

Abstract
Methylation, which is one of the most prominent post-translational modifications on proteins, regulates many important cellular functions. Though several model-based methylation site predictors have been reported, all existing methods employ machine learning strategies, such as support vector machines and random forest, to predict sites of methylation based on a set of “hand-selected” features. As a consequence, the subsequent models may be biased toward one set of features. Moreover, due to the large number of features, model development can often be computationally expensive. In this paper, we propose an alternative approach based on deep learning to predict arginine methylation sites. Our model, which we termed DeepRMethylSite, is computationally less expensive than traditional feature-based methods while eliminating potential biases that can arise through features selection. Based on independent testing on our dataset, DeepRMethylSite achieved efficiency scores of 68%, 82% and 0.51 with respect to sensitivity (SN), specificity (SP) and Matthew’s correlation coefficient (MCC), respectively. Importantly, in side-by-side comparisons with other state-of-the-art methylation site predictors, our method performs on par or better in all scoring metrics tested.
1. Introduction
Methylation is a well-studied posttranslational modification (PTM) that occurs predominantly on arginine (Arg; R) and lysine (Lys; K) residues and, to a lesser extent, on histidine, asparagine, and cysteine residues1,2,3, 4. Though traditional methods used to identify methylation sites, such as tandem mass-spectrometry5, 6, methylation specific antibodies, and ChIP-Chip, have provided important insights into global methylation profiles, these methods are expensive, time-consuming and require a high level of technical expertise. As the number of known methylation sites has grown, computational methods have emerged as an efficient, cost-effective strategy to complement and extend traditional experimental methods of methylation site identification.
Various computational models have been built for prediction of methylation PTMs. There are two major observations through the PTM predictor. First, compared to the datasets used to train early methylation site predictors, the number of known methylation sites has increased dramatically. Secondly, the performance of predictor models has improved with the use of machine learning models, such as support vector machines (SVM)7-9 , Random Forest10 and group-based algorithms11. While it is believed that the prediction would do better by including structural features, there is a huge gap between the availability of structural information and the availability of sequence data. This knowledge gap can have a substantial impact model development and performance. As a consequence, some models, such as MeMo7, use sequential features, while others, such as the model developed by Chou et al8, use structural features. Meanwhile, still others, like GPS-MSP11, use only primary amino acid sequences. Importantly, in all cases, feature selection was based on a series of hand-selected characteristics, such as pseudo amino acid composition (PseAAC), Shannon Entropy (SE) and others, that could introduce bias into model development.
Therefore, in order to reduce bias while simultaneously decreasing the complexity and time required for model development11, we generated a deep learning-based approach that is able to replace hand-selected features and still contribute improvements in predictor performance. Though there have been a few deep learning models used in DNA methylation site prediction,12, 13 all existing protein methylation site methods are based on feature selection14, 15.
To the best of our knowledge, this work is the first to apply deep learning to predict methylation sites in proteins. Moreover, in our work, we provide (1) an improved dataset for arginine methylation PTMs; (2) an ensemble deep learning model, based on Keras16 2D Convolutional layer network, and Long Short Term Memory (LSTM) models for prediction of PTM sites; (3) parameter selection based on 10-fold cross-validation results to test the performance of the model; (4) independent test results validating the performance of our model with the state-of-the-art models. Overall, our model, which we termed DeepRMethylSite, exhibits improved performance compared to previously published Arg methylation site predictors14.
2. Material and methods
2.1. Dataset Preparation
To build our training dataset, we used the Arg methylation dataset provided by DBPTMv317, PhosphositePlusv6.5.818 and Uniprot19 databases. We queried the Uniprot database to get experimentally verified methylation sites from publications published after 2017. Overall, our dataset contained 12,976 Arg methylation sites from 5,725 unique proteins.
To construct the positive dataset, we generated a window size of 51 with the methylated Arg site in the center, flanked by 25 amino acids upstream and downstream of the methylation site. Meanwhile, the negative dataset was similarly generated around Arg sites not known to be methylated.
Next, we removed any duplicate sequences within the positive and negative datasets. Also, if we found a duplicate sequence among the positive and negative datasets, we removed the duplicate sequence from the negative sequence. We termed the new positive and negative dataset the “clean” datasets. Since Arg methylation sequences are often conserved across species and we are building a general, non-specific model, we identified many duplicate sequences that were removed during this procedure. After removing duplicates, 10,429 Arg methylation sites remained in the clean positive set and 305,700 unmethylated Arg sites remained in the clean negative dataset. Finally, we used 80% of the clean dataset for training and validation sets and set aside the remaining 20% of the clean dataset as the independent test set (Table 1). Statistical analysis using the two logo chart was carried out to confirm the methylation dataset followed the trend of experimental arginine methylation sites (Figure S1 in Supplementary Information).
Table 1.
Number of positive and negative sites in the training and test sets before (left) and after (right) balancing.
| Dataset | Positive sites (before/after) |
Negative sites (before/after) |
|---|---|---|
| Total | 10,429/10,429 | 305,700/10,429 |
| Training | 8,344/8,344 | 244,600/8,344 |
| Train: 6,676 | 6,676 | |
| Val: 1,668 | 1,668 | |
| Test | 2,085/2,085 | 61,150/2,085 |
Perhaps not surprisingly, we noticed that there is large imbalance between the positive and negative datasets. This may be due to the fact that only positive sites are reported while the negative set is composed of those Arg residues that have not been found to be methylated. Thus, in order to balance the positive and negative datasets, we used undersampling from the imblearn package20. Undersampling is a technique in which the set having the larger number of samples is pruned to create a balance with the other set. There are many ways to deal with the unbalanced nature of the dataset. The unbalanced nature of PTM datasets is prone to artefacts stemming from limited knowledge about the number of negatives compared to the number of experimentally verified positive samples. Broadly, there are two ways of balancing the dataset, either by manipulating the dataset or by using a cost function that takes into account the imbalanced nature of the dataset21. In the way of manipulating the dataset, the positive samples can either be synthetically increased to match the size of the negative dataset, known as oversampling, or the negative dataset can be reduced to match the size of the positive dataset, known as undersampling. In our case, we are using Scikit learn package for undersampling. The resulting dataset is summarized in Table 1. Compared to Arg methylation dataset used in PRmePRed, we have increased the dataset 8-fold.
2.2. Input encoding
In most machine learning algorithms, features are extracted from the sequence data and, thus, meaningful numerical representations of the sequences are fed to the model. In contrast, in deep learning, the sequences themselves are numerically represented as encodings, as follows:
One hot encoding22, where each amino acid is defined as a 20 length vector, with only one of the 20 bits as 1, thus uniquely representing the twenty amino acids. It has also been used as a feature, twenty bit feature by Wei et al10.
Embedding Integer encoding, where each amino acid is allocated random integers of d dimensions long, where d is a parameter16. We used this encoding as an input to the embedding layer. The embedding layer helps in transforming the data into specified dimension, d. Since the encoding changes with each epoch, this encoding possesses a dynamic nature to its representation compared to one hot encoding23, where the encoding is fixed. Thus, the encoding embeds the representation learned through the deep architecture/algorithms.
Deep learning thus bypasses the need for feature extraction. For comparison, we extracted methylation relevant features14, 24 from the same dataset, and fed them to the tree-based classifier XGBoost followed by several machine learning algorithms. Details are provided in supplementary material.
2.3. DeepRMethylSite: Ensemble Model
An Ensemble model aggregates two or more model predictions to improve the prediction power of a classifier. Here, we created an ensemble between an LSTM model and CNN model. An LSTM model learns through cell states, while the CNN model employs different filters to extract various features. The ensemble aggregates the predictions learned by the CNN model and the LSTM model, based on the trust attained by each classifier.
Each of the member predictions was weighted with a weight factor and the predictions were aggregated to get the ensemble results. The weights were found for each classifier using a grid search between (0,1) in steps of 0.1. To obtain proper weights to prevent overfitting during model development, a validation set was created to compare the training and validation accuracy of the models25. This was accomplished by taking the remaining 8,344 positive and negative sites after the independent test set was removed and further dividing them into the training set (composed of 6,676 distinct positive and negative sites) and the validation set (composed of 1,688 distinct positive and negative sites) (Table 1). The weights were then normalized using L1 normalization and tensor dot was used to efficiently implement the weighted vector of predictions. Thus, the predictions are tensor multiplication of weight with the predictions (Eq. 1)
| (1) |
where Wl is weight given to LSTM weights and Wc is weight given to CNN weights and ŷl and ŷc are the respective predictions.
2.3.1-. CNN Model
The model is based on Keras 16 Convolutional Neural Network (CNN). The model consists of 7 layers, including the feature processing layer and the output layer. The first layer is the embedding layer, which learns the feature representation to the input sequences. A lambda layer is then used as a transition layer to the Convolutional 2D layer, where an extra dimension is added to match the input shape for the convolutional layer. Next, two Convolutional 2D layers with ReLu activation are employed. Padding is disabled for the first Convolutional layer and enabled for the next layer. Initial filter size for the Convolutional layer was selected as ((n-1)/2,3), where n is the window size. The filter size was selected such that the center residue of window is included in every stride as the center residue target for our prediction. The dimensions of output from the first convolutional layer changes when padding is disabled and remains the same when padding is enabled for the second layer. For example, if the output from the first convolutional layer has dimensions 17×19, it remains the same for consecutive layers. Each Convolutional 2D layers was then followed by a dropout layer of 0.6 to avoid overfitting. A higher dropout rate was used in order to reduce the overfitting and to achieve a more generalized model. Dropout mitigates the overparameterization of the deep learning model by dropping out a few neurons from computation. Next, a max pooling layer calculates the maximum value for each patch of the feature map and provides a down-sampled representation of the input. Two hidden layers of size 768 and 256 were employed with each, followed by dropout of 0.5. Finally, a softmax layer with two neurons, representing the true and false prediction, acted as an output layer. The architecture is summarized in Figure 2B.
Figure 2:

A. parameters used in LSTM model. B. Parameters used in CNN model. C. Ensemble model generated by combining through grid search weights on CNN and LSTM models.
Once developed, the model was optimized using Adam26. Adam is an adaptive moment estimation-based algorithm specifically used in training deep networks. The ModelCheckpoint function in Keras was used to save the best model with respect to validation accuracy. The plot showing change in accuracy across epochs using a dropout rate of 0.6 has been provided in Figure S2A. In practice, when the training accuracy just improves slightly in comparison to validation accuracy, the model should stop learning, and thus should avoid overfitting due to a greater number of epochs. The parameters used in the model are given in Table 2.
Table 2:
Parameters for CNN.
| Parameters | Settings |
|---|---|
| Embedding Output Dimension | 21 |
| Learning Rate | 0.001 |
| Batch Size | 256 |
| Epochs | 80 |
| Conv2d_1 number of filters | 64 |
| Dropout | 0.6 |
| Conv2d_1 number of filters | 128 |
| Dropout | 0.6 |
| MaxPooling2d | 2 x 2 |
| Dense 1 | 768 |
| Dropout | 0.5 |
| Dense_2 | 256 |
| Dropout | 0.5 |
| Checkpointer | Best validation accuracy |
2.3.2-. LSTM Model
Long Short Term Memory27 models have overcome the vanishing and exploding gradient problems in RNN and are known to capture long term dependencies. LSTM consists of three gates: input, forget, and output gates, which together define the flow of data governed by the state of the cell. LSTM helps to memorize the states of the cell and has the ability to save each of the sequences through layers, with return sequences option. Further, as the use of hidden states of a cell is increased, the power of learning through LSTM is known. Here we used a stacked LSTM model in comparison with the CNN approach to model classification for Arg methylation. The model consists of four layers: 1) the input layer, which consists of an embedding layer that learns the best representation of the integer encoded sequences through subsequent epochs. The embedding layer transforms the sequence information at a dimension. Thus, the output of the layer has shape (window size, embedding dimension) in compatibility with input dimensions of the LSTM layer; 2) an LSTM layer, which consists of 128 neurons with return sequences kept as true; 3) a dropout LSTM layer with 64 neurons, with dropout and recurrent dropout sets at 0.5 each with hyperbolic tangent activation where recurrent dropouts results in dropping the horizontal connections within the cell28 and 4) an output layer, with 2 neurons and soft-max activation, where the two neurons summarize the classification as true or false (Figure 2A). The model was compiled with the Adadelta optimizer29 and binary cross-entropy as loss function. Similar to the CNN model, the ModelCheckpoint function in Keras was used to obtain the best model with respect to validation accuracy. The plot showing change in accuracy across epochs has been provided in Figure S2B.
2.4. Performance and Evaluation
To evaluate the performance of each model, we used a confusion matrix to determine Sensitivity (SN), Specificity (SP), Accuracy (ACC) and Receiver Operating Characteristic (ROC) curve as the performance metrics. We used 10-fold cross-validation on the benchmark training dataset and an independent test set to evaluate the models.
ACC defines the correctly predicted residues out of the total residues (Eq. 2). SN defines the model’s ability to distinguish positive residues (Eq. 3) whereas the SP measures the model’s ability to correctly identify the negative residues (Eq. 4). Matthews Correlation Coefficient (MCC) is the calculated score that takes into account the model’s predictive capability with respect to both positive and negative residues (Eq. 5). Likewise, the ROC curve provides a graphical representation of the diagnostic ability of the classifier. The area under the ROC curve (AUC) is used to compare various models, with the models having the highest AUC scores performing better in classification than those with lower AUC scores.
| (2) |
| (3) |
| (4) |
| (5) |
3. Results and Discussion.
3.1. Selection of window size
An initial 10-fold cross-validation was carried on each of our models (i.e., LSTM and CNN) to determine the parameters. A window size of 51 was extracted from the dataset and other window sizes were generated by flanging the windows from both ends. This kept the dataset size constant and hence the comparison fair when determining the window size.
Furthermore, the window size determines the number of residues exposed to the problem. Methylation sites have been found buried in the protein core while others have been found on the protein surface. The potential site is centered in a window with an equal number of residues on both sides. The results are tabulated in Table 4. While the model follows the same trend in different window sizes, different window sizes were optimized for different models, owing to differences in their architectures. For instance, a window size of 39 was optimized for CNN, while a window size of 21 was optimized for LSTM. Following the strategy outlined in our Succinylation site prediction work30 , the embedding dimension was fixed at 21.
Table 4:
10-fold cross-validation performance metrics for different window sizes with an embedding dimension of 21. The highest values in each category are highlighted in boldface. MCC: Matthew’s Correlation Coefficient; SN: sensitivity, SP: Specificity; ACC: Accuracy.
| Size | CNN | LSTM | ||||||
|---|---|---|---|---|---|---|---|---|
| MCC | SN | SP | AC C |
MCC | SN | SP | AC C |
|
| 51 | 0.52 | 0.71 | 0.81 | 0.76 | 0.44 | 0.66 | 0.77 | 0.72 |
| 45 | 0.52 | 0.72 | 0.80 | 0.76 | 0.44 | 0.66 | 0.77 | 0.72 |
| 39 | 0.53 | 0.73 | 0.80 | 0.76 | 0.45 | 0.66 | 0.78 | 0.72 |
| 33 | 0.52 | 0.71 | 0.81 | 0.76 | 0.46 | 0.66 | 0.79 | 0.73 |
| 27 | 0.52 | 0.70 | 0.81 | 0.76 | 0.46 | 0.67 | 0.78 | 0.73 |
| 21 | 0.50 | 0.71 | 0.79 | 0.75 | 0.46 | 0.65 | 0.80 | 0.73 |
| 15 | 0.49 | 0.70 | 0.79 | 0.74 | 0.44 | 0.66 | 0.77 | 0.72 |
| 9 | 0.46 | 0.69 | 0.77 | 0.73 | 0.42 | 0.65 | 0.76 | 0.71 |
3.2. Selection of embedding dimension
The embedding dimension can be summarized as the feature space that is able to best define the representation of the input sequences. Therefore, we used various dimensions during 10-fold cross validation of the models at their optimized window sizes, as summarized in Table 5. The embedding dimension of 33 was optimized for CNN, while LSTM was optimized at window size of 39. Nonetheless, despite the fact that we optimized the embedding dimension, there did not seem to be a substantial improvement among the dimensions for either model.
Table 5:
10-fold cross-validation results for different embedding dimensions for their optimized window size. MCC: Matthew’s Correlation Coefficient; SN: sensitivity; SP: Specificity; ACC: Accuracy.
| Dim | CNN | LSTM | ||||||
|---|---|---|---|---|---|---|---|---|
| MCC | SN | SP | ACC | MCC | SN | SP | ACC | |
| 9 | 0.52 | 0.70 | 0.82 | 0.76 | 0.43 | 0.67 | 0.76 | 0.71 |
| 15 | 0.52 | 0.70 | 0.81 | 0.76 | 0.45 | 0.68 | 0.76 | 0.72 |
| 21 | 0.52 | 0.73 | 0.79 | 0.76 | 0.46 | 0.66 | 0.79 | 0.73 |
| 27 | 0.53 | 0.71 | 0.80 | 0.76 | 0.46 | 0.67 | 0.77 | 0.73 |
| 33 | 0.53 | 0.72 | 0.80 | 0.76 | 0.46 | 0.66 | 0.79 | 0.73 |
| 39 | 0.52 | 0.70 | 0.81 | 0.76 | 0.47 | 0.65 | 0.80 | 0.73 |
| 45 | 0.52 | 0.70 | 0.81 | 0.76 | 0.46 | 0.68 | 0.78 | 0.73 |
3.3. Comparison with one hot encoding
Since embedding has be shown to increase the dynamic nature of the sequences, embedding tends to enhance model performance over one hot encoding. Thus, we conducted 10-fold cross-validation with the optimum parameters for the One hot encoding to confirm whether it is still true in our case. Owing to the dynamic nature of embedding, the training time was less for embedding. This approach also saved computational time, as training time is less for embedding compared to One hot encoding. For these reasons, we compared One hot encoding at the optimized parameters for the embedding model (Table 6).
Table 6:
Comparison of one hot encoding model to embedding models based on 10-fold cross-validation MCC: Matthew’s Correlation Coefficient; SN: sensitivity; SP: Specificity; OHE: One hot encoding; Emb: Embedding; ACC: Accuracy.
| Model | CNN | LSTM | ||||||
|---|---|---|---|---|---|---|---|---|
| MCC | SN | SP | ACC | MCC | SN | SP | ACC | |
| OHE | 0.47 | 0.69 | 0.78 | 0.73 | 0.45 | 0.69 | 0.76 | 0.73 |
| Emb | 0.53 | 0.72 | 0.80 | 0.76 | 0.47 | 0.65 | 0.80 | 0.73 |
3.4. Evaluating Ensemble Model
We used independent testing to evaluate the ensemble model. Both LSTM and CNN were trained on the training set and evaluated on the test set, as defined in Table 1. The ensemble uses a grid search method to optimize the weights for each model. Thus, the optimized weights are [0.16,0.83] for LSTM and CNN models, respectively. Figure 3 shows the receiver operator curve (ROC) for the CNN, LSTM and ensemble models. The final ensemble model, which we termed DeepRMethylSite, performed well with respect to SP, SN and MCC (Table 7). We also evaluated the ensemble model against CNN and LSTM models using Student’s t-test (Table S3).
Figure 3:

ROC Curve for LSTM, CNN and Ensemble model.
Table 7:
Independent Test Results using the CNN, LSTM and Ensemble models. MCC: Matthew’s Correlation Coefficient; SN: sensitivity; SP: Specificity’ ACC: Accuracy; AUC: Area under the receiver operator curve.
| Model | MCC | SN | SP | ACC | AUC |
|---|---|---|---|---|---|
| LSTM | 0.46 | 0.80 | 0.65 | 0.73 | 0.796 |
| CNN | 0.50 | 0.68 | 0.81 | 0.75 | 0.816 |
| DeepRMethylSite | 0.51 | 0.68 | 0.82 | 0.75 | 0.821 |
3.5. Comparison with existing models.
Next, we wanted to compare the performance of DeepRMethylSite to existing Arg methylation site predictors. The SVM-based predictor, PrmePred14, is currently the best performing method in the field based on MCC, which is often used as an indicator of overall method performance. Therefore, to evaluate our model, we used the training set and validation test set used by Kumar et al during the development of PRmePred14 to retrain our model and then used their independent test set to evaluate model performance. The independent test results of DeepRMethylSite, along with those from other predictors using the PRmePred independent set, is tabulated in Table 8. Likewise, the performance of either the CNN model or the LSTM model alone using the independent test set from PRmePred is provided in Table S1.
Table 8:
Comparison of DeepRMethylSite with other prediction methods.
| Method | Algo | MCC | SN | SP | ACC |
|---|---|---|---|---|---|
| MeMo 7 | SVM | 0.46 | 0.38 | 0.99 | 0.68 |
| MASA32 | SVM | 0.41 | 0.31 | 0.99 | 0.65 |
| BPB-PPMS33 | SVM | 0.25 | 0.12 | 1.00 | 0.56 |
| PMeS 9 | SVM | 0.16 | 0.43 | 0.73 | 0.58 |
| iMethyl-PseAAC 8 | SVM | 0.30 | 0.18 | 1.00 | 0.59 |
| PSSMe 31 | SVM | 0.44 | 0.60 | 0.83 | 0.72 |
| MePred-RF10 | RF | 0.46 | 0.41 | 0.97 | 0.69 |
| PRmePred 14 | SVM | 0.74 | 0.87 | 0.87 | 0.87 |
| DeepRMethylSite | CNN | 0.79 | 0.71 | 0.99 | 0.92 |
Because we were using the same datasets used by PRmePred, direct comparisons between the models can be made. However, results are confined to the window size of 19. Nonetheless, the results were informative. For instance, in side-by-side comparisons with PRmePred, DeepRMethylSite achieved SP, ACC, and MCC scores that were ~13.8%, ~5.8% and ~6.8% higher, respectively, than those exhibited by PRmePred14 (Table 8). On the other hand, DeepRMethylSite exhibited SN scores that were ~19.4% lower than those observed for PRmePred. Likewise, DeepRMethylSite achieved the highest MCC and ACC scores across all existing methods (Table 8). Likewise, DeepRMethylSite exhibited SP and SN scores that were on par with or better than those of the existing models7-10, 14, 31-33 (Table 8). Taken together, these data suggest that DeepRMethylSite is a robust predictor of Arg methylation sites in proteins.
5. Conclusion
Here, we describe the development and analysis of an Arg methylation site prediction tool, DeepRMethylSite, based on a deep learning strategy. An ensemble model was used to combine the better sensitivity of our LSTM-based model with the specificity of our CNN-based model. Interestingly, while the ensemble model exhibited significant improvements in MCC and SN compared to the LSTM model and generally outperformed the CNN model with respect to MCC and SP, it did not achieve significant performance improvements compared to CNN (Table S3).
Unlike other machine learning algorithms, deep learning does not require feature extraction. Not only does this reduce the potential for intrinsic bias in feature selection, but it also substantially reduces the computational cost required for model development. Importantly, in side-by-side comparisons, our model outperforms PRmePred—the current gold standard in Arg methylation site prediction—with respect to SP, ACC and MCC using their independent test set. Therefore, the use of a deep learning-based model has not only avoided the need for feature extraction, but it has also improved the prediction performance for arginine site prediction. These predictions, which will complement the list of experimentally identified Arg methylation sites, will be useful for understanding how Arg methylation affects cellular processes such as transcriptional regulation, RNA metabolism, apoptosis and DNA repair34. Currently, our model does not distinguish between mono-methylated, symmetrically-dimethylated and asymmetrically-dimethylated residues, which can have important implications for the cellular consequences of a given Arg methylated site35. In the future, it will be interesting to explore whether our model can be enhanced to distinguish between these methylation states, as well. Also, it is important to note that, in this study, we increased the size of the dataset used for training and evaluation by ~8-fold compared to the dataset used during the development of PRmePred. We hope that these datasets, which we have made freely available to the community at https://github.com/dukkakc/DeepRMethylSite, will facilitate the development of improved methylation site prediction methods. Likewise, to facilitate the use of our predictor by the cell signaling and bioinformatics communities, the method and all code used for its development are freely available at https://github.com/dukkakc/DeepRMethylSite.
Supplementary Material
Figure 1:
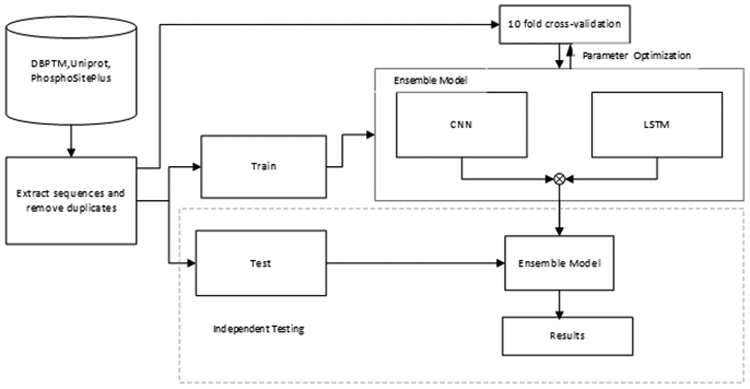
Flow Diagram depicting steps carried to create our model. Sequences were extracted from public databases and the dataset was divided into training, validation and test sets. 10-fold cross-validation was used in optimizing the parameters on each model, and then independent testing was used to evaluate the models.CNN: Convolutional Neural Network; LSTM: Long-term short-term memory.
Table 3:
Parameters used in LSTM Model
| Parameters | Settings |
|---|---|
| Embedding Output Dimension | 39 |
| Learning Rate | 0.01 |
| Batch Size | 256 |
| Epochs | 100 |
| LSTM_layer1_neurons | 128 |
| LSTM_layer2 | 64 |
| Dropout | 0.5 |
| Recurrent Dropout | 0.5 |
| Dense_layer_neurons | 32 |
Acknowledgements.
This work was supported by National Science Foundation (NSF) grant nos. 1901793, 2003019 and 2021734 (to DK). RHN is supported by an HBCU-UP Excellence in Research Award from NSF (1901793) and an SC1 Award from the National Institutes of Health National Institute of General Medical Science (5SC1GM130545). HS was supported by JSPS KAKENHI Grant Numbers JP18H0176 and JP19H04176.
Footnotes
Conflicts of interest.
There are no conflict to declare.
References.
- 1.Ambler RP and Rees MW, Epsilon-N-Methyl-lysine in bacterial flagellar protein, Nature, 184, 56–57 (1959). [DOI] [PubMed] [Google Scholar]
- 2.Martin C and Zhang Y, The diverse functions of histone lysine methylation, Nat Rev Mol Cell Biol, 6, 838–849 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Bedford MT and Richard S, Arginine methylation an emerging regulator of protein function, Mol Cell, 18, 263–272 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Bannister AJ and Kouzarides T, Regulation of chromatin by histone modifications, Cell Res, 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ong SE, Mittler G and Mann M, Identifying and quantifying in vivo methylation sites by heavy methyl SILAC, Nat Methods, 1, 119–126 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Wu CC, MacCoss MJ, Howell KE and Yates JR 3rd, A method for the comprehensive proteomic analysis of membrane proteins, Nat Biotechnol, 21, 532–538 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Xue Y, Huang N, Yao X and Sun Z, MeMo: a web tool for prediction of protein methylation modifications, Nucleic Acids Res, 34, W249–253 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu WR, Xiao X, Lin WZ and Chou KC, iMethyl-PseAAC: identification of protein methylation sites via a pseudo amino acid composition approach, Biomed Res Int, 2014, 947416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi SP, Qiu JD, Sun XY, Suo SB, Huang SY and Liang RP, PMeS: prediction of methylation sites based on enhanced feature encoding scheme, PLoS One, 7, e38772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei L, Xing P, Shi G, Ji Z and Zou Q, Fast Prediction of Protein Methylation Sites Using a Sequence-Based Feature Selection Technique, IEEE/ACM Transactions on Computational Biology and Bioinformatics, 16, 1264–1273 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Deng W, Wang Y, Ma L, Zhang Y, Ullah S and Xue Y, Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins, Briefings in Bioinformatics, 18, 647–658 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Tian Q, Zou J, Tang J, Fang Y, Yu Z and Fan S, MRCNN: a deep learning model for regression of genome-wide DNA methylation, BMC Genomics, 20, 192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy JJ, Titus AJ, Petersen CL, Chen Y, Salas LA and Christensen BC, MethylNet: an automated and modular deep learning approach for DNA methylation analysis, BMC Bioinformatics, 21, 108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Joy J, Pandey A and Gupta D, PRmePRed: A protein arginine methylation prediction tool, PLoS One, 12, e0183318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pejaver V, Hsu W-L, Xin F, Dunker AK, Uversky VN and Radivojac P, The structural and functional signatures of proteins that undergo multiple events of post-translational modification, Protein Sci, 23, 1077–1093 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keras CF, Keras: The Python Deep Learning library, https://github.com/fchollet/keras,
- 17.Huang KY, Su MG, Kao HJ, Hsieh YC, Jhong JH, Cheng KH, Huang HD and Lee TY, dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins, Nucleic Acids Res, 44, D435–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V and Skrzypek E, PhosphoSitePlus, 2014: mutations, PTMs and recalibrations, Nucleic Acids Res, 43, D512–520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.UniProt C, UniProt: a worldwide hub of protein knowledge, Nucleic Acids Res, 47, D506–D515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemaitre G, Nogueira F and Aridas C, K, Imbalanced-learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning, Journal of Machine Learning Research, 18, 1 – 5 (2017). [Google Scholar]
- 21.Johnson JM and Khoshgoftaar TM, Survey on deep learning with class imbalance, Journal of Big Data, 6, 27 (2019). [Google Scholar]
- 22.Wang D, Zeng S, Xu C, Qiu W, Liang Y, Joshi T and Xu D, MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction, Bioinformatics, 33, 3909–3916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulmanov M, Khan MA, Hoehndorf R and Wren J, DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier, Bioinformatics, 34, 660–668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ismail H, Smith M and Kc D, FEPS: Feature Extraction from Protein Sequences webserver,(2016).
- 25.Krogh A and Vedelsby J, Neural Network Ensembles, Cross Validation and Active Learning, Nips’ 94, 231–238 (1994). [Google Scholar]
- 26.Kingma D and Ba J, Adam: A method for stochastic optimization, presented in part at the ICLR, (2015). [Google Scholar]
- 27.Hochreiter S and Schmidhuber J, Long short-term memory, Neural Comput, 9, 1735–1780 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Gal Y and Ghahramani Z, A theoretically grounded application of dropout in recurrent neural networks, presented in part at the Proceedings of the 30th International Conference on Neural Information Processing Systems, Barcelona, Spain, (2016). [Google Scholar]
- 29.Zeiler MD, ADADELTA: An adaptive learning rate method, arXiv:1212.5701,(2012). [Google Scholar]
- 30.Thapa N, Chaudhari M, McManus S, Roy K, Newman RH, Saigo H and Kc DB, DeepSuccinylSite: a deep learning based approach for protein succinylation site prediction, BMC Bioinformatics, 21, 63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen PP, Shi SP, Xu HD, Wang LN and Qiu JD, Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization, Bioinformatics, 32, 3107–3115 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Shien D-M, Lee T-Y, Chang W-C, Hsu JB-K, Horng J-T, Hsu P-C, Wang T-Y and Huang H-D, Incorporating structural characteristics for identification of protein methylation sites, Journal of Computational Chemistry, 30, 1532–1543 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Shao J, Xu D, Tsai S-N, Wang Y and Ngai S-M, Computational Identification of Protein Methylation Sites through Bi-Profile Bayes Feature Extraction, PLOS ONE, 4, e4920 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rakow S, Pullamsetti SS, Bauer U-M and Bouchard C, Assaying epigenome functions of PRMTs and their substrates, Methods,(In Press). [DOI] [PubMed]
- 35.Bedford MT, Arginine methylation at a glance, Journal of Cell Science, 120, 4243 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.