

SUMMARY
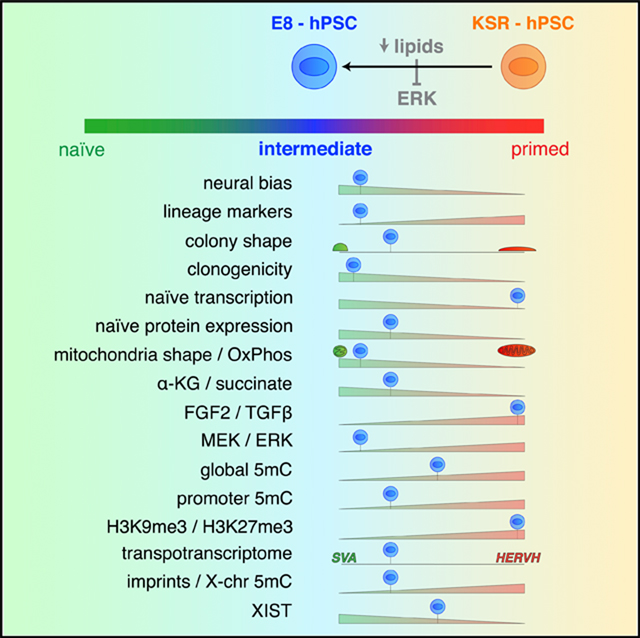
Current challenges in capturing naive human pluripotent stem cells (hPSCs) suggest that the factors regulating human naive versus primed pluripotency remain incompletely defined. Here we demonstrate that the widely used Essential 8 minimal medium (E8) captures hPSCs at a naive-to-primed intermediate state of pluripotency expressing several naive-like developmental, bioenergetic, and epigenomic features despite providing primed-state-sustaining growth factor conditions. Transcriptionally, E8 hPSCs are marked by activated lipid biosynthesis and suppressed MAPK/TGF-β gene expression, resulting in endogenous ERK inhibition. These features are dependent on lipid-free culture conditions and are lost upon lipid exposure, whereas short-term pharmacological ERK inhibition restores naive-to-primed intermediate traits even in the presence of lipids. Finally, we identify de novo lipogenesis as a common transcriptional signature of E8 hPSCs and the pre-implantation human epiblast in vivo. These findings implicate exogenous lipid availability in regulating human pluripotency and define E8 hPSCs as a stable, naive-to-primed intermediate (NPI) pluripotent state.
In Brief
The regulation of human naive pluripotency remains incompletely understood. Cornacchia et al. show that lipid-free culture conditions are sufficient to skew human pluripotent stem cells toward a naive-to-primed intermediate state via endogenous ERK inhibition. Transcriptomic analyses suggest that pluripotency regulation via de novo lipogenesis mimics in vivo regulation during pre-implantation development.
Graphical Abstract
INTRODUCTION
Human pluripotent stem cells (hPSCs) hold great promise for regenerative medicine and the study of disease. However, pluripotency is a metastable state, susceptible to multiple variables that affect downstream outcomes. Thus, elucidating the complexity of pluripotency regulation is key for realizing the full potential of stem cell technology. Two discrete pluripotent states have been reported, a naive (or ground) and a primed state, corresponding to the in vivo compartments of the pre-versus post-implantation epiblast (Boroviak et al., 2015). Extensive efforts have focused on the pursuit of human naive pluripotency (Chan et al., 2013; Gafni et al., 2013; Hanna et al., 2010; Takashima et al., 2014; Theunissen et al., 2014; Ware et al., 2014), a state that remains less defined than its mouse counterpart. Recently, an intermediate state of “formative” pluripotency has been proposed that combines loss of the naive transcriptional program with intermediate levels of DNA methylation (Smith, 2017).
Pre-implantation development can be recapitulated in vitro using relatively simple conditions (Biggers, 1998), suggesting that early cell fate decisions are primarily driven by embryo-intrinsic factors. Hence, the study of the human naive-to-primed transition has focused on endogenous cues, i.e. transcription factors (Hanna et al., 2010; Takashima et al., 2014) or cell signaling (Chan et al., 2013; Gafni et al., 2013; Theunissen et al., 2014). However, the peri-implantation window involves dramatic environmental changes that may affect cell fate specification, such as a switch in the nutrient source (Wang and Dey, 2006), to which the embryo responds by shifting its metabolic state and energy production routes (Houghton et al., 1996). A close link between cell metabolism and transcriptional-epigenetic regulation has been reported for both pluripotency and cancer, involving specific metabolites that act as co-factors for transcriptional and epigenetic effectors (Lu and Thompson, 2012; Shyh-Chang and Ng, 2017). An unresolved question is whether metabolic changes driven by alterations in the embryonic environment directly contribute to early cell fate transitions. Although a role of glucose and amino acid metabolism was established in regulating pluripotency (Carey et al., 2015; Gu et al., 2016; Shiraki et al., 2014; Villegas et al., 2019), little is known about the role of lipids in early embryonic cell fate decisions. Lipids represent a major energy source during early development (Sturmey et al., 2009), and before implantation they are derived from endogenous reserves of the oocyte and from de novo biosynthesis (Johnson et al., 2003; Leese, 2012). Thus, an important question is whether the dramatic changes in lipid availability at the peri-implantation stage affect the regulation of early cell fate decisions, including the naive-to-primed pluripotency transition, and whether such lipid-dependent effects on the pluripotent state are reflected in current hPSC culture conditions.
RESULTS
E8 hPSCs Show Increased Propensity for Neuroectodermal Differentiation
Traditional hPSC culture conditions employ knockout serum replacement (KSR) and murine embryonic fibroblasts (MEFs). These animal-derived components (xenobiotics) represent a source of experimental variability (Zimmer et al., 2016) and are unsuitable for clinical applications. Thus, the need for standardized, clinical-grade hPSC protocols has led to the formulation of chemically defined, “xeno-free” hPSC culture media. Essential 8 (E8) was one of the first of such media, designed to only include essential and defined components for the maintenance of human pluripotency (Chen et al., 2011). A widely used neural differentiation paradigm for hPSC is dual inhibition of SMAD signaling (Chambers et al., 2009), a high-efficiency strategy for deriving numerous neural cell types, including FOXG1+/TBR1+ cortical neurons. A major challenge for neural differentiation studies is the variable yield across different hPSC lines (Saha and Jae-nisch, 2009). Past work suggests that genetic background diversity of hPSC lines may drive differences in lineage propensity (Bocketal., 2011).
We observed that adaptation of KSR hPSC lines to E8 prior to differentiation enhanced neural yield in multiple human embryonic stem cell (hESC) and induced pluripotent stem cell (iPSC) lines that had shown highly divergent yields under conventional KSR conditions (Figure 1A; Figures S1A-S1D). These results indicate that some of the variability in neural efficiency across hPSC lines may be reversible and not genetically determined. Neural differentiation was overall enhanced in E8 hPSCs compared with conventional KSR hPSCs, as measured by absolute levels and kinetics of expression of the neural precursor marker PAX6 (Figure 1B). Neural induction protocols for E8 versus KSR hPSCs utilize separate base media during early differentiation (E6- versus KSR-based). We thus asked whether the increased neural yield in E8 hPSCs was dependent on differences at the pluripotent state or during differentiation. Although KSR hPSCs did not tolerate transfer into E6 during neural differentiation, E8 hPSCs differentiated in KSR-based medium yielded equal or higher levels of neural conversion compared with KSR hPSCs (Figures S1A and S1D; Figure 1A). These results indicate that pluripotent culture conditions rather than differentiation conditions were responsible for the increased neural propensity of E8 hPSCs. Neural efficiency in E8 also outperformed differentiation from hPSCs grown in other feeder-free media, such as mTeSR, regardless of whether KSR- or E6-based differentiation medium was used (Figure S1E). To assess whether improved differentiation in E8 was specific to neuroectodermal fates, we next compared the levels of mesoderm and endoderm induction between E8 and KSR hPSCs following either directed or spontaneous (embryoid body [EB]) differentiation. Although both strategies resulted in higher ectodermal marker expression in E8 hPSC cultures, as quantified in Figure 1C, the efficiency toward mesoderm or endoderm was slightly reduced in E8 versus KSR hPSCs (Figure S1F). We quantified lineage bias in E8 and KSR hPSCs using the ScoreCard assay (Bock et al., 2011), measuring a set of lineage markers expressed in EBs versus embryonic stem cells (ESCs). These results confirmed that E8 hPSCs exhibit a propensity toward neural and ectodermal fates (Figures 1D and 1E). However, despite differential germlayer bias during differentiation, at the pluripotent state, E8 hPSCs showed greatly reduced expression of markers of all three germlayers (PAX6, T, and SOX17) (Figure 1C; Figure S1F).
Figure 1. Neuroectodermal Bias of E8 hPSCs.

(A) Immunofluorescence of SOX1 and PAX6 on day 10 of neural induction in H9 and the iPSC line 036 (iPSC #1) grown in KSR or E8 medium.
(B) Kinetics of PAX6 expression relative to day 0 over 12 days of neural induction from KSR and E8 hPSCs. Mean ± SEM across 3 hESC lines (H9, HUES6, and MEL1; n = 3 replicates per line) and 3 iPSC lines (036, 706, and 731; n = 1 replicate per line) is shown. E8 versus KSR (ANOVA), p = 0.0013.
(C) Percentage of cells positive for PAX6 on day 10 of neural induction in KSR versus E8 hESCs (left). 7 or more replicate wells were quantified per line (H9, HUES6, and MEL1). Center: expression of PAX6 in day 7 embryoid bodies (EBs) generated from KSR or E8 hPSCs, n = 3 replicates per hESC line (H9, HUES6, and MEL1). Right: expression of PAX6 in undifferentiated cells, n = 3 replicates per hESC line (H9, HUES6, and MEL1). Mean ± SEM of 3 independent hESC lines is shown (H9, HUES6, and MEL1).
(D) Heatmap of lineage marker upregulation during spontaneous differentiation (EB) of KSR and E8 hPSCs generated by Nanostring nCounter(Bock et al., 2011). Values indicate log2 fold change of expression in EBs versus ESCs from three technical replicates of each line (H9, HUES6, and MEL1).
(E) Scorecard analysis of lineage propensity in KSR and E8 hPSCs (Bock et al., 2011).
In (B) and (C), gene expression was normalized to GAPDH. Scale bars, 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; two-tailed t test. See also Figure S1.
E8 hPSCs Display Enhanced Pluripotency Features and Expression of Naive Markers
Increased neuroectodermal and reduced meso- and endodermal differentiation bias combined with low lineage marker expression at pluripotency are features reported for naive mouse (Marks et al., 2012) and naive human (Kisa et al., 2017; Lee et al., 2017) pluripotent stem cells (PSCs). We thus asked whether the presence of those features in E8 hPSCs could reflect a differential pluripotency state. Although both E8 and KSR hPSCs exhibit pluripotency functions, such as teratoma formation (Figure S2A), E8 hPSCs are distinct from KSR hPSCs at multiple levels. First, E8 colonies are smaller and more compact and have a more dome-shaped appearance (Figure 2A). Second, E8 hPSCs exhibit faster proliferation rates (Figure 2B; Figures S2B and S2C) and a higher clonogenic capacity (Figures 2C and 2D; Figure S2D), indicative of improved self-renewal (Gafni et al., 2013; Takashima et al., 2014; Ware et al., 2014). Furthermore, although the general human PSC marker SSEA4 was expressed in 100% of E8 and KSR hPSCs (Figure S2E), the more stringent marker SSEA3 was enriched in E8 hPSCs (Figure 2E; Figure S2F). The mRNA levels of the core pluripotency markers OCT4 and NANOG were comparable (Figure S2G). However, the protein levels of both markers were increased in E8 hPSCs (Figures 2F–2J; Figures S2H and S2I). Particularly pronounced were the differences in NANOG protein, with KSR cells showing a heterogeneous distribution of NANOG expressing cells typical of primed PSC (Chambers et al., 2007), whereas E8-hPSC displayed uniformly high NANOG and increased global NANOG protein (Figures 2F and 2G; Figures S2H and S2I), as reported for naive PSCs (Muñoz Descalzo et al., 2012). These results were further validated by flow cytometry (Figures 2H–2J). MicroRNA (miRNA) profiling revealed a subset of repressed miRNAs in E8, including members of the let-7 family, known as negative regulators of pluripotency (Jun-Hao et al., 2016; Nguyen and Zhu, 2015; Figure 2K). Gene expression of the naive marker ESRRB (Figure 2L) was increased and that of the primed-state marker DNMT3B (Figure 2M) reduced in E8 hPSCs. Moreover, analysis of transposable elements (TEs) showed that, among the differentially regulated TEs in naive and primed states (Theunissen et al., 2016), E8 hPSCs showed decreased expression of primed state-associated HERVH (Figure 2N) and a moderate increase of naive state-associated SVA elements (Figure S2J; Table S1). We next derived naive hPSCs using published protocols (Gafni et al., 2013; Takashima et al., 2014) available as commercial kits (RSet and NaiveCult) and compared KSR, E8, RSet, and NaiveCult hPSCs for the expression of a panel of established naive markers (KLF4, KLF17, ESRRB, TFCP2L1, TFE3, STELLA). As expected, in both RSet and NaiveCult hPSCs, we identified positive colonies for most of these markers, whereas KSR cells were consistently negative (Figure S2K). Interestingly, E8 cells were positive for KLF4 (Figure 2O; Figure S2K) and showed high TFE3 expression (Figure 2Q; Figure S2K). Increased total KLF4 and TFE3 protein in E8 was confirmed by western blot analysis (Figures 2P and 2R). Importantly, as for NANOG and OCT4, increased expression of KLF4 and TFE3 in E8 was detectable at the protein level but not at the mRNA level (Figures S2G and S2L), indicating a post-transcriptional mode of regulation.
Figure 2. Naive-like Pluripotency Features of E8 hPSCs.

(A) Bright-field images of KSR and E8 hPSCs (H9). Scale bars, 250 μm.
(B) Growth curve of hESCs (H9) in KSR or E8.
(C) Representative colony formation after single-cell passaging of KSR or E8 hPSCs (HUES6), staining with crystal violet.
(D) Quantification of colony formation after single-cell passaging of KSR or E8 hPSCs with or without Y-27632 (+ROCK-i). Mean ± SEM of 3 technical replicates per line (H9, HUES6, and MEL1) is shown.
(E) Flow cytometry of SSEA3+ cells in KSR and E8 hPSCs. Single dots represent technical replicates (H9, HUES6, and MEL1).
(F) Immunofluorescence of NANOG in KSR and E8 hPSCs (MEL1).
(G) Western blot of NANOG and OCT4 in KSR and E8 hPSCs (MEL1). Loading: LAMIN B1.
(H) Representative flow cytometry plot of intracellular NANOG and OCT4 expression (top) and isotype control analysis (bottom) in KSR and E8 hESCs (H9).
(I and J) Flow cytometry of NANOGhigh/OCT4high (I) and OCT4high/SOX2high (J) populations in KSR and E8 hESCs; mean ± SEM is shown (H9, HUES6, and MEL1).
(K) Fold change of pluripotency-related miRNAs in E8 versus KSR hESC (H9, HUES6, and MEL1).
(L-N) Expression level (RNA-seq) of (L) ESRRB, (M) DNMT3B, and (N) HERVH in KSR and E8 hESCs (adjusted p value [padj] determined by differential gene expression analysis based on the negative binomial distribution [DESeq2]); mean ± SEM is shown (H9, HUES6, and MEL1).
(O-R) Immunofluorescence (O and Q) and western blot (P and R) of KLF4 (O and P) and TFE3 (Q and R) in KSR and E8 hESCs (H9). Two-tailed t test; *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2 and Table S1.
E8 Medium Converts Mitochondrial Morphology and Bioenergetics to a Naive State
Naive and primed pluripotent states have distinct mitochondrial and bioenergetic profiles (Sperber et al., 2015; Takashima et al., 2014; Zhou et al., 2012). In naive cells, mitochondrial energy production is bivalent (i.e., reliant on both oxidative phosphorylation [OxPhos] and glycolysis), whereas it is exclusively glycolytic in the primed state. Aerobic respiration is reactivated upon differentiation. In parallel, mitochondrial morphology evolves from a small and round shape in naive cells to an elongated structure with complex cristae in primed PSCs and differentiated cells (Zhang et al., 2011; Zhou et al., 2012). We examined mitochondrial morphology in E8 and KSR hPSCs by electron microscopy and observed that mitochondria in KSR hPSCs were large and elongated with distinct cristae, whereas mitochondria in E8 hPSCs were significantly smaller and round with sparse cristae (Figures 3A and 3B; Figure S3A). Immature mitochondrial morphology in E8 was associated with higher mitochondrial potential, measured by tetramethylrhodamine ethyl ester (TMRE) (Figure 3C) and oxygen consumption rates (OCRs) (Figure 3D). Base OCR and OCR after treatment with the uncoupling agent Carbonyl cyanide 4- (trifluoromethoxy)phenylhydrazone (FCCP) (Figure 3D) were significantly elevated in E8 hPSCs, consistent with higher basal levels of aerobic respiration (OxPhos) and absolute mitochondrial capacity. Higher mitochondrial activity in E8 was not associated with changes in mtDNA copy number (Figure 3E).
Figure 3. Naive Mitochondrial Morphology and Bioenergetics in E8 hPSCs.

(A) Transmission electron microscopy (TEM) of mitochondria in KSR and E8 hESCs (H9). Scale bars, 0.5 μm.
(B) Quantification of mitochondrial length in KSR and E8 hESCs.
(C) Quantification of mitochondrial potential in KSR and E8 hESCs by TMRE relative to average KSR.
(D) Quantification of oxygen consumption rate (OCR) in untreated KSR and E8 hESCs (base) in response to oligomycin (oligo), FCCP, and potassium cyanide (KCN).
(E) mtDNA copy number ratio to nuclear DNA in KSR and E8 hESCs.
(F) Scheme of alternative energy production pathways.
(G) Expression (RNA-seq) of LDHA in KSR and E8 (padj determined by DESeq2). Mean ± SEM (H9, HUES6, and MEL1).
(H) Ratio of intracellular lactate to pyruvate in KSR versus E8 hESCs. Mean ± SEM (H9, HUES6, and MEL1).
(I) Immunofluorescence of NANOG in KSR and E8 hESCs (H9) after 5-day treatment with 0, 2 mM, or 4 mM 2-deoxyglucose (2-DG). Scale bars, 200 μm.
Two-tailed t test; *p < 0.05, **p < 0.01, ***p < 0.001.
Consistent with elevated aerobic glycolysis in KSR, we found increased expression of lactate dehydrogenase (LDHA) (Figures 3F and 3G) and higher intracellular lactate to pyruvate ratios in KSR hPSCs (Figure 3H). Finally, E8 hPSCs showed higher tolerance for the glycolysis inhibitor 2-deoxyglucose (2-DG), similar to naive PSCs (Takashima et al., 2014; Zhou et al., 2012), with E8 hPSCs retaining expression of NANOG in the presence of 4 mM 2-DG, a concentration at which KSR hPSCs became fully differentiated (Figure 3I).
Activated Lipid Metabolism and Suppressed MAPK/TGF-β Signaling Mark the E8 Transcriptome
We performed total RNA sequencing (RNA-seq) on three independent hESC lines (H9, HUES6, and MEL1) grown in either KSR or E8 medium. Despite the genetic diversity of these lines, unsupervised hierarchical clustering segregated transcriptomes according to culture condition (Figure 4A). Pathway enrichment analysis revealed lipid metabolism and biosynthesis as the most differential categories between E8 and KSR, followed by signaling pathways, in particular mitogen-activated protein kinase (MAPK) and transforming growth factor β (TGF-β) (Figure 4B; Table S2). Transcriptional changes in lipid metabolism were corroborated by electron microscopy and oil red O staining, showing that E8 hPSCs are devoid of intracellular lipid storage, in contrast to the abundant lipid vesicles visible in KSR hPSCs (Figure 4C; Figure S3B). Accumulation of fat droplets has been reported in primed mouse epiblast stem cells (EpiSCs) and hESCs but not in naive PSCs (Sperber et al., 2015). Lipid biosynthesis is controlled by the SREBF1/2 transcription factors, which respond to decreased levels of exogenous fatty acids and intracellular cholesterol by activating de novo lipogenesis genes (Osborne, 2000; Figure 4D). Expression of SREBF1/2 and most SREBF1/2 target genes was significantly increased in E8 hPSCs (Figures 4E and 4F), in line with a pathway analysis showing activated lipogenesis (Figure 4B). A known route of SREBF induction is via mTOR signaling (Peterson et al., 2011). We thus assayed the activation state of mTOR and of its downstream effectors AKT, S6K, p70, and 4EBP1 in E8 and KSR but observed that mTOR signaling is repressed in E8 hPSCs (Figures S3C-S3E) and, therefore, unlikely to mediate upregulation of SREBF1/2 in E8 hPSCs.
Figure 4. Activated Lipid Biosynthesis and Suppressed MAPK and TGF-β Gene Expression in E8 hPSCs.

(A) Unsupervised hierarchical clustering of total RNA-seq profiles from SSEA4+-sorted KSR or E8 hESCs.
(B) Pathway enrichment (KEGG) of the top 500 differentially expressed genes (DEGs) in KSR versus E8 hESCs.
(C) TEM images of KSR and E8 hESCs (MEL1). Red arrows indicate fat droplets (top). Scale bar 2 μm. Oil Red staining of intracellular lipids (bottom) in KSR and E8 hESC (MEL1). Scale bars, 20 μm.
(D) Illustration of lipogenic gene regulation through SREBF1/2 in response to low-lipid conditions.
(E) Expression (RNA-seq) of SREBF1 and SREBF2 in KSR and E8 hESCs. Mean transcripts per million (TPM) values ± SEM of (H9, HUES6, and MEL1).
(F) Heatmap of log2-based fold change E8 versus KSR of SREBF1/2 target genes that are differentially expressed in E8 versus KSR hESCs (H9, HUES6, and MEL1). Blue indicates upregulation in E8 and orange upregulation in KSR.
(G) Heatmap of log2-based fold change E8 versus KSR for MAPK and TGF-β signaling. Blue indicates upregulation in E8 and orange upregulation in KSR.
(H) Expression (RNA-seq) of the MAPK inhibitor phosphatases DUSP5, DUSP6, DUSP7, and SPRY4 in KSR and E8 hESCs (padj determined by DESeq2). Mean values ± SEM of hESC H9, HUES6, and MEL1 are shown.
(I) Western blot of phospho- and total ERK in KSR and E8 hESCs.
See also Figure S3.
The second key transcriptional signature of E8 hPSCs was decreased MAPK and TGF-β signaling (Figure 4B), with most MAPK- and TGF-β-related genes being downregulated in E8 (Figure 4G). In vivo, fibroblast growth factor (FGF)-MAPK signaling is associated with exit from pluripotency and initiation of lineage priming (Lanner et al., 2010). In vitro, FGF-MAPK and TGF-β signaling have been shown to sustain primed pluripotency, and, conversely, repression of FGF-MAPK signaling through ERK inhibition is key for reverting PSCs to a naive state (Hackett and Surani, 2014). In addition, we found significant upregulation of DUSP-family and SPRY4 MAPK inhibitory phosphatases (MPKs) in E8 (Figure 4H), including ERK-specific DUSP6. Increased DUSP expression has been shown to mark the naive state (Gafni et al., 2013). We next assessed the levels of ERK activation by western blot and found that, in E8, phospho-ERK was nearly undetectable compared with KSR (Figure 4I). These findings were surprising because E8 contains a 10-fold higher concentration of FGF2 (100 ng/mL) than KSR medium (10 ng/mL), and FGF signaling is thought to maintain primed pluripotency via ERK activation (Lanner and Rossant, 2010). Our results thus indicate that, despite exposure to high levels of primed-state-sustaining growth factors, E8 hPSCs repress TGF-β and FGF-MAPK-ERK signaling, paradoxically mimicking naive cytokine requirements.
The Epigenetic State of E8 hPSCs Reflects Adaptations toward Increased Lipid Biogenesis
The main molecular precursor for fatty acid and cholesterol biosynthesis is acetyl-coenzyme A (CoA), generated from glycolytic pyruvate in the mitochondria. Lipogenic acetyl-CoA is derived from citrate of the tricarboxylic acid (TCA) cycle exported to the cytosol and cleaved to acetyl-CoA and oxaloacetate (Figure 5A; Figure S3F). Alternatively, acetyl-CoA for lipid synthesis can be obtained from glutamine via IDH1 through reductive carboxylation (Metallo et al., 2011; Figure S3G). In line with increased lipogenic activity in E8 hPSCs, genes involved in acetyl-CoA production, such as the citrate shuttle SLC25A1, the ATP-citrate lyase ACLY, as well as ACSS2 and ACAT2, were increased in E8 (Figure S3H). Similarly, IDH1 and ACO1, key enzymes for reductive carboxylation, were upregulated in E8 (Figure S3I). These expression patterns suggest increased acetyl-CoA metabolism in E8, which was confirmed by quantification of intracellular acetyl-CoA (Figure 5B). Acetyl-CoA and other TCA metabolites have additional roles as epigenetic regulators (Lu and Thompson, 2012; Ryall et al., 2015). As the main acetyl-group donor, acetyl-CoA intracellular levels have been shown to regulate histone acetylation (Cai et al., 2011). Analysis of global histone acetylation revealed hyperacetylation in E8 hPSCs for H3K27Ac, H3K9Ac, and H4K8Ac (Figure 5C; Figure S4A), concomitant with increased histone acetyltransferase KAT7 and decreased histone deacetylases SIRT1 and HDAC1 expression (Figures S4B and S4C).
Figure 5. The Epigenome of E8 hPSCs Reflects Increased Lipogenesis.

(A) Scheme of crosstalk between metabolism and epigenetics. Lipogenic metabolites (acetyl-CoA) and their precursors (α-KG) epigenetically regulate chromatin acetylation and methylation.
(B) Quantification of acetyl-CoA in KSR and E8 hESCs by Liquid-Chromatography-Mass Spectrometry (LC-MS). Single dots represent technical replicates (H9, HUES6, and MEL1).
(C) Western blot of H3K27Ac, H4K8Ac, and H3K9Ac in KSR and E8 hESCs (H9).
(D) Intracellular ratios of α-KG to succinate in KSR versus E8 hESCs.
(E) ELISA quantification of global 5mC in KSR and E8 hESCs as a ratio to KSR.
(F) Scheme of the methionine cycle of one-carbon metabolism. The methyl group donor S-adenosylmethionine (SAM) is converted to S-adenosylhomocysteine (SAH) upon donation of a methyl group for protein and DNA methylation.
(G) Intracellular ratios of SAM to SAH in KSR and E8 hESCs.
(H) Whole-genome bisulfite sequencing (WGBS) quantification of the average global 5mC rate (H9, HUES6, and MEL1).
(I) Violin plots of the 5mC distribution rate at CpGi promoter regions and the 5′ UTR in KSR and E8 hESCs.
(J) 5mC rate at the KLF4 promoter in KSR and E8 hPSCs.
(K) 5mC rate at X chromosome CpGi promoters in each line, E8, and KSR (XX, female; XY, male).
(L) Heatmap of 5mC differences between E8 and KSR at differentially methylated imprinted genes; values represent the average of H9, HUES6, and MEL1.
(M and N) Principal-component analysis (PCA) of H3K27Ac (M) and H3K27me3 (N) ChIP-seq profiles in KSR and E8 hESCs; each dot represents one line (H9, Hues6, and MEL1).
(O and P) Top enriched functional categories (Kyoto Encyclopedia of genes and genomes [KEGG]/Gene Ontology [GO]) of differentially H3K27Ac (O) and H3K27me3 (P) occupied gene promoters between KSR and E8.
(Q and R) Snapshot of H3K27Ac (Q) and H3K27me3 (R) enrichment at the DUSP6 gene in KSR and E8 hESCs.
In (D), (E), and (G), mean values ± SEM (H9, HUES6, and MEL1) are shown. Two-tailed t test; *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4 and Table S3.
Another epigenetically active TCA metabolite with roles in pluripotency is α-ketoglutarate (α-KG), which acts as co-substrate for the JMJD and TET chromatin demethylases (Loenarz and Schofield, 2008). Because α-KG is converted to succinate, shifts in α-KG or succinate availability were shown to affect histone and DNA methylation (Figure 5A), where high α-KG to succinate ratios promote naive pluripotency in mESCs (Carey et al., 2015). Our measurements of intracellular α-KG and succinate showed an increased α-KG to succinate ratio in E8 (Figure 5D). We thus analyzed global DNA and histone methylation. ELISA quantification of total 5mC revealed partial DNA hypomethylation in E8 (Figure 5E). In contrast, we did not observe any significant alterations in overall histone methylation (Figure S4D).
Chromatin methylation is also metabolically regulated by the methionine cycle of one-carbon metabolism, a source of the universal methylation donor S-adenosylmethionine (SAM). SAM is converted by donation of a methyl group to S-adenosylhomocysteine (SAH) (Figure 5F), a methyltransferase inhibitor. Intracellular ratios of SAM to SAH have been shown to reflect cellular methylation potential, with low ratios predicting hypomethylation (Chen et al., 2010; Iglesias Gonzalez et al., 2015). We observed a significantly reduced SAM to SAH ratio in E8 hPSCs, consistent with partial DNA hypomethylation (Figure 5G).
DNA hypomethylation in naive PSC is enriched at promoter regions and gene bodies (Ficz et al., 2013; Habibi et al., 2013). We performed whole-genome bisulfite sequencing (WGBS) to identify hypomethylated genomic regions in E8. WGBS confirmed reduced genome-wide CpG methylation rates in E8 PSCs (Figure 5H) and showed that the most hypomethylated genomic compartments were CpG island promoters and 5’ UTRs (Figure 5I; Figure S4E). Other hypomethylated regions in E8 encompassed the KLF4 promoter (Figure 5J), CpGi promoters of the X chromosome (Figures 5K and S4F), and imprinted genes (Figure 5L), partly matching the reported epigenomic features of naive hPSCs (Theunissen etal., 2016). Interestingly, we detected increased expression of XIST in the female hPSC lines H9 and HUES6 in E8, although the fold changes between E8 and KSR were strongly divergent between the two lines (Figure S4G).
Next we performed chromatin immunoprecipitation sequencing (ChIP-seq) for H3K4me3, H3K9Ac, H3K9me3, H3K27Ac, H3K27me3, and H4K8Ac. H3K27Ac and H3K27me3 showed the most divergent profiles between E8 and KSR hPSCs (Figures 5M and 5N). Pathway analysis of differential H3K27Ac- and H3K27me3-marked genes (Figures 5O and 5P) showed a strong concordance with transcriptional categories (Figure 4B). Specifically, TGF-β- and MAPK-related genes were enriched forH3K27me3in E8 but for H3K27Acin KSR hPSCs. Conversely, MAPK inhibitors were marked by H3K27Ac in E8 but H3K27me3 in KSR (Table S3). In line with its expression pattern, DUSP6 was marked by H3K27Ac in E8 (Figure 5Q) and by H3K27me3 in KSR (Figure 5R). Finally, functional analysis of differential 5mC-, H3K27Ac-, and H3K27me3- enriched genes revealed metabolism and development as top categories for all three marks (Figure S4H). These results indicate that, in E8 hPSCs, epigenetic and transcriptional processes act in concert to establish an alternative metabolic and developmental state.
Naive-like Features in E8 Are Induced by Lipid Deprivation
Because our findings establish that hPSC cultured in E8 acquire several naive-like traits, we asked how the formulation of E8 could trigger these changes. Two critical differences between E8 and KSR media are (1) the presence in KSR of lipid-rich BSA (Albumax) and (2) a 10-fold higher concentration of FGF2 in E8 (Figure S5A). FGF-mediated ERK signaling is the main pathway to sustain primed pluripotency (Lanner and Rossant, 2010). We thus tested the effects of FGF2 concentration on ERK signaling. Changes in FGF2 concentration did not affect ERK activation (Figure S5B) despite E8 hPSCs requiring FGF2 to maintain high proliferation rates (data not shown). Albumax (AX) is the most abundant component of the KSR supplement and an extrinsic source of lipids. E8 does not contain KSR and is essentially lipid-free. Supplementation of E8 medium with 1.6% w/v AX led to a rapid (<12 h) change in colony shape that was fully reversible upon AX withdrawal (Figure 6A). Cells maintained similar expression of pluripotency markers (Figure S5C) and could be serially passaged in the presence of AX. However, AX treatment led to downregulation of SREBF1 (Figure 6B), indicating that SREBF1 in E8 is likely induced by low extrinsic lipids. Importantly, AX treatment reduced DUSP6 expression, suggesting that ERK signaling also responds to exogenous lipids (Figure 6C). In fact, ERK was re-activated in E8+AX because phospho-ERK levels equaled those in KSR hPSCs (Figure 6D; Figure S5D).
Figure 6. Lipid Deprivation in E8 Induces Naive-like Features.

(A) Bright-field images of colony morphology in E8 (E8), after AX treatment (E8+AX) and AX withdrawal (E8+AX → E8). Scale bars, 250 μm.
(B) qRT-PCR of SREBF1 in E8 and E8+AX relative to SMU (Eisenberg and Levanon, 2013). The E8+AX bar represents mean ± SEM of H9, HUES6, and MEL1; values are shown as a ratio to E8 for each line.
(C) qRT-PCR of DUSP6 expression in E8 and E8+AX relative to GAPDH. The E8+AX bar represents mean ± SEM of H9, HUES6, and MEL1; values are shown as a ratio to E8 within each line.
(D) Western blot of phospho-ERK in KSR, E8, and E8 + AX (MEL1). Loading controls: ERK and γ–tubulin.
(E) Western blot of H3K27Ac and H4K8Ac in KSR, E8, and E8+AX hESCs (H9). Loading controls: total H3, H4, and LAMIN B1.
(F) Kinetics of SOX1 expression during neural induction in KSR and E8 and E8+AX hPSCs. Mean values ± SEM of H9, HUES6, and MEL1 are shown.
(G) Top: bright-field images. Bottom: oil red O staining of E8 hESCs (E8) after treatment with AX (+AX), fatty-acid free BSA (+ffBSA), deproteinized AX (+dep AX), and chemically defined lipid concentrate (+CDLC). Top scale bars, 250 μm; bottom scale bars, 50 μm.
(H) qRT-PCR of SREBF1 in E8, E8+AX, E8+ffBSA, E8 +dep AX, and E8 +CDLC. Mean values and SEM of H9, HUES6, and MEL1 are shown, normalized to the E8 control within each cell line.
(I) Western blot of NANOG phospho-ERK and TFE3 levels in E8, E8+AX, E8+ffBSA, E8 +dep AX, and E8 +CDLC.
(J) Western blot of phospho-ERK, H4K8Ac, and TFE3 in KSR and KSR + 1 μM PD0325901.
(K) Western blot of NANOG, H4K8Ac, and TFE3 in E8, E8+AX (EAX), and E8+AX+1 μM PD0325901 (EAX+PD).
(A–H) Length of treatment and/or withdrawal from treatment: 3 days.
Two-tailed t test; ANOVA for (F): *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
AX treatment also reversed histone hyperacetylation, reducing E8 levels of H3K27Ac and H3K8Acto match those in KSR hPSCs (Figure 6E; Figure S5E). In contrast, we could not detect any effect of AX on global DNA methylation (data not shown). Finally, AX treatment affected lineage propensity in E8 hPSCs, abrogating neural differentiation bias (Figure 6F).
To discern whether the effects on pluripotency were mediated by the protein or lipid fraction of AX, we tested protein-only or lipid-only fractions of AX and chemically defined lipid concentrate (CDLC). Treatment of E8 hPSCs with fatty acid-free BSA was unable to trigger the morphological changes elicited by both AX and lipid-only (deproteinized) AX, implicating the lipid (i.e., fatty acid fraction) as the active agent. Unexpectedly, treatment with CDLC did not affect colony morphology. However, staining with the intracellular lipid dye oil red O revealed that the morphological phenotype of AX only appeared under conditions where lipids were absorbed, which did not occur upon CDLC treatment (Figure 6G). The lack of intracellular lipids and of a phenotype in E8+CDLC cells indicates the need for a lipid carrier such as BSA for lipid import and suggested that intra-rather than extracellular lipids alter cell state. Accordingly, expression of the lipogenic factor SREBF1 was only reduced after AX and deproteinized AX treatment (Figure 6H), as were other AX-mediated changes in naive-like features of E8 hPSCs, such as high NANOG and TFE3 expression and ERK inhibition (Figure 6I). Thus, our results implicate the lipid rather than protein fraction of AX in repressing naive-like traits in E8+AX cells. Conversely, the naive-like state in E8 hPSCs, including suppression of ERK signaling, depends on the lack of an extrinsic lipid source. Finally, we asked whether naive-like traits under lipid-deprived conditions were driven by ERK inhibition. Indeed, treatment with the ERK inhibitor PD0325901 at concentrations typically used for establishing and maintaining the naive state (1 μM) induced histone hyperacetylation in both KSR and E8+AX hPSCs (Figures 6J and 6K; Figure S5F) and restored high NANOG and TFE3 expression (Figure 6K). These data indicate that naive traits in E8 are mediated at least in part via intrinsic ERK inhibition. Moreover, the effect of ERK inhibition on histone acetylation implies that ERK suppression may be upstream of the generation of lipogenic precursors such as acetyl-CoA. Interestingly, we found that, under lipid-deprived conditions (E8), but not under high-lipid conditions (KSR/E8+AX), hPSCs were highly susceptible to pharmacological inhibition of SREBF1 and loss of ERK inhibition (Figures S5G and S5H).
Activated Lipid Metabolism Is a Conserved Transcriptional Signature of Naive Pluripotency
Our data implicate exogenous lipid deprivation in reverting hPSCs to a naive-to-primed intermediate state of pluripotency. We thus asked whether activated lipid metabolism is a common trait of naive pluripotency in vitro and in vivo. We correlated transcriptional data of E8 and KSR hPSCs with four existing datasets of naive versus primed mouse and human pluripotency, comprising 2 inhibitor (2i) versus serum-LIF mESCs (Marks et al., 2012), “reset” versus conventional hESCs (Takashima et al., 2014), pre-versus post-implantation mouse epiblast (Boroviak et al., 2015), and late human blastocysts versus hESCs (Yan et al., 2013). First, we analyzed differentially expressed genes (DEGs) between the corresponding naive versus primed datasets and identified functional categories of DEGs upregulated in either naive or primed states (Table S4). In all datasets, genes associated with lipid biosynthesis and metabolism were upregulated in naive cells (Tables S4 and S7). Next we identified common DEGs upregulated in E8 and each of the 4 naive datasets or upregulated in KSR and 4 primed datasets (Table S5). Strikingly, shared DEGs between E8 and any of the four analyzed naive datasets matched almost exclusively to lipid biosynthesis and metabolism categories (Figure S6A). In contrast, shared DEGs between KSR and primed datasets were enriched for cell signaling, differentiation, and developmental terms (Figure S6B). We next asked whether regulation of lipid metabolism is a transcriptional signature diagnostic of distinct pluripotent states. We generated a list of lipid-related genes that were differentially expressed in at least four of the five datasets (E8 versus KSR and four naive versus primed studies) (Table S6), irrespective of the direction of regulation (143 genes). Unsupervised hierarchical clustering of these genes showed clear segregation of naive and primed datasets, where E8 hPSCs clustered with naive and KSR hPSCs with primed samples (Figure 7A). Furthermore, we asked whether expression of signaling-related genes could segregate transcriptional profiles of naive and primed pluripotency. Again, we selected signaling-related DEGs (214 genes) differentially expressed in at least four of the five analyzed datasets, regardless of the direction of expression. Unsupervised clustering segregated naive-E8 hPSC versus primed-KSR hPSC datasets, indicating that transcriptional regulation of cell signaling-related genes can define distinct pluripotent states (Figure 7B).
Figure 7. Activated Lipid Biosynthesis Is a Shared Signature of E8 hPSCs and the Human Pre-EPI.

(A) Unsupervised hierarchical clustering of lipid-related DEGs (143 genes) in 4 of 5 naive versus primed studies (E8 versus KSR; Boroviak et al., 2015; Marks et al., 2012; Takashima et al., 2014; Yan et al., 2013).
(B) Unsupervised hierarchical clustering of signaling-related DEGs (214 genes) in 4 of the 5 naive versus primed studies (E8 versus KSR).
(C) Correlation plot of GO/KEGG pathway regulation in E8 versus KSR (y axis) and in human pre-EPI versus hESCs (x axis) (Yan et al., 2013). Each dot represents the average t-score of pathway enrichment for each GO term or KEGG pathway. Colored dots are pathways associated with lipid metabolism or cell signaling.
(D) Model of in vitro and in vivo regulation of human pluripotency through exogenous lipids. Low exogenous lipids (yellow) during pre-implantation lead to depletion of zygote-derived storage (purple), promoting de novo lipogenesis (blue). Low intracellular lipids endogenously inhibit ERK signaling, instructing naïve features (green). A lipid-rich post-implantation environment through maternal exchange (yellow) leads to intracellular lipid accumulation (purple) and ERK activation, suppressing de novo lipogenesis and promoting primed features (red).
We next analyzed specific pathways that are similarly regulated in E8 versus KSR and naive versus primed datasets. For lipid metabolism, we separately analyzed lipid anabolism-biosynthesis versus lipid catabolism-consumption and response to lipids. We performed differential gene expression analysis for each of the four datasets and intersected the resulting list of DEGs with the list of DEGs in E8 versus KSR. Next we conducted gene function-pathway enrichment analysis using DEG values from E8 versus KSR and from each naive versus primed study. We calculated the average t-scores with positive values representing pathway upregulation in E8 or naive and positive values representing upregulation in KSR or primed states (Figure 7C; Figure S7). The comparison between E8 versus KSR and the human pre- and post-implantation study (Yan et al., 2013) revealed a strong correlation for increased lipid biosynthesis in both E8 and naive and increased MAPK/TGF-β signaling in KSR and the primed state (Figure 7C). Similar trends were observed when comparing E8 versus KSR with each of the other 3 studies (Figure S7), albeit to lesser extent. This analysis indicates that activated lipid anabolism-biosynthesis is a shared transcriptional trait of E8 and the human pre-implantation epiblast (pre-EPI) (Figure 7C). In contrast, lipid catabolism-consumption and response to lipids are downregulated in human pre-EPI and E8 (Figure 7C).
These results suggest that the human pre-implantation embryo performs extensive de novo lipogenesis in response to low levels of exogenous lipids. We propose that lipid availability contributes to regulating the transition from naive-to-primed pluripotency when switching from de novo lipid biosynthesis before implantation to a rich extrinsic maternal lipid supply following implantation (Johnson et al., 2003; Leese, 2012). The acquisition of naive-like features in a low-lipid environment appears to be mediated in part by intrinsic ERK suppression (Figure 7D).
DISCUSSION
Extrinsic ERK and GSKβ inhibition (2i condition) is dominant in establishing a naive pluripotent state even in the presence of serum (Carey et al., 2015; Finley et al., 2018; Habibi et al., 2013). Complete suppression of ERK signaling via small molecules does not reflect normal physiology. In contrast, ERK inhibition via modulation of environmental cues may more closely reflect the conditions in the developing embryo at the peri-implantation stage. Future studies will have to address whether intrinsic ERK inhibition can mediate all of the pluripotent characteristics observed in E8 hPSCs, including mitochondrial and bioenergetic phenotypes, and whether manipulations of lipid availability can be used to affect pluripotent development in vivo. Moreover, it will be important to better define the specific subclasses of fatty acids and other lipids in modulating ERK signaling and acquisition of naive-to-primed intermediate features in E8 hPSCs.
Our results indicate that lipid scarcity stimulates the SREBF pathway in E8 hPSCs and that the resulting production of lipid precursors includes epigenetically active metabolites, such as α-KG and acetyl-CoA. These compounds are strong candidates for mediating the changes in the global epigenetic landscape of E8 PSCs, such as DNA hypomethylation and histone hyperacetylation. Glycolytic flux of acetyl-CoA to fuel histone acetylation is also crucial for maintaining pluripotency because its suppression reflects the metabolic switch demarcating the exit from pluripotency toward differentiation (Moussaieff et al., 2015). It should be noted that α-KG to succinate ratios appear to have contrasting roles in primed versus naive PSCs, with high ratios promoting differentiation of human PSCs and mouse EpiSCs (TeSlaa et al., 2016) and promoting self-renewal in mESC (Carey et al., 2015). In the current study, we did not directly assess the effects of α-KG in E8 PSCs but, rather, report on its regulation in response to low extrinsic lipid availability. Future studies should address whether E8-mediated effects on pluripotency are related to increased α-KG levels. Our metabolic data are compatible with the distinct bioenergetic state of hPSCs grown in E8 medium described previously (Zhang et al., 2016) in a study reporting a central role for enhanced lipid biosynthesis in driving E8-related metabolic changes. Our current study supports those findings and demonstrates that the cascade initiated by exogenous lipid scarcity has far-reaching repercussions for the human pluripotent state.
Our transcriptional analysis of in vitro and in vivo pluripotent datasets confirms that activated lipid metabolism is a conserved feature in naive mouse and human PSCs and human pre-implantation embryos. Moreover, we observe enrichment of functional terms related to increased lipid biogenesis and decreased lipid oxidation and response to lipids in human pre-EPI data. However, we did not find similar changes in the mouse pre-EPI. One possible explanation for this discrepancy is the differences in the amount of initial lipid storage in the zygote and oocyte across species, with very high lipid levels in porcine, bovine, and ovine oocytes, intermediate levels in human oocytes, and very low levels in murine oocytes (Dunning et al., 2014; McKeegan and Sturmey, 2011). It is tempting to speculate that such species-specific differences contribute to the challenges in capturing the naive pluripotent state in human or bovine embryos. Nevertheless, we find that lipid metabolism is differentially regulated between all mouse and human naive and primed state datasets in vitro, indicating a considerable degree of conservation under comparable culture conditions. Our work indicates that manipulating lipid availability may represent an alternative strategy for modulating pluripotent states, complementing transcription factor- and signaling-based manipulations.
It is important to note that E8-hPSCs do not phenocopy all features of naive pluripotency as defined for 2i naive mESCs, thus reflecting a naive-to-primed “intermediate” (NPI) state. Key differences between E8/NPI and naive PSCs include the moderate state of DNA hypomethylation, which does not match the extent of demethylation reported for 2i mESCs or naive hPSCs. However, E8 display low DNMT3B expression, critical for de novo DNA methylation in the primed state and whose expression is positively regulated by FGF/ERK signaling, with ERK inhibition (2i) shown to downregulate Dnmt3B and induce hypomethylation in mESCs (Ficz et al., 2013; Leitch et al., 2013). We do not find a global reduction in H3K27me3 and H3K9me3 levels, a known feature of naive mouse and human PSCs (Weinberger et al., 2016). Also, E8/NPI PSCs recapitulate several but not all recently reported (Theunissen et al., 2016) naive molecular features (Table S1). E8/NPI hPSCs show increased NANOG, KLF4, and TFE3 expression, whereas naive PSCs express additional markers more robustly, such as KLF17, TFCP2L1, ESRRB, and STELLA (Figure S2J). TFE3 staining in E8 hPSCs is mostly cytoplasmic, in contrast to the nuclear and cytoplasmic signal reported for mouse naive PSCs (Betschinger et al., 2013). In mouse PSCs, nuclear exclusion of TFE3 has been reported to associate with exit from naive pluripotency (Betschinger et al., 2013). However, Tfe3 has also been shown to transcriptionally upregulate Esrrb; thus, high levels of TFE3 in E8 may contribute to increased ESRRB gene expression. Interestingly, the robust upregulation of key naive markers such as NANOG, KLF4, and TFE3 in E8 appears to be limited to expression at the protein level, in contrast to naive PSCs, which also show transcriptional upregulation (Takashima et al., 2014; Theunissen et al., 2014; Weinberger et al., 2016). Altogether, our results suggest that E8 hPSCs represent a later developmental stage than bona fide naive cells while clearly preceding the primed state of conventional KSR hPSCs or mouse EPISCs. An intermediate state of formative pluripotency has been recently postulated as an obligate transition state within the developmental continuum between naive to primed pluripotency (Smith, 2017), raising the intriguing question as to what extent E8 hPSCs may capture aspects of such a formative state.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lorenz Studer, Sloan Kettering Institute New York (studerl@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human embryonic stem cell (hESC) lines employed for this study were H9 (WA-09), HUES6 and MEL1 passages 25–55. Induced pluripotent stem cell (iPSC) lines 036, 731 were generated from primary fibroblasts obtained from Coriell using CytoTune 2.0 Sendai reprogramming kit according to the manufacturer’s instructions. Line 706 was previously published in Miller et al. (2013). Cells were maintained in either KSR-based (see below) or Essential 8 (see below) or mTeSR hESC media according to manufacturer’s guidelines. SeV6 human induced pluripotent stem cell lines (hiPSC) were derived in the Studer lab from MRC5 fibroblasts and previously described in Kriks et al. (2011) and TLR3−/− iPSC were previously described in Lafaille et al. (2012). The identify of all the cell lines was periodically confirmed by STR analysis. Cell lines were routinely tested for mycoplasma and periodically assessed for genomic integrity by karyotyping. All hESC and hiPSC studies were approved by the Tri-institutional (MSKCC, Weill-Cornell, Rockefeller University) Embryonic Stem Cell Research Oversight (ESCRO) Committee.
METHOD DETAILS
Culture of hPSC in Knockout serum replacement (KSR)-based hPSC medium
hPSC were maintained on mouse embryonic fibroblasts (MEF, density: 12–13,000 cells/cm2) in KSR hPSC-medium (DMEM-F12, 20% KSR, 55 μM 2-Mercaptoethanol, 1mM L-Glutamine, 1% MEM Non-Essential amino acids, 1% Penicillin/Streptomycin, and supplemented with 10 ng/ml FGF2. Cells were passaged every 5–7 days using Dispase. In brief, cells were washed with DMEM/F12 then incubated in 1:1 DMEM/F12-Dispasefor5–7 min at 37°C, washed twice with DMEM/F12 then colonies were scraped using a cell lifter (Corning) and dissociated gently by pipetting and replated into KSR medium at a 1:2–1:7 ratio.
Culture of hPSC in Essential 8 (E8) hPSC medium
hPSC were cultured in Essential 8 (E8) medium onto plates coated with Vitronectin according to product instructions. In brief, Vitronectin was diluted 1:100 in DPBS, and incubated onto dishes for 1h at room temperature then aspirated before replating cells. E8 hPSC were passaged every 2–3 days using EDTA dissociation buffer (0.5 mM EDTA + 1.8g/l (30.8 mM) NaCl (Sigma-Aldrich) in DPBS). In brief, cells were washed twice with DPBS, then incubated with EDTA dissociation buffer for 1–2 min at 37°C, then EDTA was aspirated, cells were collected in E8 medium by gentle pipetting and replated at a 1:10–1:20 ratio.
Culture of hPSC in mTeSR hPSC medium
H9 (WA-09) HUES6 and MEL1 hESC (passages 40–50) were adapted from KSR to mTeSR medium and maintained in mTeSR onto Matrigel according to product’s instructions. Neural induction of mTeSR cells was performed > 3 passages after adaptation to mTeSR using either KSR-based differentiation medium or E6-based differentiation medium.
Directed differentiation toward neuroectoderm (Neural induction)
KSR-based neural induction protocol (10–12 days)
KSR differentiation medium: Knockout DMEM, 15% Knockout Serum Replacement, 1% Pen Strep, 1% L-Glutamine, 1% MEM Non-Essential Amino Acids, 0.1% 2-mercaptoethanol.
N2 medium: DMEM/F12, 0.1% 2-mercaptoethanol, 0.2% w/v Sodium Bicarbonate, 1.56 g (per liter of medium) D-(+)- Glucose, and 20ul (per liter of medium) progesterone (Stock: dissolve 0.032 g Progesterone in 100ml 100% ethanol) supplemented with 1% N2 supplement B.
Day −2 of differentiation
Coat tissue culture dishes with Matrigel diluted 1:50 in DMEM/F12 and store overnight at 4°C.
Day −1
Start differentiation when hPSC culture is 70%−80% confluent. Detach cells off the plate with Accutase (30 min at 37°C) and dissociate them to single cells by gentle pipetting. Pellet the cells (200 × g for 5 min). Wash cells with PBS and pellet again. Plate 250–300,000 cells/cm2 in KSR-based hESC medium with 10 μM Y-27642.
Day 0
hPSCs should appear as monolayer. Wash cells with PBS or KSR differentiation medium, then add KSR differentiation medium supplemented with 500nM LDN + 10 μM SB.
Day 1–2
Change medium to 100% KSR differentiation media supplemented with 500nM LDN + 10 μM SB
Day 3–4
Change medium to 75% KSR differentiation medium and 25% N2 medium supplemented with 500nM LDN + 10 μM SB
Day 5–6:
Change medium to 50% KSR differentiation medium and 50% N2 medium supplemented with 500nM LDN + 10 μM SB
Day 7–8
Change medium to 25% KSR differentiation media and 75% N2 media supplemented with 500nM LDN + 10 μM SB.
Day 9–12
Change medium to 100% N2 media supplemented with 500nM LDN + 10 μM SB.
Essential 6 (E6)-based neural induction protocol (10–12 days): Day −2 of differentiation
Coat tissue culture dishes with Matrigel diluted 1:50 in DMEM/F12 or Vitronectin diluted 1:100 in PBS and store overnight at 4°C.
Day −1
Start differentiation when hPSC culture is 70%−80% confluent. Detach cells off the plate with EDTA dissociation buffer (5 min at37°C) and dissociate them to single cells by gentle pipetting. Pellet the cells (200 × g for 5 min). Wash cells with PBS and pellet again. Plate 250–300,000 cells/cm2 in E8 hESC medium with 10 μM Y-27642.
Day 0
hPSCs should appear as monolayer. Wash cells with PBS or E6 medium, then add E6 medium supplemented with 500nM LDN + 10 μM SB.
Day 3
Change medium to E6 media supplemented with 500nM LDN + 10 μM SB
Day 4–12
Change medium every other day with E6 medium supplemented with 500nM LDN + 10 μM SB.
For more details on differentiation procedures refer to Tchieu et al. (2017)
To quantify differentiation efficiency, cells were fixed in 4% paraformaldehyde (Affymetrix) at day 10 of differentiation for immune-staining. Percentage of PAX6 positive cells was assessed using an Operetta high content imaging microscope (Perkin Elmer). Alternatively, cells were collected in TRIzol at days 0, 3, 6, 9, 12 for RT-qPCR analysis of PAX6 and SOX1 mRNA expression kinetics.
Treatments and inhibitors
E8+Albumax cultures were maintained with Essential 8 media supplemented with AlbuMAX (1.6% w/v) and passaged gently with EDTA. AlbuMAX deproteinization was performed as follows and as described in Garcia-Gonzalo and Izpisua Belmonte (2008): 0.5 mL of a 100X solution of Trypsin from bovine pancreas (Sigma) was added to 50 mL of a 16% w/v solution of Albumax in E8 base medium without supplement and incubated for 30 minutes at 37°C (waterbath). Then, 0.5ml of a 100X solution of Soybean Trypsin inhibitor was added to the AX-Trypsin solution and incubated at room temperature for 30 minutes. The deproteinized AlbuMAX solution was sterile filtered and added to E8 medium to a final concentration of 1.6% w/v. Fatty acid-free BSA was added to E8 medium to a final concentration of 1% w/v. Chemically defined lipid concentrate was added at a concentration of 1:100 to E8. BCI hydrochloride (DUSP6 inhibitor) was added at concentrations of 0,0.1μM, 0.5μM or1μM for 2 days. Fatostatin (SREBF1 inhibitor) was added at concentrations of 0, 2μM or 5μM for 2 days. PD0325901 (MEK/ERK inhibitor) was added at a concentration of 1 μM unless otherwise stated.
Generation of naive hPSC
Bona fide naive hPSC were generated from H9 hESC grown in mTeSR using RSet™ and NaiveCult™ kits according to manufacturer’s instructions.
E8 with FGF2 titration
E8 medium was replaced one day after passaging with E6 medium with 2 mg/ml TGF-β 1 and different concentrations of FGF2 (as shown in Figure S5B). Cells were kept in medium with different FGF2 concentrations for three days and the medium was changed every day.
Growth Curve
Cells were cultured and passaged in their respective conditions (KSR and E8) as described above. Passaging was done at 70%−80% cell confluence, splitting dates and ratios were documented to generate a growth curve.
Immunostaining
Cultured cells were fixed with 4% PFA, then blocked and permeabilized in PBS+ 1% BSA and 0.3% Triton X-100 (Block-perm buffer). Primary antibodies were diluted according to the manufacturer’s recommendation in block-perm buffer + 5% normal donkey serum and incubated overnight at 4°C. Primary antibody was washed out with PBS+0.01% Tween-20 (PBS-T). Cells were subsequently incubated in secondary antibody conjugated with Alexa Fluor 488– 555-, or 647- diluted at 1:400 in block-perm buffer + 5% normal donkey serum for 1 hour at room temperature. The secondary antibody was washed out 3 times with PBS-T and cells were imaged with 4’, 6-diamidino-2-phenylindole (DAPI) nuclear counterstain using an Olympus inverted fluorescence microscope.
Directed differentiations toward endo- and mesoderm
Directed differentiation of hPSC into definitive endoderm or mesoderm were performed using the PSC Definitive Endoderm Induction Kit or STEMdiff Mesoderm Induction Medium, respectively, according to the manufacturer’s manual. Percentage of SOX17 and BRACHUYRY (T) positive cells was assessed by immunofluorescence at day 2 or 5 of differentiation, respectively, using an Operetta high content imaging microscope (Perkin Elmer).
Embryoid body formation
Embryoid bodies (EBs) were generated, with minor modifications for KSR-MEF maintained hESCs, using the self-aggregation protocol described in Lin and Chen (2008). In brief, KSR or E8 hPSC at 70%−80% confluence were detached using either dipase (KSR) as described above or EDTA (E8) as described above and replated into Corning low attachment plates. The day after replating, medium was changed to KSR hESC medium without FGF2 or E6 medium every day for 7 days. To avoid aspiration, for medium changes, forming EBs were collected into 15 mL conical tubes, sedimented for 5 minutes under the hood, then medium was aspirated and replaced with fresh medium before replating into the same dish. EBs were collected at day 7 and RNA was extracted using TRIzol.
RNA extraction and RT-qPCR
RNA was prepared from samples collected with TRIzol (Invitrogen) using the Direct-zol RNA MiniPrep kit. cDNA was generated using the iScript Reverse Transcription Supermix for RT-qPCR. For qPCR analysis, primers were obtained from QIAGEN (Quantitect Primer assays) and the reactions were performed per manufacturers’ instructions using SsoFast EvaGreen® Supermix. Results were normalized to GAPDH or SMU housekeeping genes.
Nanostring analysis
Nanostring analysis was performed as previously described (Paull et al., 2015) and according to Nanostring nCounter XT assay manual. In brief, 100 ng of purified RNA (per sample) was hybridized overnight with a custom probe set containing markers for each of the three germ layers (Kahler et al., 2013). Following hybridization, the samples were prepared for counting using the Nanostring Prep Station and analyzed using a Nanostring nCounter analysis system. Subsequent analysis, including gene normalization was performed using NSolver v.3.0 according to the software’s manual.
Germlayer assignment of lineage markers was as shown in Figure 1D and according to Bock et al. (2011) with exception of TDGF1 (excluded because better suited as pluripotency marker) and PAX6 (only assigned to NEURO and ECTO categories).
Scorecard analysis
For each gene quantified via Nanostring platform, a Student’s t test was performed between three replicates of EB and three replicates of ESC for each cell line. Based on the assignment of lineage markers, the average f-score of genes mapped to the same lineage was calculated as the quantification of differentiation potentials for each lineage. Positive values indicate upregulation of a group of lineage-specific genes in EB against ESC and higher differentiation propensity for the corresponding lineage.
Teratoma formation
Approximately 3×106 cells embedded in Matrigel and supplemented with Y-27632 at a final concentration of 10 μM were injected double flanks subcutaneously into 6–8 weeks old NSG female mice. Tumor growth was assessed twice weekly from day 17 to day 52 post injection. Mice were sacrificed, and tumors were collected between day 41 and 52, fixed in formalin and stained with hematoxylin eosin.
Clonogenicity assay
KSR hESC were MEF-depleted by trypsinization (incubate cells with 0.05% Trypsin at room temperature for 2 minutes, tap dish and check under microscope for MEF detachment, wash cells 2x with DMEM F12). Cells were dissociated to single cells using Accutase (KSR, mTeSR) or EDTA (E8). Cells were counted using an automated cell counter (Nexcelom) and 2000 single cells per well of a 12 well plate were plated in triplicate wells for each cell line (H9, HUES6, MEL1) into the respective media and onto MEF- (KSR), Matrigel-(mTeSR) or VTN- (E8) coated plates. In addition, one well per line was supplemented with 10 μM Y-27642 as control. Colonies were fixed 4 (E8), 7 (mTeSR) or 6 days (KSR) after seeding and stained with crystal violet (Harel et al., 2012). Colonies were counted manually for KSR and mTeSR and using the ImageJ cell counter plugin for E8.
miRNA analysis
miRNA was extracted using the miRNeasy Mini Kit according to the manufacturer’s manual. miRNA expression was analyzed using the miFinder miRNA PCR Array (QIAGEN) according to the manufacturer’s manual. PCR reaction was performed on a BioRad CFX96 Touch Real-Time PCR Detection System. miRNA expression levels were calculated using the Web-based analysis platform from QIAGEN (https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/).
Flow Cytometry
Intracellular analysis of pluripotency markers (OCT4, NANOG, SOX2) was performed using the BD Human PluripotentStem Cell Transcription Factor Analysis Kit according to manufacturer’s protocol. For surface marker (SSEA3, SSEA4) staining, cells were detached and dissociated as described above respectively to their culture conditions. 1×106 cells were incubated in primary antibody at the recommended concentrations for 1h on ice and protected from light. Cells were subsequently washed in PBS and counterstained with DAPI to exclude dead cells. Flow cytometric analysis and sorting was performed on a BD FACS Aria III.
Western blotting
Cultured cells were detached as described above according to culture conditions, cell numbers were counted. Cells were lysed with 2X Laemmli Sample Buffer (Bio-Rad) at 5000 cells/se. Cell lysates were separated by NuPAGE 4%−12% Bis-Tris Protein Gel (Invitrogen) in NuPAGE MOPS SDS Running Buffer (Invitrogen) or NuPAGE MES SDS Running Buffer (Invitrogen) according to different protein size. Proteins were electrophoretically transferred to a nitrocellulose membrane (Thermo) with NuPAGE Transfer Buffer (Invitrogen). Blots were blocked for 30 min at room temperature in TBS-T + 5% BSA or 5% fat-free milk according to antibody requirements and probed overnight at 4°C with primary antibodies. After washing three times in TBS-T, signals were detected by using secondary mouse or rabbit antibodies conjugated to horseradish peroxidase. Secondary antibodies were incubated for 1 hour at room temperature. After three times washes, an enhanced chemiluminescence (ECL) detection kit (PerkinElmer) was used to develop the films and Blue Autography films (Crystalgen) were used to record the signals.
Oil Red staining
Oil Red staining was performed using the Lipid (Oil Red O) staining kit from Biovision according to manufacturer’s instructions. Hematoxylin staining was omitted as it masked the signal from the oil red lipid dye.
Electron microscopy
Cell pellets were fixed with a modified Karnovsky’s fix and a secondary fixation in reduced osmium tetroxide. Following en-bloc staining with uranyl acetate and graded ethanol dehydration, samples were embedded in an Epon analog resin. Ultrathin sections (65 nm) were contrasted with lead citrate. A simplified lead citrate stain for use in electron microscopy and viewed on a JEM 1400 electron microscope (JEOL) operated at 100 kV. Digital images were captured on a Veleta 2Kx2KCCD camera (Olympus-SIS). Mitochondrial length was quantified using ImageJ.
Mitochondrial potential (TMRE)
10 minutes prior to the assay (TMRE addition), 20 μM FCCP in complete cell culture medium was added and used as background control in the analysis. After the initial FCCP incubation, human pluripotent stem cells were washed once with PBS. After the washing step, cells were incubated with 200 nM TMRE diluted in complete cell culture media for 20 minutes in the incubator. After a single PBS washing step, cells were incubated with Accutase for 15 minutes yielding a single cell suspension after dissociation. Cells were washed once with PBS and resuspended in 0.2% BSA in PBS for immediate FACS analysis using the PE channel (BD LSR Fortessa). A 40μm cell strainer was used to remove potential clumps.
mtDNA quantification
Mitochondrial DNA copy number was quantified using the Human mitochondrial to nuclear DNA ratio kit (Takara) according to manufacturer’s instructions.
Oxygen Consumption Rate (OCR)
Cells were detached with Accutase and resuspended in 400 μL experimental medium (DMEM, no glucose, no phenol red, no glutamine, Thermo Fisher Scientific) + 1 mM pyruvate + 2 mM glutamine. Oxygen consumption was measured polarographically using an O2Koxygraph (Oroboros instruments) in a 2 mL volume of experimental medium. Roughly 2×106 cells were detached with Accutase and added to the chambers before sealing and starting recording oxygen concentration. Respiration rates under baseline condition, oligomycin (1 μM), Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 0.3 μM) and KCN (1mM) were each recorded for 4 minutes. Protein concentrations of the cell suspensions were measured for normalization by Lowry assay (DC protein assay, BioRad).
2-Deoxy-D-glucose (2-DG)
One day after passaging, cell culture medium (E8 or KSR-containing medium) was supplemented with 0mM, 2mM or 4 mM of 2-Deoxy-D-glucose (2-DG, Sigma). Medium was changed to fresh medium supplemented with fresh 2-DG every day. Cells were fixed after 5 days. Pluripotency was assessed by NANOG immunostaining.
Metabolite profiling
Metabolomic profiling was conducted on three technical replicates of each H9, HUES6 and MEL1 hESC grown in either E8 or KSR conditions. Before collection, cells were washed once with PBS to remove residual culture medium. Cells were then extracted with 1 mL of 80:20 methanol:water. After overnight incubation at −80°C, cell extract was scraped and transferred to a 1.5 mL tube and centrifuged at 21,000 g for 20 min at 4°C. Extracts were then dried in an evaporator (Genevac EZ-2 Elite). Samples were re-suspended in 60 mL of 60:40 acetonitrile:water. LC separation was using an Agilent 1290 UPLC system and ZIC-pHILIC column (150 mm x 2.1 mm, 5 μm; Merck Sequant). Mobile phase A was 90:10 water:acetonitrile, mobile phase B was 90:10 acetonitrile:water, both containing 10 mM ammonium bicarbonate (pH 9.4). The injection volume was 5 mLand LC gradient conditions were: 0 min: 95% B; 1 min: 95%B; 10 min: 50% B; 13 min: 50% B; 14 min: 30% B; 17 min 30% B, with 6 min of re-equilibration time. MS detection was using an Agilent 6545Q-TOF mass spectrometer with Dual JetStream source operating in negative ionization mode. MS parameters were: gas temp: 300°C; gas flow: 10 l/min; nebulizer pressure: 40 psig; sheath gas temp: 300°C; sheath gas flow: 12 l/min; VCap:3,000 V; Fragmentor: 125 V; Skimmer: 45 V; Octopole RF: 750 V. Active reference mass correction was through a second nebulizer using masses with m/z: 119.03632 and 980.016375. Data was acquired from m/z 50 – 1700 at 1 Hz. Data analysis was performed within Mass Hunter Profinder software (Agilent) with statistical analysis in Mass Profiler Professional (Agilent) and R.
5mC ELISA
Cells were detached and genomic DNA was extracted using the Quick-DNA Miniprep Plus Kit (Zymo research) according to the product’s instructions. Genomic 5mC rates were quantified using the MethylFlash Methylated DNA 5-mC Quantification Kit (EpiGentek) according to the manufacturer’s protocol.
RNA profiling
For RNA-seq cells were sorted by flow cytometry for the SSEA4+ fraction as described above. RNA was extracted from 2–5×106 SSEA4+ cells using the Direct-zol™ kit (Zymo) according to manufacturer’s instructions.
RNA-seq library preparation was performed at the Weill Cornell Epigenomics Core Facility using the TruSeq Stranded Total RNA library prep kit (Illumina) according to manufacturer’s instructions. Libraries were sequenced on an Illumina HiSeq 2500 platform with 100bp paired end reads.
RNA-seq analysis
Reads were aligned to the hg19 Human transcripts using STAR (version 2.5.21b) (Dobin et al., 2013) using default parameters and resulting bam files were sorted and indexed using samtools. Gene counts were obtained using featureCounts (version 1.4.3) (Liao et al., 2014) to sorted bam files, and only unique-mapping reads were used. Genes without any expression counts in any sample were discarded. Differential gene expression analysis was performed using DESeq2 (version 1.4.5) R package (Love et al., 2014) that normalize gene count data to transcription per million (TPM), and then detect differentially expressed genes (DEG) between E8 and KSR groups with (FDR < 0.1). Mosaic version 1.1 was used to retrieve gene ontology (GO) information for all genes of the Human genome (Zhang et al., 2012). Functional analysis was performed on DEG with DAVID (Huang et al., 2009) (version 6.8) and biological process GO terms and KEGG pathways with enrichment p < 0.05 were selected as overrepresented functions. To remove the redundancy of GO terms, up to top 350 GO terms were used as the input of REVIGO (http://revigo.irb.hr/) to generate the list of intelligible functions. We applied the same procedure to external data from four studies (Boroviak et al., 2015; Marks et al., 2012; Takashima et al., 2014; Yan et al., 2013).
2D plots
Each 2D plot represents a comparison of gene function/pathway regulation between E8/KSR and one of the four mentioned naive versus primed studies (Boroviak et al., 2015; Marks et al., 2012; Takashima et al., 2014; Yan et al., 2013). Common DEG between E8/KSR and the respective naive versus primed study were calculated based on published RNA-seq data. The same gene function/pathway enrichment analysis was applied to the common DEG list. For each gene, a Student’s t test was performed between naive and primed samples and between E8 and KSR samples separately based on normalized read counts. For each enriched function or pathway, the average t-score was calculated using the genes in the DEG list that are annotated with this specific function for E8/KSR and naive versus primed study separately. Positive values indicate upregulation of functional terms in E8 or naive samples, and negative values indicate upregulation in KSR or primed samples.
Whole Genome Bisulfite Sequencing (WGBS) Library preparation
Genomic DNA was isolated using a DNeasy Blood and tissue kit (QIAGEN). Library preparation was performed at the Integrated Genomics Operation (IGO) at MSKCC. Briefly, 250ng of genomic DNA was prepared using the NextFlex Bisulfite-Seq (Perkin Elmer). DNA was fragmented using a Covaris E220. After end repair and tailing, barcoded methylated adapters were ligated. Fragments between 200 and 1000bp were size selected and bisulfate converted. Amplification underwent 16 cycles. Sequencing libraries were clustered using a cBot and sequenced on a Hiseq 4000. An average of 87 million paired reads was generated (PE100) per sample.
WGBS analysis
FASTQ files were generated by bcl2fastq (V2.17) and filtered for pass filter reads based on Illumina’s chastity filter. Sequencing adapters were trimmed by FLEXBAR (V2.4) (Dodt et al., 2012), genomic alignments using Bismark (V0.14.4) (Krueger and Andrews, 2011) and Bowtie2 (V2.2.5) (Langmead and Salzberg, 2012) to reference human genome hg19, and per base CpG methylation metrics were calculated with a custom PERL script. CpGs at a minimum threshold coverage of 5 reads were used for downstream analysis.
ChIP-seq
ChIP-seq was performed as follows: Cells were harvested from 10cm dishes. For KSR hESC, MEF were eliminated by brief trypsinization prior to collection as described above. Chromatin was prepared using a truChIP™ Chromatin Shearing Kit following the manufacturer’s High Cell protocol for suspension cells. Chromatin was sheared to a target fragment size of 200–700bp at the Epigenomics Core facility at WCMC using a Covaris S220 sonicator. Immunoprecipitation was performed as follows and as described in Bulut-Karslioglu et al. (2012) with some modifications: 10 μg of chromatin was diluted at a ratio of 9:1 in Dilution Buffer (1% Triton, 0.1% SDS, 2mM EDTA, 167 mM NaCl, 20 mM Tris HCl pH8.1), 1% of the diluted sample was set aside as Input. 2 μg of anti-H3K27me3 or anti-H3K27Ac per 10 μg of chromatin was added to the reaction and incubated under rotation overnight at 4°C. After incubation, Protein G Dynabeads were added to the ChIP reaction as per product’s instructions and incubated for 2 hours at 4°C under rotation. Then, beads were collected using a magnetic rack and washed 3 times in Low Salt Buffer (0.1% SDS, 1% Triton, 2mM EDTA, 150 mM NaCl, 20 mM Tris-HCl pH 8.1), once in High Salt Buffer (0.1% SDS, 1% Triton, 2mM EDTA, 500 mM NaCl, 20 mM Tris-HCl pH 8.1) once in LiCl buffer (0.25M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1mM EDTA, 10mM Tris HCl pH 8.1) and once in TE buffer (10 mM Tris pH 8.0,1mM EDTA). All buffers were freshly supplemented with protease inhibitors prior to washes. For the washes, beads were incubated for 5–10 minutes at 4°C under rotation. Before elution, beads were transferred to DNA LoBind tubes. 200 ml of Elution buffer (1% SDS, 100 mM NaHCO3) was added to each tube and incubated at room temperature for 1 hour under rotation. Eluates were collected and transferred to new DNA LoBind tubes, Inputs volume was brought to 200μl with Elution buffer and the same following procedure was applied to ChIP and Input samples: 1μl RNase A was added to each tube and incubated at 37°C for 1 hour. 4.1 μl of Proteinase K was added to each tube and incubated at 65°C overnight. ChIP’ed DNA was purified using the ChIP DNA Clean and Concentrator kit. ChIP was validated by RT-qPCR using primers for constitutively active genes (H3K27me7H3K27Ac+) or tissue-specific genes (H3K27me+/H3K27Ac− in hESC) (see Key Resources Table for ChIP validation primers). Recovery was calculated over 1% Input by ΔCt method.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-4EBP1, phospho (Thr37/Thr46) | Cell Signaling | 2855; RRID:AB_560835 |
| Rabbit anti-AKT | Cell Signaling | 9272; RRID:AB_329827 |
| Rabbit anti-Phospho-AKT (Ser473) | Cell Signaling | 9271; RRID:AB_329825 |
| Goat anti-BRACHUYRY (T) | R&D | AF2085; RRID:AB_2200235 |
| Rabbit anti-p44/42 MAPK (ERK1/2) | Cell Signaling | 9102; RRID:AB_330744 |
| Rabbit anti-p44/42 MAPK (Phospho-ERK1/2) | Cell Signaling | 9101; RRID:AB_331646 |
| Rabbit anti-ESRRB | Millipore | ABD101 |
| Mouse anti-H3 | Abcam | ab24834; RRID:AB_470335 |
| Rabbit anti-H3K4me3 | Abcam | ab8580; RRID:AB_306649 |
| Rabbit anti-H3K9Ac | Abcam | ab4441; RRID:AB_2118292 |
| Rabbit anti-H3K9me3 | Abcam | ab8898; RRID:AB_306848 |
| Rabbit anti-H3K27Ac | Abcam | ab4729; RRID:AB_2118291 |
| Rabbit anti-H3K27me3 | Millipore | 07-449; RRID:AB_310624 |
| Rabbit anti-H4 | Abcam | ab70701; RRID:AB_1209472 |
| Rabbit anti-H4K8Ac | Abcam | ab15823; RRID:AB_880455 |
| Rabbit anti-LAMIN B1 | Abcam | ab16048; RRID:AB_443298 |
| Mouse anti-KLF4 | Santa Cruz | Sc-166238; RRID:AB_2130234 |
| Rabbit anti-KLF17 | Atlas Antibodies | HPA024629; RRID:AB_1668927 |
| Rabbit anti-mTOR (7C10) | Cell Signaling | 2983; RRID:AB_2105622 |
| Rabbit anti-Phospho-mTOR (Ser2448 D9C2) | Cell Signaling | 5536; RRID:AB_10691552 |
| Rabbit anti-NANOG (D73G4) | Cell Signaling | 4903; RRID:AB_10559205 |
| Mouse anti-OCT3/4 (C-10) | Santa Cruz | SC-5279; RRID:AB_628051 |
| Rabbit anti-PAX6 | Biolegend | PRB-278P; RRID:AB_291612 |
| Rabbit anti-Phospho-S6K p70 (Thr389) (108D2) | Cell Signaling | 9234; RRID:AB_2269803 |
| Rabbit anti-Phospho-S6K p70 (Ser371) | Cell Signaling | 9208; RRID:AB_330990 |
| Rat anti-SSEA3 | BD | 560237; RRID:AB_1645542 |
| Mouse anti-SSEA4 (MC813-70) | BD | 560796; RRID:AB_2033991 |
| Goat anti-SOX1 | R&D | AF3369; RRID:AB_2239879 |
| Mouse anti-SOX17 | R&D | MAB1924; RRID:AB_2195646 |
| Mouse anti-STELLA | Millipore | MAB4388; RRID:AB_2094156 |
| Goat anti-TFCP2L1 | R&D | AF5726; RRID:AB_2202564 |
| Rabbit anti-TFE3 | Sigma | HPA023881; RRID:AB_1857931 |
| Mouse anti-γ TUBULIN | Sigma | T6557; RRID:AB_477584 |
| AlexaFluor Donkey Anti-Goat 568 | Thermo Fisher Scientific | A-11057; RRID:AB_142581 |
| AlexaFluor Donkey Anti-Rabbit 488 | Thermo Fisher Scientific | A-21206; RRID:AB_141708 |
| AlexaFluor Donkey Anti-Rabbit 555 | Thermo Fisher Scientific | A-31572; RRID:AB_162543 |
| AlexaFluor Donkey Anti-Mouse 488 | Thermo Fisher Scientific | R37114; RRID:AB_2556542 |
| AlexaFluor Donkey Anti-Mouse 555 | Thermo Fisher Scientific | A-31570; RRID:AB_2536180 |
| Biological Samples | ||
| Mouse embryonic fibroblasts (MEF) | Thermo Fisher Scientific | A34959 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Knockout Serum Replacement (KSR) | Thermo Fisher Scientific | 10828-028 |
| DMEM-F12 | Thermo Fisher Scientific | 11320-032 |
| Essential 8 (E8) | Thermo Fisher Scientific | A1517001 |
| Essential 6 (E6) | Thermo Fisher Scientific | A1516401 |
| Knockout DMEM | Thermo Fisher Scientific | 10829-018 |
| mTeSR | StemCell Technologies, Inc. | 85850 |
| DMEM no glucose, no phenol red, no glutamine | Thermo Fisher Scientific | A14430 |
| AlbuMAX I Lipid-Rich BSA | Thermo Fisher Scientific | 11020021 |
| BSA fatty-acid free | Sigma Aldrich | A3803 |
| Chemically defined lipid concentrate | Thermo Fisher Scientific | 11905031 |
| L-Glutamine (100X) | Thermo Fisher Scientific | 25030-081 |
| Non-essential Amino Acids (100X) | Thermo Fisher Scientific | 11140-050 |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15140-122 |
| 2-mercaptoethanol | Thermo Fisher Scientific | 21985-023 |
| Sodium Bicarbonate | Sigma Aldrich | S5761 |
| D-(+)-Glucose | Sigma Aldrich | G7021 |
| 2-Deoxy-D-glucose | Sigma Aldrich | D6134 |
| N2 supplement B | Stem Cell Technologies | 07156 |
| Progesterone | Sigma Aldrich | P8783 |
| Recombinant human FGF2 | R&D | 233-FB-001MG/CF |
| Recombinant human TGF-beta 1 | R&D | 240-B-002 |
| 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575-020 |
| Dispase | Stem Cell Technologies | 07913 |
| Accutase | Innovative Cell Technologies | AT104-500 |
| Vitronectin (VTN-N) | Thermo Fisher Scientific | A14700 |
| Matrigel | Corning | 354234 |
| Normal Donkey Serum | Sigma Aldrich | S30 |
| Trypsin from Bovine Pancreas | Sigma Aldrich | T9935 |
| Soybean Trypsin inhibitor | Thermo Fisher Scientific | 17075029 |
| Y-27632 (ROCKi) | R&D | 1254 |
| SB431542 (SB) | R&D | 1614 |
| LDN193189 (LDN) | Stemgent | 04-0074 |
| PD0325901 | Stemgent | 04-0006 |
| BCI hydrochloride | Sigma Aldrich | B4313 |
| Fatostatin A | Tocris | 4444 |
| Critical Commercial Assays | ||
| Direct-zol RNA preparation kit | Zymo | R2062 |
| SsoFast Eva Green Supermix | Bio-Rad | 172-5200 |
| iScript Reverse Transcription Supermix | Bio-Rad | 170-8891 |
| nCounter custom codeset | Nanostring | N/A |
| Quick-DNA miniprep plus kit | Zymo Research | D4069 |
| DNeasy Blood and tissue kit | QIAGEN | 69504 |
| miRNeasy Mini Kit | QIAGEN | 217004 |
| miFinder miRNA PCR Array | QIAGEN | MIHS-122Z |
| ChIP DNA Clean and Concentrator | Zymo Research | D5205 |
| TMRE - Mitochondrial Membrane Potential Assay Kit | Abcam | ab113852 |
| CytoTune-iPS 2.0 Sendai Reprogramming Kit | Thermo Fisher Scientific | A16517 |
| Human Pluripotent Stem Cell Transcription Factor Analysis Kit | BD | 560589 |
| Human Mitochondrial DNA (mtDNA) Monitoring Primer Set | Takara | 7246 |
| Methylflash methylated DNA 5mC quantification kit | Epigentek | P1034 |
| RSet naive | Stem Cell Technologies | 05970 |
| NaiveCult | Stem Cell Technologies | 05580, 05590 |
| PSC Definitive Endoderm Induction Kit | Thermo Fisher Scientific | A3062601 |
| STEMdiff Mesoderm Induction Medium | Stem Cell Technologies | 05220 |
| Lipid (Oil Red O) staining kit | Biovision | K580-24 |
| NextFlex Bisulfite-Seq | Perkin Elmer | 5119-01 |
| truChIP Chromatin Shearing Kit | Covaris | 520154 |
| TruSeq Stranded Total RNA | Illumina | RS-122-2301 |
| Deposited Data | ||
| RNA Seq, ChIP Seq, WGBS | NCBI SRA | SRA: SRP104789 |
| Experimental Models: Cell Lines | ||
| H9 (WA-09) hESC line | WiCell Research Institute | NIHhESC-10-0062 |
| MEL1 hESC line | Stem Cells Ltd. | NIHhESC-11-0139 |
| HUES 6 hESC line | HSCI iPS Core | NIHhESC-09-0019 |
| 706 iPSC line | MSKCC Stem Cell Core | (Miller et al., 2013) |
| 036 iPSC line (parental line: primary fibroblasts) | iPSC: This study; pFIB: Coriell Institute | GM02036 |
| 731 iPSC line (parental line: primary fibroblasts) | iPSC: This study; pFIB: Coriell Institute | GM00731 |
| SeV6 iPSC line | MSKCC Stem Cell Core | (Kriksetal., 2011) |
| TLR3 iPSC line | Studer lab | (Lafaille et al., 2012) |
| Oligonucleotides – ChIP qPCR control primers | ||
| H3K27Ac+, H3K27me3− | N/A | |
| hsGAPDH E1 F: TCGACAGTCAGCCGCATCT; hsGAPDH I1 R: CTAGCCTCCCGGGTTTCTCT |
This study | N/A |
| hsRPL30 E1 F: CAAGGCAAAGCGAAATTGGT; hsRPL30 E1 R: GCCCGTTCAGTCTCTTCGATT |
This study | N/A |
| hsALDOA I2 F: TCCTGGCAAGATAAGGAGTTGAC; hsALDOA I2 R: ACACACGATAGCCCTAGCAGTTC |
This study | N/A |
| H3K27Ac−, H3K27me3+ | N/A | |
| hsMYOD E1 F: CCGCCTGAGCAAAGTAAATGA; hsMYOD E1 R: GGCAACCGCTGGTTTGG |
This study | N/A |
| hsSERPINA1 E1 F: GGCTCAAGCTGGCATTCCT; hsSERPINAI E1 R: GGCTTAATCACGCACTGAGCTTA |
This study | N/A |
| hsAFM I1 F: GCAGAACCTAGTTCCTCCTTCAAC; hsAFM I1 R: AGTCATCCCTTCCTACAGACTGAGA |
This study | N/A |
| Software and Algorithms | ||
| FIJI – ImageJ | (Schindelin et al., 2012) | https://fiji.sc/ |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR/ |
| featureCounts | (Liao et al., 2014) | http://bioinf.wehi.edu.au/featureCounts/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DAVID | (Huang et al., 2009) | https://david.ncifcrf.gov/ |
| REVIGO | http://revigo.irb.hr/ | N/A |
| FLEXBAR | (Dodtetal., 2012) | https://github.com/seqan/flexbar |
| Bismark | (Krueger and Andrews, 2011) | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2 |
| MACS | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| DiffBind | http://bioconductor.org/packages/release/bioc/html/DiffBind.html | N/A |
| Other | ||
| Complete mini EDTAfree protease inhibitor | Roche | 11836170001 |
| Protein G Dynabeads | Thermo Fisher Scientific | 10003D |
| RNase A | Thermo Fisher Scientific | EN0531 |
| Proteinase K | Thermo Fisher Scientific | 25530-031 |
| DNA LoBind tubes | Eppendorf | 0030108051 |
ChIP-seq library preparation
ChIP-seq library preparation was performed at the Weill Cornell Epigenomics Core Facility (New York) using the TruSeq ChIP library preparation kit (Illumina IP-202–1012) according to manufacturer’s instructions. Libraries were sequenced on an Illumina HiSeq 2500 platform with 50 bp single end reads.
ChIP-seq analysis
Reads were aligned to the hg19 Human genome using Bowtie2 (V2.2.5) (Langmead and Salzberg, 2012) with default parameters and resulting BAM files sorted and index using samtools. MACS (V 2.1.0) (Zhang et al., 2008) with default parameters used to detect statistically significant peaks by identifying genomic regions with enriched coverage in ChIP samples relative to input control. The comparative ChIP-seq analysis was applied to the peaks with q-value < 0.01 to detect the differential binding sites between E8 and KSR samples using DiffBind (http://bioconductor.org/packages/release/bioc/html/DiffBind.html). Corresponding genes that were mapped to differential ChIP sites were selected for subsequent analysis. The functional enrichment analysis was performed similarly as for DEG in RNA-seq analysis.
DATA AND SOFTWARE AVAILABILITY
RNA-seq, ChIP-seq and WGBS data used in this paper are available at NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under accession SRA: SRP104789.
Supplementary Material
Highlights.
Essential 8 (E8) hPSCs exhibit naive-to-primed intermediate (NPI) pluripotency features
NPI traits are dependent on lipid-free E8 composition
Lipid deprivation triggers endogenous ERK inhibition despite exposure to high FGF2
De novo lipogenesis marks transcriptomes of E8 hPSCs and human pre-implantation epiblast
ACKNOWLEDGMENTS
We thank members of the Studer lab for discussions and M. Tomishima and S. Irion for feedback on the manuscript. We thank G. Rehbein and V. Tabar for providing SREBF1 reagents. RNA-seq and ChIP-seq library preparation and sequencing were performed at the Epigenomics Core at Weill Cornell Medicine. WGBS was performed by the Integrated Genomics Operation of MSKCC. Electron microscopy was performed by Lee Cohen-Gould at the CLC Imaging Core Facility at Weill Cornell Medicine. Teratoma formation assays were conducted by the Antitumor Assessment and Pathology Cores at MSKCC. Flow cytometry was performed by Kiran Ramnarine at the Stem Cell Core of MSKCC. We thank F. Lafaille from the Casanova lab at Rockefeller University for providing the SeV6 and TLR3 iPSC lines. The work was supported in part by grant R01AG054720 from the National Institute of Aging (to L.S. and D.B.) and by core grant P30CA008748 from the National Cancer Institute.
Footnotes
DECLARATION OF INTERESTS
L.S. is a scientific co-founder and paid consultant of Bluerock Therapeutics. B. Z. is a full-time employee of Evotec SE. S.M.C. is employed by and owns stock in Amgen.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.05.001.
REFERENCES
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, and Smith A (2013). Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggers JD (1998). Reflections on the culture of the preimplantation embryo. Int. J. Dev. Biol. 42, 879–884. [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. (2011). Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, and Bertone P (2015). Lineage-Specific Profiling Delineates the Emergence and Progression of Naive Pluripotency in Mammalian Embryogenesis. Dev. Cell 35, 366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, Perrera V, Scaranaro M, de la Rosa-Velazquez IA, van de Nobelen S, Shukeir N, Popow J, Gerle B, Opravil S, Pagani M, et al. (2012). A transcription factor-based mechanism for mouse heterochromatin formation. Nat. Struct. Mol. Biol. 19, 1023–1030. [DOI] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, and Tu BP (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular a-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, and Smith A (2007). Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–1234. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YS, Goke J, Ng JH, Lu X, Gonzales KA, Tan CP, Tng WQ, Hong ZZ, Lim YS, and Ng HH (2013). Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 13, 663–675. [DOI] [PubMed] [Google Scholar]
- Chen NC, Yang F, Capecci LM, Gu Z, Schafer AI, Durante W, Yang XF, and Wang H (2010). Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J. 24, 2804–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, et al. (2011). Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M, Roehr JT, Ahmed R, and Dieterich C (2012). FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology (Basel) 1, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning KR, Russell DL, and Robker RL (2014). Lipids and oocyte developmental competence: the role of fatty acids and ß-oxidation. Reproduction 148, R15–R27. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, and Levanon EY (2013). Human housekeeping genes, revisited. Trends Genet. 29, 569–574. [DOI] [PubMed] [Google Scholar]
- Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, et al. (2013). FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell 13, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LWS, Vardhana SA, Carey BW, Alonso-Curbelo D, Koche R, Chen Y, Wen D, King B, Radler MR, Rafii S, et al. (2018). Pluripotency transcription factors and Tet1/2 maintain Brd4-independent stem cell identity. Nat. Cell Biol. 20, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viukov S, Maza I, Zviran A, et al. (2013). Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, and Izpisiua Belmonte JC (2008). Albumin-associated lipids regulate human embryonic stem cell self-renewal. PLoS ONE 3, e1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, Braas D, Plath K, Lowry WE, and Christofk HR (2016). Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 19, 476–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, Lepikhov K, Gut M, Brun-Heath I, Hubner NC, et al. (2013). Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 13, 360–369. [DOI] [PubMed] [Google Scholar]
- Hackett JA, and Surani MA (2014). Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell 15, 416–430. [DOI] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, and Jaenisch R (2010). Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 107, 9222–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel S, Tu EY, Weisberg S, Esquilin M, Chambers SM, Liu B, Carson CT, Studer L, Reizis B, and Tomishima MJ (2012). ZFX controls the self-renewal of human embryonic stem cells. PLoS ONE 7, e42302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton FD, Thompson JG, Kennedy CJ, and Leese HJ (1996). Oxygen consumption and energy metabolism of the early mouse embryo. Mol. Reprod. Dev. 44, 476–485. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Iglesias Gonzalez T, Cinti M, Montes-Bayon M, Fernandez de la Campa MR, and Blanco-Gonzalez E (2015). Reversed phase and cation exchange liquid chromatography with spectrophotometric and elemental/molecular mass spectrometric detection for S-adenosyl methionine/S-adenosyl homocysteine ratios as methylation index in cell cultures of ovarian cancer. J. Chromatogr. A 1393, 89–95. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Mahmood S, and Patel MS (2003). Intermediary metabolism and energetics during murine early embryogenesis. J. Biol. Chem. 278, 31457–31460. [DOI] [PubMed] [Google Scholar]
- Jun-Hao ET, Gupta RR, and Shyh-Chang N (2016). Lin28 and let-7 in the Metabolic Physiology of Aging. Trends Endocrinol. Metab. 27, 132–141. [DOI] [PubMed] [Google Scholar]
- Kahler DJ, Ahmad FS, Ritz A, Hua H, Moroziewicz DN, Sproul AA, Dusenberry CR, Shang L, Paull D, Zimmer M, et al. (2013). Improved methods for reprogramming human dermal fibroblasts using fluorescence activated cell sorting. PLoS ONE 8, e59867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisa F, Shiozawa S, Oda K, Yoshimatsu S, Nakamura M, Koya I, Kawai K, Suzuki S, and Okano H (2017). Naive-like ESRRB+ iPSCs with the Capacity for Rapid Neural Differentiation. Stem Cell Reports 9, 1825–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, et al. (2012). Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner F, and Rossant J (2010). The role of FGF/Erk signaling in pluripotent cells. Development 137, 3351–3360. [DOI] [PubMed] [Google Scholar]
- Lanner F, Lee KL, Sohl M, Holmborn K, Yang H, Wilbertz J, Poellinger L, Rossant J, and Farnebo F (2010). Heparan sulfation-dependent fibroblast growth factor signaling maintains embryonic stem cells primed for differentiation in a heterogeneous state. Stem Cells 28, 191–200. [DOI] [PubMed] [Google Scholar]
- Lee JH, Laronde S, Collins TJ, Shapovalova Z, Tanasijevic B, McNicol JD, Fiebig-Comyn A, Benoit YD, Lee JB, Mitchell RR, and Bhatia M (2017). Lineage-Specific Differentiation Is Influenced by State of Human Pluripotency. Cell Rep. 19, 20–35. [DOI] [PubMed] [Google Scholar]
- Leese HJ (2012). Metabolism of the preimplantation embryo: 40 years on. Reproduction 143, 417–427. [DOI] [PubMed] [Google Scholar]
- Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, Grabole N, Mansfield W, Nashun B, Knezovich JG, Smith A, et al. (2013). Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 20,311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin Y, and Chen G (2008). Embryoid body formation from human pluripotent stem cells in chemically defined E8 media. https://www.ncbi.nlm.nih.gov/books/NBK424234/. DOI: 10.3824/stembook.1.98.1. [DOI] [PubMed]
- Loenarz C, and Schofield CJ (2008). Expanding chemical biology of 2-ox-oglutarate oxygenases. Nat. Chem. Biol. 4, 152–156. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, and Thompson CB (2012). Metabolic regulation of epigenetics. Cell Metab. 16, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, and Stunnenberg HG (2012). The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeegan PJ, and Sturmey RG (2011). The role of fatty acids in oocyte and early embryo development. Reprod. Fertil. Dev. 24, 59–67. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402. [DOI] [PubMed] [Google Scholar]
- Muñoz Descalzo S, Rue P, Garcia-Ojalvo J, and Martinez Arias A (2012). Correlations between the levels of Oct4 and Nanog as a signature for naive pluripotency in mouse embryonic stem cells. Stem Cells 30, 2683–2691. [DOI] [PubMed] [Google Scholar]
- Nguyen LH, and Zhu H (2015). Lin28 and let-7 in cell metabolism and cancer. Transl. Pediatr. 4, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne TF (2000). Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 275, 32379–32382. [DOI] [PubMed] [Google Scholar]
- Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, Tsankov A, Shang L, Krumholz K, Jagadeesan P, et al. (2015). Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat. Methods 12, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, and Sabatini DM (2011). mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Cliff T, Dalton S, and Sartorelli V (2015). Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 17, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K, and Jaenisch R (2009). Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 5, 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9,676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, and Kume S (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, and Ng HH (2017). The metabolic programming of stem cells. Genes Dev. 31, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017). Formative pluripotency: the executive phase in a developmental continuum. Development 144, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, et al. (2015). The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 17, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturmey RG, Reis A, Leese HJ, and McEvoy TG (2009). Role of fatty acids in energy provision during oocyte maturation and early embryo development. Reprod. Domest. Anim. 44, 50–58. [DOI] [PubMed] [Google Scholar]
- Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, et al. (2014). Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158, 1254–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchieu J, Zimmer B, Fattahi F, Amin S, Zeltner N, Chen S, and Studer L (2017). A Modular Platform for Differentiation of Human PSCs into All Major Ectodermal Lineages. Cell Stem Cell 21, 399–410.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, and Teitell MA (2016). a-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 24, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen TW, Powell BE, Wang H, Mitalipova M, Faddah DA, Reddy J, Fan ZP, Maetzel D, Ganz K, Shi L, et al. (2014). Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 15, 471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen TW, Friedli M, He Y, Planet E, O’Neil RC, Markoulaki S, Pontis J, Wang H, louranova A, Imbeault M, et al. (2016). Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 19, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas F, Lehalle D, Mayer D, Rittirsch M, Stadler MB, Zinner M, Olivieri D, Vabres P, Duplomb-Jego L, De Bont E, et al. (2019). Lysosomal Signaling Licenses Embryonic Stem Cell Differentiation via Inactivation of Tfe3. Cell Stem Cell 24, 257–270.e8. [DOI] [PubMed] [Google Scholar]
- Wang H, and Dey SK (2006). Roadmap to embryo implantation: clues from mouse models. Nat. Rev. Genet. 7, 185–199. [DOI] [PubMed] [Google Scholar]
- Ware CB, Nelson AM, Mecham B, Hesson J, Zhou W, Jonlin EC, Jimenez-Caliani AJ, Deng X, Cavanaugh C, Cook S, et al. (2014). Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA 111, 4484–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger L, Ayyash M, Novershtern N, and Hanna JH (2016). Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169. [DOI] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. (2013). Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1131–1139. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. (2011). UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30, 4860–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Hanspers K, Kuchinsky A, Salomonis N, Xu D, and Pico AR (2012). Mosaic: making biological sense of complex networks. Bioinformatics 28, 1943–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Badur MG, Divakaruni AS, Parker SJ, Jäger C, Hiller K, Murphy AN, and Metallo CM (2016). Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Rep. 16, 1536–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, and Ruohola-Baker H (2012). HIF1a induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 31, 2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer B, Piao J, Ramnarine K,Tomishima MJ, Tabar V, and Studer L (2016). Derivation of Diverse Hormone-Releasing Pituitary Cells from Human Pluripotent Stem Cells. Stem Cell Reports 6, 858–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ChIP-seq and WGBS data used in this paper are available at NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under accession SRA: SRP104789.