

Abstract
Stroke is the 2nd leading cause of death worldwide and the leading cause of physical disability and cognitive issues. Although we have made progress in certain aspects of stroke treatment, the consequences remain substantial and new treatments are needed. Hypertension has long been recognised as a major risk factor for stroke, both haemorrhagic and ischaemic. The renin angiotensin system (RAS) plays a key role in blood pressure regulation and this, plus local expression and signalling of RAS in the brain, both support the potential for targeting this axis therapeutically in the setting of stroke. While historically, focus has been on suppressing classical RAS signalling through the angiotensin type 1 receptor (AT1R), the identification of a counter-regulatory axis of the RAS signalling via the angiotensin type 2 receptor (AT2R) and Mas receptor has renewed interest in targeting the RAS. This review describes RAS signalling in the brain and the potential of targeting the Mas receptor and AT2R in preclinical models of ischaemic stroke. The animal and experimental models, and the route and timing of intervention, are considered from a translational perspective.
Keywords: Renin angiotensin system, Ischaemic stroke, AT2R, Mas receptor, Ang-(1–7), Ang-(1–9), C21
Abbreviations: AD, Alzheimer's disease; Aβ), amyloid β; Ang I), angiotensin I; Ang II), angiotensin II; Ang-(1–7)), angiotensin-(1–7); Ang-(1–9)), angiotensin-(1–9); ACE), angiotensin converting enzyme; ACE2), angiotensin converting enzyme 2; ACEi), angiotensin converting enzyme inhibitor; ARB), angiotensin receptor blocker; AT1R), angiotensin II type 1 receptor; AT2R), angiotensin II type 2 receptor; BBB), blood brain barrier; BP), blood pressure; B2R), bradykinin type 2 receptor; BDNF), brain derived neurotrophic factor; CBF), cerebral blood flow; C21), compound 21; DIZE), diminazene aceturate; eNOS), endothelial nitric oxide synthase; ET-1), endothelin-1; ERK 1/2), extracellular signal related kinase 1/2; GPCR), G-protein coupled receptor; MrgD), Mas related GPCR type D; MAPKs), mitogen activated protein kinases; NOX), NAD(P)H oxidases; nNOS), neuronal nitric oxide synthase; NO), nitric oxide; pMCAO), permanent middle cerebral artery occlusion; ROS), reactive oxygen species; RAS), renin angiotensin system; SHR), spontaneously hypertensive rat; tPA), tissue plasminogen activator; tMCAO), transient middle cerebral artery occlusion
Highlights
-
•
There is an unmet need for novel therapies for treating ischaemic stroke.
-
•
The brain renin angiotensin system (RAS) provides a target for stroke treatment.
-
•
Cellular signalling via the counter regulatory RAS axis may aid brain repair.
-
•
Stimulating counter regulatory RAS is beneficial in preclinical stroke models.
-
•
Timing and route of delivery require to be addressed for clinical translation.
1. Introduction
Stroke is a leading cause of death and disability worldwide [1], and can result in the development of dementia in 30% of cases [2,3], yet treatment options for this condition are limited. Stroke can be caused by a ruptured cerebral blood vessel, known as haemorrhagic stroke, or more commonly by the blockage of a blood vessel within the brain, known as ischaemic stroke [4]. Starvation of the brain tissue of oxygen and glucose during ischaemia results in a pathophysiological cascade of damage consisting of ionic dysregulation, excitotoxicity, oxidative stress and inflammation [5]. Recanalization of the vessel, either pharmacologically using a clot busting drug known as tissue plasminogen activator (tPA) or by mechanical clot removal known as thrombectomy remain the only available clinical interventions. Different aspects of stroke pathophysiology have been previously targeted as neuroprotective strategies [6], but so far, aside from improvements achieved utilising thrombectomy [[7], [8], [9]] there has been no progress towards improving patient outcome following ischaemic stroke. It is recognised that reperfusion paradoxically results in further injury by reperfusion injury [10] but the benefit achieved through recanalization outweighs the cost associated with failure to restore blood flow. There is an ongoing quest to develop neuroprotective strategies to increase tissue salvage and improve functional outcome for patients post stroke and the hope is that the efforts of the preclinical stroke community to improve methodological rigor in experimental stroke research [[11], [12], [13]] will bring a novel neuroprotectant to fruition.
The renin angiotensin system (RAS) is a physiological system that maintains cardiovascular homeostasis through maintenance of arterial blood pressure (BP). Classical RAS signalling mediated by Angiotensin II (Ang II) via the Angiotensin II type 1 receptor (AT1R) increases systemic BP; however it also has further effects on a tissue level that are implicated in disease [[14], [15], [16]]. Hypertension is the key modifiable risk factor for stroke [17], systemic RAS blockade, with ACE inhibitors (ACEi) or Ang II type 1 receptor (AT1R) antagonists (angiotensin receptor blockers; ARBs), is a common therapy for treating hypertension [18] and with efficient BP control the risk of stroke is reduced [19,20]. However, the benefits of modulation of BP acutely post-stroke remains controversial [21]. Evidence of the existence of local expression and signalling of RAS within the brain may provide a potential therapeutic target for neurological or cerebrovascular disorders including ischaemic stroke, although whether this will be independent of BP modulation will be dependent on many factors including dose and route of delivery. This review will outline RAS receptor signalling and summarise the current available evidence for the potential benefit of counter-regulatory RAS receptor agonism in ischaemic stroke.
2. The Renin Angiotensin System
In the classical systemic RAS, the biologically active peptide, Ang II, is produced through the actions of the enzymes renin and angiotensin converting enzyme (ACE) and exerts its effects primarily through the AT1R causing increased BP via vasoconstriction, increased water and sodium uptake in the kidney directly and via aldosterone and vasopressin release, and stimulation of the thirst response. (Fig. 1 and associated references).
Fig. 1.

The classical renin angiotensin system. The protein angiotensinogen is constitutively released from the liver into the circulation [22]. In response to detection of reduced BP, reduced electrolytes or sympathetic innervation in the kidney [23], the enzyme renin is secreted from the kidney (red) which cleaves angiotensinogen to the decapeptide angiotensin I (Ang I). Angiotensin converting enzyme (ACE) is present on the endothelial wall of blood vessels in particular in the lung [24,25]. ACE cleaves Ang I to the octapeptide Ang II, which acts upon the angiotensin II type 1 receptor (AT1R) to increase BP and blood volume by vasoconstriction of blood vessel [26], stimulating aldosterone release from the adrenal gland (on top of the kidney) and hence increasing sodium and water uptake [27], and actions on the brain increasing arterial pressure by sympathetic innervation or to increase the thirst response and release vasopressin to increase water uptake [28,29]. ACE inhibitors (ACEi) or angiotensin receptor blockers (ARB) (orange) are BP lowering medications which block these elements of the RAS to prevent increased BP. Letters in peptides indicate the amino acid sequence. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
A counter-regulatory axis of this system exists [30], consisting of an alternative enzyme, ACE2, which produces the peptides, Ang-(1–9) and Ang-(1–7), and alternative receptors for these to act through, the Ang II type 2 receptor (AT2R) and Mas receptor respectively. This produces antagonistic effects to AT1R signalling. This is a simplistic summary of the RAS and counter-regulatory RAS but realistically the system is much more complex consisting of various other enzymes, peptides and receptors as outlined in Fig. 2.
Fig. 2.
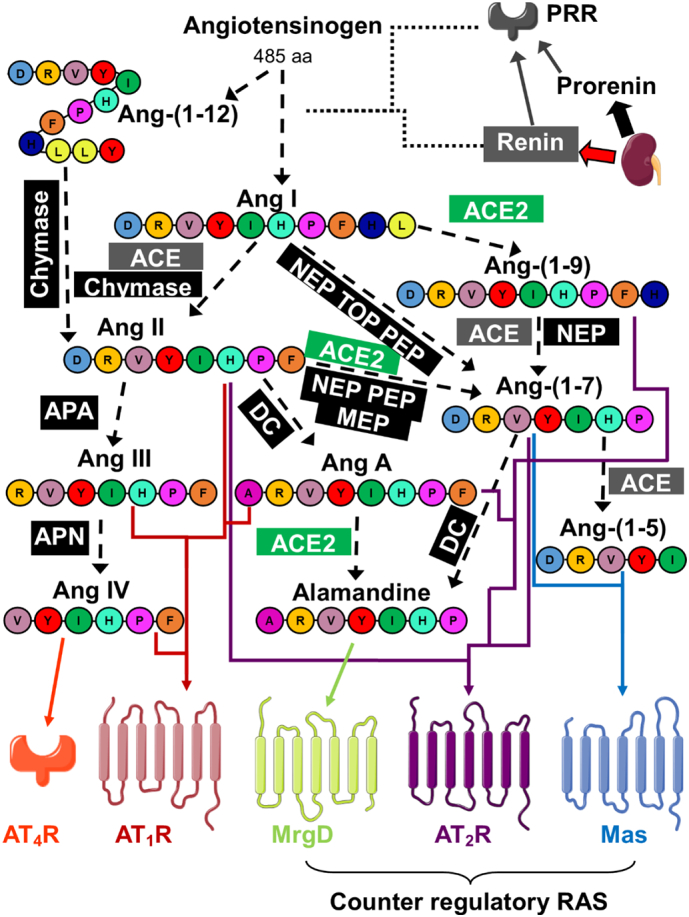
The extended RAS and counter-regulatory axis. Diagram illustrating the additional discoveries in the RAS. Prorenin is constitutively secreted from the kidney (black block arrow) while cleaved renin is secreted in response to stimuli (red block arrow) [31]. Renin acts directly on angiotensinogen to cleave it to Ang I, while both prorenin and renin can bind to the (pro)renin receptor (PRR) allowing increased cleavage activity of angiotensinogen [32]. The classic and counter-regulatory enzymes for angiotensin peptide cleavage, angiotensin converting enzyme (ACE) (grey) [33] and ACE2 (green) [34] are shown along with additional enzymes which result in peptide cleavage (black) [[35], [36], [37], [38], [39], [40], [41], [42], [43], [44]]. Dashed lines represent cleavage while coloured arrows indicate action of the peptide upon the colour coded receptors. Ang-(1–5), Ang III, Ang IV, Ang-(1–7) and Ang-(1–9) are all active peptides in the RAS [[45], [46], [47], [48], [49]]. Ang A shows a reduced vasoconstrictive effect through AT1R compared to Ang II and also acts upon the AT2R [50]. The angiotensin II type 2 receptor (AT2R), Mas receptor and Mas related GPCR type D (MrgD) receptor form the counter-regulatory axis of the RAS, opposing the signalling effects of AT1R [30]. Furthermore, AT1R and Mas [51], AT1R and AT2R [52], and AT2R and Mas [53] have been found to form signalling heterodimers. AT4R is not a GPCR like the other angiotensin receptors, but an enzyme, insulin-regulated membrane aminopeptidase (IRAP) [54]. Abbreviations: NEP, neprilysin; TOP, thimet oligopeptidase; PEP, prolyl-endopeptidases; MEP, metalloendopeptidases; APA, aminopeptidase A; APN, aminopeptidase N; DC, decarboxylase enzyme. Letters in peptides indicate the amino acid sequence. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. The RAS in the brain
The existence of RAS signalling in the brain has long been known; indeed early research on Ang II and Ang-(1–7) implied direct actions in the brain [[55], [56], [57]]. The central/brain RAS plays a role in the control of systemic BP mediated by sympathetic innervation [58], vasopressin release [59] and increased thirst [60], and inhibition of the central RAS, by intracerebroventricular injection of the prototype ARB saralasin, has been shown to reduce hypertension [61]. Although the majority of RAS research focusses on Ang II, it has been shown that conversion of Ang II to Ang III in the brain is responsible for the pressor response, where Ang II was ineffective when its conversion to Ang II was blocked with an aminopeptidase A (APA) inhibitor (Fig. 2), and thus Ang III may be the active peptide of classical RAS signalling in the central RAS [62]. Indeed, APA inhibitors are in clinical development for the treatment of hypertension (reviewed in [63]). In the brain, angiotensin immunoreactive nerve fibers have been mapped to demonstrate that they originate in circumventricular organs (CVO) (such as the subfornical organ, organum vasculosum of the lamina terminalis and area postrema). The CVO are areas of the brain which have no blood brain barrier (BBB) and therefore can directly interact with the circulation and circulating RAS peptides. The angiotensinergic neurons project to the paraventricular nucleus, supraoptic nucleus and the nucleus of the solitary tract [64,65] which are all neural circuits involved in fluid regulation, vasopressin release, sympathetic innervation and thirst response [66].
Furthermore, expression of the RAS components has been demonstrated within the brain, with renin activity demonstrated in the brain independent of circulating renin [67], angiotensinogen mRNA detected across the rat brain [68] and in both astrocytes [69] and neurons [70], ACE detected by radioligand binding in various regions of the human brain [71], and detection of Ang II and other angiotensin peptides (including Ang-(1–9) and Ang-(1–7)) in rat and sheep brains [72]. Following the discovery of ACE2, its expression was also confirmed within the brain at both a mRNA and protein level [73], and expression of the RAS receptors, AT1R, AT2R and Mas receptor, in the brain have been demonstrated by numerous studies [[74], [75], [76], [77], [78], [79], [80], [81], [82], [83]]. Recently, however, the existence of a locally expressed brain RAS has been questioned when van Thiel et al. demonstrated that, despite angiotensinogen mRNA expression in mouse brains, angiotensinogen protein was not detected, and perfusion of brains before analysis resulted in a marked reduction in renin activity, suggesting that the detected renin in brain tissues may just be circulating renin trapped within blood vessels in the tissues [84]. In contrast, using a newly developed microanalytical assay, coupling a laboratory-built capillary electrophoresis nano-electrospray ionization platform to a high-resolution mass spectrometer, several of the Ang peptides, including Ang I, Ang II, Ang-(1–7) and Ang-(1–9), were detected in the subfornical organ and the paraventricular nucleus of the hypothalamus in mice [85].
4. RAS receptor signalling
4.1. AT1R
The AT1R is a G-protein coupled receptor (GPCR) expressed throughout tissues of the body. In humans, a single gene exists for the receptor, however in rodents two pharmacologically indistinguishable isoforms exist, AT1AR and AT1BR [86]. The tissue expression of these isoforms varies, with AT1AR being the predominant isoform in all tissues, including brain, vasculature, lung and liver, except the adrenal and pituitary gland where AT1BR is the predominant isoform [87]. AT1R is regulated at a transcriptional level by being downregulated by Ang II [88] and can also be modified by numerous other factors such as insulin, glucose, estrogen, chemokines, nitric oxide (NO), reactive oxygen species (ROS) or low-density lipoprotein (LDL) cholesterol [89] with insulin, for example, increasing AT1R expression in vascular smooth muscle cells [90] and estrogen [91] and NO [92] leading to a down-regulation of AT1R in the hypothalmus or in neurons respectively. AT1R is also regulated by desensitisation to further activation by being rapidly internalised [93] or phosphorylated [94,95] upon activation, or, as is a common feature of GPCR signalling [96], AT1R can also be regulated by the formation of heterodimers with other receptors such as the bradykinin receptor (B2R) [97], the β-adrenergic receptor [98], the Mas receptor [51] or the AT2R [52].
Downstream signalling from Ang II at the AT1R involves activation of phospholipase C (PLC), phospholipase D (PLD) and phospholipase A2 (PLA2) along with activation of mitogen-activated protein kinases (MAPKs) and NADPH oxidases (NOXs), non-receptor tyrosine kinases (NRTKs) or receptor tyrosine kinases (RTKs) (Fig. 3 and associated references). In the systemic circulation, these signalling events result in contraction of vascular smooth muscle cells but also reactive oxygen species (ROS) production, inflammation, apoptosis, proliferation, protein synthesis, cell growth and migration influencing cell survival and pathological effects such as hypertrophy and fibrosis [16,99]. Indeed, much of the research on downstream signalling from AT1R has been conducted in vascular or cardiac cells, however Ang II mediated effects in the brain have also been attributed to some of these signalling pathways. For example, AT1R stimulation with Ang II in neuronal cultures results in activation of MAPKs [100] while AT1R blockade results in the reduction of MAPK activation, with MAPK activation being associated with microglial or astrocyte activation [101]. Within the brain, classical RAS signalling through AT1R leads to: vasopressin release [59] and suppression of the baroreflex, which has been shown to be mediated via PLC activation [102]; a switch towards the proinflammatory microglia phenotype with increased levels of proinflammatory cytokines such as tissue necrosis factor α (TNFα) or interleukin-1β (IL-1β) [103]; increased astrogliosis [104]; or in neurons, increased mitochondria-dependent apoptosis [105], increased production of ROS leading to apoptosis [106,107] or autophagy activation contributing to Ang II induced apoptosis via the AT1R [108]. Ang II induces ROS production in neurons via NOX activation which has been demonstrated to be mediated by protein kinase C (PKC) [109], but neuronal NO synthase (nNOS) is also upregulated by Ang II stimulation of neurons, contributing to oxidative stress and subsequent apoptosis induction [107]. PKC mediated activation of NOX leading to ROS production also results in microglial activation via activation of Rho-kinase by NFκB which additionally feeds back to further upregulate AT1R expression [110,111]. There is also evidence to suggest that Ang II stimulation of AT1R inhibits iron uptake in neurons [112,113] and iron metabolism dysregulation is linked to neurodegeneration [114].
Fig. 3.

AT1R signalling. Diagram depicting summary of intracellular signalling cascades induced by angiotensin II type 1 receptor (AT1R) activation by Ang II, or blocked by angiotensin receptor blockers (ARBs) or heterodimerisation with angiotensin II type 2 receptor (AT2R) or Mas receptor. Arrows indicate activation while dashed line arrows indicate production or cleavage to form a product. Signalling information obtained from [16,51,[121], [122], [123],52,107,[115], [116], [117], [118], [119], [120]]. Abbreviations: AA mets, arachidonic acid metabolites; AA, arachidonic acid; ASK1, apoptosis signal regulating kinase 1; DAG, diacylglycerol; ERK 1/2, extracellular signal related kinase 1/2; FAK, focal adhesion kinase; IP3, inositol trisphosphate; JAK, Janus kinase; JNK, c-Jun N-terminal kinase; MAPKs, mitogen activated protein kinases; NOX, NAD(P)H oxidases; nNOS; neuronal nitric oxide synthacse; NRTKs, non-receptor tyrosine kinases; p38, p38 MAPK; PA, phosphatidic acid; PC, phosphatidylcholine; PDGFR, platelet derived growth factor receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PLA2, phospholipase A2; PLC, phospholipase C; PLD, phospholipase D; ROS, reactive oxygen species; RTKs, receptor tyrosine kinases.
4.2. Mas receptor
The Mas receptor is a GPCR coded for by a gene originally identified as a proto-oncogene [124] but suggested early to code for an angiotensin receptor [125]. Like AT1R and other GPCRs, upon activation by Ang-(1–7), the Mas receptor is internalised and therefore desensitised to further activation [126]. Additionally, the Mas receptor can form heterodimers with other GPCRs, for example with AT2R which is necessary for signalling in some cell types [53], or with AT1R causing an inhibitory, regulatory effect on AT1R [51]. Ang-(1–7) is the heptapeptide product of ACE2 cleavage of Ang II or of ACE cleavage of Ang-(1–9) [34,39] (Fig. 2), which shows counter-regulatory effects to AT1R signalling [46] and mediates its effects through the Mas receptor [127]; although there have also been reports of effects mediated via the AT2R [128]. AVE 0991 is a nonpeptide, commercially available agonist at the Mas receptor, originally characterised in endothelial cells [129] while conversely, A779 acts as an antagonist of the Mas receptor [130].
Downstream signalling following Mas receptor activation by Ang-(1–7) includes activation of the phosphoinositide-3-kinase (PI3K) signalling pathway and endothelial NO synthase (eNOS) activation along with activation of tyrosine phosphatases, PLA2, protein kinase A (PKA) or MAPKs (Fig. 4 and associated references). The production of NO and arachidonic acid (AA) metabolites causes vasodilation in opposition to Ang II AT1R mediated vasoconstriction [127,131,132], while inhibition of MAPKs or Src attenuates remodelling in heart [[133], [134], [135]] or vasculature [136]. Conversely, however, the activation of MAPKs, such as ERK1/2 or p38 MAPK, by Mas receptor activation has been implicated in vasodilation and angiogenesis [137].
Fig. 4.
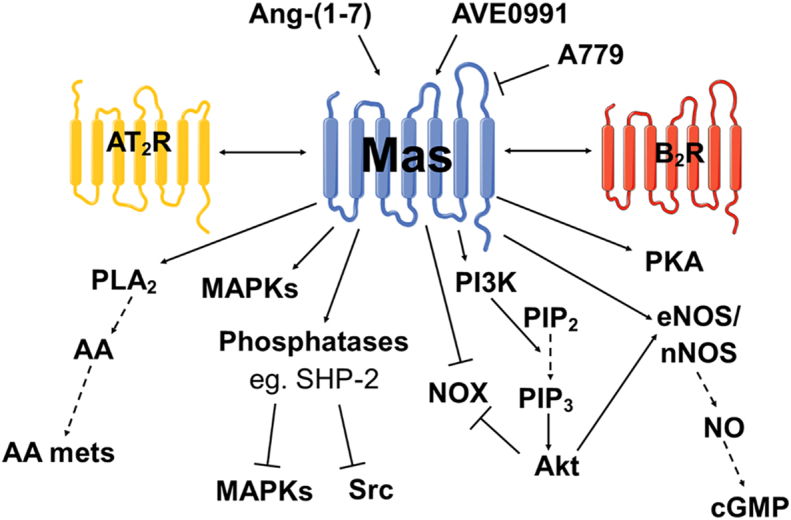
Mas receptor signalling. Diagram depicting summary of intracellular signalling cascades induced by Mas receptor (Mas receptor) activation. Agonists and antagonists are also shown along with receptors with which functional heterodimerisation can occur (AT2R and the bradykinin 2 receptor (B2R)). Arrows indicate activation while dashed line arrows indicate production or cleavage to form a product. Signalling information obtained from [127,131,166,167,[132], [133], [134],137,140,156,164,165]. Abbreviations: AA mets, arachidonic acid metabolites; AA, arachidonic acid; Akt, protein kinase B; cGMP, cyclic guanidine monophosphate; eNOS, endothelial nitric oxide synthase; MAPKs, mitogen activated protein kinases; NO, nitric oxide; PI3K, phosphoinositide-3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PKA: protein kinase A; PLA2, phospholipase A2; SHP-2, SH2 domain-containing protein tyrosine phosphatase-2.
Specifically in the brain, Ang-(1–7)/Mas has been shown to modulate central cardiovascular regulation with a BP and heart rate lowering effect when Ang-(1–7) was injected into the nucleus of the solitary tract (NTS) region within the dorsal medulla of rats [57]. This hypotensive effect is mediated by NO production [138,139], or more specifically the bradykinin dependent NO pathway of Mas signalling stimulating cyclic GMP (cGMP) and cGMP-dependent protein kinase (PKG), resulting in reduced norepinephrine release and reduced sympathetic innervation [140]. Conversely, however, central Ang-(1–7) administration into the ventrolateral medulla (VLM) of rats, either the rostral VLM (RVLM) [[141], [142], [143]] or the caudal pressor area (CPA) of the VLM [141,143], or the RVLM, but not caudal VLM, of rabbits [144] resulted in an increase in BP. Additionally, Ang-(1–7) can induce vasopressin release, although this may not be mediated by the Mas receptor [145]. It has also been suggested that Ang-(1–7)/Mas signalling in astrocytes rather than neurons may play an important role in cardiovascular regulation [146]. Taken together, these studies may indicate divergent effects of Ang-(1–7) depending on the brain region or angiotensin receptor expression in that area.
Aside from cardiovascular regulation however, Ang-(1–7) and Mas receptor signalling have demonstrated specific effects on brain cells such as: reduced nuclear and mitochondrial superoxide production [147]; improved neuronal survival and reduced oxidative stress in the brain mediated via PKA [148], or improved survival and reduced levels of ROS in rotenone induced injury of cultured neurons mediated by increased antioxidant activity and reduced NOX activity [149]; reduced microglial activation and astrogliosis allowing for improved neuronal density following traumatic brain injury [150]; reduced inflammation in astrocytes mediated by the MAPK inhibition signalling pathway [151]; reduced Shiga toxin 2 (Stx2) induced neuronal, astrocyte and oligodendrocyte damage [152]; or a switch to the anti-inflammatory M2 microglia phenotype with a reduction in pro-inflammatory cytokines (TNFα and IL1β) and increased levels of anti-inflammatory IL-10 [153]. Moreover, central Ang-(1–7) administration has demonstrated an increase in bradykinin levels and expression of bradykinin receptors [154] which may itself have neuroprotective effects [155]; or protection against cerebral endothelial cell dysfunction and oxidative stress via NOX inhibition and the PI3K/NO signalling pathway [156]. Additionally, Mas receptor signalling pathways (Fig. 4) have been demonstrated specifically in neurons with increased NO, AA release, MAPK activation and protein kinase B (PKB/Akt) activation with Ang-(1–7) treatment [157], or the downregulation of p38 MAPK and NOX coupled with the activation of PI3K and PKB/Akt resulting in increased levels of brain derived neurotrophic factor (BDNF), a neuronal survival promoting protein [158].
Finally, Ang-(1–7)/Mas signalling has also been implicated in learning and memory, with Ang-(1–7) increasing long-term potentiation in the hippocampus or amygdala via NO production [159,160], improving cognitive function in diabetic rats [161], or ACE2 activation improving cognition, coupled with reduced neuronal apoptosis, reduced inflammation and increased levels of BDNF, mediated via PI3K, in an Alzheimer's disease (AD) model [162]. Moreover Mas activation has demonstrated reduced anxiety like behaviour [163].
4.3. AT2R
The AT2R shares 34% homology with AT1R with a similar seven transmembrane GPCR structure [168]. Its expression is highest in fetal tissues and declines after birth [169] although more recently this concept was challenged with a study showing higher levels of AT2R protein in brainstem, liver and kidney in adult rats compared to fetal or neonatal rats [170]. The gene coding for AT2R has been mapped to the X chromosome [171] and studies in rodents have indicated a higher expression of AT2R in females compared to males due to upregulation by estrogen in the brain and heart [172,173], however, others have recently reported no overall sex related differences in AT2R, or AT1R, expression in the mouse brain [82]. Other regulators of AT2R expression include glucocorticoids and cytokines [174] or specifically, the nuclear protein poly(ADP-ribose) polymerase-1 (PARP1) [175] or transcription factor promyelocytic zinc finger protein (PLZF) [176] which regulate AT2R transcription. Further, the AT2R binding protein (ATBP)/AT2R interacting protein (ATIP) interacts with the C terminus of the receptor to promote its expression on the cell membrane [177,178]. Various other angiotensin peptides are reported to be ligands at the AT2R, namely Ang III, Ang IV, Ang-(1–7) and Ang-(1–9), although the affinity for Ang II is the highest [47,179]. Additionally, peptide (CGP42112 [180], β-Pro [7] Ang III [181]) and non-peptide (Compound 21, C21 [182]) agonists of AT2R have been developed which have further aided the understanding of AT2R signalling.
Unlike the AT1R and other GPCRs, the AT2R does not display internalization and desensitisation in response to ligand binding [183] and can therefore induce long term signalling effects. However, similar to other GPCRs, AT2R can form functional heterodimers for example with Mas receptor [53] or the B2R [184], and acts as an antagonist of AT1R by forming a heterodimer with this receptor independent of AT2R signalling [52]. In addition, AT2R forms homodimers causing constitutive activation without Ang II stimulation [185].
AT2R signalling is mediated through activation of protein phosphatases, or, similar to Mas receptor signalling, regulation of NO-cGMP, and activation of PLA2 leading to systemic effects opposing AT1R signalling such as vasodilation or anti-fibrotic or anti-hypertrophic effects [30] (Fig. 5 and associated references). Additionally, however, inhibition of MAPK activation due to phosphatase activity via AT2R signalling can result in apoptosis induction and this has been demonstrated in neuronal cells [186,187]. The induction of apoptosis by AT2R signalling occurs in tumor cells, thus implicating this receptor in possible cancer treatments [188,189], and also in Chinese hamster ovary and vascular smooth muscle cells [190]. In primary neuronal cultures, AT2R contributed to apoptosis induction only when in combination with other insults such as zinc treatment [191] or ultraviolet light exposure [187] suggesting additional complexities to AT2R signalling depending on the environment. Conversely, cell death was reduced in vitro in neurons subjected to glucose deprivation treated with the AT2R agonist, CGP42112 but not C21 [192]. Interestingly, however, in vivo delivery of CGP42112 (ip) [192] or C21 (daily ip injections) [193] resulted in reduced numbers of apoptotic neurons after experimental stroke in mice. Combined, these studies demonstrate AT2R stimulation may exert a beneficial effect through both apoptotic and anti-apoptotic mechanisms, perhaps dependent on the cell type affected or timing in terms of the balance between neuro-injury and neurorepair.
Fig. 5.

AT2R signalling. Diagram depicting summary of intracellular signalling cascades induced by angiotensin II type 2 receptor (AT2R) activation. Agonists and antagonists are also shown along with receptors with which functional heterodimerisation or homodimerization can occur. Arrows indicate activation while dashed line arrows indicate production or cleavage to form a product. Signalling information obtained from [53,100,219,[221], [222], [223], [224], [225], [226], [227],128,174,186,187,[203], [204], [205], [206]]. Abbreviations: AA mets, arachidonic acid metabolites; AA, arachidonic acid; Bcl-2, B-cell lymphoma 2; cGMP, cyclic guanidine monophosphate; eNOS, endothelial nitric oxide synthase; JNK; c-Jun N-terminal kinase; MAPKs, mitogen activated protein kinases; MKP-1, MAPK phosphatase-1; NO, nitric oxide; PLA2, phospholipase A2; PP2A, protein phosphatase 2A; SHP-1, Src homology region 2 domain-containing phosphatase.
Other effects of AT2R signalling in the brain are reduced ROS production [194], improved neuronal cell survival [195,196], a switch to the anti-inflammatory M2 microglia phenotype [110], and a reduction in the pro-inflammatory cytokine, TNFα, and increased levels of anti-inflammatory IL-10 [197,198]. Moreover, AT2R signalling in brain cells has been shown to directly oppose AT1R signalling; for example reducing Ang II induced NOX activity and subsequent ROS production in neuronal cultures [199], or activation of protein phosphatase 2A (PP2A) causing inhibition of PKC and the subsequent NOX activation and ROS production which would cause microglial activation [110]. In addition, the phosphatase mediated inhibition of MAPK signalling pathway of AT2R, namely PP2A inhibition of ERK1/2 activity, has been confirmed in neuronal cultures [100,200]. Interestingly, AT2R stimulation also modulates vasopressin release in the brain via synapse interaction of AT2R positive neurons with vasopressin secreting neurons, counteracting Ang II induced vasopressin release by AT1R [201].
Further effects of AT2R signalling in neurons include the modulation of neuronal excitability, through reduction of T-type calcium channel currents via phosphotyrosine phosphatase activity [202,203]. or increased potassium channel currents via PLA2, PP2A activation or NO production [[204], [205], [206]]; induction of neurite outgrowth and differentiation via NO production [[207], [208], [209], [210]] which may be via the PI3K/Akt pathway [211] or increased MAPK (ERK1/2) activity [209,212,213]; increased neuronal migration via PP2A activation [214]; or induction of neurogenesis and neural stem cell proliferation via Akt activation, ERK activation and modulation of potassium channels [215]. Conversely however, other neuronal cell lines have demonstrated reduced proliferation with AT2R stimulation [213].
The demonstration of local effects of both AT2R and Mas receptor signalling in the brain has led to the exploration of targeting the RAS for many neurological disorders, for example Parkinson's disease [216], depression [217], Alzheimer's Disease (AD) [218], cognitive impairment [219] or as reviewed here and elsewhere, the cerebrovascular disorder, stroke [220].
5. The classical RAS in stroke
Blockade of the systemic RAS is routinely used for the treatment of hypertension through the use of ARBs and ACEis [18] and therefore this allows for the analysis of the effect of these systemic drugs on stroke outcome by acute BP modulation or risk through long-term BP control. In the LIFE trial, treatment for hypertension with the ARB losartan resulted in 25% fewer strokes than with the β1 receptor antagonist (β-blocker) atenolol despite similar BP reductions [228], while the MOSES trial demonstrated fewer cerebrovascular and cardiovascular events over a 2.5 year follow-up period when patients who had suffered a stroke in the past two years were treated with the ARB eprosartan compared to the calcium channel blocker nitrendipine, despite similar BP reductions with both drugs [229]. In a randomized controlled trial comparing treatment with the ARB candesartan immediately following stroke or delayed until seven days later (ACCESS trial) there was no significant difference in functional outcome at three months, however early ARB treatment did significantly reduce the number of cardiovascular events [230]. An observational study of French stroke cohorts demonstrated no benefit of on-going ACEi or ARB treatment on stroke outcomes at 3 months [231]. However, in contrast to those studied described above, these patients also received thrombolysis with rt-PA. A meta-analysis of 26 clinical trials indicated that ARBs and ACEis offer no BP independent risk reduction for stroke [232].
Despite these varied results in clinical trials, there remains significant evidence that the brain RAS plays a role in ischaemic stroke pathophysiology and may offer potential as a therapeutic target. In experimental stroke models, intracerebroventricular infusion of an ARB resulted in beneficial outcomes of improved neurological score or decreased infarct size following transient middle cerebral artery occlusion (tMCAO), a commonly used model of ischaemia-reperfusion stroke injury, but did not lower the systemic BP due to the low dose utilised [233,234]. The dose was chosen to allow any BP-independent effects of the ARB to be identified and to avoid worsening outcome through aggressive BP lowering in the acute period after stroke [233,234]. Similarly, with systemic administration of an ARB dose too low to exert a BP lowering effect, pre- or post-tMCAO, reduced infarct volumes and improved functional outcomes were observed [[235], [236], [237], [238]]. The ACEi, ramipril, however, demonstrated no neuroprotective effects with systemic delivery prior to tMCAO [235]. Furthermore, vaccination with an anti-Ang II antibody demonstrated neuroprotective effects with reduced infarct volume following permanent occlusion of the middle cerebral artery (pMCAO), another commonly used stroke animal model [239]. Although the antibody did not appear to penetrate the brain tissue of control rats, it could penetrate the ischaemic lesion to block AT1R signalling in the brain, but did not affect BP [239]. Studies with transgenic mice have further corroborated the detrimental effect of AT1R signalling in ischaemic stroke. Mice over-expressing human renin and angiotensinogen genes displayed higher levels of Ang II in the circulation and the brain and, as a result, worse neurological deficit following pMCAO compared to wild-type which was prevented with prior ARB treatment [240]. Moreover, over-expression of angiotensinogen resulted in a smaller area of salvageable brain tissue (penumbra) at one hour and larger infarct volume at 24 h following pMCAO, while knockout (KO) of the AT1R resulted in the opposite effect [241]. Although BP was affected by the overexpression of angiotensinogen (increased) or AT1R KO (decreased) which may have impacted the resultant infarct, the authors further demonstrated beneficial effects of AT1R KO on cell death in an oxygen glucose deprivation in vitro model suggesting BP independent effects [241]. Together, these results indicate a detrimental effect of AT1R signalling in the setting of ischaemic stroke through mechanisms including neuroinflammation, ROS generation, apoptosis and neurodegeneration, all of which are independent of BP yet clinically, systemic blockade of AT1R has not been shown to consistently improve stroke outcome or risk. Therefore, perhaps the counter-regulatory axis of the RAS provides an alternative therapeutic target and indeed, there is increasing evidence that activation of the counter-regulatory RAS receptors is beneficial for stroke outcome (Table 1 & Table 2).
Table 1.
Summary of studies utilising Mas receptor agonism in experimental stroke. Abbreviations: AVE0991: Mas antagonist; BBB: blood brain barrier, BP: blood pressure, CBF: cerebral blood flow, COX-2: cyclooxygenase-2; DIZE: diminazene aceturate; eNOS: endothelial nitric oxide synthase ET-1: endothelin-1 injection, HPβCP-Ang-(1–7): Ang-(1–7) complexed with hydroxypropyl-β-cyclodextrins; ICV: intracerebroventricular, IL-1α/1β/6: interleukin-1α/1β/6; iNOS: inducible nitric oxide synthase; IP: intraperitoneal, IV: intravenous; MDA: malondialdehyde; NFκB: nuclear factor κ-light-chain-enhancer of activated B cells; NO: nitric oxide; NOX1: NADPH oxidase 1; SD: Sprague Dawley, SOD: superoxide dismutase; t/pMCAO: transient/permanent middle cerebral artery occlusion, TNF-α: tumor necrosis factor-α; VEGF: vascular endothelial growth factor.
| Animal. | Drug delivery dose, route & timing | Stroke model | Outcome | Mechanistic insight (if any) | Reference |
|---|---|---|---|---|---|
| Male SD rats | Ang-(1–7) 1.1 nM/0.5 μL/h or DIZE 5 μg/ 5 μL/h (ACE2 activator) ICV minipump infusion 7 days prior & 3 days post | ET-1 | Ang-(1–7): No effect BP No effect CBF ↓ infarct volume at 3 days ↑ neurological score & fine motor function at 3 days DIZE: ↓ BP with 7 days of treatment ↓ infarct volume at 3 days ↑ neurological score at 3 days |
Positive stroke outcomes blocked by Mas inhibition. Ang-(1–7) attenuated stroke induced increase in iNOS expression |
244 |
| Male SD rats | Ang-(1–7) 1.11 nM/1 μL/h ICV minipump infusion 48 h prior & 24 h post | Filament pMCAO | No effect CBF ↓ infarct volume at 24 h ↑ neurological score at 24 h |
Reduced oxidative stress (reduced MDA & increased SOD levels) Reduced NFκB activation Reduced expression of pro-inflammatory cytokines (TNF-α and IL-1β) and COX-2 Effects blocked by Mas inhibition but not AT2R inhibition |
246 |
| Male SD rats | Ang-(1–7) 1.1 nM/0.5 μL/h ICV minipump infusion 7 days prior & post until sacrifice | ET-1 | ↓ infarct volume at 24 h | Reduced expression of iNOS Blunting inflammatory response - reduced expression pro-inflammatory cytokines IL-1α & IL-6, & chemokine receptors CXCL12 & CXCR4, reduced expression of CD11b (marker of macrophage/microglial activation) In vitro evidence of reduction of NO production in glia, prevented by Mas inhibition |
248 |
| Male SD rats | Ang-(1–7) 1.1 nM/0.25 μL/h ICV minipump infusion 4 weeks prior to | Filament pMCAO | No effect BP prior to stroke ↑ peripheral CBF ↓ infarct volume at 24 h ↑ neurological score at 24 h |
Increased eNOS activation, NO production, VEGF levels and angiogenesis Positive stroke outcomes and signalling effects abolished by Mas or eNOS inhibition |
247 |
| Male C57BL6/J mice, 8 week old | AVE0991 10 mg/kg or 20 mg/kg IP at reperfusion & 4 h post | Filament tMCAO 60 min | No effect CBF No effect neurological deficit, or locomotor and motor coordination at 24 h No effect infarct volume at 24 h |
Reduced glucose deprivation induced neuronal death in vitro | 252 |
| Male SD rats, 9–13 weeks | HPβCP-Ang-(1–7) 125 μg/kg orally at 90 min, 4 h, 24 h & 48 h post hydroxypropyl-β-cyclodextrins, | ET-1 | ↓ infarct volume at 3 days ↑ neurological function at 3 days No effect CBF No effect BP |
Not investigated | 250 |
| Male Wistar rats | Ang-(1–7) 1.1 nmol/μL/h ICV minipump infusion post-reperfusion | Filament tMCAO, 90 min | ↑ tissue salvage at 7 days No effect infarct volume at 7 days No effect neurological score at 7 days No effect BP |
Upregulation of NOX1 Prevention of ‘steal phenomenon’ in contralateral hemisphere perfusion No effect on BBB permeability, microglia activation or inflammatory markers |
249 |
Table 2.
Summary of studies utilising AT2R agonism in experimental stroke. Abbreviations: Akt: protein kinase B; Aβ: β-amyloid; BBB: blood brain barrier, BDNF: brain derived neurotrophic factor; BP: blood pressure, C21: compound 21, CBF: cerebral blood flow, CGP42112: peptide agonist of AT2R; eNOS: endothelial nitric oxide synthase; ET-1: endothelin-1 injection, GAP43: growth associated protein B; ICV: intracerebroventricular, IL-1β/10: interleukin 1β/10; iNOS: inducible nitric oxide synthase; IP: intraperitoneal, IV: intravenous; KO: knockout; MCP-1: monocyte chemoattractant protein-1; mTOR: mammalian target of rapamycin; PI3K: phosphoinositide-3-kinase; PPAR-γ: peroxisome proliferator-activated receptor-γ; SD: Sprague Dawley, SHR: spontaneously hypertensive rat, t/pMCAO: transient/permanent middle cerebral artery occlusion, TNF-α: tumor necrosis factor-α; tPA: tissue plasminogen activator; TrkB: tropomyosin receptor kinase B; ZO-1: zona occludens protein 1.
| Animal. | Drug delivery dose, route & timing | Stroke model | Outcome | Mechanistic insight (if any) | Reference |
|---|---|---|---|---|---|
| Male SHR (15–16 week) | CGP42112, 0.1, 1 or 10 ng/kg/min, ICV minipump infusion 5 days prior +3 days post | ET-1, conscious | No effect BP. ↓ infarct volume at 3 days (1 & 10 ng doses). ↑ motor function at 1 & 3 days. |
Increased neuronal survival Increased AT2R expression Reduced superoxide production All effects abolished by AT2R blockade |
260 |
| Male SHR | CGP42112 3 μg/kg/dose, ICV bolus at 6, 24, 48 & 72 h post stroke | ET-1, conscious | No effect BP. ↓ infarct volume at 3 days. ↑ motor function at 1 & 3 days. |
Increased neuronal survival Reduced apoptosis Increased microglia activation All effects abolished by AT2R blockade |
261 |
| Male C57BL/6 mice (8–12 week) | CGP42112, 1 mg/kg, IP at reperfusion | Filament tMCAO, 30 min | ↑ CBF at reperfusion. ↑ neurological score and motor function at 24 h. ↓ infarct volume at 24 h. |
Reduced neuronal apoptosis in vitro prevented by AT2R blockade | 192 |
| Male SD (8 week) | C21, ICV 7.5 ng/μl/h minipump infusion for 7 days prior +3 days post OR IP 0.03 or 0.1 mg/kg prior to (2 h) and post(4, 24 & 48 h) OR IP 0.03 mg/kg 4, 24 & 48 h post |
ET-1 | ↓ infarct volume with all delivery schemes at 3 days. ↑ neurological score with all delivery schemes at 3 days. IP delivery no effect on non-stroked BP or CBF. |
Positive stroke outcome effects abolished by AT2R blockade Reduced expression of some pro-inflammatory genes: iNOS, CCL2 and CCR2 (chemokines) |
264 |
| Male SHR | C21, 50 ng/kg/min ICV minipump infusion 5 days prior & 3 days post OR 144 μg/kg ICV bolus at 6, 24, 48 & 72 h post |
ET-1, conscious | No effect on BP ↓ infarct at 3 days with pre- and post delivery. ↑ motor deficit at 1 day (pre-treatment only). |
Positive stroke outcome effects abolished by AT2R blockade Increased neuronal survival* Reduced apoptosis Increased microglia activation Suggestion of link between AT2R positive microglia and BDNF production Ex vivo vasorelaxation of cerebral arteries* *these effects also abolished by AT2R blockade |
263 |
| Male C57BL/6 mice (10–12 week) | C21, 10 μg/kg/day, IP after pMCAO, 24 h and daily thereafter | Electrocoagulation (pMCAO) | ↓ infarct volume at days 1–5. ↑ neurological score at days 3–7. ↑ CBF at days 1 & 3 ↓ BBB disruption at 3 days. |
AT2R KO mice had larger infarcts and infarcts not reduced by C21 Reduced superoxide production Reduced expression of proinflammatory cytokines TNF-α and MCP-1 |
262 |
| Male Wistar rats | C21, 0.03 mg/kg IP at reperfusion | Filament tMCAO 90 min or 3 h | No effect BP ↓ infarct volume at 24 h ↑ neurological score & motor function at 1–7 days |
Positive stroke outcome effects abolished by AT2R blockade Upregulation of AT2R and downregulation of AT1R expression Increased Akt and eNOS phosphorylation (pro-survival) Increased BDNF and IL-10 expression (neuroprotective) Decreased cleaved caspase-3 (apoptosis) Decreased nNOS, iNOS and nitrotyrosine (nitrative stress) Increased vascular density in vivo plus In vitro evidence of BDNF and AT2R dependent endothelial cell migration (pro-angiogenic) |
267 |
| Male C57BL/6 mice, WT or AT2R KO | C21 0.03 mg/kg IP 45 min post and daily thereafter | Filament tMCAO 30 min | No effect BP. No effect CBF. No effect infarct volume at 4 days. ↑ survival ↑ neurological score at day 1, 2 & 4 |
Increased levels of BDNF, its receptor TrkB and GAP43 (marker of neuronal outgrowth) Decreased neuronal apoptosis These effects plus improved neurological score were not seen with C21 treatment in AT2R KO mice |
193 |
| Male Wistar rats (8–12 week) | C21, 0.3 mg/kg/day, IP 6 h, 1, 2, 3, 4 & 5 days post | Filament pMCAO | ↑ neurological score at days 3 & 4 ↓ infarct volume at days 5 & 21 |
Increased VEGF expression mediated by mTOR dependent mechanism Confirmed in vitro neuronal cultures to be mediated by PI3K/Akt/mTOR pathway |
266 |
| Male SD rats (12 weeks) | CGP42112 IP 1 mg/kg per day post (specific timing not stated) | Filament tMCAO 2 h | ↓ infarct volume at 7 days | Increased AT2R expression Reduced expression of pro-inflammatory cytokines IL-1β & TNF-α. All effects abolished by AT2R blockade |
79 |
| Male SD (18–20 month) | C21 0.03 mg/kg IP 90 min, 1 & 2 days post | Filament tMCAO 45 min | ↑ neurological score and motor function from 1 to 21 days. ↓ infarct volume at 3 weeks. |
Not investigated | 268 |
| Male Wistar rats | C21 0.03 mg/kg IP at reperfusion | Filament tMCAO 3 h | ↓ infarct volume at 21 h. ↑ neurological score and motor function at 21 h. |
Positive outcomes partially mediated by IL-10 Reduced expression of pro-inflammatory TNF-α In vitro evidence of increased neuronal survival and reduced neuronal apoptosis |
195 |
| Male SHR (4 month) | C21 0.03 mg/kg/day IP at 2 h post | Filament tMCAO 60 min | No effect on BP. No effect neurological score and motor function at day 1 or 28. ↑ cognition at 21 days. |
Reduced Aβ accumulation In vitro evidence of Aβ toxicity to neurons and endothelial cells Reduced sustained (30 days post stroke) microglial activation |
265 |
| Male SD (10–13 week) | C21 1.5 μg/kg intranasal delivery 1.5, 4, 24 & 48 h post | ET-1 | No effect on BP. ↓ infarct volume at 3 days. ↑ neurological score at 1% 3 days. |
Not investigated | 271 |
| Male C57BL6/J mice | C21 10 μg/kg/day IP for 2 weeks prior | Filament pMCAO | No effect on BP. ↓ infarct volume at 24 h. ↑ neurological score at 24 h. ↑ CBF at 24 h. |
Positive outcomes partially mediated by PPAR-γ activation Reduced superoxide production and increased superoxide dismutase activity Increased expression of BBB stabilisation genes, occludin, claudin-5 and ZO-1 |
194 |
| Male Wistar rats (8–10 week) | C21 0.01, 0.03 or 0.06 mg/kg IV at 3 h OR 0.01 mg/kg at 3 h & 0.04 mg/kg/day orally days 2–5 alone or in combination with tPA |
Embolic | No effect on BP. ↑ neurological and motor function at days 3 or 5 (0.01 mg.kg dose). No effect infarct size. Trend towards ↑ cognition at 7 days. No effect when used in combination with tPA. |
Not investigated | 273 |
| Male Wistar rats, healthy or diabetic (12–15 weeks) | C21 0.12 mg/kg orally at day 3 post | Filament tMCAO 60 min | ↑ sensorimotor deficit at 1–8 weeks. ↑ cognition of diabetic rats at 1–8 weeks. Preserved brain volume at 8 weeks. |
No effect on number of activated microglia but shift towards the M2 anti-inflammatory phenotype In vitro evidence suggests microglial may be independent of AT2R |
270 |
| Male Wistar (14 month) | C21 0.12 mg/kg orally at 24 h post and daily thereafter | Distal pMCAO, electrocoagulation | ↓ weight loss No effect on neurological score or sensorimotor function at 28 days. ↑ cognition at 21 & 28 days. |
Reduced Aβ accumulation No effect on BDNF concentration |
272 |
| Female Wistar rats (3–6 month) | C21 0.03 mg/kg/day IP at reperfusion followed by daily IP or 0.12 mg/kg/day orally | Filament tMCAO 1, 2 or 3 h | Trend towards ↓ infarct volume at 24 h and 14 days ↑ neurological score and trends with sensorimotor function at 24 h. |
Trend towards increased PPAR-γ expression | 269 |
6. Mas receptor and stroke
Experimental models have demonstrated that Mas receptor and ACE2 mRNA are upregulated in the brain during cerebral ischaemia and, as a result, brain and plasma levels of Ang-(1–7) are also increased, suggesting that the Mas receptor/ACE2/Ang-(1–7) axis plays a role in ischaemic injury [75]. Additionally, further studies indicated that neuronal-specific ACE2 overexpression reduces infarct volume and improves neurological score after pMCAO and this is mediated by the Mas receptor [242]. Although ACE2 overexpression did result in a reduction in BP, titrating the systemic pressure to a similar level to that of control mice with norepinephrine demonstrated that the beneficial effects were independent of BP [242]. Similarly, activation of ACE2 by systemic injections [243] or intracerebroventricular infusion [244] of the ACE2 activator, diminazene aceturate (DIZE), pre- or post-stroke resulted in beneficial effects without affecting BP or cerebral blood flow (CBF) but the effect was abolished with Mas receptor blockade [244]. Interestingly, these studies with ACE2 only utilised the Mas receptor blocker A779 and did not assess the effect of AT2R blockade despite knowledge that Ang-(1–9) is also produced by ACE2 [34]; although reports of Ang-(1–9) being an active peptide of the counter-regulatory RAS were only beginning to emerge around this time [47,245]. Use of the AT2R antagonist PD123,319 might indicate whether Ang-(1–9) is involved in any of the effects of ACE2 activation, although interpreting results might be challenging in this setting due to an ACE2 activator altering levels of many different angiotensin peptides. For example, Ang-(1–7) is also reported to utilise the AT2R [128] while PD123,319 is also known to block MrgD, the receptor for the alternative angiotensin peptide alamandine [48]. Coupled with the knowledge of heterodimerization of different angiotensin receptors [[51], [52], [53]] interpretation of data following use of PD123,319 might be complex.
Beneficial effects of delivery of Ang-(1–7), acting via Mas receptor and not AT2R, have also been demonstrated post-stroke in experimental models with both delivery prior to stroke induction [244,[246], [247], [248]] and after reperfusion [249,250] (Table 1). Time of delivery varied from 48 h [246], 7 days [244,248] to 4 weeks [247] prior to stroke, and continued to the study endpoint, typically 24 h [[246], [247], [248]] out to 3 days [244]. In those studies using post-stroke administration, treatment began as early as 90 min after occlusion (to coincide with reperfusion) [249] or at 90 min, 4/24/48 h after stroke [250]. Moreover, beyond the setting of ischaemic stroke, Ang-(1–7) treatment also improved survival and reduced haemorrhages in the stroke prone spontaneously hypertensive rat which is a model of spontaneous haemorrhagic stroke [251]. The non-peptide Mas receptor agonist, AVE 0991, has had less consistent results with improved outcome when delivered intranasally in a subarachnoid hemorrhage model attributed to reduced oxidative stress and reduced apoptosis [148] but no benefits observed with systemic delivery after stroke in an ischaemic stroke model [252]. In the latter study, the authors attribute the failure to dosing or timing issues considering they observed neuroprotective effects in an in vitro glucose deprivation model [252] and previous studies have demonstrated its ability to cross the blood brain barrier due to its hydrophobic nature [253].
The mechanisms behind Mas receptor neuroprotection in ischaemia reperfusion has been attributed to increased levels of bradykinin or the bradykinin receptors [154], reduced levels of ROS [246]; mediation of anti-inflammatory effects for example by inhibition of NFκB resulting in reduction of IL1β or TNFα [153,246,248]; improved BBB stability due to increased tight junction protein expression and reduced expression of MMP9 via TIMP1 [254]; reduced levels of iNOS [244]; and production of NO which, aside from vasodilatory effects, can also induce pro-angiogenic signalling [247] (Table 1). Although activation of eNOS resulting in NO production is a downstream signalling pathway of Mas in the brain [255] that can have a beneficial effect on stroke outcome [247], activation of iNOS may be detrimental to stroke outcome [256] and Ang-(1–7)/Mas signalling has demonstrated a reduction in iNOS expression [244,248].
These studies suggest an encouraging potential for Mas agonism as a neuroprotective treatment following ischaemic stroke, however the majority of promising results were obtained with intracerebral delivery of Ang-(1–7) (Table 1) with just one study using a more translationally relevant approach through oral administration [250]. Furthermore, all studies used only male healthy rodent animal models (SD or Wistar rats or C57BL6/J mice) and so co-morbidity and sex were not considered. That said, several stroke models have been studied including permanent and transient MCAO using endothelin-1 (ET-1) or the intraluminal filament models. Therefore, collectively, these studies fall short of many of the STAIR criteria for stroke studies [11] and there is much further work required before targeting this receptor could become a reality as a treatment for stroke patients.
7. AT2R and stroke
The importance of AT2R signalling in cerebral ischaemia was demonstrated with AT2R KO mice which displayed larger infarct volumes, worsened neurological score and reduced CBF, following pMCAO, than wild-type (WT) mice [257]. AT2R KO did not affect BP in these mice and inhibition of AT1R was not sufficient to rescue the effects of AT2R KO but rather demonstrates that the beneficial effects of AT1R inhibition in stroke is, in part, due to indirect AT2R stimulation [257]. Transient ischaemia also resulted in larger infarcts in AT2R KO mice compared to WT suggesting an important role in protection of IR injury [193]. Furthermore, numerous studies have shown, via different methods, that AT2R is upregulated following both permanent or transient MCAO which further suggests an upregulation as an endogenous protective response to ischaemia [77,258,259].
Agonism of AT2R has also demonstrated promising results in experimental stroke models (Table 2). Initial proof of concept studies utilised intracerebroventricular delivery of the peptide agonist of AT2R, CGP42112. Delivery five days prior to tMCAO significantly reduced infarct volume and improved motor deficits coupled with increased AT2R expression, improved neuronal survival and reduced ROS production in the infarct area, all of which were prevented by AT2R blockade [260]. More encouragingly, commencement of intracerebroventricular delivery of CGP42112 after tMCAO resulted in similar beneficial effects on infarct volume, motor deficit and neuronal survival as before, and also reduced apoptosis in the infarct and peri-infarct regions and increased microglia activation which may be a protective mechanism to remove cellular debris [261]. In both studies, CGP42112 had no effect on the BP of the spontaneously hypertensive rats (SHR) utilised. Systemic administration of CGP42112 at the commencement of reperfusion also had beneficial effects on infarct volume and functional outcome in a mouse stroke model suggesting the peptide does not need to be delivered directly to the brain to have an effect [192].
Most of the research on the benefit of AT2R agonism in stroke has been conducted using the non-peptide agonist, C21 (Table 2). Once daily intraperitoneal (IP) injections of C21 in the two weeks prior to pMCAO in WT mice and AT2R KO mice demonstrated that C21 reduced infarct volume in WT but not KO mice [262]. Further, C21 delivery only after pMCAO also improved neurological score and infarct volume out to seven days post-stroke, with improved CBF, reduced ROS, BBB permeability and proinflammatory cytokines three days after stroke [262]. These promising results were further corroborated in hypertensive rats, where continuous intracerebroventricular delivery of C21 five days prior to tMCAO, or as four bolus doses beginning 6 h after tMCAO, resulted in reduced infarct volume and improved neuronal survival which was prevented by AT2R blockade [263]. However only the pre-treatment arm and not the post-treatment arm also demonstrated improved motor deficit at 24 h and increased microglial activation [263]. It was further demonstrated that C21, via AT2R, induced vasorelaxation of basilar arteries in ex vivo myography experiments [263]. This may translate to an in vivo protective mechanism to improve CBF, although there was no effect of C21 on the systemic BP of the SHR [263]. Furthermore, both before and after stroke, a high proportion of BDNF positive cells were also positive for AT2R implying that AT2R signalling is involved in BDNF release [263]. Studies have since had conflicting results with regard to the effect of C21 on CBF with some demonstrating no effect [193,264] while others suggest improvement [194], but BDNF mRNA and protein levels have been shown to be increased with C21 treatment post-stroke and this effect is absent with AT2R KO [193]. Further studies demonstrated that IP delivery of C21 after stroke induction, whether permanent or transient, improved outcome after stroke including in co-morbid animals with hypertension or advanced age [193,195,[265], [266], [267], [268]] (Table 2). Additionally, AT2R signalling with C21 has demonstrated further translational potential with promising results achieved with post-stroke oral delivery in female rats [269] or in a type 2 diabetes animal model [270], or with an intranasal delivery approach resulting in high levels detectable in the cortex and striatum, and improved outcome following stroke [271].
Mechanisms behind the beneficial effects observed with C21 induced AT2R signalling in experimental stroke have been attributed to reduced proinflammatory cytokines [262,264], reduced apoptosis [193,265,267], reduced ROS and oxidative stress [194,262,267], increased VEGF production [266], increased BDNF production [193,267], a switch from the M1 to M2 microglia phenotype [270], BBB stabilisation [194,262], and increased angiogenesis [267] (Table 2). Some of these effects have been demonstrated to be dependent on IL-10 [195] or peroxisome proliferator-activated receptor-gamma (PPARγ) activation [194] (Table 2).
In addition, C21 AT2R signalling has also demonstrated reduced haemorrhagic transformation [267] and decreased β-amyloid (Aβ) deposition following stroke [265,272]. Indeed, Aβ deposition is implicated in cognitive impairment and C21 treatment has demonstrated beneficial effects on post-stroke cognitive impairment (PSCI) in both hypertensive [265] and aged rats [272], and in an embolic model of stroke [273]. Although results were promising in the embolic model, only C21 alone induced sensorimotor and cognitive improvements but not tPA alone or in combination with C21, suggesting translatability issues with the model considering tPA is the only clinically approved drug for stroke treatment [273]. Interestingly however, a recent clinical trial for the potentially neuroprotective compound NA-1 had similar results where benefits were seen only in patients who did not receive tPA [274]. This raises the possible potential for the use of drugs that offer benefits in the absence of tPA as a new treatment option for those patients who are ineligible for tPA treatment.
Further to the benefits to PSCI mentioned above, AT2R agonism with C21 has also demonstrated beneficial effects on cognition in animals models utilising chronic hypoperfusion [[275], [276], [277]], Aβ injections in the brain (AD model) [278] or in a type 2 diabetes model [279]. C21 produced increased CBF [276,278,279], reduced proinflammatory cytokines [276], increased levels of BDNF in the brain [279] and reduced Aβ deposition [277] in these models and although no effect on overall vascular remodelling was observed, one study did demonstrate increased size of the basilar artery which could be responsible for increased blood supply to the hippocampus [275]. Furthermore, combination of C21 with memantine, an NMDA antagonist, and therefore glutamate toxicity modulator, used in AD, resulted in even greater levels of BDNF in the brain but no additive effect on CBF or cognition was observed [279].
Interestingly, although the brain RAS plays an important role in regulation of systemic BP and AT2R signalling can oppose vasoconstrictive AT1R signalling, AT2R agonism by CGP4112 or C21 does not affect BP when the agonist is delivered before [262] or after [261,267] MCAO either by systemic [264], intracerebral [260] or intranasal [271] delivery nor after embolic stroke [273]. However, contrasting studies have been reported a marked hypotensive response following central C21 administration into conscious normo- [280,281] or hypertensive [281] rats. Therefore, given the uncertainty surrounding BP lowering in acute ischaemic stroke care, the lack of BP effect by AT2R agonism in stroke is encouraging and reassuring but should be considered cautiously.
Clearly there is strong evidence to support the role of protective effects of AT2R signalling in the brain and the potential of targeting this receptor as a treatment for ischaemic stroke and possibly PSCI. Collectively, these studies demonstrate consideration of many of the STAIR guidelines criteria for preclinical stroke studies [11] arguably placing this potential novel therapeutic, C21, ahead of other strategies to target the counter regulatory axis of the RAS in the setting of stroke. Furthermore, recently C21 was shown to be safe and well-tolerated when administered orally in healthy adult male volunteers [282] and further ongoing trials with C21 in Raynaud's phenomenon (ClinicalTrials.gov Identifier: NCT04388176), idiopathic pulmonary fibrosis (ClinicalTrials.gov Identifier: NCT04533022) and COVID-19 (ClinicalTrials.gov Identifier: NCT04452435) will provide further evidence on its safety in human subjects.
8. Conclusion
Cellular signalling via the Mas receptor and AT2R of the counter regulatory axis of the RAS has been well established and provides multiple mechanisms to oppose negative signalling effects of the AT1R within the cardiovascular system. Additionally, many of these signalling mechanisms have been confirmed within the brain and could potentially aid neuroprotection and brain repair following stroke. Indeed, preclinical stroke studies utilising agonism of the counter regulatory axis of the RAS demonstrate consistently encouraging results across several experimental and animal models, although often the cellular signalling mechanism mediating the beneficial effect is not confirmed. Encouragingly, many of these studies have used dosing protocols which would align with the window of therapeutic intervention afforded through existing stroke treatments, tPA or intra-arterial thrombectomy. Further well designed preclinical studies, for example utilising mixed sex cohorts with co- or multi-morbidities and considering the polypharmacy associated with stroke patients may see progress in the targeting of the brain counter regulatory RAS axis in stroke patients.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
A. McFall was supported by a British Heart Foundation PhD studentship (FS/15/64/32035).
References
- 1.Johnson C.O. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:439–458. doi: 10.1016/S1474-4422(19)30034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hénon H. Poststroke dementia: incidence and relationship to prestroke cognitive decline. Neurology. 2001;57:1216–1222. doi: 10.1212/wnl.57.7.1216. [DOI] [PubMed] [Google Scholar]
- 3.Pohjasvaara T. Clinical determinants of poststroke dementia. Stroke. 1998;29:75–81. doi: 10.1161/01.str.29.1.75. [DOI] [PubMed] [Google Scholar]
- 4.Hankey G.J. Stroke. Lancet. 2017;389:641–654. doi: 10.1016/S0140-6736(16)30962-X. [DOI] [PubMed] [Google Scholar]
- 5.Moskowitz M.A., Lo E.H., Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamorro Á., Dirnagl U., Urra X., Planas A.M. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016;15:869–881. doi: 10.1016/S1474-4422(16)00114-9. [DOI] [PubMed] [Google Scholar]
- 7.Badhiwala J.H. Endovascular Thrombectomy for Acute Ischemic Stroke. JAMA. 2015;314:1832. doi: 10.1001/jama.2015.13767. [DOI] [PubMed] [Google Scholar]
- 8.Nogueira R.G. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018;378:11–21. doi: 10.1056/NEJMoa1706442. [DOI] [PubMed] [Google Scholar]
- 9.Albers G.W. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N. Engl. J. Med. 2018;378:708–718. doi: 10.1056/NEJMoa1713973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nour M., Scalzo F., Liebeskind D.S. Ischemia-Reperfusion Injury in Stroke. Interv. Neurol. 2013;1:185–199. doi: 10.1159/000353125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher M. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lapchak P.A., Zhang J.H., Noble-Haeusslein L.J. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl. Stroke Res. 2013;4:279–285. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Percie du Sert N. The IMPROVE Guidelines (Ischaemia Models: Procedural Refinements Of in Vivo Experiments) Journal of Cerebral Blood Flow and Metabolism. 2017;37 doi: 10.1177/0271678X17709185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volpe M. The renin-angiotensin system as a risk factor and therapeutic target for cardiovascular and renal disease. J. Am. Soc. Nephrol. 2002;13(Suppl. 3):S173–S178. doi: 10.1097/01.asn.0000032549.36050.78. [DOI] [PubMed] [Google Scholar]
- 15.Skov J., Persson F., Frøkiær J., Christiansen J.S. Tissue renin-angiotensin systems: A unifying hypothesis of metabolic disease. Front. Endocrinol. (Lausanne). 2014;5 doi: 10.3389/fendo.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta P.K., Griendling K.K. Angiotensin II cell signalling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. - Cell Physiol. 2007;292 doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 17.O’Donnell M.J. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. 2016;388:761–775. doi: 10.1016/S0140-6736(16)30506-2. [DOI] [PubMed] [Google Scholar]
- 18.NICE . NICE guideline [NG136] 2019. Hypertension in adults: diagnosis and management.https://www.nice.org.uk/guidance/ng136/chapter/Recommendations#choosing-antihypertensive-drug-treatment-for-people-with-or-without-type-2-diabetes Available at. (Accessed: 1st April 2020) [Google Scholar]
- 19.Law M.R., Morris J.K., Wald N.J. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: Meta-analysis of 147 randomized trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338(1245) doi: 10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss J. Benefits and harms of intensive blood pressure treatment in adults aged 60 years or older: A systematic review and meta-analysis. Annals of Internal Medicine. 2017;166:419–429. doi: 10.7326/M16-1754. [DOI] [PubMed] [Google Scholar]
- 21.Bath P.M., Appleton J.P., Krishnan K., Sprigg N. Blood Pressure in Acute Stroke: To Treat or Not to Treat: That Is Still the Question. Stroke. 2018;49:1784–1790. doi: 10.1161/STROKEAHA.118.021254. [DOI] [PubMed] [Google Scholar]
- 22.Deschepper C.F. Angiotensinogen. Kidney International. Vol. 46. Nature Publishing Group; 1994. Hormonal regulation and relative importance in the generation of angiotensin II; pp. 1561–1563. [DOI] [PubMed] [Google Scholar]
- 23.Davis J.O., Freeman R.H. Mechanisms regulating renin release. Physiological Reviews. 1976;56:1–56. doi: 10.1152/physrev.1976.56.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Riordan J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003;4:225. doi: 10.1186/gb-2003-4-8-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth M., Weitzman A.F., Piquilloud Y. Converting enzyme content of different tissues of the rat. Experientia. 1969;25:1247. doi: 10.1007/BF01897474. [DOI] [PubMed] [Google Scholar]
- 26.Touyz R.M., Schiffrin E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacological Reviews. 2000;52:639–672. [PubMed] [Google Scholar]
- 27.Mulrow P.J. Angiotensin II and aldosterone regulation. Regul. Pept. 1999;80:27–32. doi: 10.1016/s0167-0115(99)00004-x. [DOI] [PubMed] [Google Scholar]
- 28.McKinley M.J. The brain renin-angiotensin system: Location and physiological roles. International Journal of Biochemistry and Cell Biology. 2003;35:901–918. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 29.Coble J.P., Grobe J.L., Johnson A.K., Sigmund C.D. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: Importance of the subfornical organ. American Journal of Physiology - Regulatory Integrative and Comparative Physiology. 2015;308:R238–R249. doi: 10.1152/ajpregu.00486.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ocaranza M.P. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nature Reviews Cardiology. 2020;17:116–129. doi: 10.1038/s41569-019-0244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krop M., Lu X., Danser A.H.J., Meima M.E. The (pro)renin receptor. A decade of research: What have we learned? Pflugers Archiv European Journal of Physiology. 2013;465:87–97. doi: 10.1007/s00424-012-1105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen G. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skeggs L.T., Kahn J.R., Shumway N.P. The preparation and function of the hypertensin-converting enzyme. J. Exp. Med. 1956;103:295–299. doi: 10.1084/jem.103.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donoghue M. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 35.Pereira M.G.A.G. Angiotensin II-independent angiotensin-(1–7) formation in rat hippocampus: involvement of thimet oligopeptidase. Hypertens.Dallas Tex. 2013;1979(62):879–885. doi: 10.1161/HYPERTENSIONAHA.113.01613. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto K., Chappell M.C., Brosnihan K.B., Ferrario C. 1992. vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertens. (Dallas, Tex. 1979)19, 692–6. [DOI] [PubMed] [Google Scholar]
- 37.Welches W.R. Evidence that prolyl endopeptidase participates in the processing of brain angiotensin. J. Hypertens. 1991;9:631–638. doi: 10.1097/00004872-199107000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Rice G.I., Thomas D.A., Grant P.J., Turner A.J., Hooper N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004;383:45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vickers C. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277(14) doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 40.Zini S. Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: Predominant role of angiotensin III in the control of vasopressin release. Proc. Natl. Acad. Sci. U. S. A. 1996;93 doi: 10.1073/pnas.93.21.11968. 11,968–11,973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chappell M.C., Pirro N.T., Sykes A., Ferrario C.M. Metabolism of angiotensin-(1–7) by angiotensin-converting enzyme. Hypertens. Dallas Tex. 1998;1979(31):362–367. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- 42.Urata H., Nishimura H., Ganten D. Chymase-dependent angiotensin II forming system in humans. Am. J. Hypertens. 1996;9:277–284. doi: 10.1016/0895-7061(95)00349-5. [DOI] [PubMed] [Google Scholar]
- 43.Ahmad S. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1−12) metabolizing enzyme. Biochem. Biophys. Res. Commun. 2016;478:559–564. doi: 10.1016/j.bbrc.2016.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagata S. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem. Biophys. Res. Commun. 2006;350:1026–1031. doi: 10.1016/j.bbrc.2006.09.146. [DOI] [PubMed] [Google Scholar]
- 45.Yu L., Yuan K., Phuong H.T.A., Park B.M., Kim S.H. Angiotensin-(1–5), an active mediator of renin-angiotensin system, stimulates ANP secretion via Mas receptor. Peptides. 2016;86:33–41. doi: 10.1016/j.peptides.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 46.Santos R.A.S. Angiotensin-(1–7) and its receptor as a potential targets for new cardiovascular drugs. Expert Opinion on Investigational Drugs. 2005;14:1019–1031. doi: 10.1517/13543784.14.8.1019. [DOI] [PubMed] [Google Scholar]
- 47.Flores-Muñoz M., Smith N.J., Haggerty C., Milligan G., Nicklin S.A. Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J. Physiol. 2011;589:939–951. doi: 10.1113/jphysiol.2010.203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lautner R.Q. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013;112:1104–1111. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- 49.Le M.T. Angiotensin IV is a potent agonist for constitutive active human AT1 receptors: Distinct roles of the N- and C-terminal residues of angiotensin II during AT1 receptor activation. J. Biol. Chem. 2002;277 doi: 10.1074/jbc.C200201200. 23,107–23,110. [DOI] [PubMed] [Google Scholar]
- 50.Jankowski V. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007;27:297–302. doi: 10.1161/01.ATV.0000253889.09765.5f. [DOI] [PubMed] [Google Scholar]
- 51.Kostenis E. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–1813. doi: 10.1161/01.CIR.0000160867.23556.7D. [DOI] [PubMed] [Google Scholar]
- 52.AbdAlla S., Lother H., Abdel-tawab A.M., Quitterer U. The Angiotensin II AT2 Receptor Is an AT1 Receptor Antagonist. J. Biol. Chem. 2001;276(39):721–39,726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 53.Leonhardt J. Evidence for Heterodimerization and Functional Interaction of the Angiotensin Type 2 Receptor and the Receptor MAS. Hypertension. 2017;69:1128–1135. doi: 10.1161/HYPERTENSIONAHA.116.08814. [DOI] [PubMed] [Google Scholar]
- 54.Chai S.Y. The angiotensin IV/AT4 receptor. Cellular and Molecular Life Sciences. 2004;61:2728–2737. doi: 10.1007/s00018-004-4246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taquini A.C., Blaquier P.C., Bohr D.F. Neurogenic factors and angiotensin in etiology of hypertension. Am. J. Physiol. 1961;201:1173–1175. doi: 10.1152/ajplegacy.1961.201.6.1173. [DOI] [PubMed] [Google Scholar]
- 56.Scroop G.C., Lowe R.D. Central pressor effect of angiotensin mediated by the parasympathetic nervous system. Nature. 1968;220:1331–1332. doi: 10.1038/2201331a0. [DOI] [PubMed] [Google Scholar]
- 57.Campagnole-Santos M.J. Cardiovascular effects of angiotensin-(1–7) injected into the dorsal medulla of rats. Am. J. Physiol. - Hear. Circ. Physiol. 1989:257. doi: 10.1152/ajpheart.1989.257.1.H324. [DOI] [PubMed] [Google Scholar]
- 58.Allen A.M., Dampney R.A., Mendelsohn F.A. Angiotensin receptor binding and pressor effects in cat subretrofacial nucleus. Am. J. Physiol. 1988;255:H1011–H1017. doi: 10.1152/ajpheart.1988.255.5.H1011. [DOI] [PubMed] [Google Scholar]
- 59.Hogarty D.C., Speakman E.A., Puig V., Ian Phillips M. The role of angiotensin, AT1 and AT2 receptors in the pressor, drinking and vasopressin responses to central angiotensin. Brain Res. 1992;586:289–294. doi: 10.1016/0006-8993(92)91638-u. [DOI] [PubMed] [Google Scholar]
- 60.Jensen L.L., Harding J.W., Wright J.W. Role of paraventricular nucleus in control of blood pressure and drinking in rats. Am. J. Physiol. - Ren. Fluid Electrolyte Physiol. 1992;262 doi: 10.1152/ajprenal.1992.262.6.F1068. [DOI] [PubMed] [Google Scholar]
- 61.Phillips M.I. Lowering of hypertension by central saralasin in the absence of plasma renin. Nature. 1977;270:445–447. doi: 10.1038/270445a0. [DOI] [PubMed] [Google Scholar]
- 62.Reaux A. Aminopeptidase A inhibitors as potential central antihypertensive agents. Proc. Natl. Acad. Sci. U. S. A. 1999;96 doi: 10.1073/pnas.96.23.13415. 13,415–13,420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Llorens-Cortes C., Touyz R.M. Evolution of a New Class of Antihypertensive Drugs: Targeting the Brain Renin-Angiotensin System. Hypertension. 2020;75:6–15. doi: 10.1161/HYPERTENSIONAHA.119.12675. [DOI] [PubMed] [Google Scholar]
- 64.Llorens-Cortes C., Mendelsohn F.A.O. Organization and functional role of the brain angiotensin system. JRAAS - Journal of the Renin-Angiotensin-Aldosterone System. 2002;3 doi: 10.3317/jraas.2002.029. [DOI] [PubMed] [Google Scholar]
- 65.Lind W., Swanson L.W., Ganten D. Organization of Angiotensin II Immunoreactive Cells and Fibers in the Rat Central Nervous System. Neuroendocrinology. 1985;40:2–24. doi: 10.1159/000124046. [DOI] [PubMed] [Google Scholar]
- 66.Zimmerman C.A., Leib D.E., Knight Z.A. Neural circuits underlying thirst and fluid homeostasis. Nature Reviews Neuroscience. 2017;18:459–469. doi: 10.1038/nrn.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ganten D., Boucher R., Genest J. Renin activity in brain tissue of puppies and adult dogs. Brain Res. 1971;33:557–559. doi: 10.1016/0006-8993(71)90137-5. [DOI] [PubMed] [Google Scholar]
- 68.Lynch K.R., Hawelu-Johnson C.L., Guyenet P.G. Localization of brain angiotensinogen mRNA by hybridization histochemistry. Mol. Brain Res. 1987 doi: 10.1016/0169-328X(87)90008-8. [DOI] [PubMed] [Google Scholar]
- 69.Stornetta R.L., Hawelu-Johnson C.L., Guyenet P.G., Lynch K.R. Astrocytes synthesize angiotensinogen in brain. Science. 1988;242:1444–1446. doi: 10.1126/science.3201232. [DOI] [PubMed] [Google Scholar]
- 70.Kumar A., Rassoli A., Raizada M.K. Angiotensinogen gene expression in neuronal and glial cells in primary cultures of rat brain. J. Neurosci. Res. 1988;19:287–290. doi: 10.1002/jnr.490190302. [DOI] [PubMed] [Google Scholar]
- 71.Chai S.Y., McKenzie J.S., McKinley M.J., Mendelsohn F.A.O. Angiotensin converting enzyme in the human basal forebrain and midbrain visualized by in vitro autoradiography. J. Comp. Neurol. 1990;291:179–194. doi: 10.1002/cne.902910203. [DOI] [PubMed] [Google Scholar]
- 72.Lawrence A.C., Clarke L.J., Campbell D.J. Angiotensin Peptides in Brain and Pituitary of Rat and Sheep. J. Neuroendocrinol. 1992;4:237–244. doi: 10.1111/j.1365-2826.1992.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 73.Doobay M.F. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2007;292:R373. doi: 10.1152/ajpregu.00292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Young D., O’Neill K., Jessell T., Wigler M. Characterization of the rat mas oncogene and its high-level expression in the hippocampus and cerebral cortex of rat brain. Proc. Natl. Acad. Sci. U. S. A. 1988;85:5339–5342. doi: 10.1073/pnas.85.14.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu J. The expression of angiotensin-converting enzyme 2–angiotensin-(1–7)–Mas receptor axis are upregulated after acute cerebral ischemic stroke in rats. Neuropeptides. 2013;47:289–295. doi: 10.1016/j.npep.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 76.Lenkei Z., Palkovits M., Corvol P., Llorens-Cortès C. Expression of Angiotensin Type-1 (AT1) and Type-2 (AT2) Receptor mRNAs in the Adult Rat Brain: A Functional Neuroanatomical Review. Front. Neuroendocrinol. 1997;18:383–439. doi: 10.1006/frne.1997.0155. [DOI] [PubMed] [Google Scholar]
- 77.Kagiyama T., Kagiyama S., Phillips M.I. Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regul. Pept. 2003;110:241–247. doi: 10.1016/s0167-0115(02)00223-9. [DOI] [PubMed] [Google Scholar]
- 78.de Kloet A.D. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct. Funct. 2016;221:891–912. doi: 10.1007/s00429-014-0943-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma C.-Y., Yin L. Neuroprotective effect of angiotensin II type 2 receptor during cerebral ischemia/reperfusion. Neural Regen. Res. 2016;11:1102–1107. doi: 10.4103/1673-5374.187044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garrido-Gil P., Valenzuela R., Villar-Cheda B., Lanciego J.L., Labandeira-Garcia J.L. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: An intracellular renin-angiotensin system in the nigra. Brain Struct. Funct. 2013;218:373–388. doi: 10.1007/s00429-012-0402-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez-Pallares J. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008;31:58–73. doi: 10.1016/j.nbd.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 82.Sumners C. Brain angiotensin type-1 and type-2 receptors: cellular locations under normal and hypertensive conditions. Hypertens. Res. 2020;43:281–295. doi: 10.1038/s41440-019-0374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Freund M., Walther T. Von Bohlen Und Halbach, O. Immunohistochemical localization of the angiotensin-(1–7) receptor Mas in the murine forebrain. Cell Tissue Res. 2012;348:29–35. doi: 10.1007/s00441-012-1354-3. [DOI] [PubMed] [Google Scholar]
- 84.van Thiel B.S. Brain Renin-Angiotensin System: Does It Exist? Hypertens. DallasTex. 2017;1979(69):1136–1144. doi: 10.1161/HYPERTENSIONAHA.116.08922. [DOI] [PubMed] [Google Scholar]
- 85.Lombard-Banek C., Yu Z., Swiercz A.P., Marvar P.J., Nemes P. A microanalytical capillary electrophoresis mass spectrometry assay for quantifying angiotensin peptides in the brain. Anal. Bioanal. Chem. 2019;411:4661–4671. doi: 10.1007/s00216-019-01771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Greindling K.K., Lassegue B., Alexander R.W. Angiotensin Receptors and Their Therapeutic Implications. Annu. Rev. Pharmacol. Toxicol. 1996;36:281–306. doi: 10.1146/annurev.pa.36.040196.001433. [DOI] [PubMed] [Google Scholar]
- 87.Kitami Y., Okura T., Marumoto K., Wakamiya R., Hiwada K. Differential gene expression and regulation of type-1 angiotensin II receptor subtypes in the rat. Biochem. Biophys. Res. Commun. 1992;188:446–452. doi: 10.1016/0006-291x(92)92405-m. [DOI] [PubMed] [Google Scholar]
- 88.Lassègue B. Angiotensin II down-regulates the vascular smooth muscle AT1 receptor by transcriptional and post-transcriptional mechanisms: evidence for homologous and heterologous regulation. Mol. Pharmacol. 1995;48 [PubMed] [Google Scholar]
- 89.Nickenig G., Harrison D.G. The AT1-type angiotensin receptor in oxidative stress and atherogenesis. Part II: AT1 receptor regulation. Circulation. 2002;105:530–536. doi: 10.1161/hc0402.102619. [DOI] [PubMed] [Google Scholar]
- 90.Paukku K. Regulation of AT1R expression through HuR by insulin. Nucleic Acids Res. 2012;40:5250. doi: 10.1093/nar/gks170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O’Hagan T.S., Wharton W., Kehoe P.G. Interactions between estrogen and the renin angiotensin system - Potential mechanisms for gender differences in Alzheimer’s disease. American. Journal of Neurodegenerative Diseases. 2012;1:266–279. [PMC free article] [PubMed] [Google Scholar]
- 92.Sharma N.M., Zheng H., Li Y.F., Patel K.P. Nitric oxide inhibits the expression of AT 1 receptors in neurons. Am. J. Physiol. - Cell Physiol. 2012;302:C1162. doi: 10.1152/ajpcell.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Griendling K.K., Delafontaine P., Rittenhouse S.E., Gimbrone M.A., Alexander R.W. Correlation of receptor sequestration with sustained diacylglycerol accumulation in angiotensin II-stimulated cultured vascular smooth muscle cells. J. Biol. Chem. 1987;262(14):555–562. [PubMed] [Google Scholar]
- 94.Kai H. Agonist-induced phosphorylation of the vascular type 1 angiotensin II receptor. Hypertens. Dallas Tex. 1994;1979(24):523–527. doi: 10.1161/01.hyp.24.4.523. [DOI] [PubMed] [Google Scholar]
- 95.Oppermann M., Freedman N.J., Alexander R.W., Lefkowitz R.J. Phosphorylation of the type 1A angiotensin II receptor by G protein-coupled receptor kinases and protein kinase C. J. Biol. Chem. 1996;271(13):266–13,272. doi: 10.1074/jbc.271.22.13266. [DOI] [PubMed] [Google Scholar]
- 96.Terrillon S., Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.AbdAlla S., Lother H., Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407:94–98. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 98.Barki-Harrington L., Luttrell L.M., Rockman H.A. Dual inhibition of β-adrenergic and angiotensin II receptors by a single antagonist: A functional role for receptor-receptor interaction in vivo. Circulation. 2003;108:1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- 99.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circulation Research. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 100.Huang X.C., Richards E.M., Sumners C. Mitogen-activated protein kinases in rat brain neuronal cultures are activated by angiotensin II type 1 receptors and inhibited by angiotensin II type 2 receptors. J. Biol. Chem. 1996;271(15):635–15,641. doi: 10.1074/jbc.271.26.15635. [DOI] [PubMed] [Google Scholar]
- 101.Bhat S.A., Goel R., Shukla S., Shukla R., Hanif K. Angiotensin Receptor Blockade by Inhibiting Glial Activation Promotes Hippocampal Neurogenesis Via Activation of Wnt/β-Catenin Signalling in Hypertension. Mol. Neurobiol. 2018;55:5282–5298. doi: 10.1007/s12035-017-0754-5. [DOI] [PubMed] [Google Scholar]
- 102.Wong L., Polson J.W., Murphy D., Paton J.F.R., Kasparov S. Genetic and pharmacological dissection of pathways involved in the angiotensin II-mediated depression of baroreflex function. FASEB J. 2002;16:1595–1601. doi: 10.1096/fj.02-0099com. [DOI] [PubMed] [Google Scholar]
- 103.Torika N., Asraf K., Danon A., Apte R.N., Fleisher-Berkovich S. Telmisartan modulates glial activation: In vitro and in vivo studies. PLoS One. 2016;11 doi: 10.1371/journal.pone.0155823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ongali B. Transforming growth factor-β1 induces cerebrovascular dysfunction and astrogliosis through angiotensin II type 1 receptor-mediated signalling pathways. Can. J. Physiol. Pharmacol. 2018;96:527–534. doi: 10.1139/cjpp-2017-0640. [DOI] [PubMed] [Google Scholar]
- 105.Ou Z. Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. JRAAS - J. Renin-Angiotensin-Aldosterone Syst. 2016;17(1–8) doi: 10.1177/1470320316672349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao H.R. Angiotensin II Triggers Apoptosis Via Enhancement of NADPH Oxidase-Dependent Oxidative Stress in a Dopaminergic Neuronal Cell Line. Neurochem. Res. 2015;40:854–863. doi: 10.1007/s11064-015-1536-y. [DOI] [PubMed] [Google Scholar]
- 107.Chen J. Angiotensin II-induced mouse hippocampal neuronal HT22 cell apoptosis was inhibited by propofol: Role of neuronal nitric oxide synthase and metallothinonein-3. Neuroscience. 2015;305:117–127. doi: 10.1016/j.neuroscience.2015.07.076. [DOI] [PubMed] [Google Scholar]
- 108.Gao Q. Activation of Autophagy Contributes to the Angiotensin II-Triggered Apoptosis in a Dopaminergic Neuronal Cell Line. Mol. Neurobiol. 2016;53:2911–2919. doi: 10.1007/s12035-015-9177-3. [DOI] [PubMed] [Google Scholar]
- 109.Wang, G. et al. Nox2, Ca2+, and Protein Kinase C Play a Role in Angiotensin II-Induced Free Radical Production in Nucleus Tractus Solitarius. Hypertension 48, 482–489 (2006). [DOI] [PubMed]
- 110.Bhat S.A., Sood A., Shukla R., Hanif K. AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47 phox Phosphorylation by PP2A. Mol. Neurobiol. 2019;56:3005–3023. doi: 10.1007/s12035-018-1272-9. [DOI] [PubMed] [Google Scholar]
- 111.Rodriguez-Perez A.I., Borrajo A., Rodriguez-Pallares J., Guerra M.J., Labandeira-Garcia J.L. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia. 2015;63:466–482. doi: 10.1002/glia.22765. [DOI] [PubMed] [Google Scholar]
- 112.Chen Y.J., Qian Z.M., Sheng Y., Zheng J., Liu Y. Angiotensin II down-regulates transferrin receptor 1 and ferroportin 1 expression in Neuro-2a cells via activation of type-1 receptor. Neurosci. Lett. 2020;716:134,684. doi: 10.1016/j.neulet.2019.134684. [DOI] [PubMed] [Google Scholar]
- 113.Liu Y. Angiotensin II inhibits iron uptake and release in cultured neurons. Neurochem. Res. 2014;39:893–900. doi: 10.1007/s11064-014-1285-3. [DOI] [PubMed] [Google Scholar]
- 114.Ke Y., Qian Z.M. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurology. 2003;2:246–253. doi: 10.1016/s1474-4422(03)00353-3. [DOI] [PubMed] [Google Scholar]
- 115.Alexander R.W., Brock T.A., Gimbrone M.A.J., Rittenhouse S.E. Angiotensin increases inositol trisphosphate and calcium in vascular smooth muscle. Hypertens. Dallas, Tex. 1979;7:447–451. [PubMed] [Google Scholar]
- 116.Griendling K.K. Sustained diacylglycerol formation from inositol phospholipids in angiotensin II-stimulated vascular smooth muscle cells. J. Biol. Chem. 1986;261:5901–5906. [PubMed] [Google Scholar]
- 117.Lassegue B., Alexander R.W., Clark M., Akers M., Griendling K.K. Phosphatidylcholine is a major source of phosphatidic acid and diacylglycerol in angiotensin II-stimulated vascular smooth-muscle cells. Biochem. J. 1993;292:509–517. doi: 10.1042/bj2920509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Olson E.R., Shamhart P.E., Naugle J.E., Meszaros J.G. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertens. DallasTex. 2008;1979(51):704–711. doi: 10.1161/HYPERTENSIONAHA.107.098459. [DOI] [PubMed] [Google Scholar]
- 119.Barnett R.L. Angiotensin-mediated phosphatidylcholine hydrolysis and protein kinase C activation in mesangial cells. Am. J. Physiol. - Cell Physiol. 1993;265 doi: 10.1152/ajpcell.1993.265.4.C1100. [DOI] [PubMed] [Google Scholar]
- 120.Zafari A.M. Arachidonic Acid Metabolites Mediate Angiotensin II-Induced NADH/NADPH Oxidase Activity and Hypertrophy in Vascular Smooth Muscle Cells. Antioxidants Redox Signal. 1999;1:167–179. doi: 10.1089/ars.1999.1.2-167. [DOI] [PubMed] [Google Scholar]
- 121.Griendling K.K., Ushio-Fukai M., Lassègue B., Alexander R.W. Angiotensin II signalling in vascular smooth muscle: New concepts. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 122.Rao G.N., Lassegue B., Alexander R.W., Griendling K.K. Angiotensin II stimulates phosphorylation of high-molecular-mass cytosolic phospholipase A2 in vascular smooth-muscle cells. Biochem. J. 1994;299:197–201. doi: 10.1042/bj2990197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rao G.N. Activation of mitogen-activated protein kinases by arachidonic acid and its metabolites in vascular smooth muscle cells. J. Biol. Chem. 1994;269(32):586–591. [PubMed] [Google Scholar]
- 124.Young D., Waitches G., Birchmeier C., Fasano O., Wigler M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell. 1986;45:711–719. doi: 10.1016/0092-8674(86)90785-3. [DOI] [PubMed] [Google Scholar]
- 125.Jackson T.R., Blair L.A.C., Marshall J., Goedert M., Hanley M.R. The mas oncogene encodes an angiotensin receptor. Nature. 1988;335:437–440. doi: 10.1038/335437a0. [DOI] [PubMed] [Google Scholar]
- 126.Gironacci M.M. Angiotensin (1–7) induces MAS receptor internalization. Hypertens. Dallas Tex. 2011;1979(58):176–181. doi: 10.1161/HYPERTENSIONAHA.111.173344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Santos R.A.S. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. U. S. A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walters P.E., Gaspari T.A., Widdop R.E. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertens. Dallas Tex. 2005;1979(45):960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- 129.Wiemer G., Dobrucki L.W., Louka F.R., Malinski T., Heitsch H. AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1–7) on the endothelium. Hypertension. 2002;40:847–852. doi: 10.1161/01.hyp.0000037979.53963.8f. [DOI] [PubMed] [Google Scholar]
- 130.Peiró C. Endothelial dysfunction through genetic deletion or inhibition of the G protein-coupled receptor Mas: a new target to improve endothelial function. J. Hypertens. 2007;25:2421–2425. doi: 10.1097/HJH.0b013e3282f0143c. [DOI] [PubMed] [Google Scholar]
- 131.Sampaio W.O. Angiotensin-(1–7) Through Receptor Mas Mediates Endothelial Nitric Oxide Synthase Activation via Akt-Dependent Pathways. Hypertension. 2007;49:185–192. doi: 10.1161/01.HYP.0000251865.35728.2f. [DOI] [PubMed] [Google Scholar]
- 132.Muthalif M.M., Benter I.F., Uddin M.R., Harper J.L., Malik K.U. Signal transduction mechanisms involved in angiotensin-(1–7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. J. Pharmacol. Exp. Ther. 1998;284:388–398. [PubMed] [Google Scholar]
- 133.Tallant E.A., Ferrario C.M., Gallagher P.E. Angiotensin-(1–7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am. J. Physiol. - Hear. Circ. Physiol. 2005;289 doi: 10.1152/ajpheart.00941.2004. [DOI] [PubMed] [Google Scholar]
- 134.Mercure C. Angiotensin(1–7) blunts hypertensive cardiac remodelling by a direct effect on the heart. Circ. Res. 2008;103:1319–1326. doi: 10.1161/CIRCRESAHA.108.184911. [DOI] [PubMed] [Google Scholar]
- 135.McCollum L.T., Gallagher P.E., Ann Tallant E. Angiotensin-(1–7) abrogates mitogen-stimulated proliferation of cardiac fibroblasts. Peptides. 2012;34:380–388. doi: 10.1016/j.peptides.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tallant E.A., Diz D.I., Ferrario C.M. Antiproliferative actions of angiotensin-(1–7) in vascular smooth muscle. Hypertension. 1999;34:950–957. doi: 10.1161/01.hyp.34.4.950. [DOI] [PubMed] [Google Scholar]
- 137.Hoffmann B.R. Mechanisms of Mas1 Receptor-Mediated Signalling in the Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2017;37:433–445. doi: 10.1161/ATVBAHA.116.307787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu Z.T. The PI3K signalling-mediated nitric oxide contributes to cardiovascular effects of angiotensin-(1–7) in the nucleus tractus solitarii of rats. Nitric Oxide - Biol. Chem. 2016;52:56–65. doi: 10.1016/j.niox.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 139.Feng Y. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ. Res. 2010;106:373–382. doi: 10.1161/CIRCRESAHA.109.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gironacci M.M., Valera M.S., Yujnovsky I., Peña C. Angiotensin-(1–7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension. 2004;44:783–787. doi: 10.1161/01.HYP.0000143850.73831.9d. [DOI] [PubMed] [Google Scholar]
- 141.Fontes M.A.P. Evidence that angiotensin-(1–7) plays a role in the central control of blood pressure at the ventro-lateral medulla acting through specific receptors. Brain Res. 1994;665:175–180. doi: 10.1016/0006-8993(94)91171-1. [DOI] [PubMed] [Google Scholar]
- 142.Silva-Barcellos N.M., Frézard F., Caligiorne S., Santos R.A.S. Long-Lasting Cardiovascular Effects of Liposome-Entrapped Angiotensin-(1–7) at the Rostral Ventrolateral Medulla. Hypertension. 2001;38:1266–1271. doi: 10.1161/hy1201.096056. [DOI] [PubMed] [Google Scholar]
- 143.Silva L.C.S. Cardiovascular effects produced by micro-injection of angiotensin-(1–7) on vasopressor and vasodepressor sites of the ventrolateral medulla. Brain Res. 1993;613:321–325. doi: 10.1016/0006-8993(93)90920-i. [DOI] [PubMed] [Google Scholar]
- 144.Potts P.D., Horiuchi J., Coleman M.J., Dampney R.A.L. The cardiovascular effects of angiotensin-(1–7) in the rostral and caudal ventrolateral medulla of the rabbit. Brain Res. 2000;877:58–64. doi: 10.1016/s0006-8993(00)02626-3. [DOI] [PubMed] [Google Scholar]
- 145.Qadri F., Wolf A., Waldmann T., Rascher W., Unger T. Sensitivity of hypothalamic paraventricular nucleus to C- and N-terminal angiotensin fragments: vasopressin release and drinking. J. Neuroendocrinol. 1998;10:275–281. [PubMed] [Google Scholar]
- 146.Guo F. Astroglia are a possible cellular substrate of angiotensin(1–7) effects in the rostral ventrolateral medulla. Cardiovasc. Res. 2010;87:578–584. doi: 10.1093/cvr/cvq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Costa-Besada M.A. Paracrine and Intracrine Angiotensin 1-7/Mas Receptor Axis in the Substantia Nigra of Rodents, Monkeys, and Humans. Mol. Neurobiol. 2018;55:5847–5867. doi: 10.1007/s12035-017-0805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mo J. AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats. Redox Biol. 2019;20:75–86. doi: 10.1016/j.redox.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhou Y. Neuroprotective effect of angiotensin-(1–7) against rotenone-induced oxidative damage in CATH.a neurons. Toxicol. Vitr. 2018;50:373–382. doi: 10.1016/j.tiv.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 150.Janatpour Z.C., Korotcov A., Bosomtwi A., Dardzinski B.J., Symes A.J. Subcutaneous Administration of Angiotensin-(1–7) Improves Recovery after Traumatic Brain Injury in Mice. J. Neurotrauma. 2019;36:3115–3131. doi: 10.1089/neu.2019.6376. [DOI] [PubMed] [Google Scholar]
- 151.Moore E.D., Kooshki M., Metheny-Barlow L.J., Gallagher P.E., Robbins M.E. Angiotensin-(1–7) prevents radiation-induced inflammation in rat primary astrocytes through regulation of MAP kinase signalling. Free Radic. Biol. Med. 2013;65:1060–1068. doi: 10.1016/j.freeradbiomed.2013.08.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goldstein J. Angiotensin-(1–7) protects from brain damage induced by shiga toxin 2-producing enterohemorrhagic Escherichia coli. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2016;311:R1173–R1185. doi: 10.1152/ajpregu.00467.2015. [DOI] [PubMed] [Google Scholar]
- 153.Liu M., Shi P., Sumners C. Direct anti-inflammatory effects of angiotensin-(1–7) on microglia. J. Neurochem. 2016;136:163–171. doi: 10.1111/jnc.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lu J., Zhang Y., Shi J. Effects of intracerebroventricular infusion of angiotensin-(1–7) on bradykinin formation and the kinin receptor expression after focal cerebral ischemia-reperfusion in rats. Brain Res. 2008;1219:127–135. doi: 10.1016/j.brainres.2008.04.057. [DOI] [PubMed] [Google Scholar]
- 155.Ji B. Neuroprotection of bradykinin/bradykinin B2 receptor system in cerebral ischemia. Biomedicine and Pharmacotherapy. 2017;94:1057–1063. doi: 10.1016/j.biopha.2017.08.042. [DOI] [PubMed] [Google Scholar]
- 156.Xiao X. Angiotensin-(1–7) counteracts angiotensin II-induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp. Cell Res. 2015;336:58–65. doi: 10.1016/j.yexcr.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cerniello F.M., Silva M.G., Carretero O.A., Gironacci M.M. Mas receptor is translocated to the nucleus upon agonist stimulation in brainstem neurons from spontaneously hypertensive rats but not normotensive rats. Cardiovasc. Res. 2019 doi: 10.1093/cvr/cvz332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rabie M.A., Abd El Fattah M.A., Nassar N.N., El-Abhar H.S., Abdallah D.M. Angiotensin 1–7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-κB axis. Biochem. Pharmacol. 2018;151:126–134. doi: 10.1016/j.bcp.2018.01.047. [DOI] [PubMed] [Google Scholar]
- 159.Albrecht D. Angiotensin-(1–7)-induced plasticity changes in the lateral amygdala are mediated by COX-2 and NO. Learn. Mem. 2007;14:177–184. doi: 10.1101/lm.425907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hellner K., Walther T., Schubert M., Albrecht D. Angiotensin-(1–7) enhances LTP in the hippocampus through the G-protein-coupled receptor Mas. Mol. Cell. Neurosci. 2005;29:427–435. doi: 10.1016/j.mcn.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 161.Zhang D., Xiao Q., Luo H., Zhao K. Effects of angiotensin-(1–7) on hippocampal expressions of GFAP and GDNF and cognitive function in rats with diabetes mellitus. Nan Fang Yi Ke Da Xue Xue Bao. 2015;35:646–651. [PubMed] [Google Scholar]
- 162.Kamel A.S. Stimulation of ACE2/ANG(1–7)/Mas Axis by Diminazene Ameliorates Alzheimer’s Disease in the D-Galactose-Ovariectomized Rat Model: Role of PI3K/Akt Pathway. Mol. Neurobiol. 2018;55:8188–8202. doi: 10.1007/s12035-018-0966-3. [DOI] [PubMed] [Google Scholar]
- 163.Wang L. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology. 2016;105:114–123. doi: 10.1016/j.neuropharm.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Burghi V. Participation of gai-adenylate cyclase and ERK1/2 in mas receptor signalling pathways. Front. Pharmacol. 2019;10 doi: 10.3389/fphar.2019.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Meng Y. Angiotensin-Converting Enzyme 2/Angiotensin-(1–7)/Mas Axis Protects against Lung Fibrosis by Inhibiting the MAPK/NF-κB Pathway. Am. J. Respir. Cell Mol. Biol. 2014;50:723–736. doi: 10.1165/rcmb.2012-0451OC. [DOI] [PubMed] [Google Scholar]
- 166.Gallagher P.E., Ferrario C.M., Tallant E.A. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am. J. Physiol. - Cell Physiol. 2008;295(C1169) doi: 10.1152/ajpcell.00145.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lara L.S. PKA-mediated effect of MAS receptor in counteracting angiotensin II-stimulated renal Na + -ATPase. Arch. Biochem. Biophys. 2010;496:117–122. doi: 10.1016/j.abb.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 168.Mukoyama M. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 1993;268 24,539–24,542. [PubMed] [Google Scholar]
- 169.Shanmugam S., Corvol P., Gasc J.M. Angiotensin II type 2 receptor mRNA expression in the developing cardiopulmonary system of the rat. Hypertens. Dallas Tex. 1996;1979(28):91–97. doi: 10.1161/01.hyp.28.1.91. [DOI] [PubMed] [Google Scholar]
- 170.Yu L. Developmental changes in AT1 and AT2 receptor-protein expression in rats. JRAAS - J. Renin-Angiotensin-Aldosterone Syst. 2010;11:214–221. doi: 10.1177/1470320310379065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Koike G. Human type 2 angiotensin II receptor gene: Cloned, mapped to the X chromosome, and its mRNA is expressed in the human lung. Biochem. Biophys. Res. Commun. 1994;203:1842–1850. doi: 10.1006/bbrc.1994.2402. [DOI] [PubMed] [Google Scholar]
- 172.Liu Y. Unique expression of angiotensin type-2 receptor in sex-specific distribution of myelinated ah-type baroreceptor neuron contributing to sex-dimorphic neurocontrol of circulation. Hypertension. 2016;67:783–791. doi: 10.1161/HYPERTENSIONAHA.115.06815. [DOI] [PubMed] [Google Scholar]
- 173.Xue Q., Xiao D., Zhang L. Estrogen Regulates Angiotensin II Receptor Expression Patterns and Protects the Heart from Ischemic Injury in Female Rats1. Biol. Reprod. 2015;93 doi: 10.1095/biolreprod.115.129619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circulation Research. 1998;83:1182–1191. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- 175.Reinemund J. Poly(ADP-ribose) polymerase-1 (PARP-1) transcriptionally regulates angiotensin AT2 receptor (AT2R) and AT2R binding protein (ATBP) genes. Biochem. Pharmacol. 2009;77:1795–1805. doi: 10.1016/j.bcp.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 176.Senbonmatsu T. A novel angiotensin II type 2 receptor signalling pathway: possible role in cardiac hypertrophy. EMBO J. 2003;22:6471–6482. doi: 10.1093/emboj/cdg637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wruck C.J. Regulation of transport of the angiotensin AT2 receptor by a novel membrane-associated Golgi protein. Arterioscler. Thromb. Vasc. Biol. 2005;25:57–64. doi: 10.1161/01.ATV.0000150662.51436.14. [DOI] [PubMed] [Google Scholar]
- 178.Nouet, S. et al. trans-Inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J. Biol. Chem.279, 28,989–28,997 (2004). [DOI] [PubMed]
- 179.Bosnyak S. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. 2011;121:297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- 180.Israel A. Angiotensin II receptor subtypes and phosphoinositide hydrolysis in rat adrenal medulla. Brain Res. Bull. 1995;38:441–446. doi: 10.1016/0361-9230(95)02011-f. [DOI] [PubMed] [Google Scholar]
- 181.Del Borgo M. β-Pro7Ang III is a novel highly selective angiotensin II type 2 receptor (AT2R) agonist, which acts as a vasodepressor agent via the AT2R in conscious spontaneously hypertensive rats. Clin. Sci. 2015;129:505–513. doi: 10.1042/CS20150077. [DOI] [PubMed] [Google Scholar]
- 182.Wan Y. Design, Synthesis, and Biological Evaluation of the First Selective Nonpeptide AT2 Receptor Agonist. J. Med. Chem. 2004;47:5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 183.Hunyady L., Bor M., Balla T., Catt K.J. Identification of a cytoplasmic Ser-Thr-Leu motif that determines agonist- induced internalization of the AT1 angiotensin receptor. J. Biol. Chem. 1994;269(31):378–31,382. [PubMed] [Google Scholar]
- 184.Abadir P.M., Periasamy A., Carey R.M., Siragy H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertens. Dallas Tex. 2006;1979(48):316–322. doi: 10.1161/01.HYP.0000228997.88162.a8. [DOI] [PubMed] [Google Scholar]
- 185.Miura S.I., Karrik S.S., Saku K. Constitutively active homo-oligomeric angiotensin II type 2 receptor induces cell signalling independent of receptor conformation and ligand stimulation. J. Biol. Chem. 2005;280(18):237–18,244. doi: 10.1074/jbc.M500639200. [DOI] [PubMed] [Google Scholar]
- 186.Horiuchi M., Hayashida W., Kambe T., Yamada T., Dzau V.J. Angiotensin type 2 receptor dephosphorylates Bcl-2 by activating mitogen-activated protein kinase phosphatase-1 and induces apoptosis. J. Biol. Chem. 1997;272(19):022–19,026. doi: 10.1074/jbc.272.30.19022. [DOI] [PubMed] [Google Scholar]
- 187.Shenoy U.V., Richards E.M., Huang X.-C., Sumners C., Angiotensin I.I. Type 2 Receptor-Mediated Apoptosis of Cultured Neurons from Newborn Rat Brain. Endocrinology. 1999;140:500–509. doi: 10.1210/endo.140.1.6396. [DOI] [PubMed] [Google Scholar]
- 188.Ishiguro S. Involvement of angiotensin II type 2 receptor (AT2R) signalling in human pancreatic ductal adenocarcinoma (PDAC): a novel AT2R agonist effectively attenuates growth of PDAC grafts in mice. Cancer Biol. Ther. 2015;16:307–316. doi: 10.1080/15384047.2014.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li H. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol. Cancer Ther. 2009;8:3255–3265. doi: 10.1158/1535-7163.MCT-09-0237. [DOI] [PubMed] [Google Scholar]
- 190.Miura S.I., Karnik S.S. Ligand-independent signals from angiotensin II type 2 receptor induce apoptosis. EMBO J. 2000;19:4026–4035. doi: 10.1093/emboj/19.15.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Park M.H., Kim H.N., Lim J.S., Ahn J.S., Koh J.Y. Angiotensin II potentiates zinc-induced cortical neuronal death by acting on angiotensin II type 2 receptor. Mol. Brain. 2013;6:50. doi: 10.1186/1756-6606-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee S. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp. Transl. Stroke Med. 2012;4:16. doi: 10.1186/2040-7378-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schwengel K. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J. Mol. Med. 2016;94:957–966. doi: 10.1007/s00109-016-1406-3. [DOI] [PubMed] [Google Scholar]
- 194.Shan B.S. Attenuation of stroke damage by angiotensin II type 2 receptor stimulation via peroxisome proliferator-activated receptor-gamma activation. Hypertens. Res. 2018;41:839–848. doi: 10.1038/s41440-018-0082-9. [DOI] [PubMed] [Google Scholar]
- 195.Fouda A.Y., Pillai B., Dhandapani K.M., Ergul A., Fagan S.C. Role of interleukin-10 in the neuroprotective effect of the Angiotensin Type 2 Receptor agonist, compound 21, after ischemia/reperfusion injury. Eur. J. Pharmacol. 2017 doi: 10.1016/j.ejphar.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Grammatopoulos T.N. Angiotensin II protects cultured midbrain dopaminergic neurons against rotenone-induced cell death. Brain Res. 2005;1045:64–71. doi: 10.1016/j.brainres.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 197.Dai S.Y., Zhang Y.P., Peng W., Shen Y., He J.J. Central infusion of angiotensin II Type 2 receptor agonist compound 21 attenuates DOCA/NaCl-induced hypertension in female rats. Oxid. Med. Cell. Longev. 2016;2016 doi: 10.1155/2016/3981790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Khorooshi R. J. 2019. Angiotensin AT2 receptor–induced interleukin-10 attenuates neuromyelitis optica spectrum disorder–like pathology. Mult. Scler. [DOI] [PubMed] [Google Scholar]
- 199.Lu J. Angiotensin AT2 receptor stimulation inhibits activation of NADPH oxidase and ameliorates oxidative stress in rotenone model of Parkinson’s disease in CATH.a cells. Neurotoxicol. Teratol. 2015;47:16–24. doi: 10.1016/j.ntt.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 200.Huang X.-C., Richards E.M., Sumners C., Angiotensin I.I. Type 2 Receptor-Mediated Stimulation of Protein Phosphatase 2A in Rat Hypothalamic/Brainstem Neuronal Cocultures. J. Neurochem. 1995;65:2131–2137. doi: 10.1046/j.1471-4159.1995.65052131.x. [DOI] [PubMed] [Google Scholar]
- 201.De Kloet A.D. Angiotensin type-2 receptors influence the activity of vasopressin neurons in the paraventricular nucleus of the hypothalamus in male mice. Endocrinology. 2016;157:3167–3180. doi: 10.1210/en.2016-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Buisson B., Bottari S.P., de Gasparo M., Gallo-Payet N., Payet M.D. The angiotensin AT2 receptor modulates T-type calcium current in non-differentiated NG108-15 cells. FEBS Lett. 1992;309:161–164. doi: 10.1016/0014-5793(92)81086-2. [DOI] [PubMed] [Google Scholar]
- 203.Buisson B. A G protein is involved in the angiotensin AT2 receptor inhibition of the T-type calcium current in non-differentiated NG108-15 cells. J. Biol. Chem. 1995;270:1670–1674. doi: 10.1074/jbc.270.4.1670. [DOI] [PubMed] [Google Scholar]
- 204.Kang J., Posner P., Sumners C. Angiotensin II type 2 receptor stimulation of neuronal K+ currents involves an inhibitory GTP binding protein. Am. J. Physiol. - Cell Physiol. 1994;267 doi: 10.1152/ajpcell.1994.267.5.C1389. [DOI] [PubMed] [Google Scholar]
- 205.Gao J., Zhang H., Le K.D., Chao J., Gao L. Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am. J. Hypertens. 2011;24:724–730. doi: 10.1038/ajh.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhu M., Gelband C.H., Moore J.M., Posner P., Sumners C. Angiotensin II type 2 receptor stimulation of neuronal delayed-rectifier potassium current involves phospholipase A2 and arachidonic acid. J. Neurosci. 1998;18:679–686. doi: 10.1523/JNEUROSCI.18-02-00679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Côté F., Laflamme L., Payet M.D., Gallo-Payet N. Endocrine Research24, 403–407 Marcel Dekker Inc. 1998. Nitric oxide, a new second messenger involved in the action of angiotensin II on neuronal differentiation of NG108-15 cells. [DOI] [PubMed] [Google Scholar]
- 208.Tsutsumi Y. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gendron L., Côté F., Payet M.D., Gallo-Payet N. Nitric Oxide and Cyclic GMP Are Involved in Angiotensin II AT2 Receptor Effects on Neurite Outgrowth in NG108-15 Cells. Neuroendocrinology. 2002;75:70–81. doi: 10.1159/000048222. [DOI] [PubMed] [Google Scholar]
- 210.Zhao Y. Contribution of bradykinin and nitric oxide to AT2 receptor-mediated differentiation in PC12 W cells. J. Neurochem. 2003;85:759–767. doi: 10.1046/j.1471-4159.2003.01719.x. [DOI] [PubMed] [Google Scholar]
- 211.Hashikawa-Hobara N. Candesartan cilexetil improves angiotensin II type 2 receptor-mediated neurite outgrowth via the PI3K-Akt pathway in fructose-induced insulin-resistant rats. Diabetes. 2012;61:925–932. doi: 10.2337/db11-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gendron L., Oligny J.F., Payet M.D., Gallo-Payet N. Cyclic AMP-independent involvement of Rap1/B-Raf in the angiotensin II AT2 receptor signalling pathway in NG108-15 cells. J. Biol. Chem. 2003;278:3606–3614. doi: 10.1074/jbc.M202446200. [DOI] [PubMed] [Google Scholar]
- 213.Guimond M.O., Wallinder C., Alterman M., Hallberg A., Gallo-Payet N. Comparative functional properties of two structurally similar selective nonpeptide drug-like ligands for the angiotensin II type-2 (AT2) receptor. Effects on neurite outgrowth in NG108-15 cells. Eur. J. Pharmacol. 2013;699:160–171. doi: 10.1016/j.ejphar.2012.11.032. [DOI] [PubMed] [Google Scholar]
- 214.Kilian P. Angiotensin II type 2 receptor stimulation increases the rate of NG108-15 cell migration via actin depolymerization. Endocrinology. 2008;149:2923–2933. doi: 10.1210/en.2007-0313. [DOI] [PubMed] [Google Scholar]
- 215.Chao J., Yang L., Buch S., Gao L. Angiotensin II Increased Neuronal Stem Cell Proliferation: Role of AT2R. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Perez-Lloret S., Otero-Losada M., Toblli J.E., Capani F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson’s disease. Expert Opinion on Investigational Drugs. 2017;26:1163–1173. doi: 10.1080/13543784.2017.1371133. [DOI] [PubMed] [Google Scholar]
- 217.Vian J. The renin-angiotensin system: A possible new target for depression. BMC Medicine. 2017;15 doi: 10.1186/s12916-017-0916-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gebre A.K., Altaye B.M., Atey T.M., Tuem K.B., Berhe D.F. Targeting Renin-Angiotensin System Against Alzheimer’s disease. Frontiers in Pharmacology. 2018;9(440) doi: 10.3389/fphar.2018.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jackson L., Eldahshan W., Fagan S.C., Ergul A. Within the brain: The renin angiotensin system. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19030876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Arroja M.M.C., Reid E., McCabe C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Transl. Stroke Med. 2016;8:8. doi: 10.1186/s13231-016-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bedecs K. Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem. J. 1997;325:449–454. doi: 10.1042/bj3250449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nouet S., Nahmias C. Signal transduction from the angiotensin II AT2 receptor. Trends in Endocrinology and Metabolism. 2000;11:1–6. doi: 10.1016/s1043-2760(99)00205-2. [DOI] [PubMed] [Google Scholar]
- 223.Gohlke P., Pees C., Unger T. AT2 Receptor Stimulation Increases Aortic Cyclic GMP in SHRSP by a Kinin-Dependent Mechanism. Hypertension. 1998;31:349–355. doi: 10.1161/01.hyp.31.1.349. [DOI] [PubMed] [Google Scholar]
- 224.Ritter O. AT2 receptor activation regulates myocardial eNOS expression via the calcineurin-NF-AT pathway. FASEB J. 2003;17:283–285. doi: 10.1096/fj.02-0321fje. [DOI] [PubMed] [Google Scholar]
- 225.Savoia C. Angiotensin II/AT2 receptor-induced vasodilation in stroke-prone spontaneously hypertensive rats involves nitric oxide and cGMP-dependent protein kinase. J. Hypertens. 2006;24:2417–2422. doi: 10.1097/01.hjh.0000251902.85675.7e. [DOI] [PubMed] [Google Scholar]
- 226.Carrillo-Sepúlveda M.A. Emerging Role of Angiotensin Type 2 Receptor (AT2R)/Akt/NO Pathway in Vascular Smooth Muscle Cell in the Hyperthyroidism. PLoS One. 2013;8(61):982. doi: 10.1371/journal.pone.0061982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang G. Angiotensin ii type 2 receptor-coupled nitric oxide production modulates free radical availability and voltage-gated Ca2+ currents in NTS neurons. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2012;302:R1076. doi: 10.1152/ajpregu.00571.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dahlöf B. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): A randomized trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 229.Schrader J. Morbidity and Mortality After Stroke, Eprosartan Compared with Nitrendipine for Secondary Prevention: principal results of a prospective randomized controlled study (MOSES) Stroke. 2005;36:1218–1226. doi: 10.1161/01.STR.0000166048.35740.a9. [DOI] [PubMed] [Google Scholar]
- 230.Schrader J. The ACCESS Study: evaluation of Acute Candesartan Cilexetil Therapy in Stroke Survivors. Stroke. 2003;34:1699–1703. doi: 10.1161/01.STR.0000075777.18006.89. [DOI] [PubMed] [Google Scholar]
- 231.Gilliot S. Influence of on-going treatment with angiotensin-converting enzyme inhibitor or angiotensin receptor blocker on the outcome of patients treated with intravenous rt-PA for ischemic stroke. J. Neurol. 2018;265:1166–1173. doi: 10.1007/s00415-018-8827-6. [DOI] [PubMed] [Google Scholar]
- 232.Agodoa L. Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system. J. Hypertens. 2007;25:951–958. doi: 10.1097/HJH.0b013e3280bad9b4. [DOI] [PubMed] [Google Scholar]
- 233.Lou M. Sustained Blockade of Brain AT1 Receptors before and after Focal Cerebral Ischemia Alleviates Neurologic Deficits and Reduces Neuronal Injury, Apoptosis, and Inflammatory Responses in the Rat. J. Cereb. Blood Flow Metab. 2004;24:536–547. doi: 10.1097/00004647-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 234.Dai, W. J., Funk, A., Herdegen, T., Unger, T. & Culman, J. Blockade of central angiotensin AT(1) receptors improves neurological outcome and reduces expression of AP-1 transcription factors after focal brain ischemia in rats. Stroke30, 2391–8; discussion 2398–9 (1999). [DOI] [PubMed]
- 235.Krikov M., Thone-Reineke C., Müller S., Villringer A., Unger T. Candesartan but not ramipril pretreatment improves outcome after stroke and stimulates neurotrophin BNDF/TrkB system in rats. J. Hypertens. 2008;26:544–552. doi: 10.1097/HJH.0b013e3282f2dac9. [DOI] [PubMed] [Google Scholar]
- 236.Hosomi N. Angiotensin type 1 receptor blockage improves ischemic injury following transient focal cerebral ischemia. Neuroscience. 2005;134:225–231. doi: 10.1016/j.neuroscience.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 237.Mecca A.P., O’Connor T.E., Katovich M.J., Sumners C. Candesartan pretreatment is cerebroprotective in a rat model of endothelin-1-induced middle cerebral artery occlusion. Exp. Physiol. 2009;94:937–946. doi: 10.1113/expphysiol.2009.047936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kim C.K. Effect of Long-Term Treatment with Fimasartan on Transient Focal Ischemia in Rat Brain. Biomed Res. Int. 2015 doi: 10.1155/2015/295925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wakayama K. Angiotensin II Peptide Vaccine Protects Ischemic Brain Through Reducing Oxidative Stress. Stroke. 2017;48 doi: 10.1161/STROKEAHA.116.016269. [DOI] [PubMed] [Google Scholar]
- 240.Inaba S. Exaggeration of focal cerebral ischemia in transgenic mice carrying human renin and human angiotensinogen genes. Stroke. 2009;40:597–603. doi: 10.1161/STROKEAHA.108.519801. [DOI] [PubMed] [Google Scholar]
- 241.Walther T. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2002;16:169–176. doi: 10.1096/fj.01-0601com. [DOI] [PubMed] [Google Scholar]
- 242.Chen J. Neuronal over-expression of ACE2 protects brain from ischemia-induced damage. Neuropharmacology. 2014;79:550–558. doi: 10.1016/j.neuropharm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bennion D.M. Activation of the Neuroprotective Angiotensin-Converting Enzyme 2 in Rat Ischemic Stroke. Hypertens. Dallas Tex. 2015;1979(66):141–148. doi: 10.1161/HYPERTENSIONAHA.115.05185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mecca A.P. Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp. Physiol. 2011;96:1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ocaranza M.P. Angiotensin-(1–9) regulates cardiac hypertrophy in vivo and in vitro. J. Hypertens. 2010;28:1054–1064. doi: 10.1097/hjh.0b013e328335d291. [DOI] [PubMed] [Google Scholar]
- 246.Jiang T. Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br. J. Pharmacol. 2012;167:1520–1532. doi: 10.1111/j.1476-5381.2012.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Jiang T. Angiotensin-(1–7) induces cerebral ischaemic tolerance by promoting brain angiogenesis in a Mas/eNOS-dependent pathway. Br. J. Pharmacol. 2014;171:4222–4232. doi: 10.1111/bph.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Regenhardt R.W. Anti-inflammatory effects of angiotensin-(1–7) in ischemic stroke. Neuropharmacology. 2013;71:154–163. doi: 10.1016/j.neuropharm.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Arroja M.M.C. Assessing the effects of Ang-(1–7) therapy following transient middle cerebral artery occlusion. Sci. Rep. 2019;9:3154. doi: 10.1038/s41598-019-39102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Bennion D.M. Neuroprotection by post-stroke administration of an oral formulation of angiotensin-(1–7) in ischaemic stroke. Exp. Physiol. 2018;103:916–923. doi: 10.1113/EP086957. [DOI] [PubMed] [Google Scholar]
- 251.Regenhardt R.W. Centrally administered angiotensin-(1–7) increases the survival of stroke-prone spontaneously hypertensive rats. Exp. Physiol. 2014;99:442–453. doi: 10.1113/expphysiol.2013.075242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lee S. Effect of a Selective Mas Receptor Agonist in Cerebral Ischemia In Vitro and In Vivo. PLoS One. 2015;10 doi: 10.1371/journal.pone.0142087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Savergnini S.Q. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the mas receptor. Hypertension. 2010;56:112–120. doi: 10.1161/HYPERTENSIONAHA.110.152942. [DOI] [PubMed] [Google Scholar]
- 254.Wu J., Zhao D., Wu S., Wang D. Ang-(1–7) exerts protective role in blood-brain barrier damage by the balance of TIMP-1/MMP-9. Eur. J. Pharmacol. 2015;748:30–36. doi: 10.1016/j.ejphar.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 255.Zhang Y. Central administration of angiotensin-(1–7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats. Neuropeptides. 2008;42:593–600. doi: 10.1016/j.npep.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 256.Parmentier S. Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br. J. Pharmacol. 1999;127:546–552. doi: 10.1038/sj.bjp.0702549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Iwai M. Possible Inhibition of Focal Cerebral Ischemia by Angiotensin II Type 2 Receptor Stimulation. Circulation. 2004;110 doi: 10.1161/01.CIR.0000138848.58269.80. [DOI] [PubMed] [Google Scholar]
- 258.Li J. Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J. 2005;19:617–619. doi: 10.1096/fj.04-2960fje. [DOI] [PubMed] [Google Scholar]
- 259.Zhu Y.Z. Expression of angiotensin II AT2 receptor in the acute phase of stroke in rats. Neuroreport. 2000;11:1191–1194. doi: 10.1097/00001756-200004270-00009. [DOI] [PubMed] [Google Scholar]
- 260.McCarthy C.A., Vinh A., Callaway J.K., Widdop R.E. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;40:1482–1489. doi: 10.1161/STROKEAHA.108.531509. [DOI] [PubMed] [Google Scholar]
- 261.McCarthy C.A. Angiotensin II Type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension. 2012;60:1531–1537. doi: 10.1161/HYPERTENSIONAHA.112.199646. [DOI] [PubMed] [Google Scholar]
- 262.Min L.J. Direct stimulation of angiotensin II type 2 receptor initiated after stroke ameliorates ischemic brain damage. Am. J. Hypertens. 2014;27:1036–1044. doi: 10.1093/ajh/hpu015. [DOI] [PubMed] [Google Scholar]
- 263.McCarthy C.A. Direct angiotensin AT2 receptor stimulation using a novel AT2 receptor agonist, compound 21, evokes neuroprotection in conscious hypertensive rats. PLoS One. 2014;9 doi: 10.1371/journal.pone.0095762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Joseph J.P. The angiotensin type 2 receptor agonist Compound 21 elicits cerebroprotection in endothelin-1 induced ischemic stroke. Neuropharmacology. 2014;81:134–141. doi: 10.1016/j.neuropharm.2014.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ahmed H.A. RAS modulation prevents progressive cognitive impairment after experimental stroke: A randomized, blinded preclinical trial. J. Neuroinflammation. 2018;15 doi: 10.1186/s12974-018-1262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Mateos L., Perez-Alvarez M.J., Wandosell F. Angiotensin II type-2 receptor stimulation induces neuronal VEGF synthesis after cerebral ischemia. Biochim. Biophys. Acta - Mol. Basis Dis. 2016;1862:1297–1308. doi: 10.1016/j.bbadis.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 267.Alhusban A. Compound 21 is pro-angiogenic in the brain and results in sustained recovery after ischemic stroke. J. Hypertens. 2015;33:170–180. doi: 10.1097/HJH.0000000000000364. [DOI] [PubMed] [Google Scholar]
- 268.Bennion D.M. Post-stroke angiotensin II type 2 receptor activation provides long-term neuroprotection in aged rats. PLoS One. 2017;12 doi: 10.1371/journal.pone.0180738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Eldahshan W. Angiotensin II type 2 receptor stimulation with compound 21 improves neurological function after stroke in female rats: A pilot study. Am. J. Physiol. - Hear. Circ. Physiol. 2019;316:H1192–H1201. doi: 10.1152/ajpheart.00446.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Jackson L. Delayed Administration of Angiotensin II Type 2 Receptor (AT2R) Agonist Compound 21 Prevents the Development of Post-stroke Cognitive Impairment in Diabetes Through the Modulation of Microglia Polarization. Transl. Stroke Res. 2019 doi: 10.1007/s12975-019-00752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bennion D.M. Protective effects of the angiotensin II AT 2 receptor agonist compound 21 in ischemic stroke: A nose-to-brain delivery approach. Clin. Sci. 2018;132:581–593. doi: 10.1042/CS20180100. [DOI] [PubMed] [Google Scholar]
- 272.Ahmed H.A. Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals—A randomized double- blind pre-clinical study. Behav. Brain Res. 2019;359:560–569. doi: 10.1016/j.bbr.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ishrat T. Dose–response, therapeutic time-window and tPA-combinatorial efficacy of compound 21: A randomized, blinded preclinical trial in a rat model of thromboembolic stroke. J. Cereb. Blood Flow Metab. 2018 doi: 10.1177/0271678X18764773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hill M.D. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomized controlled trial. Lancet. 2020;395:878–887. doi: 10.1016/S0140-6736(20)30258-0. [DOI] [PubMed] [Google Scholar]
- 275.Füchtemeier M. Vascular change and opposing effects of the angiotensin type 2 receptor in a mouse model of vascular cognitive impairment. J. Cereb. Blood Flow Metab. 2015;35:476–484. doi: 10.1038/jcbfm.2014.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Iwanami J. Direct angiotensin II type 2 receptor stimulation by compound 21 prevents vascular dementia. J. Am. Soc. Hypertens. 2015;9:250–256. doi: 10.1016/j.jash.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 277.Ahmed H.A. Role of angiotensin system modulation on progression of cognitive impairment and brain MRI changes in aged hypertensive animals – A randomized double- blind pre-clinical study. Behav. Brain Res. 2018;346:29–40. doi: 10.1016/j.bbr.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Jing F. Direct stimulation of angiotensin II type 2 receptor enhances spatial memory. J. Cereb. Blood Flow Metab. 2012;32:248–255. doi: 10.1038/jcbfm.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Iwanami J. Possible synergistic effect of direct angiotensin II type 2 receptor stimulation by compound 21 with memantine on prevention of cognitive decline in type 2 diabetic mice. Eur. J. Pharmacol. 2014;724:9–15. doi: 10.1016/j.ejphar.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 280.Légat L., Brouwers S., Smolders I.J., Dupont A.G. Hypotensive response to angiotensin II type 2 receptor stimulation in the rostral ventrolateral medulla requires functional GABA-A receptors. Front. Neurosci. 2017;11:346. doi: 10.3389/fnins.2017.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Brouwers S., Smolders I., Wainford R.D., Dupont A.G. Hypotensive and sympathoinhibitory responses to selective central AT2 receptor stimulation in spontaneously hypertensive rats. Clin. Sci. 2015;129:81–92. doi: 10.1042/CS20140776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Steckelings U. Successful completion of a phase I, randomized, double-blind, placebo controlled, single ascending dose trial for the first in class angiotensin AT2-receptor agonist compound 21. J. Hypertens. 2017;35:e105–e106. [Google Scholar]