

The yeast prion [URE3] propagates as a misfolded amyloid form of the Ure2 protein. Propagation of amyloid-based yeast prions requires protein quality control (PQC) factors, and altering PQC abundance or activity can cure cells of prions. Yeast antiprion systems composed of PQC factors act at normal abundance to restrict establishment of the majority of prion variants that arise de novo. While these systems are well described, how they or other PQC factors interact with prion proteins remains unclear.
KEYWORDS: Btn2, Hsp42, amyloid, prion, protein quality control, yeast
ABSTRACT
The yeast prion [URE3] propagates as a misfolded amyloid form of the Ure2 protein. Propagation of amyloid-based yeast prions requires protein quality control (PQC) factors, and altering PQC abundance or activity can cure cells of prions. Yeast antiprion systems composed of PQC factors act at normal abundance to restrict establishment of the majority of prion variants that arise de novo. While these systems are well described, how they or other PQC factors interact with prion proteins remains unclear. To gain insight into such interactions, we identified mutations outside the Ure2 prion-determining region that destabilize [URE3]. Despite residing in the functional domain, 16 of 17 mutants retained Ure2 activity. Four characterized mutations caused rapid loss of [URE3] yet allowed [URE3] to propagate under prion-selecting conditions. Two sensitized [URE3] to Btn2, Cur1, and Hsp42, but in different ways. Two others reduced amyloid formation in vitro. Of these, one impaired prion replication and the other apparently impaired transmission. Thus, widely dispersed sites outside a prion’s amyloid-forming region can contribute to prion character, and altering such sites can disrupt prion propagation by altering interactions with PQC factors.
INTRODUCTION
Ure2 is a key transcriptional regulator of nitrogen catabolite repression and the determinant of the yeast prion [URE3] (1, 2). It is composed of a globular C-terminal domain (CTD) homologous to glutathione S-transferases that performs the transcriptional regulation function and an unstructured N-terminal domain (NTD), which forms the amyloid core of Ure2 fibers that are the basis of the [URE3] prion (1, 3–7). The NTD is not required for Ure2 function, but it contributes to Ure2 function and stability (8).
Propagation of amyloid-based yeast prions requires growth, replication, and transmission. Growth entails recruitment of the soluble protein onto the ends of prion fibers, which act as templates that convert it to the particular amyloid structure of the prion (9). This process occurs spontaneously in vitro and is apparently unaided in vivo (10–13). Transmission of prions between dividing cells also seems to occur unaided by passive diffusion (12, 14), but prion replication depends on the activity of the cytosolic protein disaggregation machinery (15, 16). This machinery, which facilitates recovery from stress by extracting monomers from aggregates of stress-denatured proteins, is driven by Hsp104 and requires assistance by Hsp70 and its J-protein and nucleotide exchange factor cochaperones (17–21). It acts similarly on prions by extracting monomers from prion fibers (15, 16, 22). Doing so results in a fiber splitting into two, each of which can act as a template for continued prion propagation.
Altering normal abundance or activity of any of these chaperones, their cochaperone partners, or other protein quality control (PQC) factors can cure yeast of prions (18, 23–31). Moreover, endogenous cellular functions of some PQC factors actively eliminate prions. For example, Hsp104 has an activity under normal conditions that interferes with establishment or propagation of many variants of [PSI+] prions, composed of Sup35 protein (32). Additionally, Btn2 at normal levels disrupts propagation of [URE3] by collecting dispersed prion aggregates into one or a few large aggregates that are not distributed efficiently during cell division (28, 33). The small heat shock protein Hsp42 and Sis1, a J-protein cochaperone of Hsp70 important for propagation of [URE3] and other prions, interact with Btn2 and influence this antiprion process (29, 33–35).
While the regions of several yeast prion proteins that confer the ability to form amyloid and prions are clearly defined, our understanding of how factors act on prions to facilitate their propagation or elimination is limited. To gain insight into these processes, we identified mutations in Ure2 outside the amyloid-forming region that disrupt the ability of Ure2 to propagate as [URE3], reasoning that they might locate sites of interaction for chaperones or other factors rather than affect amyloid propagation directly. Indeed, disruption of [URE3] propagation by some of these mutations was linked directly or indirectly to PQC factors.
RESULTS
Mutations outside the Ure2 amyloid-forming region impair [URE3] but not Ure2 function.
Our wild-type strains are Ade– because Ure2 represses transcription of the DAL5 promoter that regulates ADE2 (see Materials and Methods and below). Disruption of Ure2 function by mutation or by depletion of Ure2 into insoluble [URE3] prion aggregates relieves this repression making cells Ade+. The substrate for Ade2 can form a red pigment, so cells lacking Ade2 are also red when grown on limiting adenine due to accumulation of this pigment. Thus, [URE3] cells are Ade+ and white, while [ure-o] cells (i.e., without [URE3]) are Ade– and red.
Expressing Ure2 fragments or Ure2-green fluorescent protein (GFP) fusion proteins from plasmids in wild-type cells disrupts [URE3] propagation, resulting in “curing” the cells of [URE3] (36). We presumed expression of point-mutant versions of Ure2 could similarly disrupt [URE3]. Aiming to identify such mutations, we transformed wild-type [URE3] cells with plasmids encoding randomly mutagenized URE2 alleles and looked for red ([ure-o]) cells among the transformant colonies.
We identified eight substitutions in the Ure2 amyloid-forming region (amino acids 1 to 90) that caused such a dominant inhibitory effect (Fig. 1A). Seven of them changed asparagine, a polar residue common in yeast prion-determining regions, and the other added a charge. All of these could be expected to disrupt amyloid formation directly. We were more interested in mutations outside this region (amino acids 91 to 354), expecting them to be more likely to inhibit [URE3] by affecting interactions with cellular factors involved in [URE3] propagation or elimination. Modifying our screen, we identified 17 [URE3]-impairing mutations in this CTD region (Fig. 1A). These CTD mutants inhibited [URE3] propagation to various degrees (Fig. 1B, Table 1).
FIG 1.

Mutations in Ure2 that impair [URE3] propagation. (A) Individual amino acid substitutions identified as causing Ure2 to impair [URE3] when expressed exogenously in wild-type cells are indicated at the top. Those shown in bold were characterized. (B) Examples of dominant inhibitory effects of Ure2 mutants on [URE3]. Wild-type [URE3] strain 1075 was transformed by plasmids encoding Ure2 with the indicated amino acid substitutions. Transformants were selected on plates containing limiting adenine, on which cells that have lost [URE3] grow faster and accumulate red pigment. Shown are 2.5-cm2 sections of plates incubated for 3 days at 30°C followed by 2 days at 25°C. (C) Strain MR149 (ure2Δ) was transformed by empty vector (ev) or plasmids encoding wild-type (wt) and mutant (as indicated) Ure2 proteins. Shown are 2.5-cm2 sections of transformation plates incubated for 3 days at 30°C. The extent of Ure2 function is reflected in the degree of red coloration. (D) Western analysis for abundance of Ure2 proteins (as indicated) in cells expressing Ure2 from the URE2 chromosomal locus. The panel labeled CB shows identically loaded gel stained with Coomassie blue dye.
TABLE 1.
Relative anti-[URE3] effects of Ure2p mutations
| Ure2 mutant | [ure-o] frequency (%) for: |
||
|---|---|---|---|
| Dominant inhibitiona | Adenine additionb at: |
||
| t = 0 | 1 day | ||
| None (wild type) | 0 | 0 | 0 |
| N73D | 70 | 4 | 57 |
| K104I | 63 | 5 | 24 |
| S158F | 83 | 1 | 18 |
| R164G | 13 | 1 | 12 |
| H187Q | 29 | 2 | 22 |
| T249A | 7 | 1 | 19 |
| None (empty vector) | 0 | 0 | 0 |
Wild-type [URE3] cells were transformed by plasmids encoding the indicated Ure2 mutants. Entirely red colonies were scored as a loss event. The frequency of [URE3] loss reflects the relative “strength” of the inhibitory effect on [URE3]. Values were from roughly 500 transformants.
Cells from liquid cultures lacking adenine were tested for the [URE3] phenotype immediately after adding adenine (t = 0) and one day later.
The dominant inhibition of [URE3] caused by the CTD mutations must, of course, occur in cells expressing both wild-type and mutant Ure2 proteins. Since the mutant Ure2 proteins all possess the same wild-type amyloid-forming region, we considered that they might propagate [URE3] when expressed as the only source of Ure2 protein. The CTD possesses the activity of Ure2 for repressing transcription, however, so any mutation in the CTD might inactivate Ure2. If so, then even in the nonprion form, the inactive mutant proteins would confer the same loss of function phenotype as [URE3], which would make it tedious to determine if they could propagate [URE3].
We therefore first tested if the mutations inactivated Ure2 by transforming a ure2Δ strain with plasmids encoding the mutant proteins (Fig. 1C). Active Ure2 represses expression of Ade2 and makes these cells red. Only one (P166L) of 17 CTD mutants tested inactivated Ure2, as indicated by failure to restore a red phenotype. This rarity of mutations in the functional domain that inactivate Ure2 was somewhat surprising because our selection for mutations that dominantly inhibit [URE3] did not demand that the mutant Ure2 proteins retain activity.
CTD mutants propagate [URE3] under prion-selective conditions.
We selected K104I, S158F, R164G, H187Q, and T249A as a reasonable number of mutants to study in detail because they span the CTD and inhibited [URE3] to different degrees, which we presumed could reflect differences in how they destabilize [URE3]. After we integrated the mutant alleles at the genomic URE2 locus, we found that steady-state abundance of Ure2R164G and Ure2H187Q was reduced compared with that of wild-type Ure2 (Fig. 1D). We retained Ure2H187Q for further analysis because it retained more complete Ure2 transcriptional repression activity (see Fig. 1C).
To determine if these proteins could propagate [URE3], we crossed these strains with a ure2Δ strain that propagates [URE3] from wild-type Ure2 protein expressed from a plasmid and monitored [URE3] among meiotic progeny of the diploids. Before inducing sporulation, the diploids were grown on medium without adenine to maintain [URE3] and containing uracil to allow loss of the plasmid carrying wild-type URE2. Spore clones expressing the mutant proteins that carried the plasmid had variable pink color and weak variable growth without adenine (Fig. 2A). Both phenotypes are consistent with a dominant inhibitory effect on [URE3]. Mutant clones lacking the plasmid formed entirely red and Ade– colonies, suggesting complete loss of [URE3]. After prolonged incubation, however, rare Ade+ colonies formed within the patches of these cells on the plates lacking adenine (−ade plates). These results suggest that all spore clones inherited [URE3], as expected, but the mutant prions did not propagate efficiently enough to be inherited beyond the first few cells of the growing colony.
FIG 2.

Mutant Ure2 proteins propagate [URE3] but maintain it only under selective conditions. (A) Representative meiotic progeny of [URE3] diploids heterozygous for ure2Δ and the mutant Ure2, as indicated on the left (see the text). The dissection plates (1/2YPD) were replica plated to YPAD containing G418, which selects for ure2Δ cells, −ade medium, which selects for cells lacking Ure2 or propagating [URE3], and medium lacking uracil (−ura), which selects for cells with plasmid-borne URE2. All Ure2 point mutants (G418 sensitive) that are Ura– (i.e., lacking wild-type Ure2) fail to grow on −ade plates, showing that they lose [URE3]. Point mutants that retain the plasmid encoding URE2 dominantly inhibit [URE3] as indicated by pink color and weakened Ade+ phenotype. (B) After continued incubation, rare Ade+ colonies of Ure2 mutants arose on the −ade plates. Cells from these colonies were recovered and streaked onto similar [URE3]-selective plates (−ade, left) and from these onto nonselective 1/2YPD indicator medium (right). Both plates were incubated at 30°C for 2 days. (C) [URE3] was induced de novo in cells of the indicated Ure2 mutants by galactose-induced overexpression of Ure2(1-64) followed by spreading of 106 cells on −ade medium (left, sectors labeled +). The same numbers of identically treated cells carrying an empty vector were plated on alternate sectors (labeled –). The frequency of Ade+ colonies reflects the efficiency of [URE3] induction. This plate was replica plated onto 1/2YPD (center) and onto a similar −ade plate that selects for [URE3] (right). Rare colonies on −ade plates from uninduced cultures are uncharacterized Ade+ revertants.
Presumably, the progenitor spore and a few of its daughters that inherited [URE3] were able to propagate it well enough to continue growing under conditions that select for the prion. Indeed, when these Ade+ cells of the Ure2 mutant strains were streaked onto similar −ade plates, they all formed colonies at rates faster than wild-type cells (Fig. 2B). [URE3] is toxic (37), so this faster growth could reflect a weakened prion phenotype. When the mutant cells were subsequently transferred to rich medium, they lost the prion rapidly and again formed red colonies indistinguishable from the [ure-o] controls. These red cells did not grow when restreaked onto −ade plates, confirming that [URE3] had been lost. Thus, [URE3] composed of these mutant proteins could be maintained under conditions selecting for the prion but failed to propagate stably or was eliminated quickly when selection was relieved.
The variant of [URE3] used to infect these mutant Ure2 strains was the same as that used in our initial screen. Thus, [URE3] prions being propagated by the mutant proteins could have been obliged to adopt the amyloid structure specific to this prion variant, and effects of the Ure2 mutations might be constrained to this variant. To assess if Ure2 mutants could propagate other variants of [URE3] prions more efficiently, we induced [URE3] de novo by overexpressing prion-forming amino acids 1 to 64 of Ure2, which typically generates a broad range of [URE3] variants (3, 33, 38, 39). The H187Q mutant was omitted from this experiment because its reduced expression and partial complementation of Ure2 function allowed a weak background growth on medium lacking adenine that interfered with the assay. In strains expressing Ure2K104I, Ure2S158F, or Ure2T249A, this treatment induced [URE3] readily, although induction was noticeably less efficient in cells expressing Ure2S158F (Fig. 2C). All of these colonies grew well when transferred to similar medium lacking adenine, indicating that the mutations do not have a strong inhibitory effect on the ability of [URE3] to arise or propagate under prion-selecting conditions. Among the hundreds of [URE3] colonies obtained, however, none were mitotically stable on rich medium that does not require [URE3] for growth. These results suggest that the CTD mutations have a general effect on inhibiting [URE3] that impairs propagation of a range of prion variants.
Several mechanisms could explain how the mutant proteins interfere with prions being propagated by wild-type Ure2 and form highly unstable [URE3] prions on their own. Purified Ure2 forms a dimer in solution (11, 40). Although it has not been tested, dimerization could be important for optimal [URE3] propagation. The mutations might alter dimerization properties. Alternatively, although the mutations are outside the amyloid-forming part of Ure2, they might alter intermolecular interactions elsewhere that are important for assembly or stability of amyloid formed in vivo. They also might alter the sequence or structure of the CTD in a way that perturbs or enhances interactions of Ure2 with its normal partners or any of the many protein quality control factors that promote or restrict [URE3] propagation.
CTD mutations alter Ure2 amyloid formation.
By using size exclusion chromatography, we found that purified wild-type Ure2 and all mutant Ure2 proteins formed dimers (Fig. 3A and B), which is consistent with earlier data (39). These results are in line with their ability to function in vivo. The differences in peak height reflect variability in concentrations of soluble Ure2 applied to the column. The deflection to slower elution of Ure2H187Q could indicate reduced dimer formation, or dimers are more globular.
FIG 3.

Ure2 mutations impair amyloid formation. (A) Size exclusion chromatography of purified, full-length Ure2 proteins: wild-type (wt) and CTD mutant (as indicated). (B) Elution of FPLC size standards for the column used in panel A. The expected elution of Ure2 monomers (40.3 kDal) is at 16 ml, dimers at 14.5 ml. (C to F) Kinetics of thioflavin-T binding to purified full-length Ure2 (20 μM). For each plot, the data are normalized to the maximal yield for the wild type, with wild-type Ure2 set at 100%. Results are for wild-type Ure2, indicated mutant Ure2, and mixtures of 10 μM each of wild type and mutant proteins. Due to difficulties in maintaining Ure2 in a soluble form, different preparations of each Ure2 protein had variable lag times and yields of amyloid. For clarity, representative experiments are shown.
To assess the ability of the mutant proteins to form amyloid, we used the standard approach of monitoring kinetics of thioflavin-T (Th-T) binding to purified full-length proteins (Fig. 3C to F). Wild-type Ure2 (black lines) consistently began to form amyloid after a lag of 60 to 90 min. Once initiated, incorporation of Ure2 into aggregates continued for another 60 to 90 min and then tapered off. These results are typical for this type of experiment and resemble those obtained earlier by us and others (41, 42).
Although all of the mutant proteins possess identical wild-type amyloid-forming regions, they showed differences in kinetics of amyloid assembly (Fig. 3C to F, red lines). Ure2S158F and Ure2T249A had only modest differences from wild-type Ure2 in lag time and yield, but Ure2K104I and Ure2H187Q consistently displayed obvious differences from the wild type. Ure2K104I did not form an appreciable amount of amyloid in the 2 to 3 h it took for reactions with the other proteins to approach completion, although it slowly formed a measurable amount of amyloid upon extended incubation. A higher propensity for nonamyloid aggregation might explain part of this defect. Note, however, that Ure2K104I confers the red phenotype of normally active Ure2, indicating that it must retain substantial solubility in vivo. Ure2H187Q formed amyloid after a lag similar to that of the wild type, but it assembled more slowly and reached lower yields. Overall, these results show that altering individual amino acids outside the amyloid-forming region of Ure2 can influence the amyloid-forming properties of Ure2.
These results suggest that the mutant proteins could dominantly impair [URE3] by joining preexisting prion fibers composed of wild-type Ure2 and inhibiting subsequent incorporation of soluble Ure2. To test this possibility in vitro, we repeated the Th-T assay with equimolar mixtures (10 μM each) of wild-type and mutant proteins (Fig. 3C to F, green lines). S158F had little effect, and the mixture of Ure2T249A with the wild type was somewhat less efficient at forming amyloid than either protein alone. In contrast, reaction mixtures containing Ure2K104I and Ure2H187Q had kinetics that more closely resembled those of the mutant proteins than wild-type Ure2. Thus, these two mutant Ure2 proteins can interfere with amyloid formation by wild-type Ure2, which might explain, at least in part, their inhibitory effects on [URE3].
CTD mutations enhance sensitivity of [URE3] to Btn2, Cur1, and Hsp42.
Btn2 and Cur1 were identified by their ability to cure cells of [URE3] when overexpressed (28). Btn2 collects disperse [URE3] prion seeds into larger aggregates, which reduces prion numbers and thus chances of transmission of [URE3] among dividing cells (28, 33, 35). The mechanism of [URE3] curing by Cur1 is uncertain (33–35, 43, 44). As a first simple test of whether these PQC factors might be involved in the ability of the CTD mutants to destabilize endogenous [URE3], we tested if the dominant inhibitory effects of the CTD mutants were altered in btn2Δ cur1Δ cells (Fig. 4). When overexpressed transiently from a galactose promoter, Ure2H187Q destabilized endogenous [URE3] equally well in the presence or absence of Btn2 and Cur1, but Ure2K104I, Ure2S158F and Ure2T249A destabilized [URE3] less effectively in btn2Δ cur1Δ cells. The stability of [URE3] in cells lacking only Btn2 or Cur1 was like that of the wild type. These results suggest that prions composed of these latter three Ure2 mutants are more sensitive to curing by the combination of Btn2 and Cur1.
FIG 4.
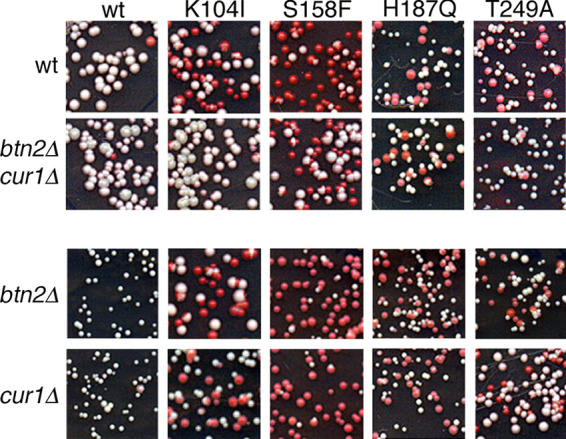
Dominant impairment of [URE3] by some mutant Ure2 proteins depends on Btn2 and Cur1. Wild type (wt), btn2Δ cur1 double mutant, and single btn2Δ and cur1Δ mutants (third and fourth rows) propagating [URE3] composed of wild-type Ure2 were transformed by plasmids (pEAD31-34) encoding the galactose-inducible Ure2 mutants indicated at the top. Transformants were grown in SGal-leu for 8 h and then plated on 1/2YPD. The frequency of red transformant colonies reflects how efficiently the mutant proteins destabilize endogenous [URE3].
To test if Btn2 and Cur1 were responsible for destabilizing prions composed solely of the mutant Ure2 proteins, we crossed Ure2 mutant strains with a btn2Δ cur1Δ [URE3] strain and monitored [URE3] in meiotic progeny of the diploids. Among over 30 btn2Δ cur1Δ double mutant spore clones for each Ure2 mutant, we did not obtain any that propagated [URE3]. Similar results were obtained for clones of the single btn2Δ and cur1Δ mutants. Thus, the mutant [URE3] prions were highly unstable even in cells lacking both Btn2 and Cur1.
Overexpression curing of [URE3] by Btn2 and Cur1 involves Hsp42 in complex ways (33). Curing by either Btn2 or Cur1 does not require the other. Efficient curing by Btn2 requires Hsp42, but curing by Cur1 does not. Yet curing by Hsp42 requires Cur1. We similarly determined if Hsp42 was driving destabilization of our mutant prions by crossing the Ure2 mutants with hsp42Δ [URE3] cells and monitoring [URE3] in meiotic progeny. Among over 20 hsp42Δ spore clones for each Ure2 mutant, none propagated [URE3]. Thus, the mutant [URE3] prions also were highly unstable in cells lacking Hsp42.
We then tested the effects of deleting all three antiprion factors. In btn2Δ cur1Δ hsp42Δ triple mutant strains, [URE3]S158F and [URE3]T249A prions propagated stably without selection (Fig. 5). Together, these results indicate that either Hsp42 (i.e., in btn2Δ cur1Δ cells) or the combination of Btn2 and Cur1 (i.e., in hsp42Δ cells) was enough to destabilize [URE3]S158F and [URE3]T249A. They also suggest that K104I and H187Q disrupt propagation of [URE3] by mechanisms unrelated to that of S158F and T249A.
FIG 5.

Collectively deleting Btn2, Cur1, and Hsp42 stabilizes mutant [URE3] prions. Wild-type (wt) and btn2Δ cur1Δ hsp42Δ triple mutant (bΔ,cΔ,42Δ) cells propagating [URE3] composed of the indicated Ure2 proteins were grown as patches of cells on −ade plates to select for [URE3] and then were replica plated onto similar plates (−ade, left) and onto 1/2YPD (right). [URE3]S158F and [URE3]T249A are stable in the cells lacking Btn2, Cur1, and Hsp42, as indicated by the white color of cells on 1/2YPD.
CTD mutations alter prion aggregation dynamics.
To obtain insight into the dynamics of prion loss, we observed prion aggregates in real time. Earlier, we described a Ure2-GFP fusion protein useful for monitoring aggregation of Ure2 in live cells (35). From an allele integrated at the RNQ1 genomic locus, it produces about 10-fold less Ure2-GFP than wild-type Ure2 and incorporates into preexisting prion fibers without destabilizing them. Our wild-type [ure-o] strains have diffuse but somewhat granular fluorescence, while [URE3] cells have up to a few hundred disperse and rapidly moving bright foci (Fig. 6, rightmost panel and top row) (35). Although relative numbers of foci can be compared, we note that the numbers of these foci cannot be determined accurately from these static images. We monitored aggregation of Ure2 in cells propagating mutant [URE3] prions by first growing cells expressing this Ure2-GFP in medium lacking adenine to maintain [URE3]. Cells were then transferred to adenine-replete medium to remove selection for [URE3], and the fluorescence pattern of Ure2-GFP was observed as the cells continued to divide.
FIG 6.

Ure2-GFP foci coalesce in cells propagating mutant [URE3] prions. Cells propagating prions composed of wild-type or mutant prions indicated on the left were shifted from medium selecting for prion maintenance to nonselective medium (+ adenine) and monitored at the times indicated at the top. Wild-type and isogenic btn2Δ cur1Δ hsp42Δ cells were monitored similarly. The rightmost column shows fluorescence of [ure-o] cells. The very large bright globules in some T249A wild-type cells at 6 h and K104I triple mutant cells at 0 h are autofluorescing vacuoles, not GFP.
Wild-type [URE3] cells grown without adenine had many small foci and occasional larger foci. Six hours after adding adenine, there were fewer larger foci, and cells displayed a more uniform pattern. This pattern did not change much during continued incubation for 2 days. Thus, growth without adenine appeared to cause formation of larger prion particles that resolved into a pattern of uniformly smaller foci after transfer to nonselective medium.
The fluorescence in [ure-o] cells of the Ure2 mutant strains was essentially indistinguishable from that of the wild-type strain. Thus, Ure2-GFP remains similarly soluble in all the [ure-o] cells. In general, cells with mutant [URE3] prions in cultures without adenine looked similar to the wild type but with an increased proportion of larger foci (Fig. 6, leftmost column). The dynamics of fluorescence changes after adding adenine was somewhat different in each mutant. Together with our other data, these observations indicate that Ure2-GFP incorporates similarly into wild-type and mutant Ure2 prions, which is expected, as they all have the identical amyloid-forming region. Thus, the Ure2 mutations do not interfere with formation of copolymers with other versions of Ure2, which aligns with their dominant inhibitory effects.
For cells with [URE3]S158F, adding adenine reduced the frequency of larger foci, but over time, foci seemed to coalesce and become increasingly larger until most cells had only one or two large foci. These changes were accompanied by an increasing proportion of cells with diffuse fluorescence resembling [ure-o] cells. For cells with [URE3]T249A, this pattern of aggregating foci was similar but showed faster kinetics. These data are consistent with loss of [URE3] from the mutant cells being caused by coalescence of dispersed prion aggregates that impairs their transmission to daughters either by reducing their numbers or by making them too large to traverse the bud neck during cell division.
The pattern of foci aggregation in these two mutants is strikingly similar to what we observe in our wild-type [URE3] cells when Btn2, Cur1, or Hsp42 is overexpressed (35). We therefore repeated the experiment using btn2Δ cur1Δ hsp42Δ triple mutant cells. In line with the stabilization of [URE3]S158F and [URE3]T249A prions in the triple mutant (Fig. 5), cells expressing Ure2S158F and Ure2T249A still had many small foci 48 h after adenine was added (Fig. 6, grid on right). Thus, the aggregation of these foci was driven primarily by these antiprion factors.
After the addition of adenine to cells propagating [URE3]K104I, the numbers of foci were reduced most rapidly until most cells showed diffuse fluorescence. There was very little difference in the fluorescence pattern for [URE3]K104I in btn2Δ cur1Δ hsp42Δ cells, suggesting that the reduction of [URE3]K104I foci in these cells was caused by something other than Btn2, Cur1, and Hsp42.
Compared with wild-type Ure2, foci in Ure2H187Q mutants were noticeably clumpier. Also, cells with diffuse fluorescence resembling [ure-o] cells were seen at all time points, and the number of such cells increased over time. In btn2Δ cur1Δ hsp42Δ cells, the foci were less clumpy, but the numbers of cells without foci did not change much. These data suggest that the instability of [URE3]H187Q is not being driven by these PQC factors and are consistent with results shown above that suggest that H187Q disrupts [URE3] propagation by a mechanism different than that of the other mutants.
CTD mutations alter prion seed numbers.
A defining characteristic of yeast prions is the average number of prion “seeds” per cell (45, 46). This number generally reflects the efficiency with which the prion amyloid assembles and divides (47). Division is driven primarily by action of the Hsp104-driven disaggregation machinery on prion fibers (16, 48, 49). Btn2 counteracts this seed production by collecting disperse prion seeds, but its ability to cure cells of [URE3] is limited by the number of prions it can collect before a cell divides (33). [URE3] variants with 1/2 to 1/5 of wild-type seed numbers per cell are cured by normal levels of Btn2.
We estimated numbers of prion seeds per cell (see Materials and Methods) and found that wild-type cells had an average of about 120 [URE3] seeds (Table 2). Isogenic cells lacking Btn2, Cur1, and Hsp42 had roughly 600 per cell, a 5-fold increase that reflects their antiprion activity. The average numbers of [URE3]S158F seeds per cell was 13, which is 10-fold less than the wild type. The chances that prions are transmitted to daughter cells should be reduced correspondingly. Depleting Btn2, Cur1, and Hsp42 increased [URE3]S158F seed numbers roughly 9-fold to a level near that of wild-type [URE3] in wild-type cells but still 5-fold less than wild-type [URE3] in btn2Δ cur1Δ hsp42Δ cells. Thus, S158F apparently reduces seed numbers to a degree that Btn2, Cur1, and Hsp42 can prevent their efficient transmission, but not enough to destabilize [URE3]S158F in cells lacking this system.
TABLE 2.
Prion seed numbers per cella
| Strain | Ure2 | Genotypeb | Avg no. (SD) | Mutant/WTc | bΔcΔ42Δ/WTd |
|---|---|---|---|---|---|
| 1976 | WT | WT | 117 (24) | ||
| 1977 | WT | bΔcΔ42Δ | 571 (444) | 5 | |
| 1979 | K104I | WT | 13 (4) | 0.1 | |
| 1980 | K104I | bΔcΔ42Δ | 26 (14) | 2 | |
| 1982 | S158F | WT | 13 (6) | 0.1 | |
| 1983 | S158F | bΔcΔ42Δ | 107 (42) | 9 | |
| 1985 | H187Q | WT | 403 (144) | 4 | |
| 1986 | H187Q | bΔcΔ42Δ | 441 (194) | 1 | |
| 1988 | T249A | WT | 12 (6) | 0.1 | |
| 1989 | T249A | bΔcΔ42Δ | 698 (492) | 60 |
Cells grown without adenine were plated on rich medium containing 3 mM guanidine-HCl and grown to colonies of <1 mm diameter. Five to ten entire colonies of each strain were suspended in water and plated on −ade medium. Values are averages of resulting Ade+ colonies (±SD). WT, wild type.
bΔcΔ42Δ, btn2::TRP1 cur1::KanMX hsp42::KanMX.
Ratio of seed numbers in Ure2 mutant cells/117 seeds per cell of Ure2 WT cells.
Fold increase in avg seed numbers of cells lacking Btn2, Cur1, and Hsp42 versus isogenic WT.
The average number of [URE3]T249A prions was 12, again 10-fold lower than the wild type and similarly explaining [URE3]T249A instability. In btn2Δ cur1Δ hsp42Δ cells, the number was 60-fold higher and similar to that of prions in wild-type cells lacking Btn2, Cur1, and Hsp42. This comparable number of seeds in cells lacking this system indicates that T249A does not simply reduce seed number but increases sensitivity of [URE3]T249A to these antiprion factors.
The average number of [URE3]K104I seeds per cell was similarly reduced to 13, but deleting Btn2, Cur1, and Hsp42 only doubled it to 26, suggesting that the reduction was not due to the action of these PQC factors. This low increase in seed numbers of the triple mutant could reflect reduced ability of the Hsp104 machinery to act on [URE3]K104I prions in a way that promotes their replication. This scenario aligns with the instability of [URE3]K104I in wild-type cells and the failure of these prions to be stabilized by depleting Btn2, Cur1, and Hsp42.
For [URE3]H187Q the average seed number (>400) was over three times higher than wild-type [URE3], and deleting Btn2, Cur1, and Hsp42 had little effect. This high number is consistent with our observations of fluorescent Ure2-GFP foci in cells propagating [URE3]H107Q and, despite the reduced ability of Ure2H187Q to form amyloid in vitro, it suggests that the mutant prions grow and replicate efficiently. Collectively, our data suggest that H187Q disrupts transmission, the remaining process necessary for yeast prion propagation.
DISCUSSION
We identified many single amino acid substitutions outside the amyloid-forming region of Ure2 that dominantly destabilized propagation of endogenous [URE3] prions and found that they prevented Ure2 from forming stably propagating prions. Of the four mutations we characterized, all allowed Ure2 to propagate as [URE3] under conditions selecting for the prion yet caused rapid and complete loss of [URE3] when selection was relieved. Despite these similarities, they impaired [URE3] propagation in mechanistically different ways. The differences could be classified loosely by whether they sensitized [URE3] to Btn2, Cur1, and Hsp42 or impaired the ability of Ure2 to form amyloid. S158F and T249A enhanced the ability of these antiprion factors to eliminate prions composed of the mutant proteins, while K104I and H187Q impaired the ability of Ure2 to form amyloid in vitro, which possibly contributed to their effects in vivo. Because the method used to select the mutants depended on the secondary effect of the dominant inhibition of endogenous wild-type [URE3], it is possible that many other point mutations in the CTD could interfere with [URE3] propagation without sharing this dominant property.
Btn2 eliminates most spontaneously occurring variants of [URE3], restricting the prions that become established to infrequent variants with average seed numbers per cell that exceed its capacity to collect them before they spread (33). Accordingly, while [URE3] variants induced in wild-type cells typically have high seed numbers, most variants arising in cells lacking Btn2 have low seed numbers. Moreover, elevating Btn2 expression more effectively cures prion variants that have lower seed numbers per cell. Although [URE3]S158F and [URE3]T249A both had 10-fold reduced seed numbers, which provides a simple explanation for their mitotic instability, our data suggest different explanations for their stabilization when Btn2, Cur1, and Hsp42 are collectively depleted.
For [URE3]S158F, when these proteins are depleted, the average number of seeds per cell was high enough for it to propagate stably but still over 5-fold less than wild-type [URE3]. This difference could reflect a propensity of individual [URE3]S158F seeds to aggregate or a reduced sensitivity to action of the disaggregation machinery needed for replication. The former seems more likely, as the relative increase in seed numbers for [URE3]S158F upon disruption of this system, which depends on the disaggregation machinery, was almost twice that of the wild type. Therefore, enhanced susceptibility of [URE3]S158F to the action of the antiprion proteins is apparently due to inherent aggregation of seeds that produces a low number of seeds per cell.
For [URE3]T249A, the similarly low seed numbers seem more likely to be explained by the prion being more sensitive to being sequestered. In the absence of Btn2, Cur1, and Hsp42, the number of [URE3]T249A seeds per cell was as high as that of wild-type [URE3], which suggests that these two prions have similar growth and replication rates. The relative reduction in prion seeds per cell caused by the action of Btn2, Cur1, and Hsp42, however, was 12-fold fold greater for [URE3]T249A than for wild-type prions, indicating that these proteins act much more efficiently on [URE3]T249A prions. T249A might therefore enhance interactions of prion aggregates with these proteins, either by presenting or exposing an attractive surface or by reducing interactions of normal Ure2 binding partners that could compete with this system for binding to Ure2.
Although a potential disruption of phosphorylation of S158 or T249 also might contribute their effects, both residues scored low by phosphorylation prediction software. Both substitutions also result in large changes in amino acid size and polarity, suggesting that their effects are a consequence of altered protein conformation.
The low seed numbers of [URE3]K104I are consistent with the considerable defect purified Ure2K104I had in forming amyloid, and its effects on prions could be due to such reduced assembly in vivo. The exceptionally extensive aggregation of Ure2K104I as observed by fluorescence could reflect a high propensity of [URE3]K104I prions to self-associate and form higher-order fibrous assemblies. Such aggregates could be expected to have reduced ability to assemble into amyloid and to be more resistant to action of the disaggregation machinery, which would result in a replication defect. This scenario would align with both poor amyloid formation by Ure2K104I in vitro and reduced [URE3]K104I seed numbers. A direct effect on reducing interaction with the Hsp104 machinery that results in a replication defect also would explain why disrupting the Btn2 antiprion system resulted in only marginally increased seed numbers of [URE3]K104I compared with wild-type prions. The numbers of [URE3]K104I prions per cell as counted and as observed by fluorescence seem low enough to cause prion loss by reducing the chances of spreading to daughter cells even when Btn2, Cur1, and Hsp42 are deleted. The dependence of Ure2K104I on Btn2 and Cur1 to have its dominant inhibitory effect could be explained if incorporation of Ure2K104I into wild-type prions interferes with replication and reduces their numbers only enough to sensitize them to Btn2 and Cur1.
In contrast, the ability of Ure2H187Q to disrupt propagation of wild-type [URE3] and the seed numbers of [URE3]H187Q were both unaffected by depleting Btn2, Cur1, and Hsp42. The anti-[URE3] effects of Ure2H187Q are therefore independent of these PQC factors. One explanation of the negative effects of H187Q that aligns with all our data is that transmission of [URE3]H187Q prions between cells during cell division is somehow disrupted. A mechanism for how that might happen remains to be determined experimentally.
Amyloid-forming regions of yeast prion proteins often are well-defined stretches of amino acids that can form amyloid and prions when separated from the rest of the protein, and they can impart prion characteristics when transferred to other proteins (4, 50–52). Thus, amyloid-forming regions of prion proteins are primary determinants of prion properties. Accessibility of PQC factors to amyloid cores of prions, which are highly ordered, tightly arranged, and partly shielded by natively folded globular domains (52–54), can be expected to be limited. Thus, the interactions of PQC factors with prions that are necessary for prion propagation or elimination can be presumed to occur mostly outside this region.
The modular properties of prion-determining and functional regions of yeast prion proteins suggest that the effects we see for Ure2 could be more general. Point mutations that disrupt prion propagation have also been identified outside the amyloid-forming regions of other yeast prion proteins. One in Sup35 and several in Rnq1 can disrupt propagation of [PSI+] and [PIN+], respectively (55, 56). The Sup35 mutation, which reduces Sup35 function, reduced amyloid-forming ability and increased nonprion aggregation, while some of the mutations in Rnq1 had effects on Sis1 interaction. Mechanisms connecting the substitutions with their effects on prion destabilization were not described. Our findings raise the possibility that altered interactions with PQC processes underlie their prion-destabilizing effects.
Lastly, despite residing in the functional domain of Ure2, the widely dispersed mutations we identified do not disrupt the function of soluble Ure2, which suggests that some aspect of normal Ure2 activity is important for the mutants to disrupt prion-forming ability. Overall, our findings demonstrate the strong influence that residues outside the amyloid-forming region can have on amyloid propagation and elimination in vivo, and they pave the way for future work to help understand the mechanisms of such prion disruption in molecular detail.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
The strains used are listed in Table 3, and the plasmids used are listed in Table 4. Mutant URE2 alleles were integrated into the genome by cotransforming MR149 (ure2Δ) with plasmid pRS316 (URA3) and linear DNA encoding the mutant genes with 500 bp of 5′ and 3′ noncoding flanking DNA. Ura+ transformants that had become G418 sensitive were isolated and verified to have integrated alleles by genomic PCR and sequencing. Strain 1660 has URE2-GFP integrated at the RNQ1 genomic locus (35). Ure2 mutant strains were backcrossed to the isogenic wild type at least twice and then to strain 1976 (isogenic to 1660) to obtain URE2 mutants that also express Ure2-GFP. Unless indicated otherwise, the [URE3] variant from our wild-type strain 1075 is the source of [URE3] in all other strains. Both deletion of URE2 and the presence of [URE3] cause slow growth of our strains, presumably due to loss of Ure2 function.
TABLE 3.
Yeast strainsa
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| 1075 | MATa kar1-1 SUQ5 PDAL5::ADE2 his3Δ200 leu2Δ1 trp1Δ63 ura3-52 [URE3] | 62 |
| MR149 | ure2::KanMX | This study |
| 1660 | PRNQ1::URE2-GFP::rnq1 | 35 |
| 1519 | URE2K104I | This study |
| 1520 | URE2R164G | This study |
| 1524 | URE2S158F | This study |
| 1521 | URE2H187Q | This study |
| 1525 | URE2T249A | This study |
| 1975 | btn2::TRP1 cur1::KanMX | This study |
| 1735 | btn2::TRP1 cur1::KanMX URE2K104 | This study |
| 1743 | btn2::TRP1 cur1::KanMX URE2S158F | This study |
| 1739 | btn2::TRP1 cur1::KanMX URE2H187Q | This study |
| 1747 | btn2::TRP1 cur1::KanMX URE2T249A | This study |
| 1976 | PRNQ1::URE2-GFP::rnq1 (1660 backcross) | This study |
| 1979 | 1976 URE2K104I | This study |
| 1982 | 1976 URE2S158 | This study |
| 1985 | 1976 URE2H187Q | This study |
| 1988 | 1976 URE2T249A | This study |
| 1977 | 1976 btn2::TRP1 cur1::KanMX hsp42::KanMX | This study |
| 1980 | 1977 URE2K104I | This study |
| 1983 | 1977 URE2S158F | This study |
| 1986 | 1977 URE2H187Q | This study |
| 1989 | 1977 URE2T249A | This study |
All are identical to 1075 except at the indicated loci.
TABLE 4.
Plasmids
| Plasmid | Relevant content | Source or reference |
|---|---|---|
| pRS316 | CEN (single copy), URA3 empty vector | 63 |
| pRS315 | CEN (single copy), LEU2 empty vector | 63 |
| p415 | CEN (single copy), LEU2 empty vector | 64 |
| pKT41 | Bacterial expression vector encoding His6-Ure2 | 6 |
| pROB155 | pRS315 + URE2 (–600 to +500; AatII-XmaI) | This study |
| pEP200 | pRS315 + URE2 (–500 to +300; SacI-SalI) | This study |
| pEP202 | pRS315 + URE2K104I (pEP200 site-directed mutant) | This study |
| pEP211 | pRS315 + URE2S158F (pEP200 site-directed mutant) | This study |
| pEP208 | pRS315 + URE2H187Q (pEP200 site-directed mutant) | This study |
| pEP217 | pRS315 + URE2T249A (pEP200 site-directed mutant) | This study |
| pEAD31 | p415 + PGAL1::URE2 (SpeI-SalI PCR from pEP200) | This study |
| pEAD32 | p415 + PGAL1::URE2K104I (pEAD31site directed mutant) | This study |
| pEAD33 | p415 + PGAL1::URE2S158 (pEAD31site directed mutant) | This study |
| pEAd34 | p415 + PGAL1::URE2H187Q (pEAD31site directed mutant) | This study |
| pEAD35 | p415 + PGAL1::URE2T249A (pEAD31site directed mutant) | This study |
| pEP300 | pKT41 with URE2 (E. coli expression vector) | This study |
| pEP302 | pKT41 with URE2K104I (pEP300 site-directed mutant) | This study |
| pEP311 | pKT41 with URE2S158F (pEP300 site-directed mutant) | This study |
| pEP308 | pKT41 with URE2H187Q (pEP300 site-directed mutant) | This study |
| pEP317 | pKT41 with URE2T249A (pEP300 site-directed mutant) | This study |
Standard yeast media and growth conditions were used (57). YPAD contains 1% yeast extract, 2% peptone, 2% dextrose, and 400 mg/liter adenine. 1/2YPD is similar but lacks adenine and contains 0.5% yeast extract. Defined glucose medium (SC) contains 6.7 mg/liter yeast nitrogen base (Difco), 2% glucose, and all nutrients except those omitted to maintain plasmids or prions. SC plates with limiting adenine contain 8 mg/liter adenine. SGal is SC with galactose in place of glucose. Cells were grown at 30°C unless indicated otherwise.
Monitoring [URE3].
When a good source of nitrogen (e.g., ammonium) is present, Ure2 represses transcription of genes involved in nitrogen catabolism, including the allantoate transporter DAL5. Our strains have ADE2 controlled by the DAL5 promoter (PDAL5::ADE2) and thus do not express Ade2 when grown on standard ammonium-containing medium (38, 58). In addition to being Ade–, cells lacking Ade2 are red when adenine is limiting enough to induce the adenine biosynthetic pathway and cause accumulation of a pigment-producing substrate of Ade2 (59, 60). In [URE3] cells, Ure2 is depleted into insoluble prion fibers, which compromises Ure2 function. We monitor [URE3] by the resulting activation of the DAL5 promoter and expression of ADE2. [URE3] cells are Ade+ and white, while [ure-o] cells (lacking [URE3]) are Ade– and are red when grown on limiting adenine.
Isolation of Ure2 mutants.
Using plasmid pROB155 as a template, URE2 was amplified using error-prone PCR, and products were recloned on AatII-XmaI fragments into pROB155 in place of wild-type URE2. We used this library to transform wild-type [URE3] cells (strain 1075) and recovered plasmids from transformants that displayed any noticeable red color, reflecting loss of [URE3] during formation of the colonies. After an initial screen identified mostly NTD mutations and nonsense mutations producing truncations of the CTD, we enriched for selection of point mutations in the CTD by repeating the screen after replacing the 1.05-kb EagI-EcoRI fragment on pROB155, which contains codons 66 to 354 of URE2, with the same fragment from the mutant library.
[URE3] induction.
[URE3] was induced de novo by transient galactose-induced overexpression of Ure2(1-64) from plasmid p680 essentially as described previously (3). Briefly, cells carrying p680 were inoculated in liquid galactose medium to an optical density at 600 nm (OD600) of 0.05 and grown for 24 h, and then 106 cells were spread onto −ade plates. More than 95% of resulting Ade+ colonies were confirmed to propagate [URE3].
Counting prion seeds.
The numbers of prion seeds per cell from cells previously grown on medium lacking adenine to select for prions were estimated as described (46). Briefly, cells were first grown to colonies on YPAD plates containing 3 mM guanidine-HCl. Entire colonies were recovered, suspended in water, and spread onto −ade plates. Resulting Ade+ colonies represent the cells from those colonies that possessed at least one prion seed. These colonies probably underestimate seed numbers, as distribution of every seed in a progenitor cell to separate individual progeny is unlikely. We typically see wide variations in [URE3] seed numbers among cells for any given strain.
Western blotting.
Whole-cell lysates were prepared in lysis buffer (phosphate-buffered saline [PBS; pH 7.5], 0.1% Triton X-100, and protease inhibitor [Roche]). Proteins (20 μg/sample) were separated on 12% SDS-PAGE gels, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with polyclonal anti-Ure2 rabbit primary antibodies raised against the entire protein. Secondary antibody was goat anti-rabbit horseradish peroxidase (HRP)-linked IgG, which was detected by using the Supersignal West Pico Chemiluminescence kit (Thermo Scientific).
Ure2 protein expression and purification.
Wild-type and mutant N-terminal His6-tagged URE2 alleles were subcloned as NdeI-SacI fragments into plasmid pKT41. Full-length Ure2 proteins were expressed in Escherichia coli [Rosetta 2(DE3)PLysS]. Cultures grown at 30°C to an OD550 of 0.7 were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 18°C for 16 h. Cells were harvested, washed, suspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, 2.4 mM imidazole, 0.12% Triton X-100, 0.2 mg/ml lysozyme, protease inhibitor), and lysed by sonication. Immediately after lysis, guanidinium chloride was added to 1 M, insoluble debris was removed by centrifugation at 15,000 × g for 30 min at 4°C, and cleared lysate was passed through a 0.2-μm filter and loaded onto preequilibrated Ni-nitrilotriacetic acid (Ni-NTA) columns. Columns were washed with wash buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl) containing 5 to 20 mM imidazole. Protein was eluted in similar buffer containing 250 mM imidazole. Fast protein liquid chromatography (FPLC) size exclusion chromatography on Superdex 200 10/300 GL columns (GE Health Care) was used to determine the oligomeric status of Ure2 proteins. Size standards (Bio-Rad) were bovine thyroglobin (670 kDa), bovine gamma globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.35 kDa).
Thioflavin-T (Th-T) assay.
The kinetics of amyloid formation by full-length wild-type and mutant Ure2 proteins purified as above was assessed by Th-T assay as described previously (61). Briefly, 20 μM purified protein was added to 20 μM Th-T in PBS with pH 7.4 to a final volume of 150 μl in clear, round-bottom 96-well plates. One 0.5-mm-diameter glass bead was added in each well, and reaction mixtures were incubated at 37°C with orbital shaking at 750 rpm. Th-T fluorescence was monitored with excitation at 450 nm and emission at 485 nm. Fluorescence was measured at 10-min intervals for 60 min and subsequently at 30-min intervals.
Fluorescence microscopy.
Ure2 was monitored in vivo by expressing a “tracer” amount of wild-type or mutant Ure2-GFP from a gene fusion regulated by the RNQ1 promoter and integrated at the RNQ1 genomic locus in place of RNQ1. Overnight cultures grown in SD medium supplemented with appropriate nutrients and lacking adenine were diluted to on OD600 of 0.025 in the evening and 0.2 in the morning using similar medium containing adenine and grown for 2 days. GFP imaging was done on the Nikon A1R confocal microscope equipped with a 60×, 1.4-numerical-aperture (NA) objective. Yeast was imaged in 8-well, 25-mm, two-chambered coverslips (Lab-Tek, Rochester, NY). Imaging of the fluorescence was always done by z-stack confocal imaging of live cells, and the images shown are the maximized z-stacks.
ACKNOWLEDGMENTS
We thank Rob Stanley, Brindar Sandhu, and Brittany-Lee Roberts for plasmid construction and mutant isolation during the early stages of the study. We thank Herman Edskes and other NIH colleagues for critical review of the manuscript and gratefully acknowledge the NHLBI microscopy core.
This work was supported by the Intramural Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases, and The National Heart, Lung, and Blood Institute.
REFERENCES
- 1.Coschigano PW, Magasanik B. 1991. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione s-transferases. Mol Cell Biol 11:822–832. doi: 10.1128/mcb.11.2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wickner RB. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 3.Masison DC, Wickner RB. 1995. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 4.Masison DC, Maddelein ML, Wickner RB. 1997. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc Natl Acad Sci U S A 94:12503–12508. doi: 10.1073/pnas.94.23.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bousset L, Belrhali H, Melki R, Morera S. 2001. Crystal structures of the yeast prion Ure2p functional region in complex with glutathione and related compounds. Biochemistry 40:13564–13573. doi: 10.1021/bi011007b. [DOI] [PubMed] [Google Scholar]
- 6.Umland TC, Taylor KL, Rhee S, Wickner RB, Davies DR. 2001. The crystal structure of the nitrogen regulation fragment of the yeast prion protein Ure2p. Proc Natl Acad Sci U S A 98:1459–1464. doi: 10.1073/pnas.041607898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ngo S, Chiang V, Ho E, Le L, Guo Z. 2012. Prion domain of yeast Ure2 protein adopts a completely disordered structure: a solid-support EPR study. PLoS One 7:e47248. doi: 10.1371/journal.pone.0047248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. 2007. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics 176:1557–1565. doi: 10.1534/genetics.107.074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickner RB, Shewmaker F, Edskes H, Kryndushkin D, Nemecek J, McGlinchey R, Bateman D, Winchester CL. 2010. Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res 10:980–991. doi: 10.1111/j.1567-1364.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. 1997. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci U S A 94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. 1999. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 12.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. 2004. Dissection and design of yeast prions. PLoS Biol 2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel BK, Liebman SW. 2007. “Prion-proof” for [PIN+]: infection with in vitro-made amyloid aggregates of Rnq1p-(132-405) induces [PIN+]. J Mol Biol 365:773–782. doi: 10.1016/j.jmb.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satpute-Krishnan P, Serio TR. 2005. Prion protein remodelling confers an immediate phenotypic switch. Nature 437:262–265. doi: 10.1038/nature03981. [DOI] [PubMed] [Google Scholar]
- 15.Lum R, Tkach JM, Vierling E, Glover JR. 2004. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 279:29139–29146. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 16.Hung GC, Masison DC. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173:611–620. doi: 10.1534/genetics.106.056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glover JR, Lindquist S. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 18.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 19.Fan Q, Park KW, Du Z, Morano KA, Li L. 2007. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetics 177:1583–1593. doi: 10.1534/genetics.107.077982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hines JK, Li X, Du Z, Higurashi T, Li L, Craig EA. 2011. [SWI+], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet 7:e1001309. doi: 10.1371/journal.pgen.1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reidy M, Miot M, Masison DC. 2012. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 192:185–193. doi:10.1534/genetics.112.142307. doi: 10.1534/genetics.112.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tessarz P, Mogk A, Bukau B. 2008. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol Microbiol 68:87–97. doi: 10.1111/j.1365-2958.2008.06135.x. [DOI] [PubMed] [Google Scholar]
- 23.Moriyama H, Edskes HK, Wickner RB. 2000. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol 20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung G, Jones G, Wegrzyn RD, Masison DC. 2000. A role for cytosolic hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics 156:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones GW, Masison DC. 2003. Saccharomyces cerevisiae Hsp70 mutations affect [PSI+] prion propagation and cell growth differently and implicate Hsp40 and tetratricopeptide repeat cochaperones in impairment of [PSI+]. Genetics 163:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones G, Song Y, Chung S, Masison DC. 2004. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol Cell Biol 24:3928–3937. doi: 10.1128/mcb.24.9.3928-3937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kryndushkin D, Wickner RB. 2007. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell 18:2149–2154. doi: 10.1091/mbc.e07-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kryndushkin DS, Shewmaker F, Wickner RB. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J 27:2725–2735. doi: 10.1038/emboj.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higurashi T, Hines JK, Sahi C, Aron R, Craig EA. 2008. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci U S A 105:16596–16601. doi: 10.1073/pnas.0808934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe KJ, Ren HY, Trepte P, Cyr DM. 2013. The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol Biol Cell 24:3588–3602. doi: 10.1091/mbc.E13-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wickner RB. 2019. Anti-prion systems in yeast. J Biol Chem 294:1729–1738. doi: 10.1074/jbc.TM118.004168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorkovskiy A, Reidy M, Masison DC, Wickner RB. 2017. Hsp104 disaggregase at normal levels cures many [PSI+] prion variants in a process promoted by Sti1p, Hsp90, and Sis1p. Proc Natl Acad Sci U S A 114:E4193–E4202. doi: 10.1073/pnas.1704016114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickner RB, Bezsonov E, Bateman DA. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci U S A 111:E2711–20. doi: 10.1073/pnas.1409582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell 23:3041–3056. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Lanz J, Steinberg D, Pease T, Ahearn JM, Bezsonov EE, Staguhn ED, Eisenberg E, Masison DC, Greene LE. 2018. Real-time imaging of yeast cells reveals several distinct mechanisms of curing of the [URE3] prion. J Biol Chem 293:3104–3117. doi: 10.1074/jbc.M117.809079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edskes HK, Gray VT, Wickner RB. 1999. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci U S A 96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwimmer C, Masison DC. 2002. Antagonistic interactions between yeast [PSI(+)] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol Cell Biol 22:3590–3598. doi: 10.1128/mcb.22.11.3590-3598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlumpberger M, Prusiner SB, Herskowitz I. 2001. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol 21:7035–7046. doi: 10.1128/MCB.21.20.7035-7046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edskes HK, McCann LM, Hebert AM, Wickner RB. 2009. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics 181:1159–1167. doi: 10.1534/genetics.108.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrett S, Freeman SJ, Butler PJ, Fersht AR. 1999. Equilibrium folding properties of the yeast prion protein determinant Ure2. J Mol Biol 290:331–345. doi: 10.1006/jmbi.1999.2872. [DOI] [PubMed] [Google Scholar]
- 41.Lian HY, Zhang H, Zhang ZR, Loovers HM, Jones GW, Rowling PJ, Itzhaki LS, Zhou JM, Perrett S. 2007. Hsp40 interacts directly with the native state of the yeast prion protein Ure2 and inhibits formation of amyloid-like fibrils. J Biol Chem 282:11931–11940. doi: 10.1074/jbc.M606856200. [DOI] [PubMed] [Google Scholar]
- 42.Reidy M, Sharma R, Roberts BL, Masison DC. 2016. Human J-protein DnaJB6b cures a subset of Saccharomyces cerevisiae prions and selectively blocks assembly of structurally related amyloids. J Biol Chem 291:4035–4047. doi: 10.1074/jbc.M115.700393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barbitoff YA, Matveenko AG, Moskalenko SE, Zemlyanko OM, Newnam GP, Patel A, Chernova TA, Chernoff YO, Zhouravleva GA. 2017. To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1. Mol Microbiol 105:242–257. doi: 10.1111/mmi.13697. [DOI] [PubMed] [Google Scholar]
- 44.Wickner RB, Bezsonov EE, Son M, Ducatez M, DeWilde M, Edskes HK. 2018. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry 57:1285–1292. doi: 10.1021/acs.biochem.7b01285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. 2000. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox B, Ness F, Tuite M. 2003. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165:23–33. doi: 10.1371/journal.pone.0004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka M, Collins SR, Toyama BH, Weissman JS. 2006. The physical basis of how prion conformations determine strain phenotypes. Nature 442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 48.Jung G, Masison DC. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol 43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 50.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. 1997. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 277:381–383. doi: 10.1126/science.277.5324.381. [DOI] [PubMed] [Google Scholar]
- 51.Li L, Lindquist S. 2000. Creating a protein-based element of inheritance. Science 287:661–664. doi: 10.1126/science.287.5453.661. [DOI] [PubMed] [Google Scholar]
- 52.Baxa U, Taylor KL, Wall JS, Simon MN, Cheng N, Wickner RB, Steven AC. 2003. Architecture of Ure2p prion filaments: the N-terminal domains form a central core fiber. J Biol Chem 278:43717–43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 53.Loquet A, Bousset L, Gardiennet C, Sourigues Y, Wasmer C, Habenstein B, Schutz A, Meier BH, Melki R, Bockmann A. 2009. Prion fibrils of Ure2p assembled under physiological conditions contain highly ordered, natively folded modules. J Mol Biol 394:108–118. doi: 10.1016/j.jmb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 54.Kryndushkin DS, Wickner RB, Tycko R. 2011. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-beta structure: evidence from solid-state NMR. J Mol Biol 409:263–277. doi: 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabani M, Cosnier B, Bousset L, Rousset JP, Melki R, Fabret C. 2011. A mutation within the C-terminal domain of Sup35p that affects [PSI+] prion propagation. Mol Microbiol 81:640–658. doi: 10.1111/j.1365-2958.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- 56.Shibata S, Kurahashi H, Nakamura Y. 2009. Localization of prion-destabilizing mutations in the N-terminal non-prion domain of Rnq1 in Saccharomyces cerevisiae. Prion 3:250–258. doi: 10.4161/pri.3.4.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sherman F. 2002. Getting started with yeast. Methods Enzymol 350:3–41. doi: 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- 58.Brachmann A, Baxa U, Wickner RB. 2005. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J 24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smirnov MN, Smirnov VN, Budowsky EI, Inge-Vechtomov SG, Serebrjakov NG. 1967. Red pigment of adenine-deficient yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun 27:299–304. doi: 10.1016/s0006-291x(67)80096-2. [DOI] [PubMed] [Google Scholar]
- 60.Silver JM, Eaton NR. 1969. Functional blocks of the ad-1 and ad-2 mutants of Saccharomyces cerevisiae. Biochem Biophys Res Commun 34:301–305. doi: 10.1016/0006-291x(69)90831-6. [DOI] [PubMed] [Google Scholar]
- 61.Giehm L, Oliveira CL, Christiansen G, Pedersen JS, Otzen DE. 2010. SDS-induced fibrillation of alpha-synuclein: an alternative fibrillation pathway. J Mol Biol 401:115–133. doi: 10.1016/j.jmb.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 62.Sharma D, Masison DC. 2008. Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179:1301–1311. doi: 10.1534/genetics.108.089458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]