

Abstract
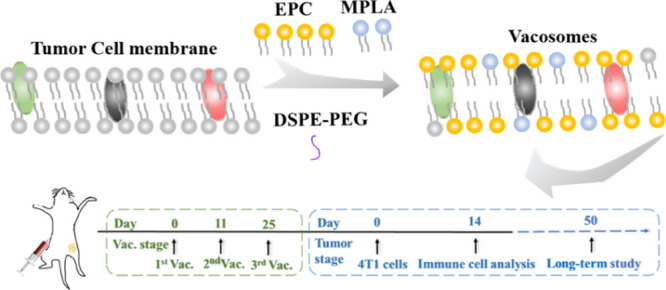
Recently, there has been an increasing interest for utilizing the host immune system to fight against cancer. Moreover, cancer vaccines, which can stimulate the host immune system to respond to cancer in the long term, are being investigated as a promising approach to induce tumor-specific immunity. In this work, we prepared an effective cancer vaccine (denoted as “vacosome”) by reconstructing the cancer cell membrane, monophosphoryl lipid A as a toll-like receptor 4 agonist, and egg phosphatidylcholine. The vacosome triggered and enhanced bone marrow dendritic cell maturation as well as stimulated the antitumor response against breast cancer 4T1 cells in vitro. Furthermore, an immune memory was established in BALB/c mice after three-time preimmunization with the vacosome. After that, the immunized mice showed inhibited tumor growth and prolonged survival period (longer than 50 days). Overall, our results demonstrate that the vacosome can be a potential candidate for clinical translation as a cancer vaccine.
Keywords: cancer vaccines, cancer immunotherapy, liposomes, cancer cell membrane
1. Introduction
Vaccines have been considered as one of the most effective approaches in controlling infectious diseases. At the same time, the scientific community also proposes to develop cancer vaccines to prevent cancer, which is expected to be similar to the conventional vaccine in the prevention of infectious diseases.1−4 The basic mechanism behind cancer vaccination relies on the activation of antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, together with processing of the cancer-related antigens provided by the vaccines.5,6 Then, these activated APCs will direct the differentiation of T cells by presenting the processed antigens. Among the various subpopulations derived from T cells, the major responsibility of CD8+ cytotoxic T lymphocytes (CTLs) is to attack tumor cells, while memory T cells are generated for the long-term protection.7,8 Because of the fundamental position of the activated APCs, it is important to consider promoting the activation of APCs and delivering the cancer-related antigens to APCs, when preventive cancer vaccines are designed.
Monophosphoryl lipid A (MPLA), a toll-like receptor 4 (TLR4) agonist, is derived from the cell wall of nonpathogenic Salmonella.9,10 MPLA has been employed in human trials in vaccine for malaria, HIV-1, and meningococcal type B disease because it can bind and activate membrane-associated TLR4 during uptake by APCs, thereby enhancing cell-mediated immunity to a variety of antigens.11,12 However, there are still some challenges in the development of MPLA as a preventive cancer vaccine, such as its hydrophobicity. As for other issues, the administration of only MPLA is not enough to promote the priming of a cancer-specific adaptive immune response because the APCs lack the antigen. Although the administration of MPLA would result into a generic activation of the immune system, which has been systematically investigated by researchers, there is still low potential against the tumor without the specificity given by the antigen. At present, different strategies have been proposed to utilize the immune activation of MPLA, for example, combination with various drug-delivery systems.10,13,14 Therefore, some formulation improvements are needed to develop MPLA-related preventive cancer vaccines.
Several tumor-specific antigens, such as tumor-specific peptides,15,16 proteins,17 mRNA,18 or DNA,19 have been utilized to develop cancer vaccines to trigger a specific antitumor immune response. Although they are specific enough, the synthesis and isolation processes for such antigens can be time-consuming and expensive. Cancer cell membranes contain plenty of tumor-specific antigens. Moreover, these antigens can be obtained directly from the isolated cell membranes retaining their bioactivity.20−22 In addition, the easy cell membrane isolation process makes this approach time effective and inexpensive.4 However, the cancer cell membrane cannot work as a preventive cancer vaccine by itself because of the limited ability in boosting immune response.23
Recently, nanomaterials have been proved to codeliver antigens and adjuvants in the same carriers and induce robust immune responses.24−28 Liposomes, a Food and Drug Administration (FDA)-approved nanomaterial, present a phospholipid bilayer structure, which is similar to the structure of the cell membrane.10 In terms of composition, liposomes, MPLA, and cell membrane mainly consist of phospholipid, which makes liposomes as an ideal nanomaterial to develop the potential ability of MPLA and cancer cell membranes as preventive cancer vaccines. In this work, we prepared a biohybrid liposome (denoted here as “vacosome,” short for “vaccine liposomes”) by reconstituting MPLA, 4T1 cancer cell membranes, and common lipid [1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)-2000], DSPE-PEG-2000, egg phosphatidylcholine, EPC, and cholesterol] (Scheme 1). We first investigated the efficacy of the vacosome in vitro by inducing BALB/c mice bone marrow dendritic cell (BMDC) maturation and by stimulating splenocytes to eliminate 4T1 cells. In vivo experiments were conducted in vacosome-immunized BALB/c mice, and the production of cytokines, such as interleukin-12 (IL-12p70), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ), was analyzed to evaluate the activity of the immune system.29 To evaluate whether the vacosome could function as a preventive cancer vaccine in vivo, a 4T1 breast cancer tumor model was selected for this study. The established in vivo immune memory after vacosome treatment was evaluated by the differentiation of the central memory T cells (TCM) and effector memory T cells (TEM).30 Additionally, the efficacy of the vacosome in protecting immunized mice against tumor challenge and in prolonging the overall survival was confirmed by analyzing the ratio of CTLs to CD 3+ cells and the ratio of regulatory T cells (Treg) to CD 4+ cells in the tumor area.
Scheme 1. Fabrication Process of the Vacosome and the Immune Response Induced by the Vacosome In Vivo.

2. Results and Discussion
The vacosome prepared in this study consisted of three major elements: an adjuvant (MPLA), an antigen (4T1 cell membrane, CM), and a lipid matrix (EPC, DSPE-PEG-2000, and cholesterol). The ability of these three individual components in inducing BMDC maturation was investigated by flow cytometry to choose the ideal amount of each component for the synthesis of the vacosome (Figure 1A–C).
Figure 1.

Preparation and characterization of the vacosome. The percentage of mature CD11c+-gated BMDCs induced with different amounts of (A) MPLA, (B) CM, and (C) lipid matrix. (D) Z-average diameter and (E) zeta potential of MPLA@Lip, CM, and vacosome. (F) TEM image of the vacosome. NC: BMDCs cultured in medium without any materials. PC: BMDCs cultured in medium with lipopolysaccharides (LPS). Data represent the mean ± SD of triplicate experiments (n = 5; *P < 0.05, ***P < 0.001).
As shown in Figure 1A, when increasing the amount of MPLA (from 0.1 to 1 μg), the percentage of matured BMDCs also increased. However, the maturation of BMDCs reached a plateau when the amount of MPLA exceeded 1 μg. As for the amount of CM (Figure 1B), the highest amount of CM (1600 μg) led to the maximum expression level of CD86 compared to the other groups tested. Additionally, the low immunogenicity of the lipid matrix was shown by coculturing BMDCs with various amounts of lipid matrix from 100 to 1000 μg (Figure 1C). According to the results, 1 μg of MPLA and 1.6 mg of CM were chosen to further fabricate the vacosome. As measured by dynamic light scattering (DLS), the diameters of MPLA@Lip, CM, and vacosome were 105.7 ± 2.3, 246.1 ± 1.2, and 122.4 ± 3.6 nm, respectively (Figures 1D and S1a). Compared with the size of MPLA@Lip, the increasing size in the vacosome could be attributed to the presence of the cell membrane.
Additionally, the long-term stability of the vacosome in 1× phosphate-buffered saline (pH 7.4) was also evaluated by DLS. Although the diameter of the vacosome increased from 109.3 ± 2.1 to 147.9 ± 3.3 nm, the size of the vacosome was still less than 200 nm (the critical diameter of nanoparticles for the enhanced permeability and retention effect) (Figure S1b). The zeta potential was also investigated (Figure 1E), and a decrease in the zeta potential was observed in the vacosome (from 7.3 ± 0.3 mV for MPLA@Lip to 4.3 ± 0.4 mV for the vacosome), which was due to the negative zeta potential of the CM (−6.25 ± 0.6 mV).
The morphologies of the vacosome, MPLA@Lip, and CM were also evaluated by transmission electron microscopy (TEM, Figures 1F and S1c,d). As shown in the TEM results, the diameter of the vacosome in the TEM experiments is slightly different from that in the DLS results, attributed to the nature of TEM imaging on dry samples as compared to the measuring size by DLS in the solution form.31 All of the above-mentioned results show that the vacosome system was successfully prepared utilizing a fixed combination of MPLA, mixture lipid, and cancer cell membrane.
After the synthesis of the vacosome, its biocompatibility and cellular uptake were further investigated in vitro. First, we evaluated the biocompatibility of the system by coculturing the vacosome with BMDCs for 1 day to 3 days (Figure 2A).
Figure 2.

In vitro investigation of the vacosome. (A) Cell viability of BMDCs treated with different formulations. NC: BMDCs cultured only with medium; PC: with 1% Triton X-100; lipid control (LC): with DSPE-PEG2000, EPC, and cholesterol; (B) Uptake of DSPE-PEG-2000-FITC labelled Lip&F, MPLA@Lip&F, and vacosome&F by BMDCs in vitro. DC maturation gated in CD11c+ by vacosome for (C) 24 h, (D) 48 h, and (E) 72 h coculture; NC: BMDCs cultured only with medium; PC: with 1 μg MPLA; LC: with DSPE-PEG2000, EPC, and cholesterol. (F) Cell viability of 4T1 cells after 24 h’ incubation. NC: 4T1 cells cultured with BMDCs and splenocytes; MPLA: with MPLA-induced BMDCs and splenocytes; LPS: with LPS-induced BMDCs and splenocytes, LC: with DSPE-PEG2000, EPC, and cholesterol-induced BMDCs and splenocytes; MPLA + CM: with MPLA and CM-induced BMDCs and splenocytes; CM: with CM-induced BMDCS and splenocytes; vacosome: with vacosome-induced BMDCs and splenocytes. Data represent the mean ± SD of triplicate experiments (n = 5; *P < 0.05, ***P < 0.001).
With the exception of the positive control (PC) groups, the other groups showed similar cell viability, which indicates that the vacosome is nontoxic in vitro over BMDCs. Then, we further evaluated whether the vacosome could have enhanced uptake in BMDCs compared with MPLA@Lip. As shown by the flow cytometry results (Figures 2B and S1e, similar percentages of fluorescein isothiocyanate (FITC+) BMDCs (which means the BMDCs taking-up FITC-labelled Lip&F, MPLA@Lip&F, or vacosome&F) were observed among the tested groups for 3 and 6 h, while more FITC+ BMDCs were found in the vacosome-treated group after 24 and 48 h.
In addition to biocompatibility and cellular uptake, the functionality, such as effective immune activation, is also a necessary part of vaccines. The composition of the vacosome, such as MPLA and 4T1 cell membrane, was proved in inducing BMDC maturation (Figure 1). To explore whether the vacosome could induce similar or enhanced immune activation, BMDCs were cocultured with various formulations from 1 day to 3 days (Figure 2C–E). After the first 24 h, PC (56.4 ± 4.5% in CD86+, 44.7 ± 4.7% in CD80+) showed effective and quick immune activation, whereas CM, MPLA@Lip, and vacosome exhibited relatively low immune stimulation on BMDCs. After 48 h, CM (44.2 ± 2.0% in CD86+, 35.4 ± 1.8% in CD80+), MPLA@Lip (51.7 ± 1.2% in CD86+, 38.9 ± 0.5% in CD80+), and vacosome (64.2 ± 2.9% in CD86+, 53.2 ± 4.0% in CD80+) groups exhibited a significant increase in the maturation percentage compared with the PC groups (57.8 ± 4.5% in CD86+, 42.7 ± 4.7% in CD80+). After 3 days incubation, the vacosome groups (82.4 ± 1.4% in CD86+, 80.8 ± 1.7% in CD80+) showed the highest maturation percentage. These results suggested that the vacosome can significantly promote immune activation compared to MPLA@Lip and CM as a result of the enhanced immune effect caused by incorporating the tumor cell antigen and immunostimulatory adjuvant simultaneously into the same carrier.24
Since the aim of using vacosome is to protect the immunized mice from tumor challenge, the vacosome is supposed to activate and educate the immune system to be prepared in fighting against cancer cells. To investigate whether the vacosome could promote the immune system to kill cancer cells in vitro, BMDCs were first incubated with different formulations for 3 days in order to mature BMDCs and process antigens. Then, these BMDCs were further cocultured with splenocytes (since spleen is one of the most important immune organs in the body) for another 3 days. Finally, the vaccination efficacy of the vacosome was demonstrated by evaluating the cell viability of 4T1 cells after 1 day of coculture with activated splenocytes and BMDCs (Figures 2F and S1f). The cell viability of 4T1 cells in the negative control (NC) group was regarded as a standard control (marked as 100%). The immune cells (BMDCs, splenocytes, and other splenocyte-derived cells) showed enhanced antitumor ability after treatment with the vacosome compared with immune cells treated with MPLA + CM (as for the control with MPLA + CM, it was used to evaluate the effect of formulating adjuvant and antigen within the same formulation compared to a simple mix of the two components). According to this result, we hypothesized that the vacosome can more effectively promote DC maturation and deliver antigen to DCs. In order to verify our hypothesis and explore the immunological mechanisms of action, we next investigated the vacosome in vivo.
With a focus on the potential clinical translation of the vaccine, the short-term safety profile of the vacosome was also evaluated in vivo. During the treatment duration, the body weight of each mouse was recorded, as shown in Figure S1g–j. The mice injected with different formulations (saline, CM, MPLA@Lip, and vacosome) showed a similar increasing trend in bodyweight. Additionally, kidney, liver, and heart were collected from the mice in each group, and then hematoxylin and eosin (H&E) staining was utilized to evaluate the tissue morphology (Figure 3E–P). According to the H&E staining results, there was no obvious inflammatory cell infiltration in the tissues, and the tissue organization was physically normal without any swelling, adhesion, or hyperplasia, which indicates that the vacosome did not show cardiotoxicity, nephrotoxicity, or liver toxicity in the normal tissue even after 28 days of treatment. Overall, these results indicated that the vacosome was safe in vivo; hence, we further explored the immunological profile of the vacosome in vivo.
Figure 3.

Biocompatibility of the vacosome in vivo. H&E staining results of kidney treated with (A) saline, (B) CM, (C) MPLA@Lip, and (D) vacosome. Liver treated with (E) saline, (F) CM, (G) MPLA@Lip, and (H) vacosome. Heart treated with (I) saline, (J) CM, (K) MPLA@Lip, and (L) vacosome.
To evaluate the immune activation state in healthy BALB/c mice after different stages of the preimmunization scheme with the formulation (Figure 4A–D), serum was collected and analyzed by cytometric bead array for the detection of the secreted IL-12p70, TNF-α, and IFN-γ (Figure S2). IL-12p70 is produced by activated DCs and is important for IFN-γ production by lymphocytes. TNF-α is also highly related to cancer immunity by causing apoptosis and inflammation. Activated CD8+ T cells can generate IFN-γ which can induce apoptosis in the target cells. Thus, these three cytokines were selected to evaluate the immune activation state.32−34 Additionally, as a Th2 related cytokine, IL-6 was also investigated (Figure S3a). At 7 days after the first immunization, there was not a significant difference in the secretion levels of IL-12p70, TNF-α, and IFN-γ in mice treated with different formulations. Three days post the second vaccination (14 days), an increase in the secretion levels of these three cytokines was detected in mice treated with the vacosome. At 28 days (3 days after the third vaccination), the mice treated with the vacosome still showed the highest levels of IL-12p70 (33.5 ± 6.5 pg/mL), TNF-α (49.9 ± 7.8 pg/mL), and IFN-γ (55.9 ± 10.3 pg/mL). Overall, for mice treated with the vacosome, from 7 to 14 days, the cytokine secretion level increased more than 3-fold. However, from 14 to 28 days, the increasing trend slowed down (Figure 4B–D). We hypothesize this could be due to the fact that the process of creating an adaptive immune response after vaccination have two different waves of immune activation. After the first immunization, the immune system takes up to 72 h to mount an adaptive immune response of small magnitude with lower levels of cytokines.35 Upon the second exposure to the antigen and adjuvant, the magnitude of the immune response reaches the peak, which will remain constant also after further rechallenge; thus, even after the stimulation, the cytokine secretion level cannot be further significantly improved.36,37 All these results revealed that the vacosome effectively stimulated the immune system and made the immune system secrete related cytokines which can fight against with cancer cells.
Figure 4.

Immune activation of the vacosome in vivo. (A) Treatment plan for immunized BALB/c mice. The concentration of (B) IL-12p70, (C) TNF-α, and (D) IFN-γ in the serum 7, 14, and 28 days after stimulation. Data represent the mean ± SD (n = 5; *P < 0.05, **P < 0.01, ***P < 0.001).
Next, we investigated whether the immunized mice can effectively reject 4T1 cells in a tumor challenge experiment. As described in Figure 5A, after three vaccinations with different formulations (saline, MPLA@Lip, CM, and vacosome), the mice were challenged by 4T1 cells at day 3 post the final vaccination (marked as day 0 in the tumor stage). The tumor growth was monitored starting from day 2 post tumor cell challenge. The CM vaccination group tumor could partially inhibit tumor progression as compared to saline groups (Figure 5B,C), and a similar phenomenon was also proved by Cheung et al.23 This is due to the fact that the administration of antigens only is not able to prime a proinflammatory immune response while often resulting into a tolerogenic effect.38 MPLA@Lip groups showed increased tumor growth inhibition efficacy compared with the CM formulation (Figure 5D). In these three groups, the tumor size exhibited obvious individual differences, although the tumor size in mice treated with MPLA (747.4 ± 454.7 mm3) was more average than those in saline (1725.3 ± 1158.2 mm3) and CM (1488.0 ± 1107.4 mm3) groups. Most importantly, the mice treated with the vacosome showed more than 10-times limited tumor size (76.0 ± 63.9 mm3) compared to all other formulations (Figures 5E,F, S4). Additionally, the long-term survival rate was investigated in the immunized mice (Figure 5G) up to 50 days, showing that 50% the mice in the vacosome group were still alive. Altogether, these data greatly support that vacosome-immunized mice can more effectively fight against cancer challenge in the aggressive 4T1 tumor model compared to TLR agonist (MPLA@Lip) and antigen (CM).
Figure 5.

Efficacy of vacosome preimmunization in 4T1-bearing BALB/c mice. (A) Schematic illustration of the experimental design for vacosome administration to inhibit tumor growth. Tumor growth of mice treated with (B) saline, (C) CM, (D) MPLA@Lip, and (E) vacosome. Each line indicates the tumor size of the individual mouse. (F) Average tumor growth curves for 4T1 tumors in mice after various treatments indicated. (G) Survival rate of different groups of 4T1 bearing mice. Data represent the mean ± SD. (B–F, n = 6; ***P < 0.001; G, n = 10; ***P < 0.001).
In order to understand the immunological mechanism preventing cancer growth in immunized mice, the mice were sacrificed to collect the spleen and tumor at day 14 post the 4T1 cells injection. Considering the immune response caused by T cells in tumor treatment, the cytotoxic T cells (CTL, CD8+ T cells) and the regulatory T cells (Treg) were evaluated in the tumor by using flow cytometry (Figure 6A–D). We found that both CM (26.9 ± 1.0%) and MPLA@Lip (31.3 ± 2.1%) promoted the differentiation of CTL compared to the saline-treated group (19.3 ± 3.5%). In addition, mice treated with vacosome (39.4 ± 5.4%) showed the highest percentages of CTL in the tumor area (Figures 6A,B). CTL plays a critical role in killing target cancer cells; thus an increased percentage of CTL in the tumor area indicates stronger immune activation and antitumor ability.39 Furthermore, Foxp3 was used as a marker to classify CD4+ T cells into effective T cells (CD3+CD4+Foxp3–) that can promote the immune response and Treg (CD3+CD4+Foxp3+), which mainly shut down the immune response.40 As shown in Figures 6C,D, saline groups showed a much higher percentage of Treg (66.2 ± 5.0%) than CM (57.2 ± 2.4%) and MPLA@Lip (48.7 ± 2.8%) groups. The amount of Treg decreased to around 38.9 ± 6.7% in the vacosome groups, which indicates that vacosome-immunized mice have reduced immune suppression in the tumor area. Moreover, the ratio between CD8+ T cells and Treg was calculated in the collected tumor tissues (Figure S3b). The BALB/c mice treated with the vacosome exhibited the highest ratio compared to the mice treated with other formulations, which indicate the more antitumor microenvironment in the vacosome-preimmunized BALB/c model. Additionally, as the immunohistochemistry staining of vacosome groups exhibited (Figure S5), CD3+, CD4+, and CD8+ T cells can be observed in the tumor tissue, which could be related to the prolonged survival period and limited tumor growth.
Figure 6.

Immune response caused by the vacosome in the tumor and spleen area. Representative flow cytometry data of (A) CD8+ T cells in the tumor areas (B). Statistical data of CD8+ in the tumor areas. (C) Treg in the tumor areas. (D) Statistical data of Treg in the tumor areas. (E) TCM and TEM in the spleen. (F) Statistical data of TCM and TEM in the spleen. Data represent the mean ± SD (n = 5; ***P < 0.001).
An important role of vaccination is the ability to make the immune system acquire immune memory, which is critical for cancer prevention. Thus, it is meaningful to evaluate the immune memory induced by the various formulations prepared. According to the functionality, proliferation, and migration capabilities, memory T cells are classified into central memory T cells (TCM, CD3+CD8+CD62L+CD44+) and effector memory T cells (TEM, CD3+CD8+CD62L–CD44+).41 As an important immune organ, spleens were collected to measure the ratios of both TCM and TEM cells (Figures 6E,F). We found that mice treated with the vacosome showed a higher TEM percentage (70.4 ± 6.0%) than mice treated with CM (38.8 ± 4.4%), MPLA@Lip groups (49.5 ± 5.4%), and saline (27.8 ± 6.0%) groups. TEM are differentiated from the CD8+ T cells that are stimulated by antigens for long-term memory. In addition, the large number of TEM are quickly expanded when they are re-exposed to their cognate antigen. TEM can appear in both lymphoid and nonlymphoid tissues and generate immediate protection by secreting cytokines, such as IFN-γ.42 All these indicate that after 28 days of vaccination by the vacosome, the immunized BALB/c mice generated immune memory. Thereby, when these mice were challenged by the 4T1 cells, TEM were quickly expanded against 4T1 cells. Overall, these results indicate that the effective prophylactic effects of the vacosome are attributed to a well-established immune memory and full-activated immune response.
3. Conclusions
In this study, we showed that the vacosome is able to effectively enhance immune response and establish immune memory against a 4T1 challenge. The administration of the vacosome can improve the activation of APCs, leading to an increased priming of CD8+ T cells. Furthermore, immunization with the vacosome resulted in increased priming of the TEM and reduction in the intratumoral Treg, which improve the antitumor efficiency compared to CM or MPLA@Lip alone. The vacosome platform provides a promising candidate for the clinical translation of cancer vaccine, for example, the vacosome system could be developed as a postoperative cancer vaccine in preventing tumor recurrence. Specifically, after the surgery, the cancer cell membranes could be isolated from the patient tumor tissues and then synthesized with MPLA to develop the personalized vacosome in preventing tumor recurrence. As a result, the synthesis process is convenient, and the materials are accessible and safe, for example, MPLA has been approved by FDA as an adjuvant and the lipids used in this study are commercially available and generally recognized as safe.
Acknowledgments
R.C. acknowledges the China Scholarship Council for a grant. P.F. acknowledges the Finnish Cultural Foundation for a research grant (decision no. 00190246). S.W. acknowledges the financial support from Jenny and Antti Wihuri Foundation. W.C. acknowledges the financial support from the National Natural Science Foundation of China (81930051), Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (20171906), and Shanghai talent development fund (2018099). Y.L. acknowledges the financial support from the Project of the Action Plan of Major Diseases Prevention and Treatment (2017ZX01001-S12). M.-A. Shahbazi acknowledges the financial support from the Academy of Finland (grant no. 317316). Prof. H.A. Santos acknowledges the financial support from the HiLIFE Research Funds, the Sigrid Jusélius Foundation, and the European Research Council Proof-of-Concept Research Grant (grant no. 825020). The authors also acknowledge the following core facilities funded by Biocenter Finland: Electron Microscopy Unit for TEM and FACS Biocore for the use of FACS instruments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c15057.
Z-averages of the hydrodynamic size of MPLA@Lip, CM, and vacosome; Z-averages of the vacosome for long-term stability investigation; TEM images of CM and MPLA@Lip; cytometry results of FITC + BMDCs cocultured with different formulations; antitumor ability of T cells induced by the vacosome; bodyweight of mice treated with saline, CM, MPLA@Lip, and vacosome; heatmap illustrating cytokine concentrations at different preimmunization stages; immunological profiles; concentration of IL-6 in the serum after different vaccinations; ratio of CD8+ T cells to Treg cells in the collected tumor tissues; collected tumor tissues in 4T1-bearing balb/c after 21 days’ treatment with different formulations; immunohistochemistry staining of vacosome groups, CD3+, CD4+, and CD8+ T cells in the tumor tissue; and materials and methods (PDF)
Author Contributions
The manuscript was completed through contributions of all authors. R.C., F.F., and H.A. initiated the topic and designed the entire content of this paper. R.C., F. F., J.X., Z.L., P.F., M.A., S.W., J.J., G.T., J.T., H.Z., T.C., W.C., Y.L, and H.A. contributed to the experiment and writing of the paper. R.C., F.F., and H.A. then discussed and revised the paper. All authors helped with the writing and revision of the paper and gave the approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ott P. A.; Hu Z.; Keskin D. B.; Shukla S. A.; Sun J.; Bozym D. J.; Zhang W.; Luoma A.; Giobbie-Hurder A.; Peter L.; Chen C.; Olive O.; Carter T. A.; Li S.; Lieb D. J.; Eisenhaure T.; Gjini E.; Stevens J.; Lane W. J.; Javeri I.; Nellaiappan K.; Salazar A. M.; Daley H.; Seaman M.; Buchbinder E. I.; Yoon C. H.; Harden M.; Lennon N.; Gabriel S.; Rodig S. J.; Barouch D. H.; Aster J. C.; Getz G.; Wucherpfennig K.; Neuberg D.; Ritz J.; Lander E. S.; Fritsch E. F.; Hacohen N.; Wu C. J. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai R.; Ochyl L. J.; Bahjat K. S.; Schwendeman A.; Moon J. J. Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater. 2017, 16, 489–496. 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocca R. A.; Abbink P.; Peron J. P. S.; de A. Zanotto P. M.; Iampietro M. J.; Badamchi-Zadeh A.; Boyd M.; Ng’ang’a D.; Kirilova M.; Nityanandam R.; Mercado N. B.; Li Z.; Moseley E. T.; Bricault C. A.; Borducchi E. N.; Giglio P. B.; Jetton D.; Neubauer G.; Nkolola J. P.; Maxfield L. F.; De La Barrera R. A.; Jarman R. G.; Eckels K. H.; Michael N. L.; Thomas S. J.; Barouch D. H. Vaccine Protection against Zika Virus from Brazil. Nature 2016, 536, 474–478. 10.1038/nature18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusciello M.; Fontana F.; Tähtinen S.; Capasso C.; Feola S.; Martins B.; Chiaro J.; Peltonen K.; Ylösmäki L.; Ylösmäki E. Artificially Cloaked Viral Nanovaccine for Cancer Immunotherapy. Nat. Commun. 2019, 10, 1–13. 10.1038/s41467-019-13744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi J. L.; Bobisse S.; Ophir E.; Tuyaerts S.; Roberti A.; Genolet R.; Baumgartner P.; Stevenson B. J.; Iseli C.; Dangaj D.; Czerniecki B.; Semilietof A.; Racle J.; Michel A.; Xenarios I.; Chiang C.; Monos D. S.; Torigian D. A.; Nisenbaum H. L.; Michielin O.; June C. H.; Levine B. L.; Powell D. J.; Gfeller D.; Mick R.; Dafni U.; Zoete V.; Harari A.; Coukos G.; Kandalaft L. E. Personalized Cancer Vaccine Effectively Mobilizes Antitumor T cell Immunity in Ovarian Cancer. Sci. Transl. Med. 2018, 10, eaao5931 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- Wang T.; Wang D.; Yu H.; Feng B.; Zhou F.; Zhang H.; Zhou L.; Jiao S.; Li Y. A Cancer Vaccine-mediated Postoperative Immunotherapy for Recurrent and Metastatic Tumors. Nat. Commun. 2018, 9, 1–12. 10.1038/s41467-018-03915-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Billingsley M. M.; Mitchell M. J. Biomaterials for Vaccine-based Cancer Immunotherapy. J. Control. Release 2018, 292, 256–276. 10.1016/j.jconrel.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferber S.; Gonzalez R. J.; Cryer A. M.; von Andrian U. H.; Artzi N. Immunology-Guided Biomaterial’s Design as Mucosal Cancer Vaccine. Adv. Mater. 2019, 190, 38–47. [DOI] [PubMed] [Google Scholar]
- Matyas G. R.; Mayorov A. V.; Rice K. C.; Jacobson A. E.; Cheng K.; Iyer M. R.; Li F.; Beck Z.; Janda K. D.; Alving C. R. Liposomes Containing Monophosphoryl lipid A: A Potent Adjuvant System for Inducing Antibodies to Heroin Hapten Analogs. Vaccine 2013, 31, 2804–2810. 10.1016/j.vaccine.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki Y.; Yuba E.; Sakaguchi N.; Koiwai K.; Harada A.; Kono K. pH-sensitive Polymer-modified Liposome-based Immunity-inducing System: Effects of Inclusion of Cationic Lipid and CpG-DNA. Biomaterials 2017, 141, 272–283. 10.1016/j.biomaterials.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Alving C. R.; Rao M.; Steers N. J.; Matyas G. R.; Mayorov A. V. Liposomes Containing Lipid A: An Effective, Safe, Generic Adjuvant System for Synthetic Vaccines. Expert Rev. Vaccines 2012, 11, 733–744. 10.1586/erv.12.35. [DOI] [PubMed] [Google Scholar]
- Alving C. R.; Peachman K. K.; Rao M.; Reed S. G. Adjuvants for Human Vaccines. Curr. Opin. Immunol. 2012, 24, 310–315. 10.1016/j.coi.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuba E. Liposome-based Immunity-inducing Systems for Cancer Immunotherapy. Mol. Immunol. 2018, 98, 8–12. 10.1016/j.molimm.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Wang N.; Chen M.; Wang T. Liposomes used as a Vaccine Adjuvant-delivery System: From Basics to Clinical Immunization. J. Controlled Release 2019, 303, 130–150. 10.1016/j.jconrel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren W.; Zhong T.; Zhang S.; Huang D.; Guo Y.; Yao X.; Wang C.; Zhang W.-Q.; Zhang X.; Zhang Q. Tumor-specific pH-responsive Peptide-modified pH-sensitive Liposomes Containing Doxorubicin for Enhancing Glioma Targeting and Anti-tumor Activity. J. Controlled Release 2016, 222, 56–66. 10.1016/j.jconrel.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Khong H.; Volmari A.; Sharma M.; Dai Z.; Imo C. S.; Hailemichael Y.; Singh M.; Moore D. T.; Xiao Z.; Huang X.-f.; Horvath T. D.; Hawke D. H.; Overwijk W. W. Peptide Vaccine Formulation Controls the Duration of Antigen Presentation and Magnitude of Tumor-specific CD8+ T cell Response. J. Immunol. 2018, 200, 3464–3474. 10.4049/jimmunol.1700467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albershardt T. C.; Parsons A. J.; Reeves R. S.; Flynn P. A.; Campbell D. J.; ter Meulen J.; Berglund P. Therapeutic Efficacy of PD1/PDL1 Blockade in B16 Melanoma is Greatly Enhanced by Immunization with Dendritic Cell-targeting Lentiviral Vector and Protein Vaccine. Vaccine 2020, 38, 3369–3377. 10.1016/j.vaccine.2020.02.034. [DOI] [PubMed] [Google Scholar]
- Pardi N.; Hogan M. J.; Porter F. W.; Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discovery 2018, 17, 261–279. 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes A.; Vanvarenberg K.; Kos Š.; Lucas S.; Colau D.; Van den Eynde B.; Préat V.; Vandermeulen G. Combination of Immune Checkpoint Blockade with DNA Cancer Vaccine Induces Potent Antitumor Immunity Against P815 Mastocytoma. Sci. Rep. 2018, 8, 1–11. 10.1038/s41598-018-33933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Xu L.; Liang C.; Wang C.; Peng R.; Liu Z. Photothermal Therapy with Immune-adjuvant Nanoparticles Together with Checkpoint Blockade for Effective Cancer Immunotherapy. Nat. Commun. 2016, 7, 1–13. 10.1038/ncomms13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Sun W.; Ye Y.; Hu Q.; Bomba H. N.; Gu Z. In situ Activation of Platelets with Checkpoint Inhibitors for Post-surgical Cancer Immunotherapy. Nat. Biomed. Eng. 2017, 1, 1–10. 10.1038/s41551-016-0011. [DOI] [Google Scholar]
- Rodell C. B.; Arlauckas S. P.; Cuccarese M. F.; Garris C. S.; Li R.; Ahmed M. S.; Kohler R. H.; Pittet M. J.; Weissleder R. TLR7/8-agonist-loaded Nanoparticles Promote the Polarization of Tumour-associated Macrophages to Enhance Cancer Immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A. S.; Koshy S. T.; Stafford A. G.; Bastings M. M. C.; Mooney D. J. Adjuvant-Loaded Subcellular Vesicles Derived From Disrupted Cancer Cells for Cancer Vaccination. Small 2016, 12, 2321–2333. 10.1002/smll.201600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R.; Xu J.; Xu L.; Sun X.; Chen Q.; Zhao Y.; Peng R.; Liu Z. Cancer Cell Membrane-coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano 2018, 12, 5121–5129. 10.1021/acsnano.7b09041. [DOI] [PubMed] [Google Scholar]
- Paulis L. E.; Mandal S.; Kreutz M.; Figdor C. G. Dendritic Cell-based Nanovaccines for Cancer Immunotherapy. Curr. Opin. Immunol. 2013, 25, 389–395. 10.1016/j.coi.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Fontana F.; Fusciello M.; Groeneveldt C.; Capasso C.; Chiaro J.; Feola S.; Liu Z.; Mäkilä E. M.; Salonen J. J.; Hirvonen J. T.; Cerullo V.; Santos H. A. Biohybrid Vaccines for Improved Treatment of Aggressive Melanoma with Checkpoint Inhibitor. ACS Nano 2019, 13, 6477–6490. 10.1021/acsnano.8b09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauleth-Ramos T.; Shih T. Y.; Shahbazi M. A.; Najibi A. J.; Mao A. S.; Liu D.; Granja P.; Santos H. A.; Sarmento B.; Mooney D. J. Acetalated Dextran Nanoparticles Loaded into an Injectable Alginate Cryogel for Combined Chemotherapy and Cancer Vaccination. Adv. Funct. Mater. 2019, 29, 1903686. [Google Scholar]
- Li W.; Liu Z.; Fontana F.; Ding Y.; Liu D.; Hirvonen J. T.; Santos H. A. Tailoring Porous Silicon for Biomedical Applications: from Drug Delivery to Cancer Immunotherapy. Adv. Mater. 2018, 30, 1703740. 10.1002/adma.201703740. [DOI] [PubMed] [Google Scholar]
- Escribà-Garcia L.; Alvarez-Fernández C.; Tellez-Gabriel M.; Sierra J.; Briones J. Dendritic Cells Combined with Tumor Cells and α-galactosylceramide Induce a Potent, Therapeutic and NK-cell Dependent Antitumor Immunity in B cell Lymphoma. J. Transl. Med. 2017, 15, 115. 10.1186/s12967-017-1219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F.; Lenig D.; Förster R.; Lipp M.; Lanzavecchia A. Two Subsets of Memory T Lymphocytes with Distinct Homing Potentials and Effector Functions. Nature 1999, 401, 708–712. 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Kuntsche J.; Horst J. C.; Bunjes H. Cryogenic Transmission Electron Microscopy (cryo-TEM) for Studying the Morphology of Colloidal Drug Delivery Systems. Int. J. Pharm. 2011, 417, 120–137. 10.1016/j.ijpharm.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Garris C. S.; Arlauckas S. P.; Kohler R. H.; Trefny M. P.; Garren S.; Piot C.; Engblom C.; Pfirschke C.; Siwicki M.; Gungabeesoon J.; Freeman G. J.; Warren S. E.; Ong S.; Browning E.; Twitty C. G.; Pierce R. H.; Le M. H.; Algazi A. P.; Daud A. I.; Pai S. I.; Zippelius A.; Weissleder R.; Pittet M. J. Successful Anti-PD-1 Cancer Immunotherapy Requires T cell-dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann T. A. Cytokines in Cancer Immunotherapy. Cold Spring Harbor Perspect. Biol. 2018, 10, a028472. 10.1101/cshperspect.a028472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E.; Oh J.-E.; Hong J.; Chung Y.; Lee Y.; Park K. D.; Kim S.; Yun C.-O. Optimized Biodegradable Polymeric Reservoir-mediated Local and Sustained Co-delivery of Dendritic Cells and Oncolytic Adenovirus Co-expressing IL-12 and GM-CSF for Cancer Immunotherapy. J. Control. Release 2017, 259, 115–127. 10.1016/j.jconrel.2017.03.028. [DOI] [PubMed] [Google Scholar]
- Miao L.; Li J.; Liu Q.; Feng R.; Das M.; Lin C. M.; Goodwin T. J.; Dorosheva O.; Liu R.; Huang L. Transient and Local Expression of Chemokine and Immune Checkpoint Traps to Treat Pancreatic Cancer. ACS Nano 2017, 11, 8690–8706. 10.1021/acsnano.7b01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B.; Ahmed R. Translating Innate Immunity into Immunological Memory: Implications for Vaccine Development. Cell 2006, 124, 849–863. 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Church S. E.; Jensen S. M.; Twitty C. G.; Bahjat K.; Hu H.-M.; Urba W. J.; Fox B. A. Multiple Vaccinations: Friend or Foe. Cancer J. 2011, 17, 379–396. 10.1097/ppo.0b013e3182346320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D. R.; Ferguson T.; Zitvogel L.; Kroemer G. Immunogenic and Tolerogenic Cell Death. Nat. Rev. Immunol. 2009, 9, 353–363. 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. C.; Holl E. K.; Boczkowski D.; Dobrikova E.; Mosaheb M.; Chandramohan V.; Bigner D. D.; Gromeier M.; Nair S. K. Cancer Immunotherapy with Recombinant Poliovirus Induces IFN-Dominant Activation of Dendritic Cells and Tumor Antigen-Specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220 10.1126/scitranslmed.aan4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Paul W. E. Heterogeneity and Plasticity of T Helper Cells. Cell Res. 2010, 20, 4–12. 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff C. A.; Gattinoni L.; Torabi-Parizi P.; Kerstann K.; Cardones A. R.; Finkelstein S. E.; Palmer D. C.; Antony P. A.; Hwang S. T.; Rosenberg S. A.; Waldmann T. A.; Restifo N. P. Central Memory self/tumor-reactive CD8+ T cells Confer Superior Antitumor Immunity Compared with Effector Memory T cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 9571–9576. 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathaliyawala T.; Kubota M.; Yudanin N.; Turner D.; Camp P.; Thome J. J. C.; Bickham K. L.; Lerner H.; Goldstein M.; Sykes M.; Kato T.; Farber D. L. Distribution and Compartmentalization of Human Circulating and Tissue-resident Memory T cell Subsets. Immunity 2013, 38, 187–197. 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.