

Abstract
Electrophysiological approaches provide powerful tools to further our understanding of how different opioids affect signaling through opioid receptors; how opioid receptors modulate circuitry involved in processes such as pain, respiration, addiction, and feeding; and how receptor signaling and circuits are altered by physiologic challenges, such as injury, stress, and chronic opioid treatment. The use of genetic manipulations to alter or remove μ-opioid receptors (MORs) with anatomic and cell type specificity and the ability to activate or inhibit specific circuits through opto- or chemogenetic approaches are being used in combination with electrophysiological, pharmacological, and systems-level physiology experiments to expand our understanding of the beneficial and maladaptive roles of opioids and opioid receptor signaling. New approaches for studying endogenous opioid peptide signaling and release and the dynamics of these systems in response to chronic opioid use, pain, and stress will add another layer to our understanding of the intricacies of opioid modulation of brain circuits. This understanding may lead to new targets or approaches for drug development or treatment regimens that may affect both acute and long-term effects of manipulating the activity of circuits involved in opioid-mediated physiology and behaviors. This review will discuss recent advancements in our understanding of the role of phosphorylation in regulating MOR signaling, as well as our understanding of circuits and signaling pathways mediating physiologic behaviors such as respiratory control, and discuss how electrophysiological tools combined with new technologies have and will continue to advance the field of opioid research.
SIGNIFICANCE STATEMENT
This review discusses recent advancements in our understanding of μ-opioid receptor (MOR) function and regulation and the role of electrophysiological approaches combined with new technologies in pushing the field of opioid research forward. This covers regulation of MOR at the receptor level, adaptations induced by chronic opioid treatment, sites of action of MOR modulation of specific brain circuits, and the role of the endogenous opioid system in driving physiology and behavior through modulation of these brain circuits.
Introduction
In the recent past, the understanding of the action of opioids on single neurons has significantly advanced in two areas: at the level of receptor modulation and in actions on opioid-sensitive pathways in the central nervous system. At the receptor level, it has been established that phosphorylation of the C-terminal tail of the μ-opioid receptor (MOR) underlies acute agonist-dependent desensitization. Much of this has come from mutations of phosphorylation sites in MOR (Wang et al., 2002; Doll et al., 2011, 2012; Lau et al., 2011; Chen et al., 2013; Just et al., 2013; Moulédous et al., 2015; Miess et al., 2018) and the development of a selective and potent G-protein–coupled receptor kinase (GRK) inhibitor, compound101 (Thal et al., 2011; Lowe et al., 2015). Electrophysiological studies have been made in cell lines, neurons in brain slices with expressed receptors, and in brain slices from knockin animals expressing MORs lacking regulatory phosphorylation sites in the C-terminal tail of the receptor (Birdsong et al., 2013, 2015; Yousuf et al., 2015; Arttamangkul et al., 2018, 2019b; Miess et al., 2018; Kliewer et al., 2019). Both acute agonist-dependent desensitization and measures of long-term tolerance are reduced or eliminated in cells expressing phosphorylation-deficient mutant receptors. It has also become clear that acute desensitization is cell-dependent in that the degree of acute desensitization varies with the cell type and brain region. The degree of desensitization is dependent on the effector under study as well: desensitization measured using the activation of potassium conductance at the cell body is distinctly different from the inhibition of transmitter release measured at axon terminals. Knockin animals that express fluorescently tagged opioid receptors and the ability to covalently tag endogenous opioid receptors with fluorescent dyes has allowed appreciation of the extent of neurons and terminal fields that express opioid receptors (Scherrer et al., 2006; Erbs et al., 2015; Arttamangkul et al., 2019a). Selective optical activation of neuron terminals coupled with opioid receptor pharmacology has yielded a greater understanding of opioid-sensitive neural circuits. This approach, in combination with the ability to knock out receptors in various brain areas using conditional MOR knockout mice, has rapidly advanced the understanding of the central actions of opioids in controlling physiologic processes (Charbogne et al., 2017; Birdsong et al., 2019). Finally, studies on the role of endogenous opioids in the brain are now approachable with the combination of selective activation of peptide-containing neurons and the developing area of genetically expressed peptide sensors. With these tools, a better understanding of the extent and significance of the role of the opioid system in modulating physiologic and maladaptive processes, such as analgesia, respiration, feeding, reward, and addiction, will be appreciated.
Receptor Phosphorylation and Acute Desensitization.
Phosphorylation of G-protein–coupled receptors as a mechanism that underlies acute agonist-dependent desensitization has been known for decades and has now been firmly established for the MOR. Acute agonist-dependent desensitization refers to a decline in signaling in the continued presence of agonist such that the cellular response to an opioid decreases over time (Fig. 1, A and B). This occurs over a time course of minutes to tens of minutes and can be measured in real time using electrophysiological techniques. Work with phosphospecific antibodies, quantitative mass spectroscopy, and alanine mutations of the C terminus of MOR have indicated that multiple sites, including short cassettes, are the targets of phosphorylation in cell lines and cultured neurons (Lau et al., 2011; Chen et al., 2013; Just et al., 2013; Moulédous et al., 2015; Miess et al., 2018). Recent physiologic work on acute agonist-dependent desensitization has extended the understanding of the role of C terminus phosphorylation of MOR on downstream signaling (Birdsong et al., 2013, 2015; Arttamangkul et al., 2018, 2019b; Kliewer et al., 2019).
Fig. 1.
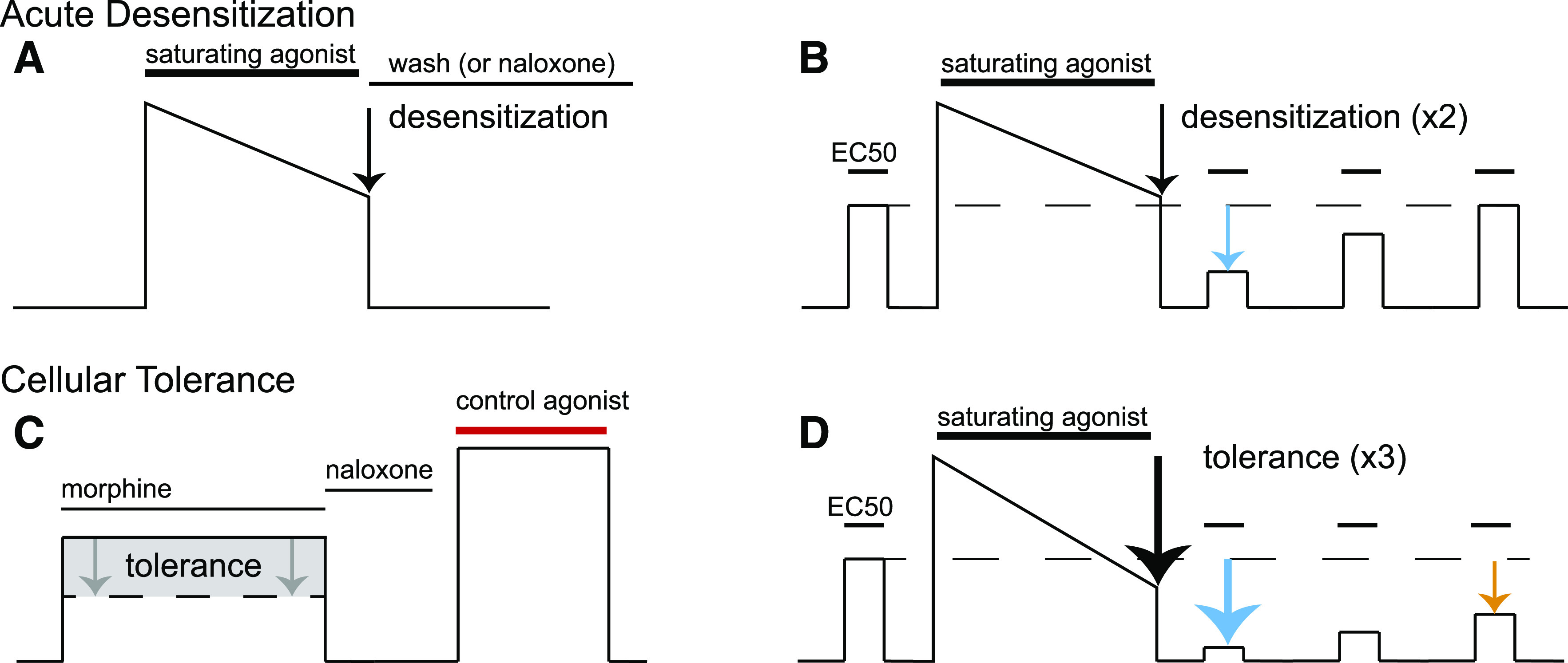
Illustrations of electrophysiological experimental measures of desensitization and cellular tolerance. Desensitization is generally measured in two ways: first as in (A), an acute decrease in the response, usually a current or voltage measurement, in the continued presence of a saturating concentration of agonist over a period of minutes (generally 5–10 minutes). The extent of this decrease in signaling is acute desensitization (black arrow). Reversal of this signaling by agonist washout or application of antagonist is used to ensure that the baseline measurement has not changed during the experiment. The second measure of desensitization (B) is used in preparations such as cell culture or with agonists that can quickly be washed out of brain slices like [Met5]enkephalin. First, a moderate concentration of agonist is applied to elicit approximately a half-maximal response (EC50). Next, a saturating concentration of agonist is applied for minutes, similar to the protocol shown in (A), to desensitize the receptor (black arrow). The saturating agonist is washed out of the preparation, and then the EC50 concentration of agonist is retested periodically to measure desensitization (cyan arrow) and the recovery from desensitization over time, which is nearly complete after 30–45 minutes. Cellular tolerance to chronic drug treatment is measured in several ways as well. (C) First, an agonist such as morphine is applied, and a response is measured; this is then reversed with an antagonist such as naloxone. Then, a control agonist that activates another receptor but ultimately activates the same downstream effector is tested (red bar). The relative response to morphine vs. the control agonist is measured. This is done in preparations from naïve animals (black line) or animals that have been chronically treated with morphine or other opioids for a period of days [generally 5–7 days (dotted line)]. The readout of cellular tolerance is a decrease in the response of morphine after chronic morphine treatment (gray shaded box, gray arrows) when normalized to the control agonist. (D) A second hallmark of opioid signaling in morphine-tolerant animals is an increase in desensitization after chronic morphine treatment. A protocol identical to that done in (B) is done on chronically morphine-treated mice; there is a characteristic increase in the decline in signaling in response to the saturating concentration of agonist (black arrow) as well as a smaller response to the EC50 concentration after acute desensitization (cyan arrow). Furthermore, the rate of recovery from desensitization is prolonged, and the recovery from desensitization is incomplete (orange arrow).
To study the regulation of opioid receptors, cultured cells have a substantial advantage over thick preparations such as brain slices because agonists and antagonists can be readily washed from the tissue, allowing rapid receptor dissociation of a variety of ligands of varying affinity and efficacy (Birdsong et al., 2013, 2015; Yousuf et al., 2015; Miess et al., 2018). The result is the ability to measure the time course of desensitization, the recovery from desensitization, and changes in ligand receptor binding kinetics. For example, a fluorescence-based assay was used to study the kinetics of ligand binding and dissociation in live cells (Birdsong et al., 2013, 2015). Fluorescently tagged ligands dermorphin-A594 and naltrexamine-A594 were used to visualize the association and dissociation rate of binding and dissociation from the plasma membrane in human embryonic kidney 293 cell line (HEK293) cells expressing an epitope-tagged MOR (FLAG-MOR). After a treatment of the cells with a saturating agonist concentration ([Met5]enkephalin) for tens of minutes to hours, a long-lasting increase in agonist, but not antagonist, affinity was observed. The increased affinity was not seen when phosphorylation sites on the C terminus were mutated to alanine. Surprisingly the increased affinity was found in cells in which arrestin2 and arrestin3 were knocked out. The conclusion was that strongly desensitizing agonists caused a gradual increase in agonist affinity that was intrinsic to the agonist-receptor interaction. The functional consequence of the increased affinity could not be determined in this assay. When examined in brain slices, there was a decrease in apparent affinity of receptors that remained functionally active after acute desensitization. This may suggest that the high-affinity state observed in HEK293 cells were less or not functional with regard to activating potassium conductances (Williams, 2014). It is also possible that the difference in the time of agonist exposure in the two experiments could account for the different results (20 and 120 minutes in HEK293 cells, 10 minutes in brain slices) such that there are rapid phosphorylation events/adaptations that occur over the time course of acute desensitization and slower phosphorylation events/adaptations that occur over hours or even days that would be more akin to cellular tolerance. Nonetheless, the increased off rate in the functional receptors measured in brain slices could contribute to the decrease in signaling associated with acute desensitization and the development of tolerance, but the precise mechanism has not been determined.
The downstream activation of potassium conductance is an ideal analog signal used to examine receptor-dependent desensitization. The desensitization induced by both [Met5]enkephalin (ME) and morphine in AtT20 cells expressing physiologically relevant levels of wild-type and mutant (11S/t-A) MORs demonstrated that alanine mutations in the C terminus eliminated desensitization induced by ME but not morphine (Yousuf et al., 2015). The desensitization induced by morphine in mice lacking all potential phosphorylation sites in the C-terminal tail (11S/T-A MOR, Fig. 2) was blocked after treatment with the C-kinase inhibitor, calphostin C. Interestingly, the desensitization induced by morphine was heterologous in that the current mediated by activation of the somatostatin receptor was reduced after desensitization with morphine. Thus, in AtT20 cells, C terminus phosphorylation of the receptor induced by ME and a secondary adaptation induced by morphine underlie acute desensitization. Similar experiments with morphine have not been possible in brain slices because of the slow washout of morphine.
Fig. 2.
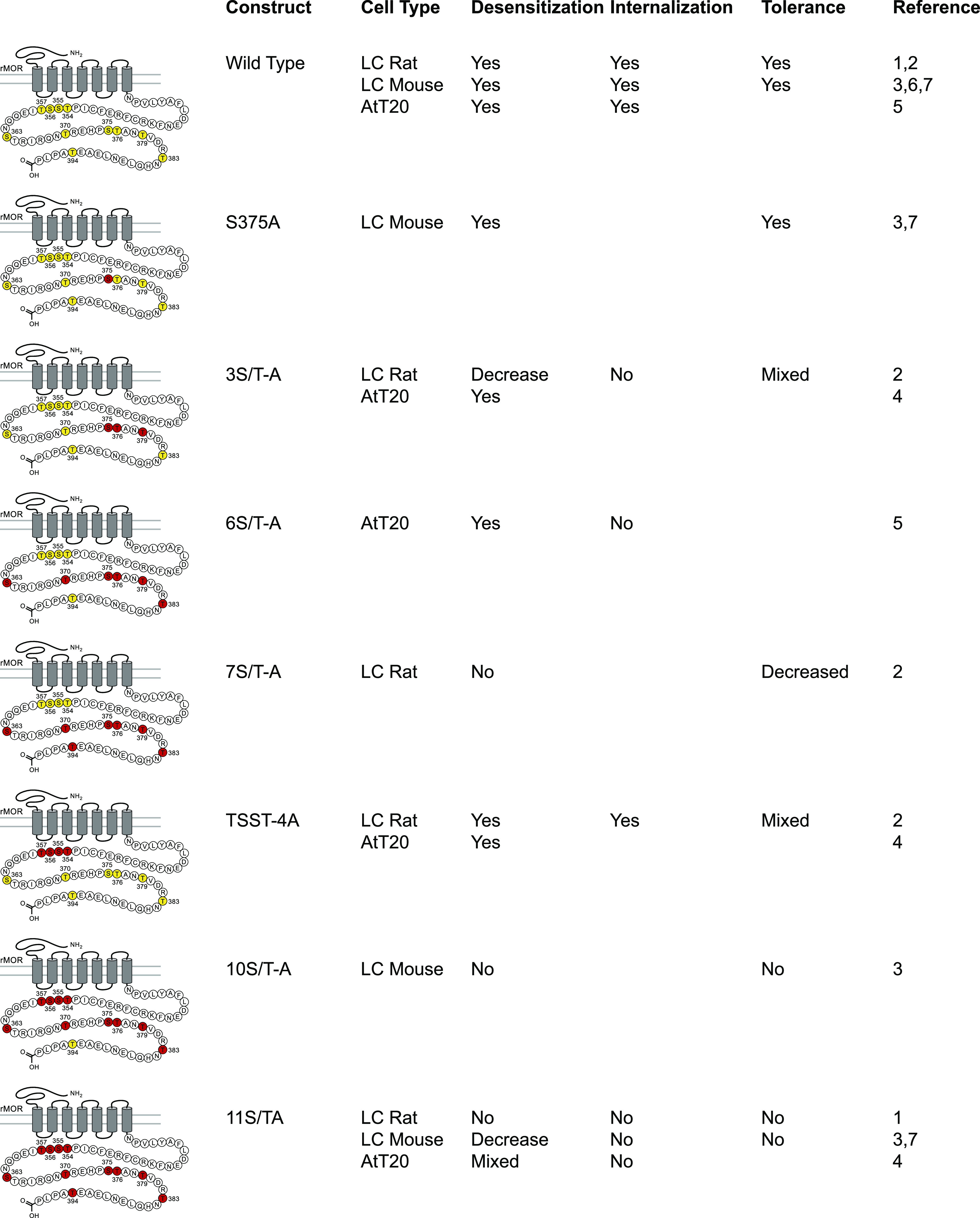
Summary of electrophysiological data examining receptor desensitization, and tolerance dependence on potential C-terminal phosphorylation sites in MOR. Eleven potential serine and threonine phosphorylation sites in the C-terminal tail of rMOR are highlighted in yellow in the wild-type rMOR (these sites are identical in rat and mouse). Potential phosphorylation sites were mutated to alanine, indicated by the red highlighting of residues, in several studies that are summarized here. These mutations are named under the Construct column. The effects of these mutations on the processes of receptor desensitization, internalization, and cellular tolerance have been measured in various systems and have been reported. Processes that remained intact in the mutant receptor are indicated with a “Yes,” whereas “No” indicates elimination of these processes, “Decreased” indicates a partial effect, and “Mixed” indicates that different assays or ligands provided differing results [sources: 1) Arttamangkul et al., 2018; 2) Arttamangkul et al., 2019b; 3) Kliewer et al., 2019; 4) Miess et al., 2018; 5) Yousuf et al., 2015; 6) Quillinan et al., 2011, 7) J. Williams, personal communication]. LC, locus coeruleus; rMOR, rat mu-opioid receptor.
The study of desensitization using neuronal recordings in brain slices has taken advantage of the ability to virally express phosphorylation-deficient mutant MOR receptors in knockout animals (mouse and rat, Birdsong et al., 2015; Arttamangkul et al., 2018, 2019b) as well as the development of knockin animals (mouse, Kliewer et al., 2019) that express alanine mutations on the MOR C terminus. Whether virally expressed or in knockin animals, desensitization was blocked when studying receptors in which all 11 phosphorylation sites were mutated to alanine. One advantage of using viral expression of mutant receptors in MOR knockout animals, both mouse and rat, is that the role of each of the phosphorylation sites in the C terminus can be determined. Although each of the phosphorylation sites play some role in acute desensitization, mutations in the sequence from S375 to T380 resulted in a much-decreased acute desensitization (Fig. 2). Additional alanine mutations resulted in greater inhibition of acute desensitization.
Cellular Tolerance and Acute Desensitization: Are They Separate Processes?
Whereas acute desensitization refers to the loss of signaling over several minutes in the continued presence of an agonist, cellular tolerance refers to adaptations at the cellular level that result in reduced sensitivity to opioids that are induced over days to weeks and can last after the drug’s removal. Both desensitization and cellular tolerance have been measured by a decrease in receptor-dependent activation of potassium conductance. The decrease in signaling has been measured in several ways. Acute desensitization has been measured first as a decline in potassium current (or membrane potential) in the continued presence of a saturating concentration of agonist (Fig. 1A). The second measure is the relative decrease in current induced by a lower concentration of agonist after the application of a saturating concentration of agonist (Fig. 1B), and these measures are not necessarily reflective of the same processes. Cellular tolerance has been measured by the decrease in the current induced by morphine (Fig. 1C). This assay was developed because morphine is a partial agonist and therefore particularly sensitive to a decrease in effector coupling (Christie et al., 1987). The current induced by morphine is normalized to the current induced by the activation of a second Gi-coupled receptor that activates the same potassium conductance. In the locus coeruleus, the α-2-adrenoceptor is most commonly used. The second assay to measure tolerance uses repeated applications of a low concentration of agonist before and after treatment with a saturating concentration (Dang and Williams, 2004). This assay compares recordings from brain slices taken from naïve and chronically treated animals (Fig. 1D). Signs of tolerance are 1) a larger decrease in the current induced by a saturating concentration of agonist, 2) a larger decrease in the current induced by a lower concentration of agonists after washout of the saturating concentration, and 3) a decrease in the rate or extent of recovery from acute desensitization (Fig. 1D).
Both knockin and virally expressed, phosphorylation-deficient MORs have been used to characterize the role of phosphorylation sites in the C-terminal tail of MOR in regulating both acute desensitization and tolerance. Acute desensitization, cellular tolerance, and analgesic tolerance are all clearly affected by mutation of MOR phosphorylation sites (Arttamangkul et al., 2018, 2019b, Kliewer et al., 2019). However, the specific phosphorylation sites or overall degree of phosphorylation may affect the relative degree of acute desensitization and cellular tolerance. For example, mutation of the sequence 354TSST357 to alanine or glutamate affected some measures of tolerance without affecting the rate of acute desensitization (Arttamangkul et al., 2019b). Likewise, alanine mutation of the 375STANT379 sequence eliminated measures of tolerance while reducing but not eliminating desensitization. Alanine mutation of other residues further decreased the degree of acute desensitization, with variable effects on cellular tolerance (Arttamangkul et al., 2018, 2019b). Thus, there may be some specificity in which phosphorylation sites regulate tolerance and desensitization, and this specificity may be agonist-dependent. For example, knockin mice harboring the S375A mutation display acute desensitization to ME and cellular and analgesic tolerance to morphine that is indistinguishable from wild-type mice, but these mice appear to not develop analgesic tolerance to fentanyl (Kliewer et al., 2019), suggesting that different phosphorylation sites may play a role in tolerance to different agonists.
Although the sequence from S375 to T379 in MOR appears to be the primary site involved in acute desensitization, the story is likely more complicated. Using phosphospecific antibodies, it has been demonstrated that deleting phosphorylation sites of S375 to T379 (and S375 alone) prevents or greatly attenuates the rate and extent of phosphorylation at other sites (Doll et al., 2011; Miess et al., 2018). Alanine mutation of the 354TSST357 cluster also modestly decreased the rate of phosphorylation of other nonmutated residues as well. Therefore, phosphorylation and/or protein binding to serine and threonine residue at these phosphorylation sites likely facilitates or catalyzes phosphorylation of other residues in the C terminus of MOR. Thus, mutation of one residue could cause changes in the extent or kinetics of phosphorylation of many residues. This may be particularly important during acute desensitization when the kinetics of receptor phosphorylation are on the same timescale as the kinetics of desensitization (Doll et al., 2011; Yousuf et al., 2015).
Because of the interconnectedness of phosphorylation in the C terminus of MOR, it has been difficult to determine whether individual phosphorylation sites regulate specific processes or whether the bulk amount of C terminus phosphorylation is responsible for receptor desensitization, internalization, and long-term tolerance. Investigating effector systems using multiple assays and timescales may be a useful approach to untangle these processes. For example, mutation of the TSST cassette blunts stable recruitment of arrestin measured with β-galactosidase complementation assay (slow timescale) but not a bioluminescent resonant energy transfer assay (rapid timescale), whereas mutation of the STANT cassette decreased β-arrestin recruitment using both assays. These results suggest that individual phosphorylation sites may play specific complementary roles in regulating opioid signaling and arrestin binding (Miess et al., 2018) depending on the time course of agonist exposure and agonist efficacy. While the exact effects that receptor phosphorylation has on opioid receptor signaling are becoming clear, progress is also being made on connecting these phosphorylation events to specific kinases.
Only recently has it been possible to pharmacologically block acute desensitization using recordings from brain slices from wild-type animals. Phosphorylation of the C-terminal tail of MOR by high-efficacy agonists has been demonstrated to depend on GRK2/3 (Doll et al., 2012). The GRK2/3 inhibitor, compound101, effectively blocked acute opioid desensitization in locus coeruleus neurons (Lowe et al., 2015). Although the inhibition of GRK was expected to block or reduce acute desensitization, there is a substantial literature that implicates several other kinases, notably PKC, JNK, and extracellular signaling-related kinase 1/2 (Bailey et al., 2004, 2009; Dang et al., 2009, reviewed Bailey et al., 2006; reviewed Williams et al., 2013). It is therefore somewhat surprising that inhibition of GRK is so effective. One potential explanation is that receptor phosphorylation by GRKs is rapid, wheras kinase activation and receptor phosphorylation by other kinases is slow relative to the time course of acute desensitization.
A role of PKC in acute desensitization was proposed in locus coeruleus neurons based on an increase in the rate and extent of desensitization induced by ME and morphine after the activation of PKC by phorbol esters and muscarinic receptors (Bailey et al., 2006, 2009). Experiments in AtT20 cells using phosphorylation-deficient receptors found that, although acute desensitization induced by peptide agonists was blocked, morphine-induced desensitization was unaffected and only blocked by the combination of inhibition of PKC and mutation of C-terminal phosphorylation sites (Yousuf et al., 2015; Miess et al., 2018). The interpretation was that PKC-mediated desensitization was mediated by phosphorylation of an accessory molecule rather than MOR. The increase in acute desensitization induced by phorbol esters has not been reproduced in the locus coeruleus; however, there is agreement that PKC contributes to the short-term tolerance observed after chronic morphine treatment (Bailey et al., 2009; Levitt and Williams, 2012; Arttamangkul et al., 2015).
Recent work with brain slices taken from morphine-treated animals (6 to 7 days) had found that compound101 is less effective at blocking one component of acute desensitization after chronic morphine treatment (Leff and Williams, unpublished observations). When the combination of kinase inhibitors to block both PKC and JNK was used, acute desensitization induced by ME was blocked in chronic morphine–treated animals, suggesting that chronic morphine treatment shifts kinase regulation of acute desensitization from predominantly GRK-mediated to GRK-, PKC-, and JNK-mediated. Unlike the heterologous morphine-induced desensitization in the AtT20 cells that was blocked by calphostin C (Yousuf et al., 2015), this mechanism was blocked in phosphorylation-deficient receptors in slices from morphine-treated animals, suggesting a direct action on the receptor. It is also possible that the continued signaling and/or the lack of internalization of the phosphorylation-deficient receptors in the brain slice experiments disrupted the activation of the PKC/JNK-dependent process. The underlying mechanism for the development of PKC/JNK-dependent tolerance remains to be determined. It is necessary to determine by what mechanism this adaptation is induced and for how long it persists after withdrawal. Also, does chronic treatment with other agonists of varying efficacy, such as fentanyl and buprenorphine, induce this remarkable adaptation? It has been known for some time that, after chronic treatment with morphine, acute desensitization is increased, and the recovery from desensitization is prolonged (Dang and Williams, 2004; Quillinan et al., 2011; Levitt and Williams, 2012). Therefore, the induction of a GRK2/3-insensitive mechanism may account for the increased acute desensitization after chronic morphine treatment.
Is Receptor Phosphorylation the Whole Story?
The activation of potassium conductance in neurons of the locus coeruleus has served as a model system for the study of MOR desensitization for many years. It is now apparent that there is considerable variation in the extent of acute receptor desensitization across brain regions. In locus coeruleus, the decline in the peak outward current is about 50% during the application of a saturating concentration of an efficacious agonist. There are examples of neurons in which the decline in current is nearly complete (cholinergic striatal interneurons, personal observation) and other examples such as neurons in the Kolliker-Fuse in which there is little or no acute decline (Levitt and Williams, 2018). Further, in the locus coeruleus, mechanisms of desensitization may change over time, as it has been observed that desensitization in older rats was homologous (occurring at MOR), whereas desensitization in younger rats appeared heterologous (affecting signaling through multiple receptors) (Llorente et al., 2012). It is not clear which mechanisms underlie the differences between neuronal subtypes or developmental stages, and there is little in the literature that has directly addressed the potential differences. From expression systems, it is clear that altering receptor number, signaling and regulatory components (such as GRKs and arrestins), or effectors can alter the efficacy, potency, and signaling induced by opioids (Whistler and von Zastrow, 1998; Zhang et al., 1998).
There is also a rapid form of apparent desensitization that has been proposed to be mediated by the GRK-dependent chelation of β/γ-subunits resulting in dissociation from the potassium channel (Raveh et al., 2010; Yousuf et al., 2015; reviewed Gurevich et al. (2012)). Thus, the extent and time course of desensitization is dependent on the expression of multiple downstream processes, which can vary widely among different cells.
Desensitization Measured with Different Effectors.
There are three electrophysiological measures that are commonly used to study the action of opioids: activation of potassium current, inhibition of calcium current, and inhibition of transmitter release. The activation of potassium current is by far the most commonly used measure to study desensitization. It is now certain that there is no detectable desensitization when one measures opioid receptor–dependent inhibition of transmitter release (Blanchet and Luscher, 2002). Manipulations such as decreasing the receptor reserve with the irreversible blockade of receptors with β-chlornaltrexamine, chronic morphine treatment, or prolonged incubation with a saturating concentration of agonist have not uncovered any evidence that the inhibition of transmitter release is affected by receptor desensitization (Fyfe et al., 2010; Pennock and Hentges, 2011, 2016; Pennock et al., 2012; Fox and Hentges, 2017).
The reason for the lack of presynaptic desensitization has been unknown. Based on recent receptor trafficking experiments, however, the lack of apparent presynaptic inhibition has been proposed to result from rapid diffusion of activated receptors along the cell surface from the axon to release sites, effectively buffering presynaptic inhibition from the loss of functional receptors due to internalization (Jullié et al., 2020). This work showed that MORs were internalized only in areas of transmitter release (axon varicosities) and not along the axons. This internalization was phosphorylation-dependent, and receptors were replenished in varicosities through local recycling and lateral diffusion of MORs from the axon to the varicosity. That work went on to show that the MOR agonist DAMGO inhibited transmitter release, measured by the imaging of synaptic vesicle exocytosis. Thus, receptor recycling and rapid lateral diffusion of receptors from the axon, where internalization does not occur, to release sites may provide a relatively constant pool of functional MOR in spite of receptor internalization and explain this apparent dichotomy between desensitization seen in postsynaptic but not presynaptic compartments (Jullié et al., 2020).
Floxed Receptors.
The understanding of the long-distance pathways affected by opioids is now possible by combining the use of the floxed MOR animals (Weibel et al., 2013) to knockout MORs in select neuronal populations (Bachmutsky et al., 2020; Varga et al., 2020). This approach has been used to increase understanding of the action of opioids that mediate respiratory depression (Bachmutsky et al., 2020; Varga et al., 2020). The regulation of respiration is mediated by multiple nuclei. Most work on the depression of respiration induced by opioids centered on the pre-Botzinger complex. Recent work found that both the Kolliker-Fuse and pre-Botzinger complex are key sites of opioid action, that the two nuclei mediated depression in different concentration ranges, and that a very small number of neurons in the pre-Botzinger complex are opioid-sensitive. Neurons in each nucleus are hyperpolarized by opioids; however, these areas are highly interconnected, indicating that presynaptic inhibition of the reciprocal connections most likely plays a key role in the overall action of opioids (Varga et al., 2020). Although the regulation of respiration involves a wide range of processes spanning multiple brain regions, the selective deletion of MORs in specific brain regions is beginning to untangle the actions of opioids in this complex system.
When MORs were selectively removed in GABA forebrain neurons, largely in the dorsal striatum and nucleus accumbens, using floxed MOR: Dlx5/6-Cre (Distal-Less Homeobox 5/6- cre recombinase) mice, locomotor effects of heroin were abolished, whereas measures of morphine analgesia and dependence were unaffected, and motivation to obtain rewards was increased (Charbogne et al., 2017). In the ventral tegmental area, GABA inhibitory postsynaptic currents measured on GABA neurons were rendered insensitive to DAMGO [whereas GABA inhibitory postsynaptic currents measured on dopamine neurons that were evoked by local stimulation remained sensitive to inhibition by DAMGO (Charbogne et al., 2017)]. These data demonstrate the ability to dissociate opioid effects on different behaviors and different subcircuits that are likely to mediate these behaviors. Although we are making progress in understanding how MOR in specific synapses and cell types is modulating drug-induced behavior, our understanding of the effects of the endogenous opioid system is still lagging behind.
Endogenous Opioids.
With the discovery of endogenous opioid peptides in the mid 1970s, there was great excitement over the determination of the physiologic role of these endogenous peptides. There is an enormity of work using exogenous application of opioid peptides to activate opioid receptors in multiple brain areas. Opioid peptides activate receptors at concentrations low enough that one would expect to observe functional consequences after the release of endogenous peptides, yet it has been incredibly difficult to detect release of endogenous opioids and study them at the cellular level, at least in brain slices or cultured neurons. One prominent hypothesis for the inability to detect the functional actions of endogenous peptides is that the method(s) used to evoke peptide release have simply not been appropriate. Studies to date have used electrical stimulation. One early study came from work at the mossy fiber synapse in the hippocampus (Weisskopf et al., 1993). In that study, selective stimulation of the mossy fibers with 10 pulses at 100 Hz four times resulted in the release of dynorphin (presumed) that was antagonized by naloxone and nor-binaltorphimine BNI (at what was most likely a concentration that blocked both μ- and κ-opioid receptors). Likewise, in the striatum, a stimulus of five pulses at 100 Hz evoked a transient release of presumably enkephalin, which inhibited glutatmate release. The inhibition of glutamate release was occluded by the MOR agonist DAMGO and blocked by the MOR antagonist CTOP (Blomeley and Bracci, 2011). A more recent study in the amygdala found release using pairs of stimuli (Winters et al., 2017). Peptidase inhibitors were used to prevent degradation of opioid peptides and thus prolong the presence of the peptides in the extracellular space to amplify the activation of receptors (Atwood et al., 2014; Winters et al., 2017). After the cocktail of peptidase inhibitors, both the amplitude and duration of endogenously released opioid was increased. Given that the distribution of peptide-containing neurons and projections is well known, as is the widespread distribution of opioid receptors, it is surprising that more has not been done at a cellular level; however, knowledge about methods to induce peptide release and systems for rapid detection of peptides has been lacking. Discovery of the functional roles of endogenous peptides in the opioid system at the cellular level is an important step in a complete understanding of opioids in the brain.
Recent technical developments may be used to foster a better understanding of peptide release and subsequent receptor activation. Difficulties in the detection of opioid peptide release using electrical stimulation may be problematic given that this form of stimulation nonselectively activates multiple pre- and postsynaptic neurons, particularly when applied at a high frequency or intensity. Recent advances in the ability to selectively activate peptide-containing neurons, and specifically opioid peptide–containing neurons, using optogenetics could potentially improve the ability to evoke peptide release. Dynorphin release has been measured using an optogenetic approach; however, the relatively low-frequency stimulation used to maintain action potential firing with channelrhodopsin necessitated long stimulation periods to measure peptide accumulation (Al-Hasani et al., 2015). The development of genetically expressed peptide sensors that increase fluorescence upon binding of peptides is a promising avenue for peptide detection. These sensors are based on G-protein–coupled receptors along with a circularly permuted GFP. The binding affinity is, for the most part, similar to that of endogenous opioid receptors (Patriarchi et al., 2018). The plasma membrane localization and the similarity in affinity between these molecules and opioid receptors makes them ideally suited for the localized detection of peptide transmitter. The sensors can be expressed in a wide area such that imaging the increase in fluorescence can be examined at low magnification, allowing both the site(s) of release and the extent of diffusion to be measured. Most importantly, with a rapid and robust detection method, the mechanisms that underlie the release of peptides can be determined.
Although this review focuses on μ-opioid receptor signaling, endogenous enkephalins and endorphins can activate both μ- and δ-opioid receptors, and it is possible for the κ-opioid selective peptide dynorphin to be metabolized to [Leu]5enkephalin. Thus, endogenous peptides can activate multiple opioid receptors, and neurons within local circuits can express various opioid receptors and serve opposing functions. δ-Opioid receptors inhibit GABA release from patches of MOR-rich neurons in striatum (Banghart et al., 2015). The activation of MORs in the striatum decreases excitatory afferents from thalamic projections, whereas δ-opioid receptor activation disinhibits neurons in the anterior cingulate cortex to increase excitatory afferent input to the striatum (Birdsong et al., 2019). Thus, the site(s) of receptor activation can have diverse effects on the final output of the medium spiny neurons in the striatum. Additionally, receptor phosphorylation, trafficking, and signaling to downstream effectors all dynamically regulate the function of opioid receptors. Therefore, understanding the temporal and spatial dynamics of how endogenous opioid peptides are sensed by both pre- and postsynaptic opioid receptors will be a key step in creating a clearer picture of how these peptides mediate their diverse physiologic effects.
To this end, the cellular distribution of opioid receptors has been explored using knockin animals that express receptors tagged with fluorescent ligands (Scherrer et al., 2006; Erbs et al., 2015). Early work imaging GFP-labeled δ-receptors in neurons cultured from knockin mice were used to characterize the internalization of those receptors (Scherrer et al., 2006). A chemistry-based approach has been developed recently called “traceless affinity labeling” (Hayashi and Hamachi, 2012). This method used naltrexamine to guide a reactive molecule to opioid receptors. Once the naltrexamine becomes bound to the receptor, the local concentration of the reactive molecule is high enough to enable a covalent reaction with the receptor. This reaction places a fluorescent tag on the receptor and at the same time cleaves the link with naltrexamine. The naltrexamine is then free to dissociate from the receptor. Thus, functional endogenous opioid receptors in wild-type animals are covalently bound to fluorescent ligands (Arttamangkul et al., 2019a). This approach is ideal for the identification of opioid-sensitive neurons in living brain slices. With the use of charged fluorescent molecules, only plasma membrane–associated receptors are labeled. Opioid receptor positive neurons can be identified in preparations of heterogeneous populations of neurons. This approach in combination with selective activation of peptide-containing neurons and the detection of peptide release with the sensors has the potential for a complete characterization of endogenous opioid communication.
Electrophysiology of Biased Agonists.
There has been intense interest in the development of biased agonists that can maintain the therapeutic actions of opioid while limiting on-target side effects. The differential activation of G-proteins and recruitment of arrestin by agonists has been the focus of considerable work and has been reviewed extensively (Rivero et al., 2012; Hill et al., 2018; Conibear and Kelly, 2019; Gurevich and Gurevich, 2019). Decades of electrophysiological work has demonstrated various mechanisms by which the excitability of neurons is decreased and neurotransmission is inhibited by opioid receptor–mediated activation of heterotrimeric G-proteins. From this body of work, G-protein–mediated activation of potassium channels, inhibition of voltage gated calcium channel, inhibition of adenylyl cyclase, and SNARE protein function have been well established (Logothetis et al., 1987; Ikeda, 1996; Blackmer et al., 2001; Zurawski et al., 2019). However, to our knowledge, there is no indication as to how arrestin signaling could inhibit neurotransmission on the timescale that is close to G-protein–dependent processes. That is not to say that arrestin signaling is not physiologically relevant, but at this time, there are no electrophysiological measures of arrestin-dependent processes. Because the phosphorylation deletion mutants of MOR do not recruit arrestin, they can be considered “G-protein–biased” receptors. Activation of these receptors still inhibits neurotransmission through activation of pre- and postsynaptic mechanisms as well as or better than the wild-type receptor. Additionally, these G-protein–biased receptors still effectively induce analgesia, respiratory depression, and withdrawal, suggesting arrestin is not involved in these processes (Kliewer et al., 2019). In agreement with data from β-arrestin2 knockout mice (Connor et al., 2015), it is clear that tolerance at the electrophysiological and behavioral level is severely impaired in these mice, supporting a role for arrestin in these processes and suggesting that tolerance could be dissociated from the other effects of opioids that are associated with G-protein signaling.
Studies of agonist bias have used a combination of electrophysiological, cell biologic, and biochemical approaches in multiple cell-based assays and have concluded that agonist efficacy, receptor reserve, and the amplification of downstream signaling are important factors in the determination of receptor bias (Rivero et al., 2012; Miess et al., 2018; reviewed Conibear and Kelly, 2019). Thus, downstream signaling and the particular assays studied impact the interpretation of agonist bias. The hypothesis that G protein-coupled receptors (GPCRs) move through multiple states that result in association with different downstream molecules has been tested recently (Stoeber et al., 2020). Using engineered molecules that mimic G-proteins (mGsi) and GRKs (Nb33), this study demonstrated that different agonists resulted in distinct association of the two molecules with opioid receptors (Stoeber et al., 2020). The engineered probes were functionally inactive and known to bind selectively to agonist-bound receptors such that probe binding directly reflected agonist-dependent GPCR conformations. In experiments with the κ-opioid receptor, there was a distinct difference in the association of the engineered molecules that was agonist-dependent. Dynorphin-bound κ-receptors increased association of both mGsi and Nb33, whereas etorphine-bound receptors only bound mGsi. Similar differential results were obtained using MOR with DAMGO and the partial agonist mitragynine pseudoindoxyl. The demonstration of agonist-dependent receptor association with downstream molecules supports the hypothesis that GPCRs move through distinct conformations that foster association with different downstream molecules. These conformations are the underlying mechanism of agonist bias (Stoeber et al., 2020). How these conformations relate to physiologic processes will be an important avenue for future research.
Summary
Electrophysiological tools have been used for decades to complement structural, molecular, pharmacological, and physiologic/behavioral methods to advance the understanding of opioid receptor function from the molecular to whole-animal level. This review focused on recent advances in the understanding of MOR function using an electrophysiological approach. It is clear that receptor phosphorylation plays a key role in mediating MOR desensitization and tolerance. This appears to be mediated by phosphorylation of the C terminus of MOR acutely by GRKs and after chronic opioid treatment by other kinases (PKC and JNK). Using optogenetics, imaging, and newly generated mouse lines, the modulation of specific neuronal populations by opioids has broadened the understanding of the opioid system. The knowledge of the cellular location, function, and dynamics of opioid receptors and the endogenous opioid pathways is key to the understanding of the actions of opioids on analgesia, respiration, feeding, reward, and addiction. These investigations have the potential to yield improved therapeutics for pain relief and addiction treatment.
Acknowledgments
We would like to thank Seksiri Arttamangkul and Sweta Adhikary for helpful comments in preparation of the manuscript.
Abbreviations
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- GPCR
G protein-coupled receptor
- GRK
Gprotein–coupled receptor kinase
- HEK293
human embryonic kidney 293 cell line
- JNK
c-Jun N-terminal kinase
- ME
[Met5]enkephalin
- MOR
μ-opioid receptor
- PKC
protein kinase C
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Birdsong, Williams.
Footnotes
This work was supported by the National Institutes of Health/National Institute of Drug Abuse Grants [R01DA042779, R01DA08136].
References
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, et al. (2015) Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Birdsong W, Williams JT. (2015) Does PKC activation increase the homologous desensitization of μ opioid receptors? Br J Pharmacol 172:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Heinz DA, Bunzow JR, Song X, Williams JT. (2018) Cellular tolerance at the µ-opioid receptor is phosphorylation dependent. eLife 7:e343989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Leff ER, Koita O, Birdsong WT, Williams JT. (2019b) Separation of acute desensitization and long-term tolerance of µ-opioid receptors is determined by the degree of C-terminal phosphorylation. Mol Pharmacol 96:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Plazek A, Platt EJ, Jin H, Murray TF, Birdsong WT, Rice KC, Farrens DL, Williams JT. (2019a) Visualizing endogenous opioid receptors in living neurons using ligand-directed chemistry. eLife 8:e49319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. (2014) Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci 17:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmutsky I, Wei XP, Kish E, Yackle K. (2020) Opioids depress breathing through two small brainstem sites. eLife 9:e52694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G. (2004) Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol 66:1592–1598. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, Henderson G. (2009) Role of protein kinase C and mu-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci 29:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G. (2006) How important is protein kinase C in mu-opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci 27:558–565. [DOI] [PubMed] [Google Scholar]
- Banghart MR, Neufeld SQ, Wong NC, Sabatini BL. (2015) Enkephalin disinhibits mu opioid receptor-rich striatal patches via delta opioid receptors. Neuron 88:1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsong WT, Arttamangkul S, Bunzow JR, Williams JT. (2015) Agonist binding and desensitization of the µ-opioid receptor is modulated by phosphorylation of the C-terminal tail domain. Mol Pharmacol 88:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsong WT, Arttamangkul S, Clark MJ, Cheng K, Rice KC, Traynor JR, Williams JT. (2013) Increased agonist affinity at the μ-opioid receptor induced by prolonged agonist exposure. J Neurosci 33:4118–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsong WT, Jongbloets BC, Engeln KA, Wang D, Scherrer G, Mao T. (2019) Synapse-specific opioid modulation of thalamo-cortico-striatal circuits. eLife 8:e45146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. (2001) G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science 292:293–297. [DOI] [PubMed] [Google Scholar]
- Blanchet C, Lüscher C. (2002) Desensitization of mu-opioid receptor-evoked potassium currents: initiation at the receptor, expression at the effector. Proc Natl Acad Sci USA 99:4674–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeley CP, Bracci E. (2011) Opioidergic interactions between striatal projection neurons. J Neurosci 31:13346–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbogne P, Gardon O, Martín-García E, Keyworth HL, Matsui A, Mechling AE, Bienert T, Nasseef MT, Robé A, Moquin L, et al. (2017) Mu opioid receptors in gamma-aminobutyric acidergic forebrain neurons moderate motivation for heroin and palatable food. Biol Psychiatry 81:778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Oldfield S, Butcher AJ, Tobin AB, Saxena K, Gurevich VV, Benovic JL, Henderson G, Kelly E. (2013) Identification of phosphorylation sites in the COOH-terminal tail of the μ-opioid receptor. J Neurochem 124:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, North RA. (1987) Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol 32:633–638. [PubMed] [Google Scholar]
- Conibear AE, Kelly E. (2019) A biased view of µ-opioid receptors. Mol Pharmacol 96:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Bagley EE, Chieng BC, Christie MJ. (2015) β-Arrestin-2 knockout prevents development of cellular μ-opioid receptor tolerance but does not affect opioid-withdrawal-related adaptations in single PAG neurons. Br J Pharmacol 172:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Napier IA, Christie MJ. (2009) Two distinct mechanisms mediate acute μ-opioid receptor desensitization in native neurons. J Neurosci 29:3322–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT. (2004) Chronic morphine treatment reduces recovery from opioid desensitization. J Neurosci 24:7699–7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Konietzko J, Pöll F, Koch T, Höllt V, Schulz S. (2011) Agonist-selective patterns of µ-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol 164:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Pöll F, Peuker K, Loktev A, Glück L, Schulz S. (2012) Deciphering µ-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol 167:1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, et al. (2015) A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct 220:677–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PD, Hentges ST. (2017) Differential desensitization observed at multiple effectors of somatic µ-opioid receptors underlies sustained agonist-mediated inhibition of proopiomelanocortin neuron activity. J Neurosci 37:8667–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe LW, Cleary DR, Macey TA, Morgan MM, Ingram SL. (2010) Tolerance to the antinociceptive effect of morphine in the absence of short-term presynaptic desensitization in rat periaqueductal gray neurons. J Pharmacol Exp Ther 335:674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJG, Mushegian A, Gurevich VV. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 133:40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. (2019) GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hamachi I. (2012) Traceless affinity labeling of endogenous proteins for functional analysis in living cells. Acc Chem Res 45:1460–1469. [DOI] [PubMed] [Google Scholar]
- Hill R, Disney A, Conibear A, Sutcliffe K, Dewey W, Husbands S, Bailey C, Kelly E, Henderson G. (2018) The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br J Pharmacol 175:2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. (1996) Voltage-dependent modulation of N-type calcium channels by G-protein β γ subunits. Nature 380:255–258. [DOI] [PubMed] [Google Scholar]
- Jullié D, Stoeber M, Sibarita JB, Zieger HL, Bartol TM, Arttamangkul S, Sejnowski TJ, Hosy E, von Zastrow M. (2020) A discrete presynaptic vesicle cycle for neuromodulator receptors. Neuron 105:663–677.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just S, Illing S, Trester-Zedlitz M, Lau EK, Kotowski SJ, Miess E, Mann A, Doll C, Trinidad JC, Burlingame AL, et al. (2013) Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol Pharmacol 83:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, Schulz S. (2019) Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 10:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau EK, Trester-Zedlitz M, Trinidad JC, Kotowski SJ, Krutchinsky AN, Burlingame AL, von Zastrow M. (2011) Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci Signal 4:ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt ES, Williams JT. (2012) Morphine desensitization and cellular tolerance are distinguished in rat locus ceruleus neurons. Mol Pharmacol 82:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt ES, Williams JT. (2018) Desensitization and tolerance of mu opioid receptors on pontine Kolliker-Fuse neurons. Mol Pharmacol 93:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente J, Lowe JD, Sanderson HS, Tsisanova E, Kelly E, Henderson G, Bailey CP. (2012) μ-Opioid receptor desensitization: homologous or heterologous? Eur J Neurosci 36:3636–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. (1987) The β γ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325:321–326. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Sanderson HS, Cooke AE, Ostovar M, Tsisanova E, Withey SL, Chavkin C, Husbands SM, Kelly E, Henderson G, et al. (2015) Role of G protein-coupled receptor kinases 2 and 3 in µ-opioid receptor desensitization and internalization. Mol Pharmacol 88:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miess E, Gondin AB, Yousuf A, Steinborn R, Mösslein N, Yang Y, Göldner M, Ruland JG, Bünemann M, Krasel C, et al. (2018) Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid µ-opioid receptor desensitization. Sci Signal 11:eaas9609. [DOI] [PubMed] [Google Scholar]
- Moulédous L, Froment C, Burlet-Schiltz O, Schulz S, Mollereau C. (2015) Phosphoproteomic analysis of the mouse brain mu-opioid (MOP) receptor. FEBS Lett 589:2401–2408. [DOI] [PubMed] [Google Scholar]
- Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, Folk RW, Broussard GJ, Liang R, Jang MJ, et al. (2018) Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360:eaat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. (2011) Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci 31:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. (2016) Desensitization-resistant and -sensitive GPCR-mediated inhibition of GABA release occurs by Ca2+-dependent and -independent mechanisms at a hypothalamic synapse. J Neurophysiol 115:2376–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Dicken MS, Hentges ST. (2012) Multiple inhibitory G-protein-coupled receptors resist acute desensitization in the presynaptic but not postsynaptic compartments of neurons. J Neurosci 32:10192–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillinan N, Lau EK, Virk M, von Zastrow M, Williams JT. (2011) Recovery from mu-opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci 31:4434–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh A, Cooper A, Guy-David L, Reuveny E. (2010) Nonenzymatic rapid control of GIRK channel function by a G protein-coupled receptor kinase. Cell 143:750–760. [DOI] [PubMed] [Google Scholar]
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, Rosethorne EM, Charlton SJ, Krasel C, Bailey CP, et al. (2012) Endomorphin-2: a biased agonist at the μ-opioid receptor. Mol Pharmacol 82:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Tryoen-Tóth P, Filliol D, Matifas A, Laustriat D, Cao YQ, Basbaum AI, Dierich A, Vonesh JL, Gavériaux-Ruff C, et al. (2006) Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci USA 103:9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber M, Jullié D, Li J, Chakraborty S, Majumdar S, Lambert NA, Manglik A, von Zastrow M. (2020) Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife 9:e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Yeow RY, Schoenau C, Huber J, Tesmer JJ. (2011) Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol Pharmacol 80:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga AG, Reid BT, Kieffer BL, Levitt ES. (2020) Differential impact of two critical respiratory centres in opioid-induced respiratory depression in awake mice. J Physiol 598:189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Chang WT, Hsu CY, Huang PC, Chow YW, Li AH. (2002) Identification of two C-terminal amino acids, Ser(355) and Thr(357), required for short-term homologous desensitization of mu-opioid receptors. Biochem Pharmacol 64:257–266. [DOI] [PubMed] [Google Scholar]
- Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D, Becker JA, Wood JN, Kieffer BL, Gaveriaux-Ruff C. (2013) Mu opioid receptors on primary afferent nav1.8 neurons contribute to opiate-induced analgesia: insight from conditional knockout mice. PLoS One 8:e74706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Zalutsky RA, Nicoll RA. (1993) The opioid peptide dynorphin mediates heterosynaptic depression of hippocampal mossy fibre synapses and modulates long-term potentiation. Nature 362:423–427. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. (1998) Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad Sci USA 95 (17):9914–9919, doi: 10.1073/pnas.95.17.9914 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT. (2014) Desensitization of functional µ-opioid receptors increases agonist off-rate. Mol Pharmacol 86:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ. (2013) Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65:223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BL, Gregoriou GC, Kissiwaa SA, Wells OA, Medagoda DI, Hermes SM, Burford NT, Alt A, Aicher SA, Bagley EE. (2017) Endogenous opioids regulate moment-to-moment neuronal communication and excitability. Nat Commun 8:14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf A, Miess E, Sianati S, Du YP, Schulz S, Christie MJ. (2015) Role of phosphorylation sites in desensitization of µ-opioid receptor. Mol Pharmacol 88:825–835. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. (1998) Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA 95 (12):7157–7162, doi: 10.1073/pnas.95.12.7157 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski Z, Yim YY, Alford S, Hamm HE. (2019) The expanding roles and mechanisms of G protein-mediated presynaptic inhibition. J Biol Chem 294:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]