

Abstract
Nucleoporins (Nups) build highly organized nuclear pore complexes (NPCs) at the nuclear envelope (NE). Several Nups assemble into a sieve‐like hydrogel within the central channel of the NPCs. In the cytoplasm, the soluble Nups exist, but how their assembly is restricted to the NE is currently unknown. Here, we show that fragile X‐related protein 1 (FXR1) can interact with several Nups and facilitate their localization to the NE during interphase through a microtubule‐dependent mechanism. Downregulation of FXR1 or closely related orthologs FXR2 and fragile X mental retardation protein (FMRP) leads to the accumulation of cytoplasmic Nup condensates. Likewise, models of fragile X syndrome (FXS), characterized by a loss of FMRP, accumulate Nup granules. The Nup granule‐containing cells show defects in protein export, nuclear morphology and cell cycle progression. Our results reveal an unexpected role for the FXR protein family in the spatial regulation of nucleoporin condensation.
Keywords: dynein, fragile X syndrome, FXR1, nucleoporins, phase separation
Subject Categories: Membrane & Intracellular Transport
Fragile X‐related proteins and dynein inhibit ectopic phase separation of nucleoporins in the cytoplasm and facilitate their localization to the nuclear envelope during G1 phase of the cell cycle.

Introduction
Formation of supramolecular assemblies and membrane‐less organelles such as the nucleolus, Cajal bodies, nuclear speckles, stress granules (SG), P‐bodies, germ granules and PML bodies are important for cellular homeostasis (Boeynaems et al, 2018). Among the factors controlling their formation and turnover is the presence of intrinsically disordered regions (IDRs) in protein components, their ability to form multivalent protein–protein, and protein–RNA interactions (Feng et al, 2019) and proteins’ local concentration. Indeed, many RNA‐binding proteins (RBPs) have the ability to demix into liquid states (liquid droplets), which can be subsequently transformed into pathological amyloids (Lin et al, 2015; Harrison & Shorter, 2017; Shorter, 2019) that have been linked to many neurological disorders (Shin & Brangwynne, 2017). One example of a large protein assembly consisting of IDR‐containing proteins is the nuclear pore complex (NPC), which plays an essential role in cellular homeostasis (Knockenhauer & Schwartz, 2016; Sakuma & D'Angelo, 2017).
NPCs are large, multisubunit protein complexes (Beck & Hurt, 2017; Hampoelz et al, 2019a) spanning the nuclear envelope (NE) that constitute the transport channels controlling the exchange of proteins and mRNA between the nucleus and the cytoplasm. They are built from roughly 30 different nucleoporins (Nups) each present in multiple copies in the NPCs. The ring‐like NPC scaffold is embedded in the NE and shows highly organized eight‐fold symmetry (Knockenhauer & Schwartz, 2016). In contrast, the central channel of the NPC is formed from Nups containing disordered elements characterized by the presence of phenylalanine–glycine (FG) repeats, the so‐called FG‐Nups. The FG‐Nups have the ability to phase separate into sieve‐like hydrogels that constitute a selective and permeable barrier for diffusing molecules and transported cargos through the NPCs (Schmidt & Görlich, 2016). This ability of the FG‐Nups to form permeable hydrogels can also be reconstituted in vitro and is highly conserved through the evolution (Frey et al, 2006; Frey & Görlich, 2007; Schmidt & Görlich, 2015). The cohesive abilities of FG‐Nups allow not only for the formation of the permeability barrier but also for building the links with the structural scaffold elements of the NPC (Onischenko et al, 2017). The non‐FG‐Nups can also form condensates in cells as they are sequestered in the SGs (Zhang et al, 2018) and in various pathological aggregates in the nucleus and in the cytoplasm (Li & Lagier‐Tourenne, 2018; Hutten & Dormann, 2020). A fraction of cytoplasmic nucleoporins was also identified in the promyelocytic leukaemia protein (PML)‐positive structures, the so‐called CyPNs (cytoplasmic accumulations of PML and nucleoporins), which could move on microtubules to dock at the NE (Jul‐Larsen et al, 2009), although the cellular roles of the CyPNs remain to be understood. This indicates that Nups have an intrinsic capacity to aberrantly assemble, suggesting protective mechanisms may exist to prevent it in the cell. Indeed, in Drosophila embryos a large excess of soluble Nups has been reported (Onischenko et al, 2004), and in cells, Nups are synthesized as soluble proteins in the cytoplasm (Davis & Blobel, 1987). How the balance of soluble Nups is controlled, and what factors regulate the localized assembly of Nups is currently unknown.
The fragile X‐related (FXR) proteins (FXR1, FXR2 and fragile X mental retardation protein [FMRP]) are a family of RNA‐binding proteins displaying a high degree of sequence and structural similarity and playing important roles in mRNA metabolism (Li & Zhao, 2014). Silencing of the FMR1 gene that encodes the FMRP protein (Santoro et al, 2012) leads to fragile X syndrome (FXS), the most common form of inherited intellectual human disability worldwide, for which no efficient therapy exists to date (Mullard, 2015). For this reason, the role of FXR proteins has been mostly investigated in brain, in the context of neurodevelopmental disorders (Bagni & Zukin, 2019), and genome‐wide association studies suggest the involvement of this family in a wide spectrum of mental illnesses (Guo et al, 2015; Khlghatyan et al, 2018).
More recent studies have linked FXR proteins to cancer progression, and in particular, FMRP and FXR1 were found overexpressed in different types of cancer (Lucá et al, 2013; Jin et al, 2016; Zalfa et al, 2017; Cao et al, 2019). Although many overlapping functions have been proposed for this protein family, different tissue, cellular and intracellular distributions of the FXR proteins suggest that they might have, in addition to their canonical role as RNA‐binding proteins, independent functions (Darnell et al, 2009). Interestingly, the protein region containing the RGG box (arginine‐ and glycine‐rich region) of FMRP has a low‐complexity sequence composition and is unfolded and flexible (Ramos, 2003), implicating its role in the membrane‐less assemblies. Here, we identify a novel role for the FXR protein family and dynein in the spatial regulation of nucleoporin condensation.
Results
FXR1 protein localizes to the NE and interacts with Nups
FXR1 co‐localizes with various cytoplasmic protein–RNA assemblies, but it is also present in the nuclear compartment in human cells (Tamanini et al, 1999; Oldenburg et al, 2014). In search for possible additional cellular functions of FXR1 independent of its role in RNA binding, we performed immunoprecipitations (IPs) of stably expressed GFP‐FXR1 protein and analysed the interacting partners by mass spectrometry. Of the interacting proteins, including the known FXR1 partners, FXR2 and FMRP, four nucleoporins (Nups), Nup210, Nup188, Nup133 and Nup85, were detected specifically in GFP‐FXR1 IPs (Dataset EV1). We confirmed the GFP‐FXR1 interaction with endogenous Nup133 and Nup85, which are components of the evolutionary conserved Nup107‐160 NPC sub‐complex also called the Y‐complex (Fig 1A) (Knockenhauer & Schwartz, 2016; Beck & Hurt, 2017). IP of stably expressed GFP‐Nup85 also demonstrated an interaction with endogenous FXR1 in HeLa cells (Fig 1B), and both Nup85 and Nup133 co‐immunoprecipitated with endogenous FXR1 in HEK293T cells (Fig 1C).
Figure 1. FXR1 protein localizes to the NE and interacts with NUPs.
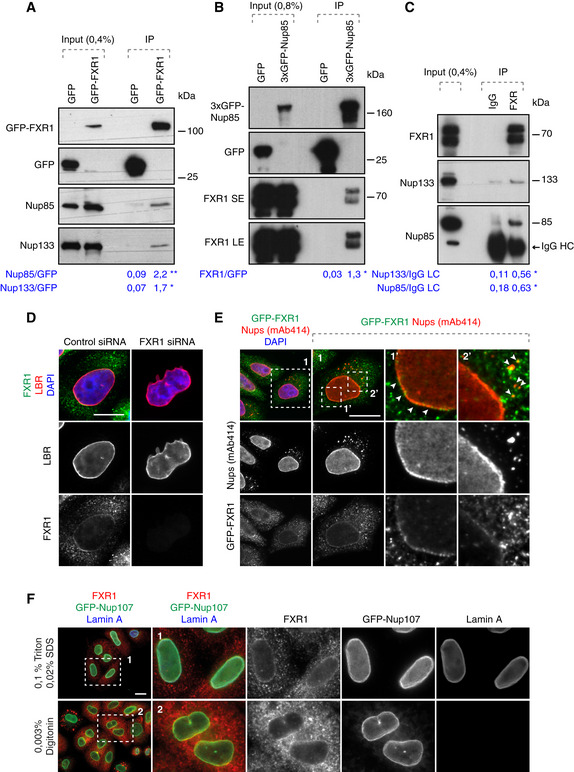
- Lysates of HeLa cells stably expressing GFP alone or GFP‐FXR1 were subjected to immunoprecipitation using GFP‐Trap beads (GFP‐IP), analysed by Western blot and quantified (shown a mean value, *P < 0.05, **P < 0.01; N = 3).
- Lysates of HeLa cells stably expressing GFP alone or 3xGFP‐Nup85 were immunoprecipitated using GFP‐Trap beads (GFP‐IP), analysed by Western blot and quantified (SE, short exposure, LE, long exposure; shown a mean value, *P < 0.05; N = 3).
- Immunoprecipitation from HEK293T cell lysates using FXR1 antibody or IgG analysed by Western blot. The arrow points to the heavy chain of IgG (IgG HC; shown a mean value, *P < 0.05; N = 3).
- HeLa cells were treated with indicated siRNAs, synchronized by double thymidine block, and released for 12 h and analysed by immunofluorescence microscopy for the lamin B receptor (LBR) to label the NE, and FXR1.
- HeLa cells stably expressing GFP‐FXR1 were analysed by immunofluorescence microscopy for GFP and mAb414, which labels FG‐Nups. The magnified framed regions are shown in the corresponding numbered panels. The arrowheads indicate NE and cytoplasmic localization of GFP‐FXR1.
- HeLa cells stably expressing GFP‐Nup107 were synchronized by double thymidine block and released for 12 h, permeabilized with Triton/SDS or digitonin for antibodies to access the nuclear and cytoplasmic or cytoplasmic side of the nucleus, respectively, and analysed by immunofluorescence microscopy.
Both endogenous FXR1 and GFP‐FXR1 localized to the nuclear envelope (Fig 1D and E) and also occasionally to small cytoplasmic foci labelled by the monoclonal antibody mAb414, which recognizes a panel of phenylalanine–glycine (FG) repeat‐containing Nups (FG‐Nups; Fig 1E). FXR1 localization to the NE and the cytoplasmic foci was abolished by treatment with FXR1 siRNA (Fig 1D), demonstrating antibody specificity. Treatment with digitonin, which in contrast to permeabilization protocol with the Triton and SDS can selectively permeabilize the plasma membrane while leaving the NE intact revealed that FXR1 localized to the outer nuclear membrane (ONM; Fig 1F). We conclude that FXR1 interacts with Nups and can localize to both the ONM and to cytoplasmic foci containing Nups.
FXR1 inhibits aberrant assembly of cytoplasmic Nups
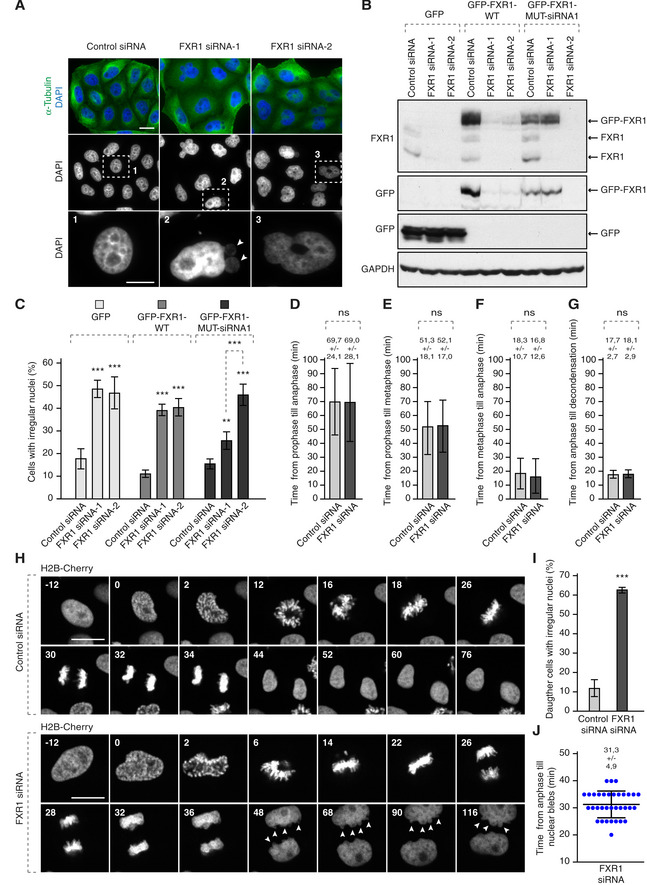
To assess the biological function of the FXR1‐Nup interactions, we treated cultured human cells with FXR1‐specific siRNA oligonucleotides. Downregulation of FXR1 in HeLa cells led to an accumulation of FG‐Nups in the cytoplasm in the form of irregular aggregate‐like assemblies of various sizes (Figs 2A and B, and EV1, Appendix Fig S1A, B, E, F and H) and in U2OS cells (Fig 2G, Appendix Fig S1J). These Nup assemblies were observed using two different siRNAs targeting FXR1 and could be rescued by stable ectopic expression of a form of GFP‐FXR1 that is resistant to one of the siRNAs used (Figs 2B and EV1).
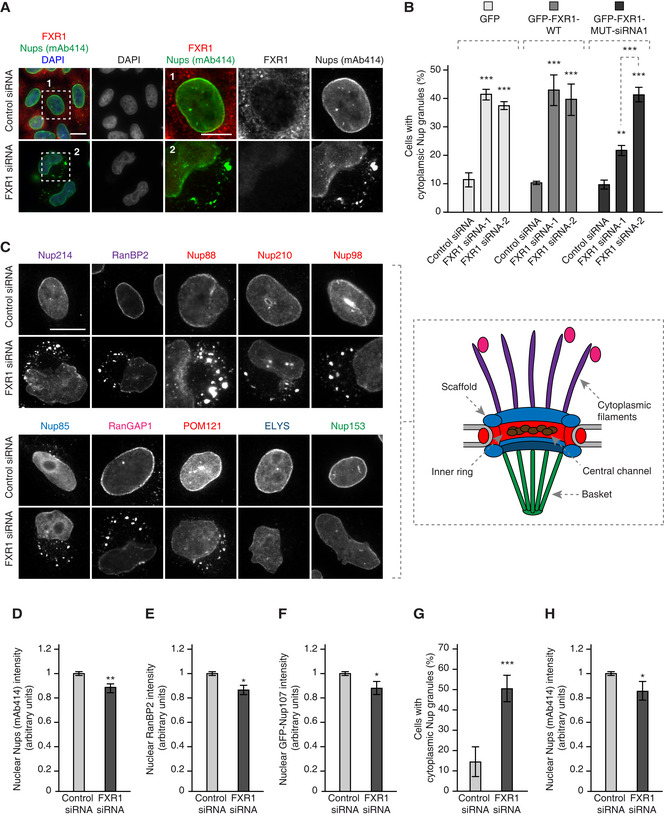
Figure 2. FXR1 inhibits aberrant assembly of cytoplasmic Nups.
-
AHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block and release for 12 h and analysed by immunofluorescence microscopy. The magnified framed regions are shown in the corresponding numbered panels.
-
BHeLa cells stably expressing GFP, GFP‐FXR1 wild type (WT) and GFP‐FXR1 mutated in the sequence recognized by FXR1 siRNA‐1 (GFP‐FXR1-MUT‐siRNA1) were treated with the indicated siRNAs, synchronized by double thymidine block, released for 24 h and then analysed by immunofluorescence microscopy. The percentage of cells with cytoplasmic nucleoporin granules was quantified, and 1,000 cells were analysed for each graph (mean ± SD, **P < 0.01; ***P < 0.001, N = 3). The corresponding representative pictures are shown in Fig EV1, and the corresponding Western blot analysis is shown in Fig 3B.
-
C–FHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h and analysed by immunofluorescence microscopy. Nups present in different NPC sub‐complexes are depicted in the colour code corresponding to the NPC scheme shown on the right. Additional or complementary representative images and channels of cells depicted in (C) are shown in Appendix Figs S1 and S2B–D. Nuclear intensity of FG‐Nups labelled by mAb414 (D), RanBP2 (E) and GFP‐Nup107 (F) was quantified. A total of 1,800 cells were analysed for each graph (mean ± SD, *P < 0.05; **P < 0.01; N = 3).
-
G, HAsynchronously proliferating U2OS cells were treated with the indicated siRNAs and analysed by immunofluorescence microscopy. The percentage of cells with cytoplasmic nucleoporin granules (G) was quantified, and nuclear intensity of FG‐Nups labelled by mAb414 (H) was quantified. A total of 1,600 cells were analysed in (G), and 2,100 cells were analysed in (H) (mean ± SD, *P < 0.05; ***P < 0.001; N = 3).
Figure EV1. FXR1 specifically controls cytoplasmic Nups and nuclear shape.
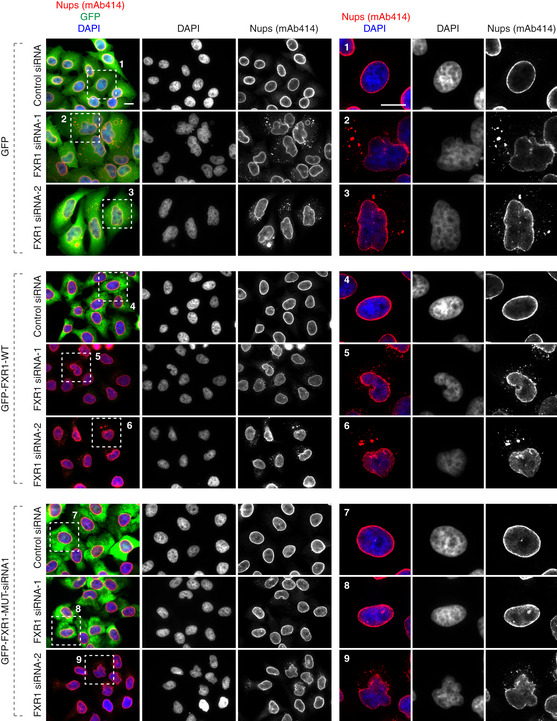
HeLa cells stably expressing GFP, GFP‐FXR1 wild type (WT) and GFP‐FXR1 mutated in the sequence recognized by FXR1 siRNA‐1 (GFP‐FXR1‐MUT‐siRNA1) were treated with the indicated siRNAs, synchronized by double thymidine block and released for 24 h and analysed by immunofluorescence microscopy for mAb414 (related to Figs 2B and 3B and C). Scale bars are 5 μm.
Downregulation of FXR1 led to the cytoplasmic retention and co‐localization in granules of at least 10 Nups spanning several functional and structural NPC groups, including FG‐Nups (Nup98, Nup214; and RanBP2); transmembrane Nups (Nup210 and POM121); Y‐complex Nups (Nup133 and Nup85, stably expressed GFP‐Nup133, stably expressed GFP‐Nup85 and stably expressed GFP‐Nup107) as well as Nup88 and NPC‐associated the RanGTPase activating protein (RanGAP1; Fig 2C; Appendix Figs S1A–H and S2C and D). Absent from the Nup granules were the nuclear ring Nup ELYS and the inner nuclear basket component Nup153, although their levels at the NE were both slightly reduced (Fig 2C; Appendix Figs S1I and S2B and C). FXR1 downregulation also moderately reduced the NE localization of FG‐Nups, RanBP2 and stably expressed GFP‐Nup107 in HeLa cells (Fig 2D–F) and of FG‐Nups in U2OS cells (Fig 2H, Appendix Fig S1J). Collectively, these data show that loss of FXR1 induces inappropriate assembly of Nups in the cytoplasm.
The FXR1 regulates nuclear morphology during G1 cell cycle phase
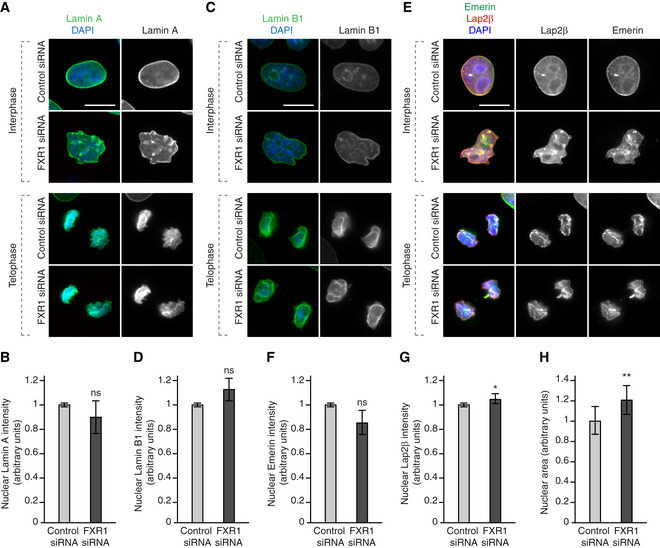
We noticed that the Nup granule‐containing cells often displayed strong nuclear atypia (Figs 1D, 2A and C, and EV1 and 2; Appendix Figs S1 and S2). Interestingly, downregulation of FXR1 did not affect the recruitment of the nuclear lamina components lamin B receptor (LBR; Fig 1D), lamin A (Fig EV2A and B), lamin B1 (Fig EV2C and D) or emerin (Fig EV2E and F) to the NE in interphase or telophase cells, while Lap2β recruitment was moderately increased upon FXR1 downregulation (Fig EV2E and G). However, these lamina and INM components displayed irregular distribution along with the misshaped nuclear rim and intranuclear foci (Fig EV2A, C and E). Moreover, the size of nucleus was moderately increased upon downregulation of FXR1 (Fig EV2H). Defects in nuclear architecture including irregular and blebbed nuclei (Fig 3A) could be largely rescued by stable ectopic expression of the siRNA‐resistant form of GFP‐FXR1 (Figs 3B and C, and EV1).
Figure EV2. FXR1 does not drive recruitment of lamina‐associated proteins.
-
A–HHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block and released for 9 (telophase) and 12 (interphase) h and analysed by immunofluorescence microscopy. The nuclear intensity of Lamin A (B), Lamin B1 (D), Emerin (F) and Lap2β (G) was quantified, and 2,000 cells were analysed (mean ± SD, *P < 0.05; ns, non‐significant; N = 3). The nuclear area was quantified (H), and 3,300 cells were analysed (mean ± SD, **P < 0.01; N = 5).
Figure 3. The FXR1 regulates nuclear morphology during G1 cell cycle phase.
-
AHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block, released for 24 h and analysed by immunofluorescence microscopy. The magnified framed regions are shown in the corresponding numbered panels. Arrowheads point to nuclear blebs observed in FXR1‐deficient cells.
-
B, CHeLa cells stably expressing GFP, GFP‐FXR1 wild type (WT) and GFP‐FXR1 mutated in the sequence recognized by FXR1 siRNA‐1 (GFP‐FXR1-MUT‐siRNA1) were treated with the indicated siRNAs, synchronized by double thymidine block, released for 24 h and analysed by Western blot (B) and immunofluorescence microscopy (C). The percentage of cells with irregular nuclei was quantified, and 1,000 cells were analysed (mean ± SD, **P < 0.01, ***P < 0.001; N = 3). The corresponding representative pictures are shown in Fig EV1.
-
D–JHeLa cells stably expressing the chromatin marker histone H2B labelled with mCherry were treated with indicated siRNAs, synchronized by double thymidine block, released for 12 h and analysed by immunofluorescence microscopy. Time from prophase till anaphase (D), from prophase till metaphase (E), from metaphase till anaphase (F) and from anaphase till chromatin decondensation (G) was quantified. The selected frames of the movies are depicted, and time is shown in minutes (H). Arrowheads point to nuclear blebs appearing during nuclear expansion of FXR1‐deficient cells. Percentage of daughter cells with irregular nuclei was quantified in (I), and time from anaphase till nuclear blebs was quantified in (J). Sixty‐six cells were analysed (mean ± SD, ***P < 0.001; N = 3).
Live video microscopy of HeLa cells stably expressing histone H2B labelled with mCherry revealed that progression and timing through different mitotic stages or fidelity of chromosome segregation was not affected in the FXR1‐deficient cells (Fig 3D–H). The nuclear morphology defects in FXR1‐deficient cells (Fig 3H and I) could first be detected approximately 30 min after the onset of chromosome segregation (Fig 3H and J), which strongly correlated with the onset of nuclear growth. Our data suggest that downregulation of FXR1 specifically affects cytoplasmic Nups and nuclear architecture during early G1.
FXR1 regulates cytoplasmic Nups during early interphase
Our data so far suggest that FXR1 regulates cytoplasmic Nups and may facilitate localization of a very small pool of soluble Nups to the NE (Fig 2D–F and H). To date, two temporally and mechanistically distinct pathways of NPC assembly at the NE have been described during the cell cycle in higher eukaryotic cells (Weberruss & Antonin, 2016). In the post‐mitotic pathway, ELYS initiates NPC assembly on segregated chromosomes, while during interphase, both Nup153 and POM121 drive de novo assembly of NPCs into an enclosed NE (D'Angelo et al, 2006; Doucet et al, 2010; Vollmer et al, 2015). ELYS assembled normally on segregating chromosomes in anaphase and on decondensing chromatin in telophase in FXR1‐deficient cells (Appendix Fig S2A). In addition, we found that ELYS and Nup153 were not recruited to the cytoplasmic Nup granules whereas POM121 was (Fig 2C; Appendix Fig S2B–D). This suggests that FXR1 affects localization of most but not all cytoplasmic Nups.
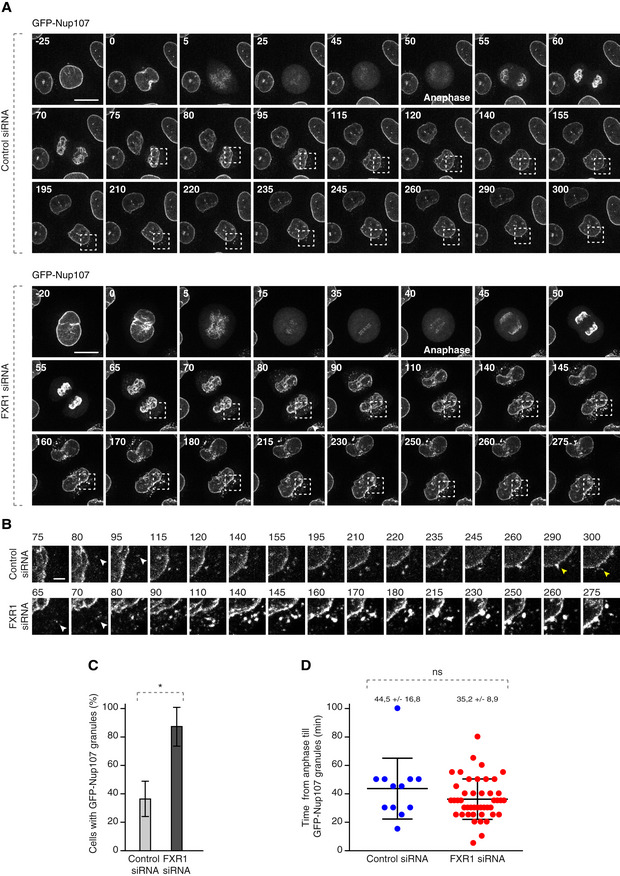
To understand the precise timing of the FXR1‐Nup pathway, we performed live video microscopy on a reporter cell line stably expressing GFP‐Nup107 (Fig 4A–D, [Link], [Link], [Link], [Link], [Link]). As expected, downregulation of FXR1 led to the accumulation of GFP‐Nup107 in cytoplasmic granules (Fig 4A–C; [Link], [Link], [Link]) with similar appearance and distribution to that observed in the fixed specimens and that had a tendency to fuse into bigger assemblies with time (Fig 4A and B; [Link], [Link]). In the control cells, smaller GFP‐Nup107 granules were occasionally observed, which had a tendency to fuse with the NE (Fig 4A and B; [Link], [Link]).
Figure 4. FXR1 regulates cytoplasmic Nups during early interphase.
-
A–DHeLa cells stably expressing GFP‐Nup107 were treated with indicated siRNAs, synchronized by double thymidine block, released and analysed by live video spinning disc confocal microscopy (A). The selected frames of the movies are depicted, and time is shown in minutes. The onset of anaphase is indicated. The magnified framed regions with time indicated in minutes are shown in (B). White arrowheads point to the cytoplasmic GFP‐NUP107 granules appearing during nuclear expansion of control and FXR1‐deficient cells, and yellow arrowheads point to the fusion events of GFP‐NUP107 granules with NE in control cells. The percentage of cells with cytoplasmic GFP‐Nup107 granules was quantified in (C). Time from anaphase till GFP‐Nup107 cytoplasmic granule formation was quantified in (D). Fifty‐seven cells were analysed (mean ± SD, *P < 0.05; ns, non‐significant; N = 3).
GFP‐Nup107‐positive granules in control and FXR1‐deficient cells became detectable in the cytoplasm on average 44 and 35 min after chromosome segregation, respectively (Fig 4D, [Link], [Link], [Link], [Link], [Link]), which strongly correlated with the timing of the nuclear morphology defects in early G1 (Fig 3H and J) described earlier.
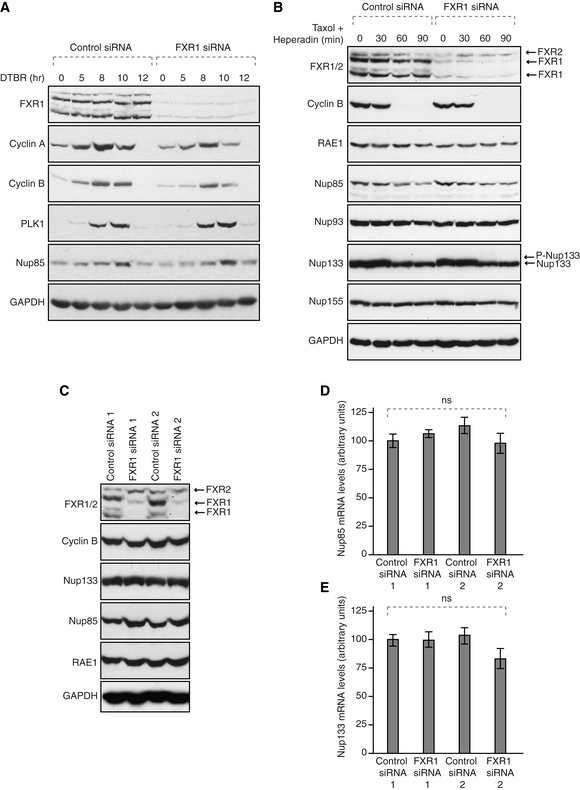
We considered that this effect may be mediated by a modulation of Nups levels. The protein levels of RAE1, Nup85, Nup93, Nup133 and Nup155 (Fig EV3A–C), and the mRNA levels of Nup85 and Nup133 (Fig EV3D and E), as well as the known cell cycle‐linked degradation of Nup85 (Fig EV3A and B) and Nup133 dephosphorylation, which occurs during mitotic exit (Fig EV3B), were unchanged upon depletion of FXR1. Consistent with the live video experiments, degradation of several mitotic factors in synchronized cells (Fig EV3A and B) was not affected by FXR1 downregulation, suggesting that Nup localization defects are likely also not due to changes in mitotic progression and exit or misregulation of the levels of the analysed Nups. However, it cannot be formally excluded that expression of other yet to be identified Nups or Nup‐associated factors is regulated by FXR1. Together, our results suggest that loss of FXR1 regulates cytoplasmic Nups during early interphase but it remains to be understood if this Nup regulation occurs in the context of any specific NPC assembly pathway.
Figure EV3. FXR1 does not regulate mitotic exit and protein levels of several Nups.
-
AHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block and released for the indicated times (hours) and analysed by Western blot.
-
BHeLa cells were treated with the indicated siRNAs, synchronized by Hesperadin release from mitotic arrest induced by Taxol and analysed by Western blot. Arrows point to FXR1 isoforms, FXR2 and phosphorylated (P‐Nup133) and unmodified form of Nup133, respectively.
-
CHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block, released in interphase and analysed by Western blot. Arrows point to FXR1 isoforms and FXR2 protein.
-
D, EHeLa cells were treated with the indicated siRNAs, and mRNA levels of Nup85 (D) and Nup133 (E) were analysed by quantitative real‐time PCR (mean ± SD, ns, non‐significant; N = 3).
Nup granules are resistant to RNA degradation but sensitive to 1,6‐hexanediol
The cytoplasmic Nup granules in FXR1‐deficient cells could correspond to annulate lamellae (AL), which are preassembled NPCs embedded in the ER membrane (Merisko, 1989), as suggested by co‐localization of various Nups in these assemblies. Indeed, cytoplasmic AL‐NPCs can be inserted “en bloc” into an intact NE during embryogenesis in Drosophila (Hampoelz et al, 2016). A closer analysis by super‐resolution microscopy revealed an amorphous organization of the Nup assemblies in the perinuclear area of FXR1‐deficient cells relative to the more regular, round shape of the small cytoplasmic Nup foci observed in the control cells (Appendix Fig S3A). Moreover, we were unable to detect any AL‐typical structures (characterized by parallel stacks of ER membranes with embedded regularly spaced NPCs), in the FXR1‐deficient cells by electron microscopy (EM; Appendix Fig S3B), and no co‐localization with the ER membranes could be observed (Appendix Fig S3C). Cytoplasmic nucleoporins were also found to be recruited to assembling SGs upon induction of cellular stress (Zhang et al, 2018). Consistently, our results demonstrated co‐localization of the Nup RanBP2 with the markers of SGs, TIA‐1 and G3BP1, in the control stress‐induced cells (Appendix Fig S4A). However, TIA‐1 and G3BP1 did not co‐localize with the Nup granules in the FXR1‐deficient cells exposed to stress, and both the SGs and Nup granules present in these cells localized to different cytoplasmic compartments. We conclude that the Nup granules in FXR1‐deficient cells are distinct from ALs and SGs.
Given the established role of the FXR protein family in RNA‐binding and the frequent role of RBPs in the formation and dynamics of membrane‐less protein assemblies, we next analysed whether Nup granules contain any RNAs or could be linked to RNA‐based processes. Hybridization with an RNA FISH probe against poly A revealed no difference in the percentage of nuclear mRNAs in the FXR1‐deficient cells relative to control cells (Appendix Fig S4B and C), suggesting that the FXR1‐Nup pathway is not implicated in mRNA export to the cytoplasm. Additionally, no cytoplasmic enrichment of mRNAs could be observed in GFP‐Nup107 granules in the FXR1‐downregulated cells (Appendix Fig S4B and D). To test whether RNAs play a role in the maintenance or dynamics of the Nup granules in the cytoplasm, we treated permeabilized, FXR‐downregulated cells with RNAse. This treatment failed to disrupt the Nup granules or change their shape and distribution relative to control cells (Appendix Fig S5A and B), suggesting that RNAs are dispensable for their maintenance and dynamics.
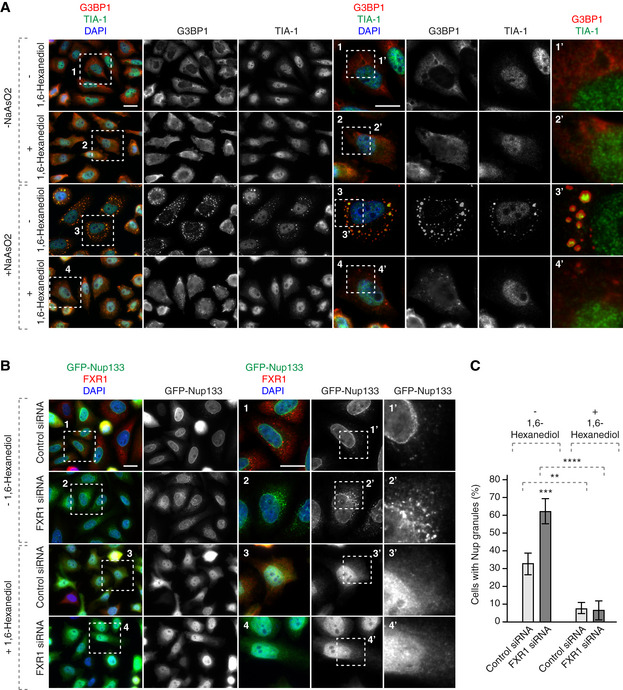
Many components of protein assemblies formed by phase separation, including FG‐Nups, are highly hydrophobic and contain very few charged amino acids (Schmidt & Görlich, 2016). For this reason, aliphatic alcohols like hexanediols are good solvents for FG‐Nups because they probably compete with the hydrophobic interactions between FG repeats (Patel et al, 2007). Indeed, hexanediols are known to disrupt FG hydrogels and the NPC permeability barrier, while FG‐derived amyloid fibres are hexanediol resistant (Ribbeck & Görlich, 2002; Kroschwald et al, 2015). Thus, hexanediols can be used to distinguish between these two types of Nup condensates. Moreover, 1,6‐hexanediol treatment can also disperse the phase‐separated condensates formed of Nup358 and the Y‐complex component Nup107 in Drosophila embryos (Hampoelz et al, 2019b) and SGs formed in HeLa cells exposed to stress (Fig 5A).
Figure 5. Cytoplasmic Nup granules are sensitive to 1,6‐hexanediol.
-
AHeLa cells stably expressing GFP‐Nup133 were synchronized by double thymidine block, released for 12 h, treated or not with NaAsO2 to induce stress granule formation and with 1,6‐hexanediol, and analysed by immunofluorescence microscopy. The magnified framed regions are shown in the corresponding numbered panels.
-
B, CHeLa cells stably expressing GFP‐Nup133 were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h, treated with or without 1,6‐hexanediol and analysed by immunofluorescence microscopy. The magnified framed regions are shown in the corresponding numbered panels. The percentage of cells with cytoplasmic GFP‐Nup133 granules was quantified in (C), and 3,100 cells were analysed (mean ± SD, **P < 0.01; ***P < 0.001; ****P < 0.0001; N = 3).
To understand if the cytoplasmic Nup granules have properties of phase‐separated condensates, we treated control and FXR1‐deficient cells with 1,6‐hexanediol using the protocol established for SGs (Fig 5A). 1,6‐Hexanediol treatment led to the dispersion of the small Nup foci present in control cells as well as the Nup granules observed upon FXR1 downregulation relative to the non‐treated cells (Fig 5B and C). Collectively, we propose that the Nup granules represent previously unknown assemblies with the properties of protein condensates that accumulate in the absence of FXR1 in human cells.
FXR1 inhibits Nup condensate formation by dynein‐based microtubule‐dependent transport
We next investigated the mechanism by which loss of FXR1 promotes the formation of Nup condensates. We noticed that the Nup granules were not scattered randomly in the cytoplasm but often formed a crescent‐like shape around the microtubule‐organizing centre (MTOC) suggesting a role for microtubules (Appendix Fig S6A). Indeed, we found that nocodazole‐mediated microtubule depolymerization also induced Nup granules (Appendix Fig S6B and C). Notably, our mass spectrometry analysis identified the cytoplasmic minus‐end‐directed motor protein dynein heavy chain (HC) co‐immunoprecipitating specifically with GFP‐FXR1, along with the Nups (Dataset EV1). We demonstrated an interaction of GFP‐FXR1 with other components of the dynein complex, specifically dynein intermediate chain (IC; which was visualized as a slower migrating band relative to the input lysate) and dynactin p150Glued (Fig 6A). Downregulation of dynein HC by two independent siRNAs, which also depleted dynein IC as reported (Splinter et al, 2010) (Appendix Fig S6D), led to the accumulation of the cytoplasmic Nup granules (Fig 6B and C; Appendix Fig S6E), and to the irregular nuclei (Appendix Fig S6F) highly reminiscent of FXR1 depletion.
Figure 6. FXR1 works together with the dynein‐BICD2 complex to inhibit cytoplasmic Nup granules formation.
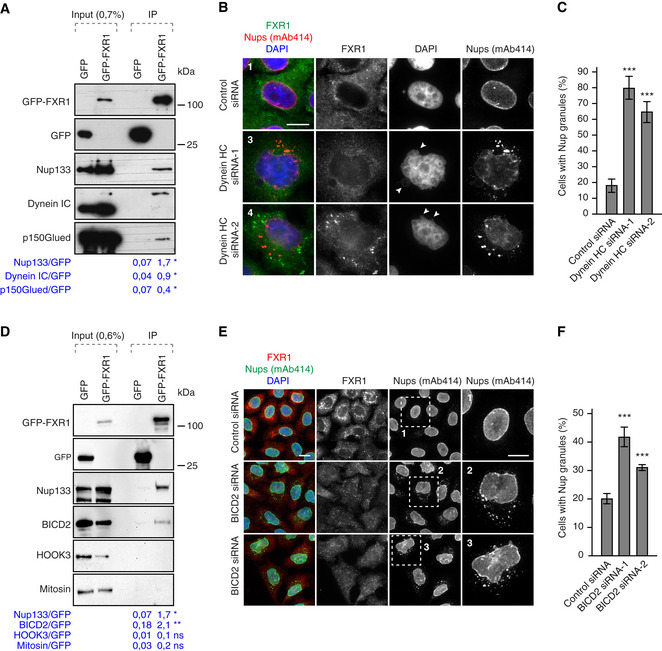
-
AHeLa cells stably expressing GFP alone or GFP‐FXR1 were immunoprecipitated using GFP‐Trap beads (GFP‐IP), analysed by Western blot and quantified (mean, *P < 0.05; N = 3).
-
B, CHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h and analysed by immunofluorescence microscopy. Images in (B) correspond to the numbered magnified framed regions indicated in the pictures shown in Appendix Fig S6E. Arrowheads indicate blebbed regions of nuclei. The percentage of interphasic cells with cytoplasmic Nup granules was quantified (C), and 900 cells were analysed (mean ± SD, ***P < 0.001; N = 3). The corresponding Western blot analysis is shown in Appendix Fig S6D.
-
DLysates of HeLa cells stably expressing GFP alone or GFP‐FXR1 were subjected to immunoprecipitation using GFP‐Trap beads (GFP‐IP), analysed by Western blot and quantified (mean, *P < 0.05; **P < 0.01; N = 3).
-
E, FHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block and released for 12 h and analysed by immunofluorescence microscopy for FXR1 and mAb414. The magnified framed regions are shown in the corresponding numbered panels in (E). The percentage of cells with cytoplasmic Nup granules was quantified in (F), and 2,700 cells were analysed (mean ± SD, ***P < 0.001; N = 3).
Analysis of several known dynein adaptor proteins in a co‐immunoprecipitation assay with GFP‐FXR1 revealed an interaction with BICD2, but not Mitosin or HOOK3 (Fig 6D). Downregulation of BICD2 by two independent siRNAs likewise led to accumulation of the cytoplasmic Nup granules (Fig 6E and F). Our results suggest that FXR1, working together with the microtubule motor dynein‐BICD2 complex, inhibits formation of cytoplasmic Nup granules.
Our earlier results in living GFP‐Nup107 cells showed cytoplasmic Nup granule formation in early interphase, which occasionally fused with the NE in control cells but became bigger in FXR1‐deficient cells (Fig 4A and B; [Link], [Link], [Link]). We also observed moderate decrease in Nup localization at the NE in FXR1‐deficient cells (Fig 2D–F and H). These results suggest that at least a very small pool of the Nups is no longer incorporated into the NE during interphase. Interestingly, in Drosophila oocytes, precursor Nup granules were observed being incorporated into membranes forming the AL‐specific NPCs (Hampoelz et al, 2019b). Thus, we considered that FXR1 together with the microtubule motor dynein‐BICD2 complex may mediate the transport of small pool of cytoplasmic Nups to the NE during interphase of human cells. To test this, we first treated the cells stably expressing GFP‐Nup133 with nocodazole to induce Nup granule formation in a reversible way (Fig 7A). Downregulation of FXR1 or dynein potentiated the effect of nocodazole and led to the increase in a number of cells with Nup granules relative to control (Fig 7A and B). Interestingly, following nocodazole washout, we observed a strong reduction of Nup granules in control cells while FXR1 and dynein downregulation did not reduce cytoplasmic nucleoporin granules to the same extent under these conditions (Fig 7A and B). To corroborate these findings, we performed live video spinning disc microscopy of cell lines stably expressing GFP‐Nup107. Following nocodazole washout, we observed the dynamics of the Nup aggregates in control, FXR1‐ and dynein‐downregulated cells (Fig 7C–E). GFP‐Nup107‐positive aggregates showed dynamic behaviour and both fusion and splitting of the granules were observed under all conditions (Fig 7C–E) supporting the condensate properties of these Nup cytoplasmic granules. As expected from the results in fixed specimens (Fig 7A and B), the percentage of cells with fusion and fission events of GFP‐Nup107 granules was increased in FXR1‐ and dynein‐downregulated cells (Fig 7D) but the frequency of these events per cell (two–three), observed in all cells positive for GFP‐Nup107 granules did not differ significantly (Fig 7E).
Figure 7. FXR1 regulates cytoplasmic Nups by dynein‐based microtubule‐dependent transport.
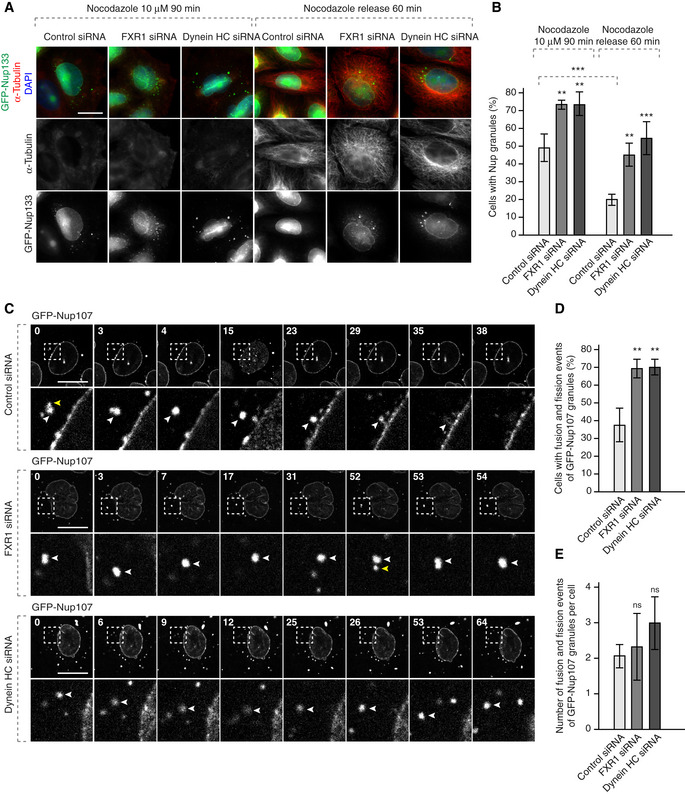
-
A, BHeLa cells stably expressing GFP‐Nup133 were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h, treated with nocodazole to induce cytoplasmic Nup granule formation and washed out as indicated and analysed by immunofluorescence microscopy. The percentage of cells with cytoplasmic GFP‐Nup133 granules was quantified in (B), and 5,200 cells were analysed (mean ± SD, **P < 0.01; ***P < 0.001; N = 3).
-
C–EHeLa cells stably expressing GFP‐Nup107 were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h, treated with nocodazole to induce granule formation and washed out as in (A) and analysed by live video spinning disc confocal microscopy. The selected frames of the movies are depicted, and time is shown in minutes. The magnified framed regions are depicted in the lower rows. White arrowheads point to individual GFP‐Nup107‐positive granules. Yellow arrowheads point to the granules undergoing fusion events. The percentage of cells with fusion/fission events of GFP‐Nup107 granules was quantified in (D), and 815 cells were analysed (mean ± SD, **P < 0.01; N = 3). The number of fusion/fission events per cell was quantified in (E), and 815 cells were analysed (mean ± SD; N = 3).
Interestingly in control cells, nocodazole washout led to NE‐directed transport and fusion of GFP‐Nup107 granules with the NE. In contrast, downregulation of FXR1 or dynein in nocodazole washed‐out cells led to retention of the GFP‐Nup107 granules in the cytoplasm. Under these conditions, the GFP‐NUP107 granules were still mobile but little NE‐directed movement was observed, and they continued to fuse and often increased in size (Fig 7C) consistent with the previous results in cells not treated with nocodazole (Fig 4A and B; [Link], [Link], [Link], [Link], [Link]). These observations suggest that microtubule‐based transport by the FXR1‐dynein complex can decrease local concentrations of cytoplasmic Nups thereby preventing their assembly into condensates.
Nup localization defects can be linked to fragile X syndrome
Next, we analysed whether all members of the FXR protein family share analogous roles in the spatial control of Nup self‐assembly. Our data show that in addition to FXR1, FXR2 and FMRP can localize at the NE in HeLa cells (Fig EV4A) and in mouse myoblasts (Fig EV4B). Interestingly, depletion of each of the three members of this protein family led to the condensation of cytoplasmic Nups relative to control cells (Fig EV4C and D). Downregulation of all three FXR proteins also led to nuclear morphology defects (Fig EV4E). Simultaneous downregulation of all three FXR proteins did not further increase the penetrance of these phenotypes (Fig EV4D and E), suggesting that FXR proteins together form a protein complex in human cells consistent with our mass spectrometry results (Dataset EV1). Interestingly, downregulation of FMRP and FXR1 led to more severe defects as compared to FXR2 (Fig EV4D and E). We speculate that higher protein sequence identity between FMRP and FXR1 (86%) (Hoogeveen et al, 2002) as compared to FMRP and FXR2 (70%), leads to sharing more common functions by these two family members.
Figure EV4. FXR protein family members localize to NE and regulate cytoplasmic Nups.
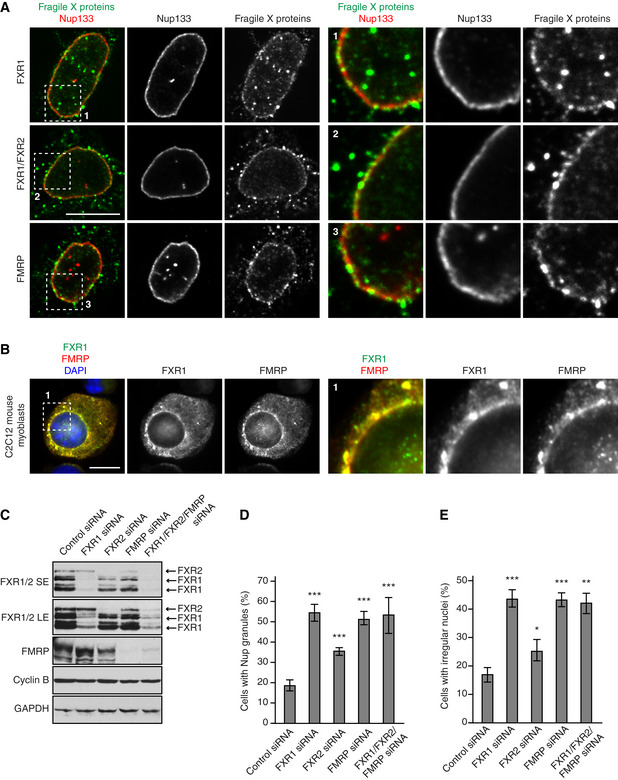
-
A, BHeLa cells (A) and mouse C2C12 myoblasts (B) were analysed by immunofluorescence microscopy for FXR1, FXR1 + 2 and FMRP. The magnified framed regions are shown in the corresponding numbered panels.
-
C–EHeLa cells were treated with the indicated siRNAs, synchronized by double thymidine block and released for 12 h and analysed by Western blot (SE—short exposure, LE—long exposure) (C) and immunofluorescence microscopy (D, E). The percentage of cells with cytoplasmic nucleoporin granules (D) and irregular nuclei (E) was quantified, and 900 cells were analysed (mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001; N = 3).
Since FMRP is absent or mutated in fragile X syndrome (Santoro et al, 2012), we asked if the Nup localization defects are observed in cellular models of this disease. We stimulated FXS patient‐derived fibroblasts which lack the FMRP protein (Fig 8A) to undergo synchronous mitotic exit and nuclear reformation. FXS fibroblasts displayed accumulation of cytoplasmic Nup granules relative to control fibroblasts (Fig 8B and C) similar to those observed in HeLa cells, and structurally abnormal nuclei (Fig 8D). To corroborate these findings, we used human induced pluripotent stem cells (iPSCs) derived from an FXS patient (FXS‐iPSCs) and the isogenic rescue cells (C1_2‐iPSCs), where reactivation of the FMR1 locus is achieved by CRISPR‐mediated excision of the expanded CGG‐repeat from the 5′UTR of the FMR1 gene (Xie et al, 2016). In the FXS‐iPSCs, accumulation of large Nup133‐positive cytoplasmic condensates was observed (Fig 8E), which were reduced in the FMRP re‐expressing cells, although they were still present (Fig 8E and F). Re‐expressed FMRP in the reactivated cell line localized to both the NE and to the cytoplasmic perinuclear region, which often also contained Nup133 (Fig 8G). We speculate that these perinuclear FMRP‐Nup133 signals could represent assembly intermediates before Nup133 is properly transferred and inserted into the NE due to lower levels of FMRP protein re‐expression in the rescue system (Xie et al, 2016) which could result in a slowdown of the process and similar to the GFP‐FXR1‐Nups signals occasionally observed in HeLa cells (Fig 1E).
Figure 8. Nup localization defects can be linked to fragile X syndrome.
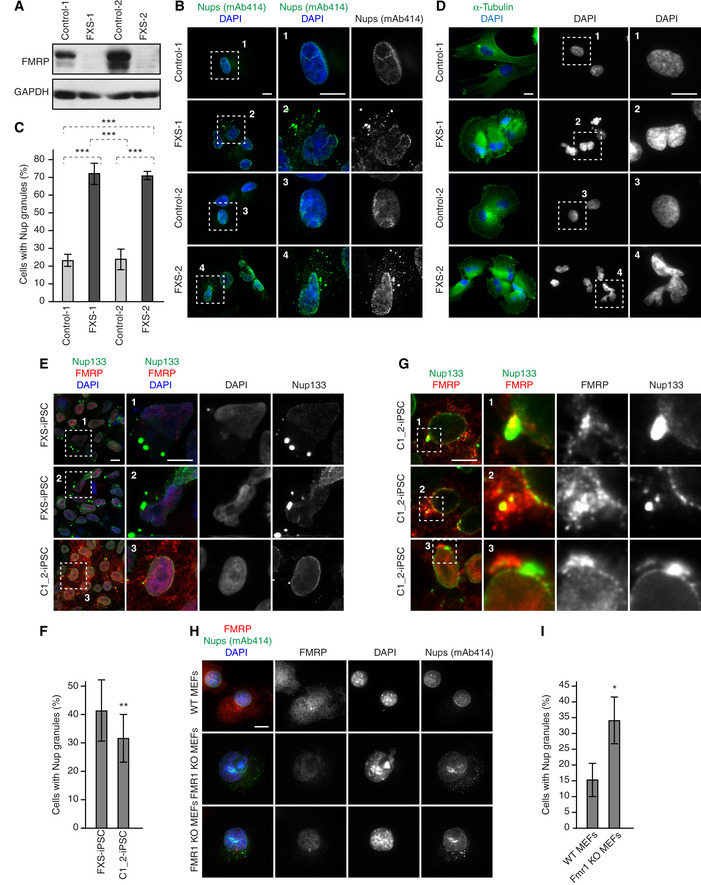
-
A–DHuman FXS patient‐derived fibroblasts (FXS‐1, FXS‐2) and control human fibroblasts were synchronized in early G1 by Monastrol release and analysed by Western blot (A) and immunofluorescence microscopy (B, D). The percentage of cells with cytoplasmic Nup granules was quantified in (C), and 283 cells were analysed (mean ± SD, ***P < 0.001; N = 3). Examples of Nup localization defects are shown in (B), and examples of nuclear architecture defects are shown in (D).
-
E–GHuman induced pluripotent stem cells (iPSCs) derived from a FXS patient (FXS‐iPSC) and the isogenic rescue cells (C1_2‐iPSC) were analysed by immunofluorescence microscopy (E, G). The percentage of cells with cytoplasmic Nup133 granules was quantified in (F), and 5,500 cells were analysed (mean ± SD, **P < 0.01; N = 3). Examples of co‐localization events of re‐expressed FMRP and Nup133 are shown in (G).
-
H, IMouse embryonic fibroblasts (MEFs) derived from the Fmr1 knockout (KO) mice and wild‐type controls were synchronized in early G1 by Monastrol release and analysed by immunofluorescence microscopy (H). The percentage of cells with cytoplasmic nucleoporin granules was quantified in (I), and 2,400 cells were analysed (mean ± SD, *P < 0.05; N = 3).
Thus, the absence of human FMRP in primary cell lines and in iPSs cells leads to the accumulation of Nup granules as also seen in the cancer cell lines.
To confirm these findings in an animal model of FXS, we used mouse embryonic fibroblasts (MEFs) derived from the Fmr1 knockout (KO) mice. Fmr1 KO MEFs also displayed accumulation of perinuclear Nup granules relative to wild type MEFs (Fig 8H and I). Taken together, our results demonstrate the presence of ectopic Nup assemblies in different cellular models of fragile X syndrome. These defects may perturb cellular homeostasis and contribute to FXS pathology.
The FXR1 regulates protein export and cell cycle progression
What could be the biological consequences of misregulation of the FXRPs‐dynein pathway and how could Nup assembly defects perturb cellular homeostasis? To understand if ectopic Nup condensation during early G1 in FXR‐deficient cells affects the function of the nuclear pores, we measured the rates of nucleocytoplasmic transport of an ectopic import/export reporter plasmid XRGG‐GFP that shuttles to the nucleus when induced with dexamethasone. FXR1 downregulation did not change the rates of nuclear import (Fig EV5A and B) relative to control cells, whereas downregulation of the Nup ELYS clearly demonstrated import defects in the same experiments, as expected (Fig EV5A and B). This indicates that, at least in the steady‐state, nucleocytoplasmic import is largely unaffected by formation of Nup granules in FXR1‐deficient cells. Interestingly, while the overall rate of protein export remained unchanged in FXR1‐deficient cells relative to controls (Fig EV5C and D), FXR1 downregulation reduced the export rate solely in early G1 cells (time points 20 and 30 min) similar to ELYS (Figs EV5D and 9A), suggesting that FXR1‐downregulation mediated Nup defects may affect the function of nuclear pores specifically during this cell cycle stage. Consistent with the observed export defects in FXR1‐deficient G1 cells, the nuclear export factor chromosomal region maintenance 1 (CRM1) protein was sequestered to Nup granules labelled with the mAb414 antibody and with GFP‐Nup133 (Fig 9B).
Figure EV5. FXR1 protein does not regulate steady‐state nucleocytoplasmic transport.
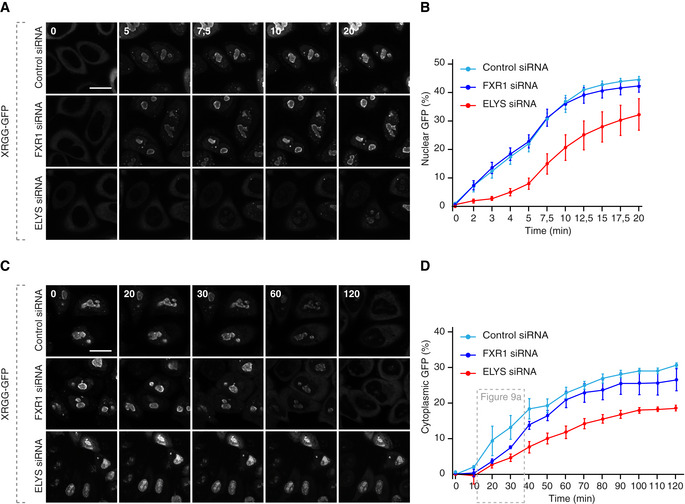
-
A, BHeLa cells were transfected with the import reporter plasmid XRGG‐GFP, treated with the indicated siRNAs and synchronized in early G1 phase by Monastrol release. Dexamethasone‐induced nuclear import of XRGG‐GFP was analysed by live video spinning disc confocal microscopy (A). The selected frames of the movies are depicted, and time is shown in min. The increase in the percentage of nuclear XRGG‐GFP over time was quantified in (B), and 247 cells were analysed (mean ± SEM, N = 3).
-
C, DHeLa cells were treated as in (A, B), and dexamethasone was added for 3 h to induce XRGG‐GFP nuclear import. Following washout, the nuclear export of XRGG‐GFP was analysed by live video spinning disc confocal microscopy (C). The selected frames of the movies are depicted, and time is shown in min. The increase in the percentage of cytoplasmic XRGG‐GFP over time was quantified in (D), and 199 cells were analysed (mean ± SEM; N = 3). The distribution of cells with cytoplasmic XRGG‐GFP at the time points corresponding to early G1 cell cycle stage (20 and 30 min) are depicted in Fig 9A.
Figure 9. FXR1 regulates G1 cell cycle progression.
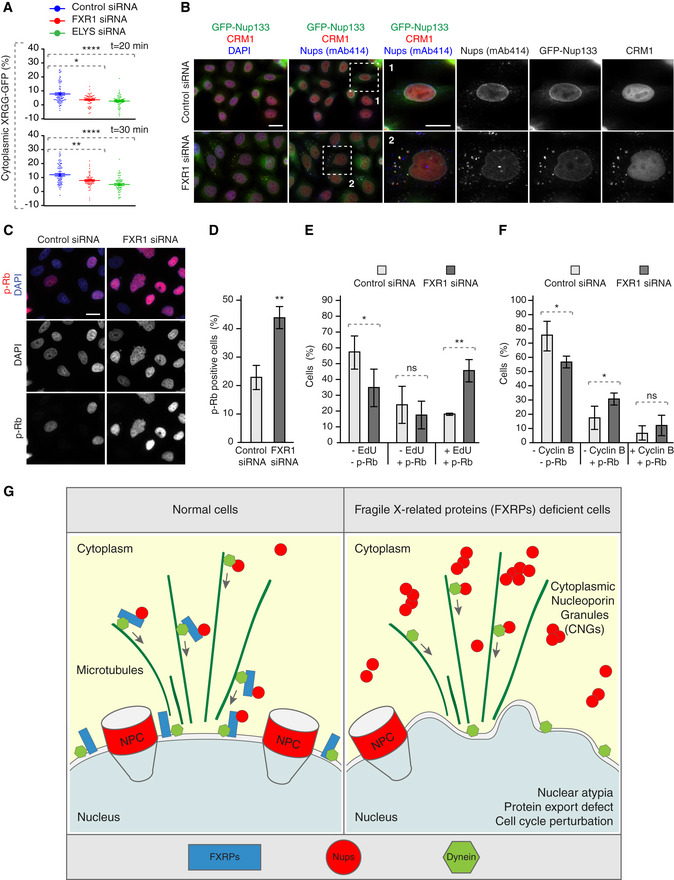
-
AHeLa cells were transfected with the import/export reporter plasmid XRGG‐GFP, treated with the indicated siRNAs and synchronized in early G1 phase by Monastrol release. Dexamethasone was added for 3 h to induce XRGG‐GFP nuclear import. Following washout, the nuclear export of XRGG‐GFP was analysed by live video spinning disc confocal microscopy. The selected frames of the movies are depicted in Fig EV5C. The percentage of cytoplasmic XRGG‐GFP over time was quantified in Fig EV5D, and quantifications of individual cells from the 20 and 30 min time points are depicted in (A), and 199 cells were analysed (mean ± SD, *P < 0.05; **P < 0.01; N = 3).
-
BHeLa cells stably expressing GFP‐Nup133 were treated with the indicated siRNAs, synchronized by double thymidine block, released for 12 h and analysed by immunofluorescence microscopy. The magnified framed regions are shown in the corresponding numbered panels.
-
C, DAsynchronously proliferating HeLa cells were treated with indicated siRNAs and analysed by immunofluorescence microscopy (C). The percentage of p‐Rb-positive cells was quantified in (D), and 2,800 cells were analysed (mean ± SD, **P < 0.01; N = 3).
-
E, FAsynchronously proliferating HeLa cells were treated with indicated siRNAs, incubated with EdU during 30 min and analysed by immunofluorescence microscopy. The percentage of p‐Rb- and/or EdU‐positive cells was quantified in (E); 2,100 cells were analysed (mean ± SD, *P < 0.05; **P < 0.01; N = 3), and the percentage of p‐Rb- and/or cyclin B‐positive cells was quantified in (F); 3,300 cells were analysed (mean ± SD, *P < 0.05; N = 3).
-
GA hypothetical model how fragile X‐related proteins spatially regulate nucleoporin condensation. FXR proteins (blue) interact with cytoplasmic soluble Nups (red circles) and dynein (green) and facilitate their localization to the NE during early G1. This function of FXR proteins inhibits formation of aberrant cytoplasmic Nup assemblies, the cytoplasmic nucleoporin granules (CNGs), contributing to the equilibrium of NE‐NPCs and driving the G1‐specific protein export and maintenance of nuclear shape and cell cycle progression. Silencing of FXR proteins, for instance in FXS patients, leads to the accumulation of CNGs, nuclear atypia, protein export defects, and defects in cell cycle progression, which may contribute to the pathology of FXS.
These observations prompted us to test whether the FXR1‐Nup pathway could also be important for cell cycle progression of cells in interphase. For this, we analysed retinoblastoma (Rb) protein which is phosphorylated during G1/S phase transition (p‐Rb) and is kept in this state until mitotic exit. Downregulation of FXR1 led to accumulation of cells with strong nuclear p‐Rb signal (Fig 9C and D), suggesting perturbations in cell cycle progression. To elucidate if this accumulation was specific to any cell cycle phases, we analysed p‐Rb together with EdU incorporation (S phase marker) or Cyclin B (G2 marker) in interphasic cells. Downregulation of FXR1 led to an increased percentage of cells positive for both p‐Rb and EdU signal (Fig 9E) but not for p‐Rb and Cyclin B signal, (Fig 9F), suggesting that upon FXR1 downregulation cells accumulate in S phase. Accordingly, the percentage of p‐Rb negative cells (corresponding to G1 phase) was decreased in all cases. Together, these data indicate that the absence of FXR1 leads to protein export defects in G1 and perturbation in cell cycle progression.
Discussion
Collectively, our data suggest a model where FXR proteins and dynein regulate the localization of a cytoplasmic pool of Nups during early G1 (Fig 9G). Absence of FXR proteins or dynein‐mediated transport leads to the formation of previously uncharacterized ectopic Nup assemblies. We speculate that the FXR‐dynein pathway regulates the pool of soluble nucleoporins either remaining in the cytoplasm after post‐mitotic NPC assembly or being translated in early interphase, which is important for functions in nuclear export and shape and in cell cycle progression. Defects in this pathway, as seen in cellular models of fragile X syndrome, may therefore compromise cellular fitness and contribute to the pathology of this human disease.
Our analysis demonstrates that FXR proteins can facilitate dispersal of Nups and reversal of cytoplasmic Nup assemblies, which we propose to name Cytoplasmic Nucleoporin Granules (CNGs; Fig 9G). Consistent with this hypothesis, re‐expressed FMRP in iPS cells (Fig 8G) and occasionally GFP‐FXR1 in cancer cells (Fig 1E) could co‐localize in foci with a pool of cytoplasmic Nups often found in the proximity of the NE. FXR1 could also interact with Nups and rescue experiments with the siRNA‐resistant form of FXR1 could reverse the formation of CNGs in cancer cells (Figs 2B and EV1). Finally, the CNGs’ fusion events observed in the live video experiments resulted in increase of the CNGs’ size in the absence of FXR1 or dynein relative to control cells (Fig 7C). Unfortunately, we did not observe an increase in co‐localization of the endogenous FXR1 and Nups in the absence of dynein/BICD2 (Fig 6B and E). We predict that either all three components are needed to form the transport complexes in the cytoplasm or that the formation of the FXR‐Nup complex is very transient and is needed for the transport of soluble Nups which are harder to visualize in the cytoplasm. The CNGs that we observe would be the result of the absence of this transport mechanism and the consequent local increase of Nups levels leading to aberrant formation of bigger (easy to visualize) Nup granules that do not necessarily contain FXR1.
The CNGs formed in the absence of FXR proteins likely represent distinct structures from ALs based on our EM analysis (Appendix Fig S3B) and lack of direct contacts with the ER membranes (Appendix Fig S3C). However, it cannot be excluded that some membranes or lipid species are part of the CNGs due to the presence of the integral membrane proteins POM121 and Nup210, both components of the NPC at the NE, and the scaffold NPC components Nup133, Nup85 and Nyp107 in these structures. The CNGs formed in the absence of FXR proteins are also distinct from SGs (Appendix Fig S4A) previously shown to recruit nucleoporins (Zhang et al, 2018) and cannot be directly linked to RNA‐based processes (Appendix Figs S4B–D and S5) at this point. We were unable to detect any changes in protein levels of several analysed Nups (Fig EV3A–C) or levels and stability of the Nups mRNAs (Fig EV3D and E). However, given that translational regulation represents one of the best‐studied roles of the FXR protein family (Darnell et al, 2009; Ascano et al, 2012), it cannot be formally excluded that expression of other, yet to be identified Nups or Nup‐associated factors, is regulated by FXR proteins.
Our data suggest a model where FXR proteins interact with the cytoplasmic pool of the scaffold NPC components Nup85 and Nup133 and act to decrease their local concentration either by their microtubule‐based, non‐NE‐directed transport and/or by transferring their small pool towards the NE. Due to the cohesive ability of many Nups and the fact that the scaffold components can directly bind to the FG‐Nups (Onischenko et al, 2017), it is therefore reasonable to predict that in the absence of the FXR proteins, the local cytoplasmic pools of Nup85 and Nup133 increase, and could result in the formation of cytoplasmic condensates containing many different NPC sub‐complexes, which is consistent with our observations (Fig 2; Appendix Figs S1 and S2). It remains to be investigated why some Nups including ELYS and Nup153 could not be detected in CNGs and if this observation could be linked to their well‐established roles in the post‐mitotic (ELYS) (Doucet et al, 2010) and interphase (Nup153) (Vollmer et al, 2015) nuclear pore complex assembly pathways. The condensate properties of the CNGs are supported by our results with the 1,6‐hexanediol treatment, which led to the dispersion of the Nup granules (Fig 5B and C) as well as the dynamic fusion and splitting events of CNGs observed in live video experiments with the GFP‐Nup107 cell line (Fig 4A and B, and 7C–E, [Link], [Link], [Link], [Link], [Link]).
What is the molecular engine for the FXR‐mediated Nup dispersal? Interestingly, FMRP was demonstrated to form a complex with the dynein motor (Ling et al, 2004; Bianco et al, 2010) and with the dynein adaptor protein BICD2 (Bianco et al, 2010) in neuronal cells. Our data are consistent with these findings and show the interaction of dynein and BICD2 with the FMRP paralog protein FXR1 (Fig 6A and D) in cultured human cancer cells. Molecular interactions of nucleoporins and dynein‐BICD2 complexes were also reported during mitotic entry (Splinter et al, 2010; Bolhy et al, 2011). Our model proposes that FXR proteins provide the molecular links between cytoplasmic Nups and the dynein‐BICD2 complex during the G1 phase of the cell cycle, allowing for the Nups dispersal and the transfer of at least a small pool of scaffold Nups towards the NE. It is interesting that FXR proteins localize to the NE (Figs 1D–F and EV4A and B), the predicted final destination of the cytoplasmic Nups. Our live video experiments are in line with the transport hypothesis and suggest that nocodazole‐induced CNGs can indeed be transferred towards intact NE (Fig 7C) in an FXR1‐ and dynein‐dependent manner. We speculate that formation of FXR1‐dynein‐Nup complexes and their transport would disperse cytoplasmic nucleoporins, thereby inhibiting formation of Nup‐containing cytoplasmic condensates. A similar function has been ascribed to the nuclear import receptors, which transfer proteins and RNAs to the nucleus across the NPCs. They were able to prevent aberrant phase separation of cytoplasmic membrane‐less organelles present in several neurological diseases (Guo et al, 2019). Thus, nuclear import receptors can also play important chaperone‐like functions by inhibiting aggregation of cargo proteins. For instance, protein fused in sarcoma (FUS) is mutated in amyotrophic lateral sclerosis (ALS) within its nuclear localization signal (NLS), which subsequently reduces its binding to the nuclear import receptor Transportin leading to cytoplasmic FUS accumulation favouring phase separation (Dormann et al, 2010). Our work also demonstrates the importance of the regulation of localized protein demixing by preventing Nup accumulation in the wrong cellular compartment (cytoplasm). In the future, it would be interesting to study if the NLS signals present in the FXR proteins are important for their functions on Nups. It also remains to be investigated if, in contrast to SGs components (Appendix Fig S4A) (Zhang et al, 2018), CNGs can sequester any other cohesive proteins known to form membrane‐less assemblies. For example, Nups can be sequestered in various pathological fibrillary amyloids, which were implicated in neurodegenerative diseases (Li & Lagier‐Tourenne, 2018; Hutten & Dormann, 2020) and in the CyPNs (cytoplasmic accumulations of PML and nucleoporins) (Jul‐Larsen et al, 2009), for which the cellular role remains unknown.
Our data suggest that the FXR‐dynein pathway is important for the maintenance of nuclear shape during early G1. Downregulation of all members of the FXR family (Fig EV4E) and dynein (Appendix Fig S6F) led to strong defects in nuclear shape in human cancer cells and nuclear atypia has been also observed in human primary fibroblasts derived from FXS patients (Fig 8D). We believe that a moderate delay in the export during G1 could (through an unknown mechanism) lead to a small yet significant difference in the nuclear area and in nuclear morphology defects. Alternatively, the nuclear size and shape could be related to the established structural roles of Nups independent of their functions in protein and RNA transport (Grossman et al, 2012). Indeed, changes in nuclear shape in cells deficient for individual Nups have been documented in various organisms (Hetzer & Wente, 2009; Mitchell et al, 2010; Ungricht et al, 2015; Onischenko et al, 2017). The first changes in nuclear morphology are observed during early G1, which correlate with the appearance of CNGs in the FXR1‐deficient cells and may perturb the progression of the cell cycle through G1 and S phases. Interestingly, the components of the Y‐complex were previously implicated in G1/S progression by regulating export of specific mRNAs of key cell cycle genes (Chakraborty et al, 2008). Furthermore, in yeast, modulation of NPCs has been reported to delay their cell cycle entry in the daughter cells (Kumar et al, 2018). Previous study in myoblasts proposed the role of FXR1 in cell cycle progression, whereby deletion of this protein led to longer G1 phase, shorter S phase and premature mitotic exit (Davidovic et al, 2013). Our results demonstrate shorter G1, longer S phases (Fig 9E and F) and no defects in mitotic progression (Fig 3D–H) in the absence of FXR1, suggesting another unrelated function of FXR1 in cancer cells.
Our data do not show global changes in the rates of export and import (Fig EV5) under normal conditions, which is expected given the small reduction of NE‐associated Nups observed in FXR1‐downregulated cells (Fig 2D–F and H). Excitingly, transient defects in protein export were observed in FXR1‐deficient cells specifically during early G1 cell cycle stage (Figs EV5D and 9A) and export factor CRM1 was sequestered to CNGs (Fig 9B). It is plausible to predict that under stress conditions or in the fast‐dividing cells of a developing embryo, this small decrease in protein export rate would significantly affect cellular homeostasis and asymmetric divisions. It is also possible that the export of CRM1‐dependent G1‐specific or more demanding cargos through NPCs is regulated by the FXR‐dynein pathway. An alternative and equally plausible explanation is that CNGs exert cytotoxic effects by sequestering yet unknown factors important for nuclear shape and cell cycle progression.
While future studies are needed to understand the precise mechanism underlying cell cycle control by the FXR‐dynein axis, and if and how it is linked to the regulation of cytoplasmic Nups, defects in this pathway are predicted to significantly perturb cellular homeostasis and may contribute to the pathology of fragile X syndrome, consistent with our observations in the cellular models of FXS (Fig 8). Collectively, our data demonstrate an unexpected role of FXR proteins and dynein in the spatial regulation of soluble Nups and provide an example of a mechanism that regulates localized protein condensate formation.
Materials and Methods
Cell lines and cell cycle synchronizations
HeLa Kyoto and derived stable cell lines (GFP, GFP‐FXR1, 3xGFP‐NUP85, GFP‐NUP107, 3xGFP‐mNup133) were cultured in Dulbecco's modified Eagle medium (DMEM; 4.5 g/l glucose, with GLUTAMAX‐I) supplemented with 10% FCS, 1% penicillin and 1% streptomycin. Cells were synchronized by two‐time addition of thymidine at 2 mM for 16 h. Cells were washed out after each thymidine addition three times with warm medium to allow for synchronous progression through cell cycle. Cells were analysed at desired time points after the release from the second thymidine block. Alternatively, cells were synchronized in early G1 by inducing an artificial mitotic exit. First, cells were treated with Taxol (paclitaxel) for 16 h at 1 μM and then subsequently released from the mitotic block by addition of Hesperadin at 100 μM for 2 h. The two primary fibroblast cell lines from healthy individuals (control cell lines) were kindly provided by Hélène Puccio. The two primary fibroblast cell lines derived from FXS individuals are described in Jacquemont et al (2018). Human primary fibroblasts were cultured in DMEM (4.5 g/l glucose) supplemented with 10% FCS and gentamicin 40 μl/ml. WT and Fmr1 KO MEFs were produced and described in Jacquemont et al, 2018. Three independent MEFs lines from control and three MEFs from Fmr1 knockout mice were cultured in DMEM (4.5 g/l glucose) supplemented with 10% FCS, 1% penicillin and 1% streptomycin. Fibroblasts and MEFs were synchronized with 100 μM Monastrol (Sigma, M8515) for 16 h, washed five times with warm medium and released into fresh medium for 2 h. HEK293T cells were cultured asynchronously in Dulbecco's modified Eagle medium (DMEM; 1 g/l glucose) supplemented with 10% FCS and 1× penicillin, and streptomycin. U2OS cells were cultured asynchronously in DMEM (4.5 g/l glucose, with GLUTAMAX‐I) supplemented with 10% FCS, 1% penicillin and 1% streptomycin. Mouse myoblasts (C2C12) were cultured asynchronously in DMEM (1 g/l glucose) supplemented with 20% FCS and gentamicin. Human induced pluripotent stem cells (iPSCs) derived from a FXS patient (FXS‐iPSCs) and the isogenic rescue cells (C1_2‐iPSCs) were grown asynchronously as indicated by (Xie et al, 2016).
EdU incorporation assay
HeLa K cells were plated on 11 mm glass coverslips (Menzel‐Glaser) in 24‐well tissue culture plates. At the end of the experiments, cells were incubated with 10 μM EdU (Lumiprobe, 20540) for 30 min at 37°C, washed once with PBS and fixed for 10 min with 4% paraformaldehyde (PFA) in PBS at RT. Subsequently, the coverslips were incubated in 100 mM Tris–HCl 100 mM pH 7.5 for 5 min at room temperature (RT) and permeabilized with 0.1% Triton X‐100 and 0.02% SDS in PBS for 5 min at RT. The coverslips were rinsed three times with PBS, and the click reaction was performed in freshly prepared label mix (Sufo‐Cy3‐Azide 8 μM [Lumiprobe, B1330], CuSO4 2 mM, ascorbic acid 20 mg/ml [Sigma, A4544] in PBS) for 30 min at RT. Coverslips were subsequently washed three times for 5 min with PBS and mounted on glass slides using Mowiol (Calbiochem) or with 0.75 μg/μl DAPI and imaged with a 63× objective using Zeiss epifluorescence microscope. When combining EdU incorporation assay with p‐Rb staining, after incubation with EdU the immunostaining was performed as for nucleoporin staining (see Immunofluorescence microscopy and sample preparation section, with the difference that post‐fixation was carried out in 4% PFA instead of 1% PFA) and then the click reaction was performed as described previously.
Immunofluorescence microscopy and sample preparation
Cells were plated on 9–15 mm glass coverslips (Menzel‐Glaser) in 12‐ or 24‐well tissue culture plates. For nucleoporin staining, at the end of the experiments cells were washed twice with PBS and fixed for 10 min with 1% PFA in PBS at RT. The coverslips were rinsed two times with PBS and permeabilized with 0.1% Triton X‐100 and 0.02% SDS in PBS for 5 min at RT, washed two times with PBS and blocked by blocking buffer 3% BSA/PBS‐T (0.01% Triton X‐100) overnight at 4°C. Coverslips were subsequently incubated with primary antibodies in blocking buffer for 1 h at RT, washed three times for 5 min with blocking buffer and incubated with secondary antibodies in blocking buffer for 30 min at RT in the dark. After incubation, coverslips were washed three times for 5 min with blocking buffer, incubated with 0.1% Triton X‐100 and 0.02% SDS in PBS for 1 min and post‐fixed for 10 min with 1% PFA in PBS at RT. Then coverslips were washed in PBS and mounted on glass slides using Mowiol or Prolong Gold reagent (Invitrogen) with 0.75 μg/μl DAPI and imaged with a 100×, 63× or 40× objectives using Zeiss epifluorescence microscope or confocal microscope Leica Spinning Disk Andor/Yokogawa. For experiments with human fibroblasts and MEFs and experiments with nocodazole or 1,6‐hexanediol treatments, cells were fixed with 4% PFA for 17 min, washed three times in PBS and permeabilized with 0.5% NP‐40 in PBS for 5 min. Cells were washed three times with PBS‐T and blocked in 3% BSA/PBS‐T for 1 h at RT or overnight at 4°C. Cells were incubated with primary antibodies for 90 min in blocking buffer, washed three times in PBS‐T for 10 min and incubated with secondary antibodies for 1 h. Cells were washed three times in PBS‐T for 10 min and mounted as previously described. For digitonin permeabilization experiments, cells with treated as indicated but permeabilized with 0.003% digitonin in PBS for 5 min and subsequent steps were performed without detergent. To induce formation of the cytoplasmic nucleoporin aggregates by microtubule depolymerization, cells were incubated with 10 μM Nocodazole or vehicle (DMSO) in culture media for 90 min at 37°C. Subsequently, nocodazole was washed five times in warm media, and 1 h after washout immunofluorescence protocol was performed as previously described. To observe the behaviour of cytoplasmic nucleoporin aggregates upon microtubule repolymerization in live video microscopy, five nocodazole washes were performed during image acquisition.
For 1,6‐hexanediol experiments, coverslips were previously coated during 1 h with fibronectin 2 μg/ml (Sigma, F1141) and collagen 20 μg/ml (Millipore, 08115) in PBS at 37°C. Subsequently, coverslips were rinsed three times with PBS before seeding cells. At the end of the experiment, cells were incubated in 10% 1,6‐hexanediol (Sigma, 88571) in medium for 70 s, washed once with PBS and immunofluorescence protocol was performed as previously described.
Poly A RNA fluorescent in situ hybridization (FISH)
HeLa K GFP‐Nup107 cells were plated on 9–15 mm glass coverslips in 12‐ or 24‐well tissue culture plates. At the end of the experiments, cells were washed once with PBS and fixed for 10 min with 4% PFA in PBS at RT. Subsequently, cells were incubated with 100% cold methanol at −20°C for 10 min. The coverslips were incubated with 70% ethanol at 4°C overnight. Cells were incubated in 1 M Tris–HCl pH 8 for 5 min at RT before proceeding to hybridization for 3 h at 37°C in hybridization buffer (2× SSC [Saline Sodium Citrate buffer], 1 mg/ml yeast tRNA, 0.005% BSA, 10% dextran sulfate, 25% formamide, 1 ng/μl oligo(dT30) fluorescent probes fused to Atto‐565 or Atto‐488 [Sigma]) protected from light. After hybridization, cells were washed once with 4× SSC, and two times with 2× SSC. Coverslips were mounted and imaged as indicated previously. For RNase treatment experiments, cells were washed three times with PBS and permeabilized with or without RNase A/T1 (0.2 mg/ml, 500 U/ml) in 0.003% digitonin PBS for 5 min, before performing the FISH protocol as described previously. All buffers were DEPC treated before use.
Microscopy and image analysis
For live‐cell microscopy, HeLa cells stably expressing indicated proteins tagged with GFP or mCherry were grown on LabTek II Chambered Slides (Thermo Scientific) or μ‐Slide VI 0.4 (IBIDI) or 35/10 mm glass bottom dishes (Greiner Bio‐One, 627871). Before filming, cells were treated with SiR‐DNA or Sir‐Tubulin probes following manufacturer's instructions when indicated. Live‐cell microscopy was carried out using 40× or 63× objective of confocal microscope Leica/Andor/Yokogawa Spinning Disk, Leica CSU‐W1 spinning disc or Nikon PFS spinning disc.
For nucleoporin cytoplasmic granule formation assay, HeLa cells stably expressing GFP‐Nup107 were treated with indicated siRNAs, synchronized by double thymidine block, released for 8 h and analysed by live video spinning disc confocal microscopy for 7 h. Z‐stacks (10 μm range, 0.5 μm step) were acquired every 5 min and movies were made with maximum intensity projection images for every time point shown at speed seven frames per second.
For protein import assay, HeLa cells were treated with the indicated siRNAs for 72 h and transfected with the reporter plasmid XRGG‐GFP (kindly provided by Jan M. van Deursen) (Love et al, 1998; Hamada et al, 2011) 30 h before filming. Cells were synchronized in early G1 phase by 100 μM Monastrol arrest for 16 h and released for 4 h. 1.25 μM dexamethasone‐induced nuclear import of XRGG‐GFP was recorded by live video spinning disc confocal microscopy for 25 min (one acquisition every 30 s).
For protein export assay, HeLa cells were treated as described above and 0.25 μM dexamethasone was added for 3 h to induce XRGG‐GFP nuclear import. Following washout, the nuclear export of XRGG‐GFP was recorded by live video spinning disc confocal microscopy for 2 h (one acquisition every 10 min).
For nucleoporin granules’ dynamics assays, HeLa cells stably expressing GFP‐Nup107 were treated with the indicated siRNAs for 72 h and synchronized in early G1 phase by 100 μM Monastrol arrest for 16 h and released for 2 h. Microtubule depolymerization and nucleoporin aggregation were induced by 10 μM nocodazole addition for 1.5 h. Following washout, nucleoporin dynamics were recorded by live video spinning disc confocal microscopy for 90 min (one acquisition every minute).
Image quantification analysis was performed using ImageJ, CellProfiler or MetaMorph software. For immunoprecipitation followed by Western blot image quantifications, protein of interest signal was normalized to GFP or IgG light chain signal (GFP‐IP and endogenous IP respectively). In all cases, the same area was used for intensity quantification and background values were subtracted. For nuclear protein intensity and nuclear area quantifications, DAPI channel was used to delimit the area and quantifications were carried out in an automated manner using Cell Profiler software. Quantifications of percentage of cells with cytoplasmic Nup granules and irregular nuclei were carried out by eye in a double‐blind manner, as well as mitotic stage timing quantifications. For protein import and export experiments, nuclear and cytoplasmic GFP signals were quantified and delta percentage for each compartment relative to time 0 was calculated. For poly A mRNA FISH experiment quantification, nuclear and cytoplasmic fluorescent oligo(dT) signal was quantified and the percentage of nuclear signal was calculated.
Super‐resolution microscopy was performed using API OMX “Blaze” with GE DeltaVision OMX stand and analysed with DeltaVision OMX softWoRx. Cells were grown on #1.5 High Precision Coverslips, fixed, permeabilized and stained according to the protocol for the fluorescent microscopy (see above). Coverslips were mounted onto the microscope slides with Vectashield H1000 mounting medium (soft setting) and sealed with a nail polish.
Experimental design, data acquisition, analysis and statistics
All experiments were done in a strictly double‐blind manner. At least three independent biological replicates were performed for each experiment and image quantifications were carried out in a blinded manner. Curves and graphs were made using GraphPad Prism and Adobe illustrator softwares.
All data were verified for normal distribution using Shapiro–Wilk test. Normal data were analysed using two‐sample two‐tailed t‐test or one‐sample two‐tailed t‐test (two‐group comparison or folds increase relative to the control, respectively) or one‐way ANOVA with Dunnett's or Sidak's correction, in case of multiple group analysis. For non‐normally distributed data, Mann–Whitney's or non‐parametric one‐way ANOVA (Kruskal–Wallis test with Dunn's correction) tests were done respectively. Data from the human induced pluripotent stem cells (iPSCs) derived from a FXS patient (FXS‐iPSCs) and the isogenic rescue cells (C1_2‐iPSCs) were analysed using paired t‐test. Error bars represent standard deviation (SD) except for live video experiments where bars represent standard error of the mean (SEM). In all cases, significance was *P < 0.05; **P < 0.01; ***P < 0.001. Details for each graph are listed in figure legends.
Plasmid and siRNA transfections
Lipofectamine 2000 (Invitrogen) was used to deliver XRGG plasmid (kindly provided by Jan M. van Deursen) (Love et al, 1998; Hamada et al, 2011) according to the manufacturer's instructions. Oligofectamine (Invitrogen) was used to deliver siRNAs for gene knockdown according to the manufacturer's instructions at a final concentration of 40–100 nM siRNA. The following siRNA oligonucleotides were used: for non‐silencing controls siGENOME Non‐targeting siRNA Pool‐1 and siGENOME Non‐targeting individual siRNA‐2 5′‐UAAGG CUAUGAAGAGAUAC‐3′ (Dharmacon), for FXR downregulation siRNA (Dharmacon) were used: FXR1 siRNA‐1 5′‐AAACGGAAUCU GAGCGUAA‐3′; FXR1 siRNA‐2 5′‐CCAUACAGCUUACUUGAUA‐3′; FXR2 5′‐CGACAAGGCUGGAUAUAGC‐3′; FMRP 5′‐AAAGCUAUGU GACUGAUGA‐3′; Dynein HC siRNA‐1 5′‐CGUACUCCCGUGAUU GAUG‐3′; Dynein HC siRNA‐2 5′‐GGAUCAAACAUGACGGAAU‐3′; BICD2 siRNA‐1 5′‐GGA GCU GUC ACA CUA CAU G‐3′; BICD2 siRNA‐2 5′‐GGU GGA CUA UGA GGC UAU C‐3′; ELYS siRNA 5′‐AUU AUC UAC AUA AUU GCU CUU TT‐3′. For endoplasmic reticulum observation experiments, cells were incubated with CellLight ER‐RFP BacMam 2.0 (Thermo Fisher, C10591) for 24 h before following the manufacturer's instructions.
Quantitative real‐time PCR
RNA of cultured cells was isolated using TRIzol reagent (Sigma) according to manufacturer's instructions. Reverse transcription was performed with random hexamer or oligodT primers using the SuperScript III First Strand cDNA Synthesis kit (Invitrogen). SYBR Green (Roche Diagnostics) based Real‐time PCR was carried out on the LightCycler 480 (Roche Diagnostics) using gene‐specific primer pairs:
NUP85: 5′‐GACTGAACAAGTTCGCAGCA‐3′ (forward), 5′‐TCAGTCGGTCACTGAGCATC‐3′ (reverse); NUP133: 5′‐TGGAGCATGAGGAGCAAGTC‐3′ (forward), 5′‐ACTTGTTGCCGTCCATGGAA‐3′ (reverse); GAPDH 5′‐ACCCAGAAGACTGTGGATGG‐3′ (forward), 5′‐TTCTAGACGGCAGGTCAGGT‐3′ (reverse); PO 5′‐GTGATGTGCAGCTGATCAAGACT‐3′ (forward) 5′‐GATGACCAGCCCAAAG GAGA‐3′ (reverse).
Primers and molecular cloning
Cloning was performed using New England Biolabs (NEB) restriction enzymes, Taq polymerase (NEB) or Fusion High‐Fidelity DNA polymerase (Thermo Scientific) according to the manufacturers’ instructions. To clone GFP‐FXR1, FXR1 was amplified from HeLa total cDNAs with primers 5′‐ttattaCTCGAGCCATGGCGGAGCTGACGGTG GAGG‐3′ (forward) and 5′‐tattatGAATTCTTATGAAACACCATTCAG GACTGC‐3′ (reverse). FXR1 cDNA was cloned into the pEGFPC1 vector using EcoRI/XhoI restriction enzymes. To obtain the plasmid used in rescue experiment expressing GFP‐FXR1‐MUT‐siRNA1, primers 5′‐CTTTCCGTTCGCTCTCTGTCTCAGAGGGGTTAGACAGC TCAGAATTTG‐3′ (forward) and 5′‐TGAGACAGAGAGCGAAC GGAAAGACGAGCTGAGTGATTGGTCATTGGC‐3′ (reverse) to mutate pEGFPC1‐FXR1 vector in the region targeted by the FXR1 siRNA1. The pEGFPC1‐NUP85 was kindly provided by Valérie Doye. For live video microscopy experiments, cDNAs were cloned into a pVITRO‐blasticidin vector (Invivogen). mCherry‐H2B was amplified with primers 5′‐AATAATGCTAGCATGCCAGAGCCAGCGAAGTCTGC‐3′ (forward) and 5′‐TAATAATCTAGATTACTTGTACAGCTCGTCCATGC‐3′ (reverse). cDNA was cloned into the pVITRO in the multi cloning site 1 using NheI/XbaI restriction enzymes. GFP‐NUP85 was amplified with primers 5′‐aataatGCTAGCGCCATCATGGTGAG CAAGGGCGAGGAGCTG‐3′ (forward) and 5′‐tattatGCTAGCTCAG GAACCTTCCAGTGAGCCTTCTC‐3′ (reverse). cDNA was cloned into the pVITRO in the multi cloning site 2 using the NheI restriction enzyme. DNA purifications were performed using commercial kits from Macherey‐Nagel according to the manufacturer's instructions.
Generation or acquisition of stable cell lines
Hela cells were transfected with pVITRO or pEGFPC1‐derived constructs using Lipofectamine 2000 according to manufacturer's instructions. Transfected cells were selected for 2–3 weeks in medium supplemented by antibiotics, either G418 (400 μg/ml) or blasticidin (5 μg/ml). Transgene‐expressing clones were then isolated by FACS (FACS ARIA, BD Biosciences). Expression was validated by Western blot and immunofluorescence analysis. GFP‐NUP107 stable cell line was purchased from CLS cell bank. HeLa Kyoto cells stably expressing 3xGFP‐mNup133 were kindly provided by Valérie Doye.
Western blotting and immunoprecipitations
HeLa cell extracts were prepared using lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X‐100, 1 mM EDTA 0.25% sodium deoxycholate, 1 mM PMSF, protease inhibitor cocktail). For GFP immunoprecipitation, GFP‐fused proteins were immunoprecipitated using GFP‐Trap A agarose beads (Chromotek). Beads were incubated with cell extracts for 1 h30 at 4 °C under constant rotation. Before elution, beads were washed 3 times for 10 min with washing buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, protease inhibitor cocktail), boiled in Laemmli SDS sample buffer and subjected to SDS–PAGE.
For endogenous immunoprecipitation experiments, protein G sepharose 4 Fast Flow beads (GE Healthcare Life Sciences) were washed three times for 1 min in washing buffer (Tris–HCl 1M pH 7,5, NaCl 150 mM, EDTA 1 mM, protease inhibitor cocktail). FXR1 protein from HEK293T cell extracts was incubated with the beads and FXR1 antibody or rabbit IgG (1.5 μl per mg of protein) for 3 h at 4°C under constant rotation. Before elution, beads were washed 5 times during 2 h with washing buffer 0.1% Tween‐20, boiled in Laemmli SDS sample buffer and subjected to SDS–PAGE. Proteins were subsequently transferred from the gel to a PVDF membrane (Millipore) for immunoblotting. Membranes were blocked in 5% non‐fat milk powder resuspended in TBS supplemented with 0.1% Tween 20 (TBS‐T) for 1 h at RT or overnight at 4°C, followed by incubation with antibodies. Membranes were developed with Luminata Forte (Millipore) or SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Electron microscopy
HeLa cells stably expressing 3xGFP‐Nup85 were grown on carbon‐coated sapphire discs and synchronized by double thymidine block and 12 h release. After synchronization, cells were high pressure frozen (HPM010, Abra Fluid) and freeze substituted with 0.1% uranyl acetate in acetone for 15 h. The temperature was then raised to ‐45°C at 5°C/h and cells were further incubated for 5 h. After rinsing in acetone, the samples were finally embedded in Lowicryl HM20 (Polysciences Inc.). Thick sections (300 nm) were cut from the UV‐polymerized resin block and picked up on carbon‐coated mesh grids. After post‐staining, 2D montages and tilt series of the areas of interest were acquired using a FEI TECNAI F30 TEM. Tomograms were reconstructed using the software package IMOD (Kremer et al, 1996).
Antibodies
The following antibodies were used: mouse monoclonal α‐tubulin (Sigma T5169, immunofluorescence microscopy 1:4,000, Western blot 1:20,000), rabbit α‐GAPDH (Sigma G9545, Western blot 1:20,000), mouse α‐GAPDH (Genetex, gtx627408, 1:20,000), mouse monoclonal α‐FXR1 + 2 (clone 2B12 from IGBMC, immunofluorescence microscopy 1:500, Western blot 1:1,000), rabbit α‐FXR1 (Sigma HPA018246, immunofluorescence microscopy 1:800, Western blot 1:1,000), mouse α‐FXR1 (Millipore 03‐176, immunofluorescence microscopy 1:800, Western blot 1:1,000), mouse α‐FMRP (clone 1C3 from IGBMC, immunofluorescence microscopy 1:250, Western blot 1:1,000), rabbit α‐FMRP (Abcam ab17722, immunofluorescence microscopy 1:250, Western blot 1:1,000), mouse α‐Nup133 (Santa cruz sc‐37673, Western blot 1:1,000), rat α‐Nup133 (kindly provided by Valérie Doye, immunofluorescence microscopy 1:250), mouse α‐FG‐Nups (Abcam mAb414, ab24609, 1:4,000), rabbit α‐Nup153 (Abcam ab84872, immunofluorescence microscopy 1:500, Western blot 1:1,000), rabbit α‐Nup85 (Bethyl, A303‐977A, immunofluorescence microscopy 1:100), mouse α‐Pericentrin‐1 (D‐4) (NUP85; Santa Cruz, sc‐376111, Western blot 1:1,000), mouse α‐PLK1 (Santa cruz sc‐17783, Western blot 1:1,000), rabbit polyclonal GFP (Abcam ab290, 1:20,000), mouse monoclonal Cyclin B1 (Santa Cruz sc‐245, clone GSN1, 1:2,000), rabbit α‐cyclin A (Santa Cruz sc‐751, 1:1,000), rabbit α‐laminA (Sigma L1293 1:500), rabbit α‐lamin B1 (Abcam, ab16048, 1:500), mouse α‐Lap2 (BD biosciences, 611000, 1:500), rabbit α‐ELYS (Bethyl, A300‐166A, 1:250), rabbit α‐POM121 (Genetex, GTX102128, 1:200), mouse α‐RanGAP1 (Cell Signalling 2365, 1:250), rabbit α‐emerin (Abcam, ab40688, 1:1,000), rabbit α‐LBR (Abcam, ab35535, 1:500), mouse α‐Dynein IC (Merck, MAB1618, 1:500), mouse α‐p150glued (BD biosciences, 610473, 1:1,000), goat α‐RanBP2 (kindly provided by Frauke Melchior, 1:2,000), rabbit α‐Nup98 (Cell Signalling, 2598S, 1:100), rabbit α‐BICD2 (Sigma HPA023013, immunofluorescence microscopy 1:250, Western blot 1:1,000), rabbit α‐G3BP1 (Genetex GTX112191, immunofluorescence microscopy 1:500), mouse α‐TIA‐1 (G‐3; Santa Cruz, sc‐166247, immunofluorescence microscopy 1:500), rabbit α‐p‐Rb (Cell Signaling 8516, immunofluorescence microscopy 1:1,600), and rabbit α‐CRM1 (Novus, NB100‐79802, immunofluorescence microscopy 1:250). Antibodies against Nup88, Nup210 and RAE1 were kindly provided by Ulrike Kutay.
Author contributions
AA‐A and SS designed and performed experiments and helped writing the manuscript. KJ, CK, AB, LP, IJB, PR and LG performed experiments. HM helped performing experiments. SJ and CB provided human patient cells and mouse embryonic fibroblasts. JLM and YS helped designing the experiments. IS supervised the project, designed experiments and wrote the manuscript with input from all authors.
Conflict of interests
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Dataset EV1
Source data for Figure 1
Source data for Figure 3
Source data for Figure 6
Source data for Figure 8
Source Data for Expanded View and Appendix
Review Process File
Acknowledgements
We thank patients and their families for their contribution. Y. Barral, S. Oliferenko, G. Sumara, O. Sumara, M. Mendoza, N. Djouder, K. Krupina, O. Bielska, Z. Zhang, Life Science Editors and the members of the Sumara group for helpful discussions on the manuscript. We are grateful to Valérie Doye for generous help with reagents. We thank Ulrike Kutay, Frauke Melchior, Hélène Puccio, Jin Peng, Stephen Warren, Romeo Ricci, Jan M. van Deursen, Matej Durik, Ioanna Mitrentsi and Nicolas Charlet‐Berguerand for help with reagents. We are grateful to Jean‐Marc Egly and Michel Labouesse for their mentorship and support. We are grateful to Giorgia Pedini for providing the WT and Fmr1 KO MEFs. We thank the Imaging Center of the IGBMC (ICI) for help on confocal microscopy and to the IGBMC core facilities for their support on this research. Alexia Loynton‐Ferrand from the Biozentrum, University of Basel for support on super‐resolution imaging. A.A.A. was supported by a Labex international PhD fellowship from IGBMC and a fellowship from the “Ligue Nationale Contre le Cancer”. S.S. was supported by a postdoctoral fellowship from the University of Strasbourg Institute of Advanced Studies (USIAS). K.J. was supported by a fellowship from Gouvernement français et L'Institut français de Prague, a LabEx international PhD fellowship from IGBMC and a fellowship from the “Ligue Nationale Contre le Cancer”. A.B. received PhD fellowships from the “Ministère de l'Enseignement Supérieur et de la Recherche” and the “Ligue Nationale contre le Cancer”. Research of L.P. and C. B. was supported by Associazione Italiana Sindrome X fragile, Telethon GGP15257, PRIN 201789LFKB and Swiss National Science Foundation (SNSF) 310030‐182651. This study was supported by the grant ANR‐10‐LABX‐0030‐INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame programme Investissements d'Avenir ANR‐10‐IDEX‐0002‐02. Research in I.S. laboratory was supported by IGBMC, CNRS, Fondation ARC pour la recherche sur le cancer, Institut National du Cancer (INCa), Agence Nationale de la Recherche (ANR), Ligue Nationale contre le Cancer, USIAS and Sanofi iAward Europe.
The EMBO Journal (2020) 39: e104467
Data availability
This study includes no data deposited in external repositories.
References
- Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M et al (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Zukin RS (2019) A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 101: 1070–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18: 73–89 [DOI] [PubMed] [Google Scholar]
- Bianco A, Dienstbier M, Salter HK, Gatto G, Bullock SL (2010) Bicaudal‐D regulates fragile X mental retardation protein levels, motility, and function during neuronal morphogenesis. Curr Biol 20: 1487–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L et al (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol 28: 420–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhy S, Bouhlel I, Dultz E, Nayak T, Zuccolo M, Gatti X, Vallee R, Ellenberg J, Doye V (2011) A Nup133‐dependent NPC‐anchored network tethers centrosomes to the nuclear envelope in prophase. J Cell Biol 192: 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Gao R, Yu C, Chen L, Feng Y (2019) The RNA‐binding protein FXR1 modulates prostate cancer progression by regulating FBXO4. Funct Integr Genomics 19: 487–496 [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Wang Y, Wei J‐H, van Deursen J, Yu H, Malureanu L, Dasso M, Forbes DJ, Levy DE, Seemann J et al (2008) Nucleoporin levels regulate cell cycle progression and phase‐specific gene expression. Dev Cell 15: 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo MA, Anderson DJ, Richard E, Hetzer MW (2006) Nuclear pores form de novo from both sides of the nuclear envelope. Science 312: 440–443 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O, Darnell RB (2009) Discrimination of common and unique RNA‐binding activities among Fragile X mental retardation protein paralogs. Hum Mol Genet 18: 3164–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovic L, Durand N, Khalfallah O, Tabet R, Barbry P, Mari B, Sacconi S, Moine H, Bardoni B (2013) A novel role for the RNA‐binding protein FXR1P in myoblasts cell‐cycle progression by modulating p21/Cdkn1a/Cip1/Waf1 mRNA stability. PLoS Genet 9: e1003367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LI, Blobel G (1987) Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci USA 84: 7552–7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IRA, Capell A, Schmid B et al (2010) ALS‐associated fused in sarcoma (FUS) mutations disrupt Transportin‐mediated nuclear import. EMBO J 29: 2841–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet CM, Talamas JA, Hetzer MW (2010) Cell cycle‐dependent differences in nuclear pore complex assembly in metazoa. Cell 141: 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Chen X, Wu X, Zhang M (2019) Formation of biological condensates via phase separation: characteristics, analytical methods, and physiological implications. J Biol Chem 294: 14823–14835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Richter RP, Görlich D (2006) FG‐rich repeats of nuclear pore proteins form a three‐dimensional meshwork with hydrogel‐like properties. Science 314: 815–817 [DOI] [PubMed] [Google Scholar]
- Frey S, Görlich D (2007) A saturated FG‐repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell 130: 512–523 [DOI] [PubMed] [Google Scholar]
- Grossman E, Medalia O, Zwerger M (2012) Functional architecture of the nuclear pore complex. Annu Rev Biophys 41: 557–584 [DOI] [PubMed] [Google Scholar]
- Guo W, Polich ED, Su J, Gao Y, Christopher DM, Allan AM, Wang M, Wang F, Wang G, Zhao X (2015) Fragile X proteins FMRP and FXR2P control synaptic GluA1 expression and neuronal maturation via distinct mechanisms. Cell Rep 11: 1651–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Fare CM, Shorter J (2019) Therapeutic dissolution of aberrant phases by nuclear‐import receptors. Trends Cell Biol 29: 308–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M, Haeger A, Jeganathan KB, van Ree JH, Malureanu L, Wälde S, Joseph J, Kehlenbach RH, van Deursen JM (2011) Ran‐dependent docking of importin‐beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J Cell Biol 194: 597–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Mackmull M‐T, Machado P, Ronchi P, Bui KH, Schieber N, Santarella‐Mellwig R, Necakov A, Andrés‐Pons A, Philippe JM et al (2016) Pre‐assembled nuclear pores insert into the nuclear envelope during early development. Cell 166: 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Andres‐Pons A, Kastritis P, Beck M (2019a) Structure and assembly of the nuclear pore complex. Annu Rev Biophys 48: 515–536 [DOI] [PubMed] [Google Scholar]
- Hampoelz B, Schwarz A, Ronchi P, Bragulat‐Teixidor H, Tischer C, Gaspar I, Ephrussi A, Schwab Y, Beck M (2019b) Nuclear pores assemble from nucleoporin condensates during oogenesis. Cell 179: 671–686.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AF, Shorter J (2017) RNA‐binding proteins with prion‐like domains in health and disease. Biochem J 474: 1417–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer MW, Wente SR (2009) Border control at the nucleus: biogenesis and organization of the nuclear membrane and pore complexes. Dev Cell 17: 606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogeveen AT, Willemsen R, Oostra BA (2002) Fragile X syndrome, the Fragile X related proteins, and animal models. Microsc Res Tech 57: 148–155 [DOI] [PubMed] [Google Scholar]
- Hutten S, Dormann D (2020) Nucleocytoplasmic transport defects in neurodegeneration ‐ cause or consequence? Semin Cell Dev Biol 99: 151–162 [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Pacini L, Jønch AE, Cencelli G, Rozenberg I, He Y, D'Andrea L, Pedini G, Eldeeb M, Willemsen R et al (2018) Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum Mol Gen 27: 2039–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Zhai B, Fang T, Guo X, Xu L (2016) FXR1 is elevated in colorectal cancer and acts as an oncogene. Tumour Biol 37: 2683–2690 [DOI] [PubMed] [Google Scholar]
- Jul‐Larsen A, Grudic A, Bjerkvig R, Bøe SO (2009) Cell‐cycle regulation and dynamics of cytoplasmic compartments containing the promyelocytic leukemia protein and nucleoporins. J Cell Sci 122: 1201–1210 [DOI] [PubMed] [Google Scholar]
- Khlghatyan J, Evstratova A, Chamberland S, Marakhovskaia A, Bahremand A, Toth K, Beaulieu J‐M (2018) Mental illnesses‐associated Fxr1 and its negative regulator Gsk3β are modulators of anxiety and glutamatergic neurotransmission. Front Mol Neurosci 11: 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer KE, Schwartz TU (2016) The nuclear pore complex as a flexible and dynamic gate. Cell 164: 1162–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three‐dimensional image data using IMOD. J Struct Biol 116: 71–76 [DOI] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nüske E, Poser I, Richter D, Alberti S (2015) Promiscuous interactions and protein disaggregases determine the material state of stress‐inducible RNP granules. eLife 4: e06807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Sharma P, Gomar‐Alba M, Shcheprova Z, Daulny A, Sanmartín T, Matucci I, Funaya C, Beato M, Mendoza M (2018) Daughter‐cell‐specific modulation of nuclear pore complexes controls cell cycle entry during asymmetric division. Nat Cell Biol 20: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhao X (2014) Concise review: fragile X proteins in stem cell maintenance and differentiation. Stem Cells 32: 1724–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lagier‐Tourenne C (2018) Nuclear pores: the gate to neurodegeneration. Nat Neurosci 21: 156–158 [DOI] [PubMed] [Google Scholar]
- Lin Y, Protter DSW, Rosen MK, Parker R (2015) Formation and maturation of phase‐separated liquid droplets by RNA‐binding proteins. Mol Cell 60: 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S‐C, Fahrner PS, Greenough WT, Gelfand VI (2004) Transport of Drosophila fragile X mental retardation protein‐containing ribonucleoprotein granules by kinesin‐1 and cytoplasmic dynein. Proc Natl Acad Sci USA 101: 17428–17433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Sweitzer TD, Hanover JA (1998) Reconstitution of HIV‐1 rev nuclear export: independent requirements for nuclear import and export. Proc Natl Acad Sci USA 95: 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucá R, Averna M, Zalfa F, Vecchi M, Bianchi F, La Fata G, Del Nonno F, Nardacci R, Bianchi M, Nuciforo P et al (2013) The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol Med 5: 1523–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merisko EM (1989) Annulate lamellae: an organelle in search of a function. Tissue Cell 21: 343–354 [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Mansfeld J, Capitanio J, Kutay U, Wozniak RW (2010) Pom121 links two essential subcomplexes of the nuclear pore complex core to the membrane. J Cell Biol 191: 505–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2015) Fragile X disappointments upset autism ambitions. Nat Rev Drug Discov 14: 151–153 [DOI] [PubMed] [Google Scholar]
- Oldenburg AR, Delbarre E, Thiede B, Vigouroux C, Collas P (2014) Deregulation of Fragile X‐related protein 1 by the lipodystrophic lamin A p. R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum Mol Genet 23: 1151–1162 [DOI] [PubMed] [Google Scholar]
- Onischenko EA, Gubanova NV, Kieselbach T, Kiseleva EV, Hallberg E (2004) Annulate lamellae play only a minor role in the storage of excess nucleoporins in Drosophila embryos. Traffic 5: 152–164 [DOI] [PubMed] [Google Scholar]
- Onischenko E, Tang JH, Andersen KR, Knockenhauer KE, Vallotton P, Derrer CP, Kralt A, Mugler CF, Chan LY, Schwartz TU et al (2017) Natively unfolded FG repeats stabilize the structure of the nuclear pore complex. Cell 171: 904–917.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Belmont BJ, Sante JM, Rexach MF (2007) Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 129: 83–96 [DOI] [PubMed] [Google Scholar]
- Ramos A (2003) G‐quartet‐dependent recognition between the FMRP RGG box and RNA. RNA 9: 1198–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbeck K, Görlich D (2002) The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J 21: 2664–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma S, D'Angelo MA (2017) The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin Cell Dev Biol 68: 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST (2012) Molecular mechanisms of fragile X syndrome: a twenty‐year perspective. Annu Rev Pathol 7: 219–245 [DOI] [PubMed] [Google Scholar]
- Schmidt HB, Görlich D (2016) Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem Sci 41: 46–61 [DOI] [PubMed] [Google Scholar]
- Schmidt HB, Görlich D (2015) Nup98 FG domains from diverse species spontaneously phase‐separate into particles with nuclear pore‐like permselectivity. eLife 4: e04251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Brangwynne CP (2017) Liquid phase condensation in cell physiology and disease. Science 357: 1253 [DOI] [PubMed] [Google Scholar]
- Shorter J (2019) Phase separation of RNA‐binding proteins in physiology and disease: an introduction to the JBC Reviews thematic series. J Biol Chem 294: 7113–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter D, Tanenbaum ME, Lindqvist A, Jaarsma D, Flotho A, Yu KL, Grigoriev I, Engelsma D, Haasdijk ED, Keijzer N et al (2010) Bicaudal D2, dynein, and kinesin‐1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol 8: e1000350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanini F, Bontekoe C, Bakker CE, van Unen L, Anar B, Willemsen R, Yoshida M, Galjaard H, Oostra BA, Hoogeveen AT (1999) Different targets for the fragile X‐related proteins revealed by their distinct nuclear localizations. Hum Mol Genet 8: 863–869 [DOI] [PubMed] [Google Scholar]
- Ungricht R, Klann M, Horvath P, Kutay U (2015) Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J Cell Biol 209: 687–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer B, Lorenz M, Moreno‐Andrés D, Bodenhöfer M, De Magistris P, Astrinidis SA, Schooley A, Flötenmeyer M, Leptihn S, Antonin W (2015) Nup153 recruits the Nup107‐160 complex to the inner nuclear membrane for interphasic nuclear pore complex assembly. Dev Cell 33: 717–728 [DOI] [PubMed] [Google Scholar]
- Weberruss M, Antonin W (2016) Perforating the nuclear boundary ‐ how nuclear pore complexes assemble. J Cell Sci 129: 4439–4447 [DOI] [PubMed] [Google Scholar]
- Xie N, Gong H, Suhl JA, Chopra P, Wang T, Warren ST (2016) Reactivation of FMR1 by CRISPR/Cas9‐mediated deletion of the expanded CGG‐repeat of the fragile X chromosome. PLoS ONE 11: e0165499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Panasiti V, Carotti S, Zingariello M, Perrone G, Sancillo L, Pacini L, Luciani F, Roberti V, D'Amico S et al (2017) The fragile X mental retardation protein regulates tumor invasiveness‐related pathways in melanoma cells. Cell Death Dis 8: e3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F et al (2018) Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173: 958–971.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Dataset EV1
Source data for Figure 1
Source data for Figure 3
Source data for Figure 6
Source data for Figure 8
Source Data for Expanded View and Appendix
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.