

Abstract

The neuronal ceroid lipofuscinoses (NCLs) are a family of rare lysosomal storage disorders. The most common form of NCL occurs in children harboring a mutation in the CLN3 gene. This form is lethal with no existing cure or treatment beyond symptomatic relief. The pathophysiology of CLN3 disease is complex and poorly understood, with current in vivo and in vitro models failing to identify pharmacological targets for therapeutic intervention. This study reports the characterization of the first CLN3 patient-specific induced pluripotent stem cell (iPSC)-derived model of the blood-brain barrier and establishes the suitability of an iPSC-derived neuron model of the disease to facilitate compound screening. Upon differentiation, hallmarks of CLN3 disease are apparent, including lipofuscin and subunit c of mitochondrial ATP synthase accumulation, mitochondrial dysfunction, and attenuated Bcl-2 expression. The model led to the identification of small molecules that cleared subunit c accumulation by mTOR-independent modulation of autophagy, conferred protective effects through induction of Bcl-2 and rescued mitochondrial dysfunction.
Keywords: drug discovery, CLN3 disease, phenotypic screening, autophagy, apoptosis, blood-brain barrier
The neuronal ceroid lipofuscinoses (NCLs) are a family of rare pediatric neurodegenerative diseases that are commonly referred to as Batten disease.1,2 The incidence of the NCLs is estimated at 1 in 12,500 in the United States.3 The disease presents with early vision problems and/or seizures which progress in severity. Children at the end-stage of the disease become blind, bedridden, spastic, and demented with poorly controlled seizures. Death follows in the teens or early twenties.2,4,5 Individual NCLs are classified by the gene that is altered in the disorder, resulting in 13 different subtypes.2−6 However, common pathological hallmarks exist across all NCLs. Storage material containing fat, but primarily protein deposits, builds up in the brain, retina, and other tissues. The storage material specifically locates to lysosomes as a secondary effect.7,8 Accelerated apoptosis, impaired autophagy, and secondary destructive inflammation have been documented.9,10 Accumulation of subunit c of mitochondrial ATP synthase in lysosome-derived organelles is a hallmark of the NCLs; however, the exact biochemical mechanism of accumulation is unknown.8,11 No cure for any of the NCLs has yet been realized.12−16 However, there is one pharmacological intervention available; the gene therapy Cerliponase (Brineura) for CLN2 disease which is an enzyme replacement therapy for tripeptidyl peptidase 1 (TPP1) that can slow or halt progression of the disease.17 There is an urgent and unmet medical need for the identification of new drug candidates that are disease-modifying, yet this is hampered by the largely unknown pathophysiology of NCL progression and a lack of phenotypic in vitro model systems for drug screening.
The CLN3 subtype of Batten disease, hereto referred to as CLN3 disease, is the most common. Mutations in the CLN3 gene result in reduced production, or dysfunction of the gene product protein, CLN3 protein, or CLN3p. This protein is primarily located in lysosomal membranes. Its exact function is unclear and how mutation in CLN3 results in expression of the CLN3 phenotype is poorly understood.18 Apoptosis, among other cellular death mechanisms, has been proposed as one of the mechanisms of neuronal death in the NCLs.19 Indeed, the role of apoptosis is implicated in neuronal cell death in many neurodegenerative diseases.20,21 One postulated function of the CLN3 protein, is modulation of the antiapoptotic protein Bcl-2,19,22,23 a protein located to the outer mitochondrial membrane which plays a key role in regulating cell death by controlling apoptotic protease (caspase) activation.24 Further, CLN3 has been proposed to modulate endogenous ceramide, resulting in suppression of apoptosis.23,25 Similarly, defects in autophagy contribute to the neurodegenerative process in many diseases, including CLN3 disease.26 Accumulation of lipofuscin and subunit c of mitochondrial ATP synthase in the lysosomes of CLN3 disease patients implicates dysfunctional autophagy.27−29 Indeed, the lysosomal storage disorders30 can be thought of as primarily autophagy disorders as lysosomes play a fundamental role in the autophagy pathway.31 We have previously disclosed compounds designed in our laboratory, based on the clinically approved nonopioid analgesic flupirtine,32 that are protective in CLN3 patient-derived lymphoblasts and healthy IMR90-c4-induced pluripotent stem cell (iPSC)-derived neurons.33−35 The compounds act on several processes,36 upregulating expression of Bcl-2 and suppressing ceramide levels while activating autophagy, directly countering two of the phenotypes of CLN3 disease.
While CLN3-patient-derived lymphoblasts, CLN3 knock-down immortalized cells, and primary neurons isolated from CLN3 mouse models provide useful in vitro models for pharmacological evaluation, they do not fully recapitulate the phenotype of human CLN3 neurons. The use of iPSCs to model neurodegenerative diseases in vitro has gained much recent interest. In particular, several studies highlighted the use of patient-derived iPSCs to model neurodegenerative diseases such as amyotrophic lateral sclerosis, adrenoleukodystrophy, the familial form of Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease.37 Such models provide a potential alternative to classical approaches. In addition to the presence of a cellular and molecular phenotype in iPSC-derived neurons, several studies have highlighted the presence of an abnormal phenotype at the blood-brain barrier (BBB) upon differentiation into brain microvascular endothelial cells (BMECs). Thus, the possible identification of a dysfunctional BBB in addition to a more established neuronal phenotype can provide an inclusive model that can account for assessing the permeability of drug candidates targeting such conditions early in the drug development process.38
Neural progenitor cells (NPCs) differentiated from CLN3 patient-derived iPSCs have been reported to display autophagy dysfunction, lysosomal dysfunction, and mitochondrial defects consistent with CLN3 disease. In a subsequent study, the CLN3 NPCs were used as a phenotypic model to validate the effect of a small molecule autophagy modifier, identified from a screen conducted in a homozygous CbCln3Δex7/8 cell line stably expressing GFP-LC3 transgene.29 Neural stem cells (NSCs) differentiated from CLN1 and CLN2 patient-derived iPSCs were found to exhibit the phenotype of reduced expression of the enzymes palmitoyl-protein thioesterase 1 (PPT1) or TPP1, respectively. These NSCs were used to evaluate the effect of two small molecules versus enzyme replacement therapy.39 Neurons differentiated from CLN5 patient-derived iPSCs expressed accumulation of autofluorescent storage material and subunit c of mitochondrial ATP synthase and are being used to further understand the mechanisms of CLN5 disease.40 An adeno-associated adenovirus serotype 2 (AAV2) carrying the full-length coding sequence of CLN3 was shown to be effective in restoring full-length CLN3 protein in CLN3 patient iPSC-derived retinal neurons.41 Despite these successes, there is a sparsity of characterized CLN3 patient-derived iPSC models that can be differentiated from NSCs to NPCs to mature neurons.
Herein, we report the differentiation and characterization of human CLN3 patient-derived iPSCs through the stages of NSCs, NPCs, and ultimately to mature neurons. The reported CLN3 patient iPSC-derived neurons recapitulate several phenotypes of CLN3 disease, including accumulation of autofluorescent lipofuscin and subunit c of mitochondrial ATP synthase, reduced Bcl-2 expression and mitochondrial dysfunction. Further, we report the differentiation and characterization of CLN3 patient-derived iPSCs to BMECs. These cells were used to generate a BBB model of CLN3 disease for the first time. Similar to reports in CLN3 mouse models,42 we show an impaired barrier function. We also observed differences in efflux transporter expression between CLN3 and control iPSC-derived BMECs and high angiogenic phenotype. Mature neurons differentiated from CLN3 patient-derived iPSCs were employed to screen previously identified lead compounds to determine their translational effect to modulate phenotypic aberrations of CLN3 disease. Three compounds significantly induced autophagy by a mammalian target of rapamycin (mTOR)-independent pathway in the highly phenotypic model system, resulting in enhanced clearance of subunit c from the neurons. Further, the compounds were shown to induce expression of the protective antiapoptotic protein Bcl-2 and rescue mitochondrial dysfunction.
Results
iPSC-Derived Neurons from a CLN3 Patient Display Neuronal Phenotype
We first characterized the neuronal differentiation of CLN3 patient iPSCs and compared such differentiation to commercially available healthy IMR90-c4 iPSCs.43 We defined our neuronal differentiation protocol following our previous publication,44 and investigated the expression of the following markers of the neuronal cell lineage as milestones in our differentiation; neuroepithelial cells (PAX6 positive cells), neural stem cells (Nestin positive cells), neural progenitor cells (DCX positive cells), and finally differentiating neurons (class III β-tubulin (TuJ1) positive cells). This classification is arbitrary and does not include the presence of a continuum in the expression of one or several markers as cells transition from one stage to another. Although these cells would qualify as NPCs based on several reports, we err on the side of caution in overstating the maturation stage of these cells. Differentiation of the CLN3-iPSC line into neuronal lineage occurred similarly with the IMR90-c4 iPSC line. The NSCs showed expression of PAX6 (a neuroepithelium marker) by day 10 of differentiation (Figure 1A), whereas NPCs showed a robust expression of nestin by day 15 of differentiation (Figure 1B). At day 35 of differentiation (Figure 1C), both iPSC-derived neuron lines showed the expression of nestin, as well as expression of TuJ1. Expression of MAP2 and NeuN were similar between the two lines, whereas NMDAR2 expression was higher in IMR90-c4-neurons. In conclusion, the presence of the CLN3 mutation appears not to impair the differentiation of iPSCs into neurons as compared to a healthy IMR90-c4 control cell line.
Figure 1.
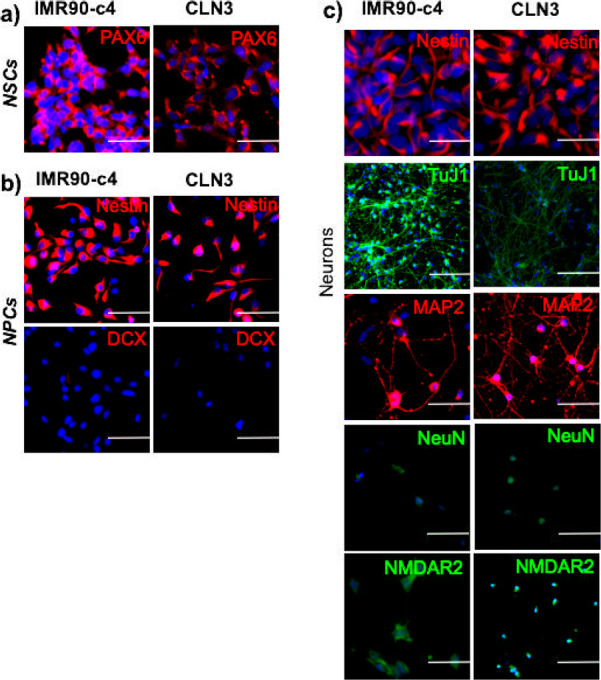
Cellular phenotyping of iPSC-derived neurons. (a) Expression of the neuroepithelial marker PAX6 of iPSC-derived neural stem cells (NSCs) from IMR90-c4 and CLN3 iPSC lines. Scale bar = 100 μm. (b) Expression profile of nestin and doublecortin (DCX) in neural progenitor cells (NPCs) derived from IMR90-c4 and CLN3 iPSC lines. Scale bar = 100 μm. (c) Neuronal phenotype of iPSC-derived neurons at 30 days of differentiation. Scale bar = 100 μm.
Neurons Derived from CLN3 Patient iPSCs Display Accumulation of Lipofuscin and Dysfunctional Lysosomes
To assess the presence and localization of lipofuscin in both iPSC-derived neuron lines, detection of autofluorescence was combined with lysosomal staining using Lysotracker. The CLN3-neurons displayed higher autofluorescence of lipofuscin, detected using the DAPI excitation wavelength (320–430 showing a maximum at 366 nm) and an emission maximum ranging from 460–630 nm (peaks from 570–605 nm) compared to IMR90-c4-derived neurons (Figure 2A). Such autofluorescence was particularly colocalized with lysosomal compartments, suggesting an aggregation and restriction of lipofuscin inclusions in lysosomal compartments. Furthermore, change in lysosomal activity was assessed in both iPSC-derived neuron lines using Lysosensor Green dye (Figure 2B). The CLN3-derived neurons displayed a higher acidification of lysosomes compared to control, as reflected by the increased fluorescence intensity. The acidity of lysosomal pH trended higher in CLN3-derived neurons compared to IMR90-c4-derived neurons, as a higher red signal (aggregated acridine orange (AO))/green (oligomeric AO) was observed (Figure 2C). Such a shift was further aggravated by induction of autophagy via either serum starvation or by treatment with rapamycin at 10 μM. Collectively these data suggest the trend of impaired lysosomal activity in CLN3-derived neurons compared to IMR90-c4-derived neurons.
Figure 2.

CLN3-neurons display lipofuscin inclusion and impaired lysosomal activity. (a) Representative micrograph picture of iPSC-derived neurons stained with Lysotracker and lipofuscin autofluorescence. The sample was detected using the DAPI-excitation wavelength (320–430 showing a maximum at 366 nm) and an emission maximum ranging from 460–630 nm (peaks from 570–605 nm). Scale bar (panel) = 300 μm. (b) CLN3-derived neurons (red bar) have a higher number of acidic lysosomes compared to IMR90-c4-derived neurons (blue bar) by whole cell fluorescence assay, N = 3/group. Representative micrograph picture of Lysosensor Green. Scale bar (panel) = 50 μm. Semiquantitative analysis of Lysosensor Green fluorescence following incubation of live cells with 100 nM Lysosensor dye. (c) Acridine orange (AO) fluorometric ratio in IMR90-c4 and CLN3-derived neurons following autophagy induction by 10 μM rapamycin or serum-starvation treatment. Values represent means ± SD of N = 3/group, ** denotes P < 0.01.
Neurons Derived from CLN3 Patient iPSCs Display Accumulation of Subunit c and Dysfunctional Mitochondria
The CLN3-derived neurons showed higher immunoreactivity to subunit c of mitochondrial ATP synthase and lipofuscin compared to IMR90-c4-derived neurons (Figure 3A). Although no changes in overall mitochondrial content was detected, a faint immunoreactivity was observed in CLN3-derived neurons (Figure 3B), suggesting possible impaired mitochondrial function. To assess the difference in overall metabolic activity, iPSC-derived neurons were challenged with increasing concentrations of rotenone, a superoxide inducer, (200 and 400 nM concentrations for 24 h), and changes in cell metabolic activity were measured using MTS reagent (Figure 3C). Unlike IMR90-c4-derived neurons that showed no differences in cell metabolic activity, CLN3-derived neurons showed a dose-dependent decrease in cell metabolic activity following treatment with rotenone. Such a difference was further demonstrated by changes in mitochondrial superoxide generation using MitoSOX assay (Figure 3D, left panel). Under resting conditions, CLN3-derived neurons showed a higher MitoSOX fluorescence intensity compared to IMR90-c4-derived neurons, suggesting higher production of superoxide free radicals at the basal level. Increase in MitoSOX fluorescence was significant in IMR90-c4-derived neurons following treatment with rotenone at 10 μM and was significant, but remained mild, in CLN3-derived neurons. Similar outcomes were observed by assessing mitochondrial potential using JC-1 staining (Figure 3D, right panel). Interestingly, the CLN3-derived neurons showed a higher depolarization status (as reflected by a decrease in red/green JC-1 fluorescence ratio) under resting conditions compared to control. However, no significant differences were observed between these two cell lines following treatment with carbonyl cyanide m-chlorophenylhydrazone (CCCP; an uncoupling agent) at 50 μM. In conclusion, CLN3 mutation impairs mitochondrial function and activity in iPSC-derived neurons.
Figure 3.

CLN3-derived neurons show impaired mitochondrial function. (a) Representative micrograph pictures of IMR90-c4 and CLN3-derived neurons staining for subunit c. Scale bar (panel) = 300 μm. (b) MitoTrackerTM (green) micrograph picture (blue = DAPI). Scale bar (panel) = 50 μm. (c) Cell metabolic activity following rotenone treatment using an MTS-based assay. Absorbance was normalized to untreated cells. (d) Mitochondrial oxidative stress and activity measured by MitoSOX and JC-1 dye fluorescence under control conditions and following treatment with 10 μM rotenone for 10 min (left) or incubation with 50 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) (right). Values represent means ± SD of N = 3/group, * and # denote P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
CLN3 Mutation Is Associated with Increased Efflux Pump Expression
In addition to the effect of CLN3 mutation on iPSC-derived neurons, the presence of a phenotype in non-neuronal cells associated with such mutation was investigated in iPSC-derived BMECs (Figure 4). Differentiation of iPSCs to BMECs was achieved employing the same differentiation protocol established by Lippmann and colleagues,45 commonly used in our studies.44 The BMECs derived from CLN3 patient iPSCs displayed a similar BMEC identity to IMR90-c4-derived BMECs (Figure 4A), with these cells showing immunoreactivity to vascular markers (PECAM-1, CDH5), as well as the expression of blood-brain barrier selective markers including GLUT1 and the tight junction proteins claudin-5 (CLDN5) and occludin (OCLDN). In addition, both IMR90-c4-derived BMECs and CLN3-derived BMECs showed positive expression (Figure 4B) for several drug efflux pumps including P-glycoprotein (p-gp), breast cancer resistant protein (BCRP) and multidrug resistance protein 1 (MRP1). Notably, CLN3-derived BMECs appeared to show higher expression of MRP1 compared to IMR90-c4-derived BMECs, as displayed by a higher immunoreactivity. In conclusion, CLN3-derived BMECs were capable to differentiate and display a BMEC phenotype similar to IMR90-c4-derived BMECs.
Figure 4.
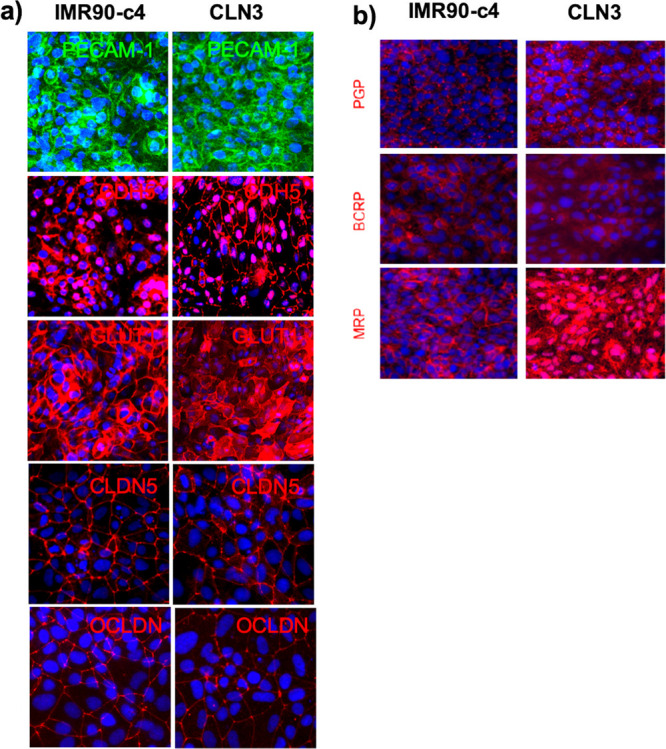
Phenotypical characteristic of iPSC-derived BMECs from the CLN3 line. (a) Representative micrograph pictures of iPSC-derived BMECs. Note the expression of vascular markers (PECAM-1, CDH5) in both iPSC lines, as well as the presence of BBB-specific markers (GLUT1, CLDN5, and OCLDN). Scale bar (panel) = 200 μm. (b) Expression profile of drug efflux transporters in both IMR90-c4-derived BMECs and CLN3-derived BMEC monolayers. DAPI staining is denoted in blue. Scale bar (panel) = 100 μm.
CLN3-Derived BMECs Display an Impaired Barrier Function Compared to IMR90-c4-Derived BMECs
Differences in barrier function between IMR90-c4-derived BMECs and CLN3-derived BMECs were assessed using transendothelial electrical resistance (TEER) (Figure 5A) and paracellular permeability using fluorescein (Figure 5B). The CLN3-derived BMECs displayed a very poor barrier function, with reported TEER in the CLN3-derived BMEC monolayers significantly lower than that in the IMR90-c4-derived BMECs (127 ± 31.44 and 1589 ± 470.6 Ω.cm2, respectively). Such discrepancy between the two monolayers was further confirmed by significant differences in paracellular permeability between CLN3-derived BMECs and IMR90-c4-derived BMECs (1.364 ± 0.49 × 10–4 cm/min and 0.64 ± 0.19 × 10–4 cm/min respectively). These results suggest that CLN3-derived BMECs have an impaired barrier function. To exclude the possible difference in the maturation process between CLN3-derived BMECs and IMR90-c4-derived BMECs as a contributor to this data, an alternative differentiation protocol was set for CLN3-derived BMECs, by extending the unconditioned medium (UM) treatment from 6 days to 7 days and by extending the endothelial medium (EM)+/+ treatment from 2 days to 6 days based on the alternative protocol described by Lippman.45 Such extended differentiation protocol also showed an impaired barrier function of CLN3-derived BMECs (Figure 5C), with no significant differences in TEER (62.14 ± 34.16 Ω·cm2 vs 48.29 ± 5.21 Ω·cm2) and fluorescein permeability ((5.335 ± 2.639) vs (13.79 ± 7.43) × 10–4 cm/min) when the two differentiation protocols are compared. The ability of CLN3-derived neurons to induce a barrier phenotype in both IMR90-c4 and CLN3-derived BMECs was assessed by the coculture method. Notably, the presence of CLN3-derived neurons in cocultures worsened the barrier outcomes in both IMR90-c4-derived BMECs and CLN3-derived BMECs monolayers compared to IMR90-c4-derived neuron cocultures, as a decrease in TEER and increased fluorescein permeability were apparent (Figure 5D). In conclusion, our data suggest that the presence of CLN3 mutation may impair BMEC differentiation and barrier induction by neurons.
Figure 5.

CLN3-derived BMECs display an impaired barrier phenotype. (a,b) Measurement of transendothelial electrical resistance (TEER) and fluorescein permeability between IMR90-c4-derived BMECs (Control, blue bar) and CLN3-derived BMECs (red bar). (c) Additional maturation time (7 days UM combined with 6 days EC) in CLN3-derived BMECs failed to improve the barrier outcomes. (d) BMEC/neuron cocultures impact the barrier outcome. (e) Protein expression profile of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance protein (MRP). (f) Drug efflux pump activity in iPSC-derived BMECs. (g) CLN3 mutation is associated with increased BSA and transferrin uptake between IMR90-c4-derived BMECs and CLN3-derived BMECs. (h) Temperature-dependent uptake of BSA and transferrin is more accentuated in CLN3-derived BMECs. (i) Immunoblot profile of clathrin and caveolin-1 (cav-1) expression in iPSC-derived BMECs. Band densities were normalized to actin. Values represent means ± SD of N = 3/group, * denotes P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
CLN3 Mutation Does Not Affect Efflux Pump Expression and Activity
To further demonstrate that such impaired barrier function in BMEC monolayers was associated with impaired drug transport activity, changes in drug efflux pump expression and activity were determined (Figure 5E,F). The CLN3-derived BMECs displayed higher P-gp expression and lower BCRP expression compared to IMR90-c4-derived BMECs. However, no differences in drug efflux pump activity were observed between the two BMEC monolayers. In addition, trafficking of larger molecules such as bovine serum albumin (BSA) and transferrin (Tf) were assessed between the two monolayers (Figure 5G,H). Notably, uptake of both BSA and Tf were higher in CLN3-derived BMECs compared to IMR90-c4-derived BMECs suggesting dysfunctional membrane trafficking in such cells. Such trafficking was temperature sensitive as a significant decrease in BSA and Tf uptake was noted following incubation at 4 °C compared to 37 °C. Finally, to assess the nature of such impaired membrane trafficking, differences in clathrin and caveolin-1 (cav-1) levels were assessed by immunoblot (Figure 5I). We noted lower levels of clathrin in CLN3-derived BMECs compared to that in IMR90-c4-derived BMECs, whereas no differences in cav-1 were noted. In conclusion, CLN3-derived BMECs barrier dysfunction was associated with impaired clathrin-mediated membrane trafficking.
CLN3-Derived BMECs Possess a Highly Angiogenic Phenotype
A previous study reported abnormal angiogenic phenotype in iPSC-derived BMECs obtained from Huntington’s disease patients.46 Changes in angiogenic phenotype were measured in our developed CLN3-derived BMEC model (Figure 6). Using a scratch assay (Figure 6A), no differences in migration were noted between IMR90-c4 and CLN3-derived BMEC monolayers, following 12 h incubation after scratch (50.02 ± 10.94 vs 52.36 ± 13.94%, respectively). However, CLN3-derived BMECs displayed a proliferative status compared to IMR90-c4-derived BMECs (100 ± 2.759 vs 197.7 ± 22.01%) after 12 h, as measured by MTT assay (Figure 6B). To further determine if such increased proliferation was associated with an angiogenic phenotype, both types of BMECs were plated on Reduced Growth Factor (RGF) basement membrane extract and assessed for changes in various angiogenic features at different time intervals following seeding (4, 6, 12, 24, 48, and 72 h; Figure 6C–E). We further compared whether the presence of basic fibroblast growth factor (bFGF) or retinoic acid in EC media can affect the outcome of tube formation by performing the assay with EC+/+ and EC–/– medium. We observed formation of tube-like structures with both media (EC+/+ and EC–/–) with no marked effect of basic fibroblast growth factor (bFGF) or retinoic acid on tube formation (Supporting Information, Figure S-1A,B). The CLN3-derived BMECs (red) contrasted IMR90-c4-derived BMECs (blue) by displaying an angiogenic phenotype marked by a higher number of nodes and junctions, as well as a higher total length of tube-like structures (Figure S-1A,B). In conclusion, CLN3-derived BMECs display an angiogenic phenotype compared to IMR90-c4-derived BMECs.
Figure 6.

CLN3-derived BMECs show an abnormal angiogenic phenotype. (a) Cell migration assay and (b) cell proliferation assay. (c–e) Angiogenic assays were performed on iPSC-derived BMECs seeded on growth-factor reduced Matrigel at a density of 10,000 cells/cm2. Total tube length (c), number of nodes (d), and number of junctions (e) were quantified from images obtained (Figure S-1A,B) at different time points. Values represent means ± SEM of N = 3/group, * denotes P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Small Molecules with Neuroprotective Activity Induce Autophagy in Neurons Differentiated from CLN3 Patient-Specific iPSCs in a mTOR-Independent Manner
Previous studies employing novel aromatic carbamate analogues demonstrated induction of autophagy in “neuron-like” PC12 cells and IMR90-c4 iPSC-derived neurons in a dose-dependent manner, with no tolerability issues observed up to 30 μM.34 A selection of aromatic carbamates, including inactive analogues as control, were screened at a concentration of 3 μM (deemed from prior studies to afford optimal effect)34 in CLN3-derived neurons to determine the translational activity of these molecules (for structures see Figure S-2). Several compounds demonstrated significant increase in acridine orange staining compared to vehicle, indicating increased acidification which is an indirect marker of inducing autophagy (Figure 7A).47 Notably, almost all of the screened compounds outperformed a rapamycin positive control (a known mTOR inhibitor and autophagy modulator). To confirm autophagy activation, the more sensitive CytoID stain was employed with three selected compounds. The three molecules showed a general trend of modulating autophagy above vehicle and rapamycin controls, albeit less pronounced than the acridine orange stain, with compound 9h providing a significant increase (Figure 7B). Additionally, expression of the autophagy marker Beclin 1 trended upward upon treatment with rapamycin and compounds 9e, 9h, and 9g by Western blot (Figure 7C), but not with rasagiline (a Bcl-2 inducer but with no known effect to modulate autophagy), further supporting the autophagy induction activity of these compounds.
Figure 7.

Neuroprotective small molecules induce autophagy in CLN3-derived neurons. (a) Aromatic carbamates induce autophagy as measured by acridine orange stain. n = 3/cell line, expressed as mean ± SD. Rapa = rapamycin. One-way ANOVA; 95% confidence interval, *p < 0.05, ****p < 0.0001. (b) Aromatic carbamates induce autophagy as measured by CytoID stain. n = 3/cell line, expressed as mean ± SD, *p < 0.05, **p < 0.01. (c) Expression of autophagy-related proteins by Western blot after treatment of CLN3-derived neurons with selected compounds (3 μM), rapamycin (Rapa, 0.5 μM), and rasagiline (Ras, 1 μM). Representative Western blot image. n = 2/cell line. Graphs represent quantification of Western blot data expressed as mean ± SD. (d) Expression of subunit c of mitochondrial ATP synthase in untreated IMR90-c4-derived neurons (IMR90 control), untreated CLN3-derived neurons (CLN3 control), and CLN3-derived neurons treated with the indicated small molecules (3 μM) for 48 h. Representative Western blot image. n = 2/cell line. Graph represents quantification of Western blot data expressed as mean ± SD. One-way ANOVA; 95% confidence interval, *p < 0.05.
To determine the mechanism of autophagy induction, mammalian target of rapamycin (mTOR) dependency was determined. It has been suggested that mTOR-independent activation of autophagy would be a more relevant therapeutic option for the treatment of neurodegenerative disorders.48 As expected, rapamycin showed a decrease in ratio of phosphorylated p70 s6 kinase to total p70 s6 kinase, a function performed by mTOR and disrupted upon mTOR inhibition. None of the three selected small molecules showed a decrease in ratio of phosphorylated p70 s6 kinase to total p70 s6 kinase, suggesting that these molecules act by mTOR-independent pathways (Figure 7C). Elevation of autophagy can be cytotoxic to cells and thus we evaluated the cytotoxic effect of aromatic carbamate analogues on CLN3-derived neurons. All compounds at 3 μM were found to be nontoxic based on propidium iodide staining (Supplemental Figure S-3). Gratifyingly, the three selected small molecule mTOR-independent autophagy inducers significantly enhanced the clearance of subunit c of mitochondrial ATP synthase from CLN3-derived neurons by immunoblot (Figure 7D). In conclusion, small molecule aromatic carbamates are mTOR-independent autophagy modulators that enhance the clearance of subunit c from CLN3-derived neurons.
Neuroprotective Small Molecules Induce Expression of the Antiapoptotic Protein Bcl-2
Previously conducted experiments using aromatic carbamates demonstrated neuroprotective activity, in part, by inducing the antiapoptotic protein Bcl-2 in a dose-dependent manner.34 To determine if the Bcl-2 induction effect translates from low passage “neuron-like” PC12 cells49 to neurons differentiated from CLN3 patient-specific iPSCs an ELISA for Bcl-2 expression was performed (Figure S-4). All three small molecules, 9e, 9g, and 9h induce significant increase in Bcl-2 expression in CLN3-derived neurons (Figure 8A), returning expression to comparable levels as IMR90-c4-derived control neurons. Notably CLN3-derived neurons express significantly lower levels of Bcl-2 than IMR90-derived neurons. In conclusion, all three small molecules significantly induce Bcl-2 expression in CLN3-derived neurons to levels greater than healthy IMR90-c4-derived neuron controls and with greater effect than the known Bcl-2 inducer rasagiline.
Figure 8.

Neuroprotective small molecules induce Bcl-2 expression in CLN3-derived neurons and rescue dysfunctional mitochondrial respiration in CLN3-derived NPCs. (a) Bcl-2 induction expressed as a percentage of Bcl-2 change upon small molecule treatment of CLN3-derived neurons at 3 μM. n = 2, expressed as mean ± SD. One-way ANOVA; 95% confidence interval; *p = < 0.05; **p = < 0.01; ***p = < 0.001; ****p = < 0.0001. (b) Mitochondrial respiration of IMR90-c4-derived NPCs (blue bar), CLN3-derived NPCs (red bar), treated with rapamycin (0.5 μM) for 48 h (green bar) or 9e (3 μM) for 48 h (purple bar) as determined by the mito stress test on a Seahorse extracellular flux analyzer. (c) Quantification of basal respiration, (d) spare respiratory capacity, and (e) ATP production as determined by the mito stress test on a Seahorse extracellular flux analyzer. n = 3, expressed as mean ± SD. One-way ANOVA; 95% confidence interval; *p = < 0.05, ***p = < 0.001, ****p = < 0.0001.
Neuroprotective Small Molecules Rescue Mitochondrial Dysfunction in CLN3-Derived NPCs
Mitochondrial dysfunction is a hallmark of the NCLs.50−52 We determined the mitochondrial respiration of CLN3-derived NPCs and IMR90-c4-derived NPCs on a Seahorse extracellular flux analyzer (Figure 8B). Basal respiration in CLN3-derived NPCs was significantly lower than in healthy IMR90-c4-derived control NPCs (Figure 8C). Basal respiration levels were increased, but not significantly, by treatment of the CLN3-derived NPCs for 48 h with the maximum nontoxic concentration of rapamycin (0.5 μM) and restored to healthy IMR90-c4-derived NPCs’ levels by compound 9e (3 μM), with similar effects observed on spare respiratory capacity (Figure 8D). Production of ATP in CLN3-derived NPCs trended lower than that of IMR90-c4-derived NPCs. Treatment of CLN3-derived NPCs with rapamycin (0.5 μM) increased ATP production, but not significantly, with levels being restored to above IMR90-c4-derived NPCs controls with 9e (3 μM) after 48 h incubation (Figure 8E). In conclusion, compound 9e rescues the dysfunctional mitochondrial phenotype of CLN3-derived NPCs.
Neuroprotective Small Molecules Inhibit the Kv7.1 Ion Channel
To begin target identification efforts for these promising compounds we screened their activity on potassium ion channels, the primary target of the flupirtine parent scaffold. Potassium channel opening indirectly inhibits the activation of NMDA receptors.53,54 We have previously reported the absence of NMDA antagonistic activity of selected analogues in IMR90-c4 iPSC-derived neurons up to 30 μM.34 Further direct studies on the ability of these compounds to open Kv7.2–5 channels will follow. Retigabine and flupirtine have been shown to be inactive against the KCNQ1 (Kv7.1) potassium channel (retigabine is a weak inhibitor with a reported IC50 ∼ 100 μM)55. Hypothesizing that a change in the differential profile of potassium channel selectivity may account for the greater neuroprotective activity of our compounds and recognizing the retention of the pharmacophoric carbamate moiety for Kv7.2–5 binding,56 we determined the effect of compound 9e on the Kv7.1 channel. The compound showed a dose-dependent inhibition activity (Table S-1). A single 10 μM concentration of 9e and 9h conferred 40% and 27% inhibition of the human KCNQ1/MinK channel, respectively. Compound 9e is comparable with the activity of the selective KCNQ1/MinK channel inhibitor control compound Chromanol 293B (43.2% inhibition at 10 μM). The KCNQ1/MinK channel is a putative target for antiarrhythmic drugs and its inhibition does not result in cardiotoxicity.57 To determine selectivity to the Mink subunit which also binds hERG, we profiled the inhibition effect of compounds 9e and 9h against the hERG channel at 10 μM, determining 28% and 34% inhibition, respectively. Further studies are planned to further document the target of action of these compounds.
Discussion
The NCLs are a set of neurodegenerative diseases marked by an irreversible and progressive onset ultimately resulting in death. To date, there are no curative treatments available. A major pitfall in developing pharmacological intervention for the NCLs is the limited repertoire of in vitro models suitable for the screening of novel neuroprotective drug candidates. The emergence of iPSC-based models has revolutionized the field of disease modeling, resulting in deeper insight into the pathological mechanisms of several neurological diseases including CLN3 disease.28,37,40 In this study, we report the successful differentiation of neurons and BMECs from a patient-derived iPSC line originating from a 21-year-old male bearing a 1kb deletion in the CLN3 gene and heterozygous for E13 c.988G>T, p.Val330Phe mutation, using established protocols.44,45
The major limitation of this study is the use of only one patient cell line which is compound heterozygous for the common 1 kb deletion in CLN3 and a rare missense mutation and comparison to IMR90-c4, an unrelated individual. Thus, the lack of an isogenic control does limit to some extent the link between loss of CLN3 and the observed phenotypes. However, the fundamental conclusions of the effect of the screened compounds are valid and represent translational potential for drug discovery. Indeed, for drug screening operations the developed in vitro model system is highly suited for phenotypic screening for which employing multiple patient lines would be cost and time restrictive. This is especially true when considering subunit c accumulation, mitochondria function, and Bcl-2 expression end points. The BMECs and neurons were fully characterized against the commercially available IMR90-c4 iPSC line which does not possess the CLN3 mutation. While an isogenic control line for the CLN3 iPSCs does not exist, the IMR90-c4 line has been extensively characterized and employed as a control line in a variety of neurodegenerative disease studies.58−61
The CLN3 patient iPSC-derived cells recapitulated several phenotypic hallmarks of CLN3 disease; the inclusion of lipofuscin within lysosomes, the accumulation of subunit c of ATP synthase, mitochondrial impairment, and alteration of lysosomal pH. Another key characteristic observed in CLN3 disease is the accelerated apoptosis of neurons.62 Cells with CLN3 knock down demonstrate diminished expression of Bcl-2, indicating a role for the CLN3 protein in Bcl-2 regulation and apoptosis control.63 Reduced expression of Bcl-2 compared with IMR90-c4-derived controls was observed in this iPSC-derived neuron model.
In addition to phenotypic neuronal features observed in CLN3-derived neurons, we also noted an impaired phenotype in BMECs derived from the CLN3 iPSCs compared to those from the IMR90-c4 iPSCs. We observed a decreased barrier function indicative of increased permeability of the BBB, in particular to the uptake of large molecules at the barrier. Such an observation is consistent with the existing literature, as Tecedor and colleagues reported impaired membrane trafficking in brain endothelial cells transfected with a mutant CLN3 gene,64 whereas Saha and colleagues reported increased permeability in an in vivo model of CLN1 disease.65 The level of tight junction proteins and abnormalities in claudin-5 levels observed in CLN3-derived BMECs partly explain reduced barrier tightness. Such studies on BBB modeling of rare diseases utilizing iPSCs provide scalable and reproducible human disease models.46,66 This report also highlights that BMECs show expression and activity of efflux transporters including P-gp, BCRP, and MRP1. Prior studies have suggested impaired membrane dynamics and endocytic pathways in Batten disease BMECs, although our model of CLN3 BMECs did not show any such phenotype and presented healthy endocytic transport. Such an observation is important and in agreement with other studies reporting an altered BBB function in other neurological diseases including adrenoleukodystrophy (ALD)67 and Huntington’s disease.46 We noted an increased angiogenic phenotype in CLN3-derived BMECs compared to control BMECs, suggesting that CLN3 may be associated with signaling pathways involved in angiogenesis and/or barrier regulation. Highly angiogenic vessels impart the phenotypes of leaky vessels and reduced BBB tightness which is detrimental in multiple diseases.46,68,69 Herein we report the first indication of increased angiogenesis in CLN3-derived BMECs. In agreement with the literature, the coculture of CLN3-derived neurons with IMR90-c4-derived BMECs was sufficient to worsen barrier function, suggesting the importance of the inclusion of neurovascular coupling when modeling neurological diseases in vitro. More investigation into the pathways responsible for increased angiogenesis may provide understanding and methods to counter barrier break down in CLN3 disease.
Presented herein, to the best of our knowledge, is the first report of using mature neurons derived from CLN3 patient iPSCs for a small molecule screen. Lojewski et al. have previously demonstrated development of CLN2 and CLN3 iPSCs using patient fibroblasts and differentiated the iPSCs to neurons. The screening of three small molecules was performed in NPCs differentiated from CLN2 iPSCs, but not mature neurons.70 We have previously reported small molecule aromatic carbamates that possess neuroprotective activity by induction of Bcl-2 and increasing autophagy.34 The neuroprotective effect of selected analogues was further demonstrated in lymphoblasts derived from CLN1, CLN2, CLN3, CLN6, and CLN8 patients.33 Herein, using neurons differentiated from CLN3 patient iPSCs, we demonstrate that selected compounds from our small molecule library translate their mechanistic effect to induce Bcl-2 and enhance autophagy in a highly phenotypic model of CLN3 disease. The selected small molecule probes significantly induce expression of the antiapoptotic protein Bcl-2, resulting in levels comparable to those observed in IMR90-c4-derived control neurons, outperforming the positive control compound rasagiline, a clinically available compound with neuroprotective activity.
We show the small molecule probes modulate autophagy in an mTOR-independent manner, a characteristic of several known neuroprotective molecules.71 Rapamycin, a known mTOR-dependent activator of autophagy, fails to deliver the same magnitude of autophagy activation in the CLN3 patient-specific iPSC-derived neurons as our developed mTOR-independent activators of autophagy. Likewise, a recent report demonstrates that rapamycin is ineffective at activating transcription factor EB (TFEB), a master regulator of lysosomal pathways, in a CLN3 mouse model due to mTOR-dependence, while mTOR-independent activation of TFEB by trehalose, a small molecule carbohydrate, promoted cellular clearance and increased survival in Cln3Δex7/8 mice.72 Furthermore, all three probe compounds conferred significant clearance of subunit c from CLN3-derived neurons supporting activation of autophagy. Thus, the discovery and optimization of mTOR-independent activators of autophagy with “drug-like” profiles of activity, BBB penetration,73 and stability (Figures S-5A–S-7B) may provide for greater therapeutic effect in this rare disease. A recent study showed that the mTOR-independent autophagy inducer, verapamil, promoted clearance of storage material from murine CLN3 disease neuronal cells.74 Autophagy and Bcl-2 are known to possess interdependency in a physiological setting; Bcl-2 and beclin1 demonstrate protein–protein interaction (PPI) and disruption of this interaction activates autophagy.75 Accumulation of lipofuscin has been shown to result in autophagy dysfunction and lower turnover of affected mitochondria. This in turn creates a feedback loop wherein defective mitochondria produce more reactive oxygen species (ROS; which is observed in our developed model system) that increase lipofuscin accumulation, questioning if activation of autophagy would necessarily clear lipofuscin accumulation.76 If the hydrolytic function of the lysosome is irreparably damaged by CLN3 mutation or other pathogenesis event, small molecule autophagy inducers would not be able to induce clearance. Such an eventuality occurs in Nieman Pick Type 1 disease, wherein enhanced autophagic flux failed to clear cholesterol levels.26 However, the observed clearance of subunit c demonstrated herein would suggest no irreparable damage.
In addition to the significant effect of 9e on inducing Bcl-2 expression and autophagy, the small molecule successfully rescued mitochondrial dysfunction of CLN3-derived NPCs. Basal respiration and ATP production have been reported to be significantly reduced in Cln3Δex7/8 mouse astrocytes.77 Basal respiration, spare respiratory capacity, and ATP function of the CLN3-derived NPCs were all significantly increased to similar or greater levels as seen in healthy IMR90-c4-derived NPC controls. While study of iPSC-derived neurons obtained directly from patients provide for a highly phenotypic model, they also pose some limitations. These include the absence of other cell types in the model system, such as astrocytes, which likely have a role to play in the disease, the effects of in vivo metabolizing enzymes on our small molecules, and the lack of a BBB to penetrate, although our studies have provided the first model of the BBB of CLN3 disease for further study.
Although our target identification efforts are in their infancy, we have shown that the effects of compound 9h and especially 9e may be due to a change in the differential effect of these compounds on potassium ion channels compared to the flupirtine parent scaffold. Compound 9e inhibits the Kv7.1 ion channel expressing the minK ß subunit by 40% at a 10 μM concentration, comparable to the selective Kv7.1 channel inhibitor Chromanol 293B. Compound 9e is a more potent inhibitor than 9h at 10 μM (27% inhibition) and correlates to greater neuroprotective activity. Potassium ion channels are known targets for neuroprotective agents, albeit the Kv7.1 channel has greater expression levels in the heart than in the CNS.78 Interestingly, the clinically approved NSAID, mefenamic acid, a selective Kv7.1 inhibitor has recently been shown to induce Bcl-2 expression in human SH-SY5Y cells.79 This observed activity provides a basis for further target identification studies on 9e.
In conclusion, we report the first iPSC-derived model of the BBB for CLN3 disease, characterizing a dysfunctional barrier. Equally, we characterize a CLN3 patient iPSC-derived neuron model that recapitulates multiple phenotypes of the disease and is used for a small molecule screen. To our knowledge, this is the first report of employing CLN3 patient iPSC-derived neurons for small molecule screening. This model can be effectively engaged to screen for autophagy modulators, antiapoptotic compounds, compounds with effects on mitochondria, or a combination of these and to determine their mechanism(s) of action. The small molecules screened herein rescue CLN3 neurons from three phenotypic consequences of the disease; upregulating Bcl-2, modulating autophagy resulting in enhanced clearance of subunit c and correction of mitochondrial dysfunction. These results identify small molecule 9e as a valuable probe compound for further study of CLN3 disease and mTOR-independent autophagy modulation and as a compound for further optimization along the drug discovery pipeline. These data provide evidence that there is no fundamental block of autophagy in CLN3 disease and pharmacological stimulation of autophagy may have a therapeutic benefit. Development of small molecule therapeutics with multiple mechanisms of action are likely to have a major impact on the treatment of complicated neurological diseases such as CLN3 disease.
Materials and Methods
Cell Lines
iPS(IMR90)-4 iPSC (RRID: RRID:CVCL_C437) line was purchased from WiCell cell repository (WiCell, Madison, WI, USA.43 CLN-3 iPSCs were obtained from the New York Stem Cell Foundation iPS Core Facility (NYSCF, New York City, NY, USA) originating from a 21-year-old male bearing a 1kb deletion in the CLN3 gene and heterozygous for E13 c.988G>T, p.Val330Phe mutation (Figure S-8). Somatic cells used for the derivation of these iPSC lines were obtained with informed consent of subjects and were approved by the respective repositories institutional review boards. Undifferentiated iPSC colonies were maintained on hPSC-grade growth factor-reduced Matrigel (C-Matrigel; Corning, Corning, MA, USA) in the presence of Essential 8 medium (E8; ThermoFisher, Waltham, MA, USA).
BMECs Differentiation
iPSCs were differentiated into BMECs following the protocol established by Lippmann and colleagues.45,80 iPSCs were seeded as single cells on T-Matrigel (Trevigen, Gaithersburg, MD, USA) at a cell density of 20 000 cells/cm2 in E8 supplemented with 10 μM Y-27632 (Tocris, Minneapolis, MN, USA). Cells were maintained in E8 for 5 days prior to differentiation. Cells were maintained for 6 days in unconditioned medium (UM: Dulbecco’s modified Eagle’s medium/F12 with 15 mM HEPES (ThermoFisher), 20% knockout serum replacement (ThermoFisher), 1% nonessential amino acids (ThermoFisher), 0.5% Glutamax (ThermoFisher), and 0.1 mM b-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA). After 6 days, cells were incubated for 2 days in the presence of EC+/+ [EC medium (ThermoFisher) supplemented with 1% platelet-poor derived serum (Alfa-Aesar, ThermoFisher, Haverhill, MA, USA), 20 ng/mL human recombinant basic fibroblast growth factor (Tocris, Abingdon, UK), and 10 μM retinoic acid (Sigma-Aldrich)]. After this maturation process, cells were dissociated by Accutase (Corning) treatment and seeded as single cells on tissue culture plastic surface coated with a solution of collagen from human placenta (Sigma-Aldrich) and bovine plasma fibronectin (Sigma-Aldrich) (80 μg/cm2 and 20 μg/cm2, respectively). At 24 h after seeding, cells were incubated in the presence of EC/(EC medium supplemented with 1% platelet-derived serum). Barrier phenotype experiments were performed 48 h after seeding. Cells were seeded at the same density between controls and CLN3 iPSCs at day eight of differentiation and photos were taken at random; images were selected at random from these photos. Consistent cell density was maintained by DAPI staining and/or by bright-field microscopy (live imaging).
iPSC-Derived BMEC Barrier Function
iPSC-derived BMECs were seeded on Transwells (polyester, 0.4 μm pore size; Corning) and coated as previously described. iPSC-derived BMECs were seeded at a seeding density of 1 × 106 cells/cm2. Barrier function was assessed 48 h after seeding for iPSC-derived BMEC monolayers; these time points were determined as the most optimal for achieving the highest barrier tightness. Barrier tightness was measured by assessing both the TEER and paracellular diffusion. TEER was measured using an EVOHM STX2 chopstick electrode (World Precision Instruments, Sarasota, FL, USA). For each experiment, three measurements were performed for each insert, and the average resistance obtained was used to determine barrier function. In experiments involving paracellular diffusion, 10 μM sodium fluorescein (Sigma-Aldrich) was added in the donor (apical) chamber, and 100 μL aliquots were sampled from the acceptor (basolateral chamber) every 15 min for up to 60 min. Permeability (Pe) for fluorescein was calculated using the clearance slopes obtained by extrapolation using the following formula:
with Pt and Pf indicative of the clearance slopes of samples and blank (empty coated) filters and S indicative of the inset surface area (cm2).
Drug Uptake Assays
Cells were incubated in the presence of 10 μM rhodamine 123 (Sigma-Aldrich), 10 μM CM-DCFDA (ThermoFisher), and 20 μM Hoechst (ThermoFisher) for 1 h in an incubator. In some experiments, cells were preincubated in the presence of 5 μM PSC833 (Sigma-Aldrich), 1 μM Ko143 (Sigma-Aldrich), or 10 μM MK571 (Sigma-Aldrich) for 1 h prior to incubation with fluorescent substrates. Following incubation, cells were quickly washed using ice-cold phosphate-buffered saline (PBS) and lysed using radioimmunoprecipitation assay buffer (ThermoFisher). Fluorescence in cell lysates was assessed using a SynergyMX2 ELISA plate reader (Bio-Tek, Winooski, VT, USA). Relative fluorescence units (RFU) were normalized against the total protein content and the protein level was determined by bicinchoninic acid assay (Pierce, Rockford, IL, USA, ThermoFisher). Fluorescence values (expressed as relative fluorescence unit or RFU) obtained from cell lysates in the absence of inhibitor (named as controls) were normalized to the protein content and expressed as RFU/μg protein.
Scratch Plate Assay (Wound Healing)
Scratch plate assay was performed as previously described.46 BMECs were plated on collagen/fibronectin-coated 12-well plates after differentiation. After 24 h, the cells reached 100% confluence and the initial scratch/wound was made. Two images/well were taken using a light microscope at times 0 and 12 h and six wells were used for each experiment/differentiation.
Proliferation Assay
Cell proliferation was assessed through the 3-[4,5-dimethylthiazol-2-yl]-2,5-dimethyltetrazolium bromide (MTT) assay, as previously described.81 Briefly, cells were seeded at a density of 2.5 × 104 cells/well in fibronectin-coated 24-well tissue culture plates. After 24 h, MTT (5 mg/mL in PBS; Fisher) was added at a volume equal to 1/10 of the medium and plates were further incubated at 37 °C for 2 h. Cells were washed once with PBS and then diluted in 100 μL/well acidified isopropyl alcohol (0.33 mL HCL in 100 mL isopropyl alcohol). After thorough agitation, the solution was transferred to a 96-well plate and read in a microplate reader at 570 nm.
In Vitro Matrigel Tube Formation Assay
The Matrigel tube formation assay was performed as previously described.82 Wells of a 96-well plate were coated with 40 μL of Growth Factor-Reduced (RGF) basement membrane extract (Trevigen) and incubated for 20 min at 37 °C to polymerize. Then, 104 cells suspended in EC medium supplemented with 1% PDS (EC–/–) were added to each well and incubated at 37 °C. Brightfield images were obtained after 4, 6, 12, 24, 48, and 72 h of incubation and were analyzed for the number of nodes, number of junctions, and total sprout length, using ImageJ software (NIH, Bethesda, MD, USA) with the “angiogenesis analyzer” plug-in.
BSA and Transferrin Uptake Assay
Differentiated BMECs were plated at confluence on collagen/fibronectin coated 12-well plates. FITC labeled bovine serum albumin (FITC-BSA, ThermoFisher) and FITC labeled transferrin (FITC-Tf, ThermoFisher) were incubated at 50 ug/mL concentration on cell monolayers for 25 min at 37 and 4 °C. Cells were lysed using RIPA buffer (ThermoFisher), and FITC fluorescence was measured in the collected buffer. Raw fluorescence intensity (RFU) was normalized against total protein content estimated in the total cell lysates.
Western Blot
Cells were washed once with PBS and lysed with RIPA buffer (ThermoFisher). Proteins were quantified via BCA assay (ThermoFisher, #PI-23221) and were then resolved by SDS-PAGE on 4–20% Tris-glycine gradient gels (BioRad). After transfer to nitrocellulose membranes, blocking was conducted for 1 h in Tris-buffered saline (10 mM Tris-HCl, 100 mM NaCl, pH7.5) containing 0.1% Tween-20 (TBST) and 1% BSA. Samples were probed overnight at 4 °C with anti-Clathrin (CST, #2410), anticaveolin (ThermoFisher, #MA3-600), anti-β-Actin (CST, #3700) (Santa Cruz Biotechnology; 155 000) anti-Subunit c (Abcam, #ab181243), anti-Bcl-2 (ThermoFisher, #PA5-27094), anti-Beclin 1 (CST, #3738S) or anti-p70 S6 kinase (CST, #2708T) antibodies diluted in TBST with 1% BSA (Table S-2). After being washed five times with TBST, samples were incubated with a peroxidase-conjugated antimouse or antirabbit secondary antibodies (Life Technologies) for 1 h at room temperature. Protein levels were detected via a Super Signal West Pico Chemiluminescent Substrate (ThermoFisher).
Neuronal Differentiation
iPSCs were differentiated into neurons using an adherent three-step differentiation method. Undifferentiated iPSCs were allowed to grow on C-Matrigel for 4 days prior to differentiation. Differentiation of these iPSCs into NSCs was induced using neural induction medium (ThermoFisher) for 11 days.83 After the induction period, NSCs were enzymatically dissociated by Accutase (Corning) and seeded as single cells at a cell density of 1 × 105 cells/cm2 in the presence of 10 μM Y-27632 on C-Matrigel-coated plates. At 24 h after seeding, NSCs were further differentiated into NPCs by incubating them in the presence of neural differentiation medium [human pluripotent stem cell serum-free medium (ThermoFisher), supplemented with 2% bovine serum albumin (ThermoFisher), 1% Glutamax I (ThermoFisher), 10 ng/mL human recombinant brain derived neurotrophic growth factor (ThermoFisher), and 10 ng/mL human recombinant glial-derived neurotrophic factor (ThermoFisher)] for 5 days.84 Neurons were seeded on poly-d-lysine (2 μg/cm2; Sigma, St Louis, MO, USA)/laminin (1 μg/cm2; Sigma)-coated plates and maintained in neuron maturation medium [NMM: Neurobasal-A medium, 2% B27 supplement (ThermoFisher)]. Medium was replaced every 2 days for 21 days.44 Cells were seeded at the same density between controls and CLN3 iPSCs at day 15 of differentiation, and photos were taken at random; images were selected at random from these photos. Consistent cell density was maintained by DAPI staining and/or by bright-field microscopy (live imaging).
Immunocytochemistry and Flow Cytometry
Cells were dissociated with Accutase and centrifuged at 200g for 5 min (BD Biosciences, San Jose, CA, USA). Cell pellets were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) or 100% cold methanol (MeOH; Sigma-Aldrich). Cells were blocked for 1 h at 25 °C in PBSG [PBS supplemented with 10% normal goat serum (Sigma-Aldrich)] with 0.2% Triton-X100. Cells were incubated overnight in primary antibodies at various dilutions (see Table S-2), washed with PBS containing 1% bovine serum album (Sigma-Aldrich), and incubated in the presence of Alexa-Fluor conjugated secondary antibodies (1:200; ThermoFisher) for 1 h at 25 °C. Cells were counter-stained with DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich) and observed on an Leica DMi-8 inverted epifluorescence microscope (Olympus, Tokyo, Japan). Micrograph pictures were acquired using Leica Suite X acquisition program (Intelligent Imaging Innovations, Denver, CO, USA) and processed using ImageJ (NIH, Bethesda, MD, USA). Flow cytometry samples were acquired and analyzed using a FACSVerse system (BD Biosciences). Relative expression was obtained by subtracting the mean fluorescence index from samples versus the mean fluorescence index from the IgG isotype control.
Lysosomal and Mitochondrial Staining
MitoTracker Deep Red FM (ThermoFisher), JC-1 dye (ThermoFisher), MitoSOX, Red Mitochondrial superoxide indicator (ThermoFisher), Lysosensor Green DND-189 (ThermoFisher), Acridine Orange (ThermoFisher) or CYTO-ID (Enzo Life Sciences) were used as per manufacturer’s protocol. Appropriate controls as detailed by the supplier were used to validate the method. Upon incubation with aforementioned dyes, cells were subjected to flow cytometric or microscopic analysis using a BD FACSVerse or Leica DMi8 fluorescence microscope. For Rotenone challenge assay, control and CLN3 neurons were incubated with 200 and 400 nM rotenone for 24 h and MTT assay (Promega) was performed to assess the metabolic rate. Data were presented as % of untreated control.
Coculture Experiments
At 24 h after purification, iPSC-derived BMECs in Transwell inserts were displaced on top of the wells containing 40-day old respective iPSC-derived neurons differentiated as described previously. The apical chamber was maintained in EC–/–, whereas the bottom chamber was replaced with fresh NMM. Cells were maintained in cocultures for 2 days. BMEC monocultures treated with similar conditions were used as controls. After 2 days of cocultures, changes in barrier function were assessed by TEER and permeability. BMEC monocultures maintained in NMM served as control monocultures to normalize such barrier function.
Acridine Orange Flow Cytometry
After 48 h of treatment with the indicated compound, neurons differentiated from CLN3 patient specific iPSCs were washed with PBS, and then incubated with 1 μg/mL of acridine orange for 30 min. The cells were washed using PBS, detached using Accutase and washed once again with PBS. The cells were resuspended in PBS and 10 000 cells were analyzed using the FITC-A and PerCP-Cy5.5A filter. Data was analyzed using FlowJo v10.2 (FlowJo, LLC) after removing cell debris.
Bcl-2 ELISA
Neurons differentiated from CLN3 patient specific iPSCs were treated with the selected chemical probe for 48 h and changes in Bcl-2 protein levels were determined by an ELISA kit (R&D Systems, DYC827-B) using the manufacturer’s recommended protocol. Neurons differentiated from IMR90-c4 iPSCs were used as control and followed the same procedure. Briefly, cells were washed once with PBS and then lysed using RIPA buffer (ThermoFisher). The proteins were quantified by BCA assay (ThermoFisher, #PI-23221). Proteins were then normalized to 30 μg of protein per sample to be used in Bcl-2 protein quantification.
Subunit c Expression
Neurons differentiated from IMR90-c4 or CLN3 patient specific iPSCs were treated with the selected chemical probe for 48 h. Cells were then washed once with PBS and lysed with RIPA buffer (Thermofisher). Proteins were quantified via BCA assay (Thermofisher, #PI-23221). Normalized protein (30 μg per sample) was then resolved by SDS-PAGE on 4–20% Tris-glycine gradient gels (BioRad), and Western blot was performed using Anti-Subunit c (Abcam, #ab181243) antibody diluted in TBST with 1% BSA (1:1000). After being washed five times with TBST, samples were incubated with a peroxidase-conjugated goat antirabbit secondary antibody (Life Technologies, #G-21234) for 1 h at room temperature. Protein levels were detected via a SuperSignal West Pico Chemiluminescent Substrate (Thermofisher, #34580). Protein band expression was then quantified using ImageJ software (NIH, Bethesda, MD, USA).
Mitochondrial Stress Test
Neural progenitor cells obtained from IMR90-c4 or CLN3 patient-specific iPSCs were separately plated at a density of 250 000 cells/well in 24-well assay plates (Seahorse Bioscience, #100850-001) using neuronal differentiation medium supplemented with StemPro hESC SFM (Life Technologies, #A1000701), BDNF (Peprotech, #450-02) and GDNF (Peprotech, #450-10) and allowed to adapt for 2 days before treatment was added. After 48 h of treatment, the neuronal differentiation medium was replaced by assay medium consisting of XF Base Medium (Seahorse Bioscience, #103575-100) supplemented with 10 mM glucose, 10 mM pyruvic acid, and 1 mM l-glutamine. Subsequently, the analysis of mitochondrial oxygen consumption rate (OCR) was performed in a Seahorse Bioscience XFe 24 flux analyzer according to the manufacturer’s instructions. The OCR values were obtained both during baseline (prior to addition of any Mito Stress Test substances), and after the addition of 1.5 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone + 0.5 μM antimycin A respectively. Prior to analysis, data were corrected by withdrawing nonmitochondrial respiration (measured after the injection of rotenone and antimycin A) from all measured OCR values. After the experiment, cells were lysed using RIPA buffer (Thermofisher), and proteins were quantified via BCA assay (Thermofisher, #PI-23221). Results were normalized to protein concentration of each well to its OCR value.
Propidium Iodide Toxicity
Neurons derived from CLN3 patient iPSCs were incubated with the indicated compound at 3 μM for 48 h. Cells were dissociated and incubated with 50 μg/mL PI solution (Life Technologies, #P3566) in NMM medium for 15 min at 37 °C. PI stained cells were analyzed by flow cytometry as previously described.33
Ion Channel Assays
KCNQ1/hminK assay was performed by Eurofins Panlabs Inc., St Charles, MO, using a cell-based automated patch clamp CiPA assay using the Qpatch platform (Assay ID: CYL8007QP2). Briefly, after whole cell configuration is achieved, a 1000 ms pulse from −80 mv to 60 mV and then a ramp for 60 mV to −80 mV over 115 ms was applied, and outward peak currents were measured upon depolarization of the cell membrane. This paradigm is delivered once every 15 s to monitor the current amplitude. The parameters measured were the maximum outward current evoked on stepping to 60 mV from the test pulse. All data were filtered for seal quality, seal drop, and current amplitude. The peak current amplitude was calculated before and after compound addition, and the amount of block was assessed by dividing the test compound current amplitude by the control current amplitude. Control is the mean hKCNQ1/hminK current amplitude collected 15 s at the end of the control; Test Compound is the mean hKCNQ1/hminK current amplitude collected in the presence of the test compound at each concentration.
hERG assay was performed by Eurofins Panlabs Inc., St Charles, MO, using a cell-based automated patch clamp CiPA assay using the Qpatch platform (Assay ID: CYL8038QP2EX).
Statistical Analysis
In this study, every N is equal to or more than 3. Each sample (N) is a biological replicate obtained from one differentiation set. Hence, we have three biological replicates obtained from three different passages, with each of them being obtained from a distinct differentiation (3 differentiation batches). In cell viability assays, three technical replicates were added to each biological replicate, as one biological replicate equated to the average of three technical replicates. Comparison of multiple groups was performed using analysis of variance (ANOVA) followed by Tukey’s post hoc test; comparisons between two groups were performed using t test. P values less than 0.05 were considered significant. Statistical calculations were performed using Microsoft Excel or GraphPad Prism Software, version 7.0 or 8.0 (Graphpad Software, La Jolla, CA). All data shown are representative of experiments from n ≥ 3 biologically independent samples (see figure legends). All error bars represent either standard deviation (SD) or standard error of measurement (SEM). See information in figure legends for specifics.
Acknowledgments
This work was funded by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award UL1TR001105 (P.C.T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors are grateful to Texas Tech University Health Sciences Center (C.M., A.A-A., and P.C.T) and the University of Nebraska Medical Center (P.C.T) for funding support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00077.
Quantification of tube length, number of nodes and number of junctions in EC++ medium and representative images of tube formation assay in EC++ and EC– medium. Structures of compounds. PI staining of CLN3 iPSC-derived neurons. Percentage change of Bcl-2 in CLN3 iPSC-derived neurons of all compounds. 1H NMR spectra of 9e, 9g, and 9h at day zero and day 10 incubation under assay conditions. Genotyping of C:N3 patient iPSCs. Kv7.1 inhibition data. Antibody source and dilution table (PDF)
Author Contributions
# N.K., A.M., and R.P. contributed equally to this work. N.K. grew and differentiated cells, synthesized compounds, performed autophagy and Bcl-2 experiments, and analyzed results. A.M. grew and differentiated cells, synthesized compounds, performed subunit c clearance experiments, Bcl-2 experiments, and the mitochondrial stress test, and analyzed results. R.P. grew, differentiated, and characterized cells, performed BBB characterization and transporter experiments, and analyzed results. A.V.C. synthesized compounds and performed NMR stability studies. M.S.S. performed angiogenic characterization. S.R. grew and differentiated cells. C.M. oversaw experimental design of the angiogenic characterization and analyzed data. A.A.-A. oversaw experimental design of stem cell studies, initiated and supervised the BBB experiments, interpreted data, and wrote the manuscript with input from all coauthors. P.C.T. conceived, initiated, and supervised the project, analyzed data, and wrote the manuscript with input from all coauthors.
The authors declare the following competing financial interest(s): N.K. and P.C.T. are inventors on a patent application (PCT Int. App. WO 2019014547) submitted by Texas Tech University that covers the composition of matter of the compounds described herein.
Supplementary Material
References
- Kollmann K.; Uusi-Rauva K.; Scifo E.; Tyynela J.; Jalanko A.; Braulke T. (2013) Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1866–1881. 10.1016/j.bbadis.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Mink J. W.; Augustine E. F.; Adams H. R.; Marshall F. J.; Kwon J. M. (2013) Classification and natural history of the neuronal ceroid lipofuscinoses. J. Child Neurol. 28 (9), 1101–1105. 10.1177/0883073813494268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santavuori P. (1988) Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 10 (2), 80–83. 10.1016/S0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- Haltia M.; Goebel H. H. (2013) The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1795–1800. 10.1016/j.bbadis.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Schulz A.; Kohlschutter A.; Mink J.; Simonati A.; Williams R. (2013) NCL diseases - clinical perspectives. Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1801–1806. 10.1016/j.bbadis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole S. E.; Anderson G.; Band H. A.; Berkovic S. F.; Cooper J. D.; Kleine Holthaus S. M.; McKay T. R.; Medina D. L.; Rahim A. A.; Schulz A.; Smith A. J. (2019) Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 18 (1), 107–116. 10.1016/S1474-4422(18)30368-5. [DOI] [PubMed] [Google Scholar]
- Anderson G. W.; Goebel H. H.; Simonati A. (2013) Human pathology in NCL. Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1807–1826. 10.1016/j.bbadis.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Palmer D. N.; Barry L. A.; Tyynela J.; Cooper J. D. (2013) NCL disease mechanisms. Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1882–1893. 10.1016/j.bbadis.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Lockhart E. M.; Warner D. S.; Pearlstein R. D.; Penning D. H.; Mehrabani S.; Boustany R.-M. (2002) Allopregnanolone attenuates N-methyl-d-aspartate-induced excitotoxicity and apoptosis in the human NT2 cell line in culture. Neurosci. Lett. 328 (1), 33–36. 10.1016/S0304-3940(02)00448-2. [DOI] [PubMed] [Google Scholar]
- Persaud-Sawin D. A.; Boustany R. M. (2005) Cell death pathways in juvenile Batten disease. Apoptosis 10 (5), 973–985. 10.1007/s10495-005-0733-6. [DOI] [PubMed] [Google Scholar]
- Boustany R. M. (2013) Lysosomal storage diseases--the horizon expands. Nat. Rev. Neurol. 9 (10), 583–598. 10.1038/nrneurol.2013.163. [DOI] [PubMed] [Google Scholar]
- Hobert J. A.; Dawson G. (2006) Neuronal ceroid lipofuscinoses therapeutic strategies: past, present and future. Biochim. Biophys. Acta, Mol. Basis Dis. 1762 (10), 945–953. 10.1016/j.bbadis.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Sands M. S. (2013) Considerations for the treatment of infantile neuronal ceroid lipofuscinosis (infantile Batten disease). J. Child Neurol. 28 (9), 1151–1158. 10.1177/0883073813495960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins-Salsbury J. A.; Cooper J. D.; Sands M. S. (2013) Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease). Biochim. Biophys. Acta, Mol. Basis Dis. 1832 (11), 1906–1909. 10.1016/j.bbadis.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. M.; Rahim A. A.; Waddington S. N.; Cooper J. D. (2010) Current therapies for the soluble lysosomal forms of neuronal ceroid lipofuscinosis. Biochem. Soc. Trans. 38 (6), 1484–1488. 10.1042/BST0381484. [DOI] [PubMed] [Google Scholar]
- Kinarivala N.; Trippier P. C. (2016) Progress in the Development of Small Molecule Therapeutics for the Treatment of Neuronal Ceroid Lipofuscinoses (NCLs). J. Med. Chem. 59 (10), 4415–4427. 10.1021/acs.jmedchem.5b01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz A.; Ajayi T.; Specchio N.; de Los Reyes E.; Gissen P.; Ballon D.; Dyke J. P.; Cahan H.; Slasor P.; Jacoby D.; Kohlschutter A. (2018) Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 378 (20), 1898–1907. 10.1056/NEJMoa1712649. [DOI] [PubMed] [Google Scholar]
- Wisniewski K. E.; Zhong N.; Kaczmarski W.; Kaczmarski A.; Sklower-Brooks S.; Brown W. T. (1998) Studies of atypical JNCL suggest overlapping with other NCL forms. Pediatr. Neurol. 18 (1), 36–40. 10.1016/S0887-8994(97)00188-4. [DOI] [PubMed] [Google Scholar]
- Lane S. C.; Jolly R. D.; Schmechel D. E.; ALroy J.; Boustany R. M. (1996) Apoptosis as the mechanism of neurodegeneration in Batten’s disease. J. Neurochem. 67 (2), 677–683. 10.1046/j.1471-4159.1996.67020677.x. [DOI] [PubMed] [Google Scholar]
- Mattson M. P. (2000) Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 1 (2), 120–9. 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Smale G.; Nichols N. R.; Brady D. R.; Finch C. E.; Horton W. E. Jr. (1995) Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol. 133 (2), 225–230. 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- Dhar S.; Bitting R. L.; Rylova S. N.; Jansen P. J.; Lockhart E.; Koeberl D. D.; Amalfitano A.; Boustany R. M. N. (2002) Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann. Neurol. 51 (4), 448–466. 10.1002/ana.10143. [DOI] [PubMed] [Google Scholar]
- Puranam K.; Qian W. H.; Nikbakht K.; Venable M.; Obeid L.; Hannun Y.; Boustany R. M. (1997) Upregulation of Bcl-2 and elevation of ceramide in Batten disease. Neuropediatrics 28 (1), 37–41. 10.1055/s-2007-973664. [DOI] [PubMed] [Google Scholar]
- Marsden V. S.; O’Connor L.; O’Reilly L. A.; Silke J.; Metcalf D.; Ekert P. G.; Huang D. C.; Cecconi F.; Kuida K.; Tomaselli K. J.; Roy S.; Nicholson D. W.; Vaux D. L.; Bouillet P.; Adams J. M.; Strasser A. (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419 (6907), 634–637. 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- Puranam K. L.; Guo W. X.; Qian W. H.; Nikbakht K.; Boustany R. M. (1999) CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol. Genet. Metab. 66 (4), 294–308. 10.1006/mgme.1999.2834. [DOI] [PubMed] [Google Scholar]
- Seranova E.; Connolly K. J.; Zatyka M.; Rosenstock T. R.; Barrett T.; Tuxworth R. I.; Sarkar S. (2017) Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61 (6), 733–749. 10.1042/EBC20170055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; Espinola J. A.; Fossale E.; Massey A. C.; Cuervo A. M.; MacDonald M. E.; Cotman S. L. (2006) Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 281 (29), 20483–20493. 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- Cotman S. L.; Staropoli J. F. (2012) The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin. Lipidol. 7 (1), 79–91. 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrachud U.; Walker M. W.; Simas A. M.; Heetveld S.; Petcherski A.; Klein M.; Oh H.; Wolf P.; Zhao W. N.; Norton S.; Haggarty S. J.; Lloyd-Evans E.; Cotman S. L. (2015) Unbiased Cell-based Screening in a Neuronal Cell Model of Batten Disease Highlights an Interaction between Ca2+ Homeostasis, Autophagy, and CLN3 Protein Function. J. Biol. Chem. 290 (23), 14361–14380. 10.1074/jbc.M114.621706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C.; Martinez-Lopez N.; Otten E. G.; Carroll B.; Maetzel D.; Singh R.; Sarkar S.; Korolchuk V. I. (2016) Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1861 (4), 269–284. 10.1016/j.bbalip.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Lieberman A. P.; Puertollano R.; Raben N.; Slaugenhaupt S.; Walkley S. U.; Ballabio A. (2012) Autophagy in lysosomal storage disorders. Autophagy 8 (5), 719–730. 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf K.; Makoukji J.; Saab S.; Makhoul N. J.; Carmona A. V.; Kinarivala N.; Ghanem N.; Trippier P. C.; Boustany R.-M. (2020) Exogenous Flupirtine as potential treatment for CLN3 disease. Cells 9, 1872. 10.3390/cells9081872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makoukji J.; Saadeh F.; Mansour K. A.; El-Sitt S.; Al Ali J.; Kinarivala N.; Trippier P. C.; Boustany R. M. (2018) Flupirtine derivatives as potential treatment for the neuronal ceroid lipofuscinoses. Ann. Clin. Transl. Neurol. 5 (9), 1089–1103. 10.1002/acn3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinarivala N.; Patel R.; Boustany R. M.; Al-Ahmad A.; Trippier P. C. (2017) Discovery of Aromatic Carbamates that Confer Neuroprotective Activity by Enhancing Autophagy and Inducing the Anti-Apoptotic Protein B-Cell Lymphoma 2 (Bcl-2). J. Med. Chem. 60 (23), 9739–9756. 10.1021/acs.jmedchem.7b01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinarivala N.; Suh J. H.; Botros M.; Webb P.; Trippier P. C. (2016) Pharmacophore elucidation of phosphoiodyn A - Potent and selective peroxisome proliferator-activated receptor beta/delta agonists with neuroprotective activity. Bioorg. Med. Chem. Lett. 26 (8), 1889–1893. 10.1016/j.bmcl.2016.03.028. [DOI] [PubMed] [Google Scholar]
- Trippier P. C.; Jansen Labby K.; Hawker D. D.; Mataka J. J.; Silverman R. B. (2013) Target- and mechanism-based therapeutics for neurodegenerative diseases: strength in numbers. J. Med. Chem. 56 (8), 3121–3147. 10.1021/jm3015926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto M. C.; Brennand K. J.; Boyer L. F.; Gage F. H. (2011) Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum. Mol. Genet. 20 (R2), R109–R115. 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatine G. D.; Barrile R.; Workman M. J.; Sances S.; Barriga B. K.; Rahnama M.; Barthakur S.; Kasendra M.; Lucchesi C.; Kerns J.; Wen N.; Spivia W. R.; Chen Z.; Van Eyk J.; Svendsen C. N. (2019) Human iPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 24 (6), 995–1005. 10.1016/j.stem.2019.05.011. [DOI] [PubMed] [Google Scholar]
- Sima N.; Li R.; Huang W.; Xu M.; Beers J.; Zou J.; Titus S.; Ottinger E. A.; Marugan J. J.; Xie X.; Zheng W. (2018) Neural stem cells for disease modeling and evaluation of therapeutics for infantile (CLN1/PPT1) and late infantile (CLN2/TPP1) neuronal ceroid lipofuscinoses. Orphanet J. Rare Dis. 13 (1), 54. 10.1186/s13023-018-0798-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusi-Rauva K.; Blom T.; von Schantz-Fant C.; Blom T.; Jalanko A.; Kyttala A. (2017) Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. Int. J. Mol. Sci. 18 (5), 955. 10.3390/ijms18050955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley L. A.; Burnight E. R.; Drack A. V.; Banach B. B.; Ochoa D.; Cranston C. M.; Madumba R. A.; East J. S.; Mullins R. F.; Stone E. M.; Tucker B. A. (2016) Using Patient-Specific Induced Pluripotent Stem Cells and Wild-Type Mice to Develop a Gene Augmentation-Based Strategy to Treat CLN3-Associated Retinal Degeneration. Hum. Gene Ther. 27 (10), 835–846. 10.1089/hum.2016.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M. L.; Tecedor L.; Lysenko E.; Ramachandran S.; Stein C. S.; Davidson B. L. (2018) Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo. Neurobiol. Dis. 115, 182–193. 10.1016/j.nbd.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Vodyanik M. A.; Smuga-Otto K.; Antosiewicz-Bourget J.; Frane J. L.; Tian S.; Nie J.; Jonsdottir G. A.; Ruotti V.; Stewart R.; Slukvin I. I.; Thomson J. A. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318 (5858), 1917–1920. 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Patel R.; Page S.; Al-Ahmad A. J. (2017) Isogenic blood-brain barrier models based on patient-derived stem cells display inter-individual differences in cell maturation and functionality. J. Neurochem. 142 (1), 74–88. 10.1111/jnc.14040. [DOI] [PubMed] [Google Scholar]
- Lippmann E. S.; Azarin S. M.; Kay J. E.; Nessler R. A.; Wilson H. K.; Al-Ahmad A.; Palecek S. P.; Shusta E. V. (2012) Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30 (8), 783–791. 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R. G.; Quan C.; Reyes-Ortiz A. M.; Lutz S. E.; Kedaigle A. J.; Gipson T. A.; Wu J.; Vatine G. D.; Stocksdale J.; Casale M. S.; Svendsen C. N.; Fraenkel E.; Housman D. E.; Agalliu D.; Thompson L. M. (2017) Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 19 (7), 1365–1377. 10.1016/j.celrep.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim W. W.; Mizushima N. (2020) Lysosome biology in autophagy. Cell Discovery 6, 6. 10.1038/s41421-020-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochfeld W. E.; Lee S.; Rubinsztein D. C. (2013) Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol. Sin. 34 (5), 600–604. 10.1038/aps.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinarivala N.; Shah K.; Abbruscato T. J.; Trippier P. C. (2017) Passage Variation of PC12 Cells Results in Inconsistent Susceptibility to Externally Induced Apoptosis. ACS Chem. Neurosci. 8 (1), 82–88. 10.1021/acschemneuro.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly R. D.; Brown S.; Das A. M.; Walkley S. U. (2002) Mitochondrial dysfunction in the neuronal ceroid-lipofuscinoses (Batten disease). Neurochem. Int. 40 (6), 565–571. 10.1016/S0197-0186(01)00128-0. [DOI] [PubMed] [Google Scholar]
- Luiro K.; Kopra O.; Blom T.; Gentile M.; Mitchison H. M.; Hovatta I.; Tornquist K.; Jalanko A. (2006) Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments. J. Neurosci. Res. 84 (5), 1124–1138. 10.1002/jnr.21015. [DOI] [PubMed] [Google Scholar]
- Wager K.; Zdebik A. A.; Fu S.; Cooper J. D.; Harvey R. J.; Russell C. (2016) Neurodegeneration and Epilepsy in a Zebrafish Model of CLN3 Disease (Batten Disease). PLoS One 11 (6), e0157365. 10.1371/journal.pone.0157365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J.; Bleich S.; Wiltfang J.; Maler M.; Parsons C. G. (1999) Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J. Neural. Transm. (Vienna) 106 (9–10), 857–867. 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- Wang J. J.; Li Y. (2016) KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol. Sin. 37 (1), 25–33. 10.1038/aps.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe M. J.; Large C. H.; Sankar R. (2012) The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53 (3), 412–424. 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- Kim R. Y.; Yau M. C.; Galpin J. D.; Seebohm G.; Ahern C. A.; Pless S. A.; Kurata H. T. (2015) Atomic basis for therapeutic activation of neuronal potassium channels. Nat. Commun. 6, 8116. 10.1038/ncomms9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C.; Bruhova I.; Lerche H.; Steinmeyer K.; Wei A. D.; Strutz-Seebohm N.; Lang F.; Busch A. E.; Zhorov B. S.; Seebohm G. (2007) Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol. Pharmacol. 71 (6), 1503–1511. 10.1124/mol.106.031682. [DOI] [PubMed] [Google Scholar]
- Qian K.; Huang H.; Peterson A.; Hu B.; Maragakis N. J.; Ming G. L.; Chen H.; Zhang S. C. (2017) Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 8 (4), 843–855. 10.1016/j.stemcr.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Qian K.; Du Z.; Cao J.; Petersen A.; Liu H.; Blackbourn L. W. t.; Huang C. L.; Errigo A.; Yin Y.; Lu J.; Ayala M.; Zhang S. C. (2014) Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14 (6), 796–809. 10.1016/j.stem.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Qiu B.; Guan W.; Wang Q.; Wang M.; Li W.; Gao L.; Shen L.; Huang Y.; Xie G.; Zhao H.; Jin Y.; Tang B.; Yu Y.; Zhao J.; Pei G. (2015) Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 17 (2), 204–212. 10.1016/j.stem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Slosarek E. L.; Schuh A. L.; Pustova I.; Johnson A.; Bird J.; Johnson M.; Frankel E. B.; Bhattacharya N.; Hanna M. G.; Burke J. E.; Ruhl D. A.; Quinney K.; Block S.; Peotter J. L.; Chapman E. R.; Sheets M. D.; Butcher S. E.; Stagg S. M.; Audhya A. (2018) Pathogenic TFG Mutations Underlying Hereditary Spastic Paraplegia Impair Secretory Protein Trafficking and Axon Fasciculation. Cell Rep. 24 (9), 2248–2260. 10.1016/j.celrep.2018.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane S. C.; Jolly R. D.; Schmechel D. E.; Alroy J.; Boustany R. M. (1996) Apoptosis as the mechanism of neurodegeneration in Batten’s disease. J. Neurochem. 67 (2), 677–683. 10.1046/j.1471-4159.1996.67020677.x. [DOI] [PubMed] [Google Scholar]
- Mao D.; Che J.; Han S.; Zhao H.; Zhu Y.; Zhu H. (2015) RNAi-mediated knockdown of the CLN3 gene inhibits proliferation and promotes apoptosis in drug-resistant ovarian cancer cells. Mol. Med. Rep. 12 (5), 6635–6641. 10.3892/mmr.2015.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecedor L.; Stein C. S.; Schultz M. L.; Farwanah H.; Sandhoff K.; Davidson B. L. (2013) CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J. Neurosci. 33 (46), 18065–18079. 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A.; Sarkar C.; Singh S. P.; Zhang Z.; Munasinghe J.; Peng S.; Chandra G.; Kong E.; Mukherjee A. B. (2012) The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: amelioration by resveratrol. Hum. Mol. Genet. 21 (10), 2233–2244. 10.1093/hmg/dds038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatine G. D.; Al-Ahmad A.; Barriga B. K.; Svendsen S.; Salim A.; Garcia L.; Garcia V. J.; Ho R.; Yucer N.; Qian T.; Lim R. G.; Wu J.; Thompson L. M.; Spivia W. R.; Chen Z.; Van Eyk J.; Palecek S. P.; Refetoff S.; Shusta E. V.; Svendsen C. N. (2017) Modeling Psychomotor Retardation using iPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell 20 (6), 831–843. 10.1016/j.stem.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. A. A.; Seo H. S.; Armien A. G.; Bates F. S.; Tolar J.; Azarin S. M. (2018) Modeling and rescue of defective blood-brain barrier function of induced brain microvascular endothelial cells from childhood cerebral adrenoleukodystrophy patients. Fluids Barriers CNS 15 (1), 9. 10.1186/s12987-018-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. G.; Zhang L.; Jiang Q.; Zhang R.; Davies K.; Powers C.; Bruggen N.; Chopp M. (2000) VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Invest. 106 (7), 829–838. 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigau V.; Morin M.; Rousset M. C.; de Bock F.; Lebrun A.; Coubes P.; Picot M. C.; Baldy-Moulinier M.; Bockaert J.; Crespel A.; Lerner-Natoli M. (2007) Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 130 (Pt 7), 1942–1956. 10.1093/brain/awm118. [DOI] [PubMed] [Google Scholar]
- Lojewski X.; Staropoli J. F.; Biswas-Legrand S.; Simas A. M.; Haliw L.; Selig M. K.; Coppel S. H.; Goss K. A.; Petcherski A.; Chandrachud U.; Sheridan S. D.; Lucente D.; Sims K. B.; Gusella J. F.; Sondhi D.; Crystal R. G.; Reinhardt P.; Sterneckert J.; Scholer H.; Haggarty S. J.; Storch A.; Hermann A.; Cotman S. L. (2014) Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 23 (8), 2005–2022. 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo S. Y.; Castoreno A. B.; Aldrich L. N.; Lassen K. G.; Goel G.; Dancik V.; Kuballa P.; Latorre I.; Conway K. L.; Sarkar S.; Maetzel D.; Jaenisch R.; Clemons P. A.; Schreiber S. L.; Shamji A. F.; Xavier R. J. (2015) Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc. Natl. Acad. Sci. U. S. A. 112 (31), E4281–4287. 10.1073/pnas.1512289112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M.; Pal R.; Nelvagal H. R.; Lotfi P.; Stinnett G. R.; Seymour M. L.; Chaudhury A.; Bajaj L.; Bondar V. V.; Bremner L.; Saleem U.; Tse D. Y.; Sanagasetti D.; Wu S. M.; Neilson J. R.; Pereira F. A.; Pautler R. G.; Rodney G. G.; Cooper J. D.; Sardiello M. (2017) mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 8, 14338. 10.1038/ncomms14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trippier P. C. (2016) Selecting Good ’Drug-Like’ Properties to Optimize Small Molecule Blood-Brain Barrier Penetration. Curr. Med. Chem. 23 (14), 1392–1407. 10.2174/0929867323666160405112353. [DOI] [PubMed] [Google Scholar]
- Petcherski A.; Chandrachud U.; Butz E. S.; Klein M. C.; Zhao W. N.; Reis S. A.; Haggarty S. J.; Ruonala M. O.; Cotman S. L. (2019) An Autophagy Modifier Screen Identifies Small Molecules Capable of Reducing Autophagosome Accumulation in a Model of CLN3-Mediated Neurodegeneration. Cells 8 (12), 1531. 10.3390/cells8121531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri M. C.; Zalckvar E.; Kimchi A.; Kroemer G. (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8 (9), 741–752. 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Terman A.; Kurz T.; Navratil M.; Arriaga E. A.; Brunk U. T. (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signaling 12 (4), 503–535. 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M. E.; Kielian T. (2019) Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities. J. Neurochem. 148 (5), 612–624. 10.1111/jnc.14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H.; Zhorov B. S. (2008) K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem. Rev. 108 (5), 1744–1773. 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. F.; Zhao M. Y.; Xu Y. J.; Su Y.; Kong X. H.; Wang Z. Y. (2020) Fenamates Inhibit Human Sodium Channel Nav1.2 and Protect Glutamate-Induced Injury in SH-SY5Y Cells. Cell. Mol. Neurobiol. 10.1007/s10571-020-00826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann E. S.; Al-Ahmad A.; Azarin S. M.; Palecek S. P.; Shusta E. V. (2015) A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 4, 4160. 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokolakis G.; Mikelis C.; Papadimitriou E.; Courty J.; Karetsou E.; Katsoris P. (2006) Effect of heparin affin regulatory peptide on the expression of vascular endothelial growth factor receptors in endothelial cells. In Vivo 20 (5), 629–635. [PubMed] [Google Scholar]
- Mikelis C.; Lamprou M.; Koutsioumpa M.; Koutsioubas A. G.; Spyranti Z.; Zompra A. A.; Spiliopoulos N.; Vradis A. A.; Katsoris P.; Spyroulias G. A.; Cordopatis P.; Courty J.; Papadimitriou E. (2011) A peptide corresponding to the C-terminal region of pleiotrophin inhibits angiogenesis in vivo and in vitro. J. Cell. Biochem. 112 (6), 1532–1543. 10.1002/jcb.23066. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Shin S.; Jha B. S.; Liu Q.; Sheng J.; Li F.; Zhan M.; Davis J.; Bharti K.; Zeng X.; Rao M.; Malik N.; Vemuri M. C. (2013) Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl. Med. 2 (11), 862–870. 10.5966/sctm.2013-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou A.; Shaltouki A.; Steiner J. P.; Jha B.; Heman-Ackah S. M.; Swistowski A.; Zeng X.; Rao M. S.; Malik N. (2014) Functional screening assays with neurons generated from pluripotent stem cell-derived neural stem cells. J. Biomol. Screening 19 (1), 32–43. 10.1177/1087057113501869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.