

Abstract
Although patients with non-small cell lung cancer harboring activating mutations in the epidermal growth factor receptor (EGFR) show good clinical response to EGFR tyrosine kinase inhibitors (TKIs), patients eventually develop acquired resistance. Previous studies have shown that several microRNAs (miRNAs) are involved in EGFR TKI resistance. Here, we aimed to investigate whether miR-146b-5p sensitizes the EGFR TKI-resistant lung cancer cells. Clinical analysis showed that miR-146b-5p expression in lung cancer cells isolated from pleural effusions of treatment-naive patients was significantly higher than that after acquiring resistance to EGFR TKI treatment. Ectopic expression of miR-146b-5p in EGFR TKI-resistant cells enhanced EGFR TKI-induced apoptosis. The same results were observed in EGFR-dependent and -independent osimertinib-resistant primary cancer cells (PE3479 and PE2988). Mechanically, miR-146b-5p suppressed nuclear factor κB (NF-κB) activity and NF-κB-related IL-6 and IL-8 production by targeting IRAK1. A negative correlation was observed between miR-146b-5p and IRAK1 in clinical specimens. In rescue experiments, restoration of IRAK1 expression reversed the effects of miR-146b-5p on EGFR TKI sensitivity and recovered NF-κB-regulated IL-6 and IL-8 production. In conclusion, miR-146b-5p/IRAK1/NF-κB signaling is important in promoting EGFR TKI resistance, and miR-146b-5p may be a useful tool for overcoming EGFR TKI resistance.
Keywords: NSCLC, EGFR TKI, resistance, IRAK1, microRNA, NF-κB
Graphical Abstract
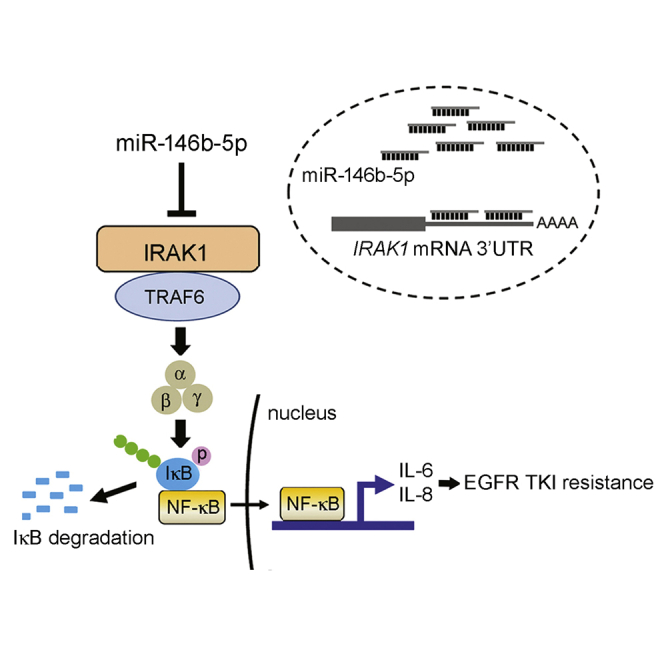
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) is a challenge in the treatment of lung cancer. Alterations in microRNA (miR)-146b-5p and interleukin-1 receptor-associated kinase 1 (IRAK1) expression are implicated in acquired resistance to EGFR-TKIs. Based on our findings, ectopic miR-146b-5p overcomes EGFR TKI resistance in human lung cancer by targeting the IRAK1/nuclear factor κB (NF-κB) signaling pathway.
Introduction
Lung cancer remains the leading cause of cancer-related deaths worldwide.1 Genetic alteration and abnormal epigenetic regulations may lead to lung carcinogenesis.2 Oncogenic activating mutations of epidermal growth factor receptor (EGFR) have been implicated in the initiation and progression of lung cancers. Approximately 10%–20% of patients with non-small cell lung cancer (NSCLC) in western countries, and 40%–50% of patients in East Asia harbor tumors with EGFR-activating mutations.3 In the past decade, EGFR tyrosine kinase inhibitors (TKIs) were developed as therapeutics for the treatment of patients with lung cancer harboring EGFR-activating mutations. Four EGFR TKIs (gefitinib, erlotinib, afatinib, and osimertinib) have been approved by the U.S. Food and Drug Administration for treating patients with NSCLC. These EGFR TKIs inhibit the phosphorylation of EGFR and interrupt downstream survival signaling. Clinically, the objective response rate is as high as 70% in EGFR TKI treatment.4,5
Despite the demonstrated benefits of EGFR TKI therapy, most patients eventually experience acquired resistance-related relapse after approximately 10 months of therapy.6 Various mechanisms of acquired resistance to EGFR TKIs have been reported, such as the appearance of new secondary mutations in EGFR, phenotype transition, aberration in downstream pathways, and activation of alternative pathways.7 New secondary mutations in EGFR are the most frequent cause of TKI resistance in patients with NSCLC. Approximately 50% of NSCLC patients with acquired resistance to EGFR TKIs developed the secondary T790M mutation within EGFR.3 Other mechanisms of acquired EGFR TKI resistance include mutations in PI3K, BRAF, or RAS; EGFR, HER2, or MET amplifications; and loss of PTEN.8 Additional mechanisms of acquired resistance shared by TKIs of all three generations include epithelial-mesenchymal transition (EMT) and small cell lung cancer phenotypic transformation, which account for approximately 10% of cases of EGFR TKI resistance in lung cancer.6 However, EGFR TKI resistance is not completely understood. Furthermore, up to 10% of patients harbor non-identified genetic or non-genetic resistant mechanisms, which require further investigation.
MicroRNAs (miRNAs) are evolutionarily conserved non-protein-coding RNAs, which negatively regulate gene expression, mainly via direct interaction with the 3′ untranslated region (3′ UTR) of corresponding target mRNAs.9 They play critical roles in multiple essential physiological processes, such as development, differentiation, proliferation, apoptosis, inflammation, and metabolism.10, 11, 12 Aberrant expression of miRNAs has been observed in lung cancer, and they act as oncogenic or tumor-suppressive miRNAs depending on the targets.13,14 Overexpression of tumor-suppressive miRNAs (miR-34a or miR-133b) in lung cancer cells inhibits tumor growth by directly binding to the 3′ UTR of EGFR and interrupting EGFR-mediated signaling.15,16 In contrast, aberrant expression of certain miRNAs was related to tumor initiation, metastasis, and progression. Previous studies have indicated that a high level of miR-137 correlated with shorter overall survival of patients with lung adenocarcinoma, and upregulation of miR-137 was found to trigger lung cancer cell invasion and progression by directly suppressing TFAP2C.17
In addition, miRNAs are involved in the development of chemotherapy and EGFR TKI resistance in patients with NSCLC.18,19 For example, miR-96 induces cisplatin chemoresistance in NSCLC cells by downregulating SAMD9. Silencing of miR-96 in NSCLC cells significantly increased SAMD9 expression and enhanced cisplatin-induced apoptosis, which could be completely reversed by knocking down SAMD9.20 Low miR-130a expression was detected in gefitinib-resistant cells, while re-expression of miR-130a restored the sensitivity to gefitinib by targeting the compensatory c-MET-mediated signaling pathway.21 Similar biological effect was observed for miR-138-5p. Downregulation of miR-138-5p contributed to gefitinib resistance, and restoration of miR-138-5p was sufficient to reverse gefitinib resistance by negatively regulating the G protein-coupled receptor.22 Accumulating evidence indicates that miRNAs may function as modulators of signaling networks in resistance to EGFR TKIs.
The aim of this study was to investigate the potential involvement of miRNAs in acquired resistance to EGFR TKIs and explore the novel molecular mechanisms related to EGFR TKI resistance in lung cancer. We analyzed miRNA expression profiles in EGFR TKI-sensitive and EGFR TKI-resistant cells and identified that miR-146b-5p was significantly downregulated in EGFR TKI-resistant cells. Low miR-146b-5p expression was also detected in primary cancer cells from the pleural effusions of patients with lung cancer after the development of acquired resistance to EGFR TKI treatment. Further experiments with EGFR TKI-resistant cell lines or primary cells revealed that miR-146b-5p enhanced the cytotoxic effects of EGFR TKIs by directly targeting interleukin-1 receptor-associated kinase 1 (IRAK1)/nuclear factor κB (NF-κB) signaling. The findings of the present study provide novel insights into the function of miR-146b-5p and IRAK1 in lung cancer, as well as into the molecular mechanisms underlying EGFR TKI resistance in NSCLC.
Results
Differential miRNA Expression between EGFR TKI-Sensitive and -Resistant NSCLC Cells
The expression profiles of miRNAs in EGFR TKI-sensitive cells (PC9 and HCC827) and EGFR TKI-resistant cells (PC9/gef and HCC827/gef) were determined using TaqMan human microRNA PCR array. In total, 754 miRNAs were profiled for each sample, and a two-fold difference in expression was applied as the cut-off criterion. These analyses revealed eight candidate miRNAs, the expression levels of which were significantly altered in EGFR TKI-resistant cells (Figure 1A; Tables S1 and S2). Among them, the expression of miR-146b-5p was significantly low (PC9/gef: 0.35-fold, and HCC827/gef: 0.35-fold) according to the results of TaqMan human microRNA PCR array analysis (Figure 1A, right). In addition, the expression levels of miR-125b-2∗ (miR-125b-2-3p), miR-23a∗ (miR-23a-5p), miR-221∗ (miR-221-5p), miR-193b∗ (miR-193b-5p), miR-139-5p, miR-29c∗ (miR-29c-5p), and miR-877 were significantly higher by more than two-fold in EGFR TKI-resistant cells than in parental EGFR TKI-sensitive cells (Figure 1A, right).
Figure 1.

miR-146b-5p Was Downregulated in Cancer Cells with Acquired Resistance to EGFR-TKIs
(A) The differential expression levels of miRNAs between EGFR TKI-sensitive (PC9, HCC827) and EGFR TKI-resistant (PC9/gef, HCC827/gef) lung cancer cells were evaluated using TaqMan microRNA PCR arrays. The differential expression of microRNAs was expressed as ΔΔCt (A, left). In total, eight significantly altered miRNAs were identified and shared in two pairs of cancer cell lines (A, middle and right). (B) qRT-PCR was performed to measure the expression levels of miR-146b-5p in the tested cell lines. The expression of miR-146b-5p was normalized to RNU6B expression. qRT-PCR data were presented as mean ± SD (Student’s t test: ∗∗p < 0.01, ∗∗∗p < 0.001). (C) The relative expression levels of miR-146b-5p were evaluated in primary cancer cells propagated from patients’ malignant pleural effusions. “Treatment-naive” indicates cancer cells collected from patients at the point of diagnosis and before taking EGFR TKIs, whereas “acquired resistance” indicates cancer cells from patients with progressive disease after EGFR TKI treatment. The expression of miR-146b-5p was normalized to RNU6B expression.
miR-146b-5p Was Downregulated in EGFR TKI-Resistant NSCLC Cells
Among the candidate miRNAs, only miR-139-5p and miR-146b-5p were confirmed to play an important role in lung cancer cell growth and progression.23,24 Recent studies have reported that miR-146b-5p functions as a tumor suppressor miRNA and prognosis predictor in NSCLC.23 Our results also indicated that miR-146b-5p was significantly downregulated in EGFR TKI-resistant cells (Figures 1A and 1B). In this study, we investigated the role of miR-146b-5p in acquired resistance to EGFR TKIs.
To evaluate whether the expression of miR-146b-5p was related to acquired EGFR TKI resistance, we collected primary cancer cells from 30 pleural effusions from patients with EGFR mutant malignant lung adenocarcinoma and determined the miR-146b-5p expression levels using quantitative reverse transcription-PCR (qRT-PCR). Among these, 15 samples were collected at the time of lung cancer diagnosis prior to treatment, while the other 15 samples were collected after the patient acquired resistance to EGFR TKIs. There were no significant differences in the clinical characteristics between treatment-naive patients and those with acquired resistance to EGFR TKIs (Table S3). As shown in Figure 1C, miR-146b-5p expression was significantly lower in lung cancer cells collected after acquisition of EGFR TKI resistance (n = 15) than in those obtained prior to treatment (n = 15; p = 0.003, by Mann-Whitney test). These results indicated that miR-146b-5p is possibly a novel biomarker associated with EGFR TKI resistance in NSCLC cells.
Ectopic Expression of miR-146b-5p Enhanced Sensitivity to EGFR TKIs
We further investigated whether miR-146b-5p contributes to EGFR TKI resistance in lung cancer. The miR-146b-5p-specific mimic was transiently transfected into EGFR TKI-resistant PC9/gef cells. After transfection, the miR-146b-5p-transfected PC9/gef cells (PC9/gef-miR-146b-5p) and the control transfectants (PC9/gef-miR-CTL) were exposed to gefitinib, and cell viability was analyzed. The results showed that cells transfected with the miR-146b-5p-specific mimic had higher miR-146b-5p expression and enhanced gefitinib-induced cell death in PC9/gef and HCC827/gef cells (Figures 2A and 2B; Figure S1A). Consistent with this finding, miR-146b-5p expression inhibited lung cell proliferation (Figure S1B). To investigate whether miR-146b-5p enhanced gefitinib sensitivity via the caspase-mediated pathway, a pan-caspase inhibitor (Z-VAD-fmk) was used to block caspase-induced apoptosis. We also observed that miR-146b-5p enhanced gefitinib-induced cleavage of caspase-3 in EGFR TKI-resistant PC9/gef (Figure 2C) and that Z-VAD-fmk reverted miR-146b-5p-induced cleavage of caspase-3 in EGFR-TKI-resistant cells (Figure 2C).
Figure 2.

Expression of miR-146b-5p Enhanced EGFR TKI-Induced Apoptosis
(A) qRT-PCR was conducted to determine the expression levels of miR-146b-5p after transient transfection of the miR-146b-5p mimic in PC9/gef cells. The expression of miR-146b-5p was normalized to RNU6B expression. qRT-PCR data were presented as mean ± SD (Student’s t test: ∗∗∗p < 0.001). (B) PC9/gef cells were transfected with miR-Ctl (scrambled control) or miR-146b-5p mimic and incubated for 24 h, followed by treatment with the indicated concentrations of gefitinib. Cell viability was assessed using the MTT assay as described in Materials and Methods. (C) After transfection with miR-Ctl (scrambled control) or miR-146b-5p mimic and incubation for 24 h, the PC9/gef cells were pre-treated with Z-VAD (50 μM) before treatment with vehicle or gefitinib (1 μM). Cleavage of caspase-3 was determined using immunoblotting. (D) qRT-PCR was performed to measure the expression levels of miR-146b-5p after transient transfection of the miR-146b-5p mimic in PE2988 or PE3479 cells. qRT-PCR data were presented as mean ± SD (Student’s t test: ∗∗∗p < 0.001). (E) After transfection of the miR-Ctl (scrambled control) or miR-146b-5p mimic and incubation for 24 h, the cells were treated with vehicle or osimertinib (0.1 μM for PE2988, 0.3 μM for PE3479). The cleavage of caspase-3 was determined using immunoblotting. (F) qRT-PCR was performed to measure the expression levels of miR-146b-5p after transient transfection of the anti-miR-146b-5p inhibitor in PC9 cells (Student’s t test: ∗∗∗p < 0.001). (G) PC9 cells were transfected with anti-miR-Ctl (scrambled control) or anti-miR-146b-5p inhibitor and incubated for 48 h, followed by treatment with the indicated concentrations of gefitinib. Cell viability was assessed using the MTT assay (Student’s t test: ∗∗p < 0.01).
miR-146b-5p expression was downregulated in primary lung cancer cells with acquired EGFR TKI resistance to first-generation EGFR TKIs (Figure 1C). In addition, we investigated whether miR-146b-5p can enhance osimertinib-induced apoptosis in osimertinib-resistant cancer cells. Osimertinib-resistant primary cancer cells (PE2988 and PE3479 cells) were isolated from malignant pleural effusions of patients with lung adenocarcinoma. PE2988 harbored the EGFR exon 19 E746-A750 deletion plus exon 20 T790M mutation, whereas PE3479 harbored exon 21 L858R, exon 20 T790M, and exon 20 C797S mutations (Table S4). PE2988 represented the EGFR C797S-independent osimertinib-resistant cancer cell, whereas PE3479 was the EGFR C797S-dependent osimertinib-resistant cancer cell. Both the primary cell lines were osimertinib resistant by determining their IC50 (half maximal inhibitory concentration), which was 4.53 μM for PE2988 and 1.31 μM for PE3479 cells. Overexpression of miR-146b-5p in both primary cancer cells promoted the cleavage of caspase-3 in the absence or presence of osimertinib (Figures 2D and 2E). We further assessed whether miR-146b-5p inhibition affects EGFR TKI sensitivity in lung cancer. After silence of miR-146b-5p with anti-miR-146b-5p inhibitor, the miR-146b-5p-depleted PC9 cells (PC9/anti-miR-146b-5p) were more viable under gefitinib treatment as compared with PC9/anti-miR-CTL cells (Figures 2F and 2G). These findings indicated that miR-146b-5p significantly increased the percentage of apoptotic cells and enhanced sensitivity to EGFR TKI in lung cancer cells.
IRAK1 Is a Direct Target of miR-146b-5p
Next, we identified the potential targets of miR-146b-5p by performing a computational screen for genes with complementary sites of miR-146b-5p in their 3′ UTR using the public TargetScan software. The predicted targets with at least 2 conserved binding sites are listed in Table S5. As shown, TRAF6 and IRAK1 were top candidate targets with conserved 8-mer sites. Although the seeding sequences of miR-146b-5p in EGFR 3′ UTR are poorly conserved in only three species (human, chimp, and rhesus), EGFR is crucial in NSCLC. Three miR-146b-5p putative targets, including EGFR, IRAK1, and TRAF6, were taken into consideration (Figure 3A). We showed that the expression levels of EGFR, IRAK1, and TRAF6 were low in miR-146b-5p-transfected EGFR TKI-resistant cells (Figure 3B). Specific small interfering RNAs (siRNAs) (siEGFR, siIRAK1, and siTRAF6) were used to knock down EGFR, IRAK1, and TRAF6 expression to examine whether these three candidate genes were involved in EGFR TKI resistance. Results showed that only transfection with the IRAK1-specific siRNA enhanced EGFR TKI-induced cell death (gefitinib, 30.1%; afatinib, 26.1%; osimertinib, 32%), whereas 20.8% of apoptotic cells were observed in the vehicle-treated siIRAK1-transfected group (Figure 3C). Moreover, suppression of miR-146b-5p with anti-miR-146b-5p inhibitor upregulated IRAK1expression (Figure S2A). EGFR TKI-resistant PC9/gef cells displayed lower levels of phospho-EGFR, suggesting an EGFR-independent pathway involved in EGFR TKI resistance (Figure S2B). These results suggested that IRAK1, but not EGFR or TRAF6, plays an important role in miR-146b-5p-mediated increase in EGFR TKI sensitivity in lung cancer.
Figure 3.

miR-146b-5p Targeted the 3′ UTR of the IRAK1 mRNA to Repress IRAK1 Expression
(A) Schematic representation of the putative miR-146b-5p binding site in the 3′ UTR of the human EGFR (2251∼2258 nt, 3193∼3199 nt, 4151∼4157 nt), IRAK1 (40∼47 nt, 56∼63 nt), and TRAF6 (473∼480 nt, 538∼545 nt, 1272∼1279 nt) mRNAs. (B) The expression levels of the EGFR, IRAK1, and TRAF6 proteins after transfection of miR-Ctl or miR-146b-5p mimics in the cell lines were assessed using immunoblotting. (C) After transfection of the scrambled siRNA (siCTL), siEGFR, siIRAK1, or siTRAF6, and incubation for 24 h, the PC9/gef cells were treated with vehicle or EGFR TKIs (gefitinib, 1 μM; afatinib, 0.25 μM; osimertinib, 0.3 μM). The percentage of cell death (annexin-V-positive cells) was measured using flow cytometry and was presented as mean ± SD (Student’s t test: ∗∗p < 0.01, ∗∗∗p < 0.001). (D) Expression of IRAK1 mRNA correlated negatively with miR-146b-5p expression in various lung cancer cell lines (PE2988, HCC827, HCC4006, A549, PC9, H1975, and CL97), as assessed using qRT-PCR. The correlation between IRAK1 and miR-146b-5p expression was assessed using Pearson’s correlation analysis (p = 0.023). (E) Expression of IRAK1 mRNA correlated negatively with miR-146b-5p expression in 17 primary lung cancer cells from the pleural effusions of patients, as assessed using qRT-PCR. The correlation between IRAK1 and miR-146b-5p expression was assessed using Pearson’s correlation analysis (p = 0.045). (F) miR-146b-5p suppressed IRAK1 via direct interaction with its 3′ UTR. The sequences encoding wild-type and mutated fragments of the IRAK1 3′ UTR are depicted. 293T cells were co-transfected with pMir-IRAK1-3′ UTR-wt reporter plasmid or pMir-IRAK1-3′ UTR-mut reporter, plus miR-Ctl (scrambled control) or miR-146b-5p mimics. The relative luciferase activities are presented as means ± SD from three independent experiments (Student’s t test: ∗∗p < 0.01). n.s., not significant.
We evaluated the correlation between miR-146b-5p and the IRAK1 mRNA in seven human lung cancer cell lines (PE2988, HCC827, HCC4006, A549, PC9, H1975, and CL97) and 17 primary cancer cells propagated from pleural effusions of patients. A significantly negative correlation was observed between the levels of IRAK1 mRNA and miR-146b-5p expression in lung cancer cell lines (Figure 3D) and primary cancer cells (Figure 3E). The negative correlation between IRAK1 mRNA and miR-146b-5p also existed in the pair of TKI-sensitive PC9 and TKI-resistant PC9/gef cells (Figure S3). Direct interaction between miR-146b-5p with the IRAK1 3′ UTR was observed using luciferase reporter assays. The luciferase activity of the wild-type (WT) IRAK1-3′ UTR was significantly suppressed by miR-146b-5p, whereas the luciferase activity of mutant IRAK1-3′ UTR was not affected (Figure 3F). Thus, we suggested that IRAK1 is one of the direct targets of miR-146b-5p in lung cancer cells.
IRAK1 Played a Crucial Role in EGFR TKI Resistance
We investigated whether suppression of IRAK1 in lung cancer cells affected EGFR TKI sensitivity. Silencing of IRAK1 in PC9/gef cells enhanced gefitinib-induced cleavage of caspase-3, and the effect of IRAK1 knockdown was also countered by Z-VAD-fmk treatment (Figure 4A). Suppression of IRAK1 in PE2988 (EGFR C797S-independent osimertinib-resistant) and PE3479 (EGFR C797S-dependent osimertinib-resistant) cells also markedly enhanced osimertinib-induced cleavage of caspase-3 and PARP (Figure 4B). Furthermore, we demonstrated that silencing of IRAK1 in PC9/gef cells significantly enhanced sensitivity to the EGFR TKIs, erlotinib, afatinib, and gefitinib (Figures 4C and 4D; Figure S4).
Figure 4.

IRAK1 Knockdown Enhanced EGFR TKI-Mediated Apoptosis
(A) After transfections of siCTL or siIRAK1 for 24 h, PC9/gef cells were pre-treated with Z-VAD (50 μM) before treatments with vehicle or gefitinib (1 μM). (B) After transfections with siCTL or siIRAK1 and incubation for 24 h, cells were treated with vehicle or osimertinib (0.1 μM for PE2988; 0.3 μM for PE3479) for 18 h. The cleavage of caspase-3 and PARP were determined using immunoblotting. (C, D) PC9/gef cells were transfected with siCTL (scrambled control) or siIRAK1 for 24 h, and then treated with the indicated concentrations of erlotinib (C) or afatinib (D) for 72 h. Cell viability was assessed using the MTT assay as described in Materials and Methods. (E) Relative expression levels of IRAK1 mRNA were evaluated in primary cancer cells from the pleural effusions of patients with malignant disease. “Treatment-naive” indicates cancer cells collected from patients at the point of diagnosis and before administration of EGFR TKIs; “acquired resistance” indicates cancer cells from patients with progressive disease after EGFR TKI treatment. The expression of IRAK1 was normalized to TBP expression.
To investigate the role of IRAK1 in EGFR TKI resistance, 74 primary cancer cells from malignant pleural effusions of patients with EGFR mutant lung adenocarcinoma were collected for IRAK1 mRNA analysis. There was no difference in the clinical characteristics between treatment-naive patients and those with acquired resistance to EGFR TKIs (Table S6). As shown in Figure 4E, IRAK1 mRNA expression in primary cancer cells obtained after the acquisition of EGFR TKI resistance (n = 34) was significantly higher than that in treatment-naive samples (n = 40, p = 0.0194, by Mann-Whitney test). These results suggested that IRAK1 suppression is critical for overcoming resistance to all generations of EGFR TKIs.
miR-146b-5p Suppressed the NF-κB Pathway via Regulation of IRAK1
IRAK1 is essential for interleukin (IL)-1R-associated NF-κB activation.25 Our results indicated that miR-146b-5p suppressed IRAK1 expression in lung cancer cells, and hence we studied the effect of miR-146b-5p on NF-κB activity using a NF-κB reporter (luc)-A549 recombinant stable cell line. The NF-κB reporter (luc)-A549 cell line containing a firefly luciferase gene driven by four copies of NF-κB response element located upstream of the minimal TATA promoter was designed for monitoring the activation of the NF-κB signal transduction pathways. The results revealed that ectopic expression of miR-146b-5p in NF-κB reporter (luc)-A549 cells significantly suppressed NF-κB activity both in the absence or presence of IL-1β (activator of NF-κB signaling) (Figures 5A and 5B). Furthermore, the nuclear expression levels of NF-κB subunits (p50 and p65) also decreased in miR-146b-5p-transfected cells as compared with miR-CTL-transfected cells both in the absence and presence of IL-1β (Figure 5C). NF-κB regulated chemokines (such as IL-6 and IL-8), which contributed to EGFR TKI resistance in lung cancer.26,27 In this study, we also observed that the NF-κB-regulated chemokines IL-6 and IL-8 were significantly suppressed in miR-146b-5p-transfected EGFR TKI-resistant cells (Figure 5D).
Figure 5.

Mir-146b-5p Suppressed NF-κB Activity in Lung Cancer Cells
(A) The expression levels of miR-146b-5p in NF-κB reporter A549 cells were measured using qRT-PCR. The expression of miR-146b-5p was normalized to RNU6B expression. qRT-PCR data were presented as the mean ± SD (Student’s t test: ∗∗∗p < 0.001). (B, C) A549-κB cells were transiently transfected with miR-Ctl or miR-146b-5p mimics in the absence or presence of IL-1β. After 48 h of transfection, the activity of the NF-κB reporter was assessed using a dual-luciferase reporter assay system (B), and the nuclear NF-κB subunits (p50 and p65) were detected using western blot (C). The relative luciferase activities are presented as means ± SD from three independent experiments (Student’s t test: ∗∗p < 0.01, ∗∗∗p < 0.001). PBS, phosphate-buffered saline. (D) Relative expression levels of IL6 (left panel) and IL8 (right panel) mRNAs were determined after transfection of miR-Ctl or miR-146b-5p mimics in the PC9/gef, PE2988, or PE3479cells using qRT-PCR. qRT-PCR data were presented as the mean ± SD (Student’s t test: ∗p < 0.05, ∗∗∗p < 0.001).
Restoration of IRAK1 Expression Reversed the Effects of miR-146b-5p on EGFR-TKI Sensitivity
To verify whether miR146b-5p overcame EGFR TKI resistance by targeting IRAK1, we evaluated whether restoration of IRAK1 expression reverted the effect of miR-146b-5p in lung cancer. IRAK1-overexpressing cells (PC9/gef-IRAK1, PE2988-IRAK1) were established by transfecting the cDNA of IRAK1 without the 3′ UTR. The expression levels of the IRAK1 mRNA and protein in IRAK1-overexpressing cells were confirmed using qRT-PCR and immunoblotting (Figure 6A). We reported that the effect of miR-146b-5p on increasing the cleavage of caspase-3 and PARP were abolished in IRAK1-overexpressing cells (Figure 6B). Our data further revealed that ectopic IRAK1 expression in the PC9/gef-miR-146b-5p cells restored the expression of the NF-κB-regulated chemokines IL-6 and IL-8 (Figure 6C). These results suggested that IRAK1 expression significantly decreased EGFR-TKI sensitivity in lung cancer and promoted the production of the NF-κB-regulated chemokines IL-6 and IL-8.
Figure 6.

miR-146b-5p Enhanced EGFR TKI-Mediated Apoptosis by Downregulating IRAK1
(A) PC9/gef-IRAK1 and PE2988-IRAK1 cells stably expressing IRAK1 were established by infecting PC9/gef or PE2988 cells with IRAK1 lentivirus particles (containing the IRAK1-encoding sequence but lacking the 3′ UTR), and IRAK1 expression was detected using qRT-PCR and immunoblotting. qRT-PCR data were presented as mean ± SD (Student’s t test: ∗∗∗p < 0.001). (B) After transfection of the miR-146b-5p mimic, the cells were treated with the vehicle of osimertinib (0.1 μM for PE2988; 0.3 μM for PE3479) for 18 h. The cleavage of caspase-3 and PARP were detected. (C) The expression levels of IL-6 and IL-8 were determined after transfection of miR-Ctl or miR-146b-5p mimics using qRT-PCR. qRT-PCR data were presented as mean ± SD (Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (D) Schematic diagram showing the role of miR-146b-5p in regulating EGFR TKI resistance.
Discussion
Several studies have suggested that miR-146b-5p plays an important role in cancer development and progression. miR-146b-5p functions as an oncogene in various human cancers, such as thyroid carcinoma, osteosarcoma, gastric cancer, and colorectal cancer. In thyroid carcinoma, miR-146b-5p promotes metastasis and induces EMT by targeting zinc and ring finger 3 (ZNRF3).28 Further studies have revealed that miR-146b-5p enhances papillary thyroid carcinoma development by suppressing coiled-coil domain-containing protein 6 (CCDC6).29 In colorectal cancer, miR-146b-5p promotes cell growth, invasion, and metabolism via directed targeting of the pyruvate dehydrogenase E1 beta subunit (PDHB).30 Elevation of miR-146b-5p expression was a strong risk factor for tumor relapse and the poor survival rate of gastric cancer due to inhibition of NOVA alternative splicing regulator 1 (NOVA1).31 However, conflicting reports support the role of miR-146b-5p as a tumor suppressor. miR-146b-5p has been suggested to inhibit cell proliferation and promote apoptosis and act as a tumor suppressor as well as a novel prognostic biomarker in glioblastoma, hepatocellular carcinoma, and lung cancer.23,32,33 NSCLC patients with high levels of miR-146b-5p had better overall survival rates than patients with low levels of miR-146b-5p.23 In this study, we first observed that the expression of miR-146b-5p was significantly downregulated in EGFR TKI-resistant cells.
Although acquired EGFR mutations (e.g., EGFR T790M, C797S) accounted for the majority of EGFR TKI-resistant cases, diverse EGFR-independent mechanisms (e.g., MET amplification, AXL upregulation, PIK3CA mutation) were associated with EGFR TKI resistance.34 Growing evidence showed non-coding RNAs contributed to EGFR TKI resistance. The long non-coding RNA small nucleolar RNA host gene 14 (SNHG14) conferred gefitinib resistance via sponging miR-206-3p.35 miR-483-3p functioning as tumor suppressor enhanced gefitinib sensitivity via reversing EMT.36 However, previous studies have not investigated whether miR-146b-5p contributes to EGFR TKI resistance in lung cancer cells. We first demonstrated that ectopic expression of miR-146b-5p enhanced the sensitivity of NSCLC to EGFR TKIs via targeting IRAK1. Overexpression of miR-146b-5p and IRAK1 silencing promoted cellular apoptosis in two primary EGFR T790M-preserved osimertinib-resistant PE2988 and PE3479 cancer cells (Figure 2). The downregulation of miR-146b-5p may provide a novel EGFR-independent mechanism for acquiring osimertinib resistance by modulating the downstream IRAK1/NF-κB/IL-6 and IL-8 in EGFR T790M-preserved cancer cells.
IRAK1 is a serine/threonine kinase of the IL-1R/TLR signaling pathway involved in NF-κB-regulated inflammation response, anti-apoptosis, and tumor progression.25 As shown in recent studies, IRAK1 drove metastasis and resistance to chemotherapy in breast cancer by increasing NF-κB activity and cytokine secretion (IL-6, IL-8, and CXCL1), suggesting that IRAK1 may be a potential therapeutic target in breast cancer.37 Other studies have revealed that the expression of IRAK1 in NSCLC correlated positively with clinical tumor-node-metastasis (TNM) stage, lymph node metastasis, tumor size, and distal metastasis.38 IRAK1 might be valuable for identifying patients with increased risk of lung cancer. Although IRAK1 has been linked to tumor progression in lung cancer, IRAK1 alteration has not been previously linked to acquired resistance to EGFR TKIs. In this study, IRAK1 suppression overcame resistance to all generations of EGFR TKIs, suggesting IRAK1 plays a pivotal role in EGFR TKI resistance in lung cancer (Figure 4).
NF-κB plays an important role in inflammation and tumor progression. Inhibition of NF-κB in vitro and in vivo resulted in apoptosis of NSCLC cells and reduced tumor development.39 Genetic loss of the NF-κB subunit p65/RelA in a K-Ras-induced lung cancer mouse model significantly inhibited tumor growth and dissemination.40 In addition, NF-κB activation is a critical adaptive survival mechanism that confers resistance to EGFR TKIs.41 The higher activity of NF-κB was observed in EGFR TKI-resistant cells as compared with EGFR TKI-sensitive cells.42 The IκB (an endogenous repressor of NF-κB) was also lower in erlotinib-resistant cancer cell lines that were resistant to erlotinib as compared with sensitive cell lines.43 A reporter assay showed that EGFR TKI treatment immediately induced NF-κB activation.44 Upon EGFR TKI treatment, NF-κB rapidly localized into the nucleus, and the engagement of NF-κB promoted tumor cell survival in patient-derived xenograft (PDX) models.41 Direct NF-κB inhibition with a pharmacologic inhibitor enhanced EGFR TKI-induced cell death and suppressed the emergence of acquired resistance in EGFR mutant NSCLC models.41 These studies indicated the persistent activation of NF-κB signaling drove adaptive resistance to EGFR TKIs. An unbiased short hairpin RNA (shRNA)-based high-throughput screen showed the involvement of NF-κB signaling pathway in modulating EGFR TKI sensitivity in lung cancer cells. In this screen, lung cancer cells were introduced with a pooled shRNA library and treated with erlotinib. Thirty-six target genes conferred erlotinib sensitivity, and 18 targets were linked to NF-κB pathway.43 Activation of NF-κB through silence of IκB conferred EGFR TKI resistance in EGFR mutant HCC827 lung cancer cells, whereas genetic inhibition of NF-κB through silencing the NF-κB/p65 subunit enhanced erlotinib-induced apoptosis in erlotinib-resistant EGFR mutant H1650 lung cancer cells.43 Decreased expression of the inhibitory IκB protein was predictive of worse progression-free survival and decreased overall survival in lung cancer patients.43 Hence, NF-κB activating cascades might be a possible therapeutic target for countering EGFR TKI resistance in cancer treatments.
The EGFR signaling pathway cross-talked with the NF-κB-dependent pathway. EGF triggers NF-κB activation via the proteasome-mediated degradation of IκBα, and transcriptional induction of NF-κB target genes exerted a positive feedback effect on EGFR signaling.45 The small molecule compound genistein enhanced the sensitivity of erlotinib by reducing the DNA-binding ability of NF-κB.46 Reports show that T790M EGFR mutant lung cancer cells (H1975) developed resistance to mutant-selective EGFR TKI by acquiring NF-κB activity.42 Furthermore, NF-κB-regulated chemokines (such as IL-6 and IL-8) have been demonstrated to contribute to EGFR TKI resistance in lung cancer.26,27 The combination of EGFR TKI plus NF-κB inhibitors significantly reduced viability and overcame the TKI resistance in TKI-resistant cells.42 Consistent with these reports, we showed here that miR-146b-5p suppresses the NF-κB pathway via regulation of IRAK1 and that the NF-κB-regulated chemokines IL-6 and IL-8 were significantly suppressed in miR-146b-5p-transfected cells (Figure 5). Restoration of IRAK1 expression reversed the effects of miR-146b-5p on EGFR TKI sensitivity and recovered IL-6 and IL-8 production (Figure 6). In addition to miR-146b-5p, miR-146a-5p has been identified as a negative regulator of IRAK1.47,48 We assessed the correlation between miR-146a-5p and IRAK1 mRNA in various lung cancer cell lines. However, there was no correlation in expression of miR-146a-5p and IRAK1 mRNA (data not shown). MicroRNA array data also revealed that miR-146a-5p was downregulated in one EGFR TKI-resistant cell line- PC9/gef cell but upregulated in the other EGFR TKI-resistant cell line HCC827/gef cell as compared with EGFR TKI-sensitive PC9 or HCC827 cells, respectively (Tables S3 and S4). These data indicated miR-146a-5p is not a consistent regulator in regulating EGFR TKI sensitivity.
Moreover, downregulation of miR-146b-5p is not the only resistant mechanism. We showed that 35%–40% of cells survived in gefitinib-treated PC9/gef-miR-146b-5p cells even in increased drug concentration. Because the spectrum of EGFR TKI resistance is wide, other resistant mechanisms may be raised in the process of establishing PC9/gef cells. A small subpopulation of cancer cells entered a “persister” state of slow growth under long-term EGFR TKI treatment. After continuous exposure to gefitinib, these persister cells gain the ability to expand and eventually develop acquired resistance. Activation of several signaling transducers were associated with persistent drug tolerance to EGFR TKI treatments, including Akt, Notch, and Casein Kinase 1.49, 50, 51 These transducers are not miR-146b-5p targets and may contribute to the survival of PC9/ge-miR-146b-5p cells in high concentration of gefitinib. The phenomenon was reported in previous study.52
In the present study, there is a limitation. It is hard to obtain the paired specimens from the same patient at diagnosis and at progression. This limits relevance of the findings presented. Molecular targeted therapy, especially EGFR TKIs, has been established as the standard therapy for NSCLC patients harboring EGFR-sensitizing mutants. However, most of these patients ultimately developed resistance to EGFR-TKI treatment. Acquired resistance to EGFR-TKIs is still a significant clinical challenge in the treatment of lung cancer. In this study, we showed that alterations in miR-146b-5p and IRAK1 expression are implicated in acquired resistance to EGFR-TKIs. Based on our findings, we concluded that miR-146b-5p overcomes EGFR TKI resistance in human lung cancer by regulating the IRAK1/NF-κB signaling pathway. miR-146b-5p acts as a potential therapeutic tool for treating EGFR TKI-resistant cancers.
Materials and Methods
Cell Lines
The human lung adenocarcinoma cell lines, HCC827, PC9, and its derivative cell line, gefitinib-resistant PC9/gef, were kindly provided by Dr. James Chih-Hsin Yang (Department of Oncology, National Taiwan University Hospital).53 Both EGFR TKI-sensitive PC9 and HCC827 cells harbored the EGFR exon 19 deletion mutation. The EGFR TKI-resistant cells (PC9/gef and HCC827/gef) were selected from parental cells after continuous exposure to the EGFR TKI gefitinib using a dose-escalation procedure.27,53 Gefitinib-resistant HCC827/gef cells were generated from HCC827 cells by stepwise escalation of the concentration of gefitinib up to 10 μM.27 293T cells were purchased from the American Type Culture Collection. PE2988 and PE3479 cells were isolated from malignant pleural fluids of EGFR-mutant lung cancer patients with acquired resistance to osimertinib and then cultured using the previously reported protocol.54 These cell lines were authenticated using short tandem repeat profiling. These cell lines were routinely tested for mycoplasma and free of mycoplasma contamination. The stable NF-κB luciferase reporter A549 cell line was cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), and other cell lines were cultured in RPMI-1640 supplemented with 10% FBS at 37°C in a humidified 5% CO2 incubator.
TaqMan Human MicroRNA Array
The TaqMan array human microRNA A+B cards set (Applied Biosystems; Waltham, MA, USA) containing 754 unique assays specific to human miRNAs was used for detecting miRNA expression. Total RNA was reverse transcribed using a multiplex RT pool set (Applied Biosystems) and loaded into the card panels as described in the manufacturer’s protocol. qRT-PCR and data analysis were performed using an ABI PRISM 7900HT sequence detection system and the standard 7900RQ software.
qRT-PCR
RNA extraction and qRT-PCR were performed as previously described.54 Briefly, total RNA was isolated using the Tri reagent (Molecular Research Center; Cincinnati, OH, USA) and reverse transcribed to cDNA using a High-Capacity cDNA reverse transcription kit (ThermoFisher Scientific; Waltham, MA, USA). qPCR was performed on an ABI 7500 system using a standard protocol, with TATA box-binding protein (TBP) or RNU6B as internal controls for mRNA or miRNA, respectively. The assay kits for miR-146b-5p (#001097) and RNU6B (#001093) were purchased from ThermoFisher Scientific. The sequences of the gene-specific primers are listed in Table S7.
Malignant Pleural Effusion and Primary Cancer Cells
This study was approved by the Institutional Review Board of National Taiwan University Hospital, and all human specimens were obtained with informed consent (201412075RINC). The malignant pleural fluid of patients with EGFR-mutant lung cancer was acquired aseptically in vacuum bottles via thoracentesis. After centrifugation, the primary cancer cells were isolated and cultured using the previously reported protocol.54 Media were replaced after every 2–3 days, and the cells were harvested after 10 days. Total RNA was extracted, and the expression levels of miR-146b-5p and IRAK1 were determined using qRT-PCR.
miRNA Expression and RNA Interference
For overexpression of miR-146b-5p, the cells were transiently transfected with a miR-146b-5p mimic using the Lipofectamine RNAiMax reagent (ThermoFisher Scientific). The miR-146b-5p-specific mimic (#C-300754-03) was obtained from Dharmacon. Gene knockdown was achieved by transfecting cells with siRNAs using the Lipofectamine RNAiMax reagent (ThermoFisher Scientific) following the manufacturer’s instructions. The miR-146b-5p-specific inhibitor (anti-miR-146b-5p, #AM10105) and siRNAs targeting human EGFR (#42833, human EGFR-siRNA), TRAF6 (#S14389, human TRAF6-siRNA), and IRAK1 (#S322, human IRAK1-siRNA) were obtained from ThermoFisher Scientific.
Cytotoxicity and Apoptosis Assays
Cytotoxicity and apoptosis assays were performed as previously described.54 Briefly, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays (Affymetrix; Santa Clara, CA, USA) were used to evaluate the cytotoxic effects of the EGFR TKIs. Briefly, 3 × 103 cells per well were plated in 96-well plates and treated with the indicated drugs. After 48−96 h, MTT (0.5 mg/mL) was added to each well and incubated for another 1 h. Finally, the formazan was dissolved in 100 μL dimethyl sulfoxide (DMSO), and the absorbance was measured at 550 nm using a SpectraMax Plus 384 microplate reader (Molecular Devices; San Jose, CA, USA).
The cytotoxic efficacies of the EGFR TKIs against PE2988 or PE3479 cells were detected using the ATPlite luminescence assay (PerkinElmer; Waltham, CA, USA). The cells were plated and treated with the indicated drugs. After 72 h, the cells were lysed and incubated with substrate solution, and the luminescent intensity was measured using a SPARK 10M (Tecan; Männedorf, Zürich, Switzerland).
Apoptosis was determined using an annexin-V-FITC detection kit (BD Biosciences; San Jose, CA, USA) according to the manufacturer’s instructions. Briefly, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated annexin-V and propidium iodide (PI) for 15 min, and analyses were performed using a flow cytometer.
Western Blot Analysis
Proteins were extracted from cells using the radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology; Danvers, MA, USA). The cytoplasmic/nuclear protein fraction was collected according to manufacturer’s procedures (Thermo Scientific Subcellular Protein Fractionation Kit, #78833). Briefly, cell pellet was dispersed with ice-cold Cytoplasmic Extraction Buffer and centrifuged at 500 × g for 5 min. The supernatant (cytoplasmic fraction) was transferred into a new tube. The residual pellet was added to ice-cold Nuclear Extraction Buffer, dispersed, centrifuged at 16,000 × g for 5 min, and supernatant (nuclear fraction) collected into a new tube. The obtained proteins were quantified, resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride (PVDF) membrane, and incubated with the indicated primary antibodies. The blots were then further incubated with horseradish peroxidase (HRP)-linked secondary antibodies, and the immunoreactive bands were visualized using enhanced chemiluminescence reagents (Millipore; Burlington, MA, USA) as previously described.54 The antibodies used in the study are listed in Table S8.
IRAK1-3′ UTR Luciferase Reporter Assay
The IRAK1-3′ UTR-WT and IRAK1-3′ UTR-Mut (mutant) reporter constructs were purchased from Addgene (Cambridge, MA, USA). The IRAK1-3′ UTR-Mut construct harbors two mutations in the site complementary to the seeding region of miR-146b-5p. Transfection was performed using Lipofectamine 2000 (ThermoFisher Scientific) according to the manufacturer’s instructions. 293T cells were co-transfected with miR-146b-5p mimic plus luciferase-IRAK1-3′ UTR-WT or luciferase-IRAK1-3′ UTR-Mut constructs. At 24 h post-transfection, luciferase activity was measured using a dual-luciferase reporter assay system (Promega; Madison, WI, USA).
NF-κB Luciferase Reporter Assay
The stable NF-κB luciferase reporter A549 cell line was purchased from Tebu-bio (Le Perray-en-Yvelines, France). The NF-κB luciferase reporter A549 cells were transfected with the miR-146b-5p mimic or scrambled control. At 48 h post-transfection, luciferase activity was measured using a dual-luciferase assay (Promega), according to the manufacturer’s instructions.
Establishment of IRAK1-Overexpressing Cells
The pReceiver-IRAK1 plasmid (#EX-K5612-Lv105) containing the coding sequence of IRAK1 but lacking the 3′ UTR was purchased from Genecopeia (Rockville, MD). For establishment of stably IRAK1-overexpressing cells, the lentivirus was generated and transduced into cancer cells, as described previously.55 The transductants were selected after 72 h growth on puromycin-containing media.
Statistical Analysis
The Student’s t test was used to compare the means of continuous variables between two groups. Clinical data were analyzed using the SPSS 15.0 statistical software (SPSS Inc.). All categorical variables were analyzed using the chi-square test, except for those with an expected frequency < 5, which were analyzed using the Fisher’s exact test. Two-sided p values < 0.05 were considered statistically significant.
Materials Availability
The materials and procedures have been attached within the article and supporting information. Original films for each western blot and original dot plots for each flow cytometry analysis are attached in the Supplementary Data. Additional information on the clinical characteristics of the involved patients is available from the corresponding author on reasonable request.
Author Contributions
Y.L., M.T., C.G., H.W., and J.S. designed the study. Y.L., S.W., T.C., and T.T. collated the data. Y.L., S.W., and T.C. carried out data analyses. Y.L., M.T., C.G., H.W., and J.S. contributed to drafting the manuscript. M.T. and J.S. supervised the entire study. All authors have read and approved the final submitted manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We acknowledge the services provided by the 3rd, 7th, and 8th core Laboratories of the Department of Medical Research at National Taiwan University Hospital. We acknowledge the service provided by the DNA Sequencing Core of the First Core Laboratory, National Taiwan University College of Medicine. This work was supported by the Ministry of Science and Technology (104-2314-B-002-172-MY3), the National Taiwan University Hospital (NTUH102-S2158), the National Taiwan University Hospital Yunlin Branch (106.M002), and the New Century Health Care Promotion Foundation (NCHCPF-107001), Taiwan.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.015.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2016. CA Cancer J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Duruisseaux M., Esteller M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 2018;51:116–128. doi: 10.1016/j.semcancer.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 4.Ramalingam S.S., Yang J.C., Lee C.K., Kurata T., Kim D.W., John T., Nogami N., Ohe Y., Mann H., Rukazenkov Y. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018;36:841–849. doi: 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- 5.Lim S.H., Lee J.Y., Sun J.M., Ahn J.S., Park K., Ahn M.J. Comparison of clinical outcomes following gefitinib and erlotinib treatment in non-small-cell lung cancer patients harboring an epidermal growth factor receptor mutation in either exon 19 or 21. J. Thorac. Oncol. 2014;9:506–511. doi: 10.1097/JTO.0000000000000095. [DOI] [PubMed] [Google Scholar]
- 6.Westover D., Zugazagoitia J., Cho B.C., Lovly C.M., Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018;29(Suppl_1):i10–i19. doi: 10.1093/annonc/mdx703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao J., Li H.R., Jin C., Jiang J.H., Ding J.Y. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin. Transl. Oncol. 2019;21:1287–1301. doi: 10.1007/s12094-019-02075-1. [DOI] [PubMed] [Google Scholar]
- 8.Wu S.G., Shih J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer. 2018;17:38. doi: 10.1186/s12943-018-0777-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iorio M.V., Croce C.M. MicroRNAs in cancer: small molecules with a huge impact. J. Clin. Oncol. 2009;27:5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwok G.T., Zhao J.T., Weiss J., Mugridge N., Brahmbhatt H., MacDiarmid J.A., Robinson B.G., Sidhu S.B. Translational applications of microRNAs in cancer, and therapeutic implications. Noncoding RNA Res. 2017;2:143–150. doi: 10.1016/j.ncrna.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rottiers V., Näär A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivey K.N., Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010;7:36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Esquela-Kerscher A., Slack F.J. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 14.Kasinski A.L., Slack F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kefas B., Godlewski J., Comeau L., Li Y., Abounader R., Hawkinson M., Lee J., Fine H., Chiocca E.A., Lawler S., Purow B. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–3572. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 16.Liu L., Shao X., Gao W., Zhang Z., Liu P., Wang R., Huang P., Yin Y., Shu Y. MicroRNA-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. FEBS J. 2012;279:3800–3812. doi: 10.1111/j.1742-4658.2012.08741.x. [DOI] [PubMed] [Google Scholar]
- 17.Chang T.H., Tsai M.F., Gow C.H., Wu S.G., Liu Y.N., Chang Y.L., Yu S.L., Tsai H.C., Lin S.W., Chen Y.W. Upregulation of microRNA-137 expression by Slug promotes tumor invasion and metastasis of non-small cell lung cancer cells through suppression of TFAP2C. Cancer Lett. 2017;402:190–202. doi: 10.1016/j.canlet.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Han F., He J., Li F., Yang J., Wei J., Cho W.C., Liu X. Emerging Roles of MicroRNAs in EGFR-Targeted Therapies for Lung Cancer. BioMed Res. Int. 2015;2015:672759. doi: 10.1155/2015/672759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L., Morselli E., Vitale I., Kepp O., Senovilla L., Criollo A., Servant N., Paccard C., Hupé P., Robert T. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–1803. doi: 10.1158/0008-5472.CAN-09-3112. [DOI] [PubMed] [Google Scholar]
- 20.Wu L., Pu X., Wang Q., Cao J., Xu F., Xu L.I., Li K. miR-96 induces cisplatin chemoresistance in non-small cell lung cancer cells by downregulating SAMD9. Oncol. Lett. 2016;11:945–952. doi: 10.3892/ol.2015.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y.M., Liu J., Sun W. miR-130a overcomes gefitinib resistance by targeting met in non-small cell lung cancer cell lines. Asian Pac. J. Cancer Prev. 2014;15:1391–1396. doi: 10.7314/apjcp.2014.15.3.1391. [DOI] [PubMed] [Google Scholar]
- 22.Gao Y., Fan X., Li W., Ping W., Deng Y., Fu X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem. Biophys. Res. Commun. 2014;446:179–186. doi: 10.1016/j.bbrc.2014.02.073. [DOI] [PubMed] [Google Scholar]
- 23.Li Y., Zhang H., Dong Y., Fan Y., Li Y., Zhao C., Wang C., Liu J., Li X., Dong M. miR-146b-5p functions as a suppressor miRNA and prognosis predictor in non-small cell lung cancer. J. Cancer. 2017;8:1704–1716. doi: 10.7150/jca.16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yong-Hao Y., Xian-Guo W., Ming X., Jin-Ping Z. Expression and clinical significance of miR-139-5p in non-small cell lung cancer. J. Int. Med. Res. 2019;47:867–874. doi: 10.1177/0300060518815379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhyasen G.W., Starczynowski D.T. IRAK signalling in cancer. Br. J. Cancer. 2015;112:232–237. doi: 10.1038/bjc.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S.M., Kwon O.J., Hong Y.K., Kim J.H., Solca F., Ha S.J., Soo R.A., Christensen J.G., Lee J.H., Cho B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012;11:2254–2264. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y.N., Chang T.H., Tsai M.F., Wu S.G., Tsai T.H., Chen H.Y., Yu S.L., Yang J.C., Shih J.Y. IL-8 confers resistance to EGFR inhibitors by inducing stem cell properties in lung cancer. Oncotarget. 2015;6:10415–10431. doi: 10.18632/oncotarget.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng X., Wu B., Xiao K., Kang J., Xie J., Zhang X., Fan Y. miR-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell. Physiol. Biochem. 2015;35:71–82. doi: 10.1159/000369676. [DOI] [PubMed] [Google Scholar]
- 29.Jia M., Shi Y., Li Z., Lu X., Wang J. MicroRNA-146b-5p as an oncomiR promotes papillary thyroid carcinoma development by targeting CCDC6. Cancer Lett. 2019;443:145–156. doi: 10.1016/j.canlet.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Y., Wu G., Yan W., Zhan H., Sun P. miR-146b-5p regulates cell growth, invasion, and metabolism by targeting PDHB in colorectal cancer. Am. J. Cancer Res. 2017;7:1136–1150. [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon S.O., Kim E.K., Lee M., Jung W.Y., Lee H., Kang Y., Jang Y.J., Hong S.W., Choi S.H., Yang W.I. NOVA1 inhibition by miR-146b-5p in the remnant tissue microenvironment defines occult residual disease after gastric cancer removal. Oncotarget. 2016;7:2475–2495. doi: 10.18632/oncotarget.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y., Wang Y., Yu L., Sun C., Cheng D., Yu S., Wang Q., Yan Y., Kang C., Jin S. miR-146b-5p inhibits glioma migration and invasion by targeting MMP16. Cancer Lett. 2013;339:260–269. doi: 10.1016/j.canlet.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 33.Li C., Miao R., Liu S., Wan Y., Zhang S., Deng Y., Bi J., Qu K., Zhang J., Liu C. Down-regulation of miR-146b-5p by long noncoding RNA MALAT1 in hepatocellular carcinoma promotes cancer growth and metastasis. Oncotarget. 2017;8:28683–28695. doi: 10.18632/oncotarget.15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le X., Puri S., Negrao M.V., Nilsson M.B., Robichaux J., Boyle T., Hicks J.K., Lovinger K.L., Roarty E., Rinsurongkawong W. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018;24:6195–6203. doi: 10.1158/1078-0432.CCR-18-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu K., Li J., Qi Y., Zhang C., Zhu D., Liu D., Zhao S. SNHG14 confers gefitinib resistance in non-small cell lung cancer by up-regulating ABCB1 via sponging miR-206-3p. Biomed. Pharmacother. 2019;116:108995. doi: 10.1016/j.biopha.2019.108995. [DOI] [PubMed] [Google Scholar]
- 36.Yue J., Lv D., Wang C., Li L., Zhao Q., Chen H., Xu L. Epigenetic silencing of miR-483-3p promotes acquired gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting integrin β3. Oncogene. 2018;37:4300–4312. doi: 10.1038/s41388-018-0276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wee Z.N., Yatim S.M., Kohlbauer V.K., Feng M., Goh J.Y., Bao Y., Lee P.L., Zhang S., Wang P.P., Lim E. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat. Commun. 2015;6:8746. doi: 10.1038/ncomms9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X., Dang Y., Li P., Rong M., Chen G. Expression of IRAK1 in lung cancer tissues and its clinicopathological significance: a microarray study. Int. J. Clin. Exp. Pathol. 2014;7:8096–8104. [PMC free article] [PubMed] [Google Scholar]
- 39.Meylan E., Dooley A.L., Feldser D.M., Shen L., Turk E., Ouyang C., Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassères D.S., Ebbs A., Levantini E., Baldwin A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blakely C.M., Pazarentzos E., Olivas V., Asthana S., Yan J.J., Tan I., Hrustanovic G., Chan E., Lin L., Neel D.S. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015;11:98–110. doi: 10.1016/j.celrep.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galvani E., Sun J., Leon L.G., Sciarrillo R., Narayan R.S., Sjin R.T., Lee K., Ohashi K., Heideman D.A., Alfieri R.R. NF-κB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget. 2015;6:42717–42732. doi: 10.18632/oncotarget.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bivona T.G., Hieronymus H., Parker J., Chang K., Taron M., Rosell R., Moonsamy P., Dahlman K., Miller V.A., Costa C. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukuoka M., Yoshioka K., Hohjoh H. NF-κB activation is an early event of changes in gene regulation for acquiring drug resistance in human adenocarcinoma PC-9 cells. PLoS ONE. 2018;13:e0201796. doi: 10.1371/journal.pone.0201796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shostak K., Chariot A. EGFR and NF-κB: partners in cancer. Trends Mol. Med. 2015;21:385–393. doi: 10.1016/j.molmed.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 46.El-Rayes B.F., Ali S., Ali I.F., Philip P.A. Potentiation of the effect of erlotinib by genistein in pancreatic cancer: the role of Akt and nuclear factor-κB. Cancer Res. 2006;66:10553–15472. doi: 10.1158/0008-5472.CAN-06-2333. [DOI] [PubMed] [Google Scholar]
- 47.Hu Q., Song J., Ding B., Cui Y., Liang J., Han S. miR-146a promotes cervical cancer cell viability via targeting IRAK1 and TRAF6. Oncol. Rep. 2018;39:3015–3024. doi: 10.3892/or.2018.6391. [DOI] [PubMed] [Google Scholar]
- 48.Wu H., Fan H., Shou Z., Xu M., Chen Q., Ai C., Dong Y., Liu Y., Nan Z., Wang Y. Extracellular vesicles containing miR-146a attenuate experimental colitis by targeting TRAF6 and IRAK1. Int. Immunopharmacol. 2019;68:204–212. doi: 10.1016/j.intimp.2018.12.043. [DOI] [PubMed] [Google Scholar]
- 49.Tetsu O., Phuchareon J., Eisele D.W., Hangauer M.J., McCormick F. AKT inactivation causes persistent drug tolerance to EGFR inhibitors. Pharmacol. Res. 2015;102:132–137. doi: 10.1016/j.phrs.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 50.Arasada R.R., Shilo K., Yamada T., Zhang J., Yano S., Ghanem R., Wang W., Takeuchi S., Fukuda K., Katakami N. Notch3-dependent β-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018;9:3198. doi: 10.1038/s41467-018-05626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lantermann A.B., Chen D., McCutcheon K., Hoffman G., Frias E., Ruddy D., Rakiec D., Korn J., McAllister G., Stegmeier F. Inhibition of Casein Kinase 1 Alpha Prevents Acquired Drug Resistance to Erlotinib in EGFR-Mutant Non-Small Cell Lung Cancer. Cancer Res. 2015;75:4937–4948. doi: 10.1158/0008-5472.CAN-15-1113. [DOI] [PubMed] [Google Scholar]
- 52.Ercan D., Zejnullahu K., Yonesaka K., Xiao Y., Capelletti M., Rogers A., Lifshits E., Brown A., Lee C., Christensen J.G. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010;29:2346–2356. doi: 10.1038/onc.2009.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang M.H., Lee J.H., Chang Y.J., Tsai H.H., Lin Y.L., Lin A.M., Yang J.C. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol. Oncol. 2013;7:112–120. doi: 10.1016/j.molonc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y.N., Tsai M.F., Wu S.G., Chang T.H., Tsai T.H., Gow C.H., Chang Y.L., Shih J.Y. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int. J. Cancer. 2019;145:1609–1624. doi: 10.1002/ijc.32487. [DOI] [PubMed] [Google Scholar]
- 55.Chang T.H., Tsai M.F., Su K.Y., Wu S.G., Huang C.P., Yu S.L., Yu Y.L., Lan C.C., Yang C.H., Lin S.B. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med. 2011;183:1071–1079. doi: 10.1164/rccm.201009-1440OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.