

Abstract
Introduction:
Neuroblastoma (NB) is the prime cancer of infancy, and accounts for 9% of pediatric cancer deaths. While children diagnosed with clinically stable NB experience a complete cure, those with high-risk disease (HR-NB) do not recover, despite intensive therapeutic strategies. Development of novel and effective targeted therapies is needed to counter disease progression, and to benefit long-term survival of children with HR-NB.
Areas covered:
Recent studies (2017–2020) pertinent to NB evolution are selectively reviewed to recognize novel and effective therapeutic targets. The prospective and promising therapeutic targets/strategies for HR-NB are categorized into (a) targeting oncogene-like and/or reinforcing tumor suppressor (TS)-like lncRNAs; (b) targeting oncogene-like microRNAs (miRs) and/or mimicking TS-miRs; (c) targets for immunotherapy; (d) targeting epithelial-to-mesenchymal transition and cancer stem cells; (e) novel and beneficial combination approaches; and (f) repurposing drugs and other strategies in development.
Expert opinion:
It is highly unlikely that agents targeting a single candidate or signaling will be beneficial for an HR-NB cure. We must develop efficient drug deliverables for functional targets, which could be integrated and advance clinical therapy. Fittingly, the looming evidence indicated an aggressive evolution of promising novel and integrative targets, development of efficient drugs, and improvised strategies for HR-NB treatment.
Keywords: Emerging therapeutics, high-risk Neuroblastoma, long non-coding RNAs, micro RNAs, immunotherapy, novel combinations, novel drugs
1. Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in infants, with the highest rate of diagnosis in the first month of life [1]. Each year, 650 new cases are diagnosed in the United States; of those, ~37% are diagnosed as infants and ~90% are younger than 5 years [2]. NB originates from actively dividing neural crest cells (NCC) that accumulate several mutations and molecular rearrangements [3], presented as tumor mass along the midline of the body [3,4]. The partial list of genetic bases for the disease includes germline mutations (e.g. ALK, Phox2B), gene aberrations (e.g. HRAS) associated with other syndromes (e.g. neurofibromatosis, Li-Fraumeni), and familial NB [5]. NB is a highly heterogeneous tumor with an array of clinical manifestations from spontaneous regression, therapy-responsive disease, clinically stable disease, and progressive disease (PD) with therapy resistance, tumor recurrence, and eventual death. Based on clinical factors (age, stage, cellular differentiation/maturation) and biological markers at the time of diagnosis, children with NB are stratified into low-risk, intermediate-risk, and high-risk (HR-NB) subsets. While low-/intermediate-risk groups display favorable prognosis with >90% overall survival (OS), the HR-NB group show poor prognosis, with <50% five-year OS and dismal long-term survival [5]. HR-NB in general is characterized with segmental chromosomal aberrations, MYCN amplification, expression of neurotrophin receptor kinases, low exonic mutations, and telomere lengthening, and is correlated with advanced stage of disease, higher risk of relapse, and poor clinical outcome [6–10]. Treatment for HR-NB is generally divided into the induction phase with dose-intensive cycles of chemotherapy (CT – cisplatin/carboplatin, etoposide, vincristine, doxorubicin, topotecan, cyclophosphamide) and load reduction surgery; the consolidation phase with myeloablative CT (busulfan/melphalan or carboplatin/etoposide/melphalan and/or cyclophosphamide/thiotepa) along with radiotherapy (external beam RT, MIBG RT) and single or tandem stem cell (SC) transplant (autologous bone marrow transplantation [ABMT]; peripheral blood SC reinfusion); and the maintenance phase with RT, retinoic acid treatment (RA, 13-cis-retinoic acid), immunotherapy (dinutuximab), and immune-activating cytokine (GM-CSF, IL-2) treatment. In addition to the current clinical therapy for HR-NB, a number of strategies are currently under investigation, with >500 studies in clinical evaluation (www.clinicaltrials.gov). Despite such colossal efforts to treat HR-NB, the long-term survival for young children is depressing and warrants novel and improved therapeutic schemes. Appropriately, basic, preclinical, and clinical research are focused on this task worldwide. Herein, we aim to compile the novel targets, drug candidates, and/or therapeutic strategies in development that could shift a paradigm in the cure of HR-NB. The developmental therapeutics field for HR-NB is evolving fast, and the advancements have been periodically reviewed [11]. Here, we focus our discussion on the emerging therapeutic targets, drugs, and strategies defined within the last three years.
2. Targeting long noncoding RNAs (lncRNAs)
LncRNAs lack protein-coding functions and are known to regulate gene expression and cell fate [12]. LncRNAs play crucial roles in cellular functions (proliferation, differentiation, cell death, migration, invasion) and immune response, and have been implicated in the pathogenesis of various malignant cancers [13–16], including NB [17,18]. Studies defining the roles and mechanisms of lncRNAs in NB development, progression, dissemination, and induced therapy resistance could permit more informed stratification and new/improved therapeutic strategies (Figure 1). Polymorphisms in lncRNAs, including H19, LAMC2, 00673, and HOTAIR, have been linked to increased susceptibility to NB [19–22]. Consistently, studies underscored the criticality of lncRNAs in NB progression [23–27]. For instance, independent studies demonstrated that higher levels of CAI2, ncRAN, SNHG1, SNHG7, SNHG16, pancEts-1, and NHEG1 act like oncogenes (ON-lncRNAs) and are associated with poor differentiation, advanced disease stage, and unfavorable clinical outcomes, designating these candidates as prognostic biomarkers for HR-NB [23,24,28–32]. Conversely, a group of tumor suppressor (TS) lncRNAs have been associated with NB evolution. For instance, loss of TS-lncRNA CASC15, its encoded isoforms (CASC15-S, CASC15–003, CASC15–004), and TS-lncRNA NBAT1, located at the NB risk-associated 6p22.3 locus [33,34], correlated with poor OS/EFS [27,33,34]. Further, the critical ON-lncRNAs to TS-lncRNAs balance in NB progression was recognized in a high-throughput study, where high expression of ON-lncRNAs (HCN3, lnc01105) and low expression of TS-lncRNA (MEG3) were associated with poor prognosis [35].
Figure 1. Schematic representation of the functional role of lncRNAs in NB pathogenesis and disease evolution.
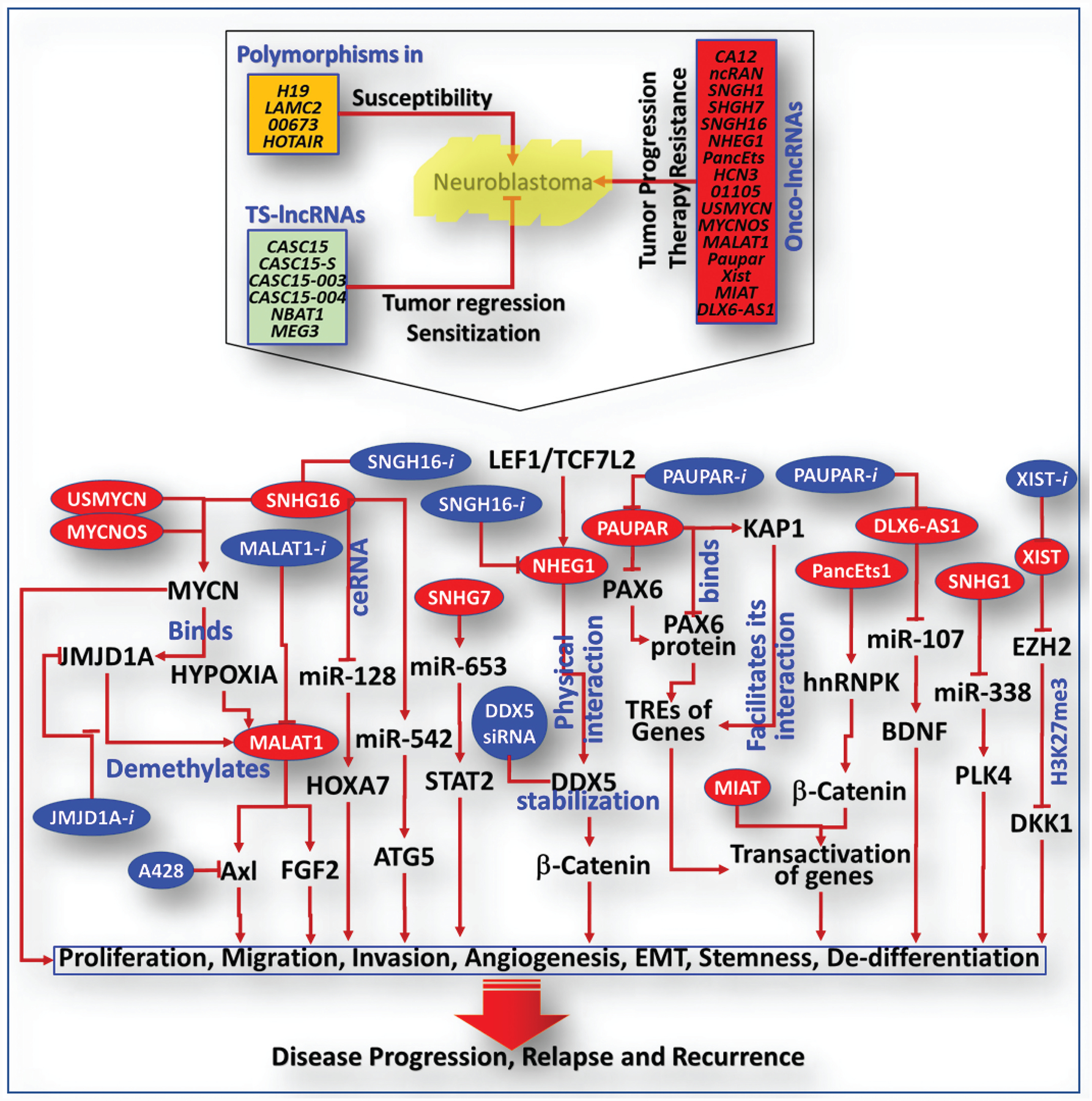
Bottom panel: Panel of oncogene-like lncRNAs and their signaling flow-through that affects malignancy characteristics (e. g., proliferation, de-differentiation, migration, invasion, angiogenesis EMT, CSC status) in NB cells, which dictates NB pathogenesis and disease evolution. Blue circles indicates the preclinically validated targeted therapeutic strategies for HR-NB.
Researchers have also documented the function and mechanism of ON-lncRNAs/TS-lncRNAs in NB evolution. For instance, USMYCN and MYCNOS have been shown to facilitate NB progression through increased MYCN expression [24–26]. MALAT1, the lncRNA involved in post-transcriptional gene regulation and mRNA splicing, activates Axl and influences NB cell invasion/migration [36]. Conversely, MYCN binds to the promoter and directly activates histone demethylase JMJD1A, which in turn demethylates histone H3K9 at the MALAT1 gene promoter and dictates NB cells’ metastatic behavior [37]. Further, the hypoxia-dependent activation of MALAT1 drives FGF2-dependent angiogenesis and contributes to NB evolution [38]. Targeting the JMJD1A/MALAT1 or MALAT1-induced Axl reverts the cellular metastatic state and prevents NB progression with metastasis [36,37]. Likewise, targeting SNHG7, which facilitates NB progression by interacting with miR-653–5p and regulating STAT2, inhibits NB cell proliferation, migration, invasion, and plasticity [28]. SNHG16 influences transcriptional/translational pathways of NB malignancy (proliferation, migration, invasion) processes [30] definitively associated with disease stage, MYCN status and poor prognosis [17,39]. Mechanistically, SNHG16 binds to mir-542–3p and activates ATG5, driving clonal expansion, migration, and invasion of NB cells [17]. Targeting SNHG16 not only suppressed clonal expansion/metastatic behavior of NB cells, but also impeded ATG5-dependent autophagy and prompted NB regression. SNHG16 also interacts with and negatively regulates miR-128–3p, which in turn activates the miR-128 direct target HOXA7, and results in activated proliferation, migration, and invasion. In parallel, SNHG16 acts as a competing endogenous RNA (ceRNA) that interacts with miR-128 and destroys its translational inhibition of HOXA7, prompting NB progression [39]. Zhao and colleagues reported that NHEG1 is a LEF1/TCF7L2-regulated lncRNA that stabilizes DDX5 through physical interaction [18]. Disrupting the LEF1/TCF7L2/NHEG1/DDX5/β-catenin positive feedback loop by targeting NHEG1 and/or DDX5 inhibits DDX5-mediated cell-cycle progression, and prompts NB regression [18].
Another potential target, Paupar, which is transcribed upstream of Pax6 that binds to the transcriptional regulatory elements across multiple chromosomes and controls the expression of distal target genes, is known to regulate NB cell proliferation/differentiation [40]. Paupar not only regulates Pax6 expression, but also directly interacts with KAP1, controls the expression of gene targets, and regulates proliferation and differentiation [41]. Silencing Paupar led to the reduction of KAP1 chromatin association and H3K9me3 at PAX6 co-bound locations, and regulated the expression of genes involved in the cell cycle, DNA replication, mitosis, and neurite outgrowth [40].
Recognizing the oncogenic functions of lncRNAs in NB offers a series of potential target(s) that could be exploited for NB therapeutics. ON-lncRNA XIST, a lncRNA that is highly expressed in NB and corresponds to poor prognosis, interacts with EZH2 and thereby increases the level of H3K27me3 in DKK1 promoter [42]. Leveling XIST increases DKK1 and inhibits cell growth, invasion, migration, and tumor progression [42]. PancEts-1, a novel ON-lncRNA, endorses growth and metastatic potential of NB cells [32]. PancEts-1 binds to hnRNPK and facilitates its interaction with β-catenin, increasing the stability/transactivation of β-catenin, transcriptional regulation of downstream targets, and orchestration of disease evolution [32]. Another study showed that silencing ON-lncRNA MIAT altered genes involved in NB malignancy (growth, survival, apoptosis, oxidative stress, migration) processes [43]. High expression of another ON-lncRNA, DLX6-AS1, which blocks miR-107-targeted inhibition of BDNF and endorses NB cell proliferation and metastasis, positively correlated with poor differentiation and advanced disease [44]. Targeting DLX6-AS1 reinforced miR-107 function (targeting BDNF) and inhibited NB growth [44]. Along this line, SNHG1, which is also an ON-lncRNA that is highly expressed in NB, activated malignancy characteristics of NB cells and tumor progression through direct regulation of miR-338–3p and activation of miR-338 target PLK4 [45].
Conversely, knockdown of TS-lncRNAs CASC15 and NBAT1 prevented NB cell differentiation through increased nucleolar localization of USP36, and activated SOX9 by the regulation of CHD7 ubiquitination [33]. Loss of NBAT-1 activated neuronal-specific transcription factor NRSF/REST, thereby affecting neuronal differentiation, and contributed to NB progression [27]. Consistently, forced silencing of NBAT-1, CASC15–003, and CASC15-S has induced NB-cell proliferation, migration, and invasion [27,33,34]. Furthermore, studies indicated that high HCN3 and 01105 and loss of MEG3 are collaborative and critical for NB disease progression [35]. Silencing 01105 and HCN3 and over-expressing MEG3 led to p53 signaling (HIF-1α, Noxa, Bid) pathway-dependent cell death. Likewise, TS-lncRNA FOXD3-AS1 is downregulated in NB, and serves as an independent good prognostic marker for NB [46]. Reinforcing FOXD3-AS1 effects differentiation and reverts NB progression. Functionally, FOXD3-AS1 interacts with PARP1, represses the activation of CTCF, and activates downstream tumor-suppressive genes [46]. Differential expression of lncRNAs (ON-lncRNA/TS-lncRNA) has been causally linked to NB pathogenesis. High-throughput approaches recognized that NB is characterized by the differential expression of lncRNAs, and further noted the criticality of ON-lncRNAs↔TS-lncRNAs expressional combinations in disease evolution. Further, the mode of action and the lncRNA-driven signaling mechanism involved in NB pathogenesis were unveiled. The recognition of their significance and function, coupled with the preclinical evidence on the efficacy of their targeting, clearly designate lncRNAs as novel and potential emerging therapeutic targets for improved treatment for HR-NB. Despite such preclinical recognition, clinical validation on the potential use of lncRNAs as therapeutic targets for NB is imminent.
3. Targeting miRNAs
MicroRNAs (miRs), a group of small (19–22 nucleotides) non-coding RNAs, regulate gene expression through DNA interaction, translational repression, or by mRNA degradation. In cancer, miRs are classified as oncomiRs, metastamiRs, and Ts-miRs, and are known to impact various cellular processes (e.g. cell cycle, apoptosis, migration, invasion, angiogenesis) [47–49]. The development of therapy resistance is another complex NB issue in which the functions of miRs have been realized. Differential expression of miRs in association with NB clinicobiological physiognomies and their functions in disease pathogenesis have been noted [50,51]. For instance, a unique embryonic stem cell miR signature in NB and its association with high-risk disease and poor prognosis were reported [52]. Recently, a study compiled the present understanding of the function of miRs in NB pathogenesis, therapy resistance, and evolution [49]. Here we limit the discussion to the recently identified miR candidates that could be potential treatment targets and could deliver therapeutic benefit for HR-NB.
Numerous studies indicated the promising therapeutic benefit of targeting oncomiRs. OncomiRs (miR-221, miR-181a/b, miR-3934, miR-223) are highly expressed in NB cells/tumors and positively correlated with clinicopathological (e.g. MYCN, metastasis) characteristics and poor prognosis [48,53–55]. Functionally, miR-221 targets NLK-dependent LEF1 phosphorylation-mediated elevation of MYCN, which consequently induces proliferation, rescues cell cycle arrest, and prompts NB evolution [48]. However, MiR-181 is known to affect autophagy, apoptosis, and GTPase activity signaling pathways, and thus drives growth and dissemination destiny of NB cells. Instrumentally, miR-181 targets TS ABI1, which endorses NB pathogenesis [53]. It has been shown that TP53INP1 is a direct target of miR-3934–5p, and targeting miR-3934 prompts TP53INP1-dependent apoptosis, indicating that the miR-3934–5p/TP53INP1 axis may serve as a potential therapeutic target for NB [54]. In contrast, targeting miR-223–3p reinforces its direct target FOXO1, resulting in increased apoptosis and decreased tumor growth and invasion [55]. Recently, 11 circulating miRs (miR-513a-5p, miR-198, miR-1280, miR-1304, miR-1308, miR-1908, miR-513b, miR-548f, miR-580, miR-1261, miR-1268) were designated as prognostic markers for PD-NB [56,57]. Along the line of targeting oncomiRs, a study with nano-hybrid technology indicated that inhibition of miR-17 could fully differentiate the cancer cells into neurons and stop their growth [58].
The functional influence of TS-miRS in NB pathogenesis/evolution, mechanism of action, and the therapeutic benefit of their reinforcement has been proposed. Studies indicated critically low levels of TS-miRs (miR-324, Let7 family, miR-193b, miR-1247, miR-149, miR-323a, miR-145) in NB, and the loss of TS-miRs was significantly associated with poor clinical outcomes [59–65]. It is evident that increasing miR-324–5p inhibited its target VDAC and thus inhibited cell viability/proliferation. Preclinical evaluation of delivering (transfection/liposomes) the miR-324–5p mimic recognized the feasibility of miR-targeted chemosensitization and/or therapeutics [59]. The transcriptional regulator LMO1, a designated NB susceptibility gene, downregulated the Let-7 family (Let-7a-5p, Let-7b-5p, Let-7c, Let-7e-5p, Let-7f-5p, Let-7g-5p, Let-7i-5p) TS-miRs and dictated disease evolution [60]. These outcomes indicate the possibility of identifying a large pool of potential therapeutic targets.
Interestingly, coordinated expression/function of TS-miR −193b-3p and oncomiRs’ (miR92-a-3p, miR-17–5p) are reported in NB [61]. miR-193b reduces NB cell proliferation, induces caspase-dependent/-independent cell death, and prompts G1 cell cycle arrest. Studies have shown that TS-miRs (miR-193b, miR-376c) target Ccnd1/MYCN, which drives G1 cell cycle arrest and prompts cell death, targeting MCL-1 [61,66]. Interestingly, acquired loss of miR-376c-3p in progressive NB after therapy is indicated [66]. Another TS-miR target, miR-129, directly inhibits MYO10, a protein that is highly expressed in NB and correlates with poor prognosis [67], consequently regulating tumor growth and promoting sensitivity to Cytoxan [67]. A plethora of recent evidence recognized the criticality of TS-miRs’ loss (and their mechanisms) in NB pathogenesis: (i) miR-146a targets the transcription/translation of BCL11a and promotes cell death [68]; (ii) miR-1247 targets ZNF346, inhibits Cyclin-D1, which dictates cell cycle progression, inhibits clonal expansion, and induces apoptosis [62]; (iii) miR-149 targets CDC42/BCL2 and inhibits proliferation, impedes colony formation, induces apoptosis, and prompts sensitization to Doxorubicin [63]; (iv) miR-323a-5p targets CCND1, CHAF1A, and INCENP, inhibits cell cycle progression, and induces apoptosis [64]; (v) miR-342–5p targets CCND1 and BCL-Xl, and thereby exerts NB regression [64]; (vi) miR-429 binds to 3′-UTR of IKKβ and inhibits clonal expansion, metastatic behavior, and tumor growth [69]; and (vii) miR-145 targets MTDH and inhibits malignancy physiognomies and tumor regression [65]. In addition, our recent studies identified a panel of 13 TS-miRs (miR-1206, miR-548–5p, miR-548f, miR-639, miR-640, miR-641, miR-647, miR-662, miR-886–3p, miR-887, miR-888, miR-576–5p, miR-600) that are completely lost in progressive disease [56,57]. Consistently, a high-throughput screening identified a large group (52 nos) of NB TS-miRs, recognizing that 7 (miR-380–5p, miR-665, miR-541–3p, miR-299–3p, miR-654–5p, miR-323–5p, miR-342–5p) shared a single locus (14q32), and the restoration of these miRs reduced NB cell viability [64]. Likewise, etoposide-resistant cells displayed monoallelic deletion of 13q14.3, where the miR-15a and miR-16–1 are located [70]. These chromosomal rearrangements corresponded to miR-15a/miR-16–1 loss-associated upregulation of BMI-1 oncoprotein and decreased TS-p16 [70].
Investigating the miR-regulatory networks for MYCN modulation, researchers showed that miR-506–3p downregulates PLAGL2, which has a dedicated binding site in the MYCN-promoter region and positively activates MYCN transcription [71]. Conversely, MYCN regulates PLAGL2 transcription through five MYCN-binding E-boxes in the PLAGL2-promoter region. PLAGL2 levels are associated with poor clinical outcomes in NB patients, and targeting PLAGL2 induces differentiation, impedes proliferation, and exerts synergism with MYCN silencing. Interestingly, RA treatment expresses miR-506 and represses MYCN and PLAGL2 [71]. Also, miR-15-a-5p, miR-15b-5p, and miR-16–5p are known to suppress MYCN mRNA by binding with the 3′UTR of MYCN, resulting in MYCN protein suppression [72]. Reinforcing these miRs inhibited proliferation, migration, and invasion of NB cells and tumor regression [72]. Moreover, the regulation of TS-miRs, at least in part, is orchestrated by the lncRNAs. LncRNAs like SNHG1, SNHG16, and DLX6-AS1 act as a ceRNA and sponge miRs, effecting negative regulation of TS-miRs (e.g. miR-107, miR-128, miR-338, miR-5423p) and destroying/depressing their target-translational inhibition capabilities [17,39,44,45]. Interestingly, certain lncRNAs (e.g. SNHG16) are known to sponge more than one miR, activating many oncogenes and drivers that converge in NB pathogenesis.
The significance of TS-miRs in NB cell differentiation is well recognized [47]. For instance, miR-124 expression influences differentiation through transcription/translational activation of β-tubulin-III, MAP2, SYN, NF-M, and Nestin, emphasizing that reinforcement of miR-124 could serve as a potential treatment strategy for NB [73]. miR-124 is known to functionally mediate RA treatment-induced NB cell differentiation [74]. More importantly, miRs have been used for targeted delivery of oncolytic virus in NB. For example, miR-124, miR-125, and miR-134 (miRs expressed in healthy central nervous system, but not in NB) de-targeted delivery of oncolytic SFV-4 virus influenced neurovirulence reductions and increased oncolytic capacity with a NB cure rate of about 50% in a mouse model [75].
4. Targets for immunotherapy
In patients with HR-NB, human-mouse chimeric anti-GD2 antibody ch14.18 (dinutuximab) increased OS, and hence became the standard, FDA-approved therapy (2015) for this subset of patients [76–78]. The contribution of gangliosides in cell differentiation, growth, adhesion, and cellular communication has been well recognized. Such influences in cellular processes, intercellular communication, and cell-matrix interactions directly translate to the invasive/metastatic behavior of the NB cells [79]. GD2 is highly expressed in NB cells (vs. stroma) and serves as a strong prognostic marker for immunotherapy [80]. In the context of dose intensity limiting toxicity, unrealized biological features of constantly evolving HR-NB, identification of novel/effective markers, and adaptability/suitability of such therapeutics with the current clinical care, it is important to identify/develop new immunotherapeutic approaches for the cure of HR-NB. Dinutuximab beta (Qarziba), the mouse-human chimeric monoclonal antibody produced in mammalian cell lines, has been developed, validated, and is in clinical use [81–83]. Qarziba increases survival (OS, EFS) and led to reduced cumulative incidence of relapse/progression in HR-NB patients [84]. However, due to its side effects, including neuropathic pain, many studies are now focused on either improving GD2-immunotherapy or developing other immunotargets [79]. Along this line, a phase II trial with anti-GD2 antibody Hu14.18K322A in combination with induction CT improved early response and reduced tumor volumes in newly diagnosed HR-NB [85]. Hu14.18K322A is 98% human (hence reduces allergic reaction), displays similar binding specificity to dinutuximab, has a single point-mutation to reduce complement-associated pain, and is synthesized in a YB2/0 rat myeloma cell line that could reduce fucosylation and enhance antibody-dependent cell-mediated cytotoxicity. Conceptually, this study addressed the feasibility/efficiency of adding GD2-therapy to the induction CT (rather than salvage therapy) in improving the outcomes for HR-NB patients. Induction CT (six courses), along with hu14.18K322A and tailed with GM-CSF/IL2; consolidation therapy (busulfan/melphalan, additional course of hu14.18K322A with parent-derived NK cells); followed by Hu14.18K322A, GM-CSF, IL2, and isotretinoin showed regimen tolerance, >75% PR, primary tumor volume reductions, no progression, and, more importantly, 85% EFS [85].
Recent investigations identifying a growing list of novel molecular targets for HR-NB immunotherapy have yielded rewards. For instance, targeting c-Kit (CD117) with antibodies has been suggested for the treatment of HR-NB. The tyrosine kinase component of c-kit facilitates the binding of stem cell factor (SCF), which activates c-kit/SCF pathway-mediated tumor progression. Despite controversial claims regarding the association of c-kit with disease stage and MYCN amplification, targeting c-kit reduced NB-cell survival and clonal expansion. The encouraging clinical outcomes of c-kit immunotargeting in other tumor systems as monotherapy or in conjunction with ICIs (antiCTLA4, anti-PD1/PDL1) clearly recognize its possible use for HR-NB [86–88].
Similarly, CD133 (prominin-1), a marker for NCC and tumors derived from NCC, plays functional roles in tumorigenesis, therapy resistance, and progression. CD133 is extensively investigated in NB; higher levels of CD133 are associated with dedifferentiation, chemoresistance, and poor prognosis [3,79]. Anti-CD133 therapeutic strategies, including CD133KDEL fusion protein, polymer nanoparticles loaded with paclitaxel and targeted to CD133, anti-CD133 immunotoxin therapy, bispecific antibodies targeting CD133 and CD3 (activation of immune cells), and CD8+ CAR-T cells specific to the AC133 epitope of CD133, displayed promising anti-tumor activity in a manifold of human tumors [79]. These favorable results and the documented insights into the role of CD133 in NB evolution designate CD133 as a promising target for HR-NB immunotherapy.
Likewise, higher levels of another NCC marker, the G-CSF receptor CD114, make NB cells highly tumorigenic and chemoresistant, with heightened self-renewal capacity and plasticity [89,90]. Despite its association with progression of NB and other tumors, and the defined role of G-CSFs in tumor-growth and chemoresistance, G-CSF is used to treat NB after myelosuppressive CT, underrating the undesirable consequences [79]. This complex and intertwined molecular process warrants a better understanding of the CD114-G-CSF axis for therapeutic benefit in NB. CD57, however, mediates invasion and migration of NCCs, and is associated with poor differentiation, bone marrow metastasis, and disease progression [79,91]. CD171 (L1CAM), an aberrantly expressed surface antigen in many tumors, including NB, plays a crucial role in cellular processes (growth, metastasis, angiogenesis), and is correlated with poor prognosis. Chimeric antibodies chCE7, iodine-labeled chCE7 targeting CD171, and the scFv fragments of chCE7 (CAR-T cells) exert anti-NB activity. With measurable effectiveness of autologous CE7R/HyTK+CD8+ cytolytic T-lymphocytes (Phase I trial) in HR-NB, additional trials are currently underway.
5. Targeting EMT and CSCs
The cellular transformation process, epithelial-to-mesenchymal transition (EMT), plays a significant role in tumor development and a conferred feature of malignancy. Upon EMT activation, cancer cells lose their epithelial characteristics (cell junctions, apical basal polarity) and acquire mesenchymal (self-renewal, migratory, invasive, sphere-forming) properties. In NB, EMT is more of an acquired process with defined molecular triggers/drivers (e.g. the WNT signaling pathway) [92]. Acquisition of EMT increases tumor-initiating capacity and confers therapy resistance. For instance, lncRNA-SNHG7 interacts with miR-653–5p, imposing STAT2 regulation, which impedes EMT and facilitates NB progression [28]. Likewise, PLK4, a regulator for centriole replication, is highly expressed in NB (vs. normal tissue) and associated with poor OS/PFS [93]. PLK4 coordinates PI3K/AKT pathway-dependent EMT, and targeting PLK4 suppresses NB progression and metastasis, qualifying as a promising therapeutic target for HR-NB [93]. An independent study showed that silencing uPAR prompts nuclear translocation of uPA, which endorses EMT with suppressed epithelial (E-cadherin, Occlidin, claudin-5) and activated mesenchymal (N-Cadherin, NFκB, Snail, α-SMA) markers [94]. Consistently, pre-clinical studies targeting uPA/uPAR with inhibitors, miRs, antibodies, peptides, and gene silencing proved to be beneficial in tumors, including NB [95]. Further, the clinical evaluation (phase I, NCT00083525) of the safety, tolerability, MTD, and pharmacokinetics/dynamics of WX-UK1 (uPA inhibitor) in combination with capecitabine were assessed in patients with non-responsive advanced malignancies.
NB is characterized with a high heterogeneity (different lineage) of CSCs with self-renewing, tumor-initiating, sphere-forming capacity and that are highly resistant to current clinical therapy [96,97]. Studies recognizing the mechanisms of stemness maintenance, CSCs function in disease evolution, and their implications in cancer therapeutics [98,99] indicate the suitability of ideal targets for HR-NB treatment. For example, IGF1R is a highly expressed receptor in NB that activates EMT by expressing vimentin, Snail, and zeb1, and endorsed CSC-status through STAT3/AKT signaling [100]. Leveling IGF1R attenuated stemness behavior by suppressing Oct4, Sox2, Nanog, EpCAM, CD44, and CD133 [100]. CD44 is highly expressed in NB-cells and associated with poor prognosis in HR-NB patients, independent of MYCN status [96]. CD44+ cells display NCC-mimicking stemness physiognomies, are highly tumorigenic, and produce progressive NB with metastasis [96]. Further, ALDH18A1 positivity was associated with MYCN amplification, advanced disease, NB progression/relapse, and poor OS/EFS [97]. ALDH18A1 is involved in RNA transport, ribosome biogenesis, DNA replication, and the cell cycle, and actuates proliferation and the self-renewal and tumor-initiating capacities of NB-cells [97]. ALDH18A1 increases MYCN expression through the miR-29b/SP1 autoregulatory loop and by disrupting the internal balance of the MYCN-inhibiting miRNA network(s) [97]. Targeting ALDH18A1 with YG1702 dictates MYCN suppression and inhibits NB growth [97]. CFC1, a recognized NB-CSC mobilizing factor, is known to control differentiation by targeting Activin-a. Leveling CFC1 suppresses NB aggravation via the blockade of stemness features.
Recent studies indicated that many drugs could be repur-posed/developed to target NB-CSCs and effect HR-NB control. The antipsychotic drug pimozide was shown to impede Wnt/β-catenin signaling-dependent EMT, inhibit the activation of STAT3 and STAT5 in CSCs, hinder the differentiation-inhibiting proteins (USP-1, WDR48), and thereby facilitate anti-neoplastic activity [101]. Dehydroeffusol (DHE) isolated from Chinese herbs promoted EMT suppression in NB by targeting Hedgehog and AKT/mTOR signaling, and consequently regulated the invasive phenotype [102]. Similarly, ginsenoside-Rk1 (saponin) treatment was shown to inhibit NB progression by blocking EMT with increased E-cadherin and decreased vimentin, MMP2, MMP9, and Snail [103].
In regard to induced drug resistance, studies have shown that targeting NB-CSCs could be an effective strategy to promote chemosensitization. Probenecid, a substrate for MRP, sensitizes NB-CSCs to cisplatin by targeting MDR1, MRP2, and BCRP [104]. Researchers documented that cisplatin-/RT-resistant NB-cells exhibit CSC characteristics (CD133, ALDH, Nestin) with stemness maintenance (Sox2, Oct4, Nonog) and heightened proliferation [105]. The AKT2/mTOR and MAPK signaling pathways were essentially involved in cisplatin-/RT-resistant NB-CSCs. Targeting this axis with AKT (CCT128930) or MEK (PD98059) inhibitor reverted their malignancy characteristics [105]. Consistently, targeting the AKT/mTOR pathway in NB-CSCs with rapamycin and triciribine inhibited proliferation/migration, suggesting that AKT/mTOR is a promising target for CSC-rich HR-NB [106].
6. Novel combination strategies
Although there are many promising molecular targets for improved therapeutic options for HR-NB, conceptually out-of-box, yet effective, strategies remain a challenge. The influence of parallel and synergistic mechanisms coupled with the regulation of compromising events in disease evolution warrants a multi-target combinatorial approach for HR-NB cure. An interesting study predicted 88 targets for HR-NB, and confirmed the function of four known (PI3K, MTOR, CDK4/6, HMGCR already in clinical trials), three in-pipeline (PPARG, CDK2, ROCK in preclinical evaluation), and four novel (MAPK8, CNR2, UGCG, TSPO) targets that coordinate NB progression [107]. A preclinical combination study with serine kinase WEE1 inhibitor AZD1775 (Adavosertib) and irinotecan displayed significant synergism and therapeutic enhancement [108]. AZD1775 forced cell cycle progression without DNA damage repair, prompting mitotic catastrophe and cellular death. More importantly, a Phase I evaluation of the AZD1775-irinotecan combination for relapsed solid tumors, including NB, showed prolonged stable disease [109]. A synthetic agonist for TLR3 efficiently synergized with RA, activated the innate immune signaling and mitochondrial stress response, and consequently augmented RA-induced apoptosis [110]. Further, the TLR3-agonist-RA combination activated RAR-β and restored ATRX, resulting in induced differentiation and reduced vessel formation that dictated tumor regression [110]. Recently, a new strategy of using autologous MSCs as cargo (to metastatic disease) to evade recognition and attack from the innate/adaptive immune system proved advantageous [111,112]. To that end, a first-in-human, first-in-child study of MSC-delivered oncolytic virus (lcovir-5) conceptually recognized the use of this strategy for HR-NB, raising the possibility of reaching metastatic sites with this delivery method [111]. Alternatively, the use of allogeneic MSCs showed an NB-specific homing ability, long-term engraftment into NB, and efficient cellular delivery of anti-tumoral agents [113]. The strategy overcame limitations such as short half-life and toxicity.
Another strategy is the use of MIBG, a norepinephrine analog that is actively taken up by norepinephrine transporter receptors that are highly localized in NB. Beyond its use in NB diagnosis, MIBG is used in the treatment of relapsed/refractory HR-NB [114–116]. Studies determining the tolerated dose/toxicity in HR-NB patients reported CR/PR rates up to 66% [115]. The availability of 131I-MIBG dosing (fixed, weight-based, radiation yield-based) options allowed the characterization of therapeutic ranges, requirements auto-SCR, toxicity, and patient stratification (age, previous treatments, soft-tissue/bone marrow) [115]. As with any radiotherapy, fractionation allows a high cumulative dose with maximal response. Researchers investigated the benefit of 131I-MIBG serial administrations. Retrospective review of stage-IV HR-NB cohort showed a 37% 5-Year OS; however, the review indicated the requirement for three sessions of 131I-MIBG treatment for prolonged survival [114]. Beyond 131I-MIBG monotherapy, researchers have also documented the tolerance, toxicity, and response rate of 131I-MIBG combination with CT (cisplatin, cyclophosphamide) and auto-SCR [115]. Coupling 131I-MIBG treatment with tandem high-dose CT and auto-SCR produced a 10% increase in OS/EFS of HR-NB patients [116]. Critically, 131I-MIBG treatment prior to the development of chemoresistance displayed a significant and lasting effect as part of induction treatment [115].
7. Drugs and strategies in development
In recent years, a manifold of promising novel targets, drugs, and strategies have been aggressively investigated in NB field. These are partially discussed below; a partial list is presented in Table 1. Third-generation ALK inhibitor repotrectinib (TPX-0005, currently in Phase I, Phase II trials) targets the active kinase conformations of ALK, ROS1, and NTRK1–3; impedes proliferation and prompts apoptosis in ALK-addicted NB cells; and inhibits ALK-driven neurite outgrowth and prompts tumor regression [117]. Vitamin K3 derivative (VK3-OH) induces apoptosis through activation of TS-p53, and downregulates Bcl-2 and Mcl-1 in MYCN-driven NB cells [118]. VK3-OH blocks MYCN transcription/translation and downregulates LIN28B in MYCN-amplified/overexpressed NB cells [118]. Sequential studies showed that therapeutic targeting (blocking peptide/sHRNA) of either MZF1-AS1 to disrupt MZF1-AS1/PAPR1/E2F1 axis-dependent proline synthesis or circ-CUX1 to deregulate circ-CUX1/EWSR1/MAZ axis-dependent glycolysis could be promising strategies for treating HR-NB [119,120]. Meriolin-1 (hybrid of meridianins/variolins) affects AKT/MAPK signaling and cell-cycle modulators (Cyclin-B1/D1, CDK1/2/4/6) that regulate cell proliferation and adhesion and prompts oxidative stress, autophagy, and death [147]. A novel antigene oligonucleotide (BGA002) that potentiates MYCN transcriptional inhibition reverts MYCN-dysregulated mitochondrial pathways in MYCN-amplified NB [148]. Likewise, the multi-kinase (RET/fusions, BRAF) inhibitor RXDX-105 targets NB cell malignancy vitals; its combination with RA produced a synergistic effect [149]. Another study showed that Isatin, an indole derivative, inhibits MAO activity and LSD1 demethylase activity, leading to increased H3K4m1 and PTEN, which negatively regulate the NB-cell metastatic state through PI3K/AKT and PTEN/p-SHC/p-FAK signaling [150]. Otsuka and colleagues demonstrated that TNIIIA2 peptide synergizes with RA treatment in increasing neuronal markers (NF-L, NF-H, GAP43) and inhibiting MYCN, dictating NB cell differentiation and tumor regression [151]. Repurposing Sildenafil for NB differentiation therapy indicated that both sildenafil and its analog IS00384 phosphorylated C3G; activated AMPK signaling; increased NeuN, NF-H, and βIII-tubulin; and induced neurite outgrowth [152]. Likewise, repurposing mefloquine for NB treatment showed that it prompts redox stress-mediated apoptosis by binding to acyl-CoA [153]. Thiopurines (small molecules, CCI52, CCI52–14) showed efficient sensitization of MYCN-spontaneous NB to 6-MP, irinotecan, and vincristine [154].
Table 1.
Partial list of emerging targets; their functions in cellular processes (malignancy characteristics), mechanism(s) of action, and signaling flow-through; and potential drugs or strategies to improve HR-NB treatment.
| Target | Function/Strategy | Targets/Signaling | Therapeutic strategy | Ref. |
|---|---|---|---|---|
| MDA-9/SYNTENIN | ↑Metastasis, ↑EMT, ↓OS | ↑Src, ↑FAK, ↑NFkB, ↑Slug, ↑MMP2, ↑MMP9. ↑integrin→SRC→RHO→RAC→CDC42 | PDZ1i, MDA-siRNA, peptide | [121,122] |
| GLS2 | ↓Proliferation, ↑Cell cycle arrest | ↓cMYC | [123] | |
| P27kip1 | ↓Progression | MYC→CKS1→SKP2→P27kip1 | Prozac | [124] |
| KLF9 | ↑Differentiation, ↓Proliferation, ↓Invasion | ↓SHH binding to the promoter, ↓GUI, | [125] | |
| WRD5 | ↑Proliferation, ↑Tumorosphere formation | ↑MYC, CARM↑, ↑H3K4me3 | [126] | |
| MZF1 | ↑Glycolysis | ↑HK2, ↑PGKI, ↑YY1 | 21-AA-MZF1-uPEP | [127] |
| SNAI2 | ↑Self-renewal, ↑EMT, ↑RA-resistance, ↓Differentiation | ↓NRCAM, ↓CDH5 | CRISPR-Cas9, shRNA | [128] |
| Stmn | ↑Migration, ↑Invasion | ↑PTPN14, miRs ↑(3935, 1281, 4682, 222, 221, 620, 488) ↓(382, 935, 4656, 132, 4492) | PTPN14-shRNA | [129] |
| ITCH | ↓Apoptosis, ↑Radioreistance | ↓TP73 | siRNA-NPs | [130] |
| PLPR1 | ↓Cell-migration, ↑Adhesion | ↓RAC1 | [131] | |
| TRKB | ↑Entrectinib resistance | ↑ERK/MAPK, ↓PTEN | [132] | |
| BARD1 | ↑DNA damage after RT, ↑Apoptosis, ↑G2/M cell cycle | ↑IGFIR, ↑P75 | BARD1-FL | [133] |
| YAP1 | ↑Trametinib resistance, ↑Cell viability, ↑Proliferation | ↑MAPK, ↑E2F, ↑MYCN, ↑P27KIP1-NL, ↑AKT | Lv-CRISPR-YAP1-null-NLF | [134,135] |
| TERT | ↓Prognosis, ↑Oncogenic pathway | ↑BRD4, H3K27AC, H3K36me3 | BRD4i (AZD5153), CDKi (dinacictib) | [136] |
| PNUTS | ↑Proliferation | ↑MYCN Protein | CDKTi (THZ1) + TKI | [137] |
| MYCN-LM01-ASCL1 | ↓Differentiation | ↓NTRK/TRKA, ↓NPY | Ponatinib, Lapatinib | [138] |
| GX15–070→↓ BCl2 | ↑Prosurvival autophagy, ↓Apoptosis | ↑LC3II | GX15–070 + HCQ | [139] |
| Hypomethylation of GATA3 | ↑Proliferation, ↑G2-M, ↓PARP cleavage | ↑GATA3 Protein, ↑CCND1 | GATA3-siRNA | [140] |
| PIM1 | ↑ALKi resistance, ↓Apoptosis | PIM1i (AZD1208) + Ceritinib | [141] | |
| GOLPH3 | ↑DNA repair, ↑Therapy resistance | ↑TPX2, ↑MY018A | [142] | |
| AURKA, AURKB | ↓Apoptosis, ↓G2-M | ↑PGAM2, ↑ACADM | AURKi - Tozasertib (VX-680) | [143] |
| PTPN1P | ↑Proliferation | PTPN1-siRNA | [144] | |
| RAD51 | ↑Chemoresistance | Lv-shRNA | [145] | |
| MCPIP1 | ↓Proliferation, ↑G1 phase | ↓Cyclins A1, b1, D1, D3, E1, E2, ↓CDK2P, ↓CDK4 P, ↓RB | [146] |
Many recent findings indicated the potential of natural phytochemicals in HR-NB treatment. As discussed above, the benefit of ginsenoside-RK1 in targeting NB EMT and malignancy/metastatic behavior has been realized [103]. Likewise, studies have extensively documented the anti-tumor and chemo/radiosensitizing potential (and mechanisms) of curcumin and its synthetic analogues in NB [155–158]. 4-hydroxy chalcone (4HC, flavonoid) treatment induced NB cell death by increasing oxidative stress and LDH activity, and prompted chemosensitization to doxorubicin and cisplatin [159]. Conversely, a polysaccharide from Angelica sinensis has been shown to attenuate BCl2, activate Bax and Casp3, and promote apoptosis [160]. This polysaccharide: (i) inhibited the TGFβ1-induced EMT by targeting N-Cad, vimentin, and Zeb1 and activating E-Cad and (ii) upregulated TS-miR-205, consequently suppressing PI3K/AKT and ERK1/2 signaling [160]. Also, S-allyl-L-cysteine from aged-garlic extract prompted mitochondrial membrane depolarization and induced NB cell death [161].
The advances in nanotechnology and its significance in targeted delivery of desired anti-cancer drugs directed focus in developing strategies for HR-NB treatment [162]. Green Zn oxide nanoparticles (ZnONPs) with skeel-peel-extract generated ROS regulated autophagy (LC3-I, LC3-II, ATG4B) and induced apoptosis (↑Bax, ↑Casp3, ↓Bcl2) [163]. A study comparing delivery of hydroalcoholic extract of marine angiosperm (Posidonia oceanica) loaded onto two different (chitosan/PEG-soluplus) nanoparticles recognized the efficiency of the approach and the benefit of the drug candidate in NB therapy [164]. Another NP approach using a combination of semi-synthetic cytotoxic retinoid and anti-angiogenic lenalidomide displayed excellent in vitro cytotoxicity and in vivo efficacy [165]. MXD3-antisense oligonucleotide-loaded SPIONPs delivered to NB without toxicity indicated its benefit as dose-reducing agent for CT [166]. In an interesting strategy, a bismuth selenide NPs core shelled with silver was coupled with RNA 3-way junction: one leg attached to the NP, the second harbored a cell-penetrating RNA+fluorescence tag, and the third was designed to target miR-17 and RA release [58]. This strategy yielded full differentiation of NB cells into neurons and stopped their growth. Furthermore, for miR-based NB treatment, many sophisticated nano-formulations (e.g. miR-542–3p-encapsulated-NPs, TS-miR-34a+GD2Ab-SilicaNPs, amino-functionalized gold NPs complexed with miR-31, NP glazed with PEG) and novel approaches were identified for efficient delivery of miRs in passive, active, or stimuli-responsive models [162].
With an unusual strategy, researchers investigated the benefit of TCDD, a dioxin with carcinogenic properties, and a toxic industrial pollutant, hexavalent chromium (CrVI), for NB treatment [167]. TCDD prompted AhR-dependent activation of pro-adhesion genes (18S, PCDHB, PCDHG, FZD2, FLOT2, DDR2) and inhibited the spontaneous movement of NB cells [167]. CrVI involves ROS-mediated Akt/ERK/AMPK-dependent cell death. However, the usefulness of these strategies in clinics is questionable.
8. Expert opinion
Despite significant advances in NB therapy over the past three decades and achieving excellent clinical outcomes for patients with stable/intermediary-risk disease, the cure rate for HR-NB is disappointing. This gap is attributed to the ongoing acquisition of genetic and molecular rearrangements, the EMT process and CSC clonal selection/enrichment, heterogeneity of CSC populations, lack of understanding on the interplay of events and compromising and/or alternating mechanisms, and equivocal claims of the linear response of a specific target or signaling. These gaps led to identification of pockets of signaling events and targets, with much less clinical translation and use. Since a complete understanding of NB biology is required for development of efficient therapy, researchers have focused on unveiling the disease blueprint through high-content GWAS, genomic analysis, whole-genome sequencing, transcriptomics, proteomics, and drug screening. The outcomes from these measures guided diagnosis, risk-stratification, and identification of clinicopathological features/therapy-response-specific molecular targets that may serve as candidate(s) for improved therapeutic strategies. Overall, the targets are grouped into: (i) oncotargets (oncogenes, oncomiRs, onco-lncRNAs) that activated corresponding to the mutations, amplification, modifications; (ii) inactivating tumor suppressors through deletion, epigenetic silencing, or mutation; (iii) NB- and/or cell-type-specific targets (e.g. GD2, NB-CSC markers, neuronal cell differentiation markers); and (iv) context (HR, therapy resistance, progressive/recurrent disease)-specific molecular determinants. However, a simultaneous, yet assenting (activated oncotargets and deactivated TS) interplay of events is known to drive disease pathogenesis and progression. In this regard, studies identifying novel molecular targets, and defining their functions and mechanism(s) are aggressively evolving (Figure 2). The new treatment protocols under current development are primarily directed to identify the diagnostic and/or prognostic relevance; mechanistic signaling flow-through; function in cellular processes of malignancy; drug delivery strategies; targeting efficacy in cellular and/or pre-clinical evaluation; combination strategies; or repurposing strategies. The promise of this focus is evident, as numerous clinical trials (>500) have been initiated to define novel therapeutic options for HR-NB, with an aim to evaluate new targets, drugs, a combination treatment, or repurposing strategies (Table 2).
Figure 2. Concentric plot showing the recent evolution of drugs and strategies for HR-NB.
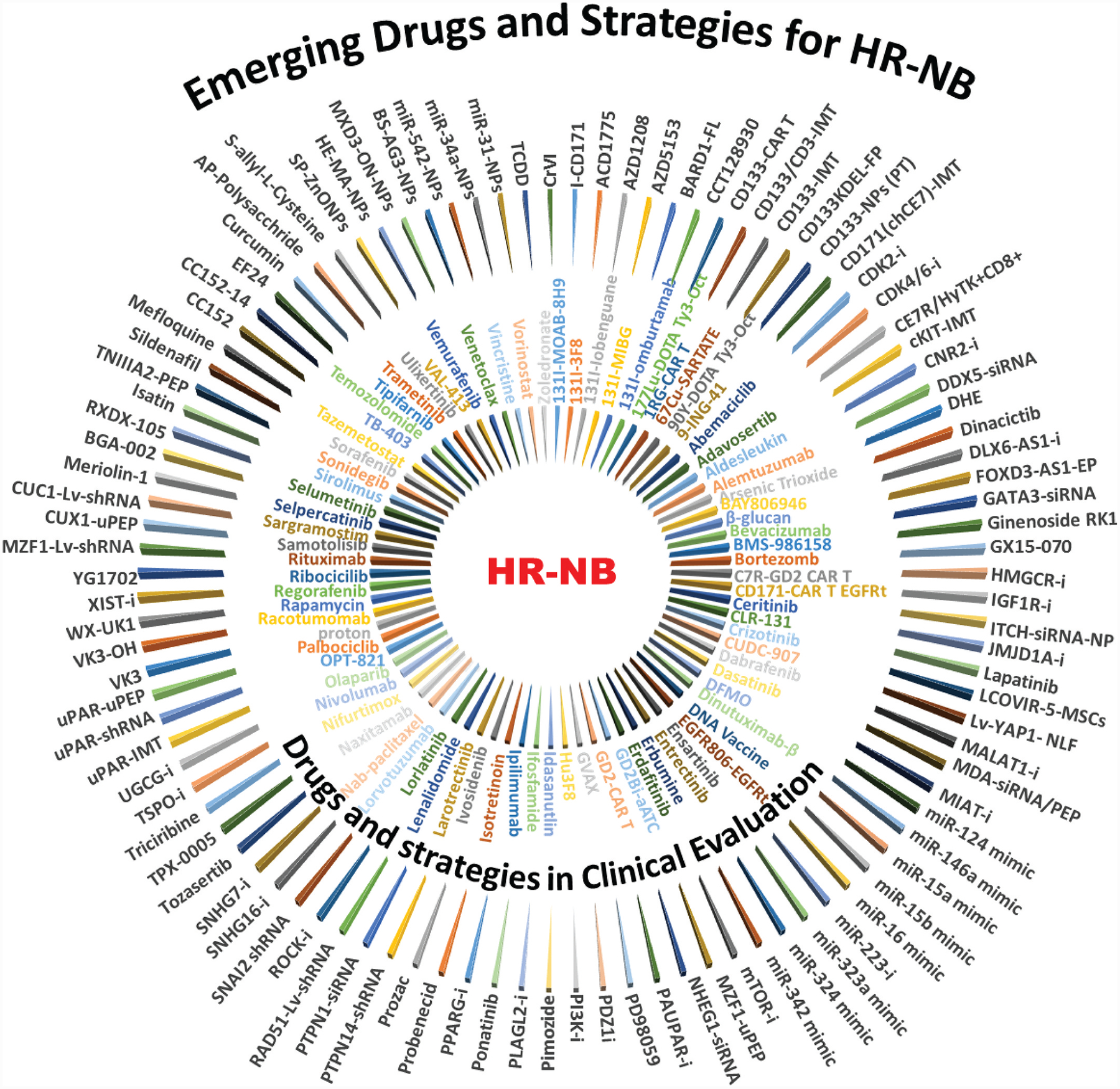
Inner circle: Partial list (from 506 current clinical trials) of targeted drugs, modalities, and/or their combination in current clinical evaluation in Phase I, II, and III trials. Outer circle: Partial list of potential and promising targeted (discussed in the text) developed or repurposed drugs (e. g., inhibitors, miRs, antibodies, peptides, gene silencing, clinical drugs) that were validated in preclinical settings, alone and/or in combination with conventional chemotherapy for HR-NB. Combination strategies revealed that these candidates integrated well with drugs in current clinical therapy, with a synergistic effect in the HR-NB setting.
Table 2.
Partial list of investigational drugs and/or strategies (and their molecular targets) that are currently under clinical evaluation. The list is limited to represent distinct drugs and strategies.
| Phase | NCT ID | Drugs / Strategy | Target |
|---|---|---|---|
| Phase 1 | NCT04049864 | DNA Vaccine | Phox2B, Survivin, MAGEA1/3, PRAME |
| Phase 1 | NCT00790413 | 131I-MIBG+CT+T-Cell−+Lymp+ +HSC+MSC+Rituximab | CD20 |
| Phase 1 | NCT02573896 | Auto-NKCs + Dinutuximab +/− Lenalidomide | GD2, TNFa, VEGF, bFGF |
| Phase 1 | NCT03294954 | GD2-CAR + IL15- Auto-NKTs + CT | GD2 |
| Phase 1 | NCT04106219 | LY3295668 Erbumine +/− CT | AURA |
| Phase 1 | NCT02298348 | Sorafenib + Topotecan, Cyclophosphamide | Tyrosine Kinase |
| Phase 1 | NCT04023331 | 67Cu-SARTATE PR-RT | SSTR |
| Phase 1 | NCT03966651 | 177Lu-DOTA0-Tyr3-Octreotate PR-RT | SSTR |
| Phase 1 | NCT02914405 | Nivolumab +Ch14.18/CHO | PD-1, GD2 |
| Phase 1 | NCT00492167 | beta-glucan + 3F8 | |
| Phase 1 | NCT04239040 | GVAX + Nivolumab, Ipilimumab | PD-1, CTLA-4 |
| Phase 1 | NCT01711554 | Lenalidomide +Dinutuximab +/− Isotretinoin | angiogenesis inhibitor |
| Phase 1 | NCT02311621 | CD171-CAR T cells expressing EGFRt | CD171, EGFR |
| Phase 1 | NCT03332667 | 131I-MIBG With Dinutuximab | GD2 |
| Phase 1 | NCT03107988 | Lorlatinib | ALK, ROS-1 |
| Phase 1 | NCT01953900 | iC9-GD2-CAR-VZV-CTLs | GD2 |
| Phase 1 | NCT02030964 | DFMO + Celecoxib + CT | ODC |
| Phase 1 | NCT02761915 | 1RG-CART | GD2 |
| Phase 1 | NCT02748135 | TB-403 | PLGF |
| Phase 1 | NCT03635632 | C7R-GD2.CART cells + CT | GD2 |
| Phase 1 | NCT00089245 | I131 MOAB 8H9 | glycoprotein 4Ig-B7-H3 |
| Phase 1 | NCT03478462 | CLR 131 | |
| Phase 1 | NCT02909777 | CUDC-907 | HDAC |
| Phase 1 | NCT04239092 | 9-ING-41 + Irinotecan | GSK-3β |
| Phase 1 | NCT03709680 | Palbociclib + Temozolomide + Irinotecan | CDK4, CDK6, DNA replication |
| Phase 1 | NCT03236857 | venetoclax + CT | BCl2 |
| Phase 1 | NCT04337177 | VAL-413 + Temozolomide | DNA Replication |
| Phase 1 | NCT04308330 | Vorinostat + Chemotherapy | HDAC |
| Phase 1 | NCT02536183 | Lyso-thermosensitive liposomal dox + MR-HIFU | |
| Phase 1 | NCT03618381 | EGFR806-EGFRt + CD19-Her2tG | EGFR |
| Phase 1 | NCT02508038 | Zoledronate + TCRαβ+/CD19+ depleted H-HSCT | |
| Phase 1 | NCT01331135 | Sirolimus | mTOR |
| Phase 1 | NCT01625351 | Sirolimus, Alemtuzumab | mTOR, CD52, |
| Phase 1 | NCT02644460 | Abemaciclib | CDK4, CDK6 |
| Phase 1 | NCT03434262 | Gemcitabine, Ribocicilib, Sonidegib, Trametinib | CyclinD1, CDK4/6, HH, MEK1, MEK2 |
| Phase 1 | NCT01121588 | Crizotinib (PF-02341066) | ALK, ROS1 |
| Phase 1 | NCT02085148 | Regorafenib + Vincristine + Irinotecan | RTK, VEGFR2-TIE2 |
| Phase 1 | NCT03936465 | BMS-986158 | BET |
| Phase 1 | NCT03507491 | Nab-paclitaxel + Gemcitabine | |
| Phase 1 | NCT04238819 | Abemaciclib + Temozolomide, Irinotecan | CDK4, CDK6 |
| Phase 1 | NCT02650401 | Entrectinib (Rxdx-101) | Trk, Ros1, Alk Fusions |
| Phase 1/2 | NCT00703222 | SNJB-JF-IL2 and SJNB-JF-Lptn + Dose Level 1 SKNLP | |
| Phase 1/2 | NCT02173093 | GD2Bi-aATC | GD2 |
| Phase 1/2 | NCT02139397 | DFMO + Bortezomib | ODC, proteasome inhibitor |
| Phase 1/2 | NCT00911560 | OPT-821 vaccine with GD2L and GD3L | GD2, GD3 |
| Phase 1/2 | NCT03860207 | Hu3F8-BsAb | GD2 |
| Phase 1/2 | NCT03923257 | PRRT-177Lu-DOTA tyr3-Octreotide | SSTR |
| Phase 1/2 | NCT04029688 | Idasanutlin + Venetoclax + CT | MDM2, BCl2 |
| Phase 1/2 | NCT02095132 | Adavosertib + Irinotecan | WEE1 |
| Phase 1/2 | NCT02124772 | Dabrafenib, Trametinib | B-RAF, MEK1, MEK2 |
| Phase 1/2 | NCT00788125 | Dasatinib + Ifosfamide + CT + surgery + RT | Bcr-ABL, Src |
| Phase 2 | NCT03363373 | GM-CSF + Naxitamab | GD2 |
| Phase 2 | NCT03503864 | Arsenic Trioxide + induction CT | |
| Phase 2 | NCT03373097 | GD2-CAR-T Cells +/− CT & lymphodepletion | GD2 |
| Phase 2 | NCT04301843 | DFMO + Etoposide | ODC, |
| Phase 2 | NCT02679144 | DFMO | ODC |
| Phase 2 | NCT04023331 | 67Cu-SARTATE PR-RT (Cohort expansion) | SSTR |
| Phase 2 | NCT02998983 | Racotumomab | anti-N-glycolyl GM3 Ab inducer |
| Phase 2 | NCT03033303 | Hu3F8/GM-CSF + Isotretinoin | GD2 |
| Phase 2 | NCT02559778 | Ceritinib + Dasatinib + Sorafenib + Vorinostat + DFMO | ODC, HDAC, ALK, VEGFR, PDGFR, RAF |
| Phase 2 | NCT02308527 | Bevacizumab + Temozolomide ± Irinotecan | VEGF-A, DNA replication |
| Phase 2 | NCT01467986 | Dasatinib + Rapamycin + Irinotecan + Temozolomide | Bcr-ABL, Src, mTOR, DNA replication |
| Phase 2 | NCT00107289 | lobenguane I 131 | |
| Phase 2 | NCT00601003 | Nifurtimox | |
| Phase 2 | NCT02035137 | 131I-MIBG +/− Viscristine, Irinotecan / Vorinostat | HDAC |
| Phase 2 | NCT02452554 | Lorvotuzumab Mertansine | CD56 binding |
| Phase 2 | NCT03273712 | PRRT-90Y-DOTA tyr3-Octreotide | SSTR |
| Phase 2 | NCT04320888 | Selpercatinib | RET |
| Phase 2 | NCT04195555 | Ivosidenib | IDH1 |
| Phase 2 | NCT04284774 | Tipifarnib | FTase |
| Phase 2 | NCT03698994 | Ulixertinib | ERK1/2 |
| Phase 2 | NCT03526250 | Palbociclib | CDK4, CDK6 |
| Phase 2 | NCT03220035 | Vemurafenib | B-RAF |
| Phase 2 | NCT03233204 | Olaparib | PARP |
| Phase 2 | NCT03213704 | Larotrectinib | TrkA, TrkB, TrkC |
| Phase 2 | NCT03213678 | Samotolisib | PI3K, mTOR, DNA-PK |
| Phase 2 | NCT03210714 | Erdafitinib | FGFR |
| Phase 2 | NCT03155620 | Ensartinib, Erdafitinib, Larotrectinib, Olaparib, Palbociclib, Samotolisib, Selpercatinib, Selumetinib Sulfate, Tazemetostat, Tipifarnib, Ulixertinib, Vemurafenib | ALK, FGFR, TrkA, TrkB, TrkC, PARP, CDK4/6, PI3K, mTOR, RET, MEK1, MEK2, EZH2, Ftase, ERK1/2, B-RAF |
| Phase 2 | NCT03213665 | Tazemetostat | EZH2 |
| Phase 2 | NCT02378428 | MIBG | NET Receptors |
| Phase 2 | NCT03458728 | BAY806946 | PI3K |
| Phase 2 | NCT02574728 | Sirolimus + Metronomic CT | mTOR |
| Phase 2/3 | NCT03275402 | 131I-omburtamab | B7-H3 |
| Phase 3 | NCT03126916 | Crizotinib/Iobenguane I131 + Auto-HSC, CT, Dinutuximab, EBRT, Isotretinoin, Sargramostim, Surgery, Vincristine | ALK, ROS-1, NET receptors |
| Phase 4 | NCT03975829 | Dabrafenib, Trametinib | B-RAF, MEK1, MEK2 |
Although a number of targets are considered for HR-NB, mechanistically the majority of these converge on a handful of key determinants (or signaling pathways), including ALK, MYCN, RAS/MAPK, cell-cycle/apoptotic regulators, EMT/CSC drivers, and differentiation modulators. Initially, it was believed that targeting activated drivers was beneficial, but the dynamic molecular rearrangements in HR-NB indicated the acquisition of compromising and/or complementing effects, warranting the reinforcement of brake (TS) systems in parallel. For example, although it is difficult to directly target MYCN, the key player in the pathogenesis and evolution of HR-NB, it is feasible to target MYCN drivers (lncUSMYCN, lncMYCNOS) or reinforce MYCN regulators TS-miRs (miR-506–3p, miR-15-a-5p, miR-15b-5p, miR-16–5p) to treat MYCN-amplified or highly expressed progressive HR-NB. Many proposed targets and approaches independently or interactively target MYCN expression and function in NB cells, resulting in tumor regression. These new pathways identify better ways of targeting MYCN that could definitively advance therapy for HR-NB. ALK activation, which acts in synergy with MYCN expression, is an oncogenic driver for NB and could be selectively druggable. Sequential generations of small molecule ALK inhibitors are developed based on their ALK-binding affinity to all mutational variants and are in clinical evaluation for HR-NB. ALK-targeted therapy alone or in conjunction with the conventional CT is a promising possibility, particularly when a targeted population that responds to ALK inhibitors could be defined through either genomic screening for mutations in the TK domain or ALK amplification/overexpression.
Targeting EMT drivers and surface antigens that are selectively/highly expressed on NB-CSCs cells is highly beneficial, especially for progressive HR-NB. Combination and/or repurposing strategies with drugs for other medical conditions are highly effective, but warrant the evaluation of clinical benefit in HR-NB. With recent dosing advances in targeted (I131MIBG) RT, this approach is exceptionally beneficial for the treatment of HR-NB. Likewise, the evolving sophistication in nanotechnology not only allows precise delivery of drugs in a safer and effective manner, but also permits a multi-target (e.g. RA, miR) approach. Beyond the betterment of strategies and drugs for the conventional targets, emerging evidence indicates a number of proteins/pathways (see Table 1) that could be promising targets for this deadly disease. These candidates fall into the categories of apoptotic, differentiation, autophagy, signal transduction, EMT, and stemness maintenance regulators. Although the functional relevance of these determinants is pre-clinically documented in HR-NB, the therapeutic benefit of targeting such candidates alongside current clinical therapy is yet to be discovered.
Taken together, the understanding of the biology of progressive HR-NB, dynamic rearrangements in response to therapy, molecular targets and mechanism(s) of action, identification/development of drug candidates, and identification of delivery tools are aggressively evolving. The continuous flow of knowledge and technical sophistication is likely to identify new, improved, and effective therapeutic strategies for specific subsets of patients with HR-NB. Though currently the progress is only halfway through, assessing the recent evolution in the field and considering the focus, the paradigm shift in HR-NB treatment could be realized in the next 5 years or so.
Article highlights.
Novel and improved therapeutic schemes are required to counter unacceptable survival rates in children with high-risk neuroblastoma (HR-NB)
Oncogene-like and tumor suppressor lncRNAs serves as emerging therapeutic targets for NB
OncomiRs - TS-miRS’ modulation and coordinated functions dictates NB pathogenesis
lncRNAs sponge multiple miRs, activating oncogenes and drivers that converge in NB pathogenesis
Novel targets for immunotherapy indicates a promising strategy for the treatment of HR-NB
Targeting EMT and CSC stemness maintenance is highly beneficial for progressive disease
Combination strategies and/or repurposing clinical drugs are highly effective for HR-NB.
This box summarizes key points contained in the article.
Acknowledgments
The authors acknowledge the OUHSC Staff Editor (Ms. Kathy Kyler) for the help in critically reviewing this manuscript.
Funding
Supported by the National Institutes of Health [NIH-P20GM103639]; and Oklahoma Center for the Advancement of Science and Technology [OCAST-HR19-045].
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975–2009 (Vintage 2009 populations). Bethesda (MD): National Cancer Institute; 2012. [cited 2020 Feb 26]. Based on November 2011 SEER data submission. Posted to the SEER web site. Available from: https://seer.cancer.gov/csr/1975_2009_pops09/ [Google Scholar]
- 2.London WB, Castleberry RP, Matthay KK, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the children’s oncology group. J Clin Oncol. 2005. September 20;23(27):6459–6465. [DOI] [PubMed] [Google Scholar]
- 3.Aravindan N, Jain D, Somasundaram DB, et al. Cancer stem cells in neuroblastoma therapy resistance. Cancer Drug Resist. 2019. December;19 (2):948–967. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Complete and recent review on the cancer stem cells in neuroblastoma therapy resistance.
- 4.Ritenour LE, Randall MP, Bosse KR, et al. Genetic susceptibility to neuroblastoma: current knowledge and future directions. Cell Tissue Res. 2018. May;372(2):287–307. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides current knowledge on the genetic susceptibility to neuroblastoma.
- 5.Board PPTE. PDQ neuroblastoma treatment: health professional version. PDQ cancer information summaries; Bethesda (MD); 2020. [cited Feb 26] Available from: https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq [Google Scholar]; • Complete information on neuroblastoma of a health care professional perspective.
- 6.Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al. Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol. 2010. July 1;28(19):3122–3130. [DOI] [PubMed] [Google Scholar]
- 7.Campbell K, Gastier-Foster JM, Mann M, et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: a report from the children’s oncology group. Cancer. 2017. November 1;123(21):4224–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreissman SG, Seeger RC, Matthay KK, et al. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol. 2013. September;14(10):999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peifer M, Hertwig F, Roels F, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015. October 29;526(7575):700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013. January;45(1):12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brodeur GM, Iyer R, Croucher JL, et al. Therapeutic targets for neuroblastomas. Expert Opin Ther Targets. 2014. March;18(3):277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Previous review on the therapeutic targets for neuroblastoma in expert opinions that serves as the base for this emerging therapeutic targets review.
- 12.Salehi S, Taheri MN, Azarpira N, et al. State of the art technologies to explore long non-coding RNAs in cancer. J Cell Mol Med. 2017. December;21(12):3120–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lian D, Amin B, Du D, et al. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017. December 12;21(1):151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han W, Du X, Liu M, et al. Increased expression of long non-coding RNA SNHG16 correlates with tumor progression and poor prognosis in non-small cell lung cancer. Int J Biol Macromol. 2019. January;121:270–278. [DOI] [PubMed] [Google Scholar]
- 15.Feng F, Chen A, Huang J, et al. Long noncoding RNA SNHG16 contributes to the development of bladder cancer via regulating miR-98/STAT3/Wnt/beta-catenin pathway axis. J Cell Biochem. 2018. November;119(11):9408–9418. [DOI] [PubMed] [Google Scholar]
- 16.Su P, Mu S, Wang Z. Long noncoding RNA SNHG16 promotes osteosarcoma cells migration and invasion via sponging miRNA-340. DNA Cell Biol. 2019. February;38(2):170–175. [DOI] [PubMed] [Google Scholar]
- 17.Wen Y, Gong X, Dong Y, et al. Long non coding RNA SNHG16 facilitates proliferation, migration, invasion and autophagy of neuroblastoma cells via sponging miR-542–3p and upregulating ATG5 expression. Onco Targets Ther. 2020;13:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Zhao X, Li D, Yang F, et al. Long noncoding RNA NHEG1 drives beta-catenin transactivation and neuroblastoma progression through interacting with DDX5. Mol Ther. 2020;28(3):946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu C, Yang T, Pan J, et al. Associations between H19 polymorphisms and neuroblastoma risk in Chinese children. Biosci Rep. 2019;39:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang T, Zhang Z, Zhang J, et al. The rs2147578 C > G polymorphism in the Inc-LAMC2–1:1 gene is associated with increased neuroblastoma risk in the Henan children. BMC Cancer. 2018. October 3;18(1):948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Zhuo ZJ, Zhou H, et al. Additional data support the role of LINC00673 rs11655237 C>T in the development of neuroblastoma. Aging (Albany NY). 2019. April 20;11(8):2369–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, He J, Chang Y, et al. HOTAIR gene polymorphisms contribute to increased neuroblastoma susceptibility in Chinese children. Cancer. 2018. June 15;124(12):2599–2606. [DOI] [PubMed] [Google Scholar]
- 23.Barnhill LM, Williams RT, Cohen O, et al. High expression of CAI2, a 9p21-embedded long noncoding RNA, contributes to advanced-stage neuroblastoma. Cancer Res. 2014. July 15;74(14):3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu M, Ohira M, Li Y, et al. High expression of ncRAN, a novel non-coding RNA mapped to chromosome 17q25.1, is associated with poor prognosis in neuroblastoma. Int J Oncol. 2009. April;34(4):931–938. [DOI] [PubMed] [Google Scholar]
- 25.Liu PY, Erriquez D, Marshall GM, et al. Effects of a novel long noncoding RNA, lncUSMycN, on N-Myc expression and neuroblastoma progression. J Natl Cancer Inst. 2014. July;106(7). DOI: 10.1093/jnci/dju113 [DOI] [PubMed] [Google Scholar]
- 26.Zhao X, Li D, Pu J, et al. CTCF cooperates with noncoding RNA MYCNOS to promote neuroblastoma progression through facilitating MYCN expression. Oncogene. 2016. July 7;35 (27):3565–3576. [DOI] [PubMed] [Google Scholar]
- 27.Pandey GK, Mitra S, Subhash S, et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell. 2014. November 10;26(5):722–737. [DOI] [PubMed] [Google Scholar]
- 28.Chi R, Chen X, Liu M, et al. Role of SNHG7-miR-653–5p-STAT2 feedback loop in regulating neuroblastoma progression. J Cell Physiol. 2019. August;234(8):13403–13412. [DOI] [PubMed] [Google Scholar]
- 29.Sahu D, Hsu CL, Lin CC, et al. Co-expression analysis identifies long noncoding RNA SNHG1 as a novel predictor for event-free survival in neuroblastoma. Oncotarget. 2016. September 6;7 (36):58022–58037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Y, Chen F, Yang Y, et al. lncRNA SNHG16 is associated with proliferation and poor prognosis of pediatric neuroblastoma. Int J Oncol. 2019. July;55(1):93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li D, Wang X, Mei H, et al. Long noncoding RNA pancEts-1 promotes neuroblastoma progression through hnRNPK-mediated beta-catenin stabilization. Cancer Res. 2018. March 1;78(5):1169–1183. [DOI] [PubMed] [Google Scholar]
- 32.Mondal T, Juvvuna PK, Kirkeby A, et al. Sense-antisense lncRNA pair encoded by locus 6p22.3 determines neuroblastoma susceptibility via the USP36-CHD7-SOX9 regulatory axis. Cancer Cell. 2018. March 12;33(3):417–434e7. [DOI] [PubMed] [Google Scholar]
- 33.Russell MR, Penikis A, Oldridge DA, et al. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. 2015. August 1;75(15):3155–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang W, Dong K, Li K, et al. MEG3, HCN3 and linc01105 influence the proliferation and apoptosis of neuroblastoma cells via the HIF-1alpha and p53 pathways. Sci Rep. 2016. November 8;6:36268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bi S, Wang C, Li Y, et al. LncRNA-MALAT1-mediated Axl promotes cell invasion and migration in human neuroblastoma. Tumour Biol. 2017. May;39(5):1010428317699796. [DOI] [PubMed] [Google Scholar]
- 36.Tee AE, Ling D, Nelson C, et al. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. 2014. April 15;5(7):1793–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tee AE, Liu B, Song R, et al. The long noncoding RNA MALAT1 promotes tumor-driven angiogenesis by up-regulating pro-angiogenic gene expression. Oncotarget. 2016. February 23;7(8):8663–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao J, Zhang S, Meng Q, et al. SNHG16 silencing inhibits neuroblastoma progression by downregulating HOXA7 via sponging miR-128–3p. Neurochem Res. 2020. April;45(4):825–836. [DOI] [PubMed] [Google Scholar]
- 39.Vance KW, Sansom SN, Lee S, et al. The long non-coding RNA paupar regulates the expression of both local and distal genes. Embo J. 2014. February 18;33(4):296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavlaki I, Alammari F, Sun B, et al. The long non-coding RNA paupar promotes KAP1-dependent chromatin changes and regulates olfac-tory bulb neurogenesis. Embo J. 2018. May 15;37(10). DOI: 10.15252/embj.201798219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Li WY, Yang Y, et al. LncRNA XIST facilitates cell growth, migration and invasion via modulating H3 histone methylation of DKK1 in neuroblastoma. Cell Cycle. 2019. August;18(16):1882–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bountali A, Tonge DP, Mourtada-Maarabouni M. RNA sequencing reveals a key role for the long non-coding RNA MIAT in regulating neuroblastoma and glioblastoma cell fate. Int J Biol Macromol. 2019. June;1(130):878–891. [DOI] [PubMed] [Google Scholar]
- 43.Zhang HY, Xing MQ, Guo J, et al. Long noncoding RNA DLX6-AS1 promotes neuroblastoma progression by regulating miR-107/BDNF pathway. Cancer Cell Int. 2019;19:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang N, Liu FL, Ma TS, et al. LncRNA SNHG1 contributes to tumorigenesis and mechanism by targeting miR-338–3p to regulate PLK4 in human neuroblastoma. Eur Rev Med Pharmacol Sci. 2019. October;23(20):8971–8983. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X, Li D, Huang D, et al. Risk-associated long noncoding RNA FOXD3-AS1 inhibits neuroblastoma progression by repressing PARP1-mediated activation of CTCF. Mol Ther. 2018. March 7;26(3):755–773. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Sousares M, Partridge V, Weigum S, et al. MicroRNAs in neuroblastoma differentiation and differentiation therapy. Adv Modern Oncol Res. 2017;3(5):213–222. [Google Scholar]
- 47.He XY, Tan ZL, Mou Q, et al. microRNA-221 enhances MYCN via targeting nemo-like kinase and functions as an oncogene related to poor prognosis in neuroblastoma. Clin Cancer Res. 2017. June 1;23 (11):2905–2918. [DOI] [PubMed] [Google Scholar]
- 48.Aravindan N, Subramanian K, Somasundaram DB, et al. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist. 2019;2:1086–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Recent review on the role of MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution.
- 49.Megiorni F, Colaiacovo M, Cialfi S, et al. A sketch of known and novel MYCN-associated miRNA networks in neuroblastoma. Oncol Rep. 2017. July;38(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nallasamy P, Chava S, Verma SS, et al. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: immuno-oncology perspective. Semin Cancer Biol. 2018. October;52(Pt 2):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanhauwaert S, Decaesteker B, De Brouwer S, et al. In silico discovery of a FOXM1 driven embryonal signaling pathway in therapy resistant neuroblastoma tumors. Sci Rep. 2018. November 30;8(1):17468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu X, Peng H, Liao W, et al. MiR-181a/b induce the growth, invasion, and metastasis of neuroblastoma cells through targeting ABI1. Mol Carcinog. 2018. September;57(9):1237–1250. [DOI] [PubMed] [Google Scholar]
- 53.Ye W, Liang F, Ying C, et al. Downregulation of microRNA-3934–5p induces apoptosis and inhibits the proliferation of neuroblastoma cells by targeting TP53INP1. Exp Ther Med. 2019. November;18(5):3729–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han LL, Zhou XJ, Li FJ, et al. MiR-223–3p promotes the growth and invasion of neuroblastoma cell via targeting FOXO1. Eur Rev Med Pharmacol Sci. 2019. October;23(20):8984–8990. [DOI] [PubMed] [Google Scholar]
- 55.Ramraj SK, Aravindan S, Somasundaram DB, et al. Serum-circulating miRNAs predict neuroblastoma progression in mouse model of high-risk metastatic disease. Oncotarget. 2016. April 5;7(14):18605–18619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gholamin S, Mirzaei H, Razavi SM, et al. GD2-targeted immunotherapy and potential value of circulating microRNAs in neuroblastoma. J Cell Physiol. 2018. February;233(2):866–879. [DOI] [PubMed] [Google Scholar]
- 57.Mohammadniaei M, Yoon J, Choi HK, et al. Multifunctional nanobiohybrid material composed of Ag@Bi2Se3/RNA three-way junction/miRNA/retinoic acid for neuroblastoma differentiation. ACS Appl Mater Interfaces. 2019. March 6;11(9):8779–8788. [DOI] [PubMed] [Google Scholar]
- 58.Curtin C, Nolan JC, Conlon R, et al. A physiologically relevant 3D collagen-based scaffold-neuroblastoma cell system exhibits chemosensitivity similar to orthotopic xenograft models. Acta Biomater. 2018. April 1;70:84–97. [DOI] [PubMed] [Google Scholar]
- 59.Saeki N, Saito A, Sugaya Y, et al. Indirect down-regulation of tumor-suppressive let-7 family microRNAs by LMO1 in neuroblastoma. Cancer Genomics Proteomics. 2018. Sep-Oct;15(5):413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roth SA, Hald OH, Fuchs S, et al. MicroRNA-193b-3p represses neuroblastoma cell growth via downregulation of cyclin D1, MCL-1 and MYCN. Oncotarget. 2018. April 6;9(26):18160–18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu T, Lin Y, Xie Z. MicroRNA-1247 inhibits cell proliferation by directly targeting ZNF346 in childhood neuroblastoma. Biol Res. 2018. May 24;51(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mao F, Zhang J, Cheng X, et al. miR-149 inhibits cell proliferation and enhances chemosensitivity by targeting CDC42 and BCL2 in neuroblastoma. Cancer Cell Int. 2019;19:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soriano A, Masanas M, Boloix A, et al. Functional high-throughput screening reveals miR-323a-5p and miR-342–5p as new tumor-suppressive microRNA for neuroblastoma. Cell Mol Life Sci. 2019. June;76(11):2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao J, Zhou K, Ma L, et al. MicroRNA-145 overexpression inhibits neuroblastoma tumorigenesis in vitro and in vivo. Bioengineered. 2020. December;11(1):219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bhavsar SP, Lokke C, Flaegstad T, et al. Hsa-miR-376c-3p targets Cyclin D1 and induces G1-cell cycle arrest in neuroblastoma cells. Oncol Lett. 2018. November;16(5):6786–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Li J, Xu X, et al. miR-129 inhibits tumor growth and potentiates chemosensitivity of neuroblastoma by targeting MYO10. Biomed Pharmacother. 2018. July;103:1312–1318. [DOI] [PubMed] [Google Scholar]
- 67.Li SH, Li JP, Chen L, et al. miR-146a induces apoptosis in neuroblastoma cells by targeting BCL11A. Med Hypotheses. 2018. August;117:21–27. [DOI] [PubMed] [Google Scholar]
- 68.Zhou X, Lu H, Li F, et al. MicroRNA-429 inhibits neuroblastoma cell proliferation, migration and invasion via the NF-kappaB pathway. Cell Mol Biol Lett. 2020;25:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marengo B, Monti P, Miele M, et al. Etoposide-resistance in a neuroblastoma model cell line is associated with 13q14.3 mono-allelic deletion and miRNA-15a/16–1 down-regulation. Sci Rep. 2018. September 13;8(1):13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Z, Shelton SD, Oviedo A, et al. The PLAGL2/MYCN/miR-506–3p interplay regulates neuroblastoma cell fate and associates with neuroblastoma progression. J Exp Clin Cancer Res. 2020. February 22;39(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chava S, Reynolds CP, Pathania AS, et al. miR-15a-5p, miR-15b-5p, and miR-16–5p inhibit tumor progression by directly targeting MYCN in neuroblastoma. Mol Oncol. 2020. January;14(1):180–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharif S, Ghahremani MH, Soleimani M. Induction of morphological and functional differentiation of human neuroblastoma cells by miR-124. J Biosci. 2017. December;42(4):555–563. [DOI] [PubMed] [Google Scholar]
- 73.You Q, Gong Q, Han YQ, et al. Role of miR-124 in the regulation of retinoic acid-induced neuro-2A cell differentiation. Neural Regen Res. 2020. June;15(6):1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramachandran M, Yu D, Dyczynski M, et al. Safe and effective treatment of experimental neuroblastoma and glioblastoma using systemically delivered triple microRNA-detargeted oncolytic semliki forest virus. Clin Cancer Res. 2017. March 15;23(6):1519–1530. [DOI] [PubMed] [Google Scholar]
- 75.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010. September 30;363(14):1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szanto CL, Cornel AM, Vijver SV, et al. Monitoring immune responses in neuroblastoma patients during therapy. Cancers (Basel). 2020. February 24;12(2). DOI: 10.3390/cancers12020519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Casey DL, Cheung NV. Immunotherapy of pediatric solid tumors: treatments at a crossroads, with an emphasis on antibodies. Cancer Immunol Res. 2020. February;8(2):161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kholodenko IV, Kalinovsky DV, Doronin II, et al. Neuroblastoma origin and therapeutic targets for immunotherapy. J Immunol Res. 2018;2018:7394268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Terzic T, Cordeau M, Herblot S, et al. Expression of disialoganglio-side (gd2) in neuroblastic tumors: a prognostic value for patients treated with anti-GD2 immunotherapy. Pediatr Dev Pathol. 2018. Jul-Aug;21(4):355–362. [DOI] [PubMed] [Google Scholar]
- 80.Pennington B, Ren S, Barton S, et al. Dinutuximab beta for treating neuroblastoma: an evidence review group and decision support unit perspective of a NICE single technology appraisal. Pharmacoeconomics. 2019. August;37(8):985–993. [DOI] [PubMed] [Google Scholar]
- 81.Ladenstein R, Potschger U, Valteau-Couanet D, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018. December;19(12):1617–1629. [DOI] [PubMed] [Google Scholar]
- 82.Greenwood KL, Foster JH. The safety of dinutuximab for the treatment of pediatric patients with high-risk neuroblastoma. Expert Opin Drug Saf. 2018. December;17(12):1257–1262. [DOI] [PubMed] [Google Scholar]
- 83.Ladenstein R, Potschger U, Valteau-Couanet D, et al. Investigation of the role of dinutuximab beta-based immunotherapy in the SIOPEN high-risk neuroblastoma 1 trial (HR-NBL1). Cancers (Basel). 2020. January 28;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Furman WL, Federico SM, McCarville MB, et al. A phase II trial of Hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin Cancer Res. 2019. November 1;25(21):6320–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.London CA, Gardner HL, Rippy S, et al. KTN0158, a humanized anti-KIT monoclonal antibody, demonstrates biologic activity against both normal and malignant canine mast cells. Clin Cancer Res. 2017. May 15;23(10):2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stahl M, Gedrich R, Peck R, et al. Targeting KIT on innate immune cells to enhance the antitumor activity of checkpoint inhibitors. Immunotherapy. 2016. June;8(7):767–774. [DOI] [PubMed] [Google Scholar]
- 87.Garton AJ, Seibel S, Lopresti-Morrow L, et al. Anti-KIT monoclonal antibody treatment enhances the antitumor activity of immune checkpoint inhibitors by reversing tumor-induced immunosuppression. Mol Cancer Ther. 2017. April;16(4):671–680. [DOI] [PubMed] [Google Scholar]
- 88.Tomolonis JA, Agarwal S, Shohet JM. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. 2018. May;372(2):245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zage PE, Whittle SB, Shohet JM. CD114: a new member of the neural crest-derived cancer stem cell marker family. J Cell Biochem. 2017. February;118(2):221–231. [DOI] [PubMed] [Google Scholar]
- 90.Schlitter AM, Dorneburg C, Barth TF, et al. CD57(high) neuroblastoma cells have aggressive attributes ex situ and an undifferentiated phenotype in patients. PLoS One. 2012;7(8):e42025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szemes M, Greenhough A, Melegh Z, et al. Wnt signalling drives context-dependent differentiation or proliferation in neuroblastoma. Neoplasia. 2018. April;20(4):335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tian X, Zhou D, Chen L, et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis. 2018. January 19;9(2):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Semina EV, Rubina KA, Shmakova AA, et al. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J Cell Physiol. 2020. January;235(9):6268–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rysenkova KD, Semina EV, Karagyaur MN, et al. CRISPR/Cas9 nickase mediated targeting of urokinase receptor gene inhibits neuroblastoma cell proliferation. Oncotarget. 2018. June 29;9 (50):29414–29430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vega FM, Colmenero-Repiso A, Gomez-Munoz MA, et al. CD44-high neural crest stem-like cells are associated with tumour aggressiveness and poor survival in neuroblastoma tumours. EBioMedicine. 2019. November;49:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guo YF, Duan JJ, Wang J, et al. Inhibition of the ALDH18A1-MYCN positive feedback loop attenuates MYCN-amplified neuroblastoma growth. Sci Transl Med. 2020. February 19;12:531. [DOI] [PubMed] [Google Scholar]
- 97.Prieto-Vila M, Takahashi RU, Usuba W, et al. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci. 2017. December 1;18 (12). DOI: 10.3390/ijms18122574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Phi LTH, Sari IN, Yang YG, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang XH, Wu HY, Gao J, et al. IGF1R facilitates epithelial-mesenchymal transition and cancer stem cell properties in neuroblastoma via the STAT3/AKT axis. Cancer Manag Res. 2019;11:5459–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goncalves JM, Silva CAB, Rivero ERC, et al. Inhibition of cancer stem cells promoted by pimozide. Clin Exp Pharmacol Physiol. 2019. February;46(2):116–125. [DOI] [PubMed] [Google Scholar]
- 101.He K, Duan G, Li Y. Dehydroeffusol inhibits viability and epithelial-mesenchymal transition through the hedgehog and Akt/mTOR signaling pathways in neuroblastoma cells. Eur J Pharmacol. 2018. June;15(829):93–101. [DOI] [PubMed] [Google Scholar]
- 102.Oh J-M, Lee J, Im W-T, et al. Ginsenoside Rk1 induces apoptosis in neuroblastoma cells through loss of mitochondrial membrane potential and activation of caspases. Int J Mol Sci. 2019. March 11;20 (5):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Campos-Arroyo D, Maldonado V, Bahena I, et al. Probenecid sensitizes neuroblastoma cancer stem cells to cisplatin. Cancer Invest. 2016;34(3):155–166. [DOI] [PubMed] [Google Scholar]
- 104.Kim KW, Kim JY, Qiao J, et al. Dual-targeting AKT2 and ERK in cancer stem-like cells in neuroblastoma. Oncotarget. 2019. September 24;10(54):5645–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bahmad HF, Mouhieddine TH, Chalhoub RM, et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget. 2018. September 11;9(71):33549–33561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Almstedt E, Elgendy R, Hekmati N, et al. Integrative discovery of treatments for high-risk neuroblastoma. Nat Commun. 2020. January 3;11(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kolb EA, Houghton PJ, Kurmasheva RT, et al. Preclinical evaluation of the combination of AZD1775 and irinotecan against selected pediatric solid tumors: a pediatric preclinical testing consortium report. Pediatr Blood Cancer. 2020;23:e28098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cole KA, Pal S, Kudgus RA, et al. Phase I clinical trial of the wee1 inhibitor adavosertib (AZD1775) with irinotecan in children with relapsed solid tumors. A COG phase I consortium report (ADVL1312). Clin Cancer Res. 2019. December;26(6):1213–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chuang HC, Lin HY, Liao PL, et al. Immunomodulator polyinosinic-polycytidylic acid enhances the inhibitory effect of 13-cis-retinoic acid on neuroblastoma through a TLR3-related immunogenic-apoptotic response. Lab Invest. 2020. April;100 (4):606–618. [DOI] [PubMed] [Google Scholar]
- 110.Ruano D, Lopez-Martin JA, Moreno L, et al. First-in-human, first-in-child trial of autologous MSCs carrying the oncolytic virus icovir-5 in patients with advanced tumors. Mol Ther. 2020. January;284:1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Franco-Luzon L, Gonzalez-Murillo A, Alcantara-Sanchez C, et al. Systemic oncolytic adenovirus delivered in mesenchymal carrier cells modulate tumor infiltrating immune cells and tumor micro-environment in mice with neuroblastoma. Oncotarget. 2020. January 28;11(4):347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maniwa J, Fumino S, Kimura K, et al. Novel mesenchymal stem cell delivery system as targeted therapy against neuroblastoma using the TH-MYCN mouse model. J Pediatr Surg. 2019. December;54 (12):2600–2605. [DOI] [PubMed] [Google Scholar]
- 113.Anongpornjossakul Y, Sriwatcharin W, Thamnirat K, et al. Iodine 131 metaiodobenzylguanidine (131I-mIBG) treatment in relapsed/refractory neuroblastoma. Nucl Med Commun. 2020. April;41 (4):336–343. [DOI] [PubMed] [Google Scholar]
- 114.Olecki E, Grant CN. MIBG in neuroblastoma diagnosis and treatment. Semin Pediatr Surg. 2019. December;28(6):150859. [DOI] [PubMed] [Google Scholar]
- 115.Suh JK, Koh KN, Min SY, et al. Feasibility and effectiveness of treatment strategy of tandem high-dose chemotherapy and autologous stem cell transplantation in combination with (131) I-MIBG therapy for high-risk neuroblastoma. Pediatr Transplant. 2020. March;24(2):e13658. [DOI] [PubMed] [Google Scholar]
- 116.Cervantes-Madrid D, Szydzik J, Lind DE, et al. Repotrectinib (TPX-0005), effectively reduces growth of ALK driven neuroblastoma cells. Sci Rep. 2019. December 18;9(1):19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yoda H, Nakayama T, Miura M, et al. Vitamin K3 derivative induces apoptotic cell death in neuroblastoma via downregulation of MYCN expression. Biochem Biophys Rep. 2019;20:100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fang E, Wang X, Yang F, et al. Therapeutic targeting of MZF1-AS1/PARP1/E2F1 axis inhibits proline synthesis and neuroblastoma progression. Adv Sci (Weinh). 2019. October 2;6(19):1900581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li H, Yang F, Hu A, et al. Therapeutic targeting of circ-CUX1/EWSR1/MAZ axis inhibits glycolysis and neuroblastoma progression. EMBO Mol Med. 2019. December;11(12):e10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Das SK, Maji S, Wechman SL, et al. MDA-9/syntenin (SDCBP): novel gene and therapeutic target for cancer metastasis. Pharmacol Res. 2020. February;13:104695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bhoopathi P, Pradhan AK, Bacolod MD, et al. Regulation of neuroblastoma migration, invasion, and in vivo metastasis by genetic and pharmacological manipulation of MDA-9/syntenin. Oncogene. 2019. October;38(41):6781–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lopez de la Oliva AR, Campos-Sandoval JA, Gomez-Garcia MC, et al. Nuclear translocation of glutaminase GLS2 in human cancer cells associates with proliferation arrest and differentiation. Sci Rep. 2020. February 10;10(1):2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bibbo S, Lamolinara A, Capone E, et al. Repurposing a psychoactive drug for children with cancer: p27(Kip1)-dependent inhibition of metastatic neuroblastomas by prozac. Oncogenesis. 2020. January 2;9(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen S, Gu S, Xu M, et al. Kruppel-like factor 9 promotes neuroblastoma differentiation via targeting the sonic hedgehog signaling pathway. Pediatr Blood Cancer. 2020. March;67(3): e28108. [DOI] [PubMed] [Google Scholar]
- 125.Wang F, Zhang J, Ke X, et al. WDR5-Myc axis promotes the progression of glioblastoma and neuroblastoma by transcriptional activating CARM1. Biochem Biophys Res Commun. 2020. March 12;523(3):699–706. [DOI] [PubMed] [Google Scholar]
- 126.Fang E, Wang X, Wang J, et al. Therapeutic targeting of YY1/MZF1 axis by MZF1-uPEP inhibits aerobic glycolysis and neuroblastoma progression. Theranostics. 2020;10(4):1555–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vrenken KS, Vervoort BMT, van Ingen Schenau DS, et al. The transcriptional repressor SNAI2 impairs neuroblastoma differentiation and inhibits response to retinoic acid therapy. Biochim Biophys Acta Mol Basis Dis. 2020. March 1;1866(3):165644. [DOI] [PubMed] [Google Scholar]
- 128.Po’uha ST, Le Grand M, Brandl MB, et al. Stathmin levels alter PTPN14 expression and impact neuroblastoma cell migration. Br J Cancer. 2020. February;122(3):434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Meng J, Tagalakis AD, Hart SL. Silencing E3 ubiqutin ligase ITCH as a potential therapy to enhance chemotherapy efficacy in p53 mutant neuroblastoma cells. Sci Rep. 2020. January 23;10(1):1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tilve S, Iweka CA, Bao J, et al. Phospholipid phosphatase related 1 (PLPPR1) increases cell adhesion through modulation of Rac1 activity. Exp Cell Res. 2020. April 15;389(2):111911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.MacFarland SP, Naraparaju K, Iyer R, et al. Mechanisms of entrectinib resistance in a neuroblastoma xenograft model. Mol Cancer Ther. 2020. March;19(3):920–926. [DOI] [PubMed] [Google Scholar]
- 132.Cimmino F, Avitabile M, Lasorsa VA, et al. Functional characterization of full-length BARD1 strengthens its role as a tumor suppressor in neuroblastoma. J Cancer. 2020;11(6):1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coggins GE, Farrel A, Rathi KS, et al. YAP1 mediates resistance to MEK1/2 inhibition in neuroblastomas with hyperactivated RAS signaling. Cancer Res. 2019. December 15;79(24):6204–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shen X, Xu X, Xie C, et al. YAP promotes the proliferation of neuroblastoma cells through decreasing the nuclear location of p27(Kip1) mediated by Akt. Cell Prolif. 2020. February;53(2):e12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang M, Zeki J, Sumarsono N, et al. Epigenetic targeting of TERT-associated gene expression signature in human neuroblastoma with TERT overexpression. Cancer Res. 2020. March 1;80(5):1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tee AE, Ciampa OC, Wong M, et al. Combination therapy with the CDK7 inhibitor and the tyrosine kinase inhibitor exerts synergistic anticancer effects against MYCN-amplified neuroblastoma. Int J Cancer. 2020. February 22 DOI: 10.1002/ijc.32936 [DOI] [PubMed] [Google Scholar]
- 137.Wang L, Tan TK, Durbin AD, et al. ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat Commun. 2019. December 9;10(1):5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cournoyer S, Addioui A, Belounis A, et al. GX15–070 (Obatoclax), a Bcl-2 family proteins inhibitor engenders apoptosis and pro-survival autophagy and increases chemosensitivity in neuroblastoma. BMC Cancer. 2019. October 29;19(1):1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Almutairi B, Charlet J, Dallosso AR, et al. Epigenetic deregulation of GATA3 in neuroblastoma is associated with increased GATA3 protein expression and with poor outcomes. Sci Rep. 2019. December 12;9(1):18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trigg RM, Lee LC, Prokoph N, et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat Commun. 2019. November 28;10(1):5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ognibene M, Podesta M, Garaventa A, et al. Role of GOLPH3 and TPX2 in neuroblastoma DNA damage response and cell resistance to chemotherapy. Int J Mol Sci. 2019. September 25;20:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hsieh CH, Cheung CHY, Liu YL, et al. Quantitative proteomics of Th-MYCN transgenic mice reveals aurora kinase inhibitor altered metabolic pathways and enhanced ACADM to suppress neuroblastoma progression. J Proteome Res. 2019. November 1;18(11):3850–3866. [DOI] [PubMed] [Google Scholar]
- 143.Nunes-Xavier CE, Aurtenetxe O, Zaldumbide L, et al. Protein tyrosine phosphatase PTPN1 modulates cell growth and associates with poor outcome in human neuroblastoma. Diagn Pathol. 2019. December 14;14(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xu Y, Chen K, Cai Y, et al. Overexpression of Rad51 predicts poor prognosis and silencing of Rad51 increases chemo-sensitivity to doxorubicin in neuroblastoma. Am J Transl Res. 2019;11(9):5788–5799. [PMC free article] [PubMed] [Google Scholar]
- 145.Boratyn E, Nowak I, Karnas E, et al. MCPIP1 overexpression in human neuroblastoma cell lines causes cell-cycle arrest by G1/S checkpoint block. J Cell Biochem. 2020. June;121(5–6):3406–3425. [DOI] [PubMed] [Google Scholar]
- 146.Su D, Wang W, Wu X, et al. Meriolin1 induces cell cycle arrest, apoptosis, autophagy and targeting the Akt/MAPKs pathways in human neuroblastoma SH-SY5Y cells. J Pharm Pharmacol. 2020. February 7;72(4):561–574. [DOI] [PubMed] [Google Scholar]
- 147.Montemurro L, Raieli S, Angelucci S, et al. A novel MYCN-specific antigene oligonucleotide deregulates mitochondria and inhibits tumor growth in MYCN-amplified neuroblastoma. Cancer Res. 2019. December 15;79(24):6166–6177. [DOI] [PubMed] [Google Scholar]
- 148.Flynn SM, Lesperance J, Macias A, et al. The multikinase inhibitor RXDX-105 is effective against neuroblastoma in vitro and in vivo. Oncotarget. 2019. October 29;10(59):6323–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cong S, Luo H, Li X, et al. Isatin inhibits SH-SY5Y neuroblastoma cell invasion and metastasis through PTEN signaling. Int J Clin Exp Pathol. 2019;12(7):2446–2454. [PMC free article] [PubMed] [Google Scholar]
- 150.Otsuka K, Sasada M, Iyoda T, et al. Combining peptide TNIIIA2 with all-trans retinoic acid accelerates N-Myc protein degradation and neuronal differentiation in MYCN-amplified neuroblastoma cells. Am J Cancer Res. 2019;9(2):434–448. [PMC free article] [PubMed] [Google Scholar]
- 151.Dar MI, Jan S, Reddy GL, et al. Differentiation of human neuroblastoma cell line IMR-32 by sildenafil and its newly discovered analogue IS00384. Cell Signal. 2020. January;65:109425. [DOI] [PubMed] [Google Scholar]
- 152.Kumar A, Ghosh DK, Ranjan A. Mefloquine binding to human acyl-CoA binding protein leads to redox stress-mediated apoptotic death of human neuroblastoma cells. Neurotoxicology. 2020. March;77:169–180. [DOI] [PubMed] [Google Scholar]
- 153.Huynh T, Murray J, Flemming CL, et al. CCI52 sensitizes tumors to 6-mercaptopurine and inhibits MYCN-amplified tumor growth. Biochem Pharmacol. 2020. February;172:113770. [DOI] [PubMed] [Google Scholar]
- 154.Aravindan S, Natarajan M, Awasthi V, et al. Novel synthetic mono-ketone transmute radiation-triggered NFkappaB-dependent TNFalpha cross-signaling feedback maintained NFkappaB and favors neuroblastoma regression. PLoS One. 2013;8(8):e72464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aravindan N, Veeraraghavan J, Madhusoodhanan R, et al. Curcumin regulates low-linear energy transfer gamma-radiation-induced NFkappaB-dependent telomerase activity in human neuroblastoma cells. Int J Radiat Oncol Biol Phys. 2011. March 15;79(4):1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aravindan N, Madhusoodhanan R, Ahmad S, et al. Curcumin inhibits NFkappaB mediated radioprotection and modulate apoptosis related genes in human neuroblastoma cells. Cancer Biol Ther. 2008. April;7(4):569–576. [DOI] [PubMed] [Google Scholar]
- 157.Caruso Bavisotto C, Marino Gammazza A, Lo Cascio F, et al. Curcumin affects HSP60 folding activity and levels in neuroblastoma cells. Int J Mol Sci. 2020. January 19;21:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alshangiti AM, Tuboly E, Hegarty SV, et al. 4-hydroxychalcone induces cell death via oxidative stress in MYCN-amplified human neuroblastoma cells. Oxid Med Cell Longev. 2019;2019:1670759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yang J, Shao X, Wang L, et al. Angelica polysaccharide exhibits antitumor effect in neuroblastoma cell line SH-SY5Y by up-regulation of miR-205. Biofactors. 2019. November 23 DOI: 10.1002/biof.1586 [DOI] [Google Scholar]
- 160.Kanamori Y, Via LD, Macone A, et al. Aged garlic extract and its constituent, S-allyl-L-cysteine, induce the apoptosis of neuroblastoma cancer cells due to mitochondrial membrane depolarization. Exp Ther Med. 2020. February;19(2):1511–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pottoo FH, Barkat MA, Harshita H, et al. Nanotechnological based miRNA intervention in the therapeutic management of neuroblastoma. Semin Cancer Biol. 2019. September 25 DOI: 10.1016/j.semcancer.2019.09.017 [DOI] [PubMed] [Google Scholar]
- 162.Li F, Song L, Yang X, et al. Anticancer and genotoxicity effect of (Clausena lansium (Lour.) skeels) peel ZnONPs on neuroblastoma (SH-SY5Y) cells through the modulation of autophagy mechanism. Journal of photochemistry and photobiology B. Biology (Basel) 2020;203:111748. [DOI] [PubMed] [Google Scholar]
- 163.Piazzini V, Vasarri M, Degl’Innocenti D, et al. Comparison of chit-osan nanoparticles and soluplus micelles to optimize the bioactivity of posidonia oceanica extract on human neuroblastoma cell migration. Pharmaceutics. 2019. December 6;11:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Orienti I, Nguyen F, Guan P, et al. A novel nanomicellar combination of fenretinide and lenalidomide shows marked antitumor activity in a neuroblastoma xenograft model. Drug Des Devel Ther. 2019;13:4305–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yoshida S, Duong C, Oestergaard M, et al. MXD3 antisense oligonucleotide with superparamagnetic iron oxide nanoparticles: a new targeted approach for neuroblastoma. Nanomedicine. 2019. November 26;24:102127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Luo Y, Xu T, Xie HQ, et al. Effects of 2,3,7,8-tetrachlorodibenzop-dioxin on spontaneous movement of human neuroblastoma cells. Sci Total Environ. 2020. May 1;715:136805. [DOI] [PubMed] [Google Scholar]
- 167.Fu SC, Liu JM, Lee KI, et al. Cr(VI) induces ROS-mediated mitochondrial-dependent apoptosis in neuronal cells via the activation of Akt/ERK/AMPK signaling pathway. Toxicol In Vitro. 2020. February;12(65):104795. [DOI] [PubMed] [Google Scholar]