

Abstract
Following two decades of more than 400 clinical trials centered on the “one drug, one target, one disease” paradigm, there is still no effective disease modifying therapy for Alzheimer’s disease (AD). The inherent complexity of AD may challenge this reductionist strategy. Recent observations and advances in network medicine further indicate that AD likely shares common underlying mechanisms and intermediate pathophenotypes, or endophenotypes, with other diseases. In this review, we consider AD pathobiology, disease comorbidity, pleiotropy, and therapeutic development, and construct relevant endophenotype networks to guide future therapeutic development. Specifically, we discuss six main endophenotype hypotheses in AD: amyloidosis, tauopathy, neuroinflammation, mitochondrial dysfunction, vascular dysfunction, and lysosomal dysfunction. We further consider how this endophenotype network framework can provide advances in computational and experimental strategies for drug-repurposing and identification of new candidate therapeutic strategies for patients suffering from or at risk for AD. We highlight new opportunities for endophenotype-informed, drug discovery in AD, by exploiting multi-omics data. Integration of genomics, transcriptomics, radiomics, pharmacogenomics, and interactomics (protein-protein interactions) are essential for successful drug discovery. We describe experimental technologies for AD drug discovery including human induced pluripotent stem cells, transgenic mouse/rat models, and population-based retrospective case-control studies that may be integrated with multi-omics in a network medicine methodology. In summary, endophenotype-based network medicine methodologies will promote AD therapeutic development that will optimize the usefulness of available data and support deep phenotyping of the patient heterogeneity for personalized medicine in AD.
Keywords: Alzheimer’s disease, amyloidosis, drug repurposing, endophenotype, systems biology, omics, network medicine, pathobiology, tauopathy
1. Introduction
1.1. Emerging challenges in Alzheimer’s disease drug discovery
Alzheimer’s disease (AD), first described in 1907 by Alois Alzheimer,1 is a highly-prevalent and progressive neurodegenerative disorder with gradual cognitive decline and memory loss. It is characterized by accumulation of extracellular amyloid plaques as well as intracellular neurofibrillary tangles (NFTs).2 AD and other dementias are a major global health challenge, with 43.8 million affected people worldwide in 2016.3 High-throughput technologies, such as next-generation sequencing (NGS), have rapidly led to a robust body of genetic and genomic data in multiple national AD genome projects, including the Alzheimer’s Disease Sequencing Project (ADSP)4 and the Alzheimer’s Disease Neuroimaging Initiative (ADNI).5 Genome-wide association studies (GWAS) have identified over 40 AD susceptibility loci.6–8 Despite this progress in understanding of AD genetic risk, there are still only six drugs approved by the U.S. Food and Drug Administration (FDA) for treatment of AD symptoms: four cholinesterase inhibitors (tacrine, donepezil, galantamine, and rivastigmine), one N-methyl-D-aspartate (NMDA) receptor antagonist (memantine), and one fixed combination of donepezil and memantine. None of these drugs are effective in modifying the underlying biology of AD. The current failure rate in AD clinical trials (2002–2012) is estimated at 99.6%.9,10 146 investigational drug candidates in clinical trials have been halted due to safety and efficacy concerns (Fig. 1A) in the past two decades (1998–2017). After examination of the mechanistic classes in AD clinical trials from the Alzforum database,10,11 agents targeting amyloidosis (23.4%), neurotransmission (27.5%), neuroinflammation (10.2%), tauopathy (6.6%), and others (32.4%) have been identified as the predominant categories for discovery (Fig. 1B). To date, no drugs in these categories have been approved by the FDA.
Figure 1. Statistics of current drug development status in Alzheimer’s disease (AD).

(A) Distribution of clinically failed drugs versus approved drugs in the past 20 years (1998–2017). The data are collected from Adis R&D Insight Database; (B) Distribution of mechanistic classes in development of AD drugs. The data are collected from U.S. AD clinical trials from the Alzforum database in March 2016.
1.2. Networks and cell systems can help therapeutic development
Researchers have long sought to uncover single molecular defects that cause human diseases, with the goal of developing “magic-bullet” targeted therapies. However, the “one drug, one disease” reductionism-informed paradigm overlooks the inherent complexity of most diseases. This reductionist paradigm has often led to treatments that are inadequate or fraught with adverse side effects (Fig. 2A). Existing data resources, including genetics, transcriptomics, proteomics, radiomics (brain imaging data), and interactomics (protein-protein interactions [PPI]), hold promise for fostering therapeutic discovery but have not yet been fully integrated into the AD field. Importantly, cellular systems and molecular interactome network perturbations underlie disease processes in unanticipated ways (Fig. 2B). The field of ‘network medicine’ is an emerging discipline that seeks to redefine human disease and therapeutics, offering a non-invasive approach to identifying novel biomarkers and therapeutic targets.12
Figure 2. Reductionist versus network medicine paradigm for AD pathogenesis and drug discovery.
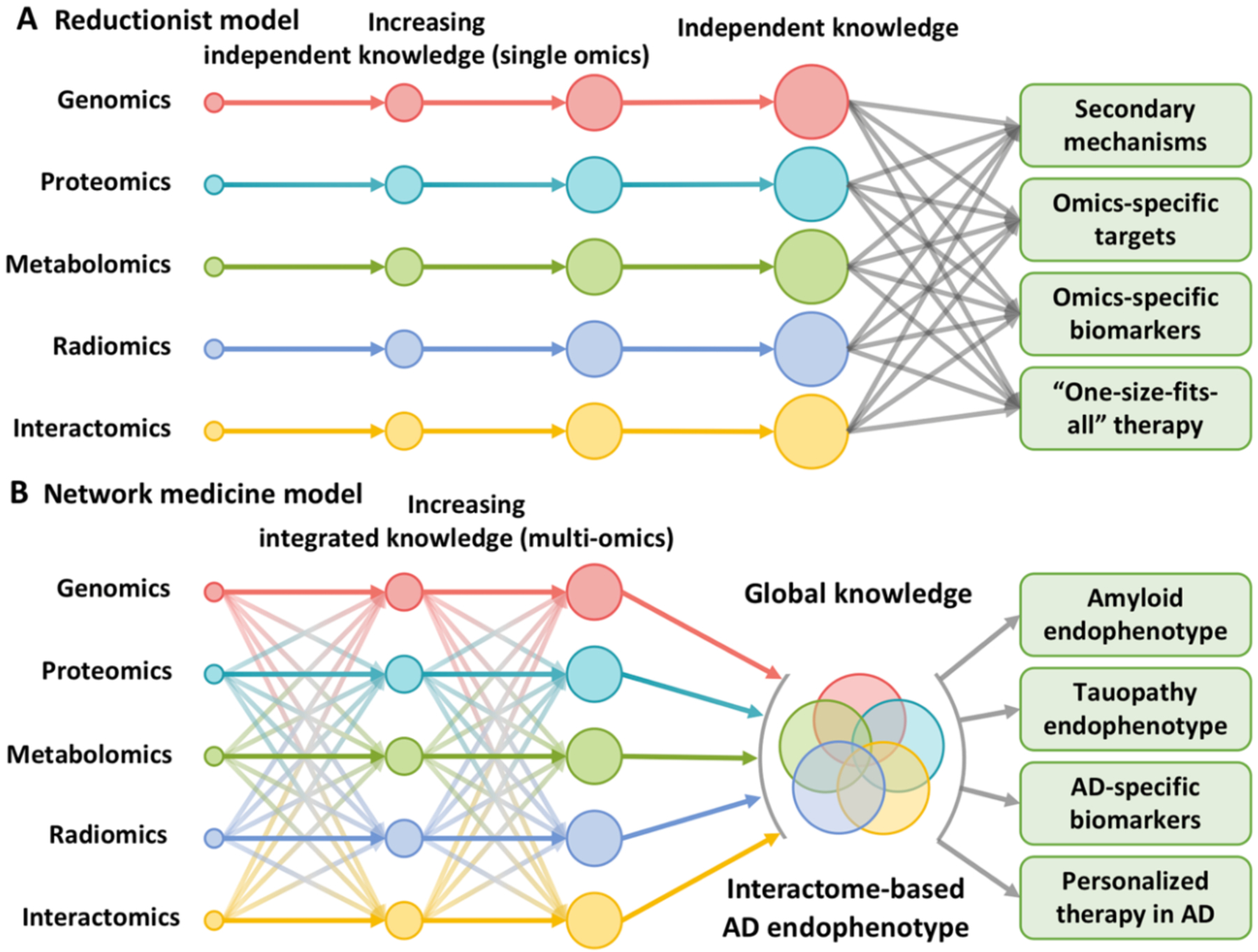
(A) The traditional reductionist paradigm that utilizes routine single omics approaches resulting in large bodies of distinct data that are not integrated. (B) Network medicine is based on the utilization of state-of-the-art network science tools and systems biology approaches to build the integrative model (AD endophenotype) from multi-omics data under the human interactome network framework.
A network medicine approach to AD rests on the underlying hypothesis that cellular networks gradually adjust to chronic processes of disease initiation and progression, leading to progressive shifts in local and global network properties and system states that underlie an evolving pathogenesis of AD. Although AD is often described as a ‘disease of the genome’, it may be more appropriately described as a ‘disease of the interactome’.13–16 Genome alterations such as amplification, deletion, translocations and mutations are the primary genetic events of AD. But such events can meaningfully manifest in human cells only if they encode disease-specific changes or perturbations in the interactome network that influences relevant systems properties of the affected brain. Thus, therapeutic interventions need to address more broad perturbations of AD systems, rather than genomic events alone.16
Emerging technologies and concepts are transforming drug discovery;17 knowledge of the network of molecular determinants of disease and drug targets in the protein-protein interaction (PPI) network (the human interactome) is essential for identification of candidate treatments including drugs approved from non-AD indications (e.g., repurposing).18 Novel recently developed approaches, such as drug-disease network proximity that shed light on the relationships between drug targets and diseases (i.e., molecular determinants in disease modules within the PPIs), may efficiently screen for novel indications19 and design of combination20 therapies from approved drugs. Drug repurposing or repositioning could significantly shorten the time and reduce the cost of drug discovery compared to lengthy and expensive de novo campaigns.21 There are nearly 30 FDA-approved drugs currently in preclinical or clinical investigations for potential repurposed treatment of AD (Table 1 and Fig. 3).
Table 1.
Summary of FDA-approved drugs that have been or being repurposed for treatment of Alzheimer’s disease.
| Drug Class | Treatment | Drug | Phase and Clinicaltrials.gov Identifier |
|---|---|---|---|
| Cardiovascular disorder | Cerebrovascular circulatory disorders | Dihydroergocristine (DB13345)258 | Investigational |
| Hypertension | Valsartan (DB00177)96 | Investigational | |
| Hypertension | Isradipine (DB00270)101 | Investigational | |
| Hypertension | Telmisartan (DB00966)259 | Phase I (NCT02471833) | |
| Hypertension | Perindopril (DB00790)260 | Phase II (NCT02085265) | |
| Hypertension | Losartan (DB00678)261 | Phase III (NCT02913664) | |
| Hypertension | Carvedilol (DB01136)99 | Phase IV (NCT01354444) | |
| Hypertension | Nilvadipine (DB06712)262 | Phase IV (NCT02017340) | |
| Hypertension | Nimodipine (DB00393)263 | Phase IV (NCT00814658) | |
| Metabolic disorder | Diabetes Mellitus | Liraglutide (DB06655)46 | Phase II (NCT01843075) |
| Diabetes Mellitus | Metformin (DB00331)264 | Phase II (NCT00620191) | |
| Diabetes Mellitus | Pioglitazone (DB01132)265 | Phase II (NCT00982202) | |
| Diabetes Mellitus | Benfotiamine (DB11748)266 | Phase II (NCT02292238) | |
| Hypercholesterolemia | Pitavastatin (DB08860)48 | Phase II (NCT00548145) | |
| Hypercholesterolemia | Atorvastatin (DB01076)48 | Phase II (NCT02913664) | |
| Hypercholesterolemia | Simvastatin (DB00641)267 | Phase II (NCT01439555) | |
| Nervous systems or mental disorders | Erectile dysfunction | Sildenafil (DB00203)102 | Investigational |
| Erectile dysfunction | Tadalaril (DB00820)268 | Phase II (NCT02450253) | |
| Aamyotrophic lateral sclerosis | Riluzole (DB00740)269 | Phase II (NCT01703117) | |
| Depressive disorder | Paroxetine (DB00715)270 | Investigational | |
| Parkinson | Rasagiline (DB01367)271 | Phase II (NCT02359552) | |
| Schizophrenia | Clozapine (DB00363)225 | Investigational | |
| Epilepsy | Levetiracetam (DB01202)272 | Phase II (NCT03489044) | |
| Urea cycle disorders | Benzoic Acid (DB03793)273 | Phase II (NCT01600469) | |
| Others | Cutaneous T cell lymphoma | Bexarotene (DB00307)274 | Phase II (NCT01782742) |
| Acute promyelocytic leukaemia | Tamibarotene (DB04942)275 | Phase II (NCT01120002) | |
| Infections | Minocycline (DB01017)50 | Phase II (NCT01463384) | |
| Malaria | Methylene blue (DB09241)276 | Phase II (NCT02380573) | |
| Mast cell tumors in animals | Masitinib (DB11526)277 | Phase III (NCT01872598) | |
| Psoriasis | Acitretin (DB00459)278 | Phase II (NCT01078168) | |
| Severe acne | Isotretinoin (DB00982)279 | Phase II (NCT01560585) |
Figure 3. Statistics of repurposed drugs in nonclinical or clinical investigations for Alzheimer therapy.
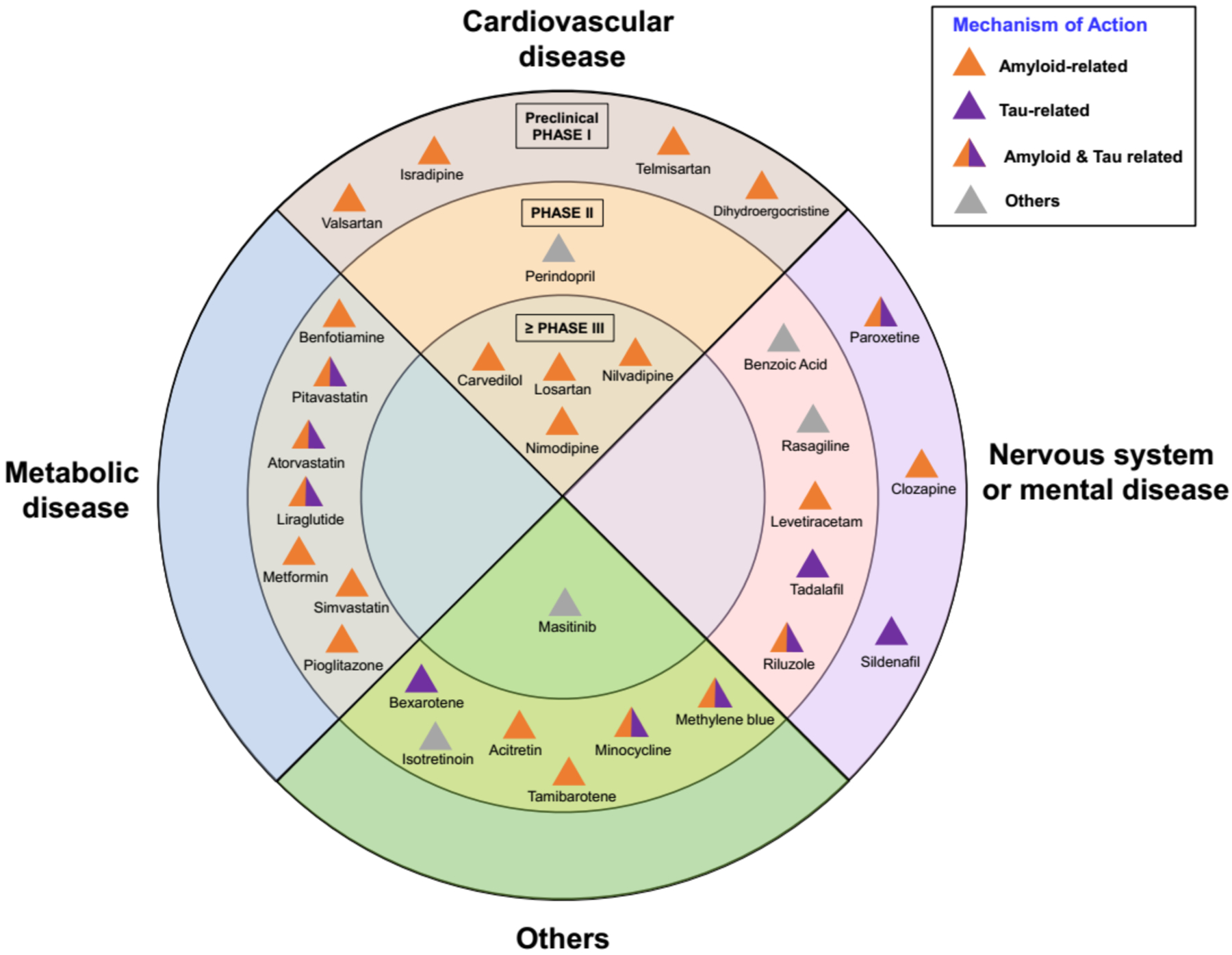
All repurposed drugs in AD clinical trials are collected from clinicaltrials.gov database as of July 31, 2019. The inner ring represents Phase III agents; the middle ring shows Phase II agents; the outer ring presents Phase I or preclinical drugs. All the drugs are classified into four types: cardiovascular drugs, metabolic drugs, nervous systems or mental drugs, and others, according to their original indication. The mechanism-of-action of drugs are classified into four types: amyloid-related, tau-related, amyloid & tau related, and others.
In this review, we discuss recent advances in computational and experimental strategies that could drive drug repurposing for treatment of individuals manifesting or at risk for AD dementia.
2. Endophenotype Networks Meet Alzheimer’s Drug Discovery
The gold standard for a definite diagnosis of AD is amyloid aggregated into plaques and hyperphosphorylated tau into tangles.22 The amyloid hypothesis, an assumption that accumulation of the peptide amyloid-beta is the main cause of the condition, has dominated research on AD for more than 25 years.23 However, several high-profile clinical trials of anti-beta-amyloid drugs for AD ended in disappointment. In addition to issues of timing of treatment with respect to the presumed pathological cascade, dosing of treatment, and adverse effects, it has been proposed that oligomers or fibrils might be insufficient or inappropriate targets.22
AD shares intermediate endophenotypes with other diseases. For example, amyloidosis and tauopathy are essential factors in many neurodegenerative diseases.24 Systematic identification of the underlying pathogenesis and disease modules within endophenotype networks across the genome, transcriptome, proteome, and human interactome, as opposed to pursuit of individual mutations (i.e., amyloid precursor protein [APP] mutations) or single mutated genes (such as SCNA), may thus serve as a foundation for predictive disease modules (Fig. 4). We have identified six endophenotype hypotheses in AD: (i) amyloidosis, (ii) tauopathy, (iii) neuroinflammation, (iv) mitochondrial dysfunction, (v) vascular dysfunction, and (vi) lysosomal dysfunction (Fig. 5). We acknowledged the existence of additional endophenotypes in AD, including oxidative stress,25 proteostasis26,27 and synaptic dysfunction28 that are also worthy of consideration, and which will be the subject of future investigation. In addition, we highlight new opportunities for endophenotype network-informed, in silico drug repurposing in AD.
Figure 4. A diagram illustrates drug repurposing approach under endophenotype networks of amyloid and tau in AD.
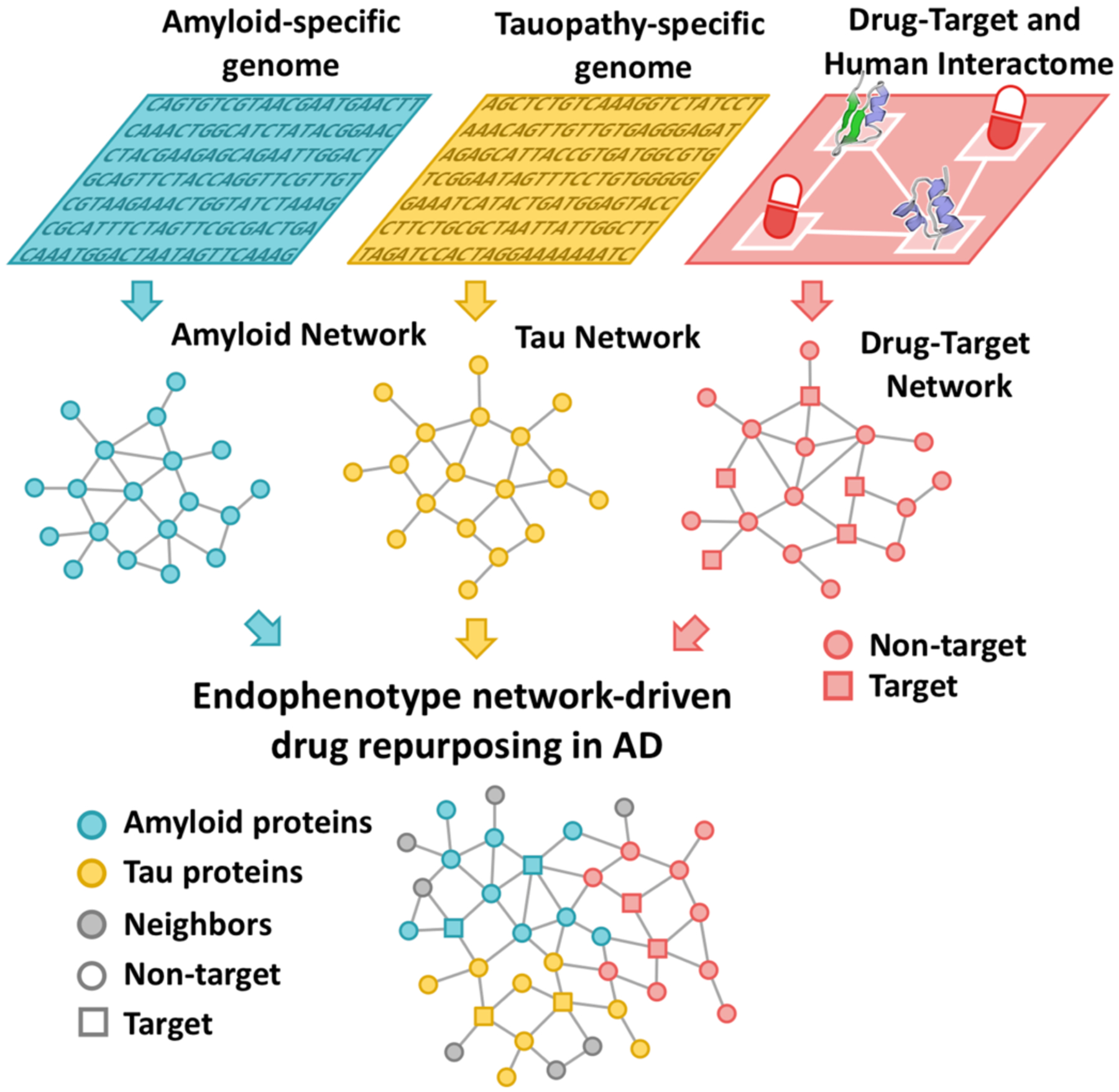
Endophenotype networks (i.e. amyloid or tau network) are generated using endophenotype-specific genetic or genomic data. Drug repurposing opportunities are revealed via integration of endophenotype network and drug-target network in human protein-protein interactome.
Figure 5. Six illustrated endophenotype hypotheses for Alzheimer’s disease.
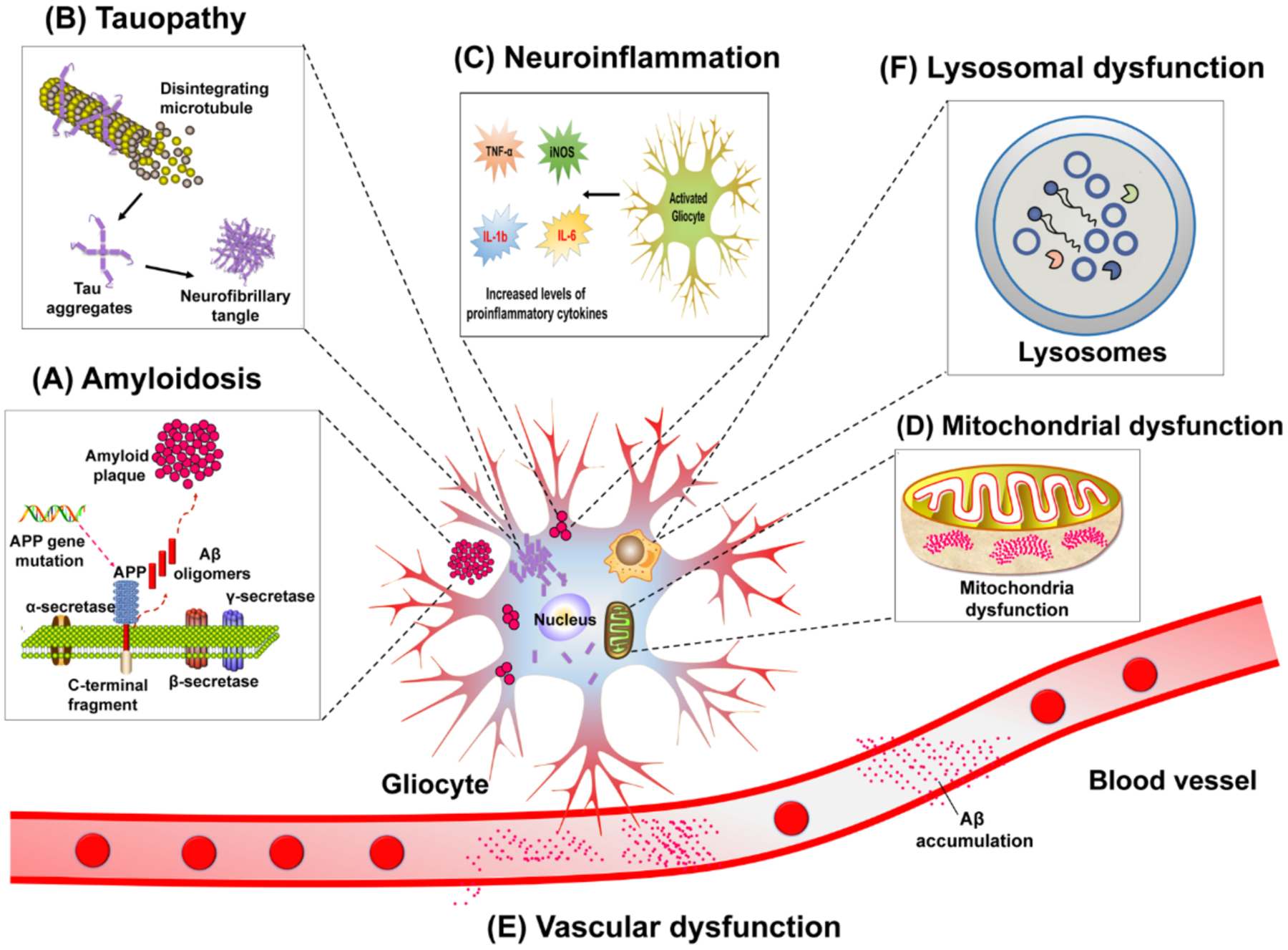
Six endophenotype hypotheses including amyloidosis (A), tauopathy (B), neuroinflammation (C), mitochondrial dysfunction (D), vascular dysfunction (E), and lysosomal dysfunction (F). TNFα: tumor necrosis factor alpha; iNOS: inducible nitric oxide synthase; IL-6: Interleukin 6; IL-1b: Interleukin 1 beta; APP: amyloid precursor protein.
2.1. Amyloidosis.
Amyloidosis proposes that an imbalance between production and clearance of brain amyloid β (Aβ) triggers a complex pathological reaction that leads to AD, including tau phosphorylation, tangle formation, synaptic loss, neuronal death, and cognitive impairment.23,29 This hypothesis is supported by evidence that nearly one percent of AD cases are caused by mutations of key genes involved in Aβ processing, including APP, presenilin-1 (PSEN1) and PSEN2.30,31 APP is cleaved by α-, β-, and γ-secretases via two pathways (non-amyloidogenic and amyloidogenic), which produce distinct peptides.32,33 In the non-amyloidogenic pathway, proteolysis of APP by α- and γ-secretases yields nonpathogenic fragments, including sAPPα. By contrast, the amyloidogenic pathway involves cleavage by β- and γ-secretases, leading to production of sAPPβ and Aβ. The production of Aβ monomers leads to the production of oligomers that are toxic to brain cells inducing synaptic damage, NFT formation, and neuronal death.34,35 Fbrils of Aβ oligomers aggregate into plaques.
Preclinical studies support the amyloid hypothesis.36,37 Amyloid-based therapeutic approaches, including decreasing Aβ production, inhibiting Aβ oligomerization or fibrillization, and accelerating Aβ degradation and clearance, reduce plaque deposition, reverse cognitive deficits, and improve other dystrophic processes in AD animal models.34,38 Although the “amyloid hypothesis” has dominated AD research for more than 20 years, no specifically-designed anti-amyloid agent is approved by the FDA. One possible reason is that amyloid may be a result, not cause, or is part of a complex network of causes of AD.39 The limited success of the anti-amyloid therapies is shifting the field focus on to other therapeutic opportunities. For example, recent novel therapeutic approaches to fostering neuronal survival have shown efficacy in TgF344-AD rats, a model of AD based on the amyloid and related inflammatory processes.40
2.2. Tauopathy
NFTs composed of hyperphosphorylated tau are another key neuropathological feature in patients with AD.41 Tau is encoded by a single gene, microtubule-associated protein tau (MAPT), on human chromosome 17, and missense mutations in the MAPT gene have been identified in inherited cases of the most common tauopathy, a key mechanism of AD.42,43 Tau is found mainly in axons of the neuron, where it binds to microtubules to stabilize their structure. Under pathological conditions, there is an aberrant increase of intraneuronal hyperphosphorylated tau in the cytosol. This blocks tau’s interaction with microtubules, leading to axonal flow disruption.44 Mechanistically, hyperphosphorylated tau polymerizes into paired helical filaments and straight filaments, preceding tau aggregation into NFTs.45 Inhibitors of kinases and activators of phosphatases that mediate tau-related processes are being considered for potential intervention. In addition, and in support of the network hypothesis, liraglutide, an approved glucagon-like peptide 1 receptor agonist, was recently shown to confer a constellation of beneficial effects on neuronal physiology that ultimately ameliorated AD-like tau pathology in mice models.46,47 Multiple anti-tau drug repurposing trials are under investigation, including pitavastatin,48 bexarotene,49 and minocycline.50
2.3. Neuroinflammation
Neuroinflammation refers to the inflammatory response in the central nervous system (CNS) secondary to neuronal insult.51 This response involves activation and recruitment of immune cells, particularly microglia and astrocytes, and it is regulated by production and release of cytokine signaling molecules.52 Increased levels of pro-inflammatory cytokines (e.g. tumor necrosis factor alpha [TNFα] and interleukin-1β [IL-1β]) have been observed in the serum and brain of AD patients in comparison to control subjects53, suggesting a significant role of glia-dependent neuroinflammation in AD progression. Microglia, the resident immune cells of the brain, represent 10%–15% of the cells of the brain and another potential target for AD. These cells play crucial roles, not only in regulating synaptic plasticity and remodeling neuronal circuits in the adult brain but also as a first line of immune defense in the face of brain injury and neurodegenerative disorders.54 In AD, microglia bind to soluble Aβ oligomers and Aβ fibrils via cell-surface receptors such as Toll-like receptors, which activate microglia and trigger the inflammatory reaction.55 Recently, it was demonstrated that microglial dysregulation in the TgF344 rat model of AD was linked to neurodegeneration and behavioral aberrations in a model of exposure to chlorpyrifos, an organophosphate pesticide that has been epidemiologically linked to increased incidence of AD.56 Microglia are important players in synaptic pruning during early brain development.57 Microglia mediate synapse loss in AD via a complement-dependent pathway,58 and C3 depletion reduced the number of phagocytic microglia, decreased synapse loss, and rescued cognitive deficits in a mouse model of AD.59 Interestingly, aberrant and sustained microglial activation was recently implicated as a potential etiologic factor in the increased risk of AD following occupational exposure to chlorpyrifos pesticide.56 Astrocytes, the most abundant type of glial cells, serve an essential role in neuronal survival and maintenance. Reactive astrocytes accumulate around senile plaques in postmortem brain tissue from subjects with AD.60 Like microglia, activated astrocytes release cytotoxic cytokines and interleukins, thereby exacerbating neuroinflammation. Accumulating evidence demonstrates that impairment in astrocyte reaction to external injury can trigger or exacerbate hyperphosphorylation of tau as well as Aβ pathologies, leading to neuronal dysfunction.61 Despite independent roles in neuroinflammation, there is growing evidence for intense interactions between microglia and astrocytes in AD.62 First, microglia-secreted cytokines can activate astrocytes, which leads to loss of physiological functions and become neuronal toxicity.63 In addition, the complement system is important in microglia-astrocyte crosstalk; astrocyte-secreted complement factor C3 interacts with microglial C3a receptor (C3aR), which mediates β-amyloid pathology and neuroinflammation in AD mouse models.64
Recent omics advances especially on single-cell or single-nucleus RNA-sequencing have also shed light on a major role of immune cells (microglia and astrocytes) in AD.65–67 For example, Michal et al. identified a population of disease-associated astrocytes (DAAs) in an AD mouse model via single-nucleus RNA sequencing. DAAs appeared at an early stage of AD and increased in abundance with disease progression.65 Using single-cell RNA-sequencing, Ido et al. found the disease-associated microglia type (DAM) from brains of the 5XFAD mouse model.66 They showed that DAMs were activated sequentially by TREM2-independent and -dependent mechanisms.66 Targeting neuroinflammation may offer future therapeutic or preventive strategies for AD. For example, perindopril, an angiotensin converting enzyme inhibitor, significantly attenuated cortical expression of the CD11b microglial markers and ameliorated microglial-mediated neuroinflammation in the 5XFAD mouse model.68 Currently, perindopril is undergoing a Phase II trial (NCT02085265) for treatment of hypertensive mild-moderate AD dementia patients.
Recent studies indicate a significant role for the brain-gut-microbiota axis in neuroinflammation in AD.69–71 Bacteria populating the gut microbiome excrete substantial lipopolysaccharides (LPSs) and amyloids. These LPSs and amyloids change the gut microbiota composition, increase permeability of the gut barrier, and propagate immune cell activation that augments inflammatory processes. This ultimately leads to impairment of the blood-brain barrier and promotes neuroinflammation.72 For example, porphyromonas gingivalis induces an inflammatory response in the liver through increased expression of the pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6) and interleukin 1 beta (IL-1β), which subsequently leads to neuroinflammation and cognitive impairment in mice.73 Another study reported that Bacteroides fragilis lipopolysaccharide exposure to human primary neurons served as a potent inducer of the pro-inflammatory transcription factor NF-kB (p50/p65) complex, a potent trigger for neuroinflammation.74 Thus, targeting neuroinflammation via modulation of gut microbiota may offer an approach to AD treatment. GV-971, a marine-derived oligosaccharide, targeting the gut microbiota with the aim of reducing neuroinflammation, was recently approved in China to improve cognition in mild to moderate AD dementia.75
2.4. Mitochondrial dysfunction
Mitochondrial homeostasis plays a key role in synaptic plasticity, learning, and memory,76 and Swerdlow and Khan have proposed a mitochondrial cascade hypothesis for AD.77 This hypothesis suggests that while regulation of mitochondrial baseline activity is genetically based, it can be influenced by environmental factors, such that the histologic changes associated with AD will be triggered once mitochondrial function falls below a critical threshold.78,79 This emphasizes that mitochondrial dysfunction lies at the core of neural degeneration, driven by metabolic abnormalities, and can lead to classic AD pathology. Mitochondrial dysfunction drives excessive production of reactive oxygen species (ROS), leading to the aberrant amyloidogenic processing of APP and subsequent formation of Aβ species and Aβ plaques.80 Aβ can initiate a destructive cycle to adversely affect mitochondrial function and integrity in vitro and in vivo.81 Mitochondrial dysfunction can trigger hyperphosphorylation of tau, microtubule depolymerization, and NFT-like pathology, while hyperphosphorylated tau can reciprocally further exacerbate mitochondrial dysfunction.76,82 Several mitochondria-targeting compounds with potential efficacy in AD have been identified, such as piracetam, simvastatin, and curcumin.83 However, preclinical data indicate beneficial effects, randomized clinical trials did not meet primary outcomes.83 Possible explanation for the high attrition rate of therapeutic candidates include introduction too late in the process or lack of efficacy at the target.
2.5. Vascular dysfunction
The vascular hypothesis of AD was first proposed by Torre in 1993.84 Vascular risk factors and normal aging lead to vascular dysfunction; with AD there is pathological accumulation of Aβ in the parenchyma and blood vessels that further potentiates neurodegeneration.85 This hypothesis is supported by subsequent epidemiological, pathological, and clinical studies.86–88 Recent in vivo studies on animals demonstrate that both Aβ and tau lead to blood vessel abnormalities and blood-brain barrier (BBB) breakdown,89,90 serving as an early biomarker of brain dysfunction independent of Aβ and tau.91 AD is strongly associated with several cardiovascular diseases, such as cerebrovascular disease,86 hypertension87 and atherosclerosis.88 Imaging studies have identified decreased blood flow prior to Aβ deposition in the brains of both AD mouse models and human patients.92 Several AD genetic risk factors are involved in vascular processes, including apolipoprotein E-4 (APOE4), PICALM, CLU, APP, and PSEN1.93 Vascular dementia, often coexisting with AD, has emerged as the leading cause of age-related cognitive impairment.94 Preventative and therapeutic prospects to repair vascular damage or to increase cerebral blood flow have shown potential in treating patients with vascular dementia.39,94
The vascular hypothesis raises the possibility that some cardiovascular drugs may be repurposed for AD. Nine FDA-approved cardiovascular drugs are currently in nonclinical studies or clinical trials for AD, including eight antihypertensive drugs and one drug for cerebral and peripheral vascular disease (Table 1 and Fig. 3). For example, angiotensin receptor blockers (ARBs) are reported to decrease the incidence of dementia.95 In vivo studies have revealed that valsartan, telmisartan, and losartan all reduce amyloid burden and alleviate spatial learning and memory deficits in AD mouse models.96–98 Carvedilol (an approved ARB for hypertension) significantly attenuated brain oligomeric Aβ levels and cognitive deterioration in two independent AD mouse models.99 However, a completed clinical trial (NCT01354444) suggested that carvedilol treatment was not significantly associated with improvement in AD. Several calcium channel blockers (e.g. nimodipine, nilvadipine, and isradipine) have been tested in nonclinical or clinical investigations for AD (Table 1). A combination of nimodipine with galantamine is currently being assessed in a clinical trial (NCT00814658) of patients with dementia. Another calcium channel blocker, isradipine, reduced the neurotoxic consequences of Aβ accumulation in vivo or in vitro.100,101 Tadalafil (Cialis) and Sildenafil (Viagra), two phosphodiesterase 5 (PDE5) inhibitors approved for erectile dysfunction and pulmonary hypertension, are being examined for treatment of dementia. Tadalaril is undergoing a Phase II trial (NCT02450253) to treat patients with vascular dementia, while sildenafil has been reported to improve cognitive function and memory and to reduce tau hyperphosphorylation in an AD mouse model.102 Other PDE types, such as PDE4 and PDE9, are considered promising targets for the treatment of AD.103,104 For example, a PDE4 inhibitor etazolate (NCT00880412) as well as a PDE9 inhibitor BI-409306 (NCT02240693) are being investigated in Clinical trials for treatment of AD. Collectively, FDA-approved cardiovascular drugs have potential for treating vascular dementia and may impact AD.
2.6. Lysosomal dysfunction
Numerous studies have shown substantial links between AD pathogenesis and lysosomal dysfunction.105–107 Lysosomes are membrane-bound organelles involved in the degradation and recycling of extracellular material via endocytosis and phagocytosis.105 The endo-lysosomal and autophagic networks (autophagy-lysosomal networks) are crucial for both the production of toxic Aβ and the clearance of misfolded or aggregated proteins.108 The lysosomal dysfunction in AD includes perturbed trafficking of lysosomal enzymes and accumulation of their substrates such as APP and its metabolites.106,109 Lysosomes, housing over 60 unique hydrolytic enzymes, are a prominent site of APP processing, Aβ uptake, and Aβ production.110 It has been reported that inhibition of lysosomal function leads to Aβ accumulation and memory deficits in AD mouse models.111 Enhancing lysosomal activity with the transcription factor EB (TFEB) reduces the amount of APP C-terminal fragments and Aβ production.109 These studies highlight the therapeutic potential of improving lysosomal activity implicated in AD pathogenesis. In addition, previous studies have shown that some lysosomal proteins are dysregulated in AD patients, including the lysosomal enzyme cathepsin D and lysosome-associated membrane protein 1 (LAMP1).106 Several genetic risk genes for late-onset AD (LOAD) are functionally related to lysosomal dysfunction, including APOE4, BIN1, CD2AP, PICALM, PLD3 and TREM2.108 Therapeutic intervention against AD by targeting lysosome dysfunction, while promising, has not yet entered a stage of being assessed in clinical trials.
2.7. Other endophenotypes
In addition to the six endophenotypes discussed, there are several additional endophenotypes in AD, including oxidative stress,25 proteostasis26,27 and synaptic dysfunction.28 Oxidative stress involves in multiple biological processes in AD, such as Aβ deposition, tau hyperphosphorylation, and tangle formation. The detailed molecular mechanisms and genetics of oxidative stress in AD were comprehensive described in a previous Review.25 Proteostasis refers to the dysfunction in protein homeostasis, leading to the accumulation of protein aggregates.26 Targeting endoplasmic reticulum acetylation to improve proteostasis in the secretory pathway can rescue AD in a mouse model.27 Synaptic dysfunction is closely related to cognitive decline in AD patients, as well as learning and memory deficits in various AD transgenic mice.28 Mechanically, Aβ induces dysfunction of neuronal synapses through distinct cell surface receptors, while tau protein leads to synaptic toxicity via pathological modification, localization, and propagation.28 In summary, elucidation of various endophenotypes, including oxidative stress, proteostasis, and synaptic dysfunction, will offer novel mechanisms and therapeutic targets for AD therapeutic development.
2.8. Deep endophenotyping
Since each endophenotype above has a contributing role in AD pathogenesis, it is unlikely that each endophenotype exists and functions independently of each other. Several lines of evidence have demonstrated synergistic roles for amyloid and tau in AD. Reducing endogenous tau levels prevents cognitive impairment in APP transgenic mice, without altering Aβ levels or plaque load.112 Kim et al. showed that the Aβ42/40 ratio, but not total Aβ amount, regulated tau pathology in a three-dimensional (3D) human neural cell culture system.113,114 An intense crosstalk exists between Aβ deposition, tau pathology and neuroinflammation. Blurton et al. found that elevated tau led to microglia-mediated Aβ clearance in an AD transgenic mouse model overexpressing hyperphosphorylated tau. Mechanically, microglia increased phagocytic capacity to accelerate the clearance of insoluble Aβ and decrease plaque load.115 Microglia are positively correlated with tau pathology, and activating microglia accelerates tau pathology in tau-transgenic mice.116 Activated microglia release substantial pro-inflammatory cytokines and further affect tau hyperphosphorylation in neurons.117 Depleting microglia blocked tau propagation in a virus-based mouse model. Specifically, microglia spread tau through exosome secretion, and inhibiting exosome synthesis reduced tau propagation in vitro and in vivo.118 In addition, mitochondrial dysfunction as well as vascular dysfunction have been strongly associated with tau pathology in AD.82,90 Collectively, deep endophenotyping approaches are urgently needed to identify the distinct endophenotypes for AD and their interactions.
In recent years, various bioinformatic or network-based approaches have provided tools for mechanistic examination of endophenotypes implicated in AD.19,119 For example, the Multi-objective Analyzer for Genetic Marker Acquisition (MAGMA) algorithm was utilized to explore whether genetic risk of AD (such as AD GWAS candidate genes) impact different endophenotypes.15 Fig. 6 presents a proof-of-concept of AD disease module (molecular determinants of disease pathobiology/physiology in the human interactome network).120 The 102 unique genes within the module include 31 amyloid genes, 11 tau genes, 12 amyloid & tau gene, and 48 other genes. This implies both amyloidosis and tauopathy are key components of the AD module. In addition, there is a significant gene overlap (12 overlapping genes, P = 1.82 × 10−27, Fisher’s exact test) between the two endophenotypes (amyloidosis and tauopathy). The above network analysis highlights the proof-of-concept of amyloid and tau endophenotypes as well as high inter-relatedness in AD pathogenesis.
Figure 6. Proof-of-concept of disease module for Alzheimer’s disease (AD).
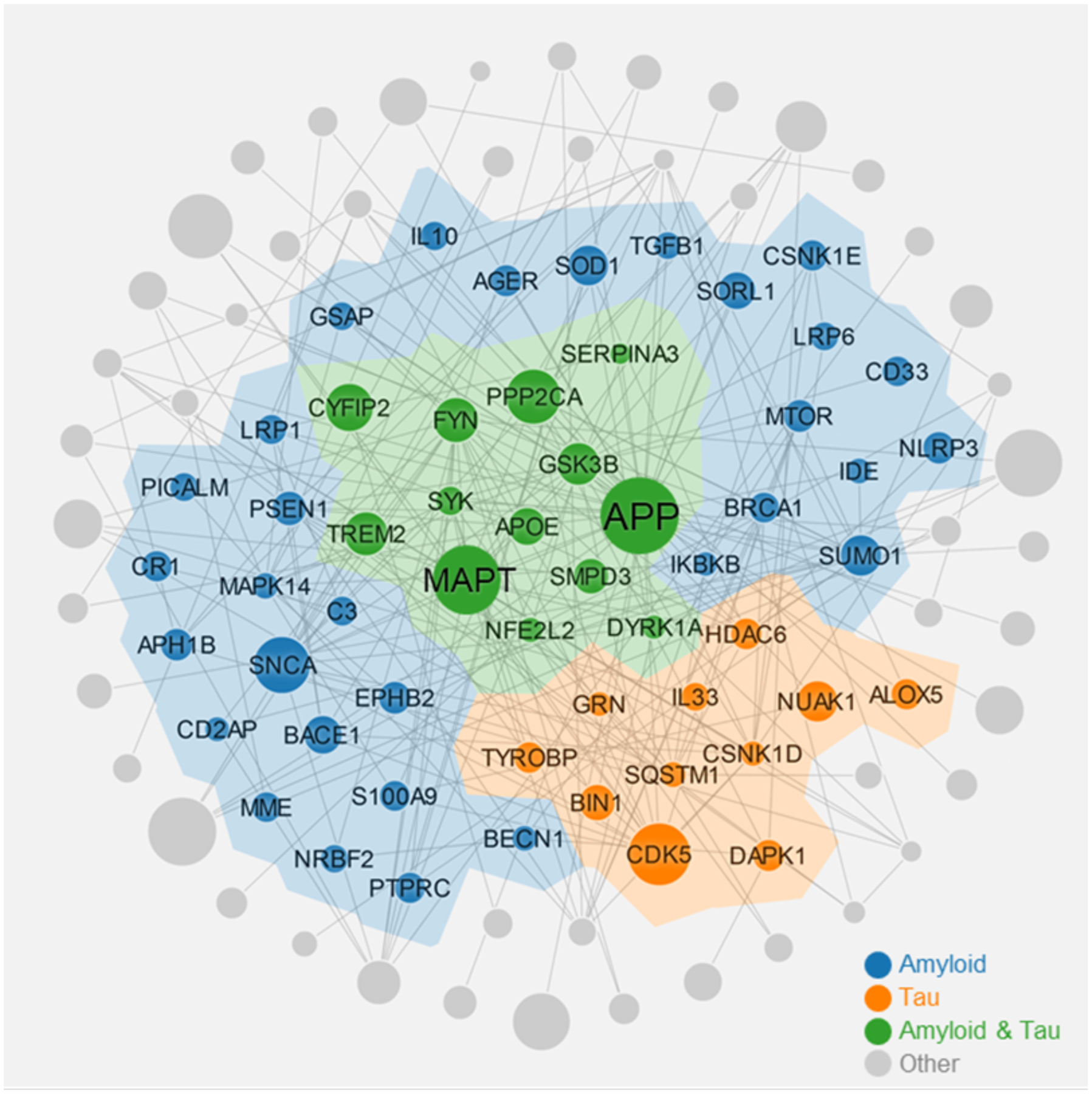
This AD module highlights several network features under the human protein-protein interactome: 1) both amyloidosis and tauopathy are the key components of AD module; 2) there is a significant gene overlap (12 overlapping genes, P = 1.82 × 10−27, Fisher’s exact test) between the two endophenotypes (amyloidosis and tauopathy). The AD module includes 227 protein-protein interactions (PPIs) (edges or links) connecting 102 unique proteins (including 31 amyloid genes, 11 tau genes, 12 amyloid & tau gene, and 48 other genes). AD module is generated based on 144 AD seed genes collected from the literatures.
2.9. Endophenotype impact on different stages of AD pathology
Emerging studies have suggested that different endophenotypes have differential impact across different stages of AD progression. For example, a recent study measured mitochondrial complex I availability quantitatively in the human brain, suggesting that mitochondrial dysfunction in the parahippocampus manifests in the early stages of AD.121 Hansson et al. found that both of neuroinflammation and cerebrovascular dysfunction were early events occurring at the presymptomatic stage of AD by measure of biomarkers in CSF.122 Hansson et al. used 18F-AV-1451 tau positron emission tomography (PET) and structural magnetic resonance imaging (MRI) to examine whether early- and late-onset AD showed differential, regional tau pathology and atrophy patterns.123 They found the early-onset AD group had greater uptake than the late-onset group in prefrontal, premotor, and inferior parietal cortex, exhibiting distinct tau PET retention patterns in different stages of AD.123
3. Omics Resources in AD
Databases that collect and store high-throughput experimental data are essential for development of network-based disease modules for better understanding of pathobiology and drug repurposing in AD. We describe five types of bioinformatics resources: (1) genomics (including GWAS, whole-genome/exome sequencing, targeted gene sequencing, and functional genomics); (2) transcriptomics (including microarrays and RNA-seq); (3) radiomics (brain imaging); (4) pharmacogenomics (including drug-target network and drug-induced transcriptome [drug-gene signatures]), and (5) interactomics (i.e., PPIs). The details of these resources are provided in Table 2.
Table 2.
Summary of bioinformatics resources for development of endophenotype-based models in Alzheimer’s disease.
| Names | Description | Website |
|---|---|---|
| Genomics resources for Alzheimer’s disease | ||
| NIAGADS124 GenomicsDB | Providing publicly available NIAGADS summary genome-wide association study (GWAS) statistics datasets for AD and related neuropathologies. | https://www.niagads.org/genomics/ |
| ADGC | GWAS studies to identify genetic variants associated with risk for LOAD | http://www.adgenetics.org/ |
| AlzGene125 | A collection of published AD GWAS studies, with random-effects meta-analyses for polymorphisms with genotype data. | http://www.alzgene.org/ |
| ADSP4 | Whole exome sequence (WES) and whole genome sequence (WGS) data covering over 62,100 individuals | https://www.niagads.org/adsp/ |
| AMP-AD126 | Genotype, WGS, WES, DNA and methylation assay covering over 15,000 individuals. | https://www.synapse.org/#!Synapse:syn2580853/wiki/409840 |
| AlzData133 | High throughput omics data collection of AD, including expression profiles/differential expression results of individual studies. | www.alzdata.org |
| GTEx127 | An atlas of the tissue-specific gene expression and regulation generated from 11,688 samples from 714 donors across 53 tissues. | http://www.gtexportal.org/http://thebiogrid.org |
| ENCODE128 | 1,678,800 sncRNA loci from 802 data sets covering 185 tissues or cell types. | https://www.encodeproject.org |
| Roadmap Epigenomics129 | 2,804 genome-wide datasets, including 1,821 histone modification datasets, 360 DNase datasets, 277 DNA methylation datasets, and 166 RNA-Seq datasets | http://www.roadmapepigenomics.org |
| FANTOM5130 | A comprehensive map of regulatory elements in the human genome. | http://fantom.gsc.riken.jp/5/ |
| DASHR131 | 1,678,800 sncRNA loci from 802 data sets covering 185 tissues or cell types. | http://dashr2.lisanwanglab.org/ |
| Transcriptomics | ||
| AMP-AD126 | Microarray and RNA-sequencing data from individuals. | https://www.synapse.org/#!Synapse:syn2580853/wiki/409840 |
| ADGC | Microarray and RNA-sequencing data from individuals. | http://www.adgenetics.org/ |
| MOUSEAC134 | Providing RNASeq and microarray differential expression gene in four lines of “amyloid” transgenic mice (mutant human APP, PSEN1, or APP/PSEN1) and “TAU” transgenic mice (mutant human MAPT gene) | http://www.mouseac.org |
| Tsai et al135 | Single-cell transcriptomics analysis cross six major brain cell-types from prefrontal cortex of 48 individuals with varying degrees of AD pathology | |
| Polo et al136 | Single-cell transcriptomic analysis of entorhinal cortex samples from control and Alzheimer’s disease brains (n = 6 per group) across six cell types | http://adsn.ddnetbio.com |
| Radomics (imaging data) | ||
| ADNI5 | Providing MRI and PET image data covering 483 elderly controls, 1,001 MCI and 437 AD patients. | http://adni.loni.usc.edu/ |
| Pharmacogenomics resources | ||
| DrugBank142 | A comprehensive database with the detailed drug and target information. | https://www.drugbank.ca/ |
| DrugCentral143 | A drug compendium containing drug information approved by FDA and other regulatory agencies. | http://drugcentral.org |
| PDSP155 | A standardized database for quantitative interaction of drugs with human receptors | http://pdspdb.unc.edu/ |
| DGIdb144 | A database containing drug-gene interactions and the druggable genome | http://www.dgidb.org/ |
| TTD145 | Information on the therapeutic targets. | http://bidd.nus.edu.sg/group/ttd |
| DSigDB146 | A resource that connects 17 389 unique compounds (1202 FDA-approved drugs) and their target genes. | http://tanlab.ucdenver.edu/DSigDB/DSigDBv1.0/ |
| CMap (v 2.0)147 | Gene-expression signatures to connect small molecules, genes, and disease | https://www.broadinstitute.org/cmap/ |
| LINCs148 | Library of integrated network-based cellular signatures | http://www.maayanlab.net/LINCS/LCB/ |
| DRUG-seq149 | A high-throughput platform for drug discovery, captures transcriptional changes profiling 433 compounds across 8 doses. | NA |
| open TG-GATEs150 | A large-scale toxicogenomics database | toxico.nibio.go.jp/english/index.html |
| Drug Gene Budger (DGB)151 | Ranking drugs to modulate a specific gene based on transcriptomic signatures | http://amp.pharm.mssm.edu/DGB/ |
| Mantra 2.0152 | A collaborative resource for drug mode of action and repurposing using transcriptional profiles | http://mantra.tigem.it/ |
| NFFinder153 | Identify connections between drugs, diseases and phenotype based on transcriptomic data. | http://nffinder.cnb.csic.es/ |
| Interactomics: human protein-protein interactome | ||
| BioGRID157 | An interaction repository containing 1,658,808 protein and genetic interactions | http://thebiogrid.org |
| HPRD158 | HPRD Release 9 contains 41,327 human PPIs | http://www.hprd.org |
| Interactome3D159 | A database for the structural annotation and modeling of PPIs | http://interactome3d.irbbarcelona.org |
| STRING160 | Functional protein association networks database (>2000 million PPIs) | http://string-db.org |
| MINT161 | Experimentally verified interaction database (126,712 PPIs) mined from refereed journals | http://mint.bio.uniroma2.it/mint |
| IntAct162 | A molecular interaction database containing over 870,000 PPIs from literature curation. | https://www.ebi.ac.uk/intact/ |
3.1. Genomics
Genome-wide association studies (GWAS).
The Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS), funded by the National Institute on Aging (NIA), is a national genetic and genomic data repository for AD. To date, NIAGADS has collected 64 datasets including 84,220 samples across 12 data types (accessed in May, 2020). NIAGADS GenomicsDB, is a sub database that provides publicly available NIAGADS summary GWAS statistics datasets for AD.124 The Alzheimer’s Disease Genetics Consortium (ADGC) is funded by NIA to conduct GWAS studies for identification of risk genes in LOAD. AlzGene is a database that comprehensively catalogs all GWAS studies in the field of AD. AlzGene covers 1,395 studies, 695 genes, 2,973 polymorphisms, and 320 meta-analyses.125
Whole-exome/genome sequencing studies.
The Alzheimer’s Disease Sequencing Project (ADSP), launched in 2012, has sequenced and analyzed genomes to identify a wide range of AD risk or protective gene variants including whole-exome sequencing (WES) data from brain tissues (i.e., hippocampus and prefrontal cortex) from nearly 11,000 individuals and whole-genome sequencing (WGS) data of brain tissues from nearly 600 individuals.4 The Accelerating Medicines Partnership - Alzheimer’s Disease (AMP-AD) concentrates on identifying novel, clinically relevant therapeutic targets and discovering biomarkers to validate existing therapeutic targets. The AMP-AD data and analysis results are stored in the AMP-AD Knowledge Portal, including various genomic, metabolomic and proteomic data from over 15,000 individuals.126
Functional genomics.
Functional genomics databases, such as The Genotype-Tissue Expression (GTEx),127 Encyclopedia of DNA Elements (ENCODE) Consortium,128 Roadmap Epigenomics,129 FANTOM5,130 and DASHR,131 provide genotype-tissue expression, molecular regulatory profiling, and epigenetic information to examine gene regulatory networks in human diseases including AD.132 The GETx (version 7) stores the tissue-specific gene expression and regulation data generated from 11,688 samples from 714 donors across 53 tissues (including 13 brain regions).127 The ENCODE Consortium aims to build a comprehensive constituent list of functional elements in the human cell for studying the epigenomic signatures of cells.128 The National Institutes of Health (NIH) Roadmap Epigenomics Consortium was launched to establish global maps of regulatory elements based on integrative analysis of 111 reference human epigenomes.129 The Human Epigenome Atlas (Release 9), a product of the NIH Roadmap Epigenomics Consortium, has thus far collected 2,804 genome-wide datasets, including 1,821 histone modification datasets, 360 DNase datasets, 277 DNA methylation datasets, and 166 RNA-Seq datasets.129 DASHR v2.0 is an integrative database of small human noncoding RNAs with detailed annotations in human tissues and cell types. Small human noncoding RNAs regulate diverse tissue-specific cellular processes by associating with transcription factor complexes or binding to micro RNAs. As of May of 2020, DASHR contains 1,678,800 small noncoding RNA loci from 802 data sets covering 185 tissues or cell types.131
3.2. Transcriptomics
AlzData, a high throughput data collection database, analyzed 20 original microarray datasets from four brain regions in 684 AD patients and 562 controls.133 The regions include entorhinal cortex, hippocampus, temporal cortex, and frontal cortex. In 2015, Matarin et al. launched a Web server (MOUSEAC) that stores both RNA-seq and microarray data across three brain regions (hippocampus, cortex, and cerebellum) in five lines of AD model transgenic mice. These mouse models include four amyloid transgenics (mutant human APP, PSEN1, or APP/PSEN1) and one tau transgenic (mutant human MAPT gene).134 Tsai et al. performed large-scale single-cell transcriptomic analysis across six major brain cell-types from prefrontal cortex of 48 individuals with varying degrees of AD pathology. The six types include excitatory neurons, inhibitory neurons, astrocytes, oligodendrocytes, microglia, and oligodendrocyte progenitor cells.135 This unique resource provided a comprehensive cellular-level description of the landscape of transcriptional alterations of AD pathology. Importantly, this single-cell study revealed cell type-specific and shared gene expression perturbations, disease-associated cellular subpopulations, and sex-biased transcriptional patterns.135 Recently, Polo et al. generated a single-cell atlas of entorhinal cortex from patients with AD across six cell types, including microglia, astrocytes, neurons, oligodendrocyte progenitor cells, oligodendrocytes, and endothelial cells. This resource further contributes to better understanding of cellular heterogeneity and defining functional changes at single-cell resolution for AD brain.136
3.3. Radiomics
Recent advances in imaging techniques, such as magnetic resonance imaging (MRI) and PET, have greatly increased our knowledge of AD pathobiology.137,138 ADNI is a longitudinal multicenter study, funded in 2004, with the goal of collecting, validating and utilizing data, including MRI, PET images, and cerebrospinal fluid (CSF)/blood biomarkers as clinical endpoints to evaluate drug responses in AD trials. According to the statistics in the AD drug development pipeline report of 2019, the most common biomarkers as outcome measures in Phase II and Phase III clinical trials included CSF amyloid, CSF tau, volumetric MRI, and amyloid PET.139 The newest ADNI update (version3) contains imaging data for 483 elderly controls, 1,001 subjects with mild cognitive impairment (MCI), and 437 subjects with AD dementia, enabling the sharing of data among researchers around the world.5,140,141
3.4. Pharmacogenomics
Pharmacogenomics resources, including physical drug-target interactions and functional drug-induced gene signatures, offer new insights into understanding molecular mechanism of drugs and also provide resources for in silico drug repurposing in AD.
Physical drug-target interactions.
There are several common physical drug-target interaction databases, such as DrugBank,142 DrugCentral,143 DGIdb,144 and TTD.145 DrugBank is a comprehensive database with detailed drug and target information. As of May of 2020, the current version of DrugBank 5.1.6 includes 13,543 drug entries containing 4,002 approved drugs (2,630 small molecules), 131 nutraceuticals and over 6,358 experimental drugs.142 DrugCentral, an open-access online drug compendium, integrates structure, target and indication information for approved drugs. DrugCentral (2018 release) contains 4,531 approved drugs, including 2,094 FDA-approved agents and 2,437 drugs approved by other regulatory agencies.143
Functional drug-gene signatures.
Functional drug-induced gene signatures, such as provided by DSigDB,146 CMap (v2.0),147 LINCS,148 DRUG-Seq,149 open TG-GATEs,150 Drug Gene Budger (DGB),151 Mantra 2.0,152 and NFFinder,153 provide connections among diseases, genetic perturbation, and drug actions. These databases are utilized to identify the molecular mechanism of given compounds for elucidation of side effects and drug repurposing. Connectivity Map (CMap2.0) is a reference collection (>7,000) of gene-expression profiles from four types of cancer cell lines treated with 1,309 bioactive small molecules.147 Given the limited number of cancer cell lines, the Library of Integrated Network-based Cellular Signatures (LINCS) project produced over 1 million gene expression profiles of more than 10,000 compounds tested in nearly 80 different cancer cell lines. This large-scale drug-induced expression data repository aims to assist investigators to better understand human disease and advance new therapies.148 DRUG-Seq, a high-throughput platform for drug discovery, captures transcriptional changes profiling 433 compounds across 8 doses with various chemical and genetic perturbations and can be used for exploring molecular mechanisms for novel and existing drugs.149 NFFinder is an online server for searching similar transcriptomic profiles in the context of drug repurposing. NFFinder integrates expression data from drug-gene signature resources, such as CMap and DrugMatrix,154 to connect relationships among drugs, diseases and phenotypes of interest.153 Drug Signatures Database (DSigDB) collects drug and small molecule-related gene sets based on quantitative inhibition and/or drug-induced gene expression profile data. As of February of 2019, DSigDB contains 22,527 gene sets, connecting 17,389 unique compounds (1,202 FDA-approved drugs) and their target genes.146
Several limitations of current pharmacogenomics resources should be acknowledged. First, most of the physical binding data from public databases, such as DrugBank,142 are tested by in vitro assays in engineered cells. It neglects the fact that drug concentrations in the human body are affected by pharmacokinetics/pharmacodynamics (PK/PD) properties. Second, high concentrations (10 uM) of compounds tested in the LINCS database are far beyond achievable ranges in human patients. Standardized databases for quantitative physical interaction of drugs with human receptors, such as the Psych-Active Drug Screening Program,155 may provide more useful drug-target resources. Finally, CMap as well as LINCS are functional drug-induced gene signature databases in cancer cell lines, not cell lines biologically relevant to AD.147,148
3.5. Interactomics: The human protein-protein interactome
Human PPIs play a crucial role in understanding human diseases and drug responses19,20 from a cellular network perspective.156 Recent human interactome studies have suggested novel network-based drug-disease or drug-drug relationships by assembling binary, physical PPIs from multiple resources.19,20 Several well-known PPI databases, including BioGRID,157 HPRD,158 Interactome3D,159 STRING,160 MINT,161 and IntAct,162 offer high-quality experimental, computationally-predicted, or literature-curated PPIs. BioGRID, a biomedical literature-curated database, consists of 1,814,182 protein and genetic interactions across 74 organisms (including 573,910 interactions in Homo sapiens, version 3.5.185) by extracting data from 72,164 publications.157 STRING assembles known and computationally predicted PPIs for a large number of organisms. STRING (v11.0) collected 97,587,721 PPIs with quantitative confidence scores higher than 0.7 (high confidence) across 2,031 organisms (accessed in February of 2019).160 IntAct is an open source database system for molecular interaction data, which curates PPI data from the literature and integrates data from 11 common PPI databases. Currently, it contains 1,050,463 PPIs connecting 115,486 proteins from 21,346 publications (accessed in May of 2020).162
3.6. Multi-omics data integration
In addition to the databases providing omics resources for AD, data analysis using bioinformatics approaches or tools remains indispensable for developing disease modules. Weighted gene co-expression network analysis (WGCNA), an R analytical package, provides systems-level insights into high-throughput omics data such as microarray or RNA-seq datasets.163 The package can be used for describing the correlation patterns among genes across omics samples. Allan et al. applied WGCNA to identify distinct modules of co-expressed microglial genes that are related to AD pathology using existing transcriptomic microarray datasets. They predicted distinct DAM sub-populations and validated them in AD mouse models. Finally, they performed a connectivity map (CMAP) analysis to prioritize selective modulators for DAM profiles using drug-induced transcriptional responses in human cell lines.164 Genome-wide Positioning Systems network (GPSnet) is a network-based algorithm for in silico drug repurposing. It includes two main components: (i) disease module identification from individual patient’s DNA and RNA sequencing profiles, and (ii) network-based drug repurposing.165 In addition to WGCNA and GPSnet, another network approach, termed the largest connected component (LCC) approach, can be used to build disease modules. The significance of a module is evaluated by counting the number of permutations greater than the number of observed LCC formed by the randomly selected proteins with a similar connectivity distribution in the human interactome network.19 Taken together, such computational methodologies offer invaluable tools for disease module generation in AD.
4. Computational Approaches for Alzheimer’s Drug Repurposing
The massive growth of bioinformatics resources creates new opportunities to apply computational approaches for drug repurposing in AD. Over the last decade, several types of computational approaches, including structure-based (e.g. molecular docking),166 ligand-based (e.g. chemical similarity),167,168 bioinformatics-based,169 network-based, and system biology-based methods, have shown potential for in silico drug repurposing in multiple complex diseases, including AD. Details about in silico drug repurposing can be found in several recent reviews.166,170–173 In this review, we present three types of approaches with potential of exploiting the wealth of publicly available multi-omics data in pursuit of AD drug repurposing: (i) Genetics-based, (ii) omics-based, and (iii) network-based approaches.
4.1. Genetics-based approaches
More than 40 GWAS studies have been published in AD, which has identified over 40 AD risk-associated loci.2,174,175 For example, a recent GWAS study identified 29 risk loci (including 13 novel loci) for AD, using meta-analysis of 71,880 cases and 383,378 controls.8 A systematic analysis of human genetic evidence suggested that GWAS data could greatly contribute to novel therapeutic targets and development of new drugs, including identifying opportunities for drug repurposing.176,177 Based on these observations, several studies have aimed to use GWAS findings to identify novel therapeutic targets for drug repurposing in AD. Kwok et al. systematically examined the relationship between existing drug targets and GWAS closest genes in AD and assessed whether these genes were targeted by existing drugs that could be repurposed for AD. They found several drugs (i.e. afatinib, glatiramer, and vandetanib) that target these GWAS-derived genes, including several immunosuppressive disease-modifying anti-rheumatic and multiple sclerosis drugs.178 In 2017, So et al. compared transcriptomes imputed from GWAS data with drug-induced gene expression profiles from CMap database.179 By examining drugs in CMap showing opposite patterns of expression to diseases in GWAS, they identified repurposable drugs for seven psychiatric disorders.179 They further showed that some predictions were validated by preclinical or clinical evidence.179 Recently, our group presented an integrated, network-based methodology to rapidly identify drug targets from GWAS findings and multi-omics data integration for AD. We found that one of the agents identified --- pioglitazone (an approved anti-diabetes drug) --- is significantly associated with decreased risk of AD in a retrospective case-control study.180 However, several limitations exist for current GWAS-based drug repurposing. First, given that many genome-wide significant loci are in noncoding regions, only a small portion of druggable genes could be mapped by GWAS analysis181 and the closest-based GWAS genes cannot represent AD-risk genes by regulatory roles of non-coding variants.182 Second, catalogs of genetic variants from GWAS that influence human disease traits are incomplete. Identification of high-risk genes from GWAS data by uniquely integrating multi-omics data and human interactome networks would offer new candidate targets for AD drug repurposing.183
4.2. Omics-based approaches
Owing to the extensive application of high-throughput, next-generation sequencing techniques, massive omics data from AD have accumulated, including transcriptomics, metabolomics, and proteomics. These complementary omics data shed new insights on the pathology expression in AD, and also offer an alternative approach to AD drug discovery.
Transcriptomics.
Transcriptomics measures the expression profiles of genes using RNA-seq or microarrays. Reversal of gene expression signatures that may translate into clinical benefit offer a means of discovering agents with the potential for drug repurposing.184,185 Several large scale transcriptome profiling studies have revealed the transcriptome landscape in AD, such as the UK Brain Expression Consortium and the Religious Order Study (ROS).186–189 In addition, Vargas et al. built a transcriptional regulatory network and conducted master regulator analysis to identify potential molecular targets in AD.190 By integrating drug-gene signatures from the CMap database with transcriptomic data of human hippocampus in AD patients and controls, they identified six FDA-approved drugs with potential for therapeutic effect in AD, including cefuroxime, cyproterone, dydrogesterone, metrizamide, trimethadione, and vorinostat.190 Siavelis et al. also integrated five AD-related microarray data sets from human hippocampus to identify differentially expressed genes using three different approaches. Based on these disease signatures, they searched drug candidates to reverse disease profiles using four drug repurposing tools, including CMap, SPIEDw, sscMap, and LINCSL1000. This effort generated a list of 27 potential anti-AD drug candidates (i.e. protein kinase C inhibitors and glycogen synthase kinase 3 inhibitors).191
Proteomics.
Compared to transcriptomics-based studies, proteomics offers more comprehensive characterization of molecular pathways in AD. Multiple proteomics studies in patients or mouse models have uncovered potential AD molecular networks.192–195 For example, Xiong et al. compared the proteomic profiles of amyloid plaques from AD patients, controls, and APP/PS1 mouse models. They found distinct protein expression changes among these three types of plaques.192 Wang et al. performed a proteome-wide screening approach to identify tau-interacting proteins, and validated Otub1 as a tau deubiquitinating enzyme in vitro and in vivo, implying the potential application of Otub1 inhibitors in tauopathies and AD.196 The analysis of the proteomics and of the sharing variations identified by proteomics could reveal pathological mechanisms of AD, which might provide novel therapeutic targets for drug repurposing.
Multi-omics.
Multi-omics data integration offers multiple molecular layers to describe AD pathogenesis compared to single omics approaches.197 Petyuk et al. integrated DNA variation, RNA expression, and proteome profiles to prioritize key targets in LOAD.198 An integrative analysis showed that heat shock protein family A member 2 (HSPA2) played a key role, validated in two cell lines.198 Swarup et al. proposed an integrative multi-omics approach to reveal the disease mechanisms in frontotemporal dementia. They identified two disease-relevant mRNA modules, including a neurodegeneration-associated synaptic [NAS] module and a neurodegeneration-associated inflammatory [NAI] module, by uniquely integrating RNA-seq and proteomics data from mouse models and AD patients. Specifically, they identified miR-203 as a hub driver of an NAS module in vitro and in vivo and showed that two histone deacetylase inhibitors (scriptaid and vorinostat) ameliorate miR-203-induced cell death in vitro.16 Zhang et al. collected a list of 524 AD-associated targets via integration of genomics, epigenomics, proteomics and metabolomics data.199 Among 19 prioritized protein targets linked to 92 existing drugs, they found that myeloid cell surface antigen CD33 (CD33) and macrophage migration inhibitory factor (MIF) were prioritized as the two highest candidate targets with seven existing drugs relevant to AD.199
4.3. Network-based approaches
Drug discovery in the modern era has become a highly integrated, systems pipeline, requiring complementary multi-omics and computational methods.200 Systematic identification and characterization of “driver interaction (edge) perturbations”, starting from genomes, exomes, transcriptomes, and the human interactome, can serve as a foundation for generating predictive, and eventually dynamic, disease network AD models.
Novel approaches, such as network-based drug-disease proximity that shed light on the relationship between drugs and diseases,19 can serve as a useful tool for efficiently screening potential new AD indications for currently marketed drugs. For example, network proximity quantifying the interplay between disease genes and drug targets in the human protein-protein interactome reveals hundreds of new drug-disease associations in various complex diseases.19 This approach aims to calculate the closest distance measured by the average shortest path length of all the disease gene nodes to the drug target nodes in the human interactome. Since this approach does not take drug affinities for the different targets into account, its performance may be further improved by utilizing drug affinity information in future studies.201 This approach can also be applied to AD drug repurposing. Here we highlight an endophenotype network-based drug repurposing infrastructure for AD drug repurposing (Fig. 7). This network framework can integrate multi-omics data (e.g. genomics, transcriptomics, proteomics, and metabolomics), drug-gene interactome/signatures, along with preclinical or patient validation. The critical infrastructure consists of four key domains: (1) an AD endophenotype-based disease module built from multi-omics data (Fig. 7A); (2) experimental or clinical validation of disease modules (Fig. 7B); (3) repurposable drugs prioritized via network proximity analysis (Fig. 7C); (4) in vitro/vivo preclinical validation as well as large scale longitudinal patient (i.e., electronic health records) data validation (Fig. 7D). Cheng et al. demonstrated that an integration of network proximity-based approaches and state-of-the-art pharmacoepidemiologic methods can facilitate drug repurposing in cardiovascular disease.19 They observed that hydroxychloroquine was associated with 24% reduced risk of coronary artery disease compared with leflunomide using large-scale patient data, an observation supported by mechanistic in vitro data.19
Figure 7. An endophenotype network-based drug repurposing infrastructure for AD.
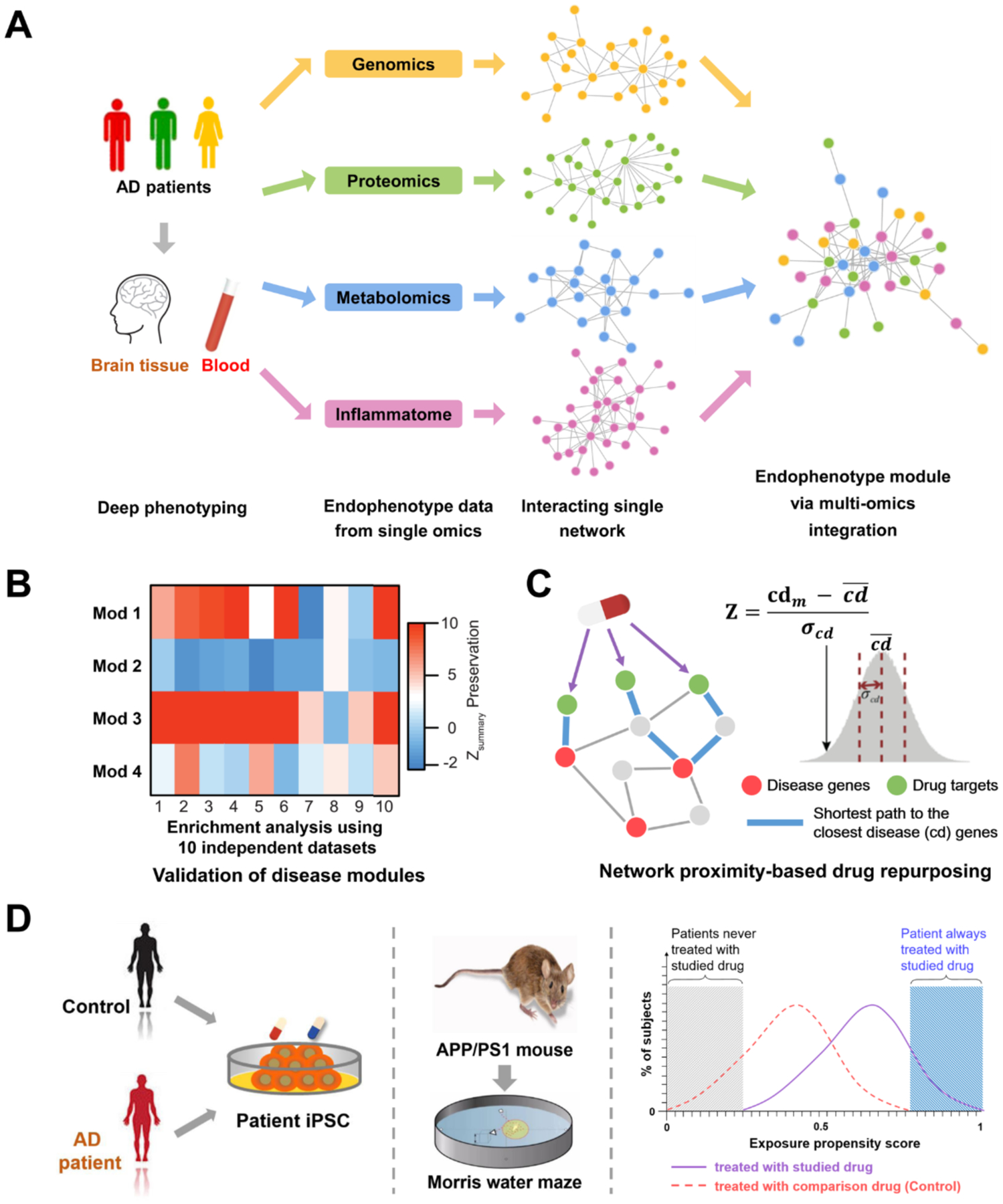
The entire infrastructure is consisted: (A) develop AD endophenotype-based disease module from multi-omics data; (B) experimental or clinical validation of disease modules; (C) in silico prediction of highly repurposable drugs via network proximity analysis; (D) in vitro/in vivo preclinical validation, and population-based validation of in silico drug repurposing.
Network-based approaches thus show potential in drug repurposing for multiple complex diseases, including AD. Several questions that form the basis of the endophenotype-based disease module warrant addressing in the future. These include: 1) how to build the detailed maps of identified and interpreted AD mutations from genomics data; 2) how to rapidly identify data-driven therapeutic targets for personalized diagnosis and treatment from brain-specific, quantitative network analysis by utilizing multi-omics data rather than single omics approaches.
There remain several challenges for CNS drug discovery and development based on current approaches. The CNS drug development pipeline should consider the following factors: quantitative and validated biomarkers, clinical endpoints, complex PK/PD relationships, and the complex interactions of different brain circuits.202 To address these issues, other approaches such as quantitative systems pharmacology (QSP) are emerging as new computational strategies in CNS drug discovery. QSP, which combines systems biology and PK/PD, offers a mechanism-based computer model of relevant neuronal circuits for CNS diseases.202,203 It can be used for evaluating drug effects and optimizing the design of clinical trials. Collectively, the computational strategies above show promise in exploiting multi-omics data in pursuit of AD drug repurposing.
5. Validation of in silico Alzheimer’s Drug Repurposing
Once repurposed drug candidates are identified, experimental validation is required to support their efficacy before human clinical trials. Several recent reviews summarize the pathological features and limitations of the major experimental models of AD.204–207 Here, we briefly discuss three types of experimental validation of drug efficacy in AD: (i) in vitro assays (i.e., human induced pluripotent stem cells [iPSCs]), (ii) in vivo assays (including transgenic mouse/rat models and other animal models), and (iii) population-based retrospective case-control studies using state-of-the-art pharmacoepidemiologic designs.
5.1. iPSC neuron models
iPSCs, generated from patient stem cell-derived neurons to recapitulate key aspects of AD pathology, are increasingly used to study AD.208 Accumulating evidence from iPSC lines from familial AD (FAD) and sporadic AD patient-derived neurons has revealed overproduction of Aβ and tau hyperphosphorylation compared to iPSCs derived from age-matched non dementia controls.209,210 iPSC neuron models may thus offer an applicable in vitro assay for AD drug discovery. For example, Kondo et al. screened and evaluated therapeutic candidates for AD using human iPSC-derived neurons, resulting in 27 anti-Aβ hits. They further identified a synergistic combination of bromocriptine, cromolyn, and topiramate as an anti-Aβ therapeutic cocktail.211 However, there are several drawbacks to the iPSC approach. First, to characterize an AD associated phenotype, these cell lines may have to be aged, which is difficult to recapitulate in vitro using differentiated neurons. Second, iPSC-neuron models do not represent the complex brain condition in vivo, even though recent advances in 3D human neural cell culture systems allow more physiological interactions between neurons and glia.114
5.2. Invertebrate animal models
Invertebrate animal models, such as Drosophila and Caenorhabditis elegans (C. elegans), have been used to recapitulate AD pathology after genetic manipulation by expressing human transgenes of APP, Aβ and/or tau.212,213 For example, Favrin et al. constructed an AD drosophila (fruit fly) model which overexpresses the human Aβ42 peptide. Transcriptome analysis of AD and control flies found 712 differentially expressed genes involved in oxidative stress and innate immunity.214 Transgenic invertebrate models are widely used in high-throughput genetic or drug screens. Shim et al. specifically employed tauP301L and tauR406W as models of tau-induced neurotoxicity in drosophila for screening compounds for the ability to ameliorate tau-induced neurotoxicity in vivo, and identified a known protein kinase C alpha (PKCα) inhibitor (Ro 31–8220), in vitro and in vivo.215 Treatment with Ro 31–8220 reversed tau-induced memory impairment and improved the motor functions in this fly model.215
C. elegans is another invertebrate model for target identification and drug screening against AD. He et al. used the transgenic C. elegans strain CL2355 expressing neural Aβ to assess the neuroprotective activity of seven 2-arylethenylquinoline derivatives. They observed that two of them (4b1 and 4c2) reduced Aβ-induced stress, inhibited the expression of neural Aβ monomers and toxic oligomers, and protected against cognitive impairment in a C. elegans AD model.216 The main advantages of using invertebrates as AD models is their easy handling, low cost, and short life span. However, results from invertebrate models need to be further validated in animal models or patient data.
5.3. Transgenic mouse/rat models
Rodents are mammals and can be identified by their teeth, including mouse, rat, squirrel, beaver, and so on. Rodents (especially mouse and rat) have been the most widely used models in biomedical research.206,217 The most commonly used experimental models are transgenic rodents that overexpress human mutations (such as APP and PSEN1), which lead to formation of amyloid plaque, NFTs, or both. In total, Alzforum lists 177 AD animal models, including 169 transgenic mouse models and 8 transgenic rat models.218 Sixty-two of the transgenic mouse models for AD overexpress human APP mutants and develop Aβ aggregation. For example, the PS2 APP mouse is an AD transgenic model harboring APP KM670/671NL and PSEN2 N141I mutations, which develops severe cerebral amyloidosis in discrete brain regions.219 However, these APP transgenic mice do not develop NFTs. To address this, several transgenic models that overexpress mutated human tau protein have been developed. For example, the rTg4510 mouse is a tauopathy model that overexpresses the human tau P301L mutation associated with familial frontotemporal dementia; the mice develop progressive age-related NFTs, neuronal loss, and behavioral impairments.220 Recently, Kim et al. reported a novel AD model, the AD-like pathology with amyloid and NFTs (ADLPAPT) mouse. This mouse harbors three human transgenes, including APP, PSEN1, and MAPT, with six mutations. ADLPAPT mice display neuronal death, an accelerated NFT pathology, amyloid plaques and memory deficits at an early age.194 One of the most rigorous and faithful rodent models of AD is the TgF344 AD rat. These animals overexpress two familial mutations that cause AD (APP K670N/M671L (Swedish) and D-exon mutant human presenilin-1 (PS1DE9), and develop a well-characterized age-dependent progressive attainment of early neurovascular dysfunction followed by cerebral amyloidosis, tauopathy, gliosis, and neuronal cell death, along with depression-like behavior preceding cognitive deficits.40,221,222
Transgenic rodent models of AD have been widely used to examine the anti-AD effect of drug candidates in vivo.56,223 The resulting measures for drug efficacy include evaluation of neuropathological characteristics such as Aβ or tau burden, as well as behavioral assays such as fear conditioning, Morris water maze, and the novel object recognition test.224 For example, long-term treatment by clozapine, an approved antipsychotic drug, ameliorates the Aβ-induced memory impairment and reduces Aβ levels in the Tg-APPswe/PS1dE9 mouse model.225 Furthermore, long-term treatment with the P7C3 class of neuroprotective agents preserves cognition and neuronal function in TgF344 AD rats without impacting other measures of brain pathology (amyloidosis, tauopathy, gliosis).40 However, many cognitive functions (e.g. language) are unique to humans and cannot be measured in transgenic experimental models. In addition, current transgenic rodents all harbor mutations associated with FAD, while most clinical cases of AD are sporadic and lack a known genetic cause. Use of the various rodent strains has been criticized as having limited clinical validity; none have accurately predicted human benefit from putative AD therapies. AD models do not recapitulate all aspects of AD and are not expected to predict complex human outcomes; they represent a valuable means of assessing drug effects on specific genetically-engineered biology and are valuable laboratory tools to assessing drug mechanisms. Given the magnitude of the public health crisis posed by AD, it is therefore imperative to press forward with evaluating, in a rigorous and thoughtful way, the efficacy of novel treatment approaches in the nonclinical models that are currently accepted by the field. At the same time, other complementary clinically-relevant approaches are warranted, such as population-based validation using large-scale patient databases.
5.4. Population-based validation
Since drug repurposing focuses on drugs that are already marketed, hypothesis testing is possible using large-scale patient-level databases such as MarketScan and Clinformatics DataMart databases. Such patient databases are regularly used to generate actionable evidence of effectiveness, harm, use, and value of medications, including computationally-predicted repurposable candidates. The MarketScan databases are a family of administrative claims databases that fully integrate many types of data for healthcare research, including de-identified records of more than 250 million patients with laboratory results, health risk assessments (HRAs), hospital discharges, and electronic medical records.226 Clinformatics DataMart Database is a de-identified claims database from a national insurance provider, containing detailed longitudinal information. This database has administrative health claims data from over 60 million patients across the United States including enrollment records, medical claims, and prescription claims.227 Columbia Open Health Data (COHD) is a database of prevalence and co-occurrence frequencies between conditions, drugs, procedures, and demographics. The lifetime dataset consists of 36,578 single concepts as well as 32,788,901 concept pairs from 5,364,781 patients.228
Recent advances in state-of-the-art pharmacoepidemiologic analyses offer powerful approaches to assess the drug user’s disease outcomes in CNS disorders.229,230 Paik et al. proposed a drug repurposing framework using both clinical signatures from large-scale patient databases and genomics signatures. They predicted and validated that terbutaline sulfate, an FDA-approved drug for treatment of asthma, had potential for treating amyotrophic lateral sclerosis (ALS) in a zebrafish model.231 In addition, Mittal et al. discovered that β2-adrenoreceptor (β2AR) ligands could modulate α-synuclein gene (SNCA) through histone 3 lysine 27 acetylation. Conducting longitudinal analyses in 4 million Norwegians, they found that salbutamol (a β2AR agonist) reduced the risk of Parkinson’s disease.232 Animal studies revealed that terbutaline can pass the blood-brain barrier of rabbits and salbutamol can pass the blood-brain barrier of rats.233,234 Future randomized controlled trials are needed to test clinical benefits of terbutaline and salbutamol in individuals with ALS or Parkinson’s disease.
Randomized controlled trials are typically limited in scope owing to a relatively modest study size, short follow-up time, and underrepresentation of relevant populations.235 A unique strength of routine healthcare data for validating drug repurposing hypotheses includes the ability to study large patient populations to detect small differences in outcomes. Another strength is the availability of a large number of patient factors recorded without any recall bias, including demographics, comorbid conditions, and medication use, which allows for high-dimensional covariate adjustment to minimize confounding.236–238 Several pitfalls exist for routine healthcare data. First, detailed clinical information is missing for health insurance claims data despite high-dimensional covariate adjustment. For example, lack of common variant genotypes, clinical diagnosis information (i.e. CSF levels of Aβ and tau), as well as some comorbidities, may affect the results of pharmacoepidemiologic analyses. Second, pharmacoepidemiologic analysis may not be suitable for testing the drugs that have fewer patient data and thus are not statistically robust. In that case, replication of the associations using multiple large population-based cohorts is warranted.
6. Discussion, Perspective, and Future Direction
6.1. Why Alzheimer’s drugs have failed in clinical trials
In the past decades, over 99% of AD clinical trials have failed. Several factors may account or contribute to this challenge. First, the transgenic rodent models used in AD preclinical studies elucidate only limited aspects of human AD pathobiology.204 For example, AD has both Aβ and NFTs in the brain. However, very few transgenic mice models that overexpress human familial AD genes have both these key pathologies. Another aspect is that Aβ can accumulate in preclinical AD without neurological symptoms, while tau tangles accumulate predominantly in the MCI phase of AD.239 The mouse with Aβ and few tau tangles may not fully represent the pathobiology of symptomatic AD onset.
Second, lack of sufficiently sensitive clinical outcome measures in clinical trials may contribute to some failures particularly in trials of asymptomatic or minimally affected individuals. Tools and analyses with greater sensitivity are required to detect drug-placebo differences.
Third, the presence of different population groups may affect the clinical results. For example, a randomized control trial demonstrated that pioglitazone showed cognitive function improvement in 42 patients with non-insulin dependent diabetes mellitus and mild AD,240 while a Phase II study (NCT00982202) of pioglitazone showed no statistically significant differences between controls and patients with mild to moderate AD.241
Varying degrees of AD pathology produce clinical syndromes ranging from asymptomatic to severely impaired. These boundaries are challenging since many past clinical trials used only cognitive tests (such as Clinical Dementia Rating Scale) to define the AD population. It cannot be excluded that the so called “MCI due to AD” includes patients without including AD pathology.242 More specific approaches, such as fluid biomarkers and brain imaging, can be used to measure the stage of disease in trial participants. Biomarkers such as concentrations of tau and Aβ proteins in CSF, as well as brain imaging to measure brain volume changes and protein deposition, should be evaluated as entry and/or end point criteria.243
Lack of drug efficacy is the most common cause of negative trials in AD drug development. Agents may lack target engagement or may be administered in too small doses or for too short periods of time.244 Factors contributing to clinical trial failures that should be accounted for in future AD trials include:
Targeting the wrong pathophysiological mechanisms
Using drugs that do not engage with the intended target
Intervening at the wrong stage of the disease
Lacking translatable pharmacodynamic and pharmacokinetic (i.e., poor brain penetration) biomarkers
Depending on animal models with poor predictive efficacy
Failing to address the complexity of AD
Failing to accurately monitor the complexity of the clinical and biological responses to therapeutic intervention
Administering treatments in too low doses or for too short periods of time during trials.
6.2. Challenges of Alzheimer’s drug repurposing
Several barriers and challenges face AD drug repurposing. Drugs predicted to impact AD pathophysiology will likely need to be able to cross the BBB. However, over 98% of all small-molecule drugs, and nearly 100% of large-molecule drugs, do not cross the BBB.245 Furthermore, considering the difference of transporter mechanisms between rodent models and humans, drugs easily crossing the BBB in animal models may not exhibit brain penetration in humans. For example, tarenflurbil showed striking effects on the AD animal models, but did not have an optimal brain penetration profile in humans and failed in its Phase III trial.246 Thus, drug delivery to the CNS is a major challenge. Development of novel drug delivery approaches to the brain may overcome poor brain penetration.247
Lack of clinical efficacy is another important concern in AD drug repurposing. In the past decade, a number of clinical trials with monotherapy have failed to affect AD disease progression or symptoms due to lack of efficacy.248 Given the complex pathophysiology of AD, combination drug therapy may be necessary for effective treatment. Memantine and donepezil are available in an FDA-approved fixed drug combination for the treatment of moderate to severe AD.249 This provides a regulatory pathway for combination drug treatment in AD treatment. Recent network proximity methodologies can identify clinically efficacious drug combinations for specific diseases by quantifying the network-based relationships among drug targets and disease modules in the human protein-protein interactome.20 Network analysis reveals that drug combinations have predicted therapeutic effects when the targets of the drugs both hit the disease module but exist in distinct biological neighborhoods.20 This approach offers a network-level view of therapeutic combinations in terms of comparative efficacy and adverse drug-drug interactions, which can be applied in rational AD drug combination design.20
The third challenge is low research investment by pharmaceutical companies in drug repurposing compared with new chemical entity-based drugs. Most repurpose-ready drugs have no or limited patent protection, and intellectual property issues decrease the interest of pharmaceutical companies in advancing these agents.250 To overcome this challenge, more academic research funds and governmental support for this national crisis are required or greater legislative protection for time-limited exclusivity for repurposed agents is required.
6.3. Network-based Rational Design of Drug Combination for Alzheimer’s Disease: Illustrative Example
Drug combinations, offer increased therapeutic efficacy and reduced toxicity, playing a crucial role in treating multiple complex diseases. However, how to identify and validate effective combinations is challenged by a combinatorial explosion, driven by both the huge number of drug pairs as well as possible dosage combinations. For a network-based approach to drug combinations to be effective, the topological relationship between two drugs must be understood. A key network-based methodology is that a drug combination is likely to be therapeutically effective only if it has a specific relationship to the disease module, as captured by the “Complementary Exposure” pattern in targets’ modules of both drugs (Fig. 8A).20 Specifically, if we are looking for therapeutically synergistic combinations, the two drugs must not only overlap with the disease module, but the two drug-target modules need to be separated in the human interactome without overlapping toxic mechanisms.251 Carvedilol (NCT01354444) and riluzole (NCT01703117) have been repurposed as AD drugs in clinical trials. A previous study showed that both of 10 μM and 20 μM of carvedilol had neuronal protective effects against endogenous Aβ neurotoxicity in mouse Neuro2a cells.252 However, in vitro findings of carvedilol tested at a high concentration of 10 μM or 20 μM are still not enough to support that it is a clinically relevant dose for central brain action in human subjects. Network proximity analysis reveals that the target modules of these two agents have significant network proximity with the APP module (Fig. 8B) yet hit separate neighborhoods, suggesting that their combination in AD may have potential efficacy without significant synergistic toxicity, based on their “Complementary Exposure” pattern. As shown in Fig. 8B, carvedilol can target a key seed gene APP, which connects to several other seed genes (i.e. FYN, GSK3B and SUMO1); riluzole can target SCN5A and SCN4B, the two neighborhood proteins of seed gene SNCA and FYN. Using these two agents (carvedilol and riluzole) show potential for combination therapy based on the network-based “Complementary Exposure” model. PXT864, a combination of acamprosate and baclofen, was reported to have potential protective effects on neurons and endothelial structures in vitro against Aβ oligomers.253 PXT864, discovered by Pharnext, is under a Phase II AD clinical trial [NCT02361424]. We found that acamprosate and baclofen were captured by the “Complementary Exposure” pattern (Fig. 8C) in the human interactome network, further supporting the proof-of-concept of “Complementary Exposure” for rational design of AD drug combinations.
Figure 8. Two case studies illustrate proof-of-concept of network-based drug combinations for anti-Alzheimer therapy.
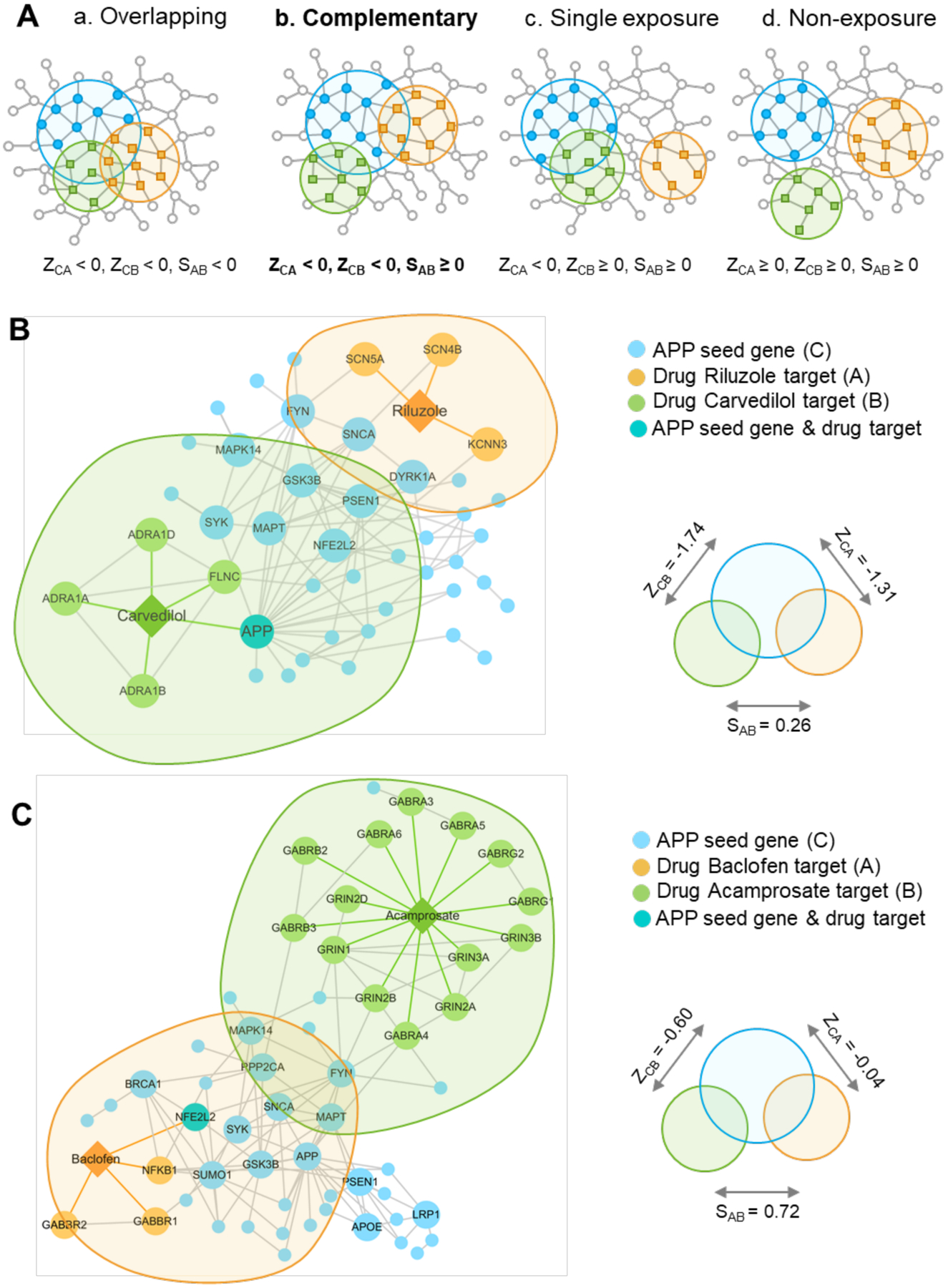
(A) The possible exposure mode of disease module to the pairwise drug combinations. An effective drug combination will be captured by the “Complementary Exposure” pattern: the targets of the drugs both hit disease module, but target separate neighborhoods in the human interactome network249. ZCA and ZCB denote the network proximity (Z-score) between targets (Drugs A and B) and disease module. SAB denotes separation score of targets between Drug A and Drug B. (B) Network proximity analysis illustrates potential drug combination (carvedilol plus riluzole) for AD. (C) Network proximity analysis illustrates a potential drug combination (acamprosate plus baclofen) for AD. Disease module is generated based on 54 genes related to Amyloid hypothesis of Alzheimer’s disease collected from the literatures.
6.4. Individualized drug repurposing in Alzheimer’s disease
The last challenge is to overcome the limitation of clinical “one-size-fits-all, magic bullet drug development”. Similar to cancer precision medicine, the way to treat AD may one day shift from traditional symptom- and sign-based therapy to individually-tailored biomarker-guided targeted therapies.254 To address this, the Alzheimer Precision Medicine Initiative (APMI) was launched in 2016, aiming to transform healthcare, conventional clinical diagnostics, and drug development research in AD.255 Recent advances in next-generation DNA/RNA sequencing have made it possible to identify new targets rapidly and to repurpose existing drugs for treating heterogeneous diseases by ‘precise’ targeting of individualized disease modules.256 For example, Cheng et al. developed a Genome-wide Positioning Systems network algorithm for drug repurposing by specifically targeting disease modules derived from individual patient DNA and RNA sequencing profiles under the human protein-protein interactome network framework.165 An individualized drug repurposing approach was proposed for prioritizing personalized drugs for type 1 diabetes mellitus.257 Development of individualized network-based drug repurposing approaches by unique integration of multi-omics data generated from patient biospecimens could improve the success rate of AD drug repurposing (Fig. 9). In addition, the individualized network-based framework can be applied to AD clinical trials and treatment decisions. For example, the patient-specific or subtype-specific disease modules (Fig. 9) could guide therapeutic evaluation during clinical trials or optimize new treatments for patients who receive these drugs after FDA approval.
Figure 9. An individualized network-based approach for personalized medicine in Alzheimer’s disease (AD).
Individualized disease network modules built from multi-omics data of individual patients under the human interatcome network model could improve the success rate of AD drug discovery, guide therapeutic evaluation during clinical trials, and optimize new treatment for patients with AD and those at risk for AD dementia.
In summary, endophenotype network-based approaches show promise in better understanding the pathobiology of AD and accelerating drug repurposing, but the process is still in its infancy. We believe that network-based drug repurposing represents a valuable and complementary approach to promoting AD therapeutic development that will optimize the usefulness of available data and support deep phenotyping of the complexity of AD.
Acknowledgments
We thank Yadi Zhou for assistance to prepare the figures and critical comments from all members of the Cheng group at the Cleveland Clinic Genomic Medicine Institute.
Funding: This work was supported by the National Institute of Aging (NIA) of the National Institutes of Health under Award Number R01AG066707 to F.C. This work has been also supported in part the National Institute of Aging (NIA) of the National Institutes of Health under Award Number R56AG063870 to F.C., L.B., and J.L. This work has been also supported in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. AAP is supported by the Brockman Foundation, as well as Project 19PABH134580006-AHA/Allen Initiative in Brain Health and Cognitive Impairment, and the Elizabeth Ring Mather & William Gwinn Mather Fund, S. Livingston Samuel Mather Trust, G.R. Lincoln Family Foundation, Wick Foundation, Gordon & Evie Safran, and the Louis Stokes VA Medical Center resources and facilities. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. J.C. acknowledges funding from the National Institute of General Medical Sciences (Grant: P20GM109025) and support from Keep Memory Alive.
Abbreviation:
- AD
Alzheimer’s disease
- ADGC
Alzheimer’s Disease Genetics Consortium
- ADNI
The Alzheimer’s Disease Neuroimaging Initiative
- ADSP
The Alzheimer’s Disease Sequencing Project
- ALS
amyotrophic lateral sclerosis
- AMP-AD
Accelerating Medicines Partnership - Alzheimer’s Disease
- APMI
The Alzheimer Precision Medicine Initiative
- APP
amyloid precursor protein
- ARB
angiotensin receptor blocker
- Aβ
amyloid‑β
- BBB
blood-brain barrier
- C. elegans
Caenorhabditis elegans
- CD33
cell surface antigen CD33
- CNS
central nervous system
- CMap
Connectivity Map
- CSF
cerebrospinal fluid
- DAM
disease-associated microglia
- DGB
Drug Gene Budger
- DSigDB
Drug Signatures Database
- ENCODE
The Encyclopedia of DNA Elements
- FAD
familial AD
- FDA
Food and Drug Administration
- GTEx
The Genotype-Tissue Expression
- GPSnet
Genome-wide Positioning Systems network
- GWAS
genome-wide association studies
- HSPA2
heat shock protein family A member 2
- IL-6
interleukin 6
- IL-1β
interleukin 1 beta
- iPSCs
human induced pluripotent stem cells
- LINCS
the Library of Integrated Network-based Cellular Signatures
- MAGMA
Multi-objective Analyzer for Genetic Marker Acquisition
- MAPT
microtubule-associated protein tau
- MCI
mild cognitive impairment
- MIF
macrophage migration inhibitory factor
- MRI
magnetic resonance imaging
- NAI
neurodegeneration-associated inflammatory
- NAS
neurodegeneration-associated synaptic
- NFTs
neurofibrillary tangles
- NGS
next-generation sequencing
- NIAGADS
The Genetics of Alzheimer’s Disease Data Storage Site
- NMDA
N-methyl-D-aspartate
- PDSP
Psych-Active Drug Screening Program
- PET
positron emission tomography
- PKCα
protein kinase C alpha
- PPI
protein-protein interaction
- PSEN1
presenilin-1
- QSP
quantitative systems pharmacology
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor alpha
- WES
whole-exome sequencing
- WGCNA
Weighted gene co-expression network analysis
- WGS
whole-genome sequencing
- β2AR
β2-adrenoreceptor
References
- 1.Alzheimer A Uber eine eigenartige Erkrankung der Hirnrinde. Zentralbl Nervenh Psych 1907;18:177–179. [Google Scholar]
- 2.Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(1):88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beecham GW, Bis JC, Martin ER, et al. The Alzheimer’s Disease Sequencing Project: study design and sample selection. Neurol Genet. 2017;3(5):e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiner MW, Aisen PS, Jack CR, et al. The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6(3):202–211.e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kodamullil AT, Zekri F, Sood M, et al. Trial watch: Tracing investment in drug development for Alzheimer disease. Nat Rev Drug Discov. 2017;16(12):819. [DOI] [PubMed] [Google Scholar]
- 11.Alteri E, Guizzaro L. Be open about drug failures to speed up research. Nature 2018;563(7731):317–319. [DOI] [PubMed] [Google Scholar]
- 12.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153(3):707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandal MJ, Haney JR, Parikshak NN, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018;359(6376):693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seyfried NT, Dammer EB, Swarup V, et al. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst. 2017;4(1):60–72 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swarup V, Hinz FI, Rexach JE, et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med. 2019;25(1):152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dugger SA, Platt A, Goldstein DB. Drug development in the era of precision medicine. Nat Rev Drug Discov. 2018;17(3):183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greene JA, Loscalzo J. Putting the patient back together - social medicine, network medicine, and the limits of reductionism. N Engl J Med. 2017;377(25):2493–2499. [DOI] [PubMed] [Google Scholar]
- 19.Cheng F, Desai RJ. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat Commun. 2018;9(1):2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng F, Kovács IA, Barabási AL. Network-based prediction of drug combinations. Nat Commun. 2019;10(1):1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corbett A, Pickett J, Burns A, et al. Drug repositioning for Alzheimer’s disease. Nat Rev Drug Discov. 2012;11(11):833–846. [DOI] [PubMed] [Google Scholar]
- 22.Espay AJ, Vizcarra JA, Marsili L, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019;92(7):329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ertekin-Taner N Gene expression endophenotypes: a novel approach for gene discovery in Alzheimer’s disease. Mol Neurodegener. 2011;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cioffi F, Adam RHI, Broersen K. Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J Alzheimers Dis. 2019;72(4):981–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421–435. [DOI] [PubMed] [Google Scholar]
- 27.Peng Y, Kim MJ, Hullinger R, et al. Improved proteostasis in the secretory pathway rescues Alzheimer’s disease in the mouse. Brain 2016;139(Pt 3):937–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Fu AKY, Ip NY. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol Ther. 2019;195:186–198. [DOI] [PubMed] [Google Scholar]
- 29.Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid-beta immunotherapy for alzheimer disease: Is it now a long shot? Ann Neurol. 2019;85(3):303–315. [DOI] [PubMed] [Google Scholar]
- 30.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68(2):270–281. [DOI] [PubMed] [Google Scholar]
- 31.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712. [DOI] [PubMed] [Google Scholar]
- 34.Barage SH, Sonawane KD. Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015;52:1–18. [DOI] [PubMed] [Google Scholar]
- 35.Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96(4):529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karran E, De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. 2016;139 Suppl 2:237–252. [DOI] [PubMed] [Google Scholar]
- 37.Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73–88. [DOI] [PubMed] [Google Scholar]
- 38.Golde TE. The Abeta hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol. 2005;15(1):84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drachman DA. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement. 2014;10(3):372–380. [DOI] [PubMed] [Google Scholar]
- 40.Voorhees JR, Remy MT, Cintron-Perez CJ, et al. (−)-P7C3-S243 protects a rat model of Alzheimer’s disease from neuropsychiatric deficits and neurodegeneration without altering amyloid deposition or reactive glia. Biol Psychiatry. 2018;84(7):488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14(7):399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12(1):15–27. [DOI] [PubMed] [Google Scholar]
- 44.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83(13):4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giacobini E, Gold G. Alzheimer disease therapy--moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9(12):677–686. [DOI] [PubMed] [Google Scholar]
- 46.McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76 Pt A:57–67. [DOI] [PubMed] [Google Scholar]
- 47.Qi L, Chen Z, Wang Y, et al. Subcutaneous liraglutide ameliorates methylglyoxal-induced Alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3beta. Am J Transl Res. 2017;9(2):247–260. [PMC free article] [PubMed] [Google Scholar]
- 48.Kurata T, Miyazaki K, Kozuki M, et al. Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 2011;1371:161–170. [DOI] [PubMed] [Google Scholar]
- 49.Huuskonen MT, Loppi S, Dhungana H, et al. Bexarotene targets autophagy and is protective against thromboembolic stroke in aged mice with tauopathy. Sci Rep. 2016;6:33176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai Z, Yan Y, Wang Y. Minocycline alleviates beta-amyloid protein and tau pathology via restraining neuroinflammation induced by diabetic metabolic disorder. Clin Interv Aging. 2013;8:1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016;12(6):719–732. [DOI] [PubMed] [Google Scholar]
- 52.White CS, Lawrence CB, Brough D, Rivers-Auty J. Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol. 2017;27(2):223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fillit H, Ding WH, Buee L, et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129(2):318–320. [DOI] [PubMed] [Google Scholar]
- 54.Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018; 217(2):459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voorhees JR, Remy MT, Erickson CM, Dutca LM, Brat DJ, Pieper AA. Occupational-like organophosphate exposure disrupts microglia and accelerates deficits in a rat model of Alzheimer’s disease. NPJ Aging Mech Dis. 2019;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. [DOI] [PubMed] [Google Scholar]
- 58.Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer’s Disease. J Neurosci. 2018;38(12):2911–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–138. [DOI] [PubMed] [Google Scholar]
- 61.Cai Z, Wan CQ, Liu Z. Astrocyte and Alzheimer’s disease. J Neurol. 2017;264(10):2068–2074. [DOI] [PubMed] [Google Scholar]
- 62.Henstridge CM, Hyman BT, Spires-Jones TL. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat Rev Neurosci. 2019;20(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lian H, Litvinchuk A, Chiang ACA, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. 2016;36(2):577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23(6):701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 67.Sala Frigerio C, Wolfs L, Fattorelli N, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019;27(4):1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Torika N, Asraf K, Roasso E, Danon A, Fleisher-Berkovich S. Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J Neuroimmune Pharmacol. 2016;11(4):774–785. [DOI] [PubMed] [Google Scholar]
- 69.Kowalski K, Mulak A. Brain-gut-microbiota axis in Alzheimer’s disease. J Neurogastroenterol Motil. 2019;25(1):48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang C, Li G, Huang P, Liu Z, Zhao B. The gut microbiota and Alzheimer’s disease. J Alzheimers Dis. 2017;58(1):1–15. [DOI] [PubMed] [Google Scholar]
- 71.Mancuso C, Santangelo R. Alzheimer’s disease and gut microbiota modifications: the long way between preclinical studies and clinical evidence. Pharmacol Res. 2018;129:329–336. [DOI] [PubMed] [Google Scholar]
- 72.Lin L, Zheng LJ, Zhang LJ. Neuroinflammation, gut microbiome, and Alzheimer’s disease. Mol Neurobiol. 2018;55(11):8243–8250. [DOI] [PubMed] [Google Scholar]
- 73.Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing. 2018;15:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front Microbiol. 2016;7:1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Sun G, Feng T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29(10):787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63(1):8–20. [DOI] [PubMed] [Google Scholar]
- 78.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842(8):1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cadonic C, Sabbir MG, Albensi BC. Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol Neurobiol. 2016;53(9):6078–6090. [DOI] [PubMed] [Google Scholar]
- 80.Leuner K, Muller WE, Reichert AS. From mitochondrial dysfunction to amyloid beta formation: novel insights into the pathogenesis of Alzheimer’s disease. Mol Neurobiol. 2012;46(1):186–193. [DOI] [PubMed] [Google Scholar]
- 81.Mao P, Manczak M, Calkins MJ, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21(13):2973–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheng Y, Bai F. The association of Tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci. 2018;12:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eckert GP, Renner K, Eckert SH, et al. Mitochondrial dysfunction--a pharmacological target in Alzheimer’s disease. Mol Neurobiol. 2012;46(1):136–150. [DOI] [PubMed] [Google Scholar]
- 84.de la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neurodegener Dis. 2010;7(1–3):116–121. [DOI] [PubMed] [Google Scholar]
- 85.Janota C, Lemere CA, Brito MA. Dissecting the contribution of vascular alterations and aging to Alzheimer’s disease. Mol Neurobiol. 2016;53(6):3793–3811. [DOI] [PubMed] [Google Scholar]
- 86.Jellinger KA. Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neurodegener Dis. 2010;7(1–3):112–115. [DOI] [PubMed] [Google Scholar]
- 87.Gabin JM, Tambs K, Saltvedt I, Sund E, Holmen J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: the HUNT Study. Alzheimers Res Ther. 2017;9(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tosto G, Bird TD, Bennett DA, et al. The role of cardiovascular risk factors and stroke in familial Alzheimer disease. JAMA Neurol. 2016;73(10):1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bennett RE, Robbins AB, Hu M, et al. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci USA. 2018;115(6):E1289–E1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nation DA, Sweeney MD. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement.2015;11(6):710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(5):887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iadecola C The pathobiology of vascular dementia. Neuron 2013;80(4):844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li NC, Lee A, Whitmer RA, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, Ho L, Chen L, et al. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117(11):3393–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Danielyan L, Klein R, Hanson LR, et al. Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. 2010;13(2–3):195–201. [DOI] [PubMed] [Google Scholar]
- 98.Torika N, Asraf K, Danon A, Apte RN, Fleisher-Berkovich S. Telmisartan modulates glial activation: In vitro and in vivo studies. PLoS One 2016;11(5):e0155823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang J, Ono K, Dickstein DL, et al. Carvedilol as a potential novel agent for the treatment of Alzheimer’s disease. Neurobiol Aging. 2011;32(12):2321.e1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Copenhaver PF, Anekonda TS, Musashe D, et al. A translational continuum of model systems for evaluating treatment strategies in Alzheimer’s disease: isradipine as a candidate drug. Dis Model Mech. 2011;4(5):634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol Dis. 2011;41(1):62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cuadrado-Tejedor M, Hervias I, Ricobaraza A, et al. Sildenafil restores cognitive function without affecting beta-amyloid burden in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2011;164(8):2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu Y, Li Z, Huang Y-Y, Wu D, Luo H-B. Novel phosphodiesterase inhibitors for cognitive improvement in Alzheimer’s disease. J Med Chem. 2018;61(13):5467–5483. [DOI] [PubMed] [Google Scholar]
- 104.Gurney ME, D’Amato EC, Burgin AB. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics. 2015;12(1):49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov. 2019;18(12):923–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Whyte LS, Lau AA, Hemsley KM, Hopwood JJ, Sargeant TJ. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer’s disease? J Neurochem. 2017;140(5):703–717. [DOI] [PubMed] [Google Scholar]
- 107.Whyte LS, Hassiotis S, Hattersley KJ, et al. Lysosomal dysregulation in the murine App model of Alzheimer’s disease. Neuroscience. 2020;429:143–155. [DOI] [PubMed] [Google Scholar]
- 108.Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener. 2019;14(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao Q, Yan P, Ma X, et al. Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Aβ generation and amyloid plaque pathogenesis. J Neurosci. 2015;35(35):12137–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(5):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gowrishankar S, Yuan P, Wu Y, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci USA. 2015;112(28):E3699–E3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. [DOI] [PubMed] [Google Scholar]
- 113.Kwak SS, Washicosky KJ, Brand E, et al. Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat Commun. 2020;11(1):1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Choi SH, Kim YH, Hebisch M, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515(7526):274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen W, Abud EA, Yeung ST, et al. Blurton-Jones M. Increased tauopathy drives microglia-mediated clearance of beta-amyloid. Acta Neuropathol Commun. 2016;4(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.De Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fang J, Zhang P, Zhou Y, et al. Endophenotype-based discovery and population-based validation of Sildenafil in reducing the incidence of Alzheimer’s disease. Nat Commun. 2020. Under revision [Google Scholar]
- 121.Terada T, Obi T, Bunai T, et al. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology. 2020;94(15):e1592–e1604. [DOI] [PubMed] [Google Scholar]
- 122.Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schöll M, Ossenkoppele R, Strandberg O, et al. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain. 2017;140(9):2286–2294. [DOI] [PubMed] [Google Scholar]
- 124.Butkiewicz M, Blue EE, Leung YY, et al. Functional annotation of genomic variants in studies of late-onset Alzheimer’s disease. Bioinformatics. 2018;34(16):2724–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23. [DOI] [PubMed] [Google Scholar]
- 126.Hodes RJ, Buckholtz N. Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) knowledge portal aids Alzheimer’s drug discovery through open data sharing. Expert Opin Ther Targets. 2016;20(4):389–391. [DOI] [PubMed] [Google Scholar]
- 127.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lizio M, Harshbarger J, Abugessaisa I, et al. Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res. 2017;45(D1):D737–D743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuksa PP, Amlie-Wolf A, Katanic Z, Valladares O, Wang LS, Leung YY. DASHR 2.0: integrated database of human small non-coding RNA genes and mature products. Bioinformatics. 2019;35(6):1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang ZT, Tan CC, Tan L, Yu JT. Systems biology and gene networks in Alzheimer’s disease. Neurosci Biobehav Rev. 2019;96:31–44. [DOI] [PubMed] [Google Scholar]
- 133.Xu M, Zhang DF, Luo R, et al. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement. 2018;14(2):215–229. [DOI] [PubMed] [Google Scholar]
- 134.Matarin M, Salih DA, Yasvoina M, et al. A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 2015;10(4):633–644. [DOI] [PubMed] [Google Scholar]
- 135.Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570(7761):332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grubman A, Chew G, Ouyang JF, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22(12):2087–2097. [DOI] [PubMed] [Google Scholar]
- 137.Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14(1):114–124. [DOI] [PubMed] [Google Scholar]
- 138.Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016;131(5):687–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Veitch DP, Weiner MW, Aisen PS, et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019;15(1):106–152. [DOI] [PubMed] [Google Scholar]
- 141.Weiner MW, Veitch DP, Aisen PS, et al. Recent publications from the Alzheimer’s Disease Neuroimaging Initiative: Reviewing progress toward improved AD clinical trials. Alzheimers Dement. 2017;13(4):e1–e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–d1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ursu O, Holmes J, Bologa CG, et al. DrugCentral 2018: an update. Nucleic Acids Res. 2019;47(D1):D963–d970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cotto KC, Wagner AH, Feng YY, et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46(D1):D1068–d1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li YH, Yu CY, Li XX, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018;46(D1):D1121–d1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. [DOI] [PubMed] [Google Scholar]
- 148.Duan Q, Flynn C, Niepel M, et al. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 2014;42(Web Server issue):W449–W460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ye C, Ho DJ, Neri M. DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery. Nat Commun. 2018;9(1):4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Igarashi Y, Nakatsu N, Yamashita T, et al. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 2015;43(Database issue):D921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang Z, He E, Sani K, Jagodnik KM, Silverstein MC, Ma’ayan A. Drug Gene Budger (DGB): an application for ranking drugs to modulate a specific gene based on transcriptomic signatures. Bioinformatics. 2019;35(7):1247–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Carrella D, Napolitano F, Rispoli R, et al. Mantra 2.0: an online collaborative resource for drug mode of action and repurposing by network analysis. Bioinformatics. 2014;30(12):1787–1788. [DOI] [PubMed] [Google Scholar]
- 153.Setoain J, Franch M, Martinez M, et al. NFFinder: an online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning. Nucleic Acids Res. 2015;43(W1):W193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ganter B, Snyder RD, Halbert DN, Lee MD. Toxicogenomics in drug discovery and development: mechanistic analysis of compound/class-dependent effects using the DrugMatrix database. Pharmacogenomics. 2006;7(7):1025–1044. [DOI] [PubMed] [Google Scholar]
- 155.Roth BL, Lopez E, Beischel S, Westkaemper RB, Evans JM. Screening the receptorome to discover the molecular targets for plant-derived psychoactive compounds: a novel approach for CNS drug discovery. Pharmacol Ther. 2004;102(2):99–110. [DOI] [PubMed] [Google Scholar]
- 156.Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chatr-Aryamontri A, Oughtred R, Boucher L, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45(D1):D369–d379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Keshava Prasad TS, Goel R, Kandasamy K, et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mosca R, Ceol A, Aloy P. Interactome3D: adding structural details to protein networks. Nat Methods. 2013;10(1):47–53. [DOI] [PubMed] [Google Scholar]
- 160.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Licata L, Briganti L, Peluso D, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40(Database issue):D857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Orchard S, Ammari M, Aranda B, et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42(Database issue):D358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cheng F, Lu W, Liu C, et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun. 2019; 10(1):3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ma DL, Chan DS, Leung CH. Drug repositioning by structure-based virtual screening. Chem Soc Rev. 2013;42(5):2130–2141. [DOI] [PubMed] [Google Scholar]
- 167.Fang J, Liu C, Wang Q, Lin P, Cheng F. In silico polypharmacology of natural products. Brief Bioinform. 2018;19(6):1153–1171. [DOI] [PubMed] [Google Scholar]
- 168.Fang J, Li Y, Liu R, et al. Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical-protein interactions. J Chem Inf Model. 2015;55(1):149–164. [DOI] [PubMed] [Google Scholar]
- 169.Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther. 2013;93(4):335–341. [DOI] [PubMed] [Google Scholar]
- 170.Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform. 2016;17(1):2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lotfi Shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR. A review of network-based approaches to drug repositioning. Brief Bioinform. 2018;19(5):878–892. [DOI] [PubMed] [Google Scholar]
- 172.Cha Y, Erez T, Reynolds IJ, et al. Drug repurposing from the perspective of pharmaceutical companies. Br J Pharmacol. 2018;175(2):168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xue H, Li J, Xie H, Wang Y. Review of Drug Repositioning Approaches and Resources. Int J Biol Sci. 2018;14(10):1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rosenberg RN, Lambracht-Washington D, Yu G, Xia W. Genomics of Alzheimer disease: a review. JAMA Neurol. 2016;73(7):867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856–860. [DOI] [PubMed] [Google Scholar]
- 178.Kwok MK, Lin SL, Schooling CM. Re-thinking Alzheimer’s disease therapeutic targets using gene-based tests. EBioMedicine. 2018;37:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.So HC, Chau CK, Chiu WT, et al. Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nat Neurosci. 2017;20(10):1342–1349. [DOI] [PubMed] [Google Scholar]
- 180.Fang J, Zhang P, Wang Q, et al. Network-based translation of GWAS findings to pathobiology and drug Repurposing for Alzheimer’s disease. medRxiv 2020;DOI: 10.1101/2020.01.15.20017160. [DOI] [Google Scholar]
- 181.Zhao J, Cheng F, Jia P, Cox N, Denny JC, Zhao Z. An integrative functional genomics framework for effective identification of novel regulatory variants in genome-phenome studies. Genome Med. 2018;10(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24(R1):R102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang Q, Chen R, Cheng F, et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat Neurosci. 2019;22(5):691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Musa A, Ghoraie LS, Zhang SD, et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform. 2018;19(3):506–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Qu XA, Freudenberg JM, Sanseau P, Rajpal DK. Integrative clinical transcriptomics analyses for new therapeutic intervention strategies: a psoriasis case study. Drug Discov Today. 2014;19(9):1364–1371. [DOI] [PubMed] [Google Scholar]
- 186.Miller JA, Guillozet-Bongaarts A, Gibbons LE. Neuropathological and transcriptomic characteristics of the aged brain. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Raj T, Li YI. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet. 2018;50(11):1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Miller JA, Ding SL, Sunkin SM, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508(7495):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vargas DM, De Bastiani MA, Zimmer ER, Klamt F. Alzheimer’s disease master regulators analysis: search for potential molecular targets and drug repositioning candidates. Alzheimers Res Ther. 2018;10(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Siavelis JC, Bourdakou MM, Athanasiadis EI, Spyrou GM, Nikita KS. Bioinformatics methods in drug repurposing for Alzheimer’s disease. Brief Bioinform. 2016;17(2):322–335. [DOI] [PubMed] [Google Scholar]
- 192.Xiong F, Ge W, Ma C. Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimers Dement. 2019;15(3):429–440. [DOI] [PubMed] [Google Scholar]
- 193.Sharma K, Schmitt S, Bergner CG, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18(12):1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim DK, Park J, Han D, et al. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol Neurodegener. 2018;13(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Savas JN, Wang YZ, DeNardo LA, et al. Amyloid accumulation drives proteome-wide alterations in mouse models of Alzheimer’s disease-like pathology. Cell Rep. 2017;21(9):2614–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang P, Joberty G, Buist A, et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017;133(5):731–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16(8):441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Petyuk VA, Chang R, Ramirez-Restrepo M, et al. The human brainome: network analysis identifies HSPA2 as a novel Alzheimer’s disease target. Brain. 2018;141(9):2721–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang M, Schmitt-Ulms G, Sato C, et al. Drug repositioning for Alzheimer’s disease based on systematic ‘omics’ data mining. PloS one. 2016;11(12):e0168812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Antman EM, Loscalzo J. Precision medicine in cardiology. Nat Rev Cardiol. 2016;13(10):591–602. [DOI] [PubMed] [Google Scholar]
- 201.Cheng F, Zhou Y, Li W, Liu G, Tang Y. Prediction of chemical-protein interactions network with weighted network-based inference method. PloS One. 2012;7(7):e41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Geerts H, Wikswo J, van der Graaf PH, et al. Quantitative systems pharmacology for neuroscience drug discovery and development: current status, opportunities, and challenges. CPT Pharmacometrics Syst Pharmacol. 2020;9(1):5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Geerts H, Spiros A, Roberts P, Carr R. Towards the virtual human patient. quantitative systems pharmacology in Alzheimer’s disease. Eur J Pharmacol. 2017;817:38–45. [DOI] [PubMed] [Google Scholar]
- 204.Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta neuropathologica. 2017;133(2):155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cavanaugh SE, Pippin JJ, Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. ALTEX. 2014;31(3):279–302. [DOI] [PubMed] [Google Scholar]
- 206.Gotz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci. 2018;19(10):583–598. [DOI] [PubMed] [Google Scholar]
- 207.Zhang L, Chen C, Mak MS, et al. Advance of sporadic Alzheimer’s disease animal models. Med Res Rev. 2020;40(1):431–458. [DOI] [PubMed] [Google Scholar]
- 208.Arber C, Lovejoy C, Wray S. Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimers Res Ther. 2017;9(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. [DOI] [PubMed] [Google Scholar]
- 210.Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kondo T, Imamura K, Funayama M, et al. iPSC-based compound screening and in vitro trials identify a synergistic anti-amyloid beta combination for Alzheimer’s disease. Cell Rep. 2017;21(8):2304–2312. [DOI] [PubMed] [Google Scholar]
- 212.Bouleau S, Tricoire H. Drosophila models of Alzheimer’s disease: advances, limits, and perspectives. J Alzheimers Dis. 2015;45(4):1015–1038. [DOI] [PubMed] [Google Scholar]
- 213.Ma L, Zhao Y, Chen Y, Cheng B, Peng A, Huang K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur J Pharmacol. 2018;819:169–180. [DOI] [PubMed] [Google Scholar]
- 214.Favrin G, Bean DM, Bilsland E, et al. Identification of novel modifiers of Abeta toxicity by transcriptomic analysis in the fruitfly. Sci Rep. 2013;3:3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shim KH, Kim SH, Hur J, Kim DH, Demirev AV, Yoon SY. Small-molecule drug screening identifies drug Ro 31–8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol Dis. 2019;130:104519. [DOI] [PubMed] [Google Scholar]
- 216.He Q, Huang G, Chen Y, Wang X, Huang Z, Chen Z. The protection of novel 2-arylethenylquinoline derivatives against impairment of associative learning memory induced by neural Abeta in C. elegans Alzheimer’s disease model. Neurochem Res. 2017;42(11):3061–3072. [DOI] [PubMed] [Google Scholar]
- 217.Ellenbroek B, Youn J. Rodent models in neuroscience research: is it a rat race? Dis Model Mech. 2016;9(10):1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Clark T, Kinoshita J. Alzforum and SWAN: the present and future of scientific web communities. Brief Bioinform. 2007;8(3):163–171. [DOI] [PubMed] [Google Scholar]
- 219.Richards JG, Higgins GA, Ouagazzal AM, et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003;23(26):8989–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ramsden M, Kotilinek L, Forster C, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci. 2005;25(46):10637–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cohen RM, Rezai-Zadeh K, Weitz TM, et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J Neurosci. 2013;33(15):6245–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Joo IL, Lai AY, Bazzigaluppi P, et al. Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Sci Rep. 2017;7:46427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wang L, Fang J, Jiang H, et al. 7-Pyrrolidinethoxy-4’-Methoxyisoflavone Prevents Amyloid beta-Induced Injury by Regulating Histamine H3 Receptor-Mediated cAMP/CREB and AKT/GSK3beta Pathways. Front Neurosci. 2019;13:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O. Behavioral assays with mouse models of Alzheimer’s disease: practical considerations and guidelines. Biochem Pharmacol. 2014;88(4):450–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Choi Y, Jeong HJ, Liu QF, et al. Clozapine improves memory impairment and reduces Abeta level in the Tg-APPswe/PS1dE9 mouse model of Alzheimer’s disease. Mol Neurobiol. 2017;54(1):450–460. [DOI] [PubMed] [Google Scholar]
- 226.Kulaylat AS, Schaefer EW, Messaris E, Hollenbeak CS. Truven Health Analytics MarketScan Databases for clinical research in colon and rectal surgery. Clin Colon Rectal Surg. 2019;32(1):54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Caram MEV, Estes JP, Griggs JJ, Lin P, Mukherjee B. Temporal and geographic variation in the systemic treatment of advanced prostate cancer. BMC cancer. 2018;18(1):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ta CN, Dumontier M. Columbia Open Health Data, clinical concept prevalence and co-occurrence from electronic health records. Sci Data. 2018;5:180273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hemingway H, Asselbergs FW, Danesh J, et al. Big data from electronic health records for early and late translational cardiovascular research: challenges and potential. Eur Heart J. 2018;39(16):1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Denny JC, Van Driest SL, Wei WQ, Roden DM. The influence of big (Clinical) data and genomics on precision medicine and drug development. Clin Pharmacol Ther. 2018;103(3):409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Paik H, Chung AY, Park HC, et al. Repurpose terbutaline sulfate for amyotrophic lateral sclerosis using electronic medical records. Sci Rep. 2015;5:8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mittal S, Bjornevik K. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science. 2017;357(6354):891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hsu CH, Robinson CP, Basmadjian GP. Tissue distribution of 3H-terbutaline in rabbits. Life Sci. 1994;54(20):1465–1469. [DOI] [PubMed] [Google Scholar]
- 234.Zhang R, Wu J, Liu S, et al. Spatial Distribution of (R)-salbutamol in Rat Brain Following Nasal and Intravenous Administration Using DESI-MS. Pharmaceutics. 2020;12(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Schneeweiss S, Eichler HG, Garcia‐Altes A, et al. Real world data in adaptive biomedical innovation: a framework for generating evidence fit for decision making. Clin Pharmacol Ther. 2016;100(6):633–646. [DOI] [PubMed] [Google Scholar]
- 236.Schneeweiss S, Avorn J. A review of uses of health care utilization databases for epidemiologic research on therapeutics. J Clin Epidemiol. 2005;58(4):323–337. [DOI] [PubMed] [Google Scholar]
- 237.Schneeweiss S, Rassen JA, Glynn RJ, Avorn J, Mogun H, Brookhart MA. High-dimensional propensity score adjustment in studies of treatment effects using health care claims data. Epidemiology. 2009;20(4):512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Brilliant MH, Vaziri K, Connor TB Jr., et al. Mining retrospective data for virtual prospective drug repurposing: L-DOPA and age-related macular degeneration. Am J Med. 2016;129(3):292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sasaguri H, Nilsson P, Hashimoto S, et al. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017;36(17):2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPARgamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32(9):1626–1633. [DOI] [PubMed] [Google Scholar]
- 241.Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol. 2011;68(1):45–50. [DOI] [PubMed] [Google Scholar]
- 242.Galimberti D, Scarpini E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs. 2017;26(1):97–101. [DOI] [PubMed] [Google Scholar]
- 243.Gauthier S, Albert M, Fox N, et al. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016;12(1):60–64. [DOI] [PubMed] [Google Scholar]
- 244.Cummings J, Feldman HH, Scheltens P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Pardridge WM. Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimers Dement. 2009;5(5):427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wan HI, Jacobsen JS, Rutkowski JL, Feuerstein GZ. Translational medicine lessons from flurizan’s failure in Alzheimer’s disease (AD) trial: Implication for future drug discovery and development for AD. Clin Transl Sci. 2009;2(3):242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Banks WA. Drug delivery to the brain in Alzheimer’s disease: consideration of the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cummings JL, Tong G, Ballard C. Treatment combinations for Alzheimer’s disease: current and future pharmacotherapy options. J Alzheimers Dis. 2019;67(3):779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Owen RT. Memantine and donepezil: a fixed drug combination for the treatment of moderate to severe Alzheimer’s dementia. Drugs Today. 2016;52(4):239–248. [DOI] [PubMed] [Google Scholar]
- 250.Cummings JL, Zhong K. Repackaging FDA-approved drugs for degenerative diseases: promises and challenges. Expert Rev Clin Pharmacol. 2014;7(2):161–165. [DOI] [PubMed] [Google Scholar]
- 251.Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Liu J, Wang M. Carvedilol protection against endogenous Aβ-induced neurotoxicity in N2a cells. Cell Stress Chaperones. 2018;23(4):695–702. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 253.Chumakov I, Nabirotchkin S, Cholet N, et al. Combining two repurposed drugs as a promising approach for Alzheimer’s disease therapy. Sci Rep. 2015;5:7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Cheng F, Liang H, Butte AJ, Eng C, Nussinov R. Personal mutanomes meet modern oncology drug discovery and precision health. Pharmacol Rev. 2019;71(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hampel H, Vergallo A, Perry G, Lista S. The Alzheimer Precision Medicine Initiative. J Alzheimers Dis. 2019;68(1):1–24. [DOI] [PubMed] [Google Scholar]
- 256.Cheng F, Hong H, Yang S, Wei Y. Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Brief Bioinform. 2017;18(4):682–697. [DOI] [PubMed] [Google Scholar]
- 257.Zheng H A novel individualized drug repositioning approach for predicting personalized candidate drugs for type 1 diabetes mellitus. Stat Appl Genet Mol Biol. 2019;9:18(5). [DOI] [PubMed] [Google Scholar]
- 258.Lei X, Yu J, Niu Q, Liu J, Fraering PC, Wu F. The FDA-approved natural product dihydroergocristine reduces the production of the Alzheimer’s disease amyloid-beta peptides. Sci Rep. 2015;5:16541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Torika N, Asraf K, Cohen H, Fleisher-Berkovich S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav Immun. 2017;64:80–90. [DOI] [PubMed] [Google Scholar]
- 260.Dong YF, Kataoka K, Tokutomi Y, et al. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J. 2011;25(9):2911–2920. [DOI] [PubMed] [Google Scholar]
- 261.Ongali B, Nicolakakis N, Tong XK, et al. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis. 2014;68:126–136. [DOI] [PubMed] [Google Scholar]
- 262.de Jong DLK, de Heus RAA, Rijpma A, et al. Effects of nilvadipine on cerebral blood flow in patients with Alzheimer disease. Hypertension. 2019;74(2):413–420. [DOI] [PubMed] [Google Scholar]
- 263.Sanz JM, Chiozzi P, Colaianna M, et al. Nimodipine inhibits IL-1beta release stimulated by amyloid beta from microglia. Br J Pharmacol. 2012;167(8):1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ou Z, Kong X, Sun X, et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun. 2018;69:351–363. [DOI] [PubMed] [Google Scholar]
- 265.Toba J, Nikkuni M, Ishizeki M, et al. PPARgamma agonist pioglitazone improves cerebellar dysfunction at pre-Abeta deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem Biophys Res Commun. 2016;473(4):1039–1044. [DOI] [PubMed] [Google Scholar]
- 266.Pan X, Gong N, Zhao J, et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. J Neurol. 2010;133(Pt 5):1342–1351. [DOI] [PubMed] [Google Scholar]
- 267.Yamamoto N, Fujii Y, Kasahara R, et al. Simvastatin and atorvastatin facilitates amyloid beta-protein degradation in extracellular spaces by increasing neprilysin secretion from astrocytes through activation of MAPK/Erk1/2 pathways. Glia. 2016;64(6):952–962. [DOI] [PubMed] [Google Scholar]
- 268.Garcia-Barroso C, Ricobaraza A, Pascual-Lucas M, et al. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology. 2013;64:114–123. [DOI] [PubMed] [Google Scholar]
- 269.Hunsberger HC, Weitzner DS, Rudy CC, et al. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J Neurochem. 2015;135(2):381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Nelson RL, Guo Z, Halagappa VM, et al. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205(1):166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Weinreb O, Badinter F, Amit T, Bar-Am O, Youdim MB. Effect of long-term treatment with rasagiline on cognitive deficits and related molecular cascades in aged mice. Neurobiol Aging. 2015;36(9):2628–2636. [DOI] [PubMed] [Google Scholar]
- 272.Sanchez PE, Zhu L, Verret L, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109(42):E2895–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Lin CH, Chen PK, Chang YC, et al. Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. 2014;75(9):678–685. [DOI] [PubMed] [Google Scholar]
- 274.Pierrot N, Lhommel R, Quenon L, et al. Targretin improves cognitive and biological markers in a patient with Alzheimer’s disease. J Alzheimers Dis. 2016;49(2):271–276. [DOI] [PubMed] [Google Scholar]
- 275.Kawahara K, Nishi K, Suenobu M, et al. Oral administration of synthetic retinoid Am80 (Tamibarotene) decreases brain beta-amyloid peptides in APP23 mice. Biol Pharm Bull. 2009;32(7):1307–1309. [DOI] [PubMed] [Google Scholar]
- 276.Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Folch J, Petrov D, Ettcheto M, et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev Neurother. 2015;15(6):587–596. [DOI] [PubMed] [Google Scholar]
- 278.Endres K, Fahrenholz F, Lotz J, et al. Increased CSF APPs-alpha levels in patients with Alzheimer disease treated with acitretin. Neurology. 2014;83(21):1930–1935. [DOI] [PubMed] [Google Scholar]
- 279.Ormerod AD, Thind CK, Rice SA, et al. Influence of isotretinoin on hippocampal-based learning in human subjects. Psychopharmacology. 2012;221(4):667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]