

Abstract
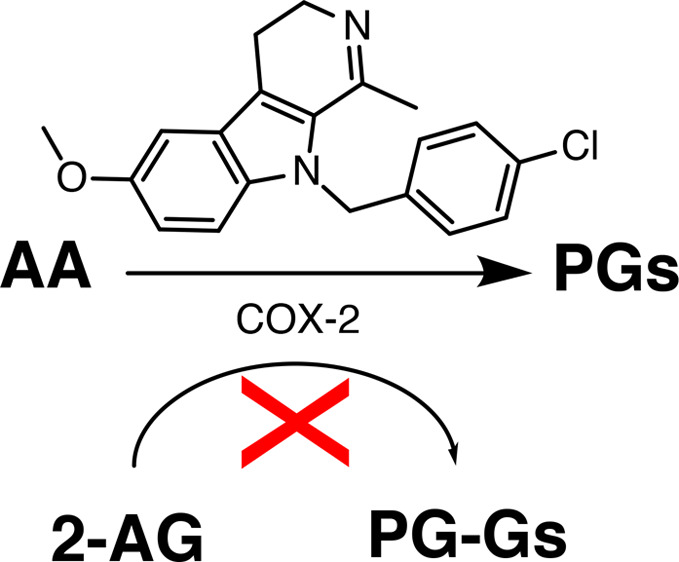
We report the design, synthesis, and evaluation of a series of harmaline analogs as selective inhibitors of 2-arachidonylglycerol (2-AG) oxygenation over arachidonic acid (AA) oxygenation by purified cyclooxygenase-2 (COX-2). A fused tricyclic harmaline analog containing a CH3O substituent at C-6 and a CH3 group at the C-1 position of 4,9-dihydro-3H-pyrido[3,4-b]indole (compound 3) was the best substrate-selective COX-2 inhibitor of those evaluated, exhibiting a 2AG-selective COX-2 inhibitory IC50 of 0.022 μM as compared to >1 μM for AA. The 2.66 Å resolution crystal complex of COX-2 with compound 3 revealed that this series of tricyclic indoles binds in the cyclooxygenase channel by flipping the side chain of L531 toward the dimer interface. This novel tricyclic indole series provides the foundation for the development of promising substrate-selective molecules capable of increasing endocannabinoid (EC) levels in the brain to offer new treatments for a variety of diseases, from pain and inflammation to stress and anxiety disorders.
Keywords: Cyclooxygenase-2, 2-arachidonylglycerol, arachidonic acid, substrate-selective inhibition
Cyclooxygenases (COXs) are key enzymes that modulate the physiology and pathophysiology of many disease processes.1 These enzymes catalyze the oxygenation of arachidonic acid (AA) into prostaglandins (PGs) and thromboxane. Of the two COX isoforms constitutively expressed, COX-1 is found in nearly all normal tissues, whereas COX-2 is induced in response to biologically active endogenous and pathogen-associated macromolecules (such as cytokines, growth factors, tumor promoters, and bacterial lipopolysaccharide) so that inflammatory, preneoplastic, and neoplastic tissues express elevated levels of this enzyme.2 Thus, nonselective COX inhibitors or selective COX-2 inhibitors exhibit anti-inflammatory, analgesic, and anticancer activities.3
In addition to their distinct expression patterns, the COX isoforms also differ with regard to substrate specificity. For example, COX-2 oxygenates neutral ester and amide derivatives of AA that are poor substrates for COX-1. Of particular interest in this regard are the endocannabinoids, 2-arachidonoylglycerol (2-AG), and arachidnoylethanolamide (AEA), which are converted to PG glyceryl ester (PG-G) and ethanolamide (PG-EA) derivatives, respectively.4 The PG-Gs originating from oxygenation of 2-AG exhibit biological properties distinct from those of free acid PGs produced from AA in a range of cell types, including neuronal, inflammatory, and cancer cells.5−16 Furthermore, as both 2-AG and AEA are high affinity endogenous ligands for the cannabinoid receptors (CB1 and CB2), oxygenation of these lipids by COX-2 may serve as an important mechanism by which cannabinoid signaling is terminated.17 Thus, COX-2-dependent endocannabinoid oxygenation may serve important physiological or pathophysiological functions. Defining these functions in vivo has remained elusive due to the difficulty of differentiating the effects of AA oxygenation from endocannabinoid oxygenation.
We have discovered that weak, competitive inhibitors of AA oxygenation by COX-2 (e.g., ibuprofen, mefenamic acid) are potent noncompetitive inhibitors of 2-AG oxygenation.18 We proposed a mechanism to explain this substrate-selective inhibition based on accumulating evidence that the COX enzymes are structural homodimers that behave as functional heterodimers with one subunit serving as the active site while the second serves an allosteric function.19−27 Current data support the hypothesis that substrate-selective inhibitors bind with high affinity in the allosteric site of COX-2 and alter the structure of the active site so that 2-AG oxygenation is inhibited but AA oxygenation is not.20,25 Inhibition of AA oxygenation requires lower affinity, reversible binding of a second molecule of inhibitor in the active site.18,28 Substrate-selective inhibition of COX-2 offers a potential means to elucidate the biological impact of endocannabinoid oxygenation in vivo; however, inhibitors that display substrate-selectivity in vitro have failed to do so in vivo due, at least in part, to inadequate selectivity or metabolic instability.29 To address this failure, we now describe the design and structure–activity relationship (SAR) studies of a series of compounds obtained from the derivatization of 4,9-dihydro-3H-pyrido[3,4-b]indole to optimize an effective and metabolically stable substrate-selective COX-2 inhibitor.
Key determinants of the substrate-selectivity of COX-2 inhibitors are unknown, which makes the design of such molecules particularly challenging. The X-ray crystal structure of 1-AG (the thermodynamically stable isomer of 2-AG) in complex with COX-2 reveals that there is a small hydrophobic pocket adjacent to Leu-531, Val-116, and Val-349, all of which are located above a constriction that demarcates the opening of the COX-2 active site. The glycerol moiety of 1-AG inserts into this pocket.30 This information suggests that compounds containing a relatively small functional group capable of binding in the hydrophobic pocket might selectively inhibit the oxygenation of 2-AG by COX-2.
Harmaline is a member of the harmala alkaloid family of naturally occurring organic compounds containing a tricyclic indole nucleus. Studies have shown that harmaline is psychoactive in humans, and rutaecarpine, a natural polycyclic analog of harmaline, binds to COX enzymes with varying degrees of isoform selectivity when tested using AA as a substrate.31−33 We hypothesized that un-natural analogs of harmaline would selectively inhibit oxygenation of 2-AG by COX-2, thereby resulting in increased endocannabinoid (EC) levels in the brain and a concomitant reduction in stress and anxiety. To test this hypothesis, we synthesized and evaluated a series of harmaline derivatives as substrate-selective inhibitors of COX-2.
We used the Bischler–Napieralski intramolecular electrophilic aromatic substitution reaction for the synthesis of substituted-4,9-dihydro-3H-pyrido[3,4-b]indoles from the cyclization of substituted N-acetyltriptamine catalyzed by phosphorus oxychloride (POCl3) in refluxing toluene. Benzylation of substituted-4,9-dihydro-3H-pyrido[3,4-b]indoles was performed using substituted benzyl bromide in good yields (compounds 1–6, Scheme 1; see Supporting Information for synthetic details).
Scheme 1. Synthesis of 4,9-Dihydro-3H-pyrido[3,4-b]indole Derivatives 1–6.

(a) CH3COCl, TEA, DCM, rt 5 h; POCl3, toluene reflux 7 h; (b) NaH, DMF 0 °C 1 h; 4-Cl-C6H4-CH2-Br, 0°C 1 h; (c) tert-butyl 2-chloro-2-oxoacetate, TEA, DCM, rt 5 h; POCl3, toluene, reflux 7 h; (d) TFA, rt 1 h,; (e) NaH, DMF 0 °C 1 h; 4-Cl-C6H4-CH2-Br, 0 °C 1 h.
The inhibitory potencies of the synthesized compounds against purified human COX-2 or ovine COX-1 were determined by a thin layer chromatography (TLC)-based assay that measures the conversion of [1-14C]-AA to radiolabeled PGs.34 Briefly, reaction mixtures of 200 μL contained hematin-reconstituted protein in 100 mM Tris-HCl, pH 8.0, 500 μM phenol, and [1-14C]AA (5 μM, ∼55–57 mCi/mmol, PerkinElmer). Reactions were terminated by solvent extraction in diethyl ether/methanol/1 M citrate buffer, pH 4.0 (30:4:1). The phases were separated by centrifugation at 2000 rpm for 2 min, and the organic phase was spotted on a TLC plate (EMD Kieselgel 60, VWR). Following development in ethyl acetate/methylene chloride/glacial acetic acid (75:25:1) at 4 °C, radiolabeled products were quantified with a radioactivity scanner (Bioscan, Inc., Washington, DC).
Compounds were next evaluated for substrate-selective COX-2 inhibitory activity using a mass spectrometry (MS)-based assay. The final assay reaction mixture contained 50 nM purified murine COX-2, 100 nM heme, 5 μM AA or 2-AG, 1 μM 5-phenyl-4-pentenyl-1-hydroperoxide (PPHP), and inhibitor or vehicle (DMSO at a final concentration of 5%) in a buffer of 50 mM Tris-HCl, pH 8.0, 0.5 mM phenol. Following addition of inhibitor, the mixture was incubated for 15 min, and then the reaction was initiated by the addition of AA or 2-AG. After 30 s, the reaction was quenched by addition of ethyl acetate or acetonitrile containing internal standards (PGE2-d4 and PGE2G-d5) at 0.3 μM. The samples were injected onto a C18 5 cm × 0.2 cm, 3 μm particle size column connected to a Shimadzu LC system coupled with an ABSCIEX MS. Elution solvents were solvent A (5 mM ammonium acetate, pH 3.6) and solvent B (94% acetonitrile with 6% solvent A) applied in a gradient from 30% to 100% B over 1.5 min followed by 100% B for 1 min. Analytes of interest were detected by selected reaction monitoring MS/MS using the following transitions: PGE2/D2m/z 370 → 317; PGE2-d4m/z 374 → 321; PGE2/D2-G m/z 444 → 391; PGE2-G-d5m/z 449 → 396. Analyte peak areas were normalized to those of their deuterated internal standards for the quantification of product formation and inhibition. Note that PPHP was included in this assay to eliminate possible effects of inhibitors on peroxide-mediated activation of COX-2, a mechanism that has been implicated in substrate-selective inhibition of 2-AG oxygenation by some inhibitors.35 The presence of PPHP tends to increase the IC50 for inhibition of both AA and 2-AG oxygenation; however, its effect is greater in the case of 2-AG because higher peroxide tone is required for activation in the case of that substrate.36
Compounds 1–6 possessed a tricyclic indole core (4,9-dihydro-3H-pyrido[3,4-b]indole) containing a methoxy substitution at the C-6 or C-7 position. The p-chlorobenzyl substitution was introduced at C-9 position of compounds 3–4 and 6 in order to achieve improved in vitro and in vivo metabolic stability. In the purified COX inhibition assay, compounds 1 and 2 (harmaline) showed no COX inhibitory activity. However, 9-(4-chlorobenzyl)-6-methoxy-1-methyl-4,9-dihydro-3H-pyrido[3,4-b]indole (compound 3) was a selective COX-2 inhibitor (IC50 = 0.2 μM), whereas its regioisomer 9-(4-chlorobenzyl)-7-methoxy-1-methyl-4,9-dihydro-3H-pyrido[3,4-b]indole (compound 4) exhibited COX-2 selectivity with a poor potency (IC50 = 2.1 μM). Although the carboxylic acid-containing compound 5 showed no COX inhibitory activity, 9-(4-chlorobenzyl)-6-methoxy-4,9-dihydro-3H-pyrido[3,4-b]indole-1-carboxylic acid (compound 6, Table 1) exhibited mild inhibitory activity against COX-1 with an IC50 value of 2.9 μM.
Table 1. Biochemical Properties of 4,9-Dihydro-3H-pyrido[3,4-b]indole Derivatives 1–6.
| IC50 (μM)a |
||
|---|---|---|
| compd | oCOX-1 | mCOX-2 |
| 1 | >4 | >4 |
| 2 | >4 | >4 |
| 3 | >4 | 0.2 |
| 4 | >4 | 2.1 |
| 5 | >4 | >4 |
| 6 | 2.9 | >4 |
IC50 values were determined by incubating several concentrations of inhibitors or DMSO vehicle with purified murine COX-2 (63 nM) or ovine COX-1 (22.5 nM) for 20 min, followed by addition of [1-14C]-AA (5 μM) at 37 °C for 30 s. Assays were run in triplicate.
We evaluated all the compounds for their ability to inhibit 2-AG oxygenation selectively over AA oxygenation by COX-2. The evaluation was performed using an MS-based assay, as described above with added inhibitor concentrations up to 1 μM. Under these conditions, no compound reached its IC50 for COX-2-dependent AA oxygenation. Only compounds 3 and 6 exhibited substrate-selective inhibitory activity against 2-AG oxygenation, with IC50 values of 0.022 μM and 0.8 μM, respectively (Table 2). In addition, we evaluated compound 3 in the presence of both AA and 2-AG (see Supporting Information for details), where it showed substrate-selective COX-2 inhibitory activity against 2-AG oxygenation with an IC50 value of 0.145 μM.
Table 2. Substrate-Selective Inhibition of COX-2 by 1–6.
| IC50 (μM)a |
||
|---|---|---|
| compd | AA | 2-AG |
| 1 | >1 | >1 |
| 2 | >1 | >1 |
| 3 | >1 | 0.022 |
| 4 | >1 | >1 |
| 5 | >1 | >1 |
| 6 | >1 | 0.8 |
IC50 values were determined by incubating five concentrations of inhibitor and a solvent control in DMSO with purified COX-2 (40 nM) for 3 min followed by addition of arachidonic acid (AA) or 2-arachidonylglycerol (2-AG) 5 μM at 37 °C for 30 s.
To explore the structural basis for substrate-selective inhibition of COX-2 by compound 3, we obtained a 2.66 Å resolution X-ray crystal structure of the inhibitor in complex with the protein (PDB code 6V3R). Statistics of X-ray data collection and structure refinement is described in the Supporting Information (Table 1s). As seen in Figure 1, compound 3 binds in the cyclooxygenase active site of COX-2, resting above a “constriction” designated by Arg-120, Tyr-355, and Glu-524, as is the case for the vast majority of COX inhibitors. Notably, however, steric hindrance between the tricyclic indole core and Leu-531 induces a movement of the side chain of this amino acid relative to the position it occupies in most COX-inhibitor crystal structures. A similar movement of Leu-531 has been observed in other COX-2:ligand complexes, including COX-2:AA (PDB files 1CVU and 3HS5, unproductive binding mode),37,38 COX-2:eicosapentaenoic acid (PDB file 3HS6),38 COX-2:1-AG (PDB file 3MDL),30 COX-2:3(S)- methylarachidonic acid (PDB file 4RUT),21 and complexes with inhibitors of the oxicam class (PDB files 4M10, 4M11, and 4O1Z).39 In each case, displacement of the Leu-531 side chain helps to accommodate a bulky ligand or otherwise unfavorable binding pose. This finding suggests that movement of Leu-531 may play a role in substrate-selective inhibition, but such a displacement was not observed in the crystal structures of other such inhibitors [e.g., ibuprofen (PDB file 4RSO)40 and mefenamic acid (PDB file 5IKR).35 Furthermore, the oxicams induce a similar displacement of Leu-531, but they are not substrate-selective inhibitors. It is also notable that in the crystal structure, compound 3 is bound to both subunits of COX-2. Thus, the structure does not provide support for the hypothesis that substrate-selective inhibition results from binding of inhibitor to the allosteric site only. This may be the result of constraints present during crystallization, however, and may not reflect the behavior of the enzyme in the cellular environment.
Figure 1.

Stereodiagram of the X-ray cocrystal structure of compound 3 bound in the active site of COX-2 (PDB code 6V3R). An omit map is contoured at 3σ in black mesh. The key residues are illustrated with a stick representation of the final model (carbon in yellow, oxygen in red, nitrogen in blue), and compound 3 is colored in green.
In conclusion, we have described the design and synthesis of a novel series of harmaline analogs that are derived from the 4,9-dihydro-3H-pyrido[3,4-b]indole core. These compounds have been evaluated for their COX inhibitory activity with respect to both isoform- and substrate-selectivity. The SAR identified a fused tricyclic rigid indole derivative, compound 3, as a promising substrate-selective COX-2 inhibitor. The crystal complex of COX-2 with 3 revealed a movement of Leu-531 in the COX active site to accommodate inhibitor binding, suggesting that subtle structural changes in this region may contribute to its substrate-selectivity. This molecule will serve as a useful tool compound for further exploration of the biological relevance of COX-2-dependent endocannabinoid oxygenation.
Acknowledgments
Spectroscopic analysis of all new molecules was conducted in the Small Molecule NMR Facility and Mass Spectrometry Research Center at the Vanderbilt Institute of Chemical Biology. In addition, we are grateful to Dr. Carol A. Rouzer of the Vanderbilt Basic Sciences for critical reading and editing of the manuscript.
Glossary
Abbreviations
- COX
cyclooxygenase
- AA
arachidonic acid
- 2-AG
2-arachidonylglycerol
- PG
prostaglandin
- PG-G
prostaglandin glyceryl ester
- PGE2
prostaglandin E2
- PGE2-G
prostaglandin E2-G
- CB1 and CB2
cannabinoid receptors
- AEA
arachidnoylethanolamide
- PG-EA
prostaglandin ethanolamide
- EC
endocannabinoid
- POCl3
phosphorus oxychloride
- PPHP
5-phenyl-4-pentenyl-1-hydroperoxide.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00555.
Full synthetic procedures and analytical and spectral characterization data of the synthesized compounds (PDF)
This work was supported by research grants from (i) the National Institutes of Health (Grant CA89450 (L.J.M.)) and (ii) Northeastern Collaborative Access Team beamlines (funded by NIGMS Grants P30 GM124165, S10 RR029205) located in Advanced Photon Source (Grant DE-AC02-06CH11357).
The authors declare no competing financial interest.
Supplementary Material
References
- Dubois R. N.; Abramson S. B.; Crofford L.; Gupta R. A.; Simon L. S.; Van De Putte L. B.; Lipsky P. E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12 (12), 1063–73. 10.1096/fasebj.12.12.1063. [DOI] [PubMed] [Google Scholar]
- Dannenberg A. J.; Lippman S. M.; Mann J. R.; Subbaramaiah K.; DuBois R. N. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J. Clin. Oncol. 2005, 23 (2), 254–66. 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- Thun M. J.; Henley S. J.; Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst 2002, 94 (4), 252–66. 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Non-redundant functions of cyclooxygenases: oxygenation of endocannabinoids. J. Biol. Chem. 2008, 283 (13), 8065–9. 10.1074/jbc.R800005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M.; Buisseret B.; Paquot A.; Guillemot-Legris O.; Muccioli G. G. The endogenous bioactive lipid prostaglandin D2-glycerol ester reduces murine colitis via DP1 and PPARgamma receptors. FASEB J. 2018, 32 (9), 5000–5011. 10.1096/fj.201701205R. [DOI] [PubMed] [Google Scholar]
- Alhouayek M.; Masquelier J.; Cani P. D.; Lambert D. M.; Muccioli G. G. Implication of the anti-inflammatory bioactive lipid prostaglandin D2-glycerol ester in the control of macrophage activation and inflammation by ABHD6. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (43), 17558–63. 10.1073/pnas.1314017110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova I. A.; Uhelski M.; Khasabov S. G.; Gupta K.; Seybold V. S.; Simone D. A. Sensitization of nociceptors by prostaglandin E2-glycerol contributes to hyperalgesia in mice with sickle cell disease. Blood 2019, 133 (18), 1989–1998. 10.1182/blood-2018-11-884346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren C. A.; Newman Z. L.; Morford J. J.; Ryan S. B.; Battani K. A.; Su Z. Cyclooxygenase-2, prostaglandin E2 glycerol ester and nitric oxide are involved in muscarine-induced presynaptic enhancement at the vertebrate neuromuscular junction. J. Physiol. 2013, 591 (19), 4749–64. 10.1113/jphysiol.2013.256727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirodi C. S.; Crews B. C.; Kozak K. R.; Morrow J. D.; Marnett L. J. The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (7), 1840–5. 10.1073/pnas.0303950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman P.; Kaplan B. L.; Kaminski N. E. 15-Deoxy-Delta(1)(2),(1)(4)-prostaglandin J(2)-glycerol, a putative metabolite of 2-arachidonyl glycerol and a peroxisome proliferator-activated receptor gamma ligand, modulates nuclear factor of activated T cells. J. Pharmacol. Exp. Ther. 2012, 342 (3), 816–26. 10.1124/jpet.112.193003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman P.; Kaplan B. L.; Thompson J. T.; Vanden Heuvel J. P.; Kaminski N. E. 15-Deoxy-delta12,14-prostaglandin J2-glycerol ester, a putative metabolite of 2-arachidonyl glycerol, activates peroxisome proliferator activated receptor gamma. Mol. Pharmacol. 2011, 80 (1), 201–9. 10.1124/mol.110.070441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richie-Jannetta R.; Nirodi C. S.; Crews B. C.; Woodward D. F.; Wang J. W.; Duff P. T.; Marnett L. J. Structural determinants for calcium mobilization by prostaglandin E2 and prostaglandin F2alpha glyceryl esters in RAW 264.7 cells and H1819 cells. Prostaglandins Other Lipid Mediators 2010, 92 (1–4), 19–24. 10.1016/j.prostaglandins.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N.; Zhang J.; Chen C. PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J. Physiol. 2006, 572 (Part 3), 735–45. 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N.; Zhang J.; Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J. Neurochem. 2007, 102 (6), 1966–1977. 10.1111/j.1471-4159.2007.04668.x. [DOI] [PubMed] [Google Scholar]
- Valdeolivas S.; Pazos M. R.; Bisogno T.; Piscitelli F.; Iannotti F. A.; Allara M.; Sagredo O.; Di Marzo V.; Fernandez-Ruiz J. The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: a potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis. 2013, 4, e862. 10.1038/cddis.2013.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Zhang J.; Andreasson K.; Chen C. COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol. Cell. Neurosci. 2008, 37 (4), 682–95. 10.1016/j.mcn.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111 (10), 5899–921. 10.1021/cr2002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Duggan K. C.; Rouzer C. A.; Marnett L. J. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 2009, 48 (31), 7353–5. 10.1021/bi900999z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Sharma N. P.; Jurban B. J.; Smith W. L. Pre-existent asymmetry in the human cyclooxygenase-2 sequence homodimer. J. Biol. Chem. 2013, 288 (40), 28641–55. 10.1074/jbc.M113.505503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Vecchio A. J.; Sharma N. P.; Jurban B. J.; Malkowski M. G.; Smith W. L. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 2011, 286 (21), 19035–46. 10.1074/jbc.M111.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar S. N.; Nikas S. P.; Kingsley P. J.; Xu S.; Galligan J. J.; Rouzer C. A.; Banerjee S.; Ji L.; Eno M. R.; Makriyannis A.; Marnett L. J. 13-Methylarachidonic acid is a positive allosteric modulator of endocannabinoid oxygenation by cyclooxygenase. J. Biol. Chem. 2015, 290 (12), 7897–909. 10.1074/jbc.M114.634014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmacz R. J.; Lands W. E. Prostaglandin H synthase. Stoichiometry of heme cofactor. J. Biol. Chem. 1984, 259 (10), 6358–63. [PubMed] [Google Scholar]
- Kulmacz R. J.; Lands W. E. Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti-inflammatory agents. J. Biol. Chem. 1985, 260 (23), 12572–8. [PubMed] [Google Scholar]
- Mitchener M. M.; Hermanson D. J.; Shockley E. M.; Brown H. A.; Lindsley C. W.; Reese J.; Rouzer C. A.; Lopez C. F.; Marnett L. J. Competition and allostery govern substrate selectivity of cyclooxygenase-2. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (40), 12366–71. 10.1073/pnas.1507307112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimon G.; Sidhu R. S.; Lauver D. A.; Lee J. Y.; Sharma N. P.; Yuan C.; Frieler R. A.; Trievel R. C.; Lucchesi B. R.; Smith W. L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (1), 28–33. 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu R. S.; Lee J. Y.; Yuan C.; Smith W. L. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 2010, 49 (33), 7069–79. 10.1021/bi1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C.; Sidhu R. S.; Kuklev D. V.; Kado Y.; Wada M.; Song I.; Smith W. L. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J. Biol. Chem. 2009, 284 (15), 10046–55. 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan K. C.; Hermanson D. J.; Musee J.; Prusakiewicz J. J.; Scheib J. L.; Carter B. D.; Banerjee S.; Oates J. A.; Marnett L. J. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 2011, 7 (11), 803–9. 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A. J.; Kingsley P. J.; Mitchener M. M.; Altemus M.; Patrick T. A.; Gaulden A. D.; Marnett L. J.; Patel S. Detection of Cyclooxygenase-2-Derived Oxygenation Products of the Endogenous Cannabinoid 2-Arachidonoylglycerol in Mouse Brain. ACS Chem. Neurosci. 2018, 9 (7), 1552–1559. 10.1021/acschemneuro.7b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchio A. J.; Malkowski M. G. The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J. Biol. Chem. 2011, 286 (23), 20736–45. 10.1074/jbc.M111.230367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraiz T.; Gonzalez D.; Ancin-Azpilicueta C.; Aran V. J.; Guillen H. beta-Carboline alkaloids in Peganum harmala and inhibition of human monoamine oxidase (MAO). Food Chem. Toxicol. 2010, 48 (3), 839–45. 10.1016/j.fct.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Dos Santos R. G.; Bouso J. C.; Hallak J. E. C. Ayahuasca, dimethyltryptamine, and psychosis: a systematic review of human studies. Ther. Adv. Psychopharmacol. 2017, 7 (4), 141–157. 10.1177/2045125316689030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S.; Hu C. Pharmacological effects of rutaecarpine as a cardiovascular protective agent. Molecules 2010, 15 (3), 1873–81. 10.3390/molecules15031873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Blobaum A. L.; Kingsley P. J.; Gorden D. L.; McIntyre J. O.; Matrisian L. M.; Subbaramaiah K.; Dannenberg A. J.; Piston D. W.; Marnett L. J. Selective visualization of cyclooxygenase-2 in inflammation and cancer by targeted fluorescent imaging agents. Cancer Res. 2010, 70 (9), 3618–27. 10.1158/0008-5472.CAN-09-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando B. J.; Malkowski M. G. Substrate-selective Inhibition of Cyclooxygeanse-2 by Fenamic Acid Derivatives Is Dependent on Peroxide Tone. J. Biol. Chem. 2016, 291 (29), 15069–81. 10.1074/jbc.M116.725713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musee J.; Marnett L. J. Prostaglandin H synthase-2-catalyzed oxygenation of 2-arachidonoylglycerol is more sensitive to peroxide tone than oxygenation of arachidonic acid. J. Biol. Chem. 2012, 287 (44), 37383–94. 10.1074/jbc.M112.381202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer J. R.; Pawlitz J. L.; Moreland K. T.; Stegeman R. A.; Hood W. F.; Gierse J. K.; Stevens A. M.; Goodwin D. C.; Rowlinson S. W.; Marnett L. J.; Stallings W. C.; Kurumbail R. G. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405 (6782), 97–101. 10.1038/35011103. [DOI] [PubMed] [Google Scholar]
- Vecchio A. J.; Simmons D. M.; Malkowski M. G. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 2010, 285 (29), 22152–63. 10.1074/jbc.M110.119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Hermanson D. J.; Banerjee S.; Ghebreselasie K.; Clayton G. M.; Garavito R. M.; Marnett L. J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J. Biol. Chem. 2014, 289 (10), 6799–808. 10.1074/jbc.M113.517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando B. J.; Lucido M. J.; Malkowski M. G. The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 2015, 189 (1), 62–6. 10.1016/j.jsb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.