

Abstract
Rationale -
There is growing evidence that common variants and rare sequence alterations in regulatory sequences can result in birth defects or predisposition to disease. Congenital heart defects (CHDs) are the most common birth defect and have a clear genetic component, yet only a third of cases can be attributed to structural variation in the genome or a mutation in a gene. The remaining unknown cases could be caused by alterations in regulatory sequences.
Objective -
Identify regulatory sequences and gene expression networks that are active during organogenesis of the human heart. Determine if these sites and networks are enriched for disease relevant genes and associated genetic variation.
Methods and Results -
We characterized chromatin state and gene expression dynamics during human heart organogenesis. We profiled seven histone modifications in embryonic hearts from each of nine distinct Carnegie stages (CS13–14, CS16–21, and CS23), annotated chromatin states, and compared these maps to over 100 human tissues and cell types. We also generated RNA-seq data, performed differential expression, and constructed weighted gene co-expression networks. We identified 177,412 heart enhancers, 12,395 had not been previously annotated as strong enhancers. We identified 92% of all functionally validated heart positive enhancers (n=281, 7.5x enrichment, p<2.2×10−16). Integration of these data demonstrated novel heart enhancers are enriched near genes expressed more strongly in cardiac tissue and are enriched for variants associated with electrocardiogram measures and atrial fibrillation. Our gene expression network analysis identified gene modules strongly enriched for heart related functions, regulatory control by heart specific enhancers, and putative disease genes.
Conclusions -
Well-connected “hub” genes with heart-specific expression targeted by embryonic heart-specific enhancers are likely disease candidates. Our functional annotations will allow for better interpretation of whole genome sequencing data in the large number of patients affected by CHDs.
Keywords: Developmental enhancer, epigenomics, transcriptomics, regulatory network, genetics, genomics, developmental biology, congenital cardiac defect, gene regulation
Subject Terms: Basic Science Research; Epigenetics; Functional Genomics; Gene Expression and Regulation; Genetic, Association Studies
Graphical Abstract
INTRODUCTION
Congenital heart defects (CHDs) are among the most common birth defects, affecting approximately 1% of live births worldwide, and remain the leading cause of infant mortality in developed nations.1–3 Despite evidence suggesting a strong genetic component, nearly 60% of CHD cases remain unexplained.4,5 Environmental causes, familial forms of CHD and de novo damaging mutations in over 400 genes each explain less than 10% of cases, while chromosomal abnormalities including aneuploidies and large structural variations are implicated in over 20% of CHD.6–10 These findings suggest that CHD arises through combinations of otherwise benign mutations in a large number of genes, unappreciated genetic-environmental interactions or disruption of regulatory sequences that control heart development. There are indications that regulatory regions are causative for CHDs. Patients homozygous for rare variation in a heart-specific regulatory sequence controlling the cardiac transcription factor TBX5 have isolated CHD.11 However, the extent to which developmental regulatory sequences could systematically contribute to CHD has not been explored.
Recent analysis of gene expression from the heart at multiple stages of human development indicates the dynamics of gene expression occur primarily during the embryonic period of human development or the first eight post-conception weeks, aligning well with known structural and functional changes in the developing heart.5,12,13 Due to the limited nature of embryonic and fetal tissues and difficulty with performing functional genomics on small amounts of primary material, several groups have employed directed differentiation approaches in human and mouse stem cells to model cardiogenesis, providing insight into global binding of multiple cardiac transcription factors (TFs), conserved cardiac regulatory networks and cooperativity amongst these specific TFs.14–18 However, these data have not been systematically compared to human cardiomyocytes and it remains unclear how faithfully these systems reflect normal human heart development at the gene regulatory level. Regulatory annotation of the human genome from the ENCODE Project and Roadmap Epigenome includes primary tissues of the human heart. However, these data are exclusively from fetal and adult stages after these major molecular and morphological changes have occurred. These two large consortia employed ChIP-seq of several histone modifications in combination with machine learning approaches in over one hundred tissues and cell types.19,20 This approach resulted in the learning of several Hidden Markov models of chromatin state (ChromHMM) using the combined tissue dataset.
The number of states, and therefore the complexity of the model, depends upon the histone modifications used. A 15-state model can be learned from the histone modifications H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3 (frequently associated with active enhancers, active promoters, stable heterochromatin, facultative heterochromatin and active transcription, respectively). When H3K27ac (associated with active chromatin with functions ranging from active transcription to active enhancers) is included, an increasingly complex model of 18 different chromatin states can be called. The addition of five other histone modifications (H2A.Z, H3K4me2, H3K9ac, H3K79me2, H4K20me1) and DNA accessibility (DNAse hypersensitivity) provide the ability to call 25 chromatin states. Through the use of such an ensemble, multi-tissue approach various types of active and repressed features are mapped onto the genome and cell type- or stage-specific enhancers can be readily distinguished.
Here we describe the systematic characterization of chromatin states from human embryonic hearts at 4–8 post-conception weeks using ChromHMM in a manner that, allows for direct comparison with over 100 tissues profiled by Roadmap Epigenome. We are able to confirm previously validated in vivo heart enhancers and identify thousands of previously unknown embryonic heart-specific regulatory sequences. Integration of chromatin and gene expression through co-expression analysis identified groups of genes that are coordinately expressed during early heart development and likely regulated by these heart specific enhancers. Our multilayered analysis also predicts new genes to be involved in CHD that warrant further study. These resources are available from the Genome Expression Omibus, as a public track hub on the UCSC Genome Browser, or directly from our laboratory website (https://cotney.research.uchc.edu/heart).
METHODS
Data Availability.
The authors declare that anonymized data have been made publicly available at the Gene Expression Omnibus (GEO) and can be accessed at via accession numbers GSE137731 (ChIP-Seq) and GSE138799 (RNA-Seq). Because of the sensitive nature of the raw sequencing data collected for this study, requests to access those data from qualified researchers trained in human subject confidentiality protocols and approved by an Institutional Review Board may be sent to the corresponding author. All other supporting data and materials presented within this article and in the Data Supplement are available from the corresponding author upon reasonable request.
Human Tissue Samples
Use of human embryonic tissue was reviewed and approved by the Human Subjects Protection Program at UConn Health (UCHC 710-2-13-14-03). Human embryonic heart tissue was collected, staged and provided by the Joint MRC/Wellcome Trust Human Developmental Biology Resource (www.hdbr.org). Tissues were flash frozen upon collection and stored at −80 °C. Hearts were homogenized by mechanical disruption and divided between samples for RNA-seq and ChIP-seq. For ChIP-seq, tissues were fixed by incubation in 1% formaldehyde for 15 minutes at room temperature with agitation before being quenched by addition of 2.5M glycine to a concentration of 150mM with rotation/agitation for 10 minutes. For RNA-Seq homogenized tissue was added to Qiazol (Qiagen) and flash frozen.
A detailed description of the methods and analysis is provided in Expanded Materials and Methods in the Data Supplement.
For a complete listing of all software, public datasets, and relevant materials please see the Major Resources Table in the Data Supplement.
RESULTS
Chromatin state profiling of human embryonic heart development
As demonstrated by Roadmap Epigenome, ensemble approaches using many different biochemical markers are required to more completely characterize how the genome is utilized in a given biological condition. In order to systematically profile chromatin states of the developing human embryonic heart, we first employed chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) using antibodies against seven histone H3 post translational modifications on individual human hearts from 18 embryos spanning the organogenesis phase of heart development (Figure 1A; Online Figure I; Online Table I). Overall these raw data were of high quality with high correlation between experiments performed with the same antibody across separate individual embryos and generally clustered by marks associated with genome activation (H3K4me1,H3K4me2,H3K4me3,H3K27ac, and H3K36me3) versus repression (H3K9me3 and H3K27me3) (Figure 1B and Online Figure IIA). We uniformly processed these data to allow direct comparison of 127 human cell types profiled by Roadmap Epigenome and 21 human developing craniofacial samples. We then employed an imputation approach to generate a complete set of 12 global epigenomic signals for each sample using ChromImpute as previously described by Roadmap Epigenome and employed by our group for human craniofacial development.19,21 The imputed signals correlated well with primary signals and predominantly clustered by known biological function as was observed in Roadmap Epigenome and human craniofacial epigenomic data (Figure 1B and Online Figure IIB).
Figure 1. Epigenomic and transcriptomic profiling of human embryonic heart development.
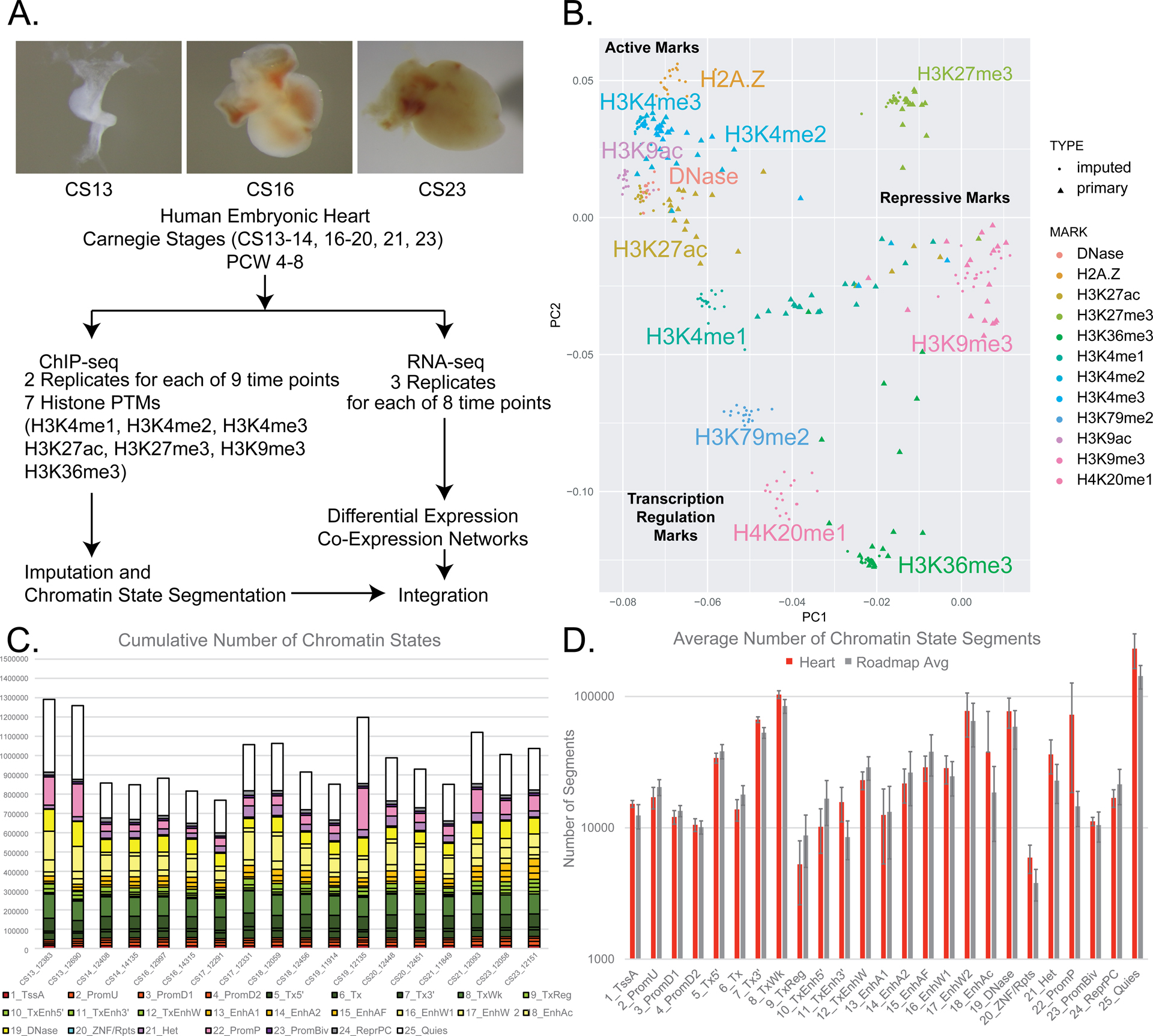
A. Top panels show representative images of primary human embryonic heart tissue at indicated Carnegie Stages. Lower panel indicates data types collects and downstream analyses performed in this study. B. Principal component analysis of genome-wide primary and imputed ChIP-Seq signals. Each mark is indicated by separate colors. Primary samples are shown as triangles and imputed data as circles. Grouping of marks and overall function are indicated in normal and bold text respectively. C. Total numbers of each chromatin state identified in segmentation of each individual embryonic tissue sample. Samples are ordered from left to right as earliest to latest timepoints. Legend of colors are located below using conventions defined by Roadmap Epigenome.19 D. Average numbers of each chromatin state for all heart samples (red) and all Roadmap Epigenome samples (grey) are shown. Error bars represent standard deviations for each chromatin state and tissue group.
Having uniform epigenomic datasets for each heart sample across the developmental series we then applied the previously generated 15, 18, and 25 state models of chromatin activity developed by Roadmap Epigenome to segment the genome into chromatin states. The individual state classifications and color coding for each model are provided in Online Figure III for easy reference. The number of segments identified for each of the 25 chromatin states was similar across all of our eighteen samples (Figure 1C). The pattern of chromatin state segments identified in our human embryonic heart samples were very similar to those identified in 127 tissues from Roadmap Epigenome, with the one exception of significantly increased numbers of poised promoter segments (state 22 from 25 state model) (Figure 1D). These findings suggest our imputation and segmentation approaches generated data globally similar to all other human tissues from Roadmap Epigenome allowing us to make direct comparisons of chromatin state utilization between tissue types and stages.
Genomic enrichments and biological features of individual chromatin states have been thoroughly explored in adult tissues, but have yet to be extensively explored during organogenesis.19,22 Two groups have recently published resources aimed at comprehensive identification of active regulatory sequences in heart development.23,24 However, they have profiled a limited set of active functional marks from tissue after human heart organogenesis has largely completed or applied a novel computational framework tuned for prediction of heart regulatory sequences that has not been extensively compared across human tissues or more generalized functional annotation tools.23–25 While these resources have both shown enrichment for in vivo developmental heart enhancers, they did not characterize other regulatory states such as repression. Therefore, we aimed to understand what the various chromatin states we have annotated mean biologically during heart development. To achieve this, we characterized structural and functional enrichments genome wide for each chromatin state and compared them to regions identified in a previously published compendium of heart enhancers and the EMERGE prediction framework.
We first characterized the positions of each chromatin state segment relative to known functional positions in the genome. We found as expected that segments of the genome annotated as state 1 from the 25 state model (1_TssA) previously classified as active transcription start sites (TSS) were located primarily within 10 base pairs of a TSS based on GENCODE version 25 annotation of the human genome (Online Figure IVA). Promoter associated states 2 through 4 (2_PromU, 3_PromD1, 4_PromD2) and bivalent promoter state 23 (PromBiv) were generally located within 1000 bp of known TSS. The remaining states from 5 to 21 were progressively more distant from known TSS with heterochromatic segment annotations (21_Het) being the most distant. Transcription associated states (5_Tx5, 6_Tx, 7_Tx3, 8_TxWk) and regulatory states located in introns of active genes (9_TxReg, 10_TxEnh5, 11_TxEnh3, 12_TxEnhW) were closer to TSS than other active regulatory states.
Amongst the seven putative enhancer states (13_EnhA1, 14_EnhA2, 15_EnhAF, 16_EnhW1, 17_EnhW2, 18_EnhAc, 19_Dnase) those segments of the genome annotated to be strong enhancer states (states 13 through 15) were closer to TSS than weak enhancer states (states 16, 17, and 19) with state 13 being systematically the closest to known TSS. State 18 (18_EnhAc), which was defined primarily by H3K27ac signals alone showed similar distributions of distances relative to TSS as other strong enhancer states (Online Figure IVA). Regions assembled in a compendium of heart enhancers by Dickel et al. showed similar distributions of distance to TSS as strong enhancer states 14, 15, and 18 but were more distant than state 13. These states were driven in the original Roadmap model by H3K27ac and H3K4me1 with the Dickel resource being heavily biases toward H3K27ac and its depositor P300. The strongest enhancer state, state 13, was identified in Roadmap Epigenome by many more features including presence of H3K27ac, H3K4me1, H3K4me2, and DNase and conversely the absence of H3K9me3, H3K27me3, H3K36me3, and H3K79me2, which are uniquely captured in our study. Interestingly, regulatory regions predicted by EMERGE showed a bimodal distribution with a significant fraction of regions located directly overlapping known TSS (Online Figure IVA).
When we analyzed the size of segments annotated as each chromatin state, we saw similar trends across all states with median values typically around 1 kilobase. Larger distributions were observed at heterochromatic regions (21_Het) and portions of the genome with no detectable activity or quiescent (25_Quies). When we analyzed the Dickel compendium and regions annotated by EMERGE we observed significant excursions from the values we observed for our chromatin state segments. The Dickel compendium identified uniformly larger regions than enhancer chromatin states identified from our data, while most regions identified by EMERGE were significantly smaller (Online Figure IVB).
Sequence conservation has been frequently used to identify functional regions of the genome particularly those with gene regulatory function in intronic or intergenic spaces of vertebrate genomes.26–29 Regions associated with active TSS or assigned active or bivalent promoter states were most conserved based on multi-species alignments from 100 vertebrate species (Online Figure IVC).30 Heterochromatic and polycomb repressed states were the least conserved amongst the 25 chromatin states. Amongst putative enhancer chromatin states, strong enhancer state 13, weak enhancer state 16, and DNase accessible regions state 19 were the most conserved. Median conservation scores of regions identified by Dickel et al. were comparable to weak enhancer state 16. Notably EMERGE regions showed amongst the lowest median conservation scores across all categories greater only than enhancer state 15.
While conservation is an indicator of selective pressure across species, a large portion of the human genome does not show significant conservation across all vertebrates and might be uniquely involved in human disease. Several groups have attempted to bridge this knowledge gap and developed machine learning techniques to predict deleteriousness of all 8.6 billion possible single nucleotide variants in the human genome.31–34 These approaches leverage many different functional genomics datasets, transcription factor binding sites predictions, and sequence conservation, among others and are informative for identifying regulatory variants linked to disease. When we interrogated our chromatin states with two of these scoring metrics (CADD and LINSIGHT), states 20 (ZNF_Repeats) and 21 (Heterochromatin) had the lowest median scores (Online Figures IVD and IVE). TSS and promoter associated states 1, 2, 3 and 23 had the highest scores predicted by these methods. Transcribed regions (states 5 through 8) despite being nearer TSS sites had uniformly depressed CADD and LINSIGHT scores. Strong enhancer state 13 had the highest median CADD and LINSIGHT scores amongst distal regulatory chromatin states, significantly higher than all other enhancer states and regions annotated by both Dickel et al. and EMERGE (Online Figures IVD and IVE).
Combined these results strongly corroborate the functional labels assigned to each of the chromatin states applied by Roadmap Epigenome. These systematic analyses revealed many expected distributions for chromatin states relative to known features of the genome and elevated sequence conservation amongst active regulatory regions. Our results also revealed a number of similarities between our annotations and previous databases of heart regulatory sequences. However, our enhancer chromatin states, particularly the strong categories, are significantly more conserved and harbor significantly more positions predicted to be involved in human disease than either of these two catalogs of putative heart regulatory sequences. While indicative of function, these metrics are largely generic and not necessarily related to the heart. Thus, we next aimed to understand the direct relevance of these chromatin state annotations for heart development and disease.
Identification of novel human embryonic heart enhancers.
Comparison of enhancers across many different tissues has shown them to be far more tissue-specific in their activation than promoters.19 To determine if the enhancer segments from our developing heart chromatin state segmentations are indeed tissue-specific, we aimed to examine analogous chromatin state annotations quantitatively across as many tissues as possible. To achieve this we first assembled all segments identified as an enhancer chromatin state based on the 25 state chromatin model in any tissues profiled by Roadmap Epigenome, this study, previously generated human craniofacial development samples, and chromatin data for nuclei isolated from fetal, infant, and adult cardiomyocytes.19,21,35 In our human embryonic heart samples alone, we identified a total of 177,412 segments that were reproducibly annotated as one of six putative enhancer states (States 13–18, EnhA1, EnhA2, EnhAF, EnhW1, EnhW2, and EnhAc) in at least two samples (Online Table II).
The chromatin state segmentations are powerful tools for genome-wide annotation, but they have limited utility on their own as the quantitative nature of the underlying data is discarded. To overcome this obstacle, we leveraged H3K27ac signals from each of these samples since it has been frequently shown to be highly tissue-specific in its distribution and generally associated with enhancer activation.19,36 Due to the overall better performance of normalized, imputed signals relative to primary ChIP-Seq data, we extracted imputed H3K27ac p-value signals from 174 samples at 444,413 enhancer segments across the genome.37 We found strongest global correlations between related tissue types, such as immune cell types, brain region tissues, and the embryonic heart samples. These data separated largely into adult versus embryonic groups and subsequently by tissue type (Figure 2A, Online Figure VA and VB). The embryonic heart samples clustered well with one another and also showed strong global correlation in H3K27ac signals with the fetal and adult heart tissue samples profiled by Roadmap Epigenome. Interestingly, the isolated cardiomyocyte nuclei formed their own distinct cluster and do not separate into adult versus developing samples. They also showed lower global correlation values with the heart tissues profiled by Roadmap Epigenome suggesting isolation of nuclei had significant effects on chromatin state and/or organization in those experiments. A tSNE projection of these data further confirmed these findings (Figure 2A) and showed the distinct nature of the isolated cardiomyocyte H3K27ac data (Online Figure VB). Given the potential for strong batch effects in the cardiomyocyte nuclei data due to isolation and sorting of nuclei prior to ChIP and the decreased correlation with fetal and adult heart samples we excluded these samples from downstream analysis. These results confirmed the early developmental stage identity and the cardiac origins of the tissues profiled in an unbiased, global fashion and further demonstrated the tissue-specific nature of the putative enhancer sequences and H3K27ac signals.
Figure 2. Multi-tissue comparisons of global enhancer activation and enrichment of heart phenotype in enhancer segments.
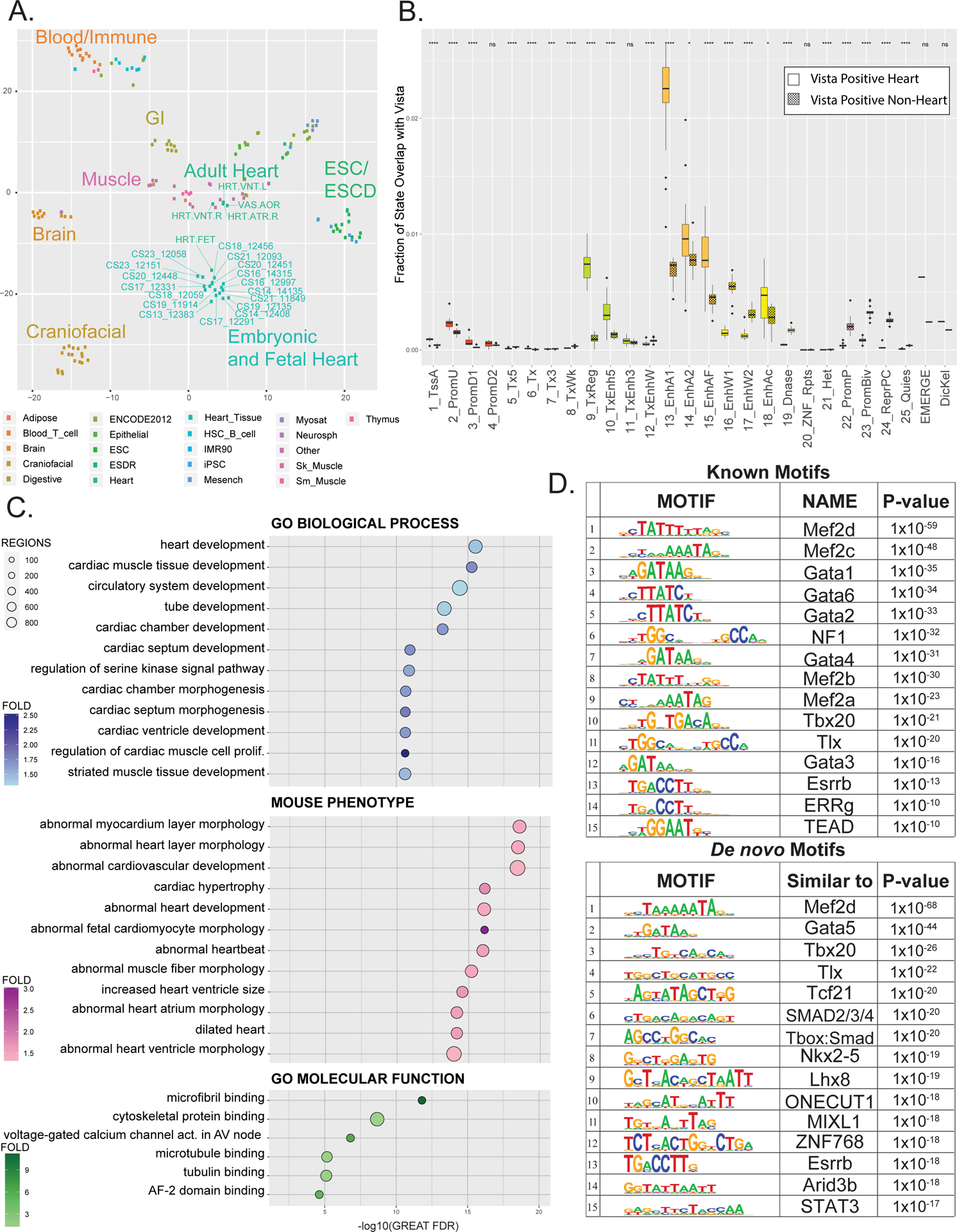
A. tSNE projection of imputed H3K27ac p-value signals at 444,413 enhancer segments from tissues profiled by Roadmap Epigenome and in this study. Dots are color coded by tissue as indicated and labelled as each individual tissue samples as profiled by Roadmap epigenome or in this study. B. Fraction of each of the 25 ChromHMM States, EMERGE, and Dickel datasets that overlap with either active heart enhancers (unshaded) or enhancers active in tissues other than heart (shaded) as tested by the Vista Enhancer Browser (enhancer.lbl.gov). Significance of difference of overlap between heart and other tissue were calculated using the Mann-Whitney test and is shown at top (p-value ≤ 0.05 = *, ≤ 0.01= **, ≤ 0.001= *** , ≤ 0.0001 = ****) C. Gene ontology enrichments for indicated functional categories for putative novel strong enhancer segments identified in human embryonic heart versus Roadmap Epigenome (n=12,395). Putative enhancers were assigned to genes and significance determined by GREAT. Position of each dot is based on -log10(Binomial FDR) and colored by binomial fold enrichment calculated by GREAT. D. Top most significantly enriched motifs in putative EHEs calculated by HOMER. Shown are position weight matrix for each motif, transcription factor predicted to bind that motif, and HOMER p-value (Upper panel) HOMER known motifs (Lower panel) de novo motifs.
To further test the validity of our annotations, particularly the putative enhancer states, we assessed the number of experimentally tested and validated developmental enhancers by the Vista Enhancer Browser captured by each of our chromatin states from the 25 state model (Figure 2B).36 Overall we identified 92% of all validated heart-positive enhancers (n=281) from the Vista Enhancer Browser, a 7.5-fold enrichment versus active enhancers lacking activity in the heart (heart-negative) (p<2.2×10−16). When we compared each of the chromatin states ability to specifically identify heart enhancers, we observed as expected that enhancer chromatin states captured a significantly larger fraction of heart-positive enhancers than other chromatin states (Figure 2B). Strong enhancer state 13 showed the greatest fraction of overlap with validated heart enhancers and the greatest fold difference relative to overlap with heart-negative enhancers. The heart specificity of state 13 was higher than all other chromatin states and significantly greater than regulatory regions annotated by EMERGE or Dickel et al.
Interestingly, weak enhancer states 16 and 17, DNase accessibility state 19, and regions harboring repressive chromatin marks including poised and bivalent promoters and polycomb repressed states showed an opposite trend in capturing heart-positive versus heart-negative enhancers. This indicated that these two weak enhancer states (16 and 17) are not informative for heart regulatory sequences and generally should be interpreted with caution in other tissues where large numbers of validated enhancers do not yet exist. When we compared the performance of chromatin states identified by the 15 and 18 state models, we found decreasing levels of specificity as number of states decreased (Online Figures VIA and VIB respectively). Strong enhancer segments identified by the 18 state model showed slightly higher levels of specificity versus EMERGE and Dickel et al (Online Figure VIB). Enhancers identified by the 15 state model performed the poorest by this metric with EMERGE showing significantly higher specificity (Online Figure VIA).
These results suggest the 25 state chromatin model is best able to identify heart specific regulatory sequences, particularly state 13 enhancers. However, this analysis does not take into account the ranking of enhancers for heart specificity provided by both Dickel and EMERGE. It is thus possible that higher ranking regulatory sequences in either resource may be better able to identify true heart-positives. To test whether this was indeed the case we measured the ability of strong enhancer states from the 25 state model to recover heart-positive versus heart-negative enhancers compared with performance of ranked lists from both EMERGE and Dickel. Area under the curve (AUC) for receiver operating characteristic curves revealed consistently higher AUC for enhancers identified by the 25 state model across all time points versus EMERGE with the exception of earliest CS13 samples. Enhancers identified by Dickel had the lowest AUC values in this analysis (Online Figure VIIC).
When we directly compared the regions identified by our chromatin states and those by EMERGE we found the most significant overlaps of EMERGE peaks with active TSS and active promoter annotations (Online Figure VIIA). The EMERGE signal was overall highly enriched near TSS and subsequently the active TSS and promoter states (states 1–4) had the highest EMERGE scores (Online Figure VIIB). When we compared overlap of all chromatin state segments with the Dickel compendium, we found much less overlap with active TSS states and higher percentages of sequences that were annotated to be weak enhancer states or even quiescent in human embryonic development (Online Figure VIIIA). Together these results suggest that the highest scoring EMERGE peaks are concentrated very close to known TSS and the 25 state chromatin model is better able to identify tissue-specific regulatory sequences than both EMERGE and Dickel compendium.
While these results suggest that our chromatin state segmentations are capable of identifying sequences with heart regulatory capacity, the Vista Enhancer Database has been largely constructed by testing elements identified with H3K27ac deposition or P300 binding. The Dickel compendium is based almost entirely on these two marks and the EMERGE framework has been tuned to find positives from this database. To determine if our chromatin state segmentations are capable of annotating regulatory sequences identified by different means we turned to a large set of sequences characterized by binding of seven cardiac transcription factors in fetal and adult mouse hearts.18 This work demonstrated that regions bound by five or more transcription factors showed significantly more activity than regions with H3K27ac signal but lacking transcription factor binding when systematically tested using a massively parallel reporter assay (MPRA).
When we interrogated regions tested by MPRA that could be identified in the human genome we found the highest median RNA to DNA ratios for enhancer state segments in our 25 state segmentations. Specifically, we found significantly higher MPRA activity relative to negative controls in strong enhancer states 14, 15, and 18 (Online Figure IVF). Weak enhancer state MPRA signals were not significantly different from negative controls. Neither the Dickel compendium nor EMERGE regions showed significantly different MPRA signals from negative controls. These findings indicate our chromatin state segmentations are capable of identifying regions that are driven by combinatorial transcription factor binding and not necessarily by H3K27ac.
Thus far our analyses have indicated the strong enhancer states (states 13 – 15 and 18) are the most specific for developing heart. We reasoned that segments annotated as strong enhancer states in the developing heart but not so in other tissues might reveal previously unknown heart-relevant gene regulatory information. When we compared our full set of strong embryonic heart enhancer segments with analogous annotations from all tissues in Roadmap Epigenome we identified 12,359 segments that had not been previously annotated as strong enhancer chromatin states. We refer to these putative enhancers hereafter as embryonic heart-specific enhancer segments (EHEs) (Online Table II). When we compared these with regions identified in the Dickel compendium with prenatal biased scoring enhancers (enhancers with a scoring ratio >2 prenatal/postnatal) or enhancers in the Dickel compendium identified in the single human fetal sample, we observed small but significant sharing of annotations (Online Figure VIIIB–E). Combined these results indicated that the vast majority of our putative embryonic heart-specific enhancer segments (75.6%) have never been previously identified in any other human tissue or stages of heart development.
Unfortunately, only a handful of the putative embryonic heart-specific enhancer segments have been functionally tested making it difficult to assess their relevance for heart development from an in vivo perspective. While such putative enhancers might be novel we reasoned they could potentially target genes known to be involved in heart development or function. To address this, we assigned embryonic heart-specific enhancer segments to the single nearest gene within 1 megabase using the Genomic Regions Enrichment of Annotations Tool (GREAT) and found significant enrichment of biological processes related to heart development and morphogenesis (Figure 2C; Online Table III). The associated mouse phenotypes are heavily fortified with those related to abnormal development, morphology, size, and function of the heart. Additionally, the enriched molecular functions contain multiple terms related to microfibril and tubulin binding along with voltage-gated channel activity in the atrioventricular node.
We next analyzed the sequence content of the EHEs for enrichment of TF binding sites using HOMER (Figure 2D; Online Table IV). We identified significantly enriched motifs that matched binding sites of transcription factors involved in heart development such as the GATA family, the MEF2 family, and TBX20 among others (Figure 2D upper panel).38,39 When we performed de novo motif enrichment using HOMER, we saw enrichment of the same families of transcription factors with the addition of factors also know to contribute to heart development, such as NKX2–5 and SMAD signaling (Figure 2D, lower panel). Through this global analysis we have identified transcription factors likely involved in human embryonic heart enhancer activation. Functional and motif enrichment within these EHEs identified from this multi-tissue, unbiased analysis demonstrated the power of these approaches and suggested that many of the novel sequences we have identified are bona fide enhancers and likely important for normal heart development.
Differential motif utilization of embryonic heart enhancers across development
Having demonstrated enrichment of heart-related TF binding sites in putative embryonic heart-specific enhancer segments, we wondered if this was a general trend across all of heart development or if there were temporal shifts in enhancer activation modulated by different transcription factors during the embryonic period of heart development. To simplify our analysis, we consolidated the developmental time course into three general periods: early (CS13), mid (CS16–17), and late (CS23) development (Figure 3A). We identified differentially activated putative enhancer segments across this trajectory by comparing H3K27ac and H3K4me2 signals at all reproducible enhancer segments between each of the three stages of embryonic heart development (Figure 3B–C and Online Figure IX).
Figure 3. Differential enhancer utilization during embryonic heart development.
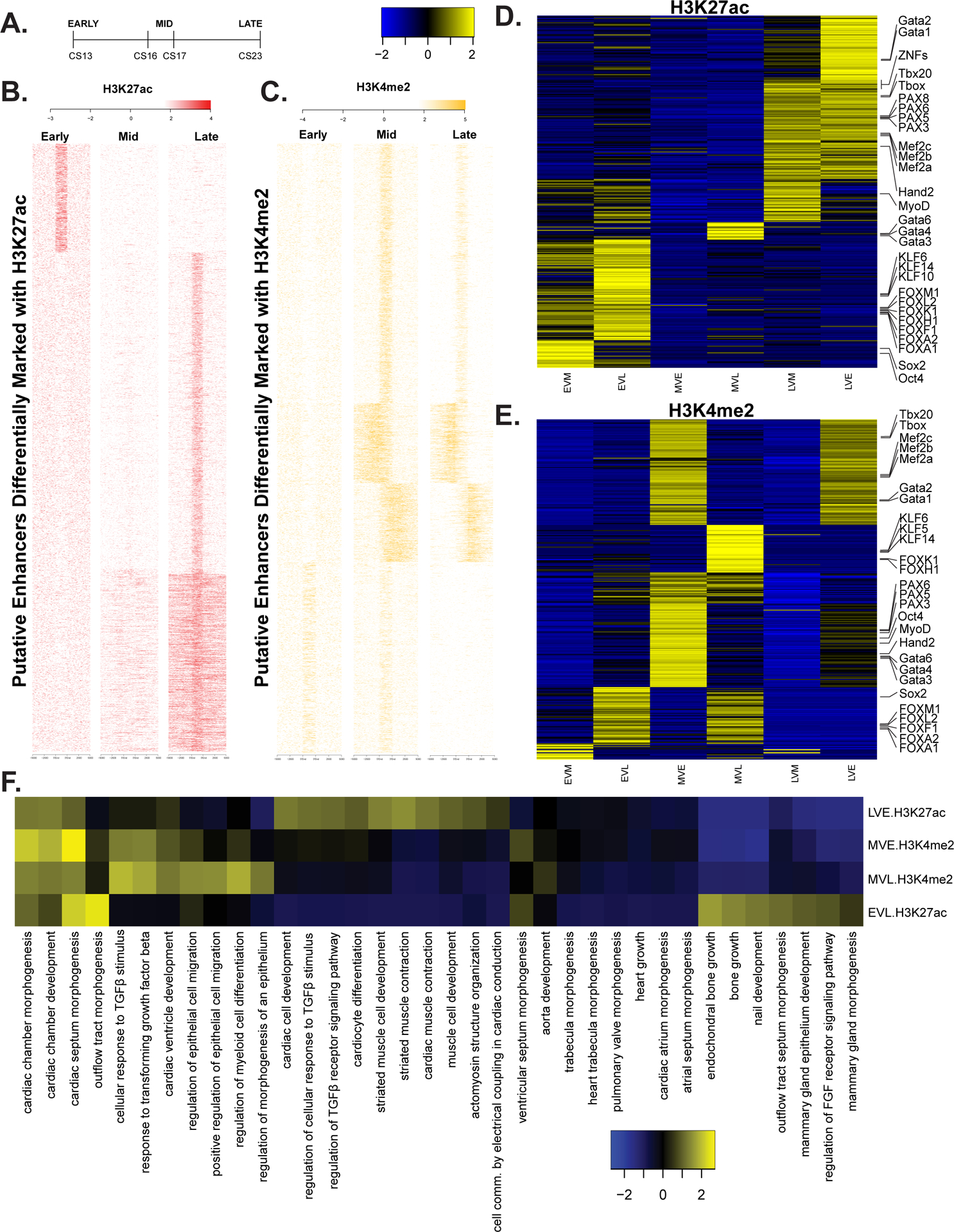
A. Delineation of three major stages of heart development during the embryonic period based on Carnegie staging. B. Heatmap of signal at putative enhancers differentially marked with H3K27ac C. Same as B but with H3K4me2. D.Heatmap of z-scores for level of significance of motifs enriched in each class of differentially regulated enhancers based on pairwise comparisons of replicates of H3K27ac signal at all embryonic heart enhancer segments using DiffBind. Comparisons are indicated as follows: early up versus mid (EVM), early up versus late (EVL), mid up versus early (MVE), mid up versus late (MVL), late up versus mid (LVM), late up versus early (LVE). The more significantly enriched motifs are colored yellow. E. Same as in D but using H3K4me2 signals. F. Heatmap of most variable z-scores for significance of enrichment of gene ontology categories for genes assigned a differentially activated enhancer by GREAT.
For putative enhancer segments differentially activated based on H3K27ac signals, we observed the greatest differences in motif enrichment between early and later stages of development (Figure 3D). Putative enhancer segments active early were specifically enriched for pluripotency related TFs like SOX2 and OCT4, and multiple members of the KLF and Forkhead families of transcription factors. Motifs enriched in late putative enhancer segments included many zinc finger containing TFs, and multiple members of the T-box, GATA, and PAX families of transcription factors. Notably, enhancer segments more strongly active in the mid period of heart embryonic development based on H3K4me2 signals showed the most pronounced enrichment of transcription factor motifs (Figure 3E). Many of the same transcription factor motifs enriched in early and late putative enhancer segments based on H3K27ac were shifted to enrichment in the mid period, suggesting dynamics in TF utilization related to chromatin state.
Based on these differences in differentially activated putative enhancer segments, we hypothesized that the location of these segments and subsequently the genes that they control might uncover distinct patterns of biological pathway utilization during this developmental series. We assigned differentially activated putative enhancer segments from each comparison to the nearest gene and identified the most significantly enriched gene ontology terms (Figure 3F). In all comparisons, terms associated with cardiac chamber and septum morphogenesis and development were highly enriched. Genes near putative enhancer segments most strongly active during the early period were uniquely enriched for pathways related to bone growth and FGF receptor signaling. Terms associated with cardiomyocyte differentiation, muscle cell development, and muscle cell contraction were observed for genes assigned putative enhancers more strongly active in the late period. These results agree with known dynamics in relative ratios of cardiomyocytes versus other cell types during these different developmental periods.40–42 Our bulk tissue experiments are incapable of disentangling such cell type specific regulatory element utilization. These differentially active putative enhancer lists are likely enriched for such sequences but requires single-cell chromatin accessibility methods to confirm.
Identification of embryonic heart specific super enhancers and long-range chromatin interactions.
To better understand the role these putative enhancer segments might play in cardiac related diseases, we started by interrogating the embryonic heart-specific enhancer segments for known heart related loci. Defects in the cardiac homeobox transcription factor NKX2–5 have been implicated in many different congenital heart defects including conotruncal malformations, septal defects, and atrioventricular conduction block.43–47 Most human patients with NKX2–5 associated CHD have heterozygous loss of function mutations and haploinsufficient mice have significantly depleted protein levels in the heart.48 Depletion or complete knockout of NKX2–5 results in dysregulation of many downstream target genes in mice and human cardiomyocytes suggesting it is a master regulator of cardiac.48,49 Therefore, identifying regulatory sequences that control NKX2–5 expression specifically in the developing heart could be valuable information for understanding the unknown genetic causes of CHDs.
When we inspected the genomic locus containing this gene, we found it was surrounded by a plethora of strong enhancer state segments including an embryonic heart-specific enhancer segment immediately downstream of the coding exons (Figure 4A). There are a number of strong enhancer segments identified uniformly from CS14 to CS23 approximately 50 kb upstream that are largely repressed in fetal and adult hearts. These regions are particularly interesting as they cannot be readily identified through sequence conservation based on comparisons of 100 vertebrate genomes (Figure 4A). Moreover, these coordinately activated strong enhancer segments are predicted as an embryonic heart super enhancer region that encompasses over 200 kbs surrounding NKX2–5 (n=4,215, Online Table II). This is nearly four times larger than any other super enhancer annotation for this region based on fetal or adult human heart samples. Such regions have been associated with tissue-specification loci, further reinforcing the central role NKX2–5 plays in heart development and the novel information our resource has identified.50–52
Figure 4. Functional annotation of cardiac phenotype associated variants and enrichment of embryonic heart enhancers in cardiac relevant long-range chromatin interactions.
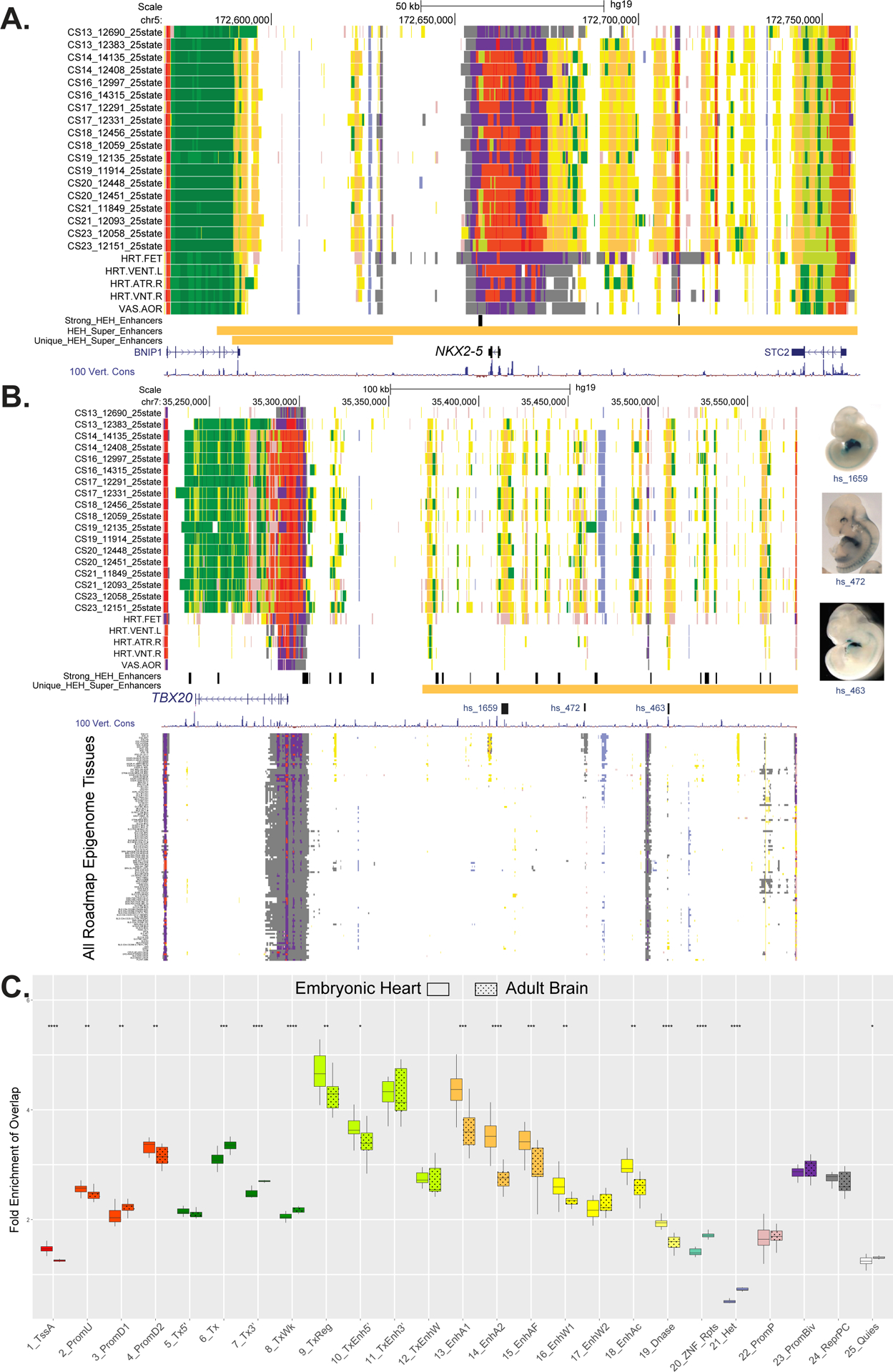
A. UCSC browser shot of NKX2.5 gene locus showing individual embryo chromatin state annotations from this study and Roadmap Epigenome. Samples are ordered from top to bottom based on developmental age, earliest to latest. Chromatin states are indicated by color segments using color convention from Figure 1C. Strong human embryonic heart (HEH) enhancers are shown in black and superenhancers and superenhancers unique to HEH are shown in orange. B. UCSC browser shot of locus near the TBX20 gene using the same conventions as in A. The region upstream of the TBX20 gene is a human embryonic heart specific super enhancer (orange bar). Of note are the strong HEH specific enhancer states track, as well as the experimentally validated enhancer elements with images to the right. In the lower panel, all the roadmap epigenome ChromHMM segmentations are stacked showing the region is not similarly active in any other profiled tissue. C. Box plots of fold enrichment of overlap of each indicated chromatin state in human embryonic heart or brain with anchor points identified by capture Hi-C interactions in iPSC-derived cardiomyocytes over matched randomly selected segments. Solid boxes represent embryonic heart chromatin segments while dotted boxes represent adult brain chromatin segments. Significance of difference between embryonic heart and adult brain fold enrichments were calculated using the Mann-Whitney test and is shown at top (p-value ≤ 0.05 = *, ≤ 0.01= **, ≤ 0.001= *** , ≤ 0.0001 = ****). The largest increases in fold enrichments for embryonic heart were identified for strong enhancer states 13 and 14.
We then inspected the locus harboring SCN5A which has been implicated in multiple cardiac diseases and specifically known to cause ~20–30% of cases of Brugada Syndrome (Online Figure XA).53,54 Here we observed several clusters of strong enhancer segments upstream, within, and downstream of the gene in heart tissues at multiple stages (adult, fetal, and embryonic tissue). The downstream cluster of strong enhancer segments has been previously annotated as a super enhancer region in mouse cardiomyocytes and deletions had significant effects on SNC5A expression in the heart.55 Our analysis confirmed this finding but also annotated a super enhancer that encompassed the strong enhancer segment cluster upstream of the gene. A strong enhancer segment in the 16th intron completely encompassed a validated enhancer with very specific activity in the mouse embryonic heart. Additionally, a 13kb LD block (R2≥0.95, D’=1, 1000G CEU) that contains multiple variants associated with cardiac phenotype related variants overlaps this tested enhancer region and adjacent loci (Online Figure XA, dashed box). These variants included rs41312411 associated with establishment of resting heart rate and P wave duration, rs3922844 associated with establishment of electrocardiogram traits and measures, and rs11708996 associated with Brugada syndrome.56–58
Variants in potential regulatory sequences across this entire locus have been tested for effects on enhancer activity in cultured cardiomyocytes.59 When we overlaid in vitro reporter assay data for putative regulatory sequences harboring alternative alleles on our chromatin state segmentations we generally observed strongest effects for sequences overlapping strong putative enhancers segments (Online Figure XA). Sequences that overlapped quiescent regions in our segmentations had minimal effects on reporter gene expression indicating they likely have limited regulatory capacity in vivo. In addition to these well studied loci we found that other genes important for normal cardiac function and development, such as HAND2 and MYOCD, were within super enhancers (Online Figure XB and Online Figure XC).
Having demonstrated that genes important for normal cardiac function reside within super enhancers and embryonic heart-specific enhancer segments are enriched for heart relevant biology, we reasoned heart-specific super enhancers could also identify important, previously unknown regulatory landscapes or important cardiac genes. When we compared super enhancer regions globally between the embryonic heart and analogous annotations from many different tissues using dbSUPER, we identified 1,611 human embryonic heart specific super enhancers (Online Table II)60. These regions were strongly enriched for genes related to transcription regulation or roles in cell junctions (Online Table II). One such example is the cardiac T-Box transcription factor TBX20, which is required for cardiomyocyte proliferation in mice and linked to dilated cardiomyopathy in humans.61–63 The large noncoding region adjacent to this gene contains 60 putative enhancers, nearly one third are EHEs, and multiple heart-positive in vivo enhancers (Figure 4B). The specific nature of our annotations is readily apparent at this location with the distal noncoding region and the sequences surrounding the TBX20 TSS are very sparsely annotated across 127 tissues and cell types in Roadmap Epigenome.
Another putative embryonic heart-specific super enhancer of note is approximately 200kb in length and resides in the large noncoding region upstream of gap junction protein GJA1. Sites throughout this approximately 1Mb region form long range interactions with GJA1 in human iPSC-derived cardiomyocytes.64 Deletion of heart-specific enhancers of Gja1 in mice are sufficient to decrease its expression, which has in turn been previously linked to arrhythmias.25,65 This set of embryonic heart-specific super enhancers includes many additional loci that are not currently known to play a role in cardiac development making them good candidates for future study.
While the examples above demonstrated cis-regulatory landscapes surrounding a single gene, as indicated for enhancers of GJA1, such regulatory sequences can interact with their targets over long distances through chromatin looping. Such loops can be difficult to predict in silico and given the tissue-specific nature of enhancers appropriate tissues or cell types have to be utilized to identify biologically relevant interactions. Although the developing heart is made up of many different cell types, recent single cell RNA-seq analysis of CS16 hearts revealed a significant proportion of cardiomyocytes.66 We therefore hypothesized that interaction data from cardiomyocytes might allow us to better understand the physical relationship between our chromatin state segmentations and target genes in a cardiac relevant context. Thus we integrated our annotations with previously published high-resolution promoter capture Hi-C (PCHi-C) data from iPSC-derived cardiomyocytes (CMs).64 We overlapped the distal anchor points from CMs with the functional annotations from our human embryonic heart samples using Roadmap adult brain samples as a control. We then calculated the fold enrichment of interactions for each chromatin state (Figure 4C). The largest enrichments in distal anchor points were in strong enhancer and transcription regulatory states (specifically states 9–11, and 13–15) for both embryonic heart and brain. However, the largest degree of specificity between embryonic heart versus brain in these states was identified for strong enhancer states 13 and 14. Little to no significant change in fold enrichment of interactions between these two tissues was seen in poised, repressed, or quiescent states (States 22–25). Overall, these findings confirmed many groups’ observations that strong, tissue-specific enhancers interact with their target gene promoters in a tissue-specific fashion and further demonstrated the relevance of our annotations for human embryonic heart development.67,68
Systematic enrichment of heart phenotype and defect associated variants in embryonic heart enhancers.
Since we observed overlap of putative enhancer segments with disease associated variants at known cardiac disease related genes, we set out to determine if this finding was generalizable across all strong putative enhancer segments regardless of their proximity to a known heart gene. We compiled variants significantly associated with variation in normal heart phenotypes and congenital heart defects from the GWAS catalog. We then assessed enrichment of these variants in strong enhancer state annotations from all tissues profiled by Roadmap Epigenome and the developing human heart. We observed significant enrichment of variants associated with systolic blood pressure, electrocardiograph traits and measures, resting heart rate, QRS characteristics, and QT interval in strong enhancer segments identified in most embryonic heart samples (Figure 5A–D, and Online Figure XIB). The earliest samples we profiled (CS13) did not show enrichment for any of these traits suggesting that gene regulatory programs influencing these phenotypes are not active until after 4.5 post-conception weeks. As a negative control we interrogated variants associated with Chron’s Disease that have been previously shown to be enriched in enhancers identified in immune cell types.69 We confirmed this finding and saw minimal enrichment of these variants in strong enhancer segments identified during human heart development (Online Figure XIC). We then turned to common variants associated with CHD incidence (Online Figure XID). Unexpectedly, we found that once we corrected p-values for multiple testing no set of tissue putative enhancer segments were significantly enriched.
Figure 5. Enrichment of cardiac phenotype associated variants in embryonic heart enhancer segments.
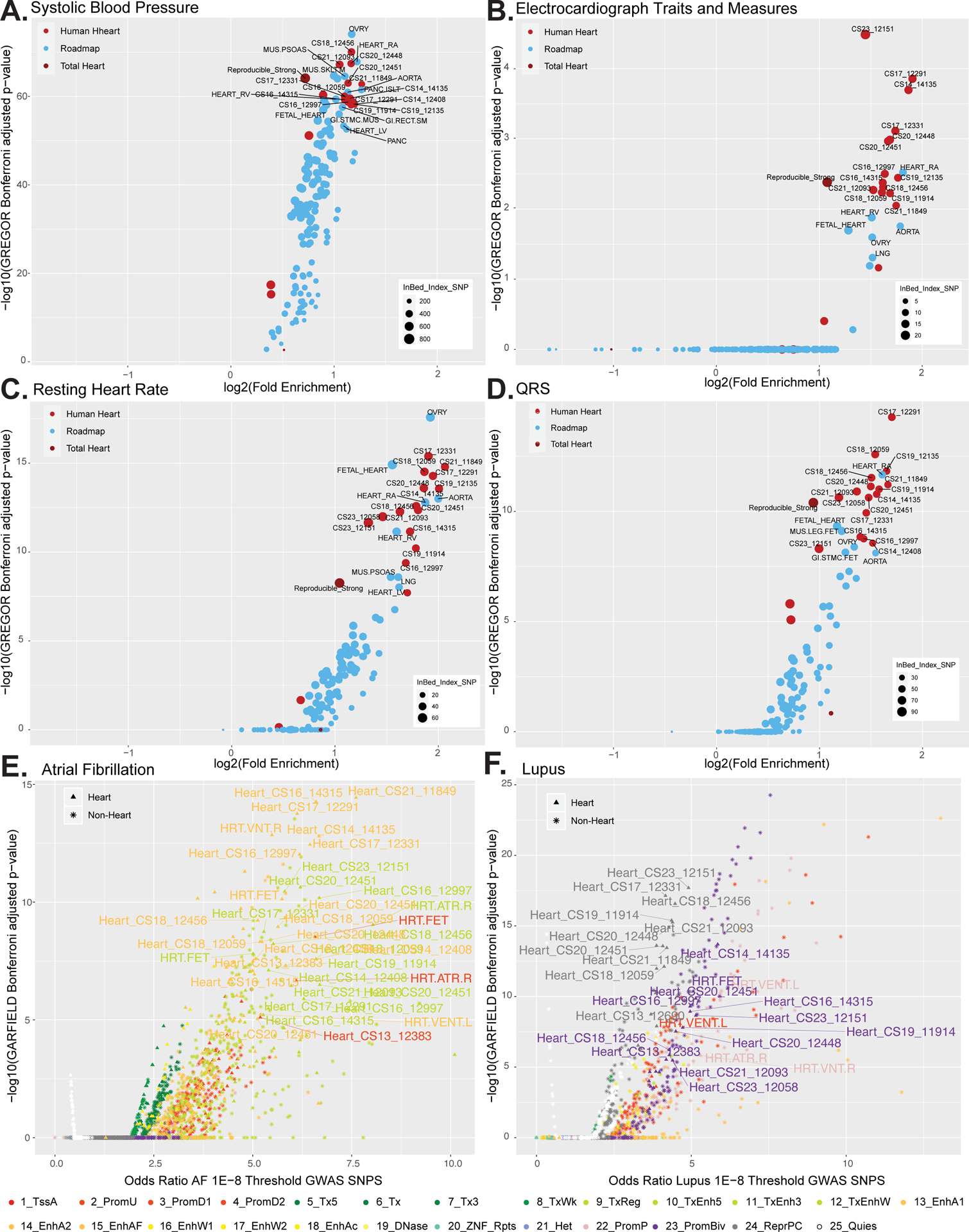
A. Scatterplot of the log2 fold enrichment and log10 Bonferroni adjusted significance level of GWAS variants associated with systolic blood pressue in all enhancers segments identified in the strong enhancer states for each embryonic heart sample (bright red), the total reproducible strong enhancers from the whole dataset (dark red) or other tissues in Roadmap Epigenome (blue). All values calculated using only variants with p values < 5×10−8 from GWAS Catalog using GREGOR. B. Same as in A using GWAS variants associated with electrocardiograph traits and measures. C. Same as in A using GWAS variants associated with resting heart rate. D. Same as in A using GWAS variants associated with QRS complex traits. E. Enrichment of GWAS analysis p-values for atrial fibrillation72 in all chromatin state annotations as determined by GARFIELD. Scatterplot of the odds ratio of atrial fibrillation GWAS SNPS using the 1E–8 Threshold by the log10 GARFIELD Bonferroni adjusted p-values. Samples from this study (triangle symbol) and Roadmap epigenome (star symbol) are colored by chromatin state as indicated by the color key. Atrial fibrillation shows greatest enrichment in strong enhancers identified in embryonic heart tissues. F. Same as in E using GWAS summary statistics for systemic lupus erythematosus105. Lupus shows greatest enrichment in strong enhancers identified in immune cell types sorted from blood. Lupus also shows enrichment in repressed and bivalent states in human embryonic heart.
Recent findings in atrial fibrillation (AF) have suggested that sites accessible during fetal heart development are enriched for variants associated with this disorder.70–72 However the functional nature of these sites is unknown, as significant enrichment in fetal heart was observed only for H3K4me1 peaks, a mark typically associated with poised enhancers.73 This could suggest that enhancers primed in fetal heart development but not yet fully active are important for AF, but it is unclear if regulatory elements active even earlier or in different chromatin states may play a role. The availability of full summary statistics for this particular disease phenotype allowed us to leverage more systematic, genome-wide analysis of more than 8 million positions in the genome in a linkage disequilibrium aware fashion.69
To address this issue, we retrieved full summary statistics for a large, multi-ethnic GWAS for AF and assessed enrichment of associated variants across all 25 chromatin states from all tissues in Roadmap Epigenome, craniofacial development, and embryonic heart development. Our findings confirm enrichment of AF associated variants in fetal and adult heart active regulatory sequences. Surprisingly, we observed enrichment of AF associated variants across virtually all embryonic heart strong enhancer states (Figure 5E, Online Figure XIA, and XIF). The greatest fold enrichment was observed in strong enhancer segments (ChromHMM state 13) active at the earliest time points we profiled (CS13, OR 6.65, p-value 8.69×10−06). Using this approach we also observed enrichment for variants associated with resting heart rate, QRS interval, and p-wave duration consistently across strong enhancer states identified in embryonic human heart (Online Figure XIH–J).
Using full GWAS summary statistics from two immune related diseases (systematic lupus erythematous and Crohn’s disorder, as negative controls), we did not observe enrichment in any embryonic heart enhancer states (Figure 5F and Online Figure XIG–K). Conversely, we observed enrichment of Lupus associated variants in active TSS (state 1) and strong enhancer state segments from immune-related cell types (Online Figure XIE).69,74 Surprisingly, when we assessed all chromatin states identified in embryonic heart samples we found significant enrichment of Lupus associated variants uniformly across polycomb repressed segments (state 24) and bivalent promoter segments (state 23) identified in the embryonic and fetal heart but not adult heart samples (Figure 5F).
Gene expression profiling of embryonic heart development
Thus far we have only analyzed putative regulatory sequences but not discerned what, if any, effects they may have on gene expression. To begin to understand how chromatin state changes during heart development influence gene expression, we profiled the transcriptomes of three biological replicates at each of 8 distinct embryonic stages (CS13, 16–21, and 23), largely overlapping the time points profiled for chromatin state. This window of time has the greatest dynamics of gene expression based on comparisons of transcriptomes of developing hearts from multiple species.12 To leverage a large number of previously published data sets, we processed our data using a uniform analysis pipeline employed by the recount2 database.75,76
Processing data in this way allowed us to directly compare our data to all tissues profiled by GTEx, including 489 samples from adult heart tissue.77 To assess the validity of this approach, we first clustered all samples from GTEx (n=9662) and our embryonic heart samples (n=24) based on expression of 36990 genes, using t-distributed stochastic neighborhood embedding.78 We observed generally good clustering of adult samples by tissue type, including sub-regions of the brain and the ventricle and atrium of the adult heart. Embryonic heart samples were tightly clustered and distinct from adult heart samples and other GTEx tissues. (Online Figure XIIA). Principal component analysis of only the embryonic heart samples showed good clustering by stage and minimal effects from gender and RNA quality in the first two components (Online Figure XIIC–E). Overall these results suggest that our expression data are of high quality and likely informative for understanding early human heart development.
Genes that are expressed in a limited number of tissues are more likely to be disease related genes than those with broader expression patterns.79,80 Based on these general trends, we hypothesized that genes expressed specifically during embryonic heart development are likely involved in cardiac defects. To evaluate this, we employed a measure of specificity based on the Gini coefficient, a metric originally used to measure income inequality, which accurately identified genes with tissue and cell-type specific expression.81–83 We identified 347 genes with elevated Gini coefficients (>0.5) and highest expression in embryonic heart (Figure 6A). These genes were strongly enriched for genes involved in heart development including TBX5, IRX4, HAND1, HAND2, and FGF12 (Figure 6B, lowest panel and Online Figure XIIB; Online Table V).
Figure 6. Transcriptional profiling of embryonic heart development.
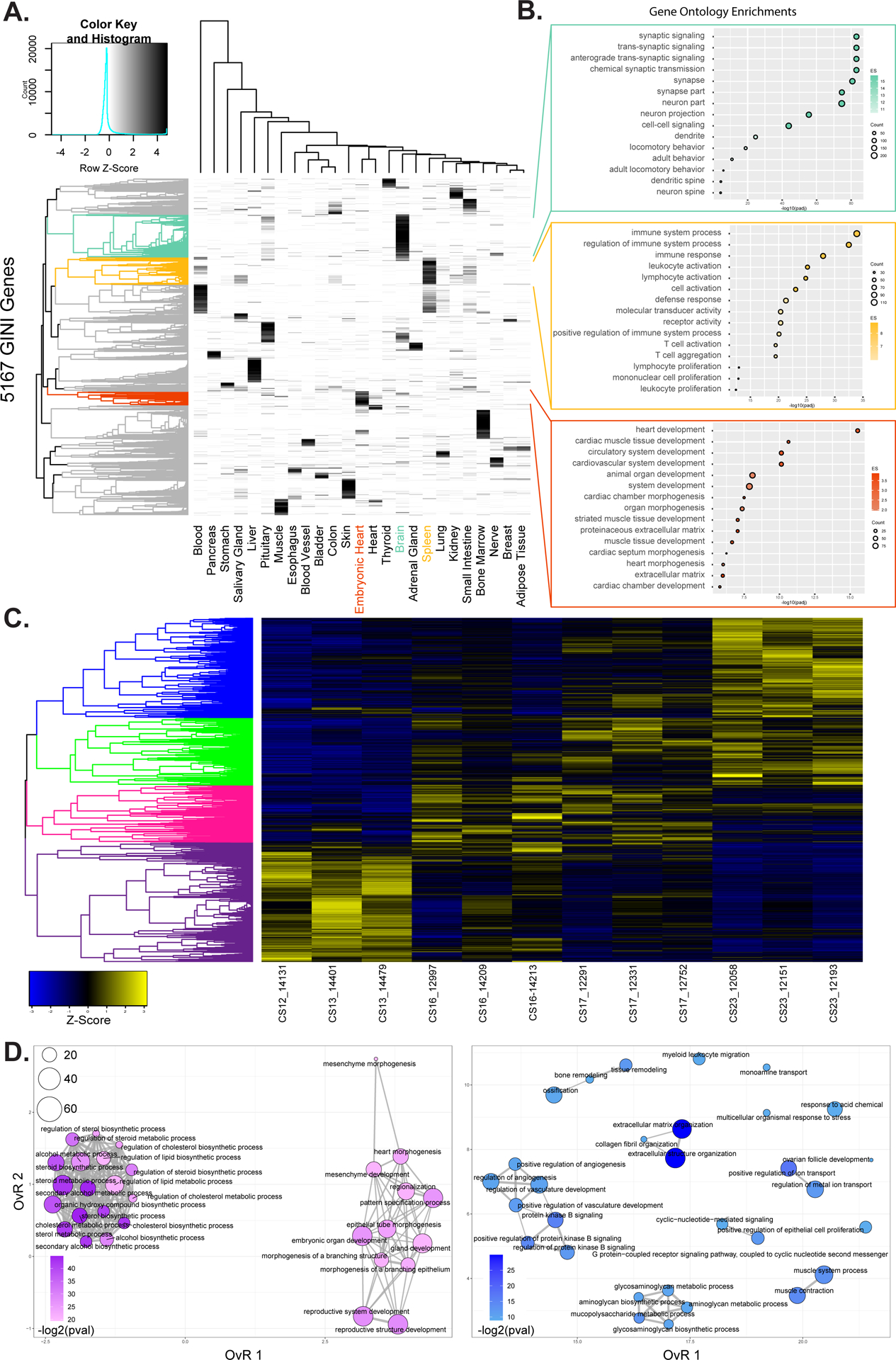
A. Heatmap showing specificity of expression for 5,167 genes identified with elevated Gini scores (>0.5) for 25 tissues from GTEx and embryonic heart. Brain, spleen, and embryonic heart specific genes are identified as colored leaves on the dendrogram along the left of the plot. B. Gene ontology enrichments for genes identified as specific for heart, spleen, and embryonic heart respectively based on genes from indicated color coded clusters in A. C. Heatmap of z-scores of normalized gene expression for genes identified as differentially expressed in pairwise comparisons of replicates from each of Carnegie Stage in our developmental series. Dendrogram on the left is hierarchical clustering of genes across a developmental series. The genes were color coded by cutting the dendrogram at a height which would result in four groups. Purple most highly expressed early. Pink and green expressed most strongly in intermediate stages of the series. Blue genes are most strongly expressed at the end of the developmental series. D. Gene ontology enrichment maps from the purple (left) and blue (right) gene sets identified in C. The size of each dot represents the number of genes and the color scale represents the -log2 transformed Benjamini & Hochberg adjusted p-value of each ontology. Darker colors indicate higher significance. The edges connect overlapping gene sets. The location of each dot is determined by the overlap ratio (OvR) calculated by enrichplot. Genes active early are enriched for functions related to embryonic patterning and morphogenesis while genes active late in embryonic heart development are enriched for vasculature development and ion-channel function.
The same tissue-specific functional trends were observed when we examined genes with elevated Gini coefficients and highest expression in either the brain or the spleen, demonstrating the unbiased nature of this analysis and the specificity of our findings (Figure 6B, upper panels). Genes with the highest degree of embryonic heart specificity (Gini ≥ 0.9) included known heart developmental transcription factors (NKX2–5, NKX2–6, and TBX20), myosin light chain genes (MYL3, MYL4, and MYL7), the long noncoding RNA BANCR and the sino-atrial node associated channel gene, HCN4 (Online Figure XIIB). The single highest Gini coefficient gene assigned to embryonic heart was LRRC10, a leucine-rich repeat containing protein previously identified as having cardiomyocyte specific expression and linked to human dilated cardiomyopathy.84,85
When we analyzed this list of genes for potential regulatory effects we found that more than half of the most significant predicted upstream regulators were genes from this list including MYOCD, TBX5, TBX20, HAND2, GATA4, NKX2–5, and NKX2–6 (Online Table VI). Consistent with these findings, we discovered that promoters of embryonic heart elevated Gini coefficient genes were significantly enriched for conserved NKX2–5 binding site motifs (Online Table VI). These findings suggest highly connected direct regulatory effects amongst these embryonic heart-specific genes. Additionally, we observed EHEs were enriched for motifs of many embryonic heart elevated Gini coefficient genes (GATA4, GATA6, TBX20, HAND2, and NKX) (Figure 2D; Online Table IV). Combined these findings show that the Gini coefficient effectively identifies many heart related disease genes, enables novel inference of regulatory relationships between these genes, and implicates new genes in heart-related disease phenotypes.
Thus far our gene expression analyses combined all of our samples into a single embryonic heart category, but dynamics of heart gene expression have been reported to be greatest during the time points we have profiled.12 To characterize these dynamics we first set out to identify genes that are differentially expressed in a pairwise fashion between each of the stages of embryonic heart development. We identified 7,167 differentially expressed between all sets of time points (Online Figure XIII). The majority of differentially expressed genes were identified relative to the CS13 time point. Very few genes were differentially expressed between time points CS17 to CS23, supporting the finding that gene expression dynamics are restricted to early heart development in humans.12 Hierarchical clustering of these differentially expressed genes identified four major groups of genes that show a wave of expression across this developmental series (Figure 6C and Online Figure XIIIA–B). Genes expressed most strongly in early samples were enriched for functions related to general embryo development/morphogenesis and cholesterol biosynthesis (Figure 6D, left). These genes were also enriched for functions related to neuron generation and differentiation suggesting similar early developmental architecture between the heart and brain (Online Table VII). Genes more strongly expressed at the end of the embryonic period were enriched for functions related to ion channel activity, growth factor binding, extracellular matrix organization, and intracellular signaling (Figure 6D, right and Online Table VII). The most significant enrichment of genes related to heart contraction and cell-cell adhesion were found in genes most strongly expressed at the CS17 time point (Online Figure XIVA, right panel). These results indicate we have captured much of the dynamics of gene expression during early heart development, but that many important events likely happen even earlier than we can currently examine.
Regulatory effects of embryonic heart enhancers on gene expression.
Since we found that EHEs are strongly enriched for binding sites of heart related transcription factors and systematically located near genes with heart related functions, we next wanted to understand the impact such regulatory elements might have on embryonic heart-specific gene expression. To address this question, we first assigned embryonic heart-specific enhancers to the single nearest gene. For genes that were assigned an EHE, most had a modest number assigned (mean = 4), but some genes had greater than 30. When we interrogated gene expression, we found that an increasing number of EHE assignments was associated with greater differences in gene expression in embryonic heart tissues relative to all other tissues from GTEx (Figure 7A). Specifically, genes with greater than 15 embryonic heart-specific enhancer segments had significantly higher gene expression within embryonic heart samples than all other GTEx tissues (Figure 7A and Online Figure XIVB). We then asked if these EHEs could be associated with embryonic heart-specific expression by measuring the distance of each enhancer to high Gini coefficient genes identified in the embryonic heart from above. We found significant enrichments of EHEs near embryonic heart-specific genes relative to random permutations of the same number of enhancers across distances ranging from 5 to 100 kb (Figure 7B).
Figure 7. Integration of chromatin state and gene expression identifies genes important for human cardiac development.
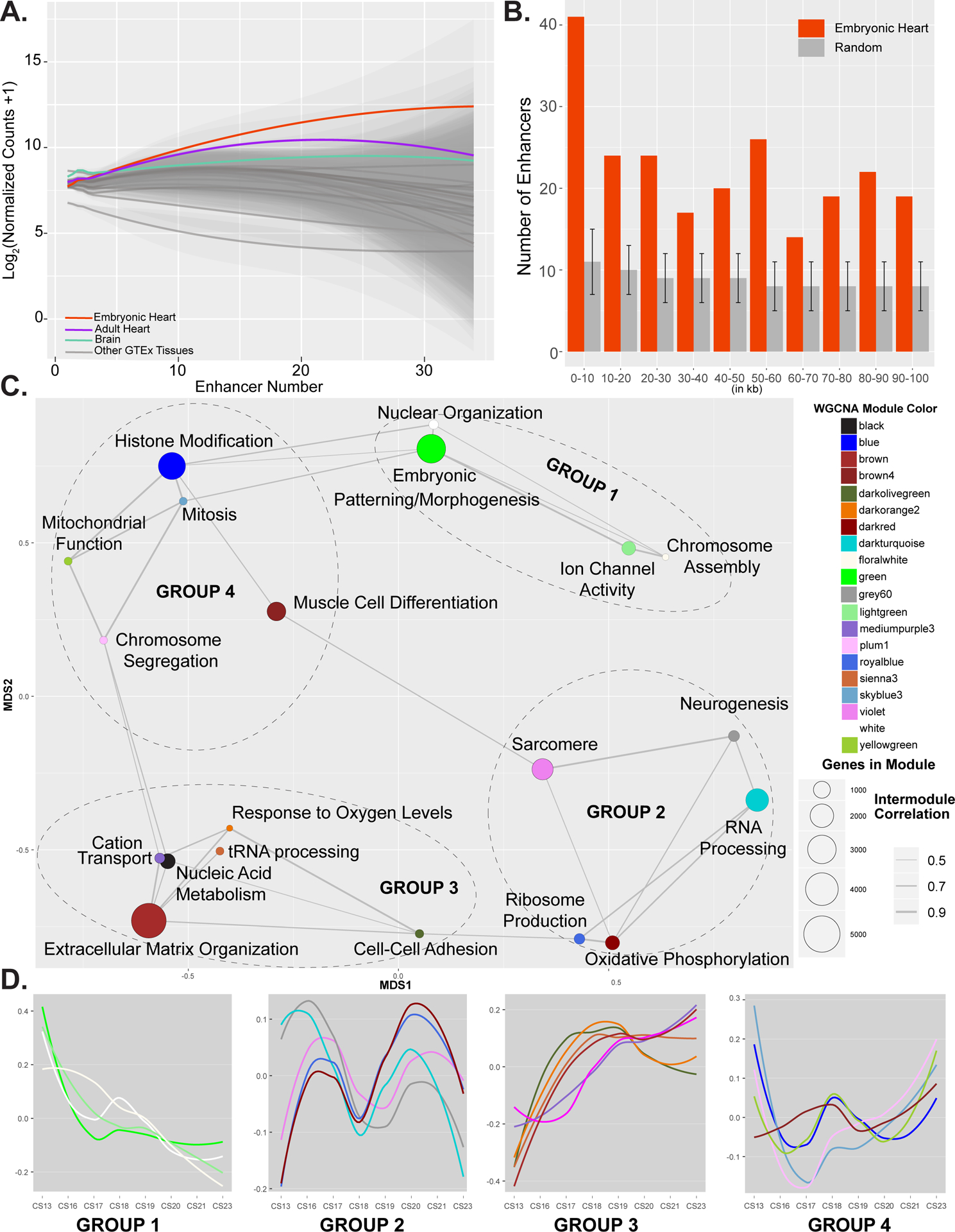
A. Plot of gene expression values from embryonic heart (red), adult heart (purple), brain (green) or all other tissues (grey) for genes assigned indicated number of EHEs as determined by GREAT. Genes assigned multiple EHEs are more strongly expressed in embryonic heart than other tissues. Significant differences in distributions of gene expression values in each comparison were determined based on Mann-Whitney test. B. Histogram of distances of EHEs (red) or randomly selected sets of enhancers (grey) to the nearest heart specific gene (GINI > 0.75) in 10 kb bins up to 100 kb. Overall EHEs are enriched near heart specific genes over all distances up to 100 kb. Error bars indicate standard deviation of 1000 random permutations of enhancers. C. Network plot of gene modules identified by WGCNA using embryonic heart gene expression data. A pearson correlation of the module eigenvectors was calculated for the edges. Positive correlations of 0.5 and greater were included. The location of each module is determined by multiple dimensional scaling of the module eigengene vectors. Modules are color coded based on names assigned by WGCNA. Size of dots indicate number of genes in each module. Each module is labelled based on the most significant biological process category gene ontology enrichment determined by DAVID, however this label is not always all encompassing. See Online Table VIII for exhaustive list. Modules are grouped based on related functional category enrichments and distance in MDS space. D. Trajectories of expression based on eigenvectors reported by WGCNA for each module across the developmental series. Groups and color coding are the same as in C. Group 1 modules have generally declining expression and include many genes involved in developmental patterning. Group 3 modules generally have increasing expression. Groups 2 and 4 have multiphasic but offset expression and contain genes involved in chromatin regulation and muscle cell differentiation and function.
These results coupled with the enrichment of physical interactions of putative embryonic heart strong enhancers with promoters active in cardiomyocytes suggested a direct role for EHEs in driving embryonic heart-specific gene expression. Additionally, the sequence content of these enhancers indicated they are in turn regulated by many of these embryonic heart-specific genes pointing to the existence of co-regulated networks of genes activated in early heart development.
Gene co-expression networks reveal trajectories of coordinated expression during early heart development.
Genes that are co-expressed across development have been proposed to share similar regulatory mechanisms and form networks that are important for normal development.79 Genes co-expressed during early brain development, particularly those that have correlations with many different genes or “hub” genes, are involved in ASD risk.86–88 We therefore hypothesized that identifying co-expression networks and resulting hub genes could reveal novel candidate CHD genes. To build a co-expression network during embryonic heart development in an unsupervised and unbiased fashion, we employed weighted gene co-expression network analysis (WGCNA) using all 24 embryonic heart samples we have profiled.89 Using this approach 26,122 genes were distributed across 29 modules, 20 of which showed enrichment for at least one gene ontology category (Online Table VIII). We identified modules with gene ontology enrichments expected for early heart development such as embryonic patterning (green, 2,573 genes), muscle cell differentiation (brown4, 945 genes), and sarcomere assembly (violet, 1,267 genes) (Figure 7C).
Multidimensional scaling (MDS) of the module eigengenes revealed inter-module co-expression, suggesting some modules were more closely related in their expression than others (Figure 7C). Comparison of trajectories of expression of eigengenes from each module revealed four groups of gene expression patterns that reflect positioning of each module in multidimensional scaling space (Figure 7D).89 Groups 1 and 3, which are diametrically opposed to one another in MDS space, showed downward and upward trends of expression throughout the embryonic period, respectively. Groups 2 and 4, which are also opposite one another in MDS space, showed multiphasic but offset patterns of expression. Group 2 showed a particularly strong wave like pattern between CS16 and CS20. When we performed gene set enrichment analyses across gene expression from CS16, CS18, and CS20 we readily observed cyclical enrichment of a number of pathways (Online Figure XV). These included heart valve development, tissue migration, mitochondrial gene expression, and several metabolic processes.
Significance tests give context to WGCNA of early developing heart.
To further characterize and validate the WGCNA network, we evaluated the enrichment of several curated gene lists. As we demonstrated above, binding sites for transcription factors expressed specifically in the embryonic heart were significantly enriched in embryonic heart-specific enhancers. This suggested that coordinated regulation between enhancer sites, the proteins that bind them, and the genes they target might be in action during early heart development. Five modules were significantly enriched for elevated Gini coefficient genes, while two modules were significantly enriched for known CHD genes (BH adjusted p < 0.05) (Figure 8A). All of these modules were significantly enriched for genes assigned at least one EHE. Analysis of our modules with single-cell RNA-seq on a 6.5 post-conception week human heart, revealed that despite performing bulk RNA experiments our modules are organized in cell-type relevant patterns.66 Analysis of genes that identify the major cell types from Carnegie Stage 16 heart revealed that many of the modules in groups 1 through 3 of our network are significantly enriched for embryonic heart cell-type specific genes (Figure 8A). For example, the brown4 and violet modules which are enriched for gene ontologies related to the sarcomere and muscle cell development (Figure 7C) concordantly have significant enrichment for the cardiomyocyte cell types (Figure 8A). The combined enrichment of both specific gene expression and specific enhancer activation in these modules suggest the network we have constructed is particularly meaningful during embryonic heart development (Figure 8A).
Figure 8. WGCNA significance tests and network for violet module.
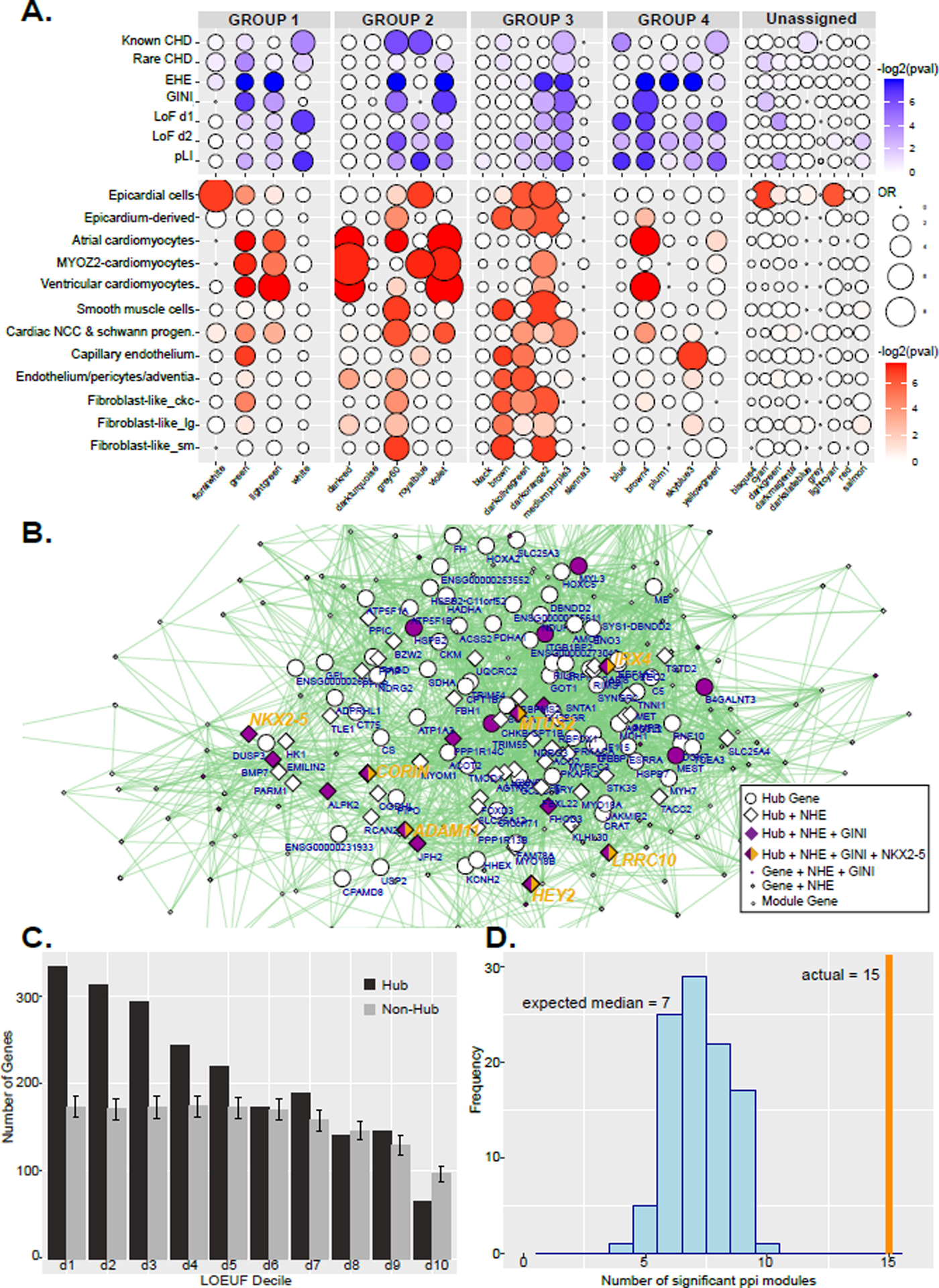
A. Dot plots of gene enrichment within the WGCNA modules. The lists of genes used are curated from multiple sources, while EHE and GINI are from this paper. The groups correspond to Figure 5. B. Network of multidimensional scaling coordinates and pairwise correlation scores for the violet module in Group 4 in D which is enlarged to show detail. All genes with correlation value greater than 0.88 with any other gene are plotted. Size of shape indicates highly connected hub genes. Diamonds represent genes assigned EHEs. Purple filled shapes indicate heart specific gene expression (GINI >= 0.5). Hub genes are labeled with gene symbol. Genes directly positively regulated by NKX2–5 binding are indicated with yellow. Several hub genes that have all these criteria are listed in larger yellow text. C. Histogram of LOEUF deciles of hub genes or randomly selected non-hub genes from all modules in the WGCNA network. Deciles range from decile 1 (d1) which represent the most constrained genes to d10, genes that are the most tolerant to putative loss-of-function (pLoF) variation. Error bars indicate standard deviation of 1000 random permutations of non-hub genes. D. Histogram of the number of gene-scrambled modules that have ppi enrichment at a Bonferroni adjusted p-value of <0.05. The vertical orange line marks the 15 modules that have significant ppi in the actual WGCNA network.
To confirm that the WGCNA network is indeed uniquely able to identify heart-relevant biology we leverage a co-expression network constructed for the developing human brain.86 Analysis of embryonic heart specific genes on this network supported this hypothesis as only two modules show enrichment for heart high Gini genes (Online Figure XVI). No modules were significantly enriched for known CHD genes. Analysis of the CS16 heart cell-type associated genes revealed modules primarily in group 5 with enrichment but overall the network lacked the cell-type specificity patterns observed on the heart WGCNA network (Online Figure XVI).
Disease relevance of WGCNA.
Functional enrichments and significance tests revealed brown4 and violet modules to be important for normal heart development however they lacked enrichment for known CHD causing genes. Together these findings indicated these modules are important for normal heart development and thus we hypothesized might harbor novel CHD genes. To address this question we leveraged measures of selection against loss-of-function variants from whole exome and whole genome sequencing of over 140 thousand healthy controls by the Genome Aggregation Database.90 Genes scoring in the lowest two deciles of the loss-of-function observed/expected upper bound fraction (LOEUF) measure were shown to be enriched for known haploinsufficient genes and genes essential for survival in cell culture models. When we analyzed our gene modules we found significant enrichment of LOEUF decile 1 and 2 genes in the violet and brown4 modules as well as many modules within expression groups 1 through 4 but not those outside these groups.
Notably, the violet module is strongly enriched for sarcomere related genes and the known CHD gene NKX2–5 was a top scoring “hub” node (Figure 8B). This transcription factor has been previously implicated in cardiac cell structure and function, particularly in sarcomere organization and falls into the second decile of LOEUF scores.91–93 Given the purported importance of hub genes from WGCNA analyses in disease and a critical cardiac TF being identified as both a hub gene and intolerant to disruption, we wondered if hub genes in our network might be generally intolerant to loss of function mutations in otherwise healthy individuals.88,94–96 When we interrogated hub genes across the entire network we found significant enrichment of low LOEUF score genes and significant depletion of genes from the tenth decile (Figure 8C). Genes that have not been implicated in CHD but are characterized by high connectivity in our network, heart-specific expression, and low LOEUF scores thus represent novel candidate CHD genes (Online Table IX).
WGCNA reveals NKX2–5 regulatory program.
Throughout this work many of our results have confirmed a central role of NKX2–5 in human heart organogenesis. Its binding sites are enriched in both embryonic heart-specific enhancer segments and the promoters of genes with heart-specific expression. In hESC-derived cardiomyocytes NKX2–5 bound the promoters and directly regulated the expression of many genes involved in heart development, specifically those involved in voltage-gated ion channel activity.49 Two prominent genes in the violet module that are directly regulated by NKX2–5 in human cardiomyocytes are the TFs HEY2 and IRX4 (Figure 8B, Online Figure XVII). Both of these have been shown to play a role in ventricular myogenesis in mice and linked to heart abnormalities in humans.97–99
The existence of these direct targets in the same module built in an unsupervised fashion from only gene expression data suggests that these modules may reflect direct, physical connections of transcription factors and their target genes. To determine if these observations were representative of larger patterns of regulation, we analyzed all WGCNA modules for enrichment of the 228 genes directly regulated by NKX2–5 in human cardiomyocytes.49 We found that indeed the violet module is enriched for direct regulatory targets, which were all well connected “hub” genes, assigned embryonic heart-specific enhancers, and had elevated heart specific expression values (Gini ≥ 0.5) (Figure 8B).
We also found enrichment of NKX2–5 target genes in four other modules (green, lightgreen, skyblue3, and mediumpurple3) that are enriched for embryonic patterning, ion channel function, and mitosis (Figure 7C, Online Figure XVIII and Online Table X). Genes that have the same characteristics as NKX2–5 in our networks, specifically high connectivity, tissue-specific expression, and potential regulation by heart-specific enhancers (Online Table X), are likely integral to normal heart development. Indeed, several genes identified in this data driven fashion have been clearly linked to CHD as reported in OMIM including NKX2–6 and TNNI3K, while others have been implicated in mouse or are pending further findings in human to be considered a CHD gene, such as IRX4, HEY2, and HCN4.
Finally, to further understand the functional relevance of the gene co-expression network we reasoned that genes might be co-expressed with one another in order to physically interact. When we assessed our modules for protein-protein interactions (ppi) we found that 15 of our modules contained significant ppi. This was a highly significant result relative to randomly constructed modules (Figure 8D) indicating this network has identified coherent connections for both gene regulation and physical interactions of proteins.
DISCUSSION
Here we have jointly profiled chromatin state and gene expression during the organogenesis phase of human heart development. Our efforts have dramatically increased the number of regulatory sequences that are predicted to be active in the developing human heart. We have validated the function and specificity of these regulatory elements by leveraging a variety of publicly available data sources including transcription factor binding, in vivo enhancer assays, genome wide association studies, and chromosome conformation.
We have identified tens of thousands of novel regulatory sequences likely active specifically in the developing human heart. Even though these are newly identified sequences, they display characteristics familiar to the heart development field. For example, they are enriched with binding sites for families of transcription factors long appreciated to play a role in heart development such as GATA, MEF2, and NKX and are located in close proximity to genes with documented roles in mammalian heart development. Our systematic comparisons of gene expression between the developing heart and 25 other human tissues has allowed us to identify a set of genes with high specificity for expression during heart organogenesis. The embryonic heart specific enhancers we have identified are enriched near these developing heart-specific genes. Furthermore, the number of heart specific enhancers that can be assigned to a gene is correlated with relative expression level compared to all available human tissues. These findings strongly support a direct role for these regulatory sequences in driving embryonic heart specific gene expression patterns.
In addition to individual regulatory sequences we have characterized localized landscapes of co-activated enhancers commonly referred to as super enhancers. Large regulatory landscapes have been implicated in tissue specification and tumorigenesis and encompass genes important for these processes. In our data we identified thousands of super enhancers across all time points of embryonic heart development, 1,611 of which had not been previously annotated (Online Table II). These include embryonic heart specific super enhancers near important heart genes, TBX20, GATA4, HAND1, HCN4, IRX3, IRX4, and IRX6, among others. Additionally, a significant majority of genes we identified with strong heart specific expression, including UNC45B, METTL11B, MYL3, MYL4, MYL7, and AFP, are directly adjacent or completely encompassed by super enhancers active in the embryonic heart. These findings demonstrate the large number of regulatory inputs that control tissue specification genes during heart development and suggest a high level of redundancy to maintain proper gene control.
Numerous studies have shown tissue-specific regulatory regions are enriched for variants associated with tissue-specific phenotypes and diseases.21,69,74 Our analysis strongly confirmed these previous results. Specifically, we showed variants associated with quantitative differences in a variety of heart phenotypes such as resting heart rate and pattern of beating were enriched most strongly in embryonic heart enhancers. Most strikingly we observed consistent enrichment of variants associated with atrial fibrillation in strong enhancer segments across all of embryonic heart development. Recent studies found significant enrichment for AF-associated index variants in accessible chromatin from fetal heart and speculated on a fetal origin of AF.70,71 In the data we have presented here, variants that confer risk of AF are in established enhancers active even earlier, specifically those active at 4.5 weeks post conception during the embryonic period of development. This suggests either AF causes a reversion to an embryonic pattern of gene regulation or predisposition to adult onset AF is established extremely early in development. Further study of this phenomenon is clearly warranted and could indicate AF should be considered a congenital heart defect.
One of the most interesting findings of the GWAS analysis came from analysis of the auto-immune disorder Lupus that we had initially intended as a control. Heart disease is a frequent complication in lupus patients and is typically attributed to inflammation of the heart due to lupus specific antibodies that direct immune cells to attack this organ. These results could suggest that inflammation activates portions of the genome that are normally repressed in heart development or that inappropriate activation of regions during heart development predisposes one to these complications. Importantly these results revealed that repressed regions can also be systematically enriched for specific disease relevant loci which has not been previously described for chromatin state segmentation data.
While we confirmed enrichment of a number of cardiac disease and phenotype associated variants in the strong enhancer segments, the original motivation of this study to identify enhancers that harbor variants for CHD risk proved untenable. Despite the common nature of the defects, our results indicate either common variants are not informative for risk of CHD with regard to regulatory element function or these GWAS studies were insufficiently powered to identify such effects. Larger numbers of whole genome sequences from CHD patients will be needed to determine if rare variants in heart-specific enhancer segments play a significant role in CHD incidence.
We have for the first time jointly analyzed gene expression and chromatin state for embryonic human heart development. Our WGCNA identified modules of genes in an unbiased way, yet when we analyzed these groups of genes we uncovered coherent biological functions and expression characteristics across this developmental trajectory. We found that a subset of modules is significantly enriched for both heart specific gene expression and heart specific enhancer activation. Many of these same modules are also enriched for known CHD genes and well connected or “hub” genes in these modules are generally intolerant to loss of function mutations in otherwise healthy individuals. When we systematically interrogated these networks with binding and functional data for the cardiac TF NKX2–5, we uncovered clear physical regulatory connections both within the NKX2–5 containing module and other modules. These NKX2–5 connected modules were enriched for functions this gene has been suggested to regulate, including activation of sarcomere and ion channel genes and repression of neurogenesis.49,92,100–103 These findings demonstrate that our networks represent real biological connections relevant to heart development. Genes with characteristics similar to NKX2–5, such as specificity of expression, highly connected to other genes in WGNCA modules, regulated by heart-specific enhancers, and low tolerance to gene disruption are therefore prime candidates for CHD genes. The gene list we provide in Online Table IX is thus of particular interest for further research and potential CHD diagnostic applications.
We provide all these data sets openly in commonly used formats that are directly comparable to other large consortia. These can be downloaded from GEO, retrieved via public track hub functionality on the UCSC Genome browser, or directly from the Cotney lab website. Our Gini index scoring for over 36,000 genes from 31 tissues is broadly useful for studies of all of these tissues and identifies both housekeeping and highly tissue-specific genes. We anticipate the regulatory regions and genes we have identified will be useful in research on the regulation of heart development, can be used as tissue-specific drivers of reporters, and provide much needed functional context for noncoding variation in clinical whole genome sequencing.104
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Gene expression during mammalian development is controlled by many tissue and time point specific regulatory sequences known as enhancers.
Heart enhancers have been identified in a single human fetal heart and several infant and adult hearts, all of which are derived after major steps in heart patterning.
Genes that are co-expressed during development of tissues and have low rates of mutations in healthy, adult humans have been implicated in developmental disorders and disease.
What New Information Does This Article Contribute?
Annotation of genome-wide chromatin states based on histone modifications during the embryonic period of human heart development revealed thousands of sequences not previously predicted to be active in the heart.
Sequences predicted to be strong enhancers during heart organogenesis are enriched with cardiac transcription factor (TF) binding sites and systematically located near genes with known roles in the developing mammalian heart or shown in this study to have highly specific expression in the developing human heart.
Putative strong enhancer sequences in the embryonic heart are enriched for common sequence variants associated with risk for atrial fibrillation (AF) but not congenital heart defects.
Comprehensive analysis of gene expression in the developing heart revealed organized patterns of co-expression that are enriched for heart relevant biology.
Highly connected “hub” genes in the co-expression network are strongly enriched for genes with low mutational rates in healthy adult humans and likely disease candidates.
Most patients affected by congenital heart defects (CHD) do not have defects in other tissues, a common feature of defects caused by damaged regulatory sequences or “enhanceropathies”. However, enhancers active during the organogenesis phase of human heart development when most CHDs are thought to arise have not been identified. We have systematically characterized chromatin state in human embryonic hearts and revealed over twelve thousand novel putative enhancers. These enhancers harbored all the hallmarks of tissue-specific enhancers: enrichment of cardiac-restricted TF binding sites, located near genes with heart-specific expression patterns, and demonstrated heart specific activity in catalogs of experimentally tested enhancers. Enhancers predicted to be strongly active in the developing human heart were significantly enriched for common variants associated with risk for atrial fibrillation demonstrating potential embryonic origins of this disorder. Construction of networks based on genes with similar trends of expression across heart development revealed a cohort of genes regulated by the canonical cardiac TF NKX2–5. These co-expression networks uncovered over two hundred genes that have similar characteristics as NKX2–5: co-expression with many other genes, specific expression in the heart, and intolerance to mutation. These genes are strong candidate disease genes warranting further study.
ACKNOWLEDGEMENTS
The human embryonic material was provided by the Joint MRC (G0700089)/Wellcome Trust (GR082557) Human Developmental Biology Resource. We thank Ion Moraru, Steven G. King, and Mike Wilson for computer resource support. We also thank John T. Hinson, Bruce Liang, Stefan Pinter, Nagham Khouri Farah, and Emma Wentworth Winchester for critical feedback during the preparation of the manuscript. J. Cotney conceived and designed the study; J. VanOudenhove and A. Wilderman performed wet lab experiments; J. VanOudenhove, T. Yankee, and J. Cotney performed statistical analysis; J. Cotney directed the project; J. VanOudenhove, T. Yankee, and J. Cotney drafted the article. All authors contributed to the final article.
SOURCES OF FUNDING
This work was supported by National Institutes of Health grant R35GM119465 (to J.C.).
Nonstandard Abbreviations and Acronyms
- AF
atrial fibrillation
- AUC
area under the curve
- BH
Benjamini-Hochberg method
- CAAD
Combined Annotation Dependent Depletion
- CHD
congenital heart defect
- ChIP-seq
chromatin immunoprecipitation followed by next-generation sequencing
- ChromHMM
chromatin state segmentation using Hidden Markov Model
- ChromImpute
chromatin signal imputation
- CS
Carnegie Stage
- CM
cardiomyocyte
- DHS
DNAse hypersensitivity site
- EHEs
embryonic heart-specific enhancer segments
- EMERGE
flexible modelling framework to predict genomic regulatory elements
- GATA
Family of transcription facts capable of binding the GATA consensus sequence
- GEO
Gene Expression Omnibus
- GJA1
Gap Junction Protein Alpha 1
- GREAT
Genomic Regions Enrichment of Annotations Tool
- GTEx
Genotype-Tissue Expression Project
- GWAS
genome-wide association study
- HAND2
Heart And Neural Crest Derivatives Expressed 2
- HOMER
Hypergeometric Optimization of Motif EnRichment
- H2A.Z
Histone 2A variant Z
- H3K4me1
Histone H3 lysine 4 monomethylation
- H3K4me2
Histone H3 lysine 4 dimethylation
- H3K4me3
Histone H3 lysine 4 trimethylation
- H3K9me3
Histone H3 lysine 9 trimethylation
- H3K9ac
Histone H3 lysine 9 acetylation
- H3K27ac
Histone H3 lysine 27 acetylation
- H3K27me3
Histone H3 lysine 27 trimethylation
- H3K36me3
Histone H3 lysine 36 trimethylation
- H3K79me2
Histone H3 lysine 79 dimethylation
- H4K20me1
Histone H4 lysine 20 monomethylation
- iPSC
induced pluripotent stem cell
- KLF
Kruppel-like factors
- LINSIGHT
Linear Model Inference of Natural Selection from Interspersed Genomically coHerent elemenTs
- LOUEF
loss-of-function observed/expected upper bound fraction
- NKX2–5
NK2 Homeobox 5
- MDS
multi-dimensional scaling
- MEF2
myocyte enhancer factor-2
- MPRA
massively parallel reporter assay
- MYOCD
Myocardin gene
- PCHi-C
promoter-capture genome wide chromatin conformation capture
- ppi
protein-protein interaction
- RNA-seq
polyA enriched cDNA coupled with next-generation sequencing
- SCN5A
Sodium Voltage-Gated Channel Alpha Subunit 5
- SMAD
Sma and mothers against decapentaplegic gene family
- TBX5
T-box transcription factor 5
- TBX20
T-box transcription factor 20
- TFs
transcription factors
- TSS
transcription start site
- WGCNA
weighted gene co-expression network analysis
- 1_TSSA
ChromHMM State 1 Active Transcription Start Site
- 2_PromU
ChromHMM State 2 Promoter Upstream TSS
- 3_PromD1
ChromHMM State 3 Promoter Downstream TSS 1
- 4_PromD2
ChromHMM State 4 Promoter Downstream TSS 2
- 5_Tx5
ChromHMM State 5 Transcribed – 5’ preferential
- 6_Tx
ChromHMM State 6 Strong transcription
- 7_Tx3
ChromHMM State 7 Transcribed – 3’ preferential
- 8_TxWk
ChromHMM State 8 Weak transcription
- 9_TxReg
ChromHMM State 9 Transcribed & regulatory
- 10_TxEnh5
ChromHMM State 10 Transcribed 5’ preferential and enhancer
- 11_TxEnh3
ChromHMM State 11 Transcribed 3’ preferential and enhancer
- 12_TxEnhW
ChromHMM State 12 Transcribed and Weak enhancer
- 13_EnhA1
ChromHMM State 13 Active enhancer 1
- 14_EnhA2
ChromHMM State 14 Active enhancer 2
- 15_EnhAF
ChromHMM State 15 Active enhancer flank
- 16_EnhW1
ChromHMM State 16 Weak enhancer 1
- 17_EnhW2
ChromHMM State 17 Weak enhancer 2
- 18_EnhAc
ChromHMM State 18 Primary H3K27ac – possible enhancer
- 19_Dnase
ChromHMM State 19 Primary DNase
- 20_ZNF/RPT
ChromHMM State 20 ZNF genes and repeats
- 21_Het
ChromHMM State 21 Heterochromatin
- 22_PromP
ChromHMM State 22 Poised Promoter
- 23_PromBiv
ChromHMM State 23 Bivalent Promoter
- 24_ReprPC
ChromHMM State 24 PolyComb Repressed
- 25_Quies
ChromHMM State 25 Quiescent
Major Resources Table
Animals (in vivo studies) - None
| Species | Vendor or Source | Background Strain | Sex | Persistent ID / URL |
|---|---|---|---|---|
Genetically Modified Animals - None
| Species | Vendor or Source | Background Strain | Other Information | Persistent ID / URL | |
|---|---|---|---|---|---|
| Parent - Male | |||||
| Parent - Female |
Antibodies
| Target antigen | Vendor or Source | Catalog # | Working concentration | Lot # (preferred but not required) | Persistent ID / URL |
|---|---|---|---|---|---|
| H3K27ac | Diagenode | C15410196 | 5ug | RRID: AB_2637079 | |
| H3K4me1 | Diagenode | C15410194 | 2.5ug | RRID: AB_2637078 | |
| H3K4me2 | Abcam | ab7766 | 2.5ug | RRID: AB_2560996 | |
| H3K4me3 | Diagenode | C1541003 | 2.5ug | RRID: AB_2616052 | |
| H3K27me3 | Diagenode | C16410195 | 5ug | RRID: AB_2753161 | |
| H3K36me3 | Diagenode | C15410192 | 5ug | RRID: AB_2744515 | |
| H3K9me3 | Diagenode | C15410193 | 5ug | RRID: AB_2616044 |
DNA/cDNA Clones - None
| Clone Name | Sequence | Source / Repository | Persistent ID / URL |
|---|---|---|---|
Cultured Cells - None
| Name | Vendor or Source | Sex (F, M, or unknown) | Persistent ID / URL |
|---|---|---|---|
Data & Code Availability
| Description | Source / Repository | Persistent ID / URL |
|---|---|---|
| ChIP-seq signals, imputed signal files and chromatin state segmentations | GEO |
GSE137731 and https://cotney.research.uchc.edu/heart/ |
| RNA-seq bigWigs, and counts matrices | GEO |
GSE138799 and https://cotney.research.uchc.edu/heart/ |
| All data tracks | UCSC Track Hubs | Human Embryonic Heart Tissue Epigenomic and Transcriptomic Data from the Cotney Lab at UConn Health |
| Generic scripts used in processing ChIP-Seq and generating chromatin states | Github | https://github.com/cotneylab/ChIP-Seq |
| Scripts used in generation of WGCNA of heart expression data and differential expression analysis | Github | https://github.com/cotneylab/RNA-Seq |
Other
| Description | Source / Repository | Persistent ID / URL |
|---|---|---|
| Human embryonic heart tissue | Joint MRC/Wellcome Trust Human Developmental Biology Resource | www.hdbr.org |
| Software and Algorithms: | ||
| Basemount | Illumina | https://basemount.basespace.illumina.com/ |
| FASTQC (v0.11.5) | Andrews, 2010 |
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ RRID:SCR_014583 |
| MultiQC (v1.1) | Ewels et al., 2016 |
http://multiqc.info/ RRID:SCR_014982 |
| Trimmomatic (v.0.36) | RRID:SCR_011848 | |
| Bowtie2 (v2.2.5) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| PhantomPeakQualTools (v.1.14) | Landt et al., 2012 |
https://code.google.com/p/phantompeakqualtools/ RRID:SCR_005331 |
| HOMER (v4.9) | Heinz et al., 2010 |
http://homer.ucsd.edu/homer/ RRID:SCR_010881 |
| BEDtools (v2.25.0) | Quinlan and Hall, 2010 |
https://github.com/arq5x/bedtools2 RRID:SCR_006646 |
| MACS2 (2.1.1.20160309) | Feng et al., 2012 | https://github.com/taoliu/MACS/ |
| Kent Source Tools (v329) | Kent et al., 2002 | https://github.com/ENCODE-DCC/kentUtils |
| deepTools2 (v2.5.0.1) | Ramírez et al., 2014 | https://github.com/fidelram/deepTools |
| ChromImpute (v1.0.3) | Ernst and Kellis, 2015 | http://www.biolchem.ucla.edu/labs/ernst/ChromImpute/ |
| ChromHMM (v1.12) | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| R Project for Statistical Computing (v3.4.1) | Team, 2017 |
http://www.r-project.org/ RRID:SCR_001905 |
| GREAT (v4.0.4) | McLean et al., 2010 |
http://great.stanford.edu/public/html/splash.php RRID:SCR_005807 |
| DiffBind | Stark and Brown, 2011 | https://doi.org/doi:10.18129/B9.bioc.DiffBind |
| rGREAT | https://doi.org/doi:10.18129/B9.bioc.rGREAT | |
| Homerkit | https://github.com/slowkow/homerkit | |
| Rtsne | https://github.com/jkrijthe/Rtsne | |
| GARFIELD (v2) | Iotchkova et al., 2019 | https://www.ebi.ac.uk/birney-srv/GARFIELD/ |
| GREGOR | RRID:SCR_009165 | |
| GWAS Studies Used: | ||
| GWAS Catalog (retrieved 2019-08-10, All associations v1.0.2) | Welter et al., 2014 |
http://www.ebi.ac.uk/gwas/home RRID:SCR_012745 |
| Systolic Blodd Pressure (2046 Associations) | EFO_0006335 | https://www.ebi.ac.uk/gwas/home |
| Atrial Fibrillation (371 Associations) | EFO_0000275 | https://www.ebi.ac.uk/gwas/home |
| Electrocardiograph Traits and Measures (38 Associations) | EFO_0004327 | https://www.ebi.ac.uk/gwas/home |
| Resting Heart Rate (129 Assocations) | EFO_0005054 | https://www.ebi.ac.uk/gwas/home |
| Congenital Heart Defects (72 Associations) | EFO_0005207, EFO_0005269 | https://www.ebi.ac.uk/gwas/home |
| QT interval (311 Associations) | EFO_0004682 | https://www.ebi.ac.uk/gwas/home |
| QRS (186 Associations) | EFO_0005055, EFO_0007742, | https://www.ebi.ac.uk/gwas/home |
| Coronary Artery Disease (936 Associations) | EFO_0000378 | https://www.ebi.ac.uk/gwas/home |
| Crohns Disease (594 Associations) | EFO_0000384 | https://www.ebi.ac.uk/gwas/home |
| QRS Interval GWAS Summary Statistics (Total SNPs with calculated p-value: 26566045) | Wojcik et al., 2019 | ftp://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/WojcikGL_31217584_GCST008054/WojcikG_PMID_qr s_interval.gz |
| Resting Heart Rate GWAS Summary Statistics (Total SNPs with calculated p-value: 5264922) | Zhu et al., 2019 | ftp://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistic s/ZhuZ_30940143_GCST007609/ZhuZ_30940143_ukbb.bolt_460K_selfRepWhite.rhrmean.assoc.gz |
| Atrial Fibrillation GWAS Summary Statistics (Total SNPs with calculated p-value: 12149979) | Roselli et al., 2018 | https://personal.broadinstitute.org/mvon/AF_HRC_GWAS _ALLv11.zip |
| P-Wave Duration GWAS Summary Statistics (Total SNPs with calculated p-value: 872325) | Christophersen et al., 2017 | ftp://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistic s/ChristophersenIE_28794112_GCST004826/harmonise d/28794112-GCST004826-EFO_0005094build37.f.tsv.gz |
| Systemic lupus erythematous GWAS Summary Statistics (Total SNPs with calculated p-value: 7915251) | Bentham et al., 2015 | ftp://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistic s/BenthamJ_26502338_GCST003156/harmonised/26502 338-GCST003156-EFO_0002690-build37.f.tsv.gz |
| Crohn’s Disease GWAS Summary Statistics (Total SNPs with calculated p-value: 154590) | Liu et al., 2015 | ftp://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistic s/LiuJZ_26192919_GCST003044/CD_trans_ethnic_asso ciation_summ_stats_b37.txt.gz |
Footnotes
DISCLOSURES
J. Cotney is listed as the inventor on a patent application submitted by the University of Connecticut related to the gene panel described in Online Table IX.
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Van Der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol 2011;58:2241–2247. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Chen S, Zühlke L, Black GC, Choy M-K, Li N, Keavney BD. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol [Internet]. 2019. [cited 2019 Nov 15];48:455–463. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30783674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC). Racial differences by gestational age in neonatal deaths attributable to congenital heart defects --- United States, 2003–2006. MMWR Morb Mortal Wkly Rep [Internet]. 2010. [cited 2019 Nov 15];59:1208–11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20864921 [PubMed] [Google Scholar]
- 4.Cowan JR, Ware SM. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol 2015;42:373–393. [DOI] [PubMed] [Google Scholar]
- 5.Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circ. Res 2017;120:923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, O’sullivan J, Winlaw DS, Bu’lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012;91:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, Depalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA, Srivastava D, Lo CW, Shen Y, Watkins WS, Yandell M, Yost HJ, Tristani-Firouzi M, Newburger JW, Roberts AE, Kim R, Zhao H, Kaltman JR, Goldmuntz E, Chung WK, Seidman JG, Gelb BD, Seidman CE, Lifton RP, Brueckner M. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glessner JT, Bick AG, Ito K et al. Increased Frequency of De Novo Copy Number Variations in Congenital Heart Disease by Integrative Analysis of SNP Array and Exome Sequence Data. Circ Res [Internet]. 2014. [cited 2019 Nov 15];115:884–896. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4209190/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA Jr, Kim R, Bilguvar K, López-Giráldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Sci (New York, NY) [Internet]. 2015;350:1262–1266. Available from: http://www.sciencemag.org/content/350/6265/1262.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe’er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, Lifton RP. De novo mutations in histone-modifying genes in congenital heart disease. Nature [Internet]. 2013;498:220–223. Available from: http://www.nature.com/doifinder/10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet [Internet]. 2012;21:3255–3263. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=22543974&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascenção K, Rummel C, Ovchinnikova S, Mazin PV, Xenarios I, Harshman K, Mort M, Cooper DN, Sandi C, Soares MJ, Ferreira PG, Afonso S, Carneiro M, Turner JMA, VandeBerg JL, Fallahshahroudi A, Jensen P, Behr R, Lisgo S, Lindsay S, Khaitovich P, Huber W, Baker J, Anders S, Zhang YE, Kaessmann H. Gene expression across mammalian organ development. Nature [Internet]. 2019. [cited 2019 Nov 15];571:505–509. Available from: http://www.nature.com/articles/s41586-019-1338-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology [Internet]. Fourth Ann Arbor, MI: Churchill Livingstone/Elsevier; 2009. Available from: http://books.google.com/books?id=56xqAAAAMAAJ&q=human+embryology+larsen&dq=human+embryology+larsen&hl=&cd=2&source=gbs_api [Google Scholar]
- 14.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry CE. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stone NR, Gifford CA, Thomas R, Pratt KJB, Samse-Knapp K, Mohamed TMA, Radzinsky EM, Schricker A, Ye L, Yu P, van Bemmel JG, Ivey KN, Pollard KS, Srivastava D. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell. 2019;25:87–102.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI, Tippens ND, Li M, Narasimha A, Radzinsky E, Moon-Grady AJ, Yu H, Pruitt BL, Snyder MP, Srivastava D. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan GC, Zhou P, Pu WT. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature [Internet]. 2015;518:317–330. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25693563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature [Internet]. 2012;489:57–74. Available from: http://www.nature.com/doifinder/10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilderman A, VanOudenhove J, Kron J, Noonan JP, Cotney J. High-Resolution Epigenomic Atlas of Human Embryonic Craniofacial Development. Cell Rep. 2018;23:1581–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chadwick LH. The NIH Roadmap Epigenomics Program data resource. Epigenomics [Internet]. 2012;4:317–324. Available from: http://www.futuremedicine.com/doi/abs/10.2217/epi.12.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, May D, Spurrell CH, Plajzer-Frick I, Pickle CS, Lee E, Garvin TH, Kato M, Akiyama JA, Afzal V, Lee AY, Gorkin DU, Ren B, Rubin EM, Visel A, Pennacchio LA. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun [Internet]. 2016;7:12923 Available from: https://www.ncbi.nlm.nih.gov/pubmed/27703156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Duijvenboden K, de Boer BA, Capon N, Ruijter JM, Christoffels VM. EMERGE: a flexible modelling framework to predict genomic regulatory elements from genomic signatures. Nucleic Acids Res [Internet]. 2015;gkv1144. Available from: http://nar.oxfordjournals.org/content/early/2015/11/02/nar.gkv1144.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Ouwerkerk AF, Bosada FM, van Duijvenboden K, Hill MC, Montefiori LE, Scholman KT, Liu J, de Vries AAF, Boukens BJ, Ellinor PT, Goumans MJTH, Efimov IR, Nobrega MA, Barnett P, Martin JF, Christoffels VM. Identification of atrial fibrillation associated genes and functional non-coding variants. Nat Commun. 2019;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer-Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A, Rubin EM. In vivo enhancer analysis of human conserved non-coding sequences. Nature [Internet]. 2006;444:499–502. Available from: http://www.nature.com/doifinder/10.1038/nature05295 [DOI] [PubMed] [Google Scholar]
- 27.Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet [Internet]. 2008;40:158–160. Available from: http://www.nature.com/ng/journal/v40/n2/abs/ng.2007.55.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, FitzPatrick DR, Afzal V, Pennacchio LA, Rubin EM, Noonan JP. Human-Specific Gain of Function in a Developmental Enhancer. Sci (New York, NY) [Internet]. 2008;321:1346–1350. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1159974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, Rosenbloom KR, Kent J, Haussler D. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res [Internet]. 2009;20:110–121. Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res [Internet]. 2018;47:D886–D894. Available from: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet [Internet]. 2014;46:310–315. Available from: https://www.ncbi.nlm.nih.gov/pubmed/24487276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YF, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet. 2017;49:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dreßen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun [Internet]. 2018. [cited 2019 Nov 15];9:391 Available from: http://www.nature.com/articles/s41467-017-02762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res [Internet]. 2007;35:D88–92. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=17130149&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol [Internet]. 2015;33:364–376. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25690853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science (80-. ) 2006;313:1922–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitcomb J, Gharibeh L, Nemer M. From embryogenesis to adulthood: Critical role for GATA factors in heart development and function. IUBMB Life [Internet]. 2019. [cited 2019 Nov 18];iub.2163. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31520462 [DOI] [PubMed] [Google Scholar]
- 40.Buijtendijk MFJ, Barnett P, Hoff MJB. Development of the human heart. Am J Med Genet Part C Semin Med Genet [Internet]. 2020. [cited 2020 Jun 15];184:7–22. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.c.31778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sylva M, van den Hoff MJB, Moorman AFM. Development of the human heart. Am J Med Genet Part A [Internet]. 2014. [cited 2020 Jun 15];164:1347–1371. Available from: http://doi.wiley.com/10.1002/ajmg.a.35896 [DOI] [PubMed] [Google Scholar]
- 42.Monaghan MG, Linneweh M, Liebscher S, Van Handel B, Layland SL, Schenke-Layland K. Endocardial-to-mesenchymal transformation and mesenchymal cell colonization at the onset of human cardiac valve development. Dev. 2016;143:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez-Roelens I, De Roy L, … CO-E journal of, 2006 undefined. A novel CSX/NKX2–5 mutation causes autosomal-dominant AV block: are atrial fibrillation and syncopes part of the phenotype? nature.com [Internet] [cited 2020 Jun 15];Available from: https://www.nature.com/articles/5201702 [DOI] [PubMed] [Google Scholar]
- 44.McElhinney D, Geiger E, … JB-J of the, 2003 undefined. NKX2. 5mutations in patients with congenital heart disease. onlinejacc.org [Internet] [cited 2020 Jun 15];Available from: http://www.onlinejacc.org/content/42/9/1650.abstract [Google Scholar]
- 45.Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. [DOI] [PubMed] [Google Scholar]
- 46.Schott J, Benson D, Basson C, … WP-, 1998 undefined. Congenital heart disease caused by mutations in the transcription factor NKX2–5 [Internet]. [cited 2020 Jun 15]. Available from: http://science.sciencemag.org/ [DOI] [PubMed] [Google Scholar]
- 47.Nakashima Y, Yanez DA, Touma M, Nakano H, Jaroszewicz A, Jordan MC, Pellegrini M, Roos KP, Nakano A. Nkx2–5 suppresses the proliferation of atrial myocytes and conduction system. Circ Res. 2014;114:1103–1113. [DOI] [PubMed] [Google Scholar]
- 48.Jay P, Rozhitskaya O, Dorfman AL, Ueyama T. Haploinsufficiency of the cardiac transcription factor Nkx2–5 variably affects the expression of putative target genes Optimal strategy for monitoring long-term performance of Fontan circulatory arrangement View project. Artic FASEB J [Internet]. 2005. [cited 2020 Jun 15];19:1495–1497. Available from: https://www.researchgate.net/publication/7770383 [DOI] [PubMed] [Google Scholar]
- 49.Anderson DJ, Kaplan DI, Bell KM, Koutsis K, Haynes JM, Mills RJ, Phelan DG, Qian EL, Leitoguinho AR, Arasaratnam D, Labonne T, Ng ES, Davis RP, Casini S, Passier R, Hudson JE, Porrello ER, Costa MW, Rafii A, Curl CL, Delbridge LM, Harvey RP, Oshlack A, Cheung MM, Mummery CL, Petrou S, Elefanty AG, Stanley EG, Elliott DA. NKX2–5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat Commun [Internet]. 2018. [cited 2019 Nov 18];9:1373 Available from: http://www.nature.com/articles/s41467-018-03714-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pott S, Lieb JD. What are super-enhancers? Nat. Genet 2015;47:8–12. [DOI] [PubMed] [Google Scholar]
- 53.Vatta M. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–345. [DOI] [PubMed] [Google Scholar]
- 54.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AAM, Brugada R, Schott JJ, Ackerman MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Hear Rhythm. 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Man JCK, Mohan RA, Boogaard M van den, Hilvering CRE, Jenkins C, Wakker V, Bianchi V, Laat W de, Barnett P, Boukens BJ, Christoffels VM. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat Commun. 2019;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eppinga RN, Hagemeijer Y, Burgess S, Hinds DA, Stefansson K, Gudbjartsson DF, Van Veldhuisen DJ, Munroe PB, Verweij N, Van Der Harst P. Identification of genomic loci associated with resting heart rate and shared genetic predictors with all-cause mortality. Nat Genet. 2016;48:1557–1563. [DOI] [PubMed] [Google Scholar]
- 57.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, Sorokin EP, Avery CL, Belbin GM, Bien SA, Cheng I, Cullina S, Hodonsky CJ, Hu Y, Huckins LM, Jeff J, Justice AE, Kocarnik JM, Lim U, Lin BM, Lu Y, Nelson SC, Park SSL, Poisner H, Preuss MH, Richard MA, Schurmann C, Setiawan VW, Sockell A, Vahi K, Verbanck M, Vishnu A, Walker RW, Young KL, Zubair N, Acuña-Alonso V, Ambite JL, Barnes KC, Boerwinkle E, Bottinger EP, Bustamante CD, Caberto C, Canizales-Quinteros S, Conomos MP, Deelman E, Do R, Doheny K, Fernández-Rhodes L, Fornage M, Hailu B, Heiss G, Henn BM, Hindorff LA, Jackson RD, Laurie CA, Laurie CC, Li Y, Lin DY, Moreno-Estrada A, Nadkarni G, Norman PJ, Pooler LC, Reiner AP, Romm J, Sabatti C, Sandoval K, Sheng X, Stahl EA, Stram DO, Thornton TA, Wassel CL, Wilkens LR, Winkler CA, Yoneyama S, Buyske S, Haiman CA, Kooperberg C, Le Marchand L, Loos RJF, Matise TC, North KE, Peters U, Kenny EE, Carlson CS. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kapoor A, Lee D, Zhu L, Soliman EZ, Grove ML, Boerwinkle E, Arking DE, Chakravarti A. Multiple SCN5A variant enhancers modulate its cardiac gene expression and the QT interval. Proc Natl Acad Sci U S A. 2019;166:10636–10645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khan A, Zhang X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res [Internet]. 2016;44:D164–71. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26438538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boogerd CJ, Zhu X, Aneas I, Sakabe N, Zhang L, Sobreira DR, Montefiori L, Bogomolovas J, Joslin AC, Zhou B, Chen J, Nobrega MA, Evans SM. Tbx20 Is Required in Mid-Gestation Cardiomyocytes and Plays a Central Role in Atrial Development. Circ Res [Internet]. 2018. [cited 2020 Jun 15];123:428–442. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.118.311339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay JP, Waddell LB, Cole AD, Hayward C, Keogh A, Macdonald P, Griffiths L, Fatkin D, Sholler GF, Zorn AM, Feneley MP, Winlaw DS, Harvey RP. Mutations in crdiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mittal A, Sharma R, Prasad R, Bahl A, Khullar M. Role of cardiac TBX20 in dilated cardiomyopathy. Mol Cell Biochem. 2016;414:129–136. [DOI] [PubMed] [Google Scholar]
- 64.Montefiori LE, Sobreira DR, Sakabe NJ, Aneas I, Joslin AC, Hansen GT, Bozek G, Moskowitz IP, McNally EM, Nóbrega MA. A promoter interaction map for cardiovascular disease genetics. Elife [Internet]. 2018. [cited 2019 Nov 18];7 Available from: https://elifesciences.org/articles/35788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jansen JA, Van Veen TAB, De Jong S, Van Der Nagel R, Van Stuijvenberg L, Driessen H, Labzowski R, Oefner CM, Bosch AA, Nguyen TQ, Goldschmeding R, Vos MA, De Bakker JMT, Van Rijen HVM. Reduced Cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ Arrhythmia Electrophysiol. 2012;5:380–390. [DOI] [PubMed] [Google Scholar]
- 66.Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, Wärdell E, Custodio J, Reimegård J, Salmén F, Österholm C, Ståhl PL, Sundström E, Åkesson E, Bergmann O, Bienko M, Månsson-Broberg A, Nilsson M, Sylvén C, Lundeberg J. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 2019;179:1647–1660.e19. [DOI] [PubMed] [Google Scholar]
- 67.Kieffer-Kwon K-R, Tang Z, Mathe E, Qian J, Sung M-H, Li G, Resch W, Baek S, Pruett N, Grontved L, Vian L, Nelson S, Zare H, Hakim O, Reyon D, Yamane A, Nakahashi H, Kovalchuk AL, Zou J, Joung JK, Sartorelli V, Wei C-L, Ruan X, Hager GL, Ruan Y, Casellas R. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell [Internet]. 2013;155:1507–1520. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=24360274&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demare LE, Leng J, Cotney JL, Reilly SK, Yin J, Sarro R, Noonan JP. The genomic landscape of cohesin-associated chromatin interactions. Genome Res [Internet]. 2013;23:1224–1234. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23704192&retmode=ref&cmd=prlinks [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iotchkova V, Ritchie GRS, Geihs M, Morganella S, Min JL, Walter K, Timpson NJ, Dunham I, Birney E, Soranzo N. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat Genet. 2019;51:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, Gabrielsen ME, Schmidt EM, Beaumont R, Wolford BN, Lin M, Brummett CM, Preuss MH, Refsgaard L, Bottinger EP, Graham SE, Surakka I, Chu Y, Skogholt AH, Dalen H, Boyle AP, Oral H, Herron TJ, Kitzman J, Jalife J, Svendsen JH, Olesen MS, Njølstad I, Løchen ML, Baras A, Gottesman O, Marcketta A, O’Dushlaine C, Ritchie MD, Wilsgaard T, Loos RJF, Frayling TM, Boehnke M, Ingelsson E, Carey DJ, Dewey FE, Kang HM, Abecasis GR, Hveem K, Willer CJ. Genome-wide Study of Atrial Fibrillation Identifies Seven Risk Loci and Highlights Biological Pathways and Regulatory Elements Involved in Cardiac Development. Am J Hum Genet. 2018;102:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, Surakka I, Mathis MR, Yamazaki M, Crawford RD, Gabrielsen ME, Skogholt AH, Holmen OL, Lin M, Wolford BN, Dey R, Dalen H, Sulem P, Chung JH, Backman JD, Arnar DO, Thorsteinsdottir U, Baras A, O’Dushlaine C, Holst AG, Wen X, Hornsby W, Dewey FE, Boehnke M, Kheterpal S, Mukherjee B, Lee S, Kang HM, Holm H, Kitzman J, Shavit JA, Jalife J, Brummett CM, Teslovich TM, Carey DJ, Gudbjartsson DF, Stefansson K, Abecasis GR, Hveem K, Willer CJ. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, Arking DE, Barnard J, Bartz TM, Benjamin EJ, Bihlmeyer NA, Bis JC, Bloom HL, Boerwinkle E, Bottinger EB, Brody JA, Calkins H, Campbell A, Cappola TP, Carlquist J, Chasman DI, Chen LY, Chen Y-DI, Choi E-K, Choi SH, Christophersen IE, Chung MK, Cole JW, Conen D, Cook J, Crijns HJ, Cutler MJ, Damrauer SM, Daniels BR, Darbar D, Delgado G, Denny JC, Dichgans M, Dörr M, Dudink EA, Dudley SC, Esa N, Esko T, Eskola M, Fatkin D, Felix SB, Ford I, Franco OH, Geelhoed B, Grewal RP, Gudnason V, Guo X, Gupta N, Gustafsson S, Gutmann R, Hamsten A, Harris TB, Hayward C, Heckbert SR, Hernesniemi J, Hocking LJ, Hofman A, Horimoto ARVR, Huang J, Huang PL, Huffman J, Ingelsson E, Ipek EG, Ito K, Jimenez-Conde J, Johnson R, Jukema JW, Kääb S, Kähönen M, Kamatani Y, Kane JP, Kastrati A, Kathiresan S, Katschnig-Winter P, Kavousi M, Kessler T, Kietselaer BL, Kirchhof P, Kleber ME, Knight S, Krieger JE, Kubo M, Launer LJ, Laurikka J, Lehtimäki T, Leineweber K, Lemaitre RN, Li M, Lim HE, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet [Internet]. 2018. [cited 2019 Nov 18];50:1225–1233. Available from: http://www.nature.com/articles/s41588-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Sci (New York, NY) [Internet]. 2012;337:1190–1195. Available from: http://www.sciencemag.org/content/337/6099/1190.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B, Leek JT. Reproducible RNA-seq analysis using recount2. Nat Biotechnol [Internet]. 2017;35:319–321. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28398307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nellore A, Collado-Torres L, Jaffe AE, Alquicira-Hernandez J, Wilks C, Pritt J, Morton J, Leek JT, Langmead B. Rail-RNA: scalable analysis of RNA-seq splicing and coverage. Bioinformatics [Internet]. 2017;33:4033–4040. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27592709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Consortium GTe. The Genotype-Tissue Expression (GTEx) project. Nat Genet [Internet]. 2013;45:580–585. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23715323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Der Maaten L, Hinton G. Visualizing Data using t-SNE. 2008. [Google Scholar]
- 79.Lage K, Hansena NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK, Szallasi Z, Jensen TS, Brunak S. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci U S A. 2008;105:20870–20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park YS, Parsana P, Segrè AV, Strober BJ, Zappala Z, Cummings BB, Gelfand ET, Hadley K, Huang KH, Lek M, Li X, Nedzel JL, Nguyen DY, Noble MS, Sullivan TJ, Tukiainen T, MacArthur DG, Getz G, Addington A, Guan P, Koester S, Little AR, Lockhart NC, Moore HM, Rao A, Struewing JP, Volpi S, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley D, Mash DC, Davis DA, Sobin L, Barcus ME, Branton PA, Abell NS, Balliu B, Delaneau O, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Han B, He AZ, Hormozdiari F, Li X, Liu B, Kang EY, McDowell IC, Ongen H, Palowitch JJ, Peterson CB, Quon G, Ripke S, Saha A, Shabalin AA, Shimko TC, Sul JH, Teran NA, Tsang EK, Zhang H, Zhou YH, Bustamante CD, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang L, Chen H, Pinello L, Yuan G-C. GiniClust: detecting rare cell types from single-cell gene expression data with Gini index. Genome Biol [Internet]. 2016. [cited 2019 Nov 18];17:144 Available from: http://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-1010-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.GINI C IL DIVERSO ACCRESCIMENTO DELLE CLASSI SOCIALI E LA CONCENTRAZIONE DELLA RICCHEZZA. G degli Econ [Internet]. 1909;38 (Anno 2:27–83. Available from: http://www.jstor.org/stable/23221859 [Google Scholar]
- 83.O’Hagan S, Wright Muelas M, Day PJ, Lundberg E, Kell DB. GeneGini: Assessment via the Gini Coefficient of Reference “Housekeeping” Genes and Diverse Human Transporter Expression Profiles. Cell Syst. 2018;6:230–244.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim KH, Kim TG, Micales BK, Lyons GE, Lee Y. Dynamic expression patterns of leucine-rich repeat containing protein 10 in the heart. Dev Dyn. 2007;236:2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qu XK, Yuan F, Li RG, Xu L, Jing WF, Liu H, Xu YJ, Zhang M, Liu X, Fang WY, Yang YQ, Qiu XB. Prevalence and spectrum of LRRC10 mutations associated with idiopathic dilated cardiomyopathy. Mol Med Rep. 2015;12:3718–3724. [DOI] [PubMed] [Google Scholar]
- 86.Werling DM, Pochareddy S, Choi J, An JY, Sheppard B, Peng M, Li Z, Dastmalchi C, Santpere G, Sousa AMM, Tebbenkamp ATN, Kaur N, Gulden FO, Breen MS, Liang L, Gilson MC, Zhao X, Dong S, Klei L, Cicek AE, Buxbaum JD, Adle-Biassette H, Thomas JL, Aldinger KA, O’Day DR, Glass IA, Zaitlen NA, Talkowski ME, Roeder K, State MW, Devlin B, Sanders SJ, Sestan N. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 2020;31:107489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell [Internet]. 2013;155:1008–1021. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867413013494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, Murtha MT, Bichsel C, Niu W, Cotney J, Ercan-Sencicek AG, Gockley J, Gupta AR, Han W, He X, Hoffman EJ, Klei L, Lei J, Liu W, Liu L, Lu C, Xu X, Zhu Y, Mane SM, Lein ES, Wei L, Noonan JP, Roeder K, Devlin B, Sestan N, State MW. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell [Internet]. 2013;155:997–1007. Available from: https://www.ncbi.nlm.nih.gov/pubmed/24267886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics [Internet]. 2008. [cited 2019 Nov 18];9:559 Available from: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature [Internet]. 2020. [cited 2020 Jun 15];581:434–443. Available from: http://www.nature.com/articles/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kasahara H, Ueyama T, Wakimoto H, Liu MK, Maguire CT, Converso KL, Kang PM, Manning WJ, Lawitts J, Paul DL, Berul CI, Izumo S. Nkx2.5 homeoprotein regulates expression of gap junction protein connexin 43 and sarcomere organization in postnatal cardiomyocytes. J Mol Cell Cardiol [Internet]. 2003. [cited 2019 Nov 18];35:243–56. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12676539 [DOI] [PubMed] [Google Scholar]
- 92.Pashmforoush M, Lu JT, Chen H St., Amand T, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR. Nkx2–5 pathways and congenital heart disease: Loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell [Internet]. 2004. [cited 2019 Nov 18];117:373–386. Available from: http://www.embase.com/search/results?subaction=viewrecord&from=export&id=L38534543 [DOI] [PubMed] [Google Scholar]
- 93.Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. Ablation of Nkx2–5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res. 2011;91:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu C, Chang H, Li XH, Qi YF, Wang JO, Zhang Y, Yang XH. Network Meta-Analysis on the Effects of DNA Damage Response-Related Gene Mutations on Overall Survival of Breast Cancer Based on TCGA Database. J Cell Biochem [Internet]. 2017;118:4728–4734. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28513990 [DOI] [PubMed] [Google Scholar]
- 95.Yin L, Cai Z, Zhu B, Xu C. Identification of key pathways and genes in the dynamic progression of HCC based on WGCNA. Genes (Basel). 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langfelder P, Mischel PS, Horvath S. When is hub gene selection better than standard meta-analysis? PLoS One [Internet]. 2013;8:e61505 Available from: https://www.ncbi.nlm.nih.gov/pubmed/23613865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hartman ME, Liu Y, Zhu WZ, Chien WM, Weldy CS, Fishman GI, Laflamme MA, Chin MT. Myocardial deletion of transcription factor CHF1/Hey2 results in altered myocyte action potential and mild conduction system expansion but does not alter conduction system function or promote spontaneous arrhythmias. FASEB J. 2014;28:3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koibuchi N, Chin MT. CHF1/Hey2 plays a pivotal role in left ventricular maturation through suppression of ectopic atrial gene expression. Circ Res. 2007;100:850–855. [DOI] [PubMed] [Google Scholar]
- 99.Bruneau BG, Bao ZZ, Tanaka M, Schott JJ, Izumo S, Cepko CL, Seidman JG, Seidman CE. Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2–5 and dHand. Dev Biol. 2000;217:266–277. [DOI] [PubMed] [Google Scholar]
- 100.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science (80- ). 1998;281:108–111. [DOI] [PubMed] [Google Scholar]
- 101.Furtado MB, Wilmanns JC, Chandran A, Tonta M, Biben C, Eichenlaub M, Coleman HA, Berger S, Bouveret R, Singh R, Harvey RP, Ramialison M, Pearson JT, Parkington HC, Rosenthal NA, Costa MW. A novel conditional mouse model for NKX2–5 reveals transcriptional regulation of cardiac ion channels. Differentiation. 2015;91:29–41. [DOI] [PubMed] [Google Scholar]
- 102.Reamon-Buettner SM, Borlak J. Somatic NKX2–5 mutations as a novel mechanism of disease in complex congenital heart disease. J Med Genet. 2004;41:684–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blankvoort S, Witter MP, Noonan J, Cotney J, Kentros C. Marked Diversity of Unique Cortical Enhancers Enables Neuron-Specific Tools by Enhancer-Driven Gene Expression. Curr Biol [Internet]. 2018;28:2103–2114 e5. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30008330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, Replogle J, Syvänen AC, Rönnblom L, Graham RR, Wither JE, Rioux JD, Alarcón-Riquelme ME, Vyse TJ. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47:1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cotney JL, Noonan JP. Chromatin immunoprecipitation with fixed animal tissues and preparation for high-throughput sequencing. Cold Spring Harb Protoc [Internet]. 2015;2015:419 Available from: https://www.ncbi.nlm.nih.gov/pubmed/25834253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods [Internet]. 2012;9:357–359. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22388286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res [Internet]. 2012;22:1813–1831. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22955991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc [Internet]. 2012;7:1728–1740. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22936215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods [Internet]. 2012;9:215–216. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22373907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res [Internet] 2014;42:W187–91. Available from: http://nar.oxfordjournals.org/content/42/W1/W187.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wilderman A, VanOudenhove J, Kron J, Noonan JP, Cotney J. High-Resolution Epigenomic Atlas of Human Embryonic Craniofacial Development. Cell Rep. 2018;23:1581–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stark R and Brown G DiffBind: differential binding analysis of ChIP-Seq peak datatle. 2016. [cited 2019 Nov 18];1–29. Available from: http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf [Google Scholar]
- 114.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell [Internet]. 2010;38:576–589. Available from: https://www.ncbi.nlm.nih.gov/pubmed/20513432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol [Internet]. 2010;28:495–501. Available from: https://www.ncbi.nlm.nih.gov/pubmed/20436461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Collado-Torres L, Nellore A, Jaffe AE. recount workflow: Accessing over 70,000 human RNA-seq samples with Bioconductor. F1000Research [Internet]. 2017. [cited 2019 Nov 18];6:1558 Available from: https://f1000research.com/articles/6-1558/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that anonymized data have been made publicly available at the Gene Expression Omnibus (GEO) and can be accessed at via accession numbers GSE137731 (ChIP-Seq) and GSE138799 (RNA-Seq). Because of the sensitive nature of the raw sequencing data collected for this study, requests to access those data from qualified researchers trained in human subject confidentiality protocols and approved by an Institutional Review Board may be sent to the corresponding author. All other supporting data and materials presented within this article and in the Data Supplement are available from the corresponding author upon reasonable request.