

Abstract
A solid understanding of the mechanisms governing ligand binding is crucial for rational design of therapeutics targeting the dopamine D2 receptor (D2R). Here, we use G protein-coupled inward rectifier potassium (GIRK) channel activation in Xenopus oocytes to measure the kinetics of D2R antagonism by a series of aripiprazole analogues, as well as the recovery of dopamine (DA) responsivity upon washout. The aripiprazole analogues comprise an orthosteric and a secondary pharmacophore and differ by the length of the saturated carbon linker joining these two pharmacophores. Two compounds containing 3- and 5-carbon linkers allowed for a similar extent of recovery from antagonism in the presence of 1 or 100 μM DA (>25 and >90% of control, respectively), whereas recovery was less prominent (∼20%) upon washout of the 4-carbon linker compound, SV-III-130, both with 1 and 100 μM DA. Prolonging the coincubation time with SV-III-130 further diminished recovery. Curve-shift experiments were consistent with competition between SV-III-130 and DA. Two mutations in the secondary binding pocket (V91A and E95A) of D2R decreased antagonistic potency and increased recovery from SV-III-130 antagonism, whereas a third mutation (L94A) only increased recovery. Our results suggest that the secondary binding pocket influences recovery from inhibition by the studied aripiprazole analogues. We propose a mechanism, supported by in silico modeling, whereby SV-III-130 initially binds reversibly to the D2R, after which the drug-receptor complex undergoes a slow transition to a second ligand-bound state, which is dependent on secondary binding pocket integrity and irreversible during the time frame of our experiments.
Keywords: drug kinetics, competitive binding, antipsychotics, PET scan, molecular dynamics simulation, Xenopus oocytes, arrestin, HEK cells
Introduction
The dopamine D2 receptor (D2R) is a G protein-coupled receptor (GPCR) and an important pharmaceutical target. D2R antagonism or weak partial agonism is the common denominator of antipsychotic drugs, and D2R agonism is a mainstay of Parkinson’s disease treatment.1−4 The time course of receptor occupancy has been suggested to have an important impact on the clinical properties of therapeutic ligands, including antipsychotics, where transient rather than continuous D2R occupancy may be associated with a more favorable profile in terms of extrapyramidal side effects.5 Ligand binding kinetics is presumed to play an important role in determining the time course of occupancy at the target receptor.6,7 For example, the action of angiotensin-1 receptor antagonists is prolonged by their long-lasting, induced-fit binding to their target receptor, reflected by insurmountable antagonism in in vitro experiments.8 Insurmountable antagonism is characterized by a decrease in the maximal agonist-induced response, which cannot be overcome by increasing the concentration of agonist.8
Similarly, differences in dissociation9 or association kinetics10 between antipsychotics have been linked to their differential side-effect profiles, although this remains a matter of debate.11 Receptor binding kinetics has also been predicted to be an important determinant of positron emission tomography (PET) tracer characteristics, such as sensitivity to endogenous neurotransmitter release.12
The recently reported D2R crystal structure reveals a deep orthosteric binding pocket, situated in the plane of the cell membrane between transmembrane segments 3, 5, and 6, as well as a secondary binding pocket, located more extracellularly and mainly comprising residues from transmembrane segment 2 and extracellular loop 1.13 In search of GPCR ligands with beneficial pharmacological properties and enhanced selectivity, attention has been drawn to bivalent ligands, consisting of two distinct pharmacophores targeting both the orthosteric and the secondary binding pocket.14 To facilitate parallel engagement of both pockets, the orthosteric- and secondary pharmacophores are covalently joined by a linker, the length of which has been found to be critical.15 At the D2R, ligand interactions with the secondary binding pocket have been reported to confer selectivity over the closely related D3R and, in some cases, to impart signaling bias or allosteric properties.16,17 In particular, a cluster of residues in transmembrane segment 2, including V91, L94, and E95, has been found to mediate important contacts between the ligand and the secondary binding pocket.18−20
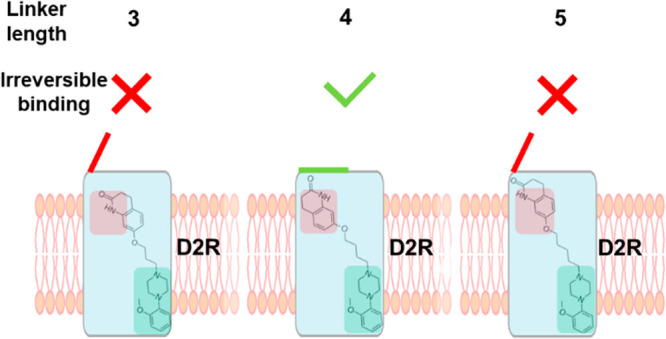
A series of candidate PET radiotracers, designed to selectively target D2R, were derived from the scaffold of the clinically used antipsychotic, aripiprazole. These compounds are composed of an orthosteric 2-methoxyphenylpiperazine pharmacophore and a secondary dihydroquinolinone pharmacophore, linked by alkyl chains of varying length.15 SWR-1-8, SV-III-130, and SWR-1-14 contain chains of 3, 4, and 5 carbon atoms, respectively (Figure 1A), resulting in different distances and relative orientations between the two pharmacophores. SV-III-130 was found to be ∼20-fold more potent at the D2R and ∼40-fold more selective for the D2R over D3R compared to SWR-1-8 and SWR-1-14, demonstrating the crucial impact of linker length on D2R affinity. [11C]-SV-III-130 has subsequently been used as a D2R selective PET tracer in nonhuman primates.21
Figure 1.

Structures, potencies, recovery from antagonism, and binding kinetics of SWR-1-8, SV-III-130, and SWR-1-14 at the D2R. A) Structures of SWR-1-8, SV-III-130, and SWR-1-14. B) Concentration–response relationship for DA-induced GIRK activation in oocytes coexpressing D2R, RGS4, and GIRK1/4 channels (EC50 = 33 nM; n = 4). C) Concentration–response curves for the inhibition of the GIRK response to 100 nM DA by SWR-1-8 (n = 3), SV-III-130 (n = 3), and SWR-1-14 (n = 3). D) Recovery of D2R-mediated GIRK activation by DA after antagonism by SWR-1-8 (n = 8 for 1 μM DA and n = 5 for 100 μM DA), SV-III-130 (n = 11 for 1 μM DA and n = 12 for 100 μM DA), and SWR-1-14 (n = 7 for 1 μM DA and n = 4 for 100 μM DA). Graphs show mean GIRK current traces normalized to the maximal response evoked by 1 μM DA (40 s), followed by 30 μM SWR-1-8 or SWR-1-14 or 1 μM SV-III-130 coapplied with 1 μM DA (125 s) and finally reversed by 1 μM (thick dotted line) or 100 μM (thick solid line) DA (400 s). Thin lines indicate SEM. E) Extent of recovery from antagonism following application of 1 or 100 μM DA (data from experiments shown in panel D). F) Rate of recovery from antagonism following application of 1 or 100 μM DA (data from experiments shown in panel D). G) Observed rates of GIRK response decay, kobs, upon application of varying concentrations of antagonist in the presence of 100 nM DA during 125 s; n = 3–7. Data shown are means ± SEM *; p < 0.05, ***; p < 0.001, and ****; p < 0.0001, Student’s t test.
While important roles of the D2R secondary binding pocket in mediating subtype selectivity and allosteric effects have been described, the putative involvement of this pocket in the reversibility of antagonism has not been examined. Here, we compared the binding kinetics, reversibility, and surmountability of the three aripiprazole analogues and examined the role of the secondary binding pocket in determining these characteristics. To this end, we used a G protein-coupled inward-rectifying potassium (GIRK) channel assay as a time-resolved readout of D2R occupancy by dopamine (DA)22,23 and developed a mechanistic model, supported by molecular dynamics simulations, explaining our observations.
Results and Discussion
Role of Linker Length in Determining Potency, Kinetics, and Surmountability of Antagonism
To determine the influence of linker length on the kinetics, reversibility, and surmountability of D2R antagonism, we studied the properties of the three aripiprazole analogues, SWR-1-8, SV-III-130, and SWR-1-14, in antagonizing DA-evoked GIRK activation in Xenopus oocytes coexpressing D2R with GIRK1/4 channels and RGS4 (Figure 1A, B). SV-III-130 was more potent compared to SWR-1-8 and SWR-1-14 (Figure 1C; Table 1). The Ki values derived from these experiments were in relative agreement with previous Ki data from [125I]-IABN radioligand binding experiments (SWR-1-8; 4.8 ± 0.9 nM, SV-III-130; 0.22 ± 0.01 nM, and SWR-1-14; 7.3 ± 1.3 nM;15). Ligand-induced GIRK channel block was observed to be <10% in all cases, as determined by application of 30 μM SWR-1-8, SWR-1-14, and SV-III-130 to oocytes expressing GIRK channels in the absence of D2R (Supplementary Figure S1). Previous investigations of SV-III-130 at the D2R reported partial agonism in an adenylate cyclase inhibition assay but antagonism in GIRK activation and ERK1/2 assays.16 In our hands, weak partial agonism at the D2R in the GIRK activation assay was observed for SV-III-130 (6.6% of the response to 1 μM DA) but not for SWR-1-8 and SWR-1-14, which behaved as antagonists/inverse agonists (Supplementary Figure S2).
Table 1. Binding Kinetics and Affinity Estimates of Ligands at the D2R WTa.
| receptor | ligand | koff (s–1) | kon (s–1 × M–1) | pKd | pKi |
|---|---|---|---|---|---|
| D2R WT | SWR-1-8 | 0.017 ± 0.002 (5) | 3.4 ± 0.4 × 105 (3–6) | 7.30 ± 0.04 | 8.01 ± 0.18 (3) |
| D2R WT | SV-III-130 | 0.007 ± 0.001 (11) | 8.6 ± 0.5 × 105 (3–6) | 8.09 ± 0.04 | 8.57 ± 0.05 (3) |
| D2R WT | SWR-1-14 | 0.021 ± 0.002 (4) | 3.0 ± 0.4 × 105 (4–7) | 7.15 ± 0.04 | 7.70 ± 0.13 (3) |
koff values were calculated from response recovery t1/2 as koff = ln (2)/t1/2. See the Methods section for derivation of kon. pKd was calculated from the koff and kon estimates as Kd = koff/kon, while pKi was calculated from the IC50 for GIRK channel inhibition using the Cheng–Prusoff equation,25 assuming a Kd of DA equaling its EC50 for GIRK activation in our assay; i.e., 33 nM. The number of oocytes is shown in parentheses; for kon, this corresponds to the number of oocytes for each data point (see Figure 1G). Data shown are means ± SEM.
Reversibility of D2R antagonism was evaluated using a protocol consisting of three steps: (i) a baseline response evoked by a maximally effective concentration (1 μM) of DA, (ii) antagonism of this response by 30 μM of SWR-1-8/SWR-1-14 or 1 μM SV-III-130, and (iii) recovery of the response upon washout of the antagonist ligand in the presence of 1 or 100 μM DA. The GIRK response is essentially saturated at 1 μM DA (Figure 1B). Therefore, any additional increase in response recovery with 100 μM DA should be an effect of increased competition with antagonist ligand which remains dissolved in the membrane and/or interior of the cell following washout, as has been reported to occur for some lipophilic compounds.22−24 For SWR-1-8, SV-III-130, and SWR-1-14, a response recovery fraction of 0.2 to 0.4 was observed when using 1 μM DA in the recovery phase, while increasing the DA concentration during the recovery phase to 100 μM significantly increased the response recovery following SWR-1-8 and SWR-1-14 application to about 0.9 to 1.0 (Figure 1D, E). Conversely, recovery from SV-III-130-induced antagonism was about 0.2 under both conditions (Figure 1D, E), indicative of insurmountable behavior. The response recovery kinetics in the presence of 100 μM DA, which we previously used as a measure of antagonist dissociation rate (koff23), was swift for SWR-1-8 and SWR-1-14 but slower for SV-III-130 (Figure 1F; Table 1). Response recovery kinetics was similar at 1 μM and 100 μM DA for SWR-1-8 and SV-III-130 but more rapid at 100 μM compared to 1 μM DA for SWR-1-14 (Figure 1F).
To estimate compound association rate constants (kons), we employed our previously published strategy,23 using the decay rate of the DA-evoked GIRK current at different concentrations of antagonist as a proxy measure of the antagonist binding rate. Plotting the observed rates of inhibition of the current response to DA against varying concentrations of competing ligand suggested a more rapid kon of SV-III-130 at the D2R, compared to SWR-1-8 and SWR-1-14 which exhibited similar kons (Figure 1G; Table 1).
SV-III-130 Behaves Competitively When Coapplied with Different Concentrations of DA
The lack of increased response recovery from SV-III-130-mediated antagonism in the face of increased concentrations of DA could be indicative of a noncompetitive or possibly allosteric binding mechanism. To further evaluate this mechanism, we performed a curve-shift assay to test whether SV-III-130 and DA compete for binding to the D2R when the two ligands are coapplied. First, 1 μM DA was applied to each cell to elicit a full agonist response which could then be compared to the pseudosteady-state response elicited by 10 nM to 100 μM DA coapplied with varying concentrations of SV-III-130, or vehicle, after 500 s coapplication (Figure 2A). The DA concentration–response curve was progressively right-shifted with increasing concentrations of SV-III-130 without any appreciable effect on the maximal response (Figure 2B), consistent with competition between DA and SV-III-130 for D2R binding. Thus, SV-III-130 displayed differential behavior (i.e., insurmountable vs competitive) after first being bound to D2R and then washed out/competed with DA (Figure 1D, E), compared to when coapplied with DA without prior D2R binding (Figure 2). GIRK currents are known to demonstrate a time-dependent decrease due to alterations of intracellular sodium levels during prolonged recording protocols.26 The effect of this rundown is evident in Figure 2B, where the GIRK current response at the end of the 500 s coapplication period is ∼60% of the instantaneous response to 1 μM DA.
Figure 2.

Curve-shift GIRK activation assay of D2R antagonism upon coapplication of SV-III-130 and DA. A) Assay principles; 1 μM DA elicits a full agonist response (left, red arrow). The subsequent response amplitude in the presence of variable concentrations of SV-III-130 and DA, following 500 s coapplication (right, green arrow), was normalized to the control response elicited by 1 μM DA. In the example shown, 1 μM SV-III-130 coapplied with 100 μM DA; n = 3 oocytes. Thick lines represent mean normalized currents, whereas thin lines indicate SEM. B) Current amplitude at the end of the 500 s coapplication period, normalized to the instantaneous maximum response amplitude in the same oocyte and plotted against DA concentration, for varying concentrations of SV-III-130 or control. n = 3–7 oocytes per data point. Data shown are means ± SEM.
Insurmountability of SV-III-130 Antagonism Depends on the D2R Secondary Binding Pocket
The high potency of SV-III-130 antagonism at the D2R and the insurmountability of this antagonism, as observed during the recovery phase when washing out the ligand in the presence of 1 or 100 μM DA (see Figure 1), likely arise from interactions between the secondary pharmacophore and the secondary binding pocket, given the differential behavior of both SWR-1-8 and SWR-1-14. Previous investigations suggested a crucial role for residues V91, L94, and E95 in contacting the secondary pharmacophore of bivalent D2R ligands, including SV-III-130.16,19 In addition, W100A in extracellular loop 1 is located at the extracellular boundary of the secondary binding pocket and has been shown to affect bivalent ligand affinity and to adopt different conformations depending on the nature of the bound ligand.13,27 To investigate the role of the secondary binding pocket in shaping SV-III-130 binding kinetics and insurmountability, V91, L94, E95, and W100 were individually mutated to alanine. First, we validated the expression of the D2R mutants by immunoblot experiments. Anti-D2R antibodies detected a broad protein band at 80–100 kDa (Supplementary Figure S3), corresponding to the expected size of fully glycosylated D2R, as previously reported.28,29 Importantly, this immunoreactive band was absent in striatal membranes from D2R–/– mice and in total membranes from uninjected oocytes (Supplementary Figure S3), thus indicating the specificity of the antibody used. Overall, these results indicated that WT D2R, V91A, L94A, E95A, and W100A receptors were properly expressed in oocytes.
Next, we assessed the surmountability of SV-III-130 at the D2R mutants in oocytes. In the GIRK activation assay, the DA potencies were similar for the WT and L94A mutant D2R and slightly higher for the V91A and E95A mutants (Figure 3A). In contrast to the other mutant receptors, the potency of DA at W100A was markedly reduced by about 10-fold compared to WT (Figure 3A). This probably reflects a loss of DA affinity as the Western blot experiments indicated relatively similar expression of W100A, WT D2R, and the other D2R mutants (Supplementary Figure S3). Further supporting a link between functional DA potency in our GIRK assay and DA binding affinity, we have previously shown that there is little or no receptor reserve under our assay conditions.23 Furthermore, the E95A and L94A mutants were shown to express at similar levels as WT and showed insignificant changes in DA potency in a previous study,18 while alanine mutation of the interaction partner of W100A, I184, significantly reduced DA potency despite similar expression. In order to achieve similar receptor occupancies by DA, we used 1 μM instead of 100 nM DA for constructing concentration–response curves for SV-III-130-mediated inhibition at the W100A mutant (Figure 3B) and 10 instead of 1 μM DA to elicit the baseline response when studying the W100A mutant in response recovery experiments (Figure 3C).
Figure 3.

Potencies, recovery from antagonism, and binding kinetics of SV-III-130 at the V91A, L94A, and E95A mutant D2R. A) DA potency at V91A (EC50 = 21 nM, n = 3–4), L94A (EC50 = 41 nM, n = 3–7), E95A (EC50 = 15 nM, n = 3–4), W100A (EC50 = 323 nM, n = 3), and WT (EC50 = 33 nM, n = 4) D2R. B) SV-III-130 potency at the V91A (n = 4), L94A (n = 3), E95A (n = 7), and W100A (n = 3) D2R. C) Recovery of activation by DA at the V91A (n = 3 for 1 μM DA and n = 4 for 100 μM DA), L94A (n = 13 for 1 μM DA and n = 13 for 100 μM DA), E95A (n = 5 for 1 μM DA and n = 3 for 100 μM DA), and W100A (n = 4 for 10 μM DA and n = 4 for 300 μM DA) mutant D2R following antagonism by SV-III-130. GIRK current traces normalized to the maximal response evoked by 1 μM (10 μM for W100A) DA. Thick lines represent mean normalized currents, whereas thin lines indicate SEM. D) Extent of recovery upon application of 1 or 100 μM (10 and 300 μM for W100A) DA following antagonism by 30 μM (V91A and E95A), 3 μM (W100A), and 1 μM SV-III-130 (L94A; data from experiments shown in panel C). E) Rate of recovery following application of 1 or 100 μM DA (10 and 300 μM for W100A; data from experiments shown in panel C). F) Observed association rates, kobs, which allowed for calculation of association rates for SV-III-130 at the V91A, L94A, and E95A D2R. n = 3–7. G) Clustering of kinetic Kd relative to Ki for SWR-1-8, SWR-1-14, and SV-III-130 at the WT receptor and SV-III-130 at the V91A, L94A, and E95A mutant receptors, as indicated. Data shown are means ± SEM *; p < 0.05, **; p < 0.01, and ***; p < 0.001, Student’s t test.
The inhibitory potency (corrected for differences in DA potency and expressed as Ki) of SV-III-130 was decreased 9-fold for V91A and W100A and 4-fold for E95A mutant D2R (Figure 3B and Table 2). In contrast, SV-III-130 was slightly more potent at the L94A mutant receptor (about 2-fold) compared to WT D2R (Figure 3B and Table 2). Partial agonist activity of SV-III-130 was lost at the V91A mutant but retained at the L94A and E95A mutants (Supplementary Figure S4).
Table 2. Binding Kinetics and Affinity Estimates of SV-III-130 at D2R Secondary Binding Pocket Mutantsa.
| receptor | ligand | koff (s–1) | kon (s–1 × M–1) | pKd | pKi |
|---|---|---|---|---|---|
| D2R V91A | SV-III-130 | 0.066 ± 0.005 (4) | 4.9 ± 0.4 × 105 (4–5) | 6.87 ± 0.05 | 7.63 ± 0.16 (4) |
| D2R L94A | SV-III-130 | 0.007 ± 0.001 (13) | 8.9 ± 0.6 × 105 (3–7) | 8.10 ± 0.05 | 8.95 ± 0.17 (3) |
| D2R E95A | SV-III-130 | 0.022 ± 0.005 (3) | 3.3 ± 0.4 × 105 (3–4) | 7.18 ± 0.05 | 7.97 ± 0.13 (7) |
| D2R W100A | SV-III-130 | 0.010 ± 0.001 (4) | n.d. | n.d. | 7.63 ± 0.07 (3) |
koff values were calculated from response recovery t1/2 as koff = ln (2)/t1/2. See Methods section for derivation of kon. pKd was calculated from the koff and kon estimates as Kd = koff/kon, while pKi was calculated from the IC50 for GIRK channel inhibition using the Cheng–Prusoff equation,25 assuming the Kd of DA at the various mutants to equal the corresponding EC50 for GIRK activation (see the legend of Figure 3A). Number of oocytes in parentheses; for kon, this corresponds to the number of oocytes for each data point (see Figure 3F). Data shown are means ± SEM. n.d., not determined.
Response recovery experiments (employing the same protocol as in Figure 1D) with SV-III-130 using 1 μM (10 μM with W100A) DA in the recovery phase demonstrated an increased recovery at V91A, L94A, E95A, and W100A mutant D2R as compared to WT D2R (Figure 3C, D). When 100 μM (300 μM with W100A) DA was used in the recovery phase, the extent of recovery at the four D2R mutants increased further, suggesting surmountable antagonism by SV-III-130 at these mutants (Figure 3C, D). Accelerated SV-III-130 recovery kinetics was observed with 1 μM and 100 μM DA for the V91A and E95A mutant D2R and with 10 and 300 μM DA for the W100A mutant, whereas for the L94A mutant, the time to half-maximal recovery was similar to that observed for WT D2R (Figure 3E). kon and kinetic Kd estimates for SV-III-130 at the V91A and E95A mutants were lower than at the WT receptor, while kon and kinetic Kd appeared similar at the L94A mutant and WT D2R (Figure 3F, G; Table 2). The preservation of SV-III-130 kon at the L94A mutant suggests that a high kon, which may affect ligand competition by causing rapid rebinding to the receptor,10 is not sufficient to explain the insurmountable antagonism of SV-III-130 at the WT receptor.
Kinetic Binding Models Support an Induced-Fit Binding Mode of SV-III-130 at the D2R
The distinct extents of recovery from antagonism and differing association and dissociation rate constants of SWR-1-8, SWR-1-14, and SV-III-130 at the D2R indicate differential binding modes. Based on the demonstrated surmountability of SWR-1-8 and SWR-1-14 antagonism at the WT D2R in the response recovery experiments (Figure 1D, E), a three-state binding model including unbound receptor (R), agonist-bound receptor (RA), and ligand-bound receptor (RL) was constructed using the experimentally determined kon and koff rate constants (Figure 4A; eq 1 in the Methods section). To account for lipophilic accumulation in the cell membrane,24 a residual ligand fraction of 2% was assumed throughout the response recovery phase. Modeling of the low extent of recovery from and insurmountable nature of SV-III-130 antagonism required the incorporation of an irreversible, second ligand-bound state (RL*; Figure 4B; eq 2 in the Methods section).
Figure 4.

Simulations of three- and four-state ligand binding at a receptor. A) Scheme depicting a surmountable antagonist ligand (L) binding to the receptor (R), in competition with the agonist (A; DA). B) Scheme depicting induced-fit insurmountable antagonist ligand binding, where RL* represents an irreversibly bound antagonist ligand. C, D, E) Simulation of response recovery from antagonism by SWR-1-8 (C), SV-III-130 (D), and SWR-1-14 (E) at WT D2R, with 1 or 100 μM DA during the recovery phase. The response is assumed to be proportional to the fraction of the agonist-bound state; RA. F) Simulated recovery of WT D2R activation by DA after prolonged, 400-s antagonism with SV-III-130. G) Experimental recovery of WT D2R activation by DA after prolonged, 400-s antagonism with SV-III-130. H) Simulated curve-shift assay with SV-III-130 at WT D2R, plotting the RA fraction after 500 s simulation time for different concentrations of DA and SV-III-130, as indicated. I) Simulated concentration–response curves for SV-III-130 antagonism at WT and L94A mutant D2R. “Normalized response” corresponds to the RA fraction after 100 s of simulation time, in the presence of 100 nM DA and varying concentrations of SV-III-130, as indicated. The L94A mutant was simulated by removing the fourth state, RL*, from the model. J) Simulation of response recovery from antagonism by SV-III-130 at the L94A mutant D2R. As in I), the three-state model is employed, using kinetic data for SV-III-130 from the L94A mutant. Simulations of SWR-1-8 (C; kon(RL) = 340 M–1 s–1, koff(RL) = 0.017 s–1), SV-III-130 (D; kon(RL) = 860 M–1 s–1, koff(RL) = 0.007 s–1, kon(RL*) = 0.01 s–1), and SWR-1-14 (E; kon(RL) = 300 M–1 s–1, koff(RL) = 0.021 s–1) at WT D2R were conducted in the presence of 1 or 100 μM DA as agonist. kon(A) = 5 × 106 M–1 s–1 and koff(A) = 0.17 s–1 for all simulations at WT D2R. Parameters for the three-state model of SV-III-130 interaction at D2R L94A; kon(RL) = 890 M–1 s–1, koff(RL) = 0.007/s, kon(A) = 5 × 106 M–1 s–1, and koff(A) = 0.21/s. koff(A) = was adjusted as necessary to yield a Kd corresponding to the DA EC50 at D2R L94A.
Assuming RA to be proportional to the experimentally observed GIRK response, simulation of the experimental response recovery data presented in Figure 1C for SWR-1-8 and SWR-1-14 using the three-state binding model recapitulated the 1 μM DA activation and blocking phases and demonstrated an increase in the extent of response recovery with increasing DA concentration (1 versus 100 μM DA; Figure 4C, E). For SV-III-130, the corresponding simulation using the extended four-state binding model recapitulated the experimental extent of recovery for both 1 and 100 μM of DA during the response recovery phase (Figure 4D). The four-state model (Figure 4B) may be thought of as modeling induced-fit binding and implies a time-dependence for SV-III-130 antagonism of the D2R, with a transition from RL to RL* defined by the fraction 1–1/(et*0.01), where t is time in seconds. The simulations predicted a RL-RL* transition of ∼98% when SV-III-130 application was prolonged from 125 to 400 s (Figure 4F). In agreement, in vitro experiments revealed a virtual elimination of response recovery following application of SV-III-130 during 400 s (Figure 4G). The four-state model also recapitulated the competitive behavior of SV-III-130 when coapplied (without prior occupancy of the receptor) with different concentrations of DA (Figure 4H), as was determined experimentally in curve-shift experiments (Figure 2B).
To simulate the L94A mutant receptor, at which SV-III-130 did not display insurmountable antagonism, the estimates of SV-III-130 kon and koff from our experiments with L94A D2R were incorporated into a three-state binding model. Simulated SV-III-130 inhibition curves demonstrated similarities between the four-state (WT) and three-state model with parameters from the L94A mutant D2R (Figure 4I). The simulated curves support the notion that the irreversibly bound state contributes very little to the Ki estimates derived from inhibition curve experiments, which would be in agreement with our experimental findings (see Figure 3G). The simulated response recovery from SV-III-130 antagonism in the three-state model (L94A mutant) was pronounced as compared to the four-state model (WT), for both 1 and 100 μM DA (Figure 4J).
L94A Mutation Induces Conformational Changes in the Receptor and Ligand Binding Mode of SV-III-130
To study the impact of the L94A mutation on ligand binding and receptor dynamics at the molecular level, we carried out all-atomistic molecular dynamics simulations for the WT D2R and the L94A mutant receptor in complex with SV-III-130 (Figure 5A). In the WT D2R, the orthosteric pharmacophore of SV-III-130 is buried deeply in the orthosteric binding pocket,16,19 whereas the secondary pharmacophore is primarily stabilized by contacts with I184 in extracellular loop 2 and W100 in extracellular loop 1 (shown as blue van der Waals radii; Figure 5A). Upon L94A mutation (orange van der Waals radii; Figure 5A), the ligand translates toward transmembrane segment 2 (yellow arrow 2; Figure 5A) while maintaining similar contact ratios with I184 and W100 compared to the WT (contact ratioW100/I184: 1.17 [mutant] vs 1.20 [WT]; Figure 5C, D). Interestingly, the similar contact ratios correlate well with the similar estimated affinities of SV-III-130 at the WT and L94A mutant receptors (Figure 3G).
Figure 5.

Structural impact of L94A mutation on SV-III-130. A) The most populated SV-III-130 binding mode in the D2R WT (blue) and the L94A (orange) receptors. Upon L94A mutation, the ligand translates toward transmembrane segment 2 as indicated by the yellow arrows. Despite translation in the L94A mutant, SV-III-130 is primarily sandwiched by W100 and I184 similar to the WT complex. B) Comparison of the position of W100 between the WT and L94A receptors in terms of distance between Cα atoms of W100 and L/A94, as well as the position of the W100 side chain center of mass in the z-dimension (perpendicular to the membrane). Both values are significantly different between the WT and the L94A receptors. C) Representative ligand binding mode in the WT receptor with W100 adopting a low z value (putative RL complex). D) Representative ligand binding mode in the L94A mutant receptor with W100 adopting a low z value (putative RL complex). E, F) Ligand binding modes in the WT receptor in which W100 adopts higher z values and stacks on top of the ligand (potentially leading to RL* complexes). In C–F), the position of the center of mass of the W100 side chain (red circle) in the z-dimension (perpendicular to the membrane) is highlighted. **; p < 0.01, Mann–Whitney U test.
We found that L94A mutation creates an empty space adjacent to transmembrane segment 2 which becomes occupied by W100, enabling tight packing of W100 against transmembrane segment 2 (Figure 5D). In contrast, the bulkier L94 impedes such tight W100 packing in the WT receptor (Figure 5C). This finding was supported by computing the distribution of the distances between the Cα atoms of W100 and L/A94 in transmembrane segment 2 over the entire simulation time (distanceW100 to TM2), which revealed significantly shorter distances in the L94A mutant compared to the WT D2R (Figure 5B, left panel). As a result of the tight W100 packing (Figure 5A; yellow arrow 1), SV-III-130 followed the movement of W100 (Figure 5A; yellow arrow 2) which in turn led to an adaption of extracellular loop 2 (Figure 5A; yellow arrow 3).
In addition to this, the distance plot of W100 to transmembrane segment 2 (Figure 5B, left panel) suggests that W100 in extracellular loop 1 is more flexible in the WT D2R compared to the L94A mutant, as reflected by its wider spatial distribution in the WT receptor. Among these conformations, we observed receptor states with W100 buried in the secondary binding pocket (Figure 5C) and conformations where W100 is exposed to the solvent forming a “lid” on top of the ligand (Figure 5E, F). The location of W100 can be approximated by its position in the z dimension: a high z value (approximately >22 Å) corresponds to the “lidlike” conformations (Figure 5E, F), whereas a low z value (approximately <22 Å) corresponds to conformations where W100 is buried in the protein (Figure 5C). Interestingly, we found that the “lidlike” conformations are only explored in the WT receptor (Figure 5E, F) as highlighted by the box plot (Figure 5B, right panel). It is tempting to speculate that such conformations obstruct the ligand exit gateway, leading to a conformation of the D2R-SV-III-130 complex which has a significantly slower koff than the conformation presented in Figure 5C. Indeed, this hypothesis is supported by recent findings that L94 interacts with W100 and I184, forming a “lid” over the secondary binding pocket in the risperidone-bound D2R crystal structure, an interaction which influences ligand residence time in radioligand binding experiments.13 A recent study by Lane et al.30 likewise highlighted the role of L94 in controlling the dynamics of W100 and also found evidence for an important role of W100 in restricting ligand access to and egress from the D2R orthosteric binding pocket. Similar findings of ligand dissociation being restricted by a “lid” formed by the extracellular loops have been reported for the serotonin 5-HT2A receptor as well.31
As described above, we directly tested the role of W100 by mutagenesis, creating a W100A D2R mutant. The loss of SV-III-130 affinity observed at this mutant (Figure 2 and Table 2) is congruent with the molecular dynamics simulations presented above, suggesting that W100 mediates important stabilizing contacts between the secondary pharmacophore of SV-III-130 and the receptor (Figure 5C, D). Notably, the rates and extents of recovery from SV-III-130 inhibition were increased at the W100A mutant, compared to WT D2R, both when using 10 μM and 300 μM DA during the recovery phase (Figure 3C–E). These findings are in agreement with our hypothesis that W100 plays a crucial role in trapping SV-III-130 in the WT D2R.
Surmountable and Insurmountable Behavior of Antagonist Ligands in the GIRK Assay Is Recapitulated upon Ligand Preapplication in a Beta-Arrestin Recruitment Assay
Using a BRET assay in transfected HEK293 cells, full concentration–response curves for DA-induced beta-arrestin2 recruitment to D2R were produced in the presence of increasing concentrations of SWR-1-8, SV-III-130, and SWR-1-14. Cells were preincubated with antagonist ligands for 5 min, after which dopamine was added.
Preincubation with SWR-1-8 or SWR-1-14 induced a progressive right-shift of the concentration–response curve for dopamine without affecting the maximum response amplitude (Figure 6A, C), consistent with competitive antagonism. On the other hand, the maximum effect of dopamine was progressively diminished in the presence of increasing concentrations of SV-III-130, while the EC50 of DA remained virtually unchanged (Figure 6B), in agreement with an insurmountable mode of antagonism.
Figure 6.

Curve-shift beta-arrestin2 recruitment assay of D2R antagonism resulting from preapplication of SWR-1-8, SV-III-130, and SWR-1-14. Curve-shift experiments for DA-induced beta-arrestin2 recruitment to D2R following 5 min preincubation with A) SWR-1-8, B) SV-III-130, and C) SWR-1-14. The normalized data represent mean values from three independent experiments (n = 3) performed in triplicate. The curves represent the best fit to the data using nonlinear regression analysis.
Our study implies the existence of two distinct binding modes of SV-III-130 at the D2R with the position of W100, relative to the bound ligand, playing an important role in creating these two states. The dynamics of W100 following ligand binding is apparently slow enough to appreciate the existence of two separate binding states. In agreement with the considerable flexibility of W100 suggested by our molecular dynamics simulations, the position of this residue differs markedly between the risperidone- and the haloperidol-bound D2R crystal structures.13,27 Whereas W100 stacks on top of risperidone, as noted above, the residue is rotated away from the secondary binding pocket in the haloperidol-bound structure.27 In future experiments, it will be interesting to further study the role of W100 for the dissociation kinetics and surmountabilities of risperidone and haloperidol. Moreover, the finding that SV-III-130 behaves as an insurmountable antagonist when investigated both in an assay of G protein-dependent signaling (GIRK activation) and in an arrestin recruitment assay may imply that the conformation of the secondary binding pocket, or at least of W100, does not differ markedly between G protein- and arrestin-coupled states of D2R. This is particularly interesting given the suggested implication of I184, which interacts with both SV-III-130 and W100, in mediating agonist bias between G protein and arrestin signaling.32
Our findings furthermore underscore how measures of ligand potency and kinetics can be heavily influenced by incubation time and order of ligand addition: In the curve-shift GIRK experiments, SV-III-130 behaved as a classic competitive agonist when the ligand was added simultaneously with DA, whereas in recovery experiments with WT D2R, there was a marked decrease in the ability of DA, even at 100 μM, to activate the receptor following washout of SV-III-130 from the extracellular medium. Interestingly, this decrease became even more pronounced with longer intervals of application of SV-III-130. We interpret this phenomenon as an example of induced-fit binding.8
The proposed induced-fit binding mechanism of SV-III-130 may be relevant for understanding the long-lasting receptor occupancy observed with clinical dosing of the structurally similar weak partial agonist antipsychotic, aripiprazole.33 The information beginning to be unraveled by the present study may also benefit PET tracer development. Whereas [11C]-SV-III-130 binding is displaced to some extent by amphetamine,21 certain other D2R PET tracers, such as [11C]-(N-methyl)benperidol, show virtually no amphetamine-induced displacement.34 A radiotracer’s receptor binding kinetics, as well as its tissue-to-plasma efflux rate, has been suggested to affect sensitivity to endogenous transmitter release,12 and the reversibility of tracer binding would also seem to be important for these characteristics. Thus, better knowledge of the mechanisms responsible for induced-fit insurmountable binding could potentially enable the rational design of PET tracers suitable for measuring D2R density vs endogenous DA release.
As with all heterologous expression systems, the Xenopus oocyte system comes with several limitations. The oocytes do not express the full complement of D2R-interacting proteins found in native cells, such as medium spiny neurons of the striatum. Furthermore, GIRK channel currents are an indirect readout of GPCR occupancy by agonist. However, the GIRK assay does allow for the study of dynamic processes with high temporal resolution and without the need for receptor modification. Importantly, we have previously shown that our affinity estimates, derived either from IC50 values or from estimates of kinetic rate constants, are in good agreement with published data from radioligand binding studies.23 Finally, it should also be noted that SV-III-130 binding could be almost completely displaced by the D2/3R antagonist, eticlopride, in a previous PET study,21 seemingly at odds with the irreversible binding implied by our model. However, this displacement took place over a longer time course (∼40 min) than studied in our experiments, suggesting that the induced-fit binding of SV-III-130 might be reversible on a longer time scale. Moreover, the ability to compete with SV-III-130 bound in the putative RL* state may be ligand-specific.
Conclusions
Our experimental and in silico findings support two distinct binding modes of SV-III-130 at D2R, where the first binding mode is competitive and surmountable by DA, whereas the second, induced-fit binding mode is irreversible and hence insurmountable. In particular, L94A mutation preserved SV-III-130 potency but abolished its insurmountability. Molecular dynamics simulations suggested that the L94A mutation might perturb the positioning of W100 in extracellular loop 1 over the ligand binding site, reducing the prevalence of “lidlike” conformations where W100 stacks on top of the ligand, preventing its egress. Thus, trapping by W100 may be the structural mechanism for insurmountable antagonism by SV-III-130. The present insights into the role of the secondary binding pocket for induced-fit, irreversible binding at the D2R may provide information for the prospective development of improved therapeutic and radiotracer ligands.
Methods
Molecular Biology
WT human dopamine D2L receptor (D2R) cDNA was subcloned into pXOOM (provided by Dr. Søren-Peter Olesen, University of Copenhagen, Denmark). Mutagenesis was performed by Genscript Biotech (Piscataway, NJ). All mutations were verified by sequencing. cDNA encoding human GIRK1 (Kir3.1), GIRK4 (Kir3.4) (provided by Dr. Terence Hebert, University of Montreal, Canada), and regulator of G protein signaling 4 (RGS4) (from the Missouri cDNA Resource Center; www.cdna.org) were in pCDNA3 (Invitrogen). Plasmids were linearized using the appropriate restriction enzymes (D2R, RGS4; XhoI and GIRK1/GIRK4; NotI), followed by in vitro transcription using the T7 mMessage mMachine kit (Ambion, Austin, TX). cRNA concentration and purity were determined by spectrophotometry.
Oocyte Preparation
Oocytes from the African clawed toad, Xenopus laevis, were isolated surgically as described previously.35 The surgical procedures were approved by the Swedish National Board for Laboratory Animals and the Stockholm Ethical Committee. Following 24-h incubation at 12 °C, oocytes were injected (using a Nanoject microinjector; Drummond Scientific) with 0.2 ng of D2L receptor cRNA, 40 ng of RGS4 cRNA, and 1 ng of each GIRK1 and GIRK4 cRNA in a volume of 50 nL per oocyte. RGS4 is a GTPase activating protein and was included in order to speed up the kinetics of G protein turnover, such that GIRK channel opening more closely follows D2R occupancy by DA.
D2R Ligands
DA was purchased from Sigma-Aldrich (St. Louis, MO). SWR-1-8, SWR-1-14, and SV-III-130 were synthesized as previously described,15 dissolved in DMSO, and diluted in the recording buffer to a maximum final DMSO concentration of 0.3% v/v.
Electrophysiology Methods
Following incubation of oocytes for 5–7 days at 12 °C, two-electrode voltage-clamp (CA-1, Dagan, Minneapolis, MN) recordings were conducted at room temperature (22 °C), as previously described.35 Data were acquired at a membrane potential of −80 mV and 134 Hz sampling frequency using pCLAMP8 (Molecular Devices, Sunnyvale, CA). To increase the inward rectifier potassium channel current at negative potentials, a high potassium concentration extracellular perfusion buffer was used (in mM: 64 NaCl, 25 KCl, 0.8 MgCl2, 0.4 CaCl2, 15 HEPES, 1 ascorbic acid, adjusted to pH 7.4), yielding a K+ reversal potential of about −40 mV. Ascorbic acid was used to prevent the spontaneous oxidation of DA. Oocytes were perfused with solutions at a rate of 1.5 mL/min using the pressure-driven, computer-controlled SmartSquirt system (Automate Scientific, Berkeley, CA).
Gel Electrophoresis and Immunoblotting
Striata from WT and D2R knockout mice (tissue from an earlier study; Taura et al., 201829) or frozen injected oocytes were homogenized in ice-cold 50 mM Tris HCl buffer (pH 7.4) containing a protease inhibitor cocktail (Roche Molecular Systems, Pleasanton, CA) using a Polytron for three periods of 10 s each. The homogenate was centrifuged for 10 min at 1000g. The resulting supernatant was centrifuged for 30 min at 12000g to pellet total membranes. The oocyte total membrane fractions were solubilized in 1% Triton X-100 to eliminate a major contaminating band of equivalent molecular mass that alters the normal migration of D2R.36
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 10% polyacrylamide gels. Proteins were transferred to a Hybond-LFP polyvinylidene difluoride (PVDF) membrane (GE Healthcare, Chicago, IL) using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA). The PVDF membrane was blocked with 5% (w/v) dry nonfat milk in phosphate-buffered saline containing 0.05% Tween-20 (PBS-T) for 45 min and immunoblotted using rabbit polyclonal anti-D2R (1 μg/mL; Frontier Institute Co. Ltd., Ishikari City, Japan) antibody in a blocking solution overnight at 4 °C. The PVDF membrane was then washed with PBS-T three times (5 min each) before incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1/30,000; Pierce Biotechnology, Rockford, IL) in a blocking solution at 20 °C during 2 h. After washing the PVDF membranes with PBS-T three times (5 min each), the immunoreactive bands were developed using a chemiluminescent detection kit (Thermo Fisher Scientific, Waltham, MA) and detected with an Amersham Imager 600 (GE Healthcare Europe GmbH, Barcelona, Spain). Subsequently, the PVDF membrane was also immunoblotted with rabbit polyclonal anti-β-tubulin-HRP (0.16 μg/mL; ab21058, Abcam, Cambridge, UK) for protein loading control.
Cell Culture
Cell culture reagents and selection antibiotics were purchased from LifeTechologies/Invitrogen (Grand Island, NY). The D2 BRET cell line was constructed as indicated below. All other buffers and compounds were purchased from Sigma-Aldrich (St Louis, MO), unless specified otherwise. Stable D2-Rluc8 – Arrestin-Venus cells (D2-BRET-Arr) were created by doubly transfecting Flp-In T-REx 293 HEK cells (Invitrogen, Carlsbad, CA) with a beta-arrestin-2-mVenus acceptor vector expressed constitutively and a D2 DAR-Rluc8 donor vector under control of a tetracycline inducible promoter. In this way, the expression of the D2 DAR can be induced and thus the receptor-donor/arrestin expression ratio controlled. D2-BRET-Arr cells were cultured in Dulbecco’s modified Eagle’s Medium containing 10% FBS, 1000 units/mL penicillin, 1000 μg/mL streptomycin, 100 mM sodium pyruvate, 1 μg/mL gentamicin, 2 μg/mL puromycin, and 100 μg/mL hygromycin.
Beta-Arrestin Recruitment Assay
Direct protein–protein interaction between D2R and beta-arrestin2 was analyzed using BRET assays where the interaction of the receptor (donor) with beta-arrestin2 (acceptor) results in a shift of the emission spectra of the Rluc8 tag (after incubation with coelenterazine-h) from 485 to 525 nm as previously described by our laboratory.37 The resulting luminescence was read and quantified on a Flexstation III multiplate reader (Molecular Devices, San Jose, CA). Results were expressed as a normalized ratio of emission observed with control agonist alone. Since the BRET signal is dependent on the donor/acceptor expression ratio, this must be optimized in order to achieve the best response by varying the expression level of the Rluc8-tagged receptor. D2-BRET-Arr cells were seeded at 3 × 106 cells/144 mm dish in culture media without puromycin or hygromycin selection. Adherent cells were then incubated with 40 nM tetracycline added directly to the culture media to induce D2R expression. The cells were then incubated for 24 h at 37 °C in 5% CO2 and 90% humidity. Following incubation, cells were centrifuged at 1000g for 10 min and resuspended (200,000 cells/mL) in Dulbecco’s Phosphate Buffered Saline containing 0.01% sucrose. Cells were dispensed into solid-bottom white 96-well plates (Greiner Bio-one, Monroe, NC) at 100 μL/well (20,000 cells/well) and incubated at room temperature for 45 min. Following incubation, cells were incubated for 5 min with 5 μM coelenterazine h (Nanolight Technology, Pinetop, AZ) in the dark. Cells were then stimulated with the appropriately diluted agonist for 5 min prior to reading. For antagonist assays, a test compound was added to the cells and incubated for 5 min prior to agonist addition.
Concentration–Response Data Analysis
Electrophysiological data were analyzed using Clampfit (Molecular Devices). Concentration–response curves were calculated using the variable-slope sigmoidal functions in GraphPad 5. For IC50 estimation, 100 nM DA was applied to an oocyte to provide a baseline response, followed by increasing concentrations of antagonist applied with 100-s intervals (for SWR-1-8 and SWR-1-14, 50-s intervals were used, as their relatively rapid kinetics allowed for faster equilibration). For each cell, the response amplitude at the end of each antagonist application interval was normalized to the current elicited by 100 nM DA at the beginning of the protocol. Data were fitted to the equation
where Y is the response as a fraction of 1, bottom is the maximal response inhibition evoked by the antagonist, and X is the logarithm of ligand concentration.
For DA potency data, increasing concentrations of DA were applied at 25-s intervals, and the response amplitudes achieved with each concentration normalized to the response evoked by 100 μM DA. The equation used for fitting agonist data was
Intrinsic efficacy of the ligands was evaluated by a 100 s application and normalization to the response of 1 μM DA in the same oocyte.38 Data points were represented as mean ± SEM.
For curve-shift experiments, a maximal response was first evoked by application of 1 μM DA, which was subsequently washed out, followed by the application of DA in the presence (or absence, to generate the control curve for DA alone) of different concentrations of SV-III-130. The current amplitude following 500 s of coapplication of DA and SV-III-130 was normalized to the initial response elicited by 1 μM DA.
Estimation of Rate Constants
Association rate constants, kon, were calculated based on the observed association rate, kobs, in accordance with previous descriptions23
where the antagonist concentration is known, and the fraction of unoccupied receptors, R0, prior to antagonist application is derived from the concentration–response curve of DA at D2R. Specifically, R0 for 100 nM DA at the WT D2R was estimated as 0.30 (Figure 1B). koff was estimated separately as
where t1/2 is the time for half-maximal response recovery.
Ligand affinity estimates based on binding kinetics were calculated as
Receptor Binding Models
Recovery from D2R antagonism was modeled as a three-state process, using experimental values for kon and koff. The three states capturing competitive binding were unbound receptor (R), agonist-bound receptor (RA), and ligand-bound receptor (RL)
 |
1 |
where kon and koff are association and dissociation rate constants for the competing ligand (L) and the agonist DA (A). The induced-fit ligand binding was modeled as
 |
2 |
where RL* denotes the ligand bound to the induced-fit state of the receptor, and k2 is the association rate of the ligand to the RL* state.
Derivation of Kinetic Parameters
Based on the previously described t1/2 for D2R-induced GIRK response termination (in the presence of RGS4) upon DA washout,39 the agonist dissociation rate koff(A) was approximated to 0.17/s. The association rate constant kon(A) was calculated from the experimental DA EC50: koff/EC50 = kon = 5 × 106 M–1 s–1. For the induced-fit model (eq 2), a constant, irreversible flux kon(RL*) from RL to RL* was selected with a time constant of 100 s to recapitulate the features of the response recovery experiments.
Molecular Dynamics Simulations
Molecular dynamics simulations have been proven valuable for shedding light on molecular mechanisms underlying ligand binding and GPCR functionality.40−42 To generate the D2R-SV-III-130 WT complex, we utilized the crystal structure of the D2R (PDB code: 6CM4). The ligand was docked into the D2R structure using the standard docking protocol from MOE (www.chemcomp.com). The final docking pose was selected based on scoring and visual inspection. The generated WT complex was aligned to the membrane using the OPM database,43 placed in a POPC membrane, and solvated with TIP3 waters, using the CHARMM-GUI server.44 The ionic strength of the system was kept at 0.15 M using NaCl ions. The L94A system was generated by introducing the mutation using the CHARMM-GUI pipeline.
Simulations were carried out similarly to previously published protocols45−47 using the ACEMD simulation package.48 Ligand parameters were assigned by ParamChem from the CGenFF force field.49,50 Parameters for other system components were obtained from CHARMM36m51 and CHARMM36 force fields.52 In the simulation protocol, we adhere to the guidelines of the GPCRmd consortium.53
The systems were first relaxed during 200 ns of simulations under constant pressure and temperature (NPT) with a time step of 2 fs, with gradually decreasing harmonic constraints applied to the protein backbone. Temperature was maintained at 310 K using the Langevin thermostat,54 and pressure was kept at 1 bar using the Berendsen barostat.55 The equilibration run was followed by four 800 ns production runs under constant volume and temperature (NVT) with a 4 fs time step. This allowed us to amass a complete simulation time of 3.2 μs for the WT and L94A systems, respectively. The temperature was maintained at 310 K using the Langevin thermostat. No harmonic constraints are applied in the NVT phase. In all simulations, we used van der Waals and short-range electrostatic interactions with a cutoff of 9 Å and the particle mesh Ewald method56 for long-range electrostatic interactions.
Ligand–receptor contacts were quantified using the “get_contacts” script.57 The computed ratio of W100 and I184 contacts was quantified by dividing the stability of W100 contacts by I184 contacts.
Acknowledgments
This study was supported by grants from Stiftelsen Lars Hiertas Minne, Åhlénstiftelsen, Karolinska Institutet Funds and Magnus Bergvalls stiftelse (to K.S.). K.S. received postdoctoral grants from the Swedish Brain Foundation and the Swedish Society of Medicine and is currently a fellow at the Wallenberg Center for Molecular Medicine at Umeå University. T.M.S. acknowledges financial support from the National Science Centre of Poland, project number 2017/27/N/NZ2/02571. J.S. received financial support from the Instituto de Salud Carlos III FEDER (PI15/00460 & PI18/00094) and the NEURON-ERANET (AC18/00030). F.C. was supported by FEDER/Ministerio de Ciencia, Innovación y Universidades–Agencia Estatal de Investigación (SAF2017-87349-R) and the Catalan government (2017 SGR 1604). R.B.F. and D.R.S. are supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke-Intramural Research Program (ZIA NS002263). We would like to thank Brittney Miller and Martin Dalefield for excellent technical assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00477.
GIRK block by tested compounds, intrinsic activities of compounds at WT and mutant D2R constructs, and expression of WT and mutant D2R constructs in Xenopus oocytes, as assessed by Western blot (PDF)
Author Contributions
R.Å. performed electrophysiology experiments and drafted the first version of the manuscript. H.Z. developed kinetic models of ligand binding and designed experiments. T.M.S. performed molecular dynamics simulations and wrote the corresponding parts of the manuscript. R.B.F. performed beta-arrestin recruitment experiments. S.W.R. synthesized the aripiprazole analogues used in the study. R.R.L. provided reagents. P.Å. provided electrophysiology resources. F.C. performed Western blot experiments. D.R.S. supervised beta-arrestin recruitment experiments. R.H.M. conceived of the study, designed the aripiprazole analogues, and supervised their synthesis. J.S. supervised molecular dynamics simulations. J.N. provided electrophysiology resources. K.S. designed and supervised the study, designed and performed electrophysiology experiments, and drafted the first version of the manuscript together with R.Å. All authors participated in manuscript writing and approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Nord M.; Farde L. (2011) Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci. Ther. 17, 97–103. 10.1111/j.1755-5949.2010.00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J. M.; Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217. 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Millan M. J.; Goodwin G. M.; Meyer-Lindenberg A.; Ove Ögren S. (2015) Learning from the past and looking to the future: Emerging perspectives for improving the treatment of psychiatric disorders. Eur. Neuropsychopharmacol. 25, 599–656. 10.1016/j.euroneuro.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Moritz A. E.; Free R. B.; Sibley D. R. (2018) Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell. Signalling 41, 75–81. 10.1016/j.cellsig.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2016) The drug–target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 15, 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Wong Y. C.; Centanni M.; de Lange E. C. M. (2019) Physiologically Based Modeling Approach to Predict Dopamine D2 Receptor Occupancy of Antipsychotics in Brain: Translation From Rat to Human. J. Clin. Pharmacol. 59, 731–747. 10.1002/jcph.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare S. R. J.; Fleck B. A.; Williams J. P.; Grigoriadis D. E. (2020) The importance of target binding kinetics for measuring target binding affinity in drug discovery: a case study from a CRF(1) receptor antagonist program. Drug Discovery Today 25, 7–14. 10.1016/j.drudis.2019.09.011. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Van Liefde I.; Swinney D. C. (2016) On the different experimental manifestations of two-state ’induced-fit’ binding of drugs to their cellular targets. Br. J. Pharmacol. 173, 1268–1285. 10.1111/bph.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S.; Seeman P. (2000) Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. Journal of Psychiatry and Neuroscience 25, 161–166. [PMC free article] [PubMed] [Google Scholar]
- Sykes D. A.; Moore H.; Stott L.; Holliday N.; Javitch J. A.; Lane J. R.; Charlton S. J. (2017) Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun. 8, 763. 10.1038/s41467-017-00716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeberg H.; Sahlholm K. (2018) Antipsychotics with similar association kinetics at dopamine D2 receptors differ in extrapyramidal side-effects. Nat. Commun. 9, 3577. 10.1038/s41467-018-04489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris E. D.; Yoder K. K. (2007) Positron emission tomography displacement sensitivity: predicting binding potential change for positron emission tomography tracers based on their kinetic characteristics. J. Cereb. Blood Flow Metab. 27, 606–617. 10.1038/sj.jcbfm.9600359. [DOI] [PubMed] [Google Scholar]
- Wang S.; Che T.; Levit A.; Shoichet B. K.; Wacker D.; Roth B. L. (2018) Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555, 269–273. 10.1038/nature25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J. R.; Sexton P. M.; Christopoulos A. (2013) Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci. 34, 59–66. 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Vangveravong S.; Zhang Z.; Taylor M.; Bearden M.; Xu J.; Cui J.; Wang W.; Luedtke R. R.; Mach R. H. (2011) Synthesis and characterization of selective dopamine D(2) receptor ligands using aripiprazole as the lead compound. Bioorg. Med. Chem. 19, 3502–3511. 10.1016/j.bmc.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedtke R. R.; Mishra Y.; Wang Q.; Griffin S. A.; Bell-Horner C.; Taylor M.; Vangveravong S.; Dillon G. H.; Huang R.-Q.; Reichert D. E.; Mach R. H. (2012) Comparison of the binding and functional properties of two structurally different D2 dopamine receptor subtype selective compounds. ACS Chem. Neurosci. 3, 1050–1062. 10.1021/cn300142q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Battiti F. O.; Bonifazi A. (2020) 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem. 63, 1779–1797. 10.1021/acs.jmedchem.9b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Michino M.; Verma R. K.; Klein Herenbrink C.; Shonberg J.; Kopinathan A.; Scammells P. J.; Capuano B.; Thal D. M.; Javitch J. A.; Christopoulos A.; Shi L.; Lane J. R. (2018) The structural determinants of the bitopic binding mode of a negative allosteric modulator of the dopamine D2 receptor. Biochem. Pharmacol. (Amsterdam, Neth.) 148, 315–328. 10.1016/j.bcp.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C.; Verma R.; Lim H. D.; Kopinathan A.; Keen A.; Shonberg J.; Draper-Joyce C. J.; Scammells P. J.; Christopoulos A.; Javitch J. A.; Capuano B.; Shi L.; Lane J. R. (2019) Molecular Determinants of the Intrinsic Efficacy of the Antipsychotic Aripiprazole. ACS Chem. Biol. 14, 1780–1792. 10.1021/acschembio.9b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R. K.; Abramyan A. M.; Michino M.; Free R. B.; Sibley D. R.; Javitch J. A.; Lane J. R.; Shi L. (2018) The E2.65A mutation disrupts dynamic binding poses of SB269652 at the dopamine D2 and D3 receptors. PLoS Comput. Biol. 14, e1005948. 10.1371/journal.pcbi.1005948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Vangveravong S.; Li S.; Fan J.; Jones L. A.; Cui J.; Wang R.; Tu Z.; Chu W.; Perlmutter J. S. (2013) Positron emission tomography imaging of dopamine D2 receptors using a highly selective radiolabeled D2 receptor partial agonist. NeuroImage 71, 168–174. 10.1016/j.neuroimage.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K.; Marcellino D.; Nilsson J.; Ogren S. O.; Fuxe K.; Arhem P. (2014) Typical and atypical antipsychotics do not differ markedly in their reversibility of antagonism of the dopamine D2 receptor. Int. J. Neuropsychopharmacol. 17, 149–155. 10.1017/S1461145713000801. [DOI] [PubMed] [Google Scholar]
- Sahlholm K.; Zeberg H.; Nilsson J.; Ogren S. O.; Fuxe K.; Arhem P. (2016) The fast-off hypothesis revisited: A functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor. Eur. Neuropsychopharmacol. 26, 467–476. 10.1016/j.euroneuro.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Packeu A.; Wennerberg M.; Balendran A.; Vauquelin G. (2010) Estimation of the dissociation rate of unlabelled ligand-receptor complexes by a ’two-step’ competition binding approach. Br. J. Pharmacol. 161, 1311–1328. 10.1111/j.1476-5381.2010.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. (Amsterdam, Neth.) 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Vorobiov D.; Levin G.; Lotan I.; Dascal N. (1998) Agonist-independent inactivation and agonist-induced desensitization of the G protein-activated K+ channel (GIRK) in Xenopus oocytes. Pfluegers Arch. 436, 56–68. 10.1007/s004240050604. [DOI] [PubMed] [Google Scholar]
- Fan L.; Tan L.; Chen Z.; Qi J.; Nie F.; Luo Z.; Cheng J.; Wang S. (2020) Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 11, 1074. 10.1038/s41467-020-14884-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R. A.; Worrall S.; Chazot P. L.; Strange P. G. (1988) Purification of brain D2 dopamine receptor. EMBO J. 7, 4129–4133. 10.1002/j.1460-2075.1988.tb03307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura J.; Valle-Leon M.; Sahlholm K.; Watanabe M.; Van Craenenbroeck K.; Fernandez-Duenas V.; Ferre S.; Ciruela F. (2018) Behavioral control by striatal adenosine A2A -dopamine D2 receptor heteromers. Genes, Brain Behav. 17, e12432. 10.1111/gbb.12432. [DOI] [PubMed] [Google Scholar]
- Lane J. R.; Abramyan A. M.; Adhikari P.; Keen A. C.; Lee K. H.; Sanchez J.; Verma R. K.; Lim H. D.; Yano H.; Javitch J. A.; Shi L. (2020) Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. eLife 9, e52189. 10.7554/eLife.52189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; Shoichet B. K.; Dror R. O.; Roth B. L. (2017) Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389.e312. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Butler K. V.; Kelly B.; Rechsteiner K.; Karpiak J.; Betz R. M.; Kormos B. L.; Shoichet B. K.; Dror R. O.; Jin J.; Roth B. L. (2018) Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 14, 126–134. 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallikaarjun S.; Salazar D. E.; Bramer S. L. (2004) Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J. Clin. Pharmacol. 44, 179–187. 10.1177/0091270003261901. [DOI] [PubMed] [Google Scholar]
- Moerlein S. M.; Perlmutter J. S.; Markham J.; Welch M. J. (1997) In Vivo Kinetics of [18F](N-Methyl)Benperidol: A Novel PET Tracer for Assessment of Dopaminergic D2-Like Receptor Binding. J. Cereb. Blood Flow Metab. 17, 833–845. 10.1097/00004647-199708000-00002. [DOI] [PubMed] [Google Scholar]
- Sahlholm K.; Barchad-Avitzur O.; Marcellino D.; Gomez-Soler M.; Fuxe K.; Ciruela F.; Arhem P. (2011) Agonist-specific voltage sensitivity at the dopamine D2S receptor--molecular determinants and relevance to therapeutic ligands. Neuropharmacology 61, 937–949. 10.1016/j.neuropharm.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Leduc-Nadeau A.; Lahjouji K.; Bissonnette P.; Lapointe J. Y.; Bichet D. G. (2007) Elaboration of a novel technique for purification of plasma membranes from Xenopus laevis oocytes. Am. J. Physiol.: Cell. Physiol. 292, C1132–1136. 10.1152/ajpcell.00136.2006. [DOI] [PubMed] [Google Scholar]
- Sanchez-Soto M.; Verma R. K.; Willette B. K. A.; Gonye E. C.; Moore A. M.; Moritz A. E.; Boateng C. A.; Yano H.; Free R. B.; Shi L.; Sibley D. R. (2020) A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity. Sci. Signaling 13, eaaw5885. 10.1126/scisignal.aaw5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agren R.; Arhem P.; Nilsson J.; Sahlholm K. (2018) The Beta-Arrestin-Biased Dopamine D2 Receptor Ligand, UNC9994, Is a Partial Agonist at G-Protein-Mediated Potassium Channel Activation. Int. J. Neuropsychopharmacol. 21, 1102–1108. 10.1093/ijnp/pyy059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K. (2011) The role of RGS protein in agonist-dependent relaxation of GIRK currents in Xenopus oocytes. Biochem. Biophys. Res. Commun. 415, 509–514. 10.1016/j.bbrc.2011.10.106. [DOI] [PubMed] [Google Scholar]
- Kaczor A. A.; Rutkowska E.; Bartuzi D.; Targowska-Duda K. M.; Matosiuk D.; Selent J. (2016) Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 132, 359–399. 10.1016/bs.mcb.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Martí-Solano M.; Schmidt D.; Kolb P.; Selent J. (2016) Drugging specific conformational states of GPCRs: challenges and opportunities for computational chemistry. Drug Discovery Today 21, 625–631. 10.1016/j.drudis.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Torrens Fontanals M.; Stępniewski T.; Rodriguez-Espigares I.; Selent J. (2018) Application of Biomolecular Simulations to G Protein-Coupled Receptors (GPCRs) 205–223. 10.1002/9783527806836.ch8. [DOI] [Google Scholar]
- Lomize A. L.; Pogozheva I. D.; Lomize M. A.; Mosberg H. I. (2006) Positioning of proteins in membranes: a computational approach. Protein Sci. 15, 1318–1333. 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. (2008) CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Azuaje J.; López P.; Iglesias A.; de la Fuente R. A.; Pérez-Rubio J. M.; García D.; Stępniewski T. M.; García-Mera X.; Brea J. M.; Selent J.; Pérez D.; Castro M.; Loza M. I.; Sotelo E. (2017) Development of Fluorescent Probes that Target Serotonin 5-HT(2B) Receptors. Sci. Rep. 7, 10765. 10.1038/s41598-017-11370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S.; Totaro A.; Scipioni L.; Dufrusine B.; Stepniewski T. M.; Selent J.; Maccarrone M.; Dainese E. (2018) Role of palmitoylation of cysteine 415 in functional coupling CB1 receptor to Galphai2 protein. Biotechnol. Appl. Biochem. 65, 16–20. 10.1002/bab.1575. [DOI] [PubMed] [Google Scholar]
- Ramirez-Anguita J. M.; Rodriguez-Espigares I.; Guixa-Gonzalez R.; Bruno A.; Torrens-Fontanals M.; Varela-Rial A.; Selent J. (2018) Membrane cholesterol effect on the 5-HT2A receptor: Insights into the lipid-induced modulation of an antipsychotic drug target. Biotechnol. Appl. Biochem. 65, 29–37. 10.1002/bab.1608. [DOI] [PubMed] [Google Scholar]
- Harvey M. J.; Giupponi G.; Fabritiis G. D. (2009) ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 5, 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 52, 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmuller H.; MacKerell A. D. Jr. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D. Jr.; Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Espigares I.; Torrens-Fontanals M.; Tiemann J. K. S.; Aranda-Garcia D.; Ramirez-Anguita J. M.; Stepniewski T. M.; Worp N.; Varela-Rial A.; Morales-Pastor A.; Medel-Lacruz B.; Pandy-Szekeres G.; Mayol E.; Giorgino T.; Carlsson J.; Deupi X.; Filipek S.; Filizola M.; Gomez-Tamayo J. C.; Gonzalez A.; Gutierrez-de-Teran H.; Jimenez-Roses M.; Jespers W.; Kapla J.; Khelashvili G.; Kolb P.; Latek D.; Marti-Solano M.; Matricon P.; Matsoukas M. T.; Miszta P.; Olivella M.; Perez-Benito L.; Provasi D.; Rios S.; I R. T.; Sallander J.; Sztyler A.; Vasile S.; Weinstein H.; Zachariae U.; Hildebrand P. W.; De Fabritiis G.; Sanz F.; Gloriam D. E.; Cordomi A.; Guixa-Gonzalez R.; Selent J. (2020) GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 17, 777–787. 10.1038/s41592-020-0884-y. [DOI] [PubMed] [Google Scholar]
- Grest G. S.; Kremer K. (1986) Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A: At., Mol., Opt. Phys. 33, 3628–3631. 10.1103/PhysRevA.33.3628. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Venkatakrishnan A. J., Fonseca R., Ma A. K., Hollingsworth S. A., Chemparathy A., Hilger D., Kooistra A. J., Ahmari R., Babu M. M., Kobilka B. K., and Dror R. O. (2019) Uncovering patterns of atomic interactions in static and dynamic structures of proteins. bioRxiv, 840694.https://www.biorxiv.org/content/10.1101/840694v1 (accessed 2020-09-09). 10.1101/840694. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.