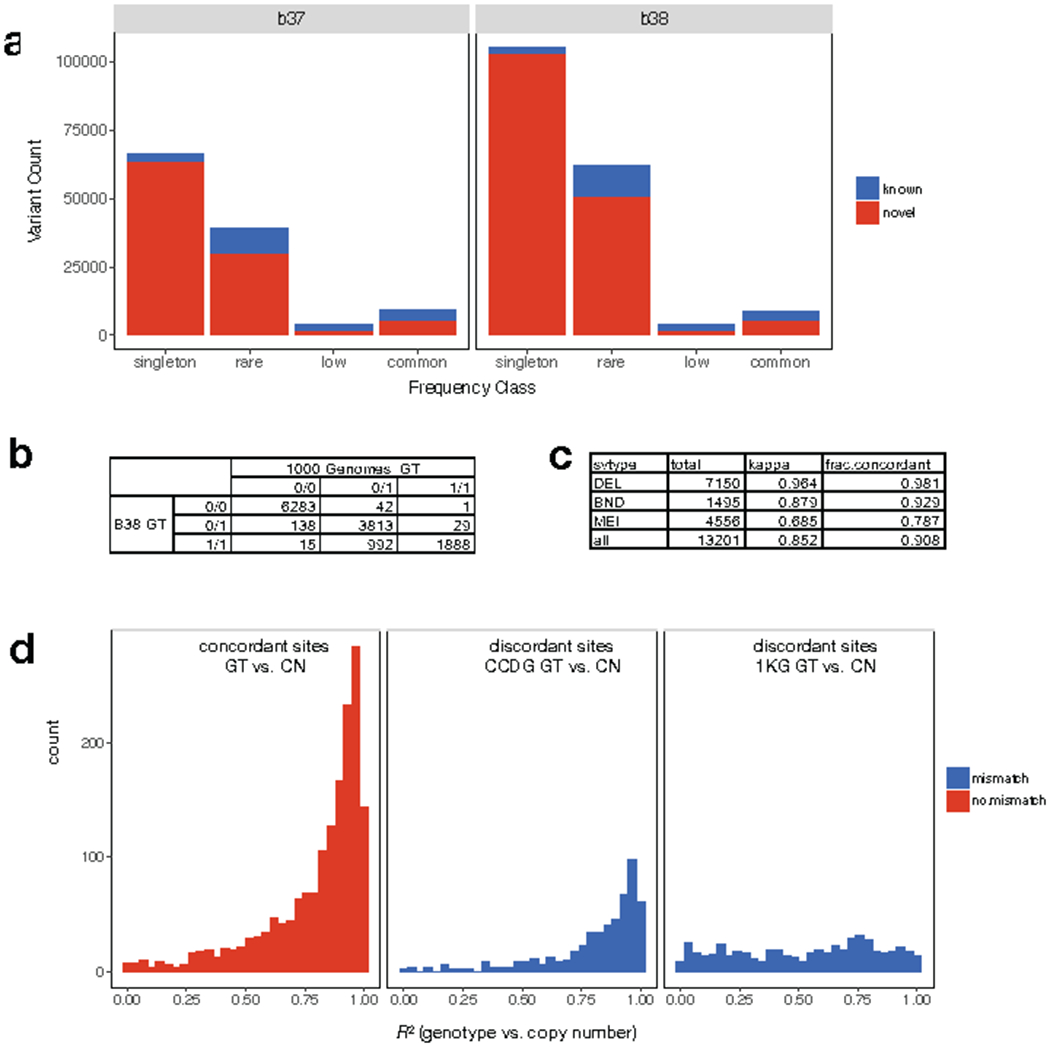
Comparison of SV calls and genotypes to the 1000 Genomes (1KG) Phase3 callset4. (a) number of known and novel SVs in the B37 (left) and B38 (right) callsets, shown by frequency class. (b) Table showing the genotypes reported in our B38 (rows) callset versus 1KG (columns) at SVs identified by both studies among the five samples included in both callsets. (c) Table showing genotype concordance by SV type including the fraction of concordant calls and Cohen’s Kappa coefficient. (d) Distribution of correlation (R2) between genotype (GT) information determined by breakpoint-spanning reads and copy number (CN) estimates determined by read-depth analysis for the SVs shown in parts (b) and (c), when genotype information between the B38 and 1KG callset are concordant (left) or discordant (middle, right). At sites with discordant genotypes, correlation with copy number information is typically higher for genotypes from the B38 callset (middle) than the 1KG callset (right).
Extended Data Figure 8.
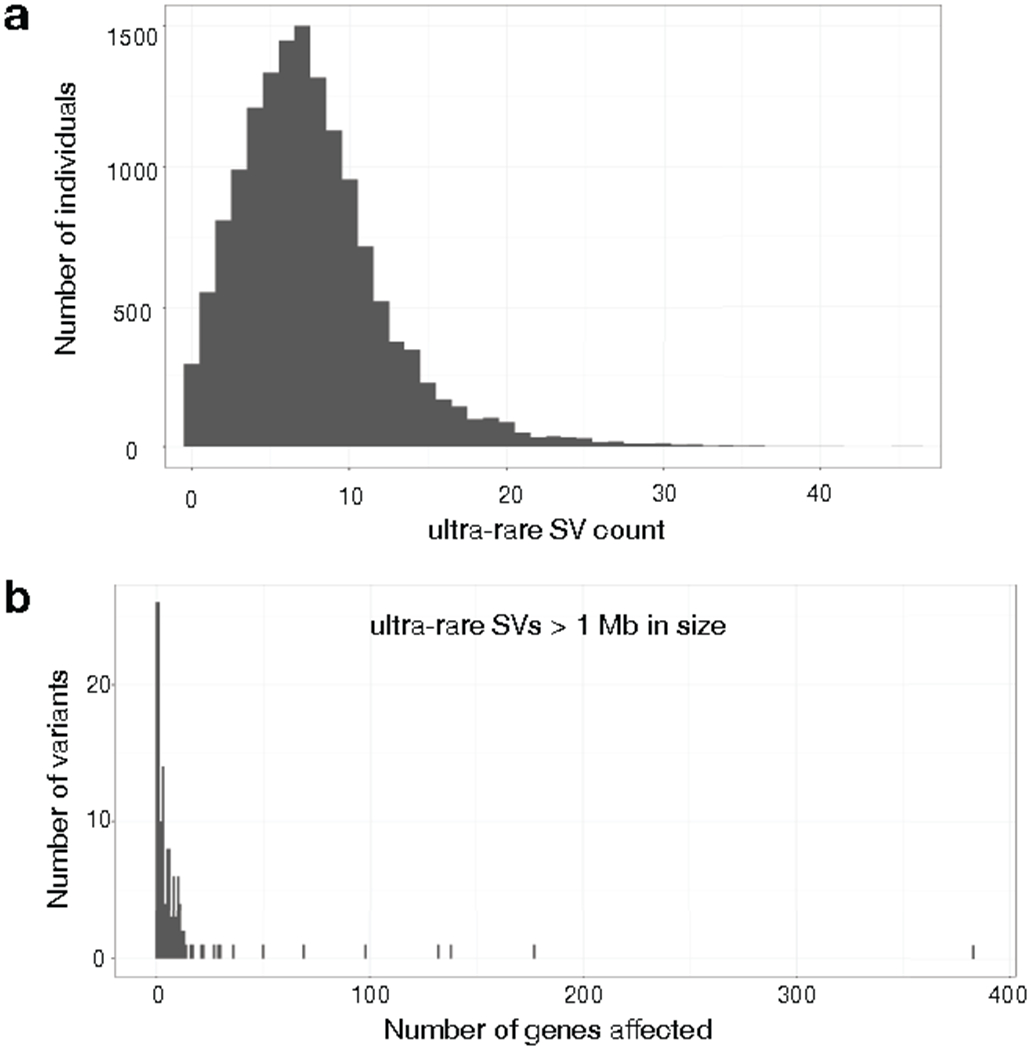
Ultra-rare SVs in the B38 callset (N=14,623). (a) Histogram showing the number of ultra-rare SVs per individual, where ultra-rare is defined as “singleton” variants private to single individual or nuclear family. (b) Histogram showing the number of genes affected by ultra-rare SVs larger than 1 Mb in size.
Extended Data Figure 9.
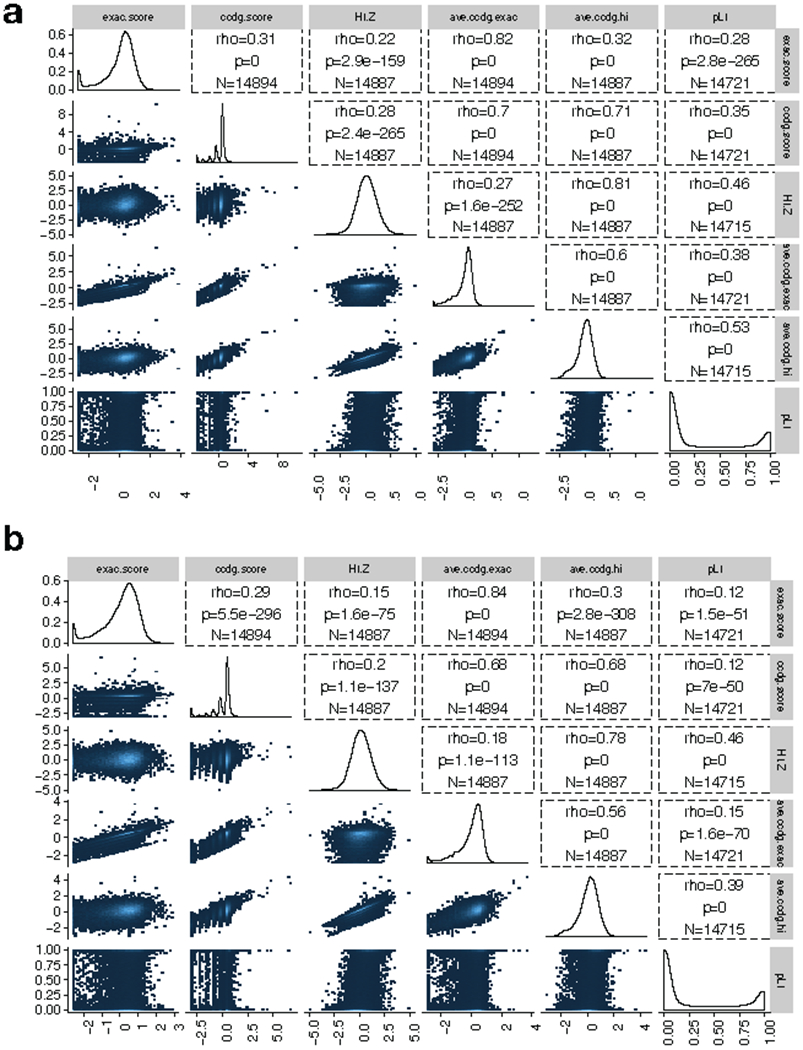
Correlations between dosage sensitivity scores for CNV in the combined callset (N=17,795). (a) Results for deletion variants. “ExAC score” is the published ExAC DEL intolerance score46. “CCDG score” is similarly calculated from our data, using CCDG deletions. “pLI” is the published loss-of-function intolerance score from ExAC28. “HI.Z” is the negative of the inverse-normal transformed haploinsufficiency score from DECIPHER47. “Ave.ccdg.exac” is the arithmetic mean of the CCDG and ExAC DEL intolerance scores. “Ave.ccdg.hi” is the arithmetic mean of the CCDG and HI-Z scores. Correlations shown are Spearman rank correlations (rho), p-values are from the 2-sided spearman rank correlation test, N represents the number of genes included in the test. (b) Results for duplication variants, using the same naming conventions as in part (a).
Supplementary Material
ACKNOWLEDGEMENTS
We thank program staff at the National Human Genome Research Institute (NHGRI) for supporting this effort. This study was funded by NHGRI CCDG awards to Washington University in St. Louis (WU) (UM1 HG008853), Broad Institute of MIT and Harvard (UM1 HG008895), Baylor College of Medicine (UM1 HG008898), and New York Genome Center (UM1 HG008901); an NHGRI Genome Sequencing Program (GSP) Coordinating Center grant to Rutgers (U24 HG008956); and a Burroughs Wellcome Fund Career Award to IMH. Additional data production at WU was funded by a separate NHGRI award (5U54HG003079). We thank Shamil Sunyaev for helpful comments on the manuscript. We gratefully acknowledge all individuals involved in the recruitment of samples analyzed for this study. Thanks to Terri Teshiba for coordinating samples for FINRISK and EUFAM sequencing. Data production for EUFAM was funded by 4R01HL113315-05. The METSIM study was supported by grants to Markku Laakso from the Academy of Finland (No. 321428), the Sigrid Juselius Foundation, the Finnish Foundation for Cardiovascular Research, Kuopio University Hospital, and the Centre of Excellence of Cardiovascular and Metabolic Diseases supported by the Academy of Finland. Data collection for the CEPH Pedigrees was funded by the George S. and Dolores Doré Eccles Foundation and NIH grants GM118335 and GM059290. Study recruitment at WU was funded by the DDRCC (NIDDK P30 DK052574) and the Helmsley Charitable Trust. Study recruitment at Cedars-Sinai was supported by the F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, NIH/NIDDK grants P01 DK046763 and U01 DK062413, and the Helmsley Charitable Trust. Study recruitment at Intermountain Medical Center was funded by the Dell Loy Hansen Heart Foundation. The Late Onset Alzheimer’s Disease Study (LOAD) study was funded by grants to T. Foroud (U24AG021886, U24AG056270, U24AG026395, R01AG041797). The Atherosclerosis Risk in Communities (ARIC) study was funded by NHLBI (HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I HHSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions. The Population Architecture Using Genomics and Epidemiology (PAGE) program is funded by NHGRI with co-funding from NIMHD (U01HG007416, U01HG007417, U01HG007397, U01HG007376, and U01HG007419). Samples from the BioMe Biobank were provided by The Charles Bronfman Institute for Personalized Medicine at the Icahn School of Medicine at Mount Sinai. The Hispanic Community Health Study/Study of Latinos was carried out as a collaborative study supported by NHLBI (N01-HC65233, N01-HC65234, N01-HC65235, N01-HC65236, N01-HC65237), with contributions from NIMHD, NIDCD, NIDCR, NIDDK, NINDS and NIH ODS. The MEC study is funded through NCI (R37CA54281,R01 CA63, P01CA33619, U01CA136792, and U01CA98758). For the Stanford Global Reference Panel, individuals from Puno, Peru were provided by Drs. Julie Baker and Carlos Bustamante, with funding from the Burroughs Welcome Fund; individuals from Rapa Nui (Easter Island) were provided by Drs. Karla Sandoval Mendoza and Andres Moreno Estrada with funding from the Charles Rosenkranz Prize for Health Care Research in Developing Countries. The WHI program is funded by NHLBI (HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C). The GALA II study and Esteban G. Burchard are supported by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, the Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, NHLBI (R01HL117004, R01HL128439, R01HL135156, X01HL134589), NIEHS (R01ES015794, R21ES24844), NIMHD (P60MD006902, R01MD010443, RL5GM118984) and the Tobacco-Related Disease Research Program (24RT-0025). The authors wish to acknowledge the following GALA II co-investigators for subject recruitment, sample processing and quality control: Celeste Eng, Sandra Salazar, Scott Huntsman, MSc, Donglei Hu, PhD, Angel C.Y. Mak, PhD, Lisa Caine, Shannon Thyne, MD, Harold J. Farber, MD, MSPH, Pedro C. Avila, MD, Denise Serebrisky, MD, William Rodriguez-Cintron, MD, Jose R. Rodriguez-Santana, MD, Rajesh Kumar, MD, Luisa N. Borrell, DDS, PhD, Emerita Brigino-Buenaventura, MD, Adam Davis, MA, MPH, Michael A. LeNoir, MD, Kelley Meade, MD, Saunak Sen, PhD and Fred Lurmann, MS. The authors also wish to thank the staff and participants who contributed to the GALA II study.
Consortia
NHGRI Centers for Common Disease Genomics
Goncalo R. Abecasis15, Elizabeth Appelbaum1, Julie Baker16, Eric Banks5, Raphael A. Bernier17, Toby Bloom9, Michael Boehnke15, Eric Boerwinkle8,18, Erwin P. Bottinger19, Steven R. Brant20, Esteban G. Burchard21, Carlos D. Bustamante16, Lei Chen1, Judy H. Cho19,22,23, Rajiv Chowdhury24, Ryan Christ1, Lisa Cook1, Matthew Cordes1, Laura Courtney1, Michael J. Cutler25, Mark J. Daly5,26,27, Scott M. Damrauer28, Robert B. Darnell9,29,30, Tracie Deluca1, Huyen Dinh8, Harsha Doddapaneni8, Evan E. Eichler31,32, Patrick T. Ellinor5,33, Andres M. Estrada34, Yossi Farjoun5, Adam Felsenfeld35, Tatiana Foroud36, Nelson B. Freimer37, Catrina Fronick1, Lucinda Fulton1, Robert Fulton1, Stacy Gabriel5, Liron Ganel1, Shailu Gargeya9, Goren Germer9, Daniel H. Geschwind38,39,40, Richard A. Gibbs8, David B. Goldstein41,42, Megan L. Grove8, Namrata Gupta5, Christopher A. Haiman43, Yi Han8, Daniel Howrigan5,27, Jianhong Hu8, Carolyn Hutter35, Ivan Iossifov44, Bo Ji1, Lynn B. Jorde45, Goo Jun18, John Kane46, Chul Joo Kang1, Hyun Min Kang15, Sek Kathiresan5,33,47, Eimear E. Kenny19,22,48,49, Lily Khaira9, Ziad Khan8, Amit Khera5,33,47, Charles Kooperberg50, Olga Krasheninina8, William E. Kraus51, Subra Kugathasan52, Markku Laakso53, Tuuli Lappalainen9,54, Adam E. Locke1,14, Ruth J.F. Loos19, Amy Ly1, Robert Maier5,27, Tom Maniatis9,55, Loic Le Marchand56, Gregory M. Marcus57, Richard P. Mayeux58, Dermot P.B. McGovern59, Karla S. Mendoza34, Vipin Menon8, Ginger A. Metcalf8, Zeineen Momin8, Guiseppe Narzisi9, Joanne Nelson1, Caitlin Nessner8, Rodney D. Newberry14, Kari E. North60, Aarno Palotie5,26,27, Ulrike Peters50, Jennifer Ponce1, Clive Pullinger46, Aaron Quinlan45, Daniel J. Rader61, Stephen S. Rich62, Samuli Ripatti5,26,27, Dan M. Roden63, Veikko Salomaa64, Jireh Santibanez8, Svati H. Shah51, M. Benjamin Shoemaker63, Heidi Sofia35, Taylorlyn Stephan35, Christine Stevens5, Stephan R. Targan59, Marja-Riitta Taskinen65, Kathleen Tibbetts5, Charlotte Tolonen5, Tychele Turner31, Paul De Vries18, Jason Waligorski1, Kimberly Walker8, Vivian Ota Wang35, Michael Wigler9,44, Richard K. Wilson1,66, Lara Winterkorn9, Genevieve Wojcik16, Jinchuan Xing11, Erica Young1,14, Bing Yu18, Yeting Zhang11
15Department of Biostatistics and Center for Statistical Genetics, University of Michigan, School of Public Health, Ann Arbor, MI, USA
16Department of Genetics, Stanford University, Stanford, CA, USA
17Department of Psychiatry & Behavioral Sciences, University of Washington, Seattle, WA, USA
18Human Genetics Center and Department of Epidemiology, University of Texas Health Science Center, Houston, TX, USA
19The Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
20Department of Medicine, Rutgers Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, New Brunswick, NJ, USA
21Department of Bioengineering, University of California, San Francisco, CA, USA
22Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mt. Sinai, New York, NY, USA
23Department of Medicine, Icahn School of Medicine at Mt. Sinai, New York, NY, USA
24MRC/BHF Cardiovascular Epidemiology Unit, Department of Public Health and Primary Care, University of Cambridge, Cambridge, UK
25Intermountain Heart Institute, Intermountain Medical Center, Murray, UT, USA
26Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Helsinki, Finland
27Analytical and Translational Genetics Unit, Psychiatric & Neurodevelopmental Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
28Department of Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
29Laboratory of Molecular Neuro-Oncology, The Rockefeller University, New York, NY, USA
30Howard Hughes Medical Institute, The Rockefeller University, New York, NY, USA
31Department of Genome Science, University of Washington, Seattle, WA, USA
32Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA
33Division of Cardiology, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
34National Laboratory of Genomics for Biodiversity (LANGEBIO), CINVESTAV, Irapuato, Guanajuato, Mexico
35National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA
36Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA
37Center for Neurobehavioral Genetics, Jane and Terry Semel Institute for Neuroscience and Human Behavior, University of California Los Angeles, Los Angeles, CA, USA
38Program in Neurogenetics, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA
39Department of Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA
40Institute of Precision Health, University of California, Los Angeles, Los Angeles, CA, USA
41Institute for Genomic Medicine, Columbia University Medical Center, New York, NY, USA
42Department of Genetics and Development, Columbia University Medical Center, New York, NY, USA
43Department of Preventative Medicine, University of Southern California, Los Angeles, CA, USA
44Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA
45Department of Human Genetics, University of Utah, Salt Lake City, UT, USA
46Cardiovascular Research Institute, University of California, San Francisco CA, USA
47Center for Genomic Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
48The Icahn Institute of Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
49Center for Statistical Genetics, Icahn School of Medicine at Mt Sinai, New York, NY, USA
50Fred Hutchinson Cancer Research Center, Seattle, WA, USA
51Department of Medicine, Duke University, Durham, NC, USA
52Department of Pediatrics, Emory University School of Medicine, Atlanta, GA, USA
53Institute of Clinical Medicine, Internal Medicine, University of Eastern Finland, Kuopio, Finland
54Department of Systems Biology, Columbia University, New York, NY, USA
55Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY, USA
56Cancer Center, University of Hawaii, Honolulu, HI, USA
57Department of Medicine, University of California, San Francisco CA, USA
58Department of Neurology, Columbia University, New York, NY, USA
59F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA
60Department of Epidemiology, University of North Carolina, Chapel Hill, NC, USA
61Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
62Center for Public Health Genomics, University of Virginia School of Medicine, Charlottesville, VA, USA
63Department of Medicine, Vanderbilt University, Nashville, TN, USA
64National Institute for Health and Welfare, Helsinki, Finland
65Research Programs Unit, Diabetes & Obesity, University of Helsinki, and Heart and Lung Centre, Helsinki University Hospital, Helsinki, Finland
66current address: Institute for Genomic Medicine, Nationwide Children’s Hospital, Columbus, OH, USA
Footnotes
Competing Interests
The authors have no competing interests.
Data Availability
The sequencing data can be accessed through dbGaP (https://www.ncbi.nlm.nih.gov/gap) under accession numbers provided in Supplementary Table 7. PacBio long read data used for SV validation can be accessed through SRA, under accession numbers provided in Supplementary Table 2. The set of high-confidence HGSVC long-read derived SV calls, validated by our independent PacBio data and used as a truth set can be found in Supplementary File 3. Supplementary Files 1–4 can be found here: https://github.com/hall-lab/sv_paper_042020.
Code Availability
Custom code used in the long-read validation can be found here: https://github.com/abelhj/long-read-validation/tree/master
References
- 1.Weischenfeldt J, Symmons O, Spitz F & Korbel JO Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet 14, 125–138, doi: 10.1038/nrg3373 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Stranger BE et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853, doi:315/5813/848 [pii] 10.1126/science.1136678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang C et al. The impact of structural variation on human gene expression. Nature genetics 49, 692–699, doi: 10.1038/ng.3834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81, doi: 10.1038/nature15394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449, doi:1138659 [pii] 10.1126/science.1138659 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss LA et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 358, 667–675, doi:NEJMoa075974 [pii] 10.1056/NEJMoa075974 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Turner TN et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722 e712, doi: 10.1016/j.cell.2017.08.047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nature genetics 50, 727–736, doi: 10.1038/s41588-018-0107-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandler WM et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331, doi: 10.1126/science.aan2261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stone JL et al. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh T et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543, doi: 10.1126/science.1155174 (2008). [DOI] [PubMed] [Google Scholar]
- 12.McCarthy SE et al. Microduplications of 16p11.2 are associated with schizophrenia. Nature genetics 41, 1223–1227, doi: 10.1038/ng.474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall CR et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature genetics 49, 27–35, doi: 10.1038/ng.3725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellcome Trust Case Control, C. et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464, 713–720, doi: 10.1038/nature08979 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myocardial Infarction Genetics, C. et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nature genetics 41, 334–341, doi: 10.1038/ng.327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacDonald JR, Ziman R, Yuen RK, Feuk L & Scherer SW The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic acids research 42, D986–992, doi: 10.1093/nar/gkt958 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bragin E et al. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic acids research 42, D993–D1000, doi: 10.1093/nar/gkt937 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lappalainen I et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic acids research 41, D936–941, doi: 10.1093/nar/gks1213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hehir-Kwa JY et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat Commun 7, 12989, doi: 10.1038/ncomms12989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maretty L et al. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548, 87–91, doi: 10.1038/nature23264 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Sudmant PH et al. Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761, doi: 10.1126/science.aab3761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson DE et al. svtools: population-scale analysis of structural variation. Bioinformatics, doi: 10.1093/bioinformatics/btz492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84, doi: 10.1186/gb-2014-15-6-r84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang C et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 12, 966–968, doi: 10.1038/nmeth.3505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Regier AA et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun 9, 4038, doi: 10.1038/s41467-018-06159-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang C et al. The impact of structural variation on human gene expression. Nature genetics 49, 692–699, doi: 10.1038/ng.3834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10, 1784, doi: 10.1038/s41467-018-08148-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, doi: 10.1038/nature19057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74, doi: 10.1038/nature15393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics 46, 310–315, doi: 10.1038/ng.2892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang YF, Gulko B & Siepel A Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nature genetics 49, 618–624, doi: 10.1038/ng.3810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaren W et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070, doi: 10.1093/bioinformatics/btq330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danecek P et al. The Variant Call Format and VCFtools. Bioinformatics, doi: 10.1093/bioinformatics/btr330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewing AD et al. Retrotransposition of gene transcripts leads to structural variation in mammalian genomes. Genome Biol 14, R22, doi: 10.1186/gb-2013-14-3-r22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrider DR et al. Gene copy-number polymorphism caused by retrotransposition in humans. PLoS Genet 9, e1003242, doi: 10.1371/journal.pgen.1003242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abyzov A et al. Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division. Genome Res 23, 2042–2052, doi: 10.1101/gr.154625.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper GM et al. A copy number variation morbidity map of developmental delay. Nature genetics 43, 838–846, doi: 10.1038/ng.909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hook EB & Hamerton JL in Population Cytogenetics: Studies in Humans (eds Hook EB & Porter LH) 63–79 (Academic Press, 1977). [Google Scholar]
- 39.Forabosco A, Percesepe A & Santucci S Incidence of non-age-dependent chromosomal abnormalities: a population-based study on 88965 amniocenteses. Eur J Hum Genet 17, 897–903, doi: 10.1038/ejhg.2008.265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malhotra A et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Research 23, 762–776, doi: 10.1101/gr.143677.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad DF et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nature genetics 42, 385–391, doi:ng.564 [pii] 10.1038/ng.564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinlan AR et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Research 20, 623–635, doi: 10.1101/gr.102970.109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills RE et al. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65, doi:nature09708 [pii] 10.1038/nature09708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kidd JM et al. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143, 837–847, doi: 10.1016/j.cell.2010.10.027 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlan AR & Hall IM Characterizing complex structural variation in germline and somatic genomes. Trends in genetics : TIG 28, 43–53, doi: 10.1016/j.tig.2011.10.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruderfer DM et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nature genetics 48, 1107–1111, doi: 10.1038/ng.3638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang N, Lee I, Marcotte EM & Hurles ME Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6, e1001154, doi: 10.1371/journal.pgen.1001154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi: 10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khurana E et al. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 1235587, doi: 10.1126/science.1235587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visel A, Minovitsky S, Dubchak I & Pennacchio LA VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic acids research 35, D88–92, doi: 10.1093/nar/gkl822 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fishilevich S et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, doi: 10.1093/database/bax028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lesurf R et al. ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic acids research 44, D126–132, doi: 10.1093/nar/gkv1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abyzov A, Urban AE, Snyder M & Gerstein M CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21, 974–984, doi: 10.1101/gr.114876.110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873, doi: 10.1093/bioinformatics/btq559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez JM et al. APPRIS: annotation of principal and alternative splice isoforms. Nucleic acids research 41, D110–117, doi: 10.1093/nar/gks1058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498, doi: 10.1038/ng.806 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan A, Abecasis GR & Kang HM Unified Representation of Genetic Variants. Bioinformatics, doi: 10.1093/bioinformatics/btv112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol 17, 122, doi: 10.1186/s13059-016-0974-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao H et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007, doi: 10.1093/bioinformatics/btt730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedman J, Hastie T & Tibshirani R Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 63.Ganel L, Abel HJ, FinMetSeq C & Hall IM SVScore: an impact prediction tool for structural variation. Bioinformatics 33, 1083–1085, doi: 10.1093/bioinformatics/btw789 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derrien T et al. Fast computation and applications of genome mappability. PLoS One 7, e30377, doi: 10.1371/journal.pone.0030377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050, doi: 10.1101/gr.3715005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffith OL et al. ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic acids research 36, D107–113, doi: 10.1093/nar/gkm967 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bejerano G et al. Ultraconserved elements in the human genome. Science 304, 1321–1325, doi: 10.1126/science.1098119 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Yip KY et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol 13, R48, doi: 10.1186/gb-2012-13-9-r48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu Y et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15, 480, doi: 10.1186/s13059-014-0480-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ashoor H, Kleftogiannis D, Radovanovic A & Bajic VB DENdb: database of integrated human enhancers. Database (Oxford) 2015, doi: 10.1093/database/bav085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380, doi: 10.1038/nature11082 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, doi: 10.1093/bioinformatics/bty191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faust GG & Hall IM YAHA: fast and flexible long-read alignment with optimal breakpoint detection. Bioinformatics 28, 2417–2424, doi: 10.1093/bioinformatics/bts456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.