

Abstract
Utrophin upregulation is considered a promising therapeutic strategy for Duchenne muscular dystrophy (DMD). A number of microRNAs (miRNAs) post-transcriptionally regulate utrophin expression by binding their cognate sites in the 3′ UTR. Previously we have shown that miRNA: UTRN repression can be alleviated using miRNA let-7c site blocking oligonucleotides (SBOs) to achieve utrophin upregulation and functional improvement in mdx mice. Here, we used CRISPR/Cas9-mediated genome editing to delete five miRNA binding sites (miR-150, miR-296-5p, miR-133b, let-7c, miR-196b) clustered in a 500 bp inhibitory miRNA target region (IMTR) within the UTRN 3′ UTR, for achieving higher expression of endogenous utrophin. Deleting the UTRN IMTR in DMD patient-derived human induced pluripotent stem cells (DMD-hiPSCs) resulted in ca. 2-fold higher levels of utrophin protein. Differentiation of the UTRN edited DMD-hiPSCs (UTRNΔIMTR) by MyoD overexpression resulted in increased sarcolemmal α-sarcoglycan staining consistent with improved dystrophin glycoprotein complex (DGC) restoration. These results demonstrate that CRISPR/Cas9-based UTRN genome editing offers a novel utrophin upregulation therapeutic strategy applicable to all DMD patients, irrespective of the dystrophin mutation status.
Keywords: utrophin, CRISPR-Cas9, IMTR, UTRNΔIMTR, DMD-hiPSC, Duchenne muscular dystrophy, stem cells, gene therapy
Graphical Abstract
Sengupta et al. report a genome editing strategy for upregulating utrophin in Duchenne muscular dystrophy patient iPSCs. Edited iPSC-derived myotubes demonstrate increased expression of utrophin and components of the dystroglycan/sarcoglycan complex. This approach represents a promising therapeutic strategy applicable for all DMD patients, irrespective of their mutation status.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease affecting approximately 1 in ∼5,000 live born males worldwide.1,2 DMD is caused by mutations in the DMD gene, resulting in the loss or extremely low expression of the dystrophin protein.3,4 Dystrophin is a 427 kDa cytoskeleton-associated protein and member of dystrophin glycoprotein complex (DGC), which functions as a linker between the extracellular matrix and the intracellular actin.5, 6, 7 Dystrophin provides structural integrity to myofibers during cycles of contraction and relaxation.8 In the absence of dystrophin, the increased sarcolemmal fragility leads to repeated cycles of muscle degeneration and regeneration, which inexorably result in replacement of contractile tissue with fibrotic and adipose tissues.9,10 DMD patients typically present by school age and a loss of ambulation by their teens. The disease continues to progress leading to increasing degrees of skeletal muscle weakness and respiratory and cardiac failure, typically by the fourth decade of life.11
Different genetic and pharmacological interventions designed to restore dystrophin expression by antisense oligonucleotide-mediated exon skipping,12, 13, 14 stop codon readthrough,15 dystrophin gene delivery,16, 17, 18 and CRISPR/Cas9 genome editing either to restore an open reading frame or to delete mutated exons19, 20, 21, 22 are currently in different stages of preclinical or clinical studies. However, there are numerous challenges associated with toxicity, the necessity for systemic delivery of these approaches, as well as, the lack of global applicability to patients due to the existence of a variety of dystrophin mutations that cause DMD (https://www.dmd.nl/database.html).
A promising therapeutic approach for DMD that circumvents many of these issues is upregulating the dystrophin-related protein, utrophin: the chromosome 6-encoded autosomal paralog of dystrophin, with a high degree of structural and functional similarity to dystrophin.23, 24, 25 The major utrophin isoform in myofibers utrophin-A, is enriched in neuromuscular and myotendinous junctions of adult muscles and at the sarcolemma of regenerating myofibers.26, 27, 28, 29 Small molecules such as heregulin,30 nabumetone,31 SMT C1100,32 and artificial transcription factors33,34 have been shown to upregulate UTRN gene expression by activating the UTRN-A promoter. In addition, utrophin can be upregulated by peptides and redistributed by biglycan-mediated protein anchoring at the post-transcriptional level.35, 36, 37 Previously, we and others have shown the existence of post-transcriptional inhibition of utrophin expression by a number of miRNAs targeting UTRN 3′ UTR.38,39 Suppressing one of those miRNA interactions with site blocking oligonucleotides (SBOs) for let-7c resulted in the utrophin upregulation-mediated functional improvement in mdx mouse.40 While providing a proof of concept, this approach is limited by the far from ideal pharmacological properties of SBO’s in vivo.41
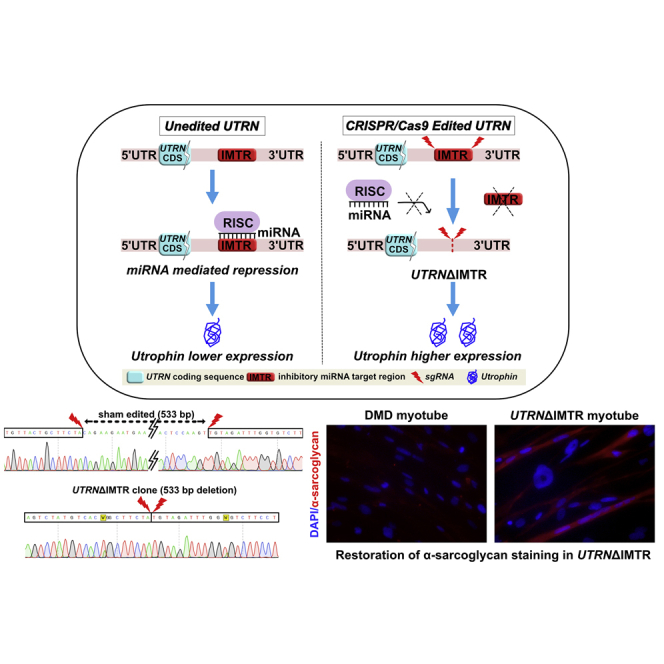
In this study, we describe a CRISPR/Cas9-based utrophin genome editing strategy designed to upregulate utrophin expression by targeting post-transcriptional miRNA-mediated inhibition for DMD (Figure 1). In the native (unedited) state, the five miRNA binding sites (for miR-150, miR-296-5p, miR-133b, miR-196b, and let-7c) that are clustered at the inhibitory miRNA target region (IMTR) repress utrophin expression. We propose that CRISPR/Cas9 genome editing-mediated deletion of the UTRN-IMTR would eliminate the miRNA-binding sites at this locale, leading to increased utrophin expression and results in amelioration of the dystrophic phenotype in DMD, in vivo.
Figure 1.

CRISPR/Cas9 Strategy for Relieving miRNA Driven Post Transcriptional Repression and Increased Expression of Utrophin
The schematic summarizes our CRISPR/Cas9 genome editing strategy to delete the IMTR from the UTRN 3′ UTR with the rationale that the edited UTRN 3′ UTR (UTRNΔIMTR) would reduce miRNA-mediated post-transcriptional repression and lead to higher expression of utrophin.
Results
CRISPR/Cas9 Genome Editing Strategy to Delete the IMTR of UTRN
We have previously shown that UTRN gene expression is regulated by five inhibitory miRNAs targeting the 3′ UTR38 (Figure 2A). To delete these miRNA-binding sites clustered in the IMTR of the UTRN 3′ UTR, we designed four compatible short guide RNAs (sgRNAs 1–4; Table S1) targeting the flanking region of IMTR in the human UTRN gene. Both SaCas9 and the sgRNA pairs were cloned in the same vector (p-UTRNΔIMTR) and transfected in Human embryonic kidney 293T (HEK293T) cells to determine the deletion efficiency and validate the editing (Figure 2B). We used a genomic PCR screening strategy to detect successful deletion of the target region using a primer pair flanking the IMTR for PCR screening (Figure 2C). Gel electrophoresis of PCR products demonstrated that the sgRNA pairs 1 and 4 deleted IMTR most efficiently (Figure 2D; Figure S1). PCR products were also subjected to DNA sequencing to confirm precision of editing (data not shown), prior to utilizing this strategy in human induced pluripotent stem cells (hiPSCs).
Figure 2.

CRISPR/Cas9 Genome Editing Targeting UTRN 3′ UTR
(A) Schematic diagram of UTRN gene showing relative positions of five inhibitory miRNA target sites (miR-150, miR-296-5p, miR-133b, let-7c, and miR-196b) located in the 3′ UTR. The IMTR is shown as red block. The SaCas9 sgRNA target sites are designed flanking the IMTR shown as red arrows. (B) Scheme of HEK293T cell transfection with a plasmid construct (p-UTRNΔIMTR) containing SaCas9 and dual sgRNAs (sg) followed by genomic DNA isolation and PCR screening for UTRNΔIMTR. (C) Scheme showing PCR screening strategy for identifying sgRNA pairs to efficiently achieve UTRNΔIMTR editing. The red lightning bolts show SaCas9 cut sites. (D) The DNA gel shows genomic PCR analysis from HEK293T cells transfected with different combinations of sgRNA pairs. The larger PCR products (800 bp) are from unedited UTRN and shorter PCR products (250–350 bp) are from UTRNΔIMTR gene.
Genome Edited UTRNΔIMTR DMD-hiPSC Lines Show Utrophin Protein Upregulation
A DMD patient fibroblast-derived hiPSC cell line22 carrying a deletion of DMD exons 46–51 (DMD-hiPSC) was subjected to sgRNA 1 and 4 pair directed CRISPR-Cas9-mediated genome editing followed by fluorescence-activated cell sorting (FACS) isolation of transfection positive population and clonal selection (Figures 3A and 3B). Stably deleted DMD-hiPSC line clones were screened for homozygous IMTR deletion (UTRNΔIMTR) using the PCR strategy described above (Figure 3C). Deletions were confirmed by sequencing PCR products from the edited clones (Figure 3D). Utrophin protein expression in UTRNΔIMTR and sham-edited DMD-hiPSC were compared by western blotting and UTRNΔIMTR clones showed up-to 2-fold utrophin upregulation (Figures 3E and 3F).
Figure 3.

UTRNΔIMTR Genome Editing in DMD-hiPSCs and Utrophin Protein Upregulation in UTRNΔIMTR Clones
(A) Fluorescence and bright-field microscopy images showing GFP expression in DMD-hiPSCs transfected with SaCas9-GFP/sgRNA1 and 4. Scale bar, 200 μm. (B) FACS sorting of GFP-positive DMD-hiPSCs gated against mock transfected DMD-hiPSCs. (C) Genomic DNA PCR gel from clonally selected, genome edited, DMD-hiPSC lines shows a 267 bp band from UTRNΔIMTR gene (UTRNΔIMTR clones a, b, and c). Whereas genomic DNA PCR from the sham edited (cells transfected with only SaCa9 and no sgRNA) cells shows only unedited 800 bp band. (D) DNA Sequencing of PCR product from UTRNΔIMTR clone shows precise (533 bp) deletion of IMTR compared to sham edited clone. The W in the chromatogram stands for T/A. (E) Representative western blot shows expression of utrophin in DMD-hiPSC sham edited and UTRNΔIMTR clones. α-tubulin was used as loading control. (F) Densitometric analysis of utrophin western blot to quantify utrophin upregulation. Bands were densitometrically quantified and utrophin normalized to α-tubulin. Error bars represent mean ± SEM (n = 4). Difference in utrophin expression between clones were statistically analyzed by the Mann-Whitney test (∗p ≤ 0.05). Significant increase in utrophin expression was observed in UTRNΔIMTR clones 2, 3, and 5 compared to sham edited clones with p value 0.028.
Validation of hiPSC Clones Post-genome Editing
Pluripotency of the wild-type, DMD, and edited UTRNΔIMTR hiPSC clones was confirmed by immunostaining for nuclear expression of the pluripotency marker Nanog42 (Figure 4). The top five potent off-target sites of the sgRNA 1 and 4 used for genome editing were determined with the COSMID bioinformatics-based tool43 (Tables S2 and S3) and sequenced by PCR amplification of the loci. No off-target mutations were observed at these sites in the selected UTRNΔIMTR clones demonstrating the precise nature of genome editing of the guide RNA pairs.
Figure 4.

Pluripotency Marker Nanog Expression in Wild-Type, DMD, and UTRNΔIMTR hiPSC Clones by Immunostaining
Immunofluorescent staining images of wild-type, DMD, and UTRNΔIMTR hiPSC clones with DAPI (blue), Nanog (red), and α-tubulin (green) for validation of pluripotency in post-genome edited lines. The merge panel at bottom shows nuclear localization of pluripotency marker Nanog in different clones. Magnification 20×, scale bar, 200 μm.
Characterization of hiPSCs Differentiated to Myogenic Lineage by MyoD Overexpression
The wild-type, DMD, and selected UTRNΔIMTR hiPSC clones were differentiated to myogenic lineage using a tamoxifen inducible MyoD expressing lentivirus22 (Figure 5A). The edited and unedited fused, multinucleated myotubes showed positive myosin heavy chain (MYHC) expression by immunostaining upon differentiation (Figure 5B). For all the three differentiated lines, 40%–50% of multinucleated myotubes were MYHC-positive myotubes (Figure 5C). Expression of the myogenic genes (MyoD1, MyoG [Myogenin], and endogenous MyoD1), as well as the pluripotency marker Nanog, were quantified in the UTRNΔIMTR cells by quantitative PCR (qPCR) at day 0, day 4, and day 8 post-tamoxifen induction. The qPCR profile showed a sharp decline in Nanog expression and a concomitant increase in MyoD1, MyoG, and endogenous MyoD1 genes, supporting differentiation of the UTRNΔIMTR cells to a myogenic lineage (Figure 5D). Utrophin expression in the differentiated myotubes were quantified by western blotting. UTRNΔIMTR myotubes showed higher utrophin expression compared to DMD myotubes (Figure S4).
Figure 5.

MyoD-Mediated Directed Differentiation of hiPSC Clones to Myogenic Lineage
(A) Schematic of myogenic differentiation of hiPSCs achieved by lentivirus mediated MyoD overexpression. (B) Differentiated wild-type, DMD, and UTRNΔIMTR myotubes were stained with DAPI (blue) and MYHC (green). Scale bar, 200 μm (C) Efficiency of myogenic differentiation determined by the fusion index (percentage of MYHC-positive myotubes with more than 2 nuclei). We counted a total of 85, 98, and 80 myotubes of wild-type, DMD, and UTRNΔIMTR, respectively. Average from three wells (three random fields from each well) with ± SEM are shown. (D) Gene-expression analysis by qPCR of MyoD infected UTRNΔIMTR clone in tamoxifen untreated (day 0) and tamoxifen treated (day 4 and day 8) point. Expression of pluripotency marker NANOG, skeletal muscle marker MyoD1, MyoG, and endogenous MyoD1 are shown (n = 3).
Utrophin Overexpression Increases Sarcolemmal α-Sarcoglycan Expression in UTRNΔIMTR hiPSC-Derived Myotubes
The absence of dystrophin protein in DMD muscles results in the disruption of the DGC and the lack of sarcolemmal staining for different components of the DGC, such as α-sarcoglycan.44,45 Restoration of individual DGC proteins expression at the sarcolemma suggests restoration of the DGC and considered a marker of improvement when evaluating dystrophin or utrophin-based therapeutic strategies.46, 47, 48, 49 We therefore tested whether upregulated utrophin could increase sarcolemmal α-sarcoglycan expression in UTRNΔIMTR DMD-hiPSC-derived myotubes, by immunostaining. The UTRNΔIMTR DMD-hiPSC-derived myotubes showed significantly higher α-sarcoglycan level compared to the DMD-hiPSC-derived myotubes, supporting the restoration of utrophin anchored DGC by genome editing in the DMD-hiPSCs (Figures 6A and 6B). The increases noted on immunostaining was independently supported by western blot data showing overall higher expression of α-sarcoglycan in UTRNΔIMTR DMD-hiPSC-derived myotubes compared with DMD-hiPSC-derived myotubes (Figure 6C). Consistent with the utrophin upregulation mediated restoration of the DGC, we showed restoration of another DGC member, β-dystroglycan in UTRNΔIMTR DMD-hiPSC-derived myotubes by immunostaining (Figure S2).
Figure 6.

α-Sarcoglycan Expression in Differentiated Myotubes
(A) Wild-type, DMD, and UTRNΔIMTR differentiated myotubes were stained with DAPI (blue), MYHC (green), and α-sarcoglycan (red). Scale bar, 200 μm. (B) α-sarcoglycan quantification in wild-type, DMD, and UTRNΔIMTR differentiated myotubes. The α-sarcoglycan intensity shown as percentage expression mean ± SEM, was calculated as median of line intensity profile (n = 20) in ImageJ and normalized with DAPI expression. Differences in α-sarcoglycan expression between individual groups were analyzed by the Kruskal-Wallis multiple comparison test. The p value is <0.0001. (C) The western blot shows α-sarcoglycan expression in wild-type, DMD, and UTRNΔIMTR myotubes. Vinculin was used as loading control.
Discussion
The rapid developments in genome editing has generated enormous excitement and hope for treating devastating diseases such as DMD.50, 51, 52, 53 In this study, we describe a CRISPR/Cas9-mediated genome editing approach for increasing utrophin expression, as a therapeutic strategy for DMD (Figure 1). We used this approach to delete a 500 bp IMTR containing five miRNA binding sites (i.e., miR-150, miR-296-5p, miR-133b, let-7c, and miR-196b) within the UTRN 3′ UTR in HEK293T cells and select appropriate sgRNAs pairs (Figure 2). To test the strategy, we used sgRNA pairs 1 and 4 to delete the IMTR from DMD-hiPSCs (Figures 3A and 3D) and validated the UTRNΔIMTR DMD-hiPSC clonal lines for utrophin upregulation by western blotting (Figures 3E and 3F) and expression of the pluripotency marker Nanog by immunofluorescence (Figure 4). Lentivirus driven MyoD-mediated myogenic differentiation was utilized to drive the hiPSCs into myotubes and differentiation validated by monitoring the fusion index, as well as reduced expression of pluripotency marker and increased levels of myogenic markers by qPCR (Figure 5). Upon differentiation to myotubes, higher α-sarcoglycan levels were noted in edited compared to unedited DMD myotubes (Figure 6), suggestive of functional improvement due the UTRN genome editing, we describe in this study.
Previously described CRISPR/Cas9-mediated genome-editing-based therapeutic strategies for DMD have largely focused on editing dystrophin and met with varying degrees of success in preclinical studies.19, 20, 21, 22,50,51,54 In common, these approaches, while extremely encouraging in preclinical studies, have fundamental limitations in that they would not be applicable to all DMD patients, need to be custom-designed for specific mutations, and would be predicted to be limited by immunity to the newly expressed dystrophin.55 Nevertheless, adeno-associated virus (AAV)-mediated CRISPR genome editing in larger animal model of DMD to correct the dystrophin mutation and express a shorter form of dystrophin supports the efficacy and promise of using genome editing for DMD.56 Dystrophin-independent CRISPR/Cas9 editing approaches have also been described for leveraging myostatin57 and transcriptional activation of utrophin58,59 as potentially therapeutic approaches. Our approach targets post-transcriptional mechanisms for increasing utrophin expression by deleting the miRNA target sites located in the IMTR of the UTRN 3′ UTR. The advantages of our approach are that other cellular targets of respective miRNAs remain unperturbed, this single editing strategy is applicable to all DMD patients, and a predicted lack of immune issues since DMD patient are not utrophin naive as they express utrophin since before birth.24
In vivo preclinical studies using these targets leveraged by genome editing have been achieved using iPSCs, as well as AAV-mediated editing with varying degrees of success. AAV-based approaches have the advantage of enabling the same therapeutic viral vector(s) to be used in a number of patients and ease of delivery. However, the AAV-based approaches have limitations related to the cloning capacity, long-term expression of Cas9, and immune reactions against the capsid or cargo (i.e., Cas9,54 Dsytrophin55). While our study was restricted to genome editing of iPSCs in vitro, editing of autologous and/or allogenic iPSCs coupled with transplantation is a promising approach that has been used in vivo in a variety of disease models including DMD.22,52,60, 61, 62 Both approaches have limitations but major efforts are ongoing to harness the potential of these approaches. Indeed, the recent demonstration that AAV9-mediated editing can transduce muscle satellite cells63 and stem cells64 exemplify the rapid pace of progress toward applying these strategies to develop therapies in DMD. Additionally, the UTRN genome editing strategy and proof-of-principle studies described here could potentially be combined with full-length utrophin, miniaturized utrophin (μUtro) upregulation,55,65 or utrophin-independent approaches for synergistic effects.
Materials and Methods
Cell Culture and Maintenance
HEK293T cells (ATCC) were maintained in standard growth condition in DMEM high glucose (GIBCO) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% Pen/Strep (GIBCO).
All hiPSCs were reprogrammed from skin fibroblast with STEMCCA cassette in Dr. April Pyle’s laboratory, UCLA, as described in Karumbayaram et al.66 We used two different hiPSC lines, one derived from a healthy individual (Wt 1002) and the other one derived from DMD patient harboring exon 46–51 deletion (CDMD1003). All the hiPSCs were grown in hESC-qualified Matrigel (Corning), fed daily with mTeSR 1 media (STEMCELL Technologies) as previously described and passaged every 4–5 days.
sgRNA Design and Cloning
All guide RNAs for generating the IMTR deletion (UTRNΔIMTR) were designed using the Benchling web tool for CRISPR design (Table S1). The CMV promoter of pX601 plasmid (Addgene plasmid # 61591) was replaced with EF1α promoter for improved expression of SaCas9 and an EGFP cassette was cloned at C-terminal of SaCas9 (pX601-EF1α::SaCas9-GFP). Individual sgRNA oligonucleotides were annealed and cloned in this modified pX601 plasmid at the BsaI restriction site before the sgRNA scaffold according to the protocols from the Zhang lab (https://www.addgene.org/crispr/zhang/; pX601-EF1α::SaCas9-GFP-U6::sgRNA). For expression of dual sgRNAs in the same plasmid, the second sgRNA under U6 promoter, were PCR amplified from the corresponding plasmid and subcloned at KpnI site of pX601-EF1α::SaCas9-GFP-U6::sgRNA. The cloned plasmids were confirmed by sequencing (Figure S3).
Genome Editing Validation of sgRNAs
HEK293T cells were transfected with plasmids containing SaCas9 and different pairs of sgRNAs using Lipofectamine 3000 (Invitrogen). The cells were suspended in DirectPCR Lysis Reagent (Viagen Biotech) and incubated with proteinase K for 6 hr at 55°C and heat inactivated at 85°C for 45 mins. 1 μL of genomic DNA (gDNA) extract were directly used for PCR screening of UTRN-IMTR deletion. In brief, a UTRN forward primer (5′-CCTTTCGGGTGAAAGATCAG-3′) and UTRN reverse primer (5′-ACTTACTTCCCATTGTTACTGC-3′) were used to amplify a fragment spanning the IMTR with GoTaq Green Master Mix (Promega), using the following cycling conditions: 95°C for 5 mins, 34 cycles at 95°C for 30 s, 60°C for 30 s, 72°C for 1 min, and final extension at 72°C for 10 mins. The PCR products and TrackIt 100 bp DNA ladder (Thermo Fisher Scientific) were electrophoresed on a 2% agarose gel. Gel images were captured using a G:Box imaging system (SYNGENE).
Electroporation of hiPSC Lines
Approximately 5 × 106 hiPSCs were harvested using Accutase solution (Sigma-Aldrich) and washed in phosphate buffered saline (PBS, without Ca+2 and Mg+2). Harvested cells were suspended in 75 μL of Resuspension Buffer R (Neon Kit, Invitrogen) and mixed with 25 μg of plasmid DNA. Cells were electroporated with three 10 ms pulses at 1,200 V (Neon Transfection System, Thermo Fisher Scientific). Post-electroporation cells were plated on Matrigel coated plates in mTeSR 1 media with 5 μM ROCK inhibitor (Y-27632, STEMCELL Technologies).
FACS of hiPSC Lines
48 h post-electroporation GFP-positive hiPSCs were FACS sorted in BD FACS Jazz System (BD Biosciences) at the FACS core of The Children Hospital of Philadelphia. Cells were harvested and suspended as single cells in FACS buffer (PBS, 1% FBS, 1 mM EDTA, 5 μM Y-27632). GFP-positive cells were gated with reference to mock electroporated GFP-negative cell population. FACS sorted GFP-positive hiPSCs were plated immediately in pre-warmed Matrigel coated 10 cm plate (5,000–10,000 cells/10 cm plate) with mTeSR™ 1 media supplemented with 10% CloneR (STEMCELL Technologies).
UTRNΔIMTR hiPSC Colony Screening
FACS sorted hiPSCs formed visible colonies by 7 days in culture. Colonies were picked and split in 96 well Matrigel coated plate with mTeSR™ 1 media. After 3 days cells were split and half harvested for genomic DNA extraction with DirectPCR Lysis Reagent (Viagen Biotech). The gDNA was used for PCR screening of UTRN-IMTR deletion with the primer pairs flanking UTRN-IMTR, as mentioned above. Positive homozygous colonies were selected for further expansion.
Western Blot
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophoshphate, 1 mM β-glycerophoshphate, 1 mM sodium orthovanadate) supplemented with complete protease inhibitor cocktail (Roche). Total protein was measured by Pierce BCA Protein assay kit (Thermo Fisher Scientific). 10 μg of total protein was resolved in 3%–8% Tris-Acetate protein gel (NuPAGE, Thermo Fisher Scientific) and transferred to nitrocellulose membrane using Trans-Blot Turbo Transfer System (Bio-Rad). For immunoblotting, membranes were first blocked with 5% non-fat dry milk in TBS with 1% Tween20 for 1 h in room temperature. After blocking, blots were incubated with the following primary antibodies; mouse monoclonal anti-utrophin (1:100, Mancho3[8A4], developed by Prof. Glenn E. Morris; DSHB, IA, USA) and mouse anti-α-tubulin (1:5,000, T6199, Sigma-Aldrich) for overnight at 4°C. Next day, blots were washed; incubated with mouse immunoglobulin Gκ (IgGκ) binding protein (m-IgGκ BP) conjugated to horseradish peroxidase (HRP) (1:2,500, sc-516102, Santa Cruz Biotechnology); washed and developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and imaged in LI-COR C-Digit Blot Scanner (LI-COR Biosciences-US).
Immunostaining
Cells were grown in Matrigel coated slide chambers, fixed with freshly prepared 4% paraformaldehyde (PFA) for 15 mins, permeabilized with 0.25% Triton X-100 for 5 mins, blocked with 4% BSA for 1 h, and stained with primary antibody overnight at 4°C. The following primary antibody dilutions were used: rabbit monoclonal anti-Sox2 (1:200, #9092, Cell Signaling Technology), rabbit monoclonal anti-Nanog (1:200, #9092, Cell Signaling Technology), mouse monoclonal anti-α-tubulin (1:200, T6199, Sigma-Aldrich), MF20c (1:50, DSHB), goat polyclonal anti-α-sarcoglycan (1:50, sc-16165, Santa Cruz Biotechnology), and mouse monoclonal anti-β-dystroglycan (1:500, NCL-b-DG, Leica Biosystems, Germany). Secondary antibody dilutions were goat anti-mouse AF488 (1:400, A11029, Thermo Fisher Scientific) and donkey anti-goat AF594 (1:400, A11058, Thermo Fisher Scientific). Finally, cells were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen). Images were obtained with the Invitrogen EVOS FL auto 2 Cell Imaging System. Quantification of α-sarcoglycan expression in differentiated myotubes were done in ImageJ software v2.0 using line intensity plot profile of individual myotubes and normalized with respective DAPI intensity. Percentage of α-sarcoglycan intensity for each group were plotted and statistical analysis was done by Kruskal-Wallis multiple comparison test.
RNA Isolation and qPCR
Total RNA was extracted from hiPSCs with TRIzol (Thermo Fisher Scientific). The yield and quality of purified RNA samples were determined using NanoDrop 2000 Spectrophotometer (Thermo Scientific). 1 μg of total RNA samples were treated with DNase I (Invitrogen) for 15 mins and then heat inactivated with 2.5 mM EDTA at 65°C for 10 mins. DNase I treated total RNA was reverse transcribed with oligo dT primer using SuperScript III reverse transcriptase (Thermo Fisher Scientific). qPCR was performed in triplicate with Power SYBR Green PCR master mix (Applied Biosystems) in QuantStudio 3 Real-Time PCR System for MyoD1, MyoG, Nanog, and GAPDH. GAPDH was used as endogenous control. Relative expression levels were calculated by the cycle threshold method. Primer sequences used in qPCR are mentioned in Table S5.
Lentivirus Generation
For tamoxifen inducible MyoD expressing 3rd generation lentivirus production, HEK293T cells were transfected at 80%–90% confluency with psPAX2, pMD2.G, and pCMVMyoD-(ERT)puro plasmids using Lipofectamine 3000 (Invitrogen). Lentiviral particles were harvested as supernatant after 48 h and 72 h of transfection. The psPAX2 and pMD2.G plasmids and the pCMVMyoD-(ERT)puro plasmid were a generous gift from Prof. Joseph A. Baur’s laboratory, UPenn, and Prof. M. Carrie Miceli’s laboratory, UCLA, respectively.
Directed Differentiation of hiPSC Lines
hiPSCs were differentiated into skeletal muscle cells by overexpression of MyoD, as described in Young et al.22 hiPSCs were plated as single cells on Matrigel in SMC4 (basal media: DMEM/F12 with 20% knock-out serum replacement (KOSR, Life Technologies), 1% Non-Essential Amino Acids Solution (NEAA, Life Technologies), 1% Glutamax (Life Technologies), 100 μM beta-mercaptoethanol, 10 ng/mL basic fibroblast growth factor (bFGF, Life Technologies), SMC4: basal media with daily addition of 5 μM ROCK inhibitor (Y27632, StemCell Technologies), 0.4 μM PD0325901 (Sigma-Aldrich), 1 μM CHIR99021 (Tocris Bioscience), and 2 μM SB431542 (Tocris Bioscience). When cells were 70%–80% confluent, they were infected with tamoxifen inducible MyoD-ERT lentivirus with 4 μg/mL protamine sulfate (Sigma-Aldrich) and spun inoculated at 1,250 rpm for 90 mins at 32°C. 48 h post-transduction cells were selected with 1 μg/mL puromycin in SMC4 for 3 days. Next day cells were split and plated on Matrigel in basal media without bFGF plus 5 μM ROCK inhibitor at approximately 1 × 105 cells/cm2. The cells were treated with 5 μM tamoxifen in DMEM with 15% FBS for 4 days for MyoD induction and then differentiated in low glucose DMEM with 5% horse serum and 1μM tamoxifen for 7 days with daily change of media.
Statistical Analysis
Data were analyzed using the GraphPad Prism v8 statistical software package. Values are presented as mean ± standard error of mean (SEM). Statistical analysis was performed using Mann-Whitney test or Kruskal-Wallis test with statistical significance set at p ≤0.05. For image quantification statistical analysis was performed using Kruskal-Wallis test with statistical significance set at p ≤0.05.
Author Contributions
T.S.K., M.K.M., and K.S. designed the study. K.S. and M.K.M. performed the experiments. K.S. and T.S.K. wrote the manuscript. T.S.K., A.D.P., M.J.S., E.L., M.K.M., and K.S. edited the paper. All authors discussed the study and approved submission.
Conflicts of Interest
T.S.K. is an inventor of several utrophin patents assigned to the University of Pennsylvania and subject to the institutional patent policies, which may include royalty distribution in the event of licensure. A.D.P. and M.J.S. are co-founders of MyoGene Bio.
Acknowledgments
This work was supported by grants from the University of Pennsylvania Orphan Disease Center and from the Muscular Dystrophy Association, USA (MDA). T.S.K, A.D.P. and M.J.S. are supported by grants (R33 NS102838, P30 AR057230 and R01 AR064327) from NINDS and NIAMS. We thank Dr. M. Carrie Miceli (University of California, Los Angeles) for providing the pCMVMyoD-(ERT) puro plasmid, Dr. Kiran Musunuru and Dr. Joseph A. Baur (University of Pennsylvania) for providing advice and the lentivirus packaging plasmids. We thank Dr. Courtney Young and Dr. Florian Barthélémy (University of California, Los Angeles) for valuable suggestions. We thank Dr. Dipankar Bhandari (Max Plank Institute for Developmental Biology, Tübingen, Germany) for critical reading.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.08.031.
Supplemental Information
References
- 1.Ryder S., Leadley R.M., Armstrong N., Westwood M., de Kock S., Butt T., Jain M., Kleijnen J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 2017;12:79. doi: 10.1186/s13023-017-0631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mah J.K., Korngut L., Dykeman J., Day L., Pringsheim T., Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014;24:482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 5.Koenig M., Monaco A.P., Kunkel L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 6.Amann K.J., Renley B.A., Ervasti J.M. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J. Biol. Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- 7.Matsumura K., Campbell K.P. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- 8.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell K.P. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 10.Brown R.H., Jr., Hoffman E.P. Molecular biology of Duchenne muscular dystrophy. Trends Neurosci. 1988;11:480–484. doi: 10.1016/0166-2236(88)90006-9. [DOI] [PubMed] [Google Scholar]
- 11.Tsuda T. Clinical Manifestations and Overall Management Strategies for Duchenne Muscular Dystrophy. Methods Mol. Biol. 2018;1687:19–28. doi: 10.1007/978-1-4939-7374-3_2. [DOI] [PubMed] [Google Scholar]
- 12.Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 13.Cirak S., Feng L., Anthony K., Arechavala-Gomeza V., Torelli S., Sewry C., Morgan J.E., Muntoni F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012;20:462–467. doi: 10.1038/mt.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao J., Kaye E.M., Mercuri E., Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016;79:257–271. doi: 10.1002/ana.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bushby K., Finkel R., Wong B., Barohn R., Campbell C., Comi G.P., Connolly A.M., Day J.W., Flanigan K.M., Goemans N., PTC124-GD-007-DMD STUDY GROUP Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50:477–487. doi: 10.1002/mus.24332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendell J.R., Rodino-Klapac L., Sahenk Z., Malik V., Kaspar B.K., Walker C.M., Clark K.R. Gene therapy for muscular dystrophy: lessons learned and path forward. Neurosci. Lett. 2012;527:90–99. doi: 10.1016/j.neulet.2012.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain J.R., Chamberlain J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017;25:1125–1131. doi: 10.1016/j.ymthe.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018;26:2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young C.S., Hicks M.R., Ermolova N.V., Nakano H., Jan M., Younesi S., Karumbayaram S., Kumagai-Cresse C., Wang D., Zack J.A. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love D.R., Hill D.F., Dickson G., Spurr N.K., Byth B.C., Marsden R.F., Walsh F.S., Edwards Y.H., Davies K.E. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339:55–58. doi: 10.1038/339055a0. [DOI] [PubMed] [Google Scholar]
- 24.Khurana T.S., Hoffman E.P., Kunkel L.M. Identification of a chromosome 6-encoded dystrophin-related protein. J. Biol. Chem. 1990;265:16717–16720. [PubMed] [Google Scholar]
- 25.Tinsley J.M., Blake D.J., Roche A., Fairbrother U., Riss J., Byth B.C., Knight A.E., Kendrick-Jones J., Suthers G.K., Love D.R. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- 26.Khurana T.S., Watkins S.C., Chafey P., Chelly J., Tomé F.M., Fardeau M., Kaplan J.C., Kunkel L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- 27.Burton E.A., Tinsley J.M., Holzfeind P.J., Rodrigues N.R., Davies K.E. A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 1999;96:14025–14030. doi: 10.1073/pnas.96.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radojevic V., Lin S., Burgunder J.M. Differential expression of dystrophin, utrophin, and dystrophin-associated proteins in human muscle culture. Cell Tissue Res. 2000;300:447–457. doi: 10.1007/s004410000213. [DOI] [PubMed] [Google Scholar]
- 29.Weir A.P., Burton E.A., Harrod G., Davies K.E. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J. Biol. Chem. 2002;277:45285–45290. doi: 10.1074/jbc.M205177200. [DOI] [PubMed] [Google Scholar]
- 30.Krag T.O., Bogdanovich S., Jensen C.J., Fischer M.D., Hansen-Schwartz J., Javazon E.H., Flake A.W., Edvinsson L., Khurana T.S. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. USA. 2004;101:13856–13860. doi: 10.1073/pnas.0405972101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moorwood C., Lozynska O., Suri N., Napper A.D., Diamond S.L., Khurana T.S. Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS ONE. 2011;6:e26169. doi: 10.1371/journal.pone.0026169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinsley J.M., Fairclough R.J., Storer R., Wilkes F.J., Potter A.C., Squire S.E., Powell D.S., Cozzoli A., Capogrosso R.F., Lambert A. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE. 2011;6:e19189. doi: 10.1371/journal.pone.0019189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattei E., Corbi N., Di Certo M.G., Strimpakos G., Severini C., Onori A., Desantis A., Libri V., Buontempo S., Floridi A. Utrophin up-regulation by an artificial transcription factor in transgenic mice. PLoS ONE. 2007;2:e774. doi: 10.1371/journal.pone.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pisani C., Strimpakos G., Gabanella F., Di Certo M.G., Onori A., Severini C., Luvisetto S., Farioli-Vecchioli S., Carrozzo I., Esposito A. Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864(4 Pt A):1172–1182. doi: 10.1016/j.bbadis.2018.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amenta A.R., Yilmaz A., Bogdanovich S., McKechnie B.A., Abedi M., Khurana T.S., Fallon J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl. Acad. Sci. USA. 2011;108:762–767. doi: 10.1073/pnas.1013067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito M., Ehara Y., Li J., Inada K., Ohno K. Protein-Anchoring Therapy of Biglycan for Mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2017;28:428–436. doi: 10.1089/hum.2015.088. [DOI] [PubMed] [Google Scholar]
- 37.Morgoulis D., Berenstein P., Cazacu S., Kazimirsky G., Dori A., Barnea E.R., Brodie C. sPIF promotes myoblast differentiation and utrophin expression while inhibiting fibrosis in Duchenne muscular dystrophy via the H19/miR-675/let-7 and miR-21 pathways. Cell Death Dis. 2019;10:82. doi: 10.1038/s41419-019-1307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basu U., Lozynska O., Moorwood C., Patel G., Wilton S.D., Khurana T.S. Translational regulation of utrophin by miRNAs. PLoS ONE. 2011;6:e29376. doi: 10.1371/journal.pone.0029376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenberg M.I., Georges S.A., Asawachaicharn A., Analau E., Tapscott S.J. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 2006;175:77–85. doi: 10.1083/jcb.200603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra M.K., Loro E., Sengupta K., Wilton S.D., Khurana T.S. Functional improvement of dystrophic muscle by repression of utrophin: let-7c interaction. PLoS ONE. 2017;12:e0182676. doi: 10.1371/journal.pone.0182676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Echevarría L., Aupy P., Goyenvalle A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018;27(R2):R163–R172. doi: 10.1093/hmg/ddy171. [DOI] [PubMed] [Google Scholar]
- 42.Silva J., Nichols J., Theunissen T.W., Guo G., van Oosten A.L., Barrandon O., Wray J., Yamanaka S., Chambers I., Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberds S.L., Anderson R.D., Ibraghimov-Beskrovnaya O., Campbell K.P. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin) J. Biol. Chem. 1993;268:23739–23742. [PubMed] [Google Scholar]
- 46.Matsumura K., Lee C.C., Caskey C.T., Campbell K.P. Restoration of dystrophin-associated proteins in skeletal muscle of mdx mice transgenic for dystrophin gene. FEBS Lett. 1993;320:276–280. doi: 10.1016/0014-5793(93)80602-q. [DOI] [PubMed] [Google Scholar]
- 47.Yuasa K., Miyagoe Y., Yamamoto K., Nabeshima Y., Dickson G., Takeda S. Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 1998;425:329–336. doi: 10.1016/s0014-5793(98)00251-8. [DOI] [PubMed] [Google Scholar]
- 48.Denti M.A., Incitti T., Sthandier O., Nicoletti C., De Angelis F.G., Rizzuto E., Auricchio A., Musarò A., Bozzoni I. Long-term benefit of adeno-associated virus/antisense-mediated exon skipping in dystrophic mice. Hum. Gene Ther. 2008;19:601–608. doi: 10.1089/hum.2008.012. [DOI] [PubMed] [Google Scholar]
- 49.Gumerson J.D., Michele D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011;2011:210797. doi: 10.1155/2011/210797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Z., Zhang Y., Gao F., Han S., Cheah K.S., Tse H.F., Lian Q. CRISPR/Cas9 Genome-Editing System in Human Stem Cells: Current Status and Future Prospects. Mol. Ther. Nucleic Acids. 2017;9:230–241. doi: 10.1016/j.omtn.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song Y., Morales L., Malik A.S., Mead A.F., Greer C.D., Mitchell M.A., Petrov M.T., Su L.T., Choi M.E., Rosenblum S.T. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat. Med. 2019;25:1505–1511. doi: 10.1038/s41591-019-0594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei Y., Chen Y., Qiu Y., Zhao H., Liu G., Zhang Y., Meng Q., Wu G., Chen Y., Cai X. Prevention of Muscle Wasting by CRISPR/Cas9-mediated Disruption of Myostatin In Vivo. Mol. Ther. 2016;24:1889–1891. doi: 10.1038/mt.2016.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wojtal D., Kemaladewi D.U., Malam Z., Abdullah S., Wong T.W., Hyatt E., Baghestani Z., Pereira S., Stavropoulos J., Mouly V. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016;98:90–101. doi: 10.1016/j.ajhg.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao H.K., Hatanaka F., Araoka T., Reddy P., Wu M.Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Núñez-Delicado E. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell. 2017;171:1495–1507. doi: 10.1016/j.cell.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maffioletti S.M., Sarcar S., Henderson A.B.H., Mannhardt I., Pinton L., Moyle L.A., Steele-Stallard H., Cappellari O., Wells K.E., Ferrari G. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018;23:899–908. doi: 10.1016/j.celrep.2018.03.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daniel-Moreno A., Lamsfus-Calle A., Raju J., Antony J.S., Handgretinger R., Mezger M. CRISPR/Cas9-modified hematopoietic stem cells-present and future perspectives for stem cell transplantation. Bone Marrow Transplant. 2019;54:1940–1950. doi: 10.1038/s41409-019-0510-8. [DOI] [PubMed] [Google Scholar]
- 62.Danisovic L., Culenova M., Csobonyeiova M. Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy. Cells. 2018;7:253. doi: 10.3390/cells7120253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nance M.E., Shi R., Hakim C.H., Wasala N.B., Yue Y., Pan X., Zhang T., Robinson C.A., Duan S.X., Yao G. AAV9 Edits Muscle Stem Cells in Normal and Dystrophic Adult Mice. Mol. Ther. 2019;27:1568–1585. doi: 10.1016/j.ymthe.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goldstein J.M., Tabebordbar M., Zhu K., Wang L.D., Messemer K.A., Peacker B., Ashrafi Kakhki S., Gonzalez-Celeiro M., Shwartz Y., Cheng J.K.W. In Situ Modification of Tissue Stem and Progenitor Cell Genomes. Cell Rep. 2019;27:1254–1264. doi: 10.1016/j.celrep.2019.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Odom G.L., Gregorevic P., Allen J.M., Finn E., Chamberlain J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karumbayaram S., Lee P., Azghadi S.F., Cooper A.R., Patterson M., Kohn D.B., Pyle A., Clark A., Byrne J., Zack J.A. From skin biopsy to neurons through a pluripotent intermediate under Good Manufacturing Practice protocols. Stem Cells Transl. Med. 2012;1:36–43. doi: 10.5966/sctm.2011-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.