

Key Points
Question
Does site-specific treatment, including molecularly targeted therapy based on profiling gene expression and gene alterations by next-generation sequencing, have clinical use for patients with cancer of unknown primary site?
Findings
This phase 2 nonrandomized clinical trial of such site-specific treatment in 97 patients with cancer of unknown primary site revealed a 1-year survival probability of 53.1%, with a durable response to targeted therapy being observed in patients with actionable genetic alterations.
Meaning
Site-specific treatment, including guided targeted therapy based on next-generation sequencing, is a promising strategy for patients with cancer of unknown primary site and warrants further investigation in a randomized clinical trial.
Abstract
Importance
Although profiling of gene expression and gene alterations by next-generation sequencing (NGS) to predict the primary tumor site and guide molecularly targeted therapy might be expected to improve clinical outcomes for cancer of unknown primary site (CUP), to our knowledge, no clinical trial has previously evaluated this approach.
Objective
To assess the clinical use of site-specific treatment, including molecularly targeted therapy based on NGS results, for patients with CUP.
Design, Setting, and Participants
This phase 2 clinical trial was conducted at 19 institutions in Japan and enrolled 111 previously untreated patients with the unfavorable subset of CUP between March 2015 and January 2018, with 97 patients being included in the efficacy analysis. Eligibility criteria included a diagnosis of unfavorable CUP after mandatory examinations, including pathological evaluation by immunohistochemistry, chest-abdomen-pelvis computed tomography scans, and a positron emission tomography scan.
Interventions
RNA and DNA sequencing for selected genes was performed simultaneously to evaluate gene expression and gene alterations, respectively. A newly established algorithm was applied to predict tumor origin based on these data. Patients received site-specific therapy, including molecularly targeted therapy, according to the predicted site and detected gene alterations.
Main Outcomes And Measures
The primary end point was 1-year survival probability. Secondary end points included progression-free survival (PFS), overall survival (OS), objective response rate, safety, efficacy according to predicted site, and frequency of gene alterations.
Results
Of 97 participants, 49 (50.5%) were women and the median (range) age was 64 (21-81) years. The cancer types most commonly predicted were lung (21 [21%]), liver (15 [15%]), kidney (15 [15%]), and colorectal (12 [12%]) cancer. The most frequent gene alterations were in TP53 (45 [46.4%]), KRAS (19 [19.6%]), and CDKN2A (18 [18.6%]). The 1-year survival probability, median OS, and median PFS were 53.1% (95% CI, 42.6%-62.5%), 13.7 months (95% CI, 9.3-19.7 months), and 5.2 months (95% CI, 3.3-7.1 months), respectively. Targetable EGFR mutations in tumor specimens were detected in 5 patients with predicted non–small-cell lung cancer (5.2%), 4 of whom were treated with afatinib; 2 of these patients achieved a durable PFS of longer than 6 months.
Conclusions and Relevance
This study’s findings suggest that site-specific treatment, including molecularly targeted therapy based on profiling gene expression and gene alterations by NGS, can contribute to treating patients with the unfavorable subset of CUP.
Trial Registration
UMIN Identifier: UMIN000016794
This clinical trial assesses the clinical use of site-specific treatment, including molecularly targeted therapy based on next-generation sequencing results, for Japanese patients with cancer of unknown primary site.
Introduction
Cancer of unknown primary site (CUP) is defined as histologically confirmed metastatic cancer for which identification of the primary site is not possible after an appropriate diagnostic approach.1,2,3,4 Most individuals with CUP, excepting the approximately 20% of patients who have favorable prognostic factors, receive empirical chemotherapy, including a platinum-taxane regimen,5,6 and have a poor survival time, with a median overall survival (OS) of approximately 6 to 12 months.7,8,9
The identification of the site of origin based on molecular profiling by gene expression analysis has been expected to allow more effective and specific treatment for patients with CUP, thereby prolonging their survival time.4,10,11,12,13,14,15 Indeed, a large prospective trial of site-specific therapy based on gene expression profiling with a 92-gene reverse-transcription polymerase chain reaction assay showed promising efficacy for patients with CUP, yielding a median OS of 12.5 months.16 Given these findings, we recently conducted a randomized study to assess whether site-specific therapy based on microarray analysis of gene expression might improve outcomes compared with empirical chemotherapy in previously untreated patients with CUP.17 However, unexpectedly, we found that site-specific treatment based on such genome-wide expression profiling did not result in an improvement in OS compared with empirical chemotherapy. It was thus possible that selection of site-specific treatment based on comprehensive profiling of gene expressions by microarray analysis is not sufficient to improve survival outcomes in CUP. More sensitive methods for genetic testing have been recently developed. RNA sequencing analysis is one of the most widely adopted and sensitive platforms for quantifying gene expressions, with the small amounts of degraded RNA extracted from formalin-fixed, paraffin-embedded (FFPE) tissue. A classifier of tumor type based on RNA sequencing data thus warrants evaluation for its ability to guide site-specific therapy.
The increasing number of effective molecularly targeted therapies available for numerous types of tumors has also increased the importance of detecting actionable genomic alterations.18 Targeted sequencing platforms are a rapid and cost-effective means with which to identify known genetic alterations in selected sets of genes, and they have recently become widely introduced into daily clinical practice.18,19 Given that CUP includes many types of advanced cancer, determination of targetable genomic tumor mutations might be expected to support the application of molecularly targeted drugs to patients with CUP.
We have now established an integrative analysis system for site prediction that combines gene expression profiling and the detection of gene alterations by targeted next-generation sequencing (NGS). To evaluate the use of this system, we performed a prospective, multi-institution phase 2 trial of site-specific therapy based on site prediction by NGS, including molecularly targeted therapy guided by the genetic findings, for patients with CUP.
Methods
Study Design and Patients
This prospective, single-arm phase 2 trial (UMIN000016794) was conducted at 19 sites in Japan and received approval from the institutional review boards at all sites. The study protocol can be found in Supplement 1. All patients provided written informed consent. Eligibility criteria included a diagnosis of CUP after a standard evaluation (medical history, physical examination, blood cell counts, chemistry profile, chest-abdomen-pelvis computed tomography scans, positron emission tomography scan, and directed assessment of all symptomatic areas).20 Patients with histologically or cytologically confirmed metastatic cancer (excluding malignant melanoma, malignant lymphoma, or neuroendocrine tumor) were eligible after adequate histological evaluation and immunohistochemical analysis were performed by certified pathologists at each participating site according to institutional standards. Sufficient FFPE tumor tissue samples were required to perform genetic analysis. Additional inclusion criteria included an Eastern Cooperative Oncology Group performance status of 0 to 2, no previous systemic therapy, and adequate organ function as defined by a hemoglobin level of ≥9.0 g/dL (to convert to grams per liter, multiply by 10), a white blood cell count of 4000/μL or greater (to convert to ×109 per liter, multiply by 0.001), a platelet count of 100 ×103/μL or greater (to convert to ×109 per liter, multiply by 1), serum transaminase levels of 2.5 or less times the upper limit of normal, serum total bilirubin level of 1.5 mg/dL or less (to convert to micromoles per liter, multiply by 17.104), and serum creatinine levels 1.5 times or less of the upper limit of normal. Patients were excluded if they belonged to the favorable CUP subset, including those with extragonadal germ cell syndrome, adenocarcinoma restricted to axillary lymph nodes (women), peritoneal carcinomatosis (women), or squamous carcinoma limited to cervical, supraclavicular, or inguinal lymph nodes.16,20 Patients for whom curative surgery or radiation therapy was suitable were also excluded.
Study Plan
On study entry, an FFPE tumor specimen was subjected to gene expression and gene mutation profiling by NGS. After the assay results became available, patients received site-specific and gene alteration–guided therapy.
The molecular assay procedures are described in the eMethods and eTables 1, 2, and 3 in Supplement 2. Standard treatments for predicted sites of origin, including dose modification and management of toxicity, were determined by the treating physician on the basis of Japanese standard practice guidelines for each type of cancer. In addition, for patients with specific targetable genetic alterations, treatment decisions regarding the application of corresponding targeted therapy by the physician were made according to variant type (pathogenic or of unknown significance), generally after discussion by the study coordinators and molecular tumor board.
Patients were evaluated for responses every 8 weeks after treatment onset until the detection of tumor progression or treatment completion, and they were all evaluated for survival. Tumor response was assessed according to Response Evaluation Criteria in Solid Tumors, version 1.1 (European Organization for Research and Treatment of Cancer). Adverse events were monitored throughout the study and were graded by investigators according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.
Statistical Analysis
The primary objective of the study was to evaluate the clinical use of site-specific therapy based on NGS analysis by determining the efficacy of such therapy for patients with CUP. The primary efficacy end point was the 1-year survival probability for all patients with the exception of those found to be ineligible after registration or those not evaluated for the outcome with regard to the study therapy. With the assumption of an expected 1-year survival probability of 50% for site-specific therapy and a threshold of 40% (corresponding to the 1-year survival probability for empirical chemotherapy based on previous trials),7,21,22,23,24 a minimum of 102 evaluable patients was required for the lower limit of the 95% confidence interval for the 1-year survival probability to be more than 40%, with a registration period of 2 years and follow-up period of 1 year. Given this requirement and the possibility of patients being subsequently found ineligible or withdrawing from the study, the target number of participants for this study was set at 110. Secondary end points included progression-free survival (PFS), OS, objective response rate, safety, and efficacy according to predicted site. The frequency of gene alterations was also included as a secondary end point. Overall survival and PFS were estimated by the Kaplan-Meier method and were compared between 2 groups classified according to responsiveness of tumors at the predicted original site as described previously,16 with the hazard ratio (HR) being derived from the Cox regression model. Analyses were conducted using SAS, version 9.4 (SAS Institute), and statistical significance was set at P < .05.
Results
Patient Characteristics
Between March 2015 and January 2018, 111 patients were enrolled. The patient flow is summarized in the eFigure in Supplement 2. The median time from enrollment to the report of a predicted site to the physician was 8 days. The NGS analysis of gene expression and gene alterations was successful for all 111 patients. Fourteen patients (12.6%) did not receive the trial therapy, 4 of whom the site of origin was revealed by further imaging or a pathologically definitive diagnosis was obtained before therapy initiation, with the identified primary tumors being biliary tract cancer, breast cancer, anal canal cancer, and non–small-cell lung cancer (NSCLC). The efficacy analysis was thus performed for 97 patients, the characteristics of whom are shown in Table 1.
Table 1. Characteristics of 97 Patients Evaluable for Efficacy.
| Characteristic | No. (%) |
|---|---|
| Age, median (range), y | 64 (21-81) |
| Male | 48 (49.5) |
| Female | 49 (50.5) |
| ECOG performance status | |
| 0 | 34 (35.1) |
| 1 | 54 (55.7) |
| 2 | 9 (9.2) |
| Histology | |
| Adenocarcinoma | 51 (52.6) |
| Poorly differentiated adenocarcinoma | 16 (16.5) |
| Undifferentiated carcinoma | 15 (15.5) |
| Squamous cell carcinoma | 9 (9.3) |
| Other | 6 (6.2) |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Prediction of Tumor Sites
An algorithm based on the gene expression and gene alteration data obtained by NGS predicted a tissue of origin for all patients, with 15 different sites predicted (Table 2). Establishment of the algorithm for predicting the primary tumor site is described in eMethods in Supplement 2. Details of the treatment regimens for site-specific therapy are also shown in Table 2, and those of subsequent therapy are presented in eTable 4 in Supplement 2.
Table 2. Results for Site Prediction.
| Predicted site | No. (%) | Treatment | |
|---|---|---|---|
| Enrolled patients (n = 111) | Patients in the efficacy analysis (n = 97) | ||
| Lung | 21 (18.9) | 21 (21.6) | Platinum + taxane (n = 9), platinum + PEM (n = 8), EGFR-TKI (n = 4) |
| Liver | 20 (18.0) | 15 (15.5) | Sorafenib |
| Kidney | 16 (14.4) | 15 (15.5) | VEGF inhibitor (n = 14), mTOR inhibitor (n = 1) |
| Colon/rectum | 14 (12.6) | 12 (12.4) | FOLFOX/CapeOX/SOX plus bevacizumab (n = 6), CapeOX (n = 2), FOLFOXIRI (n = 1), FOLFOXIRI + bevacizumab (n = 1) FOLFIRI + panitumumab (n = 1), S-1 + bevacizumab (n = 1) |
| Ovary | 9 (8.1) | 7 (7.2) | CBDCA + PTX (n = 5), CBDCA + PTX + bevacizumab (n = 2) |
| Biliary tract (gallbladder, bile ducts) | 7 (6.3) | 7 (7.2) | CDDP + GEM |
| Breast | 6 (5.4) | 5 (5.2) | AC (n = 2), PTX + bevacizumab (n = 2), DTX (n = 1) |
| Germ cell | 5 (4.5) | 5 (5.2) | BEP (n = 4), ICE (n = 1) |
| Cervix | 3 (3.6) | 3 (3.1) | CBDCA + PTX |
| Pancreas | 3 (2.7) | 2 (2.1) | GEM + nab-PTX |
| Lymphoma | 2 (1.8) | 1 (1.0) | CHOP |
| Head and neck | 2 (1.8) | 1 (1.0) | FP + cetuximab |
| Urothelium | 1 (0.9) | 1 (1.0) | CDDP + GEM |
| Stomach | 1 (0.9) | 1 (1.0) | S-1 |
| Prostate | 1 (0.9) | 1 (1.0) | Leuprorelin + bicalutamide |
Abbreviations: AC, Adriamycin (doxorubicin) and cyclophosphamide; BEP, bleomycin, etoposide, and cisplatin; CapeOX, oxaliplatin and capecitabine; CBDCA, carboplatin; CDDP, cisplatin; CHOP, Adriamycin (doxorubicin), cyclophosphamide, Oncovin (vincristine), and prednisone; DTX, docetaxel; EGFR-TKI, epidermal growth factor receptor–tyrosine kinase inhibitor; FOLFIRI, irinotecan, infusional and bolus 5-fluorouracil, and leucovorin; FOLFOXIRI, oxaliplatin, irinotecan, infusional and bolus 5-fluorouracil, and leucovorin; FOLFOX, oxaliplatin, infusional and bolus 5-fluorouracil, and leucovorin; FP, 5-fluorouracil and cisplatin; GEM, gemcitabine; ICE, ifosfamide, cisplatin, and etoposide; mTOR, mammalian target of rapamycin; nab-PTX, albumin-bound paclitaxel nanoparticle; PEM, pemetrexed; PTX, paclitaxel; SOX, oxaliplatin and S-1; S-1, tegafur, gimeracil, and oteracil potassium; VEGF, vascular endothelial growth factor.
Efficacy of Site-Specific Therapy Based on NGS
The data cutoff date for the efficacy analysis was January 31, 2019. The 1-year survival probability was 53.1% (95% CI, 42.6%-62.5%), with the study thus meeting its primary end point. At the cutoff date, there were 64 patients (66.0%) with an event (death). After a median follow-up time of 12.0 months (range, 9.6-14.5 months), the median OS was 13.7 months (95% CI, 9.3-19.7 months) (Figure 1A) and the median PFS was 5.2 months (95% CI, 3.3-7.1 months) (Figure 1B). The objective response rate calculated for the 82 patients with measurable disease was 39.0% (95% CI, 28.4%-50.4%).
Figure 1. Kaplan-Meier Plots of Overall Survival and Progression-Free Survival.
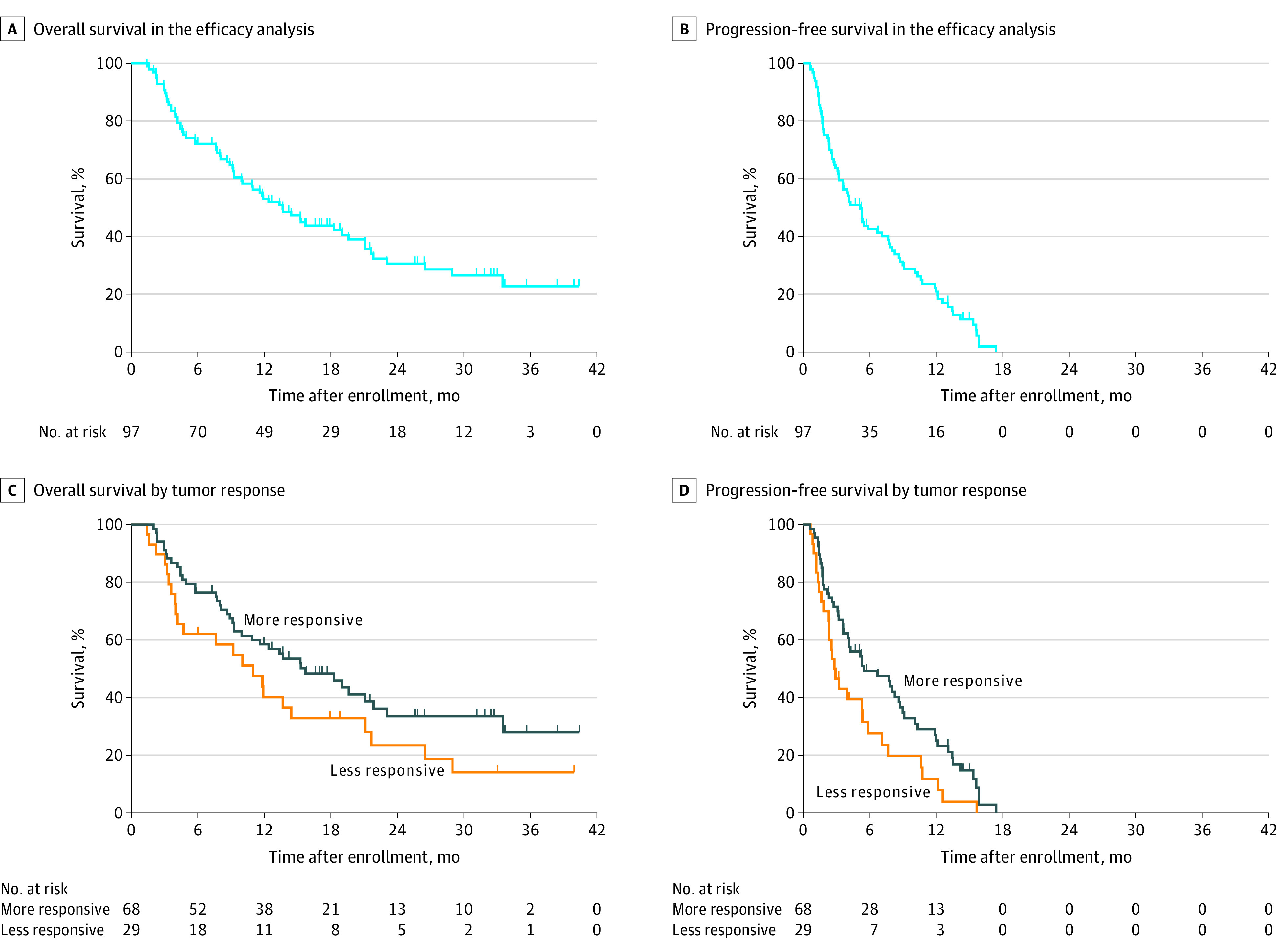
Plots of overall survival (A and C) and progression-free survival (B and D) are shown for all patients in the efficacy analysis (A and B) as well as for those with more responsive compared with less responsive tumor types (C and D).
Consistent with previous observations,16 the 68 patients (70.1%) with predicted tumor types categorized as relatively sensitive or responsive to treatment (colorectal, breast, ovarian, kidney, prostate, bladder, non–small-cell lung, and germ cell cancers, as well as lymphoma) had a median OS of 15.7 months (95% CI, 10.0-21.9 months) and median PFS of 5.5 months (95% CI, 4.1-8.6 months) compared with corresponding values of 11.0 months (95% CI, 4.0-21.2 months) and 2.8 months (95% CI, 1.8-5.9 months) for the 29 patients (29.9%) with less responsive tumor types (biliary tract, pancreatic, gastroesophageal, liver, cervical, endometrial, and head and neck cancers) (Figure 1, C and D). An effect of site-specific treatment on OS and PFS was suggested for the patients with more responsive tumor types relative to those with less responsive tumor types (OS: log-rank test P = .08; HR, 0.634; 95% CI, 0.380-1.057; PFS: P = .02; HR, 0.578; 95% CI, 0.363-0.921).
Driver Gene Alterations
Genetic alterations were evaluated by targeted sequencing in all 97 patients in the efficacy analysis (Figure 2A). The most common genetic alterations were in TP53 (45 [46.4%]), KRAS (19 [19.6%]), and CDKN2A (18 [18.6%]), consistent with previous findings.25 Several genetic driver mutations with implications for treatment selection were also detected, including those affecting EGFR and KRAS. Five patients who had tumors with activating EGFR mutations were predicted to have NSCLC. Four of these 5 patients received treatment with an EGFR tyrosine kinase inhibitor (afatinib), with 2 of them achieving a durable PFS of longer than 6 months (Figure 2B). After excluding these 4 patients treated with afatinib, the 1-year OS probability and median OS for the remaining patients were 52.1% (95% CI, 41.4%-62.8%) and 13.4 months, respectively. Regarding patients predicted to have colorectal cancer, none of the 10 individuals whose tumors had a KRAS mutation received treatment with an anti-EGFR antibody, whereas panitumumab with leucovorin calcium–fluorouracil–irinotecan hydrochloride was administered to 1 patient found to be wild type for KRAS (Figure 2C).
Figure 2. Detected Gene Alterations and Response in Patients With Tumors Having EGFR or KRAS Mutations.
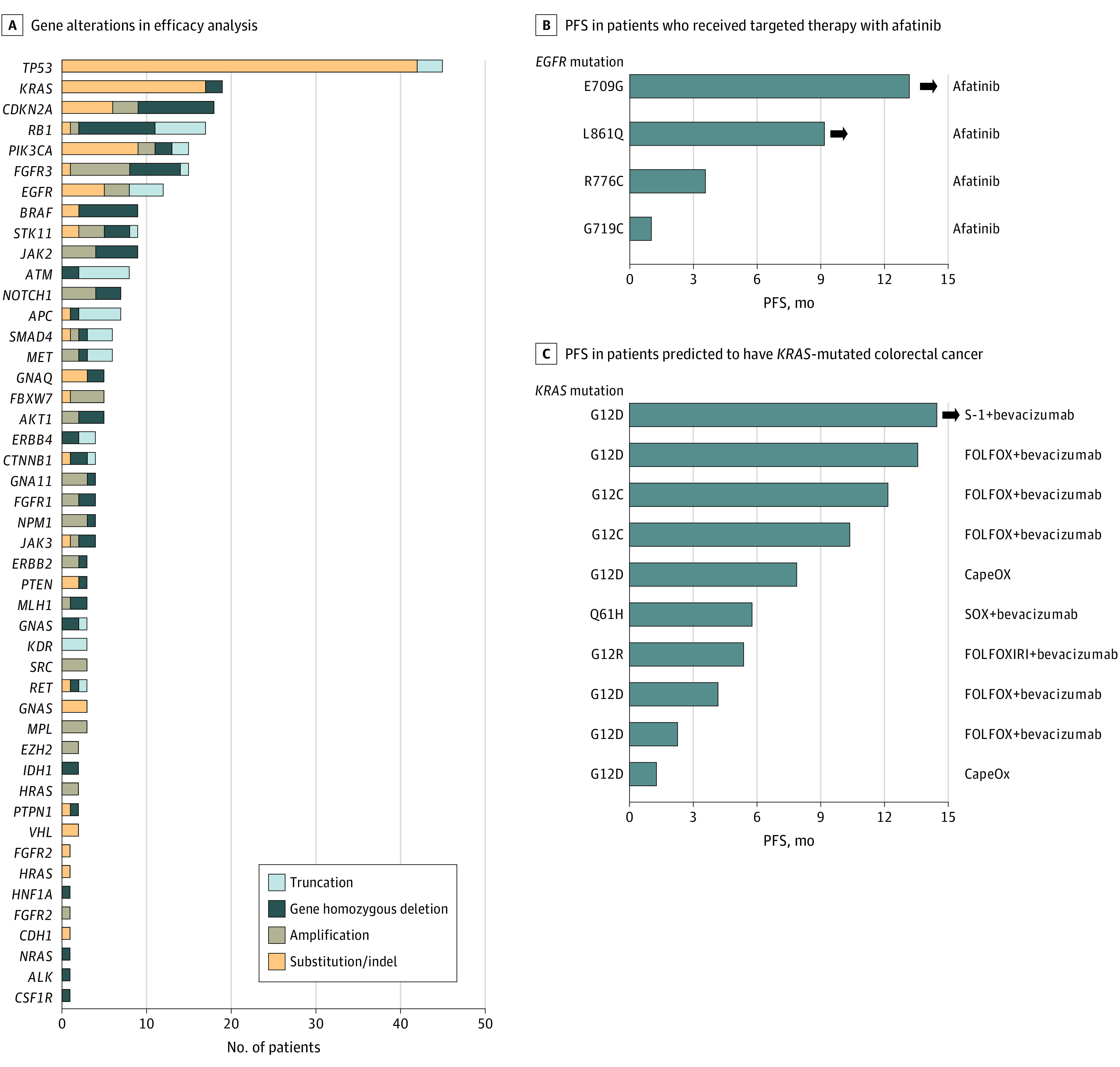
A, Number and type of detected gene alterations in patients for efficacy analysis (N = 97). B and C, Swimmer plots for progression-free survival (PFS) in patients predicted to have non–small-cell lung cancer with the indicated activating EGFR mutations and who received targeted therapy with afatinib (B) and in those predicted to have KRAS-mutated colorectal cancer and treated as indicated (C). The bar length represents the duration of PFS, with arrowheads indicating patients still experiencing tumor control.
Safety
A safety analysis was performed with 97 patients, with the adverse events being detailed in eTable 5 in Supplement 2. There were 4 treatment-related deaths from lung infection, sepsis, cerebral infarction, or pneumonitis.
Discussion
To our knowledge, this study is the first prospective trial to evaluate site-specific therapy based on profiling gene expression and gene alterations by NGS. Our finding that the median OS was 13.7 months for the study patients suggested the clinical utility of site-specific therapy determined by such molecular profiling. More than half of the patients received subsequent therapy, including molecularly targeted therapy, which may have contributed to the relatively long OS despite the relatively short PFS. In addition, a predefined subgroup analysis revealed that patients predicted to have more responsive tumor types experienced a longer OS and PFS, with a median OS of 15.7 months and PFS of 5.5 months compared with corresponding values of 11.0 and 2.8 months for those predicted to have less responsive tumor types, consistent with the findings of a previous prospective trial.16 Additionally, the number of treatment-related deaths was similar with previous randomized trials.17
Molecular tumor profiling has been under development over the last decade14,26 for predicting tumor site of origins in patients with CUP, and several assays27,28 are now available commercially. Nevertheless, our previous randomized study did not detect a survival benefit for site-specific treatment based on the results of microarray analysis compared with empirical chemotherapy.17 It was possible that this negative result was because of an insufficient predictive ability of microarray-based expression profiling. In addition, 20% of the enrolled patients in this previous study were predicted to have malignant lymphoma, which is not common in studies of CUP, whereas there was only 1 patient predicted to have lymphoma in the efficacy analysis set of the present study. This difference suggests that our NGS-based prediction system, which accounts for the expression levels of 257 genes, might be more accurate and led to the promising OS results in the current trial. Another limitation of our previous randomized trial was the requirement for a repeated biopsy to obtain frozen tissue even after the initial diagnosis, which led to a 3-week treatment delay and consequent clinical deterioration of the enrolled patients. Our present study had the advantage that nucleic acid was extracted from archival FFPE tissue and therefore had a shorter turnaround time (a median time from enrollment to report of the predicted site to the physician of 8 days), consistent with the potential application of this approach to daily clinical practice.
Our NGS-based integrative analysis system also identified several oncogenic driver gene alterations and thereby informed the selection of targeted therapy. A retrospective study of 200 CUP cases identified targetable gene alterations in receptor tyrosine kinase signaling pathways in 90 of the 125 patients with adenocarcinoma (72%).25 Some of these patients actually received targeted therapy, which showed promising clinical outcomes. A more recent study reported that 45 of 150 patients with CUP (30%) had potentially actionable genomic alterations identified by targeted genomic sequencing with the MSK-IMPACT platform (Memorial Sloan Kettering Cancer Center), and that 15 of these patients received targeted therapy, with a time to treatment failure ranging from less than 1 month to 14 months.29 In the present study, 4 of the 5 patients with predicted NSCLC positive for activating EGFR mutations actually received corresponding targeted therapy, with a durable response being achieved for 2 patients. Detection of established oncogenic driver mutations might thus contribute to improved survival for patients with CUP. Nevertheless, mutation profiling alone is not thought to be sufficient to guide the personalized treatment of CUP, although some targeted therapies, including an NTRK inhibitor, have been approved for specific genetic alterations across tumor types.30 Indeed, the mutation status of RAS genes, which are the most commonly mutated oncogenes in cancer, is considered irrelevant to treatment selection for most tumor types as a result of the limited success in targeting the mutant proteins to date,31,32 whereas evaluation of RAS mutations supports treatment optimization for colorectal cancer because of the predictive value of these mutations for treatment resistance to anti-EGFR antibodies.33 In addition, a previous basket trial of a BRAF inhibitor that focused on BRAF mutations across tumor types detected a durable response for NSCLC, Erdheim-Chester disease, and Langerhans cell histiocytosis, but not for other tumor types, including breast, colorectal, and ovarian cancer, as well as cholangiocarcinoma.34 These findings thus suggest that genetic alterations alone are not sufficient to predict the response to targeted therapy in all cancer types. Given the heterogeneous nature of their disease, the identification of genetic alterations and primary site prediction are needed to optimize targeted therapy for patients with CUP.
Limitations
Despite its promising results, this study has several limitations. First, the number of patients was limited and the study lacked a comparator group. Fourteen patients were lost between enrollment and treatment initiation, with only 97 patients being evaluated for efficacy outcome compared with the 102 required statistically for such analysis. The study was thus inadequately powered to evaluate the primary efficacy outcome as planned, despite the positive results obtained. Second, the study did not show a definite improvement in OS compared with the survival time for empirical therapy in our previous trial,17 although the primary end point of 1-year survival probability was achieved with a value of 53.1%. Given that only 1 prospective trial has evaluated the potential survival benefit of site-specific therapy directed by molecular profiling compared with empirical therapy, further well-designed randomized trials are needed. Third, only a few molecularly targeted therapies had been approved and were available during the study period, although several actionable gene alterations, including those affecting BRAF, MET, and RET, were detected in this study. In this trial, only approved and reimbursed targeted drugs were administered to patients with targetable alterations, with off-label use not being permitted. Additionally, treatment decisions by physicians were made according to variant type (pathogenic or of unknown significance), generally after discussion by the study coordinators and molecular tumor board. Actually, for some patients with tumors having EGFR mutations, cytotoxic chemotherapy was preferentially administered instead of an EGFR tyrosine kinase inhibitor when the mutation type was deemed uncommon or nonactivating. Given that treatment decisions were made by each physician, this variability might have affected the study results. Finally, the emergence of immunotherapy has substantially changed the treatment paradigm for several types of cancer, but no patients received immune-checkpoint inhibitors as study therapy because they had not yet been approved for the predicted tumor types. Given the recent finding that immune activity in CUP is similar to that for malignancies responsive to such inhibitors,35 site prediction might be expected to have an even greater role in the immunotherapy era.
Conclusions
This phase 2 study found that site-specific treatment for CUP based on profiling gene expression and gene alterations by NGS had a favorable survival outcome that was suggestive of the clinical use of this strategy. Our findings also suggest that identification of genetic mutations in tumors as a guide to selecting appropriate targeted therapy can result in a durable response even in patients with CUP. Further randomized studies comparing this treatment approach with empirical chemotherapy for patients with the unfavorable subset of CUP are warranted.
Trial Protocol
eMethods
eTable 1. Gene List for RNA Sequencing
eTable 2. The results of validation study about tumor classification
eTable 3. The example of prior probability for ERBB2 gene alteration
eTable4. Details of Poststudy Cancer Therapy
eTable5. Adverse Events of Any Grade in at Least 10% of Patients (n = 97)
eFigure. Patient Disposition
References
- 1.Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379(9824):1428-1435. doi: 10.1016/S0140-6736(11)61178-1 [DOI] [PubMed] [Google Scholar]
- 2.Varadhachary GR, Raber MN. Cancer of unknown primary site. N Engl J Med. 2014;371(8):757-765. doi: 10.1056/NEJMra1303917 [DOI] [PubMed] [Google Scholar]
- 3.Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin—diagnosis and treatment. Nat Rev Clin Oncol. 2011;8(12):701-710. doi: 10.1038/nrclinonc.2011.158 [DOI] [PubMed] [Google Scholar]
- 4.Rassy E, Pavlidis N. Progress in refining the clinical management of cancer of unknown primary in the molecular era. Nat Rev Clin Oncol. 2020. doi: 10.1038/s41571-020-0359-1 [DOI] [PubMed] [Google Scholar]
- 5.Greco FA, Erland JB, Morrissey LH, et al. Carcinoma of unknown primary site: phase II trials with docetaxel plus cisplatin or carboplatin. Ann Oncol. 2000;11(2):211-215. doi: 10.1023/A:1008369812295 [DOI] [PubMed] [Google Scholar]
- 6.Briasoulis E, Kalofonos H, Bafaloukos D, et al. Carboplatin plus paclitaxel in unknown primary carcinoma: a phase II Hellenic Cooperative Oncology Group Study. J Clin Oncol. 2000;18(17):3101-3107. doi: 10.1200/JCO.2000.18.17.3101 [DOI] [PubMed] [Google Scholar]
- 7.Greco FA, Pavlidis N. Treatment for patients with unknown primary carcinoma and unfavorable prognostic factors. Semin Oncol. 2009;36(1):65-74. doi: 10.1053/j.seminoncol.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 8.Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382. doi: 10.1016/j.jare.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrakis D, Pentheroudakis G, Voulgaris E, Pavlidis N. Prognostication in cancer of unknown primary (CUP): development of a prognostic algorithm in 311 cases and review of the literature. Cancer Treat Rev. 2013;39(7):701-708. doi: 10.1016/j.ctrv.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 10.Economopoulou P, Mountzios G, Pavlidis N, Pentheroudakis G. Cancer of unknown primary origin in the genomic era: elucidating the dark box of cancer. Cancer Treat Rev. 2015;41(7):598-604. doi: 10.1016/j.ctrv.2015.05.010 [DOI] [PubMed] [Google Scholar]
- 11.Moran S, Martínez-Cardús A, Sayols S, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016;17(10):1386-1395. doi: 10.1016/S1470-2045(16)30297-2 [DOI] [PubMed] [Google Scholar]
- 12.Pentheroudakis G, Greco FA, Pavlidis N. Molecular assignment of tissue of origin in cancer of unknown primary may not predict response to therapy or outcome: a systematic literature review. Cancer Treat Rev. 2009;35(3):221-227. doi: 10.1016/j.ctrv.2008.10.003 [DOI] [PubMed] [Google Scholar]
- 13.Pentheroudakis G, Pavlidis N, Fountzilas G, et al. Novel microRNA-based assay demonstrates 92% agreement with diagnosis based on clinicopathologic and management data in a cohort of patients with carcinoma of unknown primary. Mol Cancer. 2013;12:57. doi: 10.1186/1476-4598-12-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varadhachary G. New strategies for carcinoma of unknown primary: the role of tissue-of-origin molecular profiling. Clin Cancer Res. 2013;19(15):4027-4033. doi: 10.1158/1078-0432.CCR-12-3030 [DOI] [PubMed] [Google Scholar]
- 15.Varadhachary GR, Spector Y, Abbruzzese JL, et al. Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary. Clin Cancer Res. 2011;17(12):4063-4070. doi: 10.1158/1078-0432.CCR-10-2599 [DOI] [PubMed] [Google Scholar]
- 16.Hainsworth JD, Rubin MS, Spigel DR, et al. Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon research institute. J Clin Oncol. 2013;31(2):217-223. doi: 10.1200/JCO.2012.43.3755 [DOI] [PubMed] [Google Scholar]
- 17.Hayashi H, Kurata T, Takiguchi Y, et al. Randomized phase II trial comparing site-specific treatment based on gene expression profiling with carboplatin and paclitaxel for patients with cancer of unknown primary site. J Clin Oncol. 2019;37(7):570-579. doi: 10.1200/JCO.18.00771 [DOI] [PubMed] [Google Scholar]
- 18.Takeda M, Sakai K, Terashima M, et al. Clinical application of amplicon-based next-generation sequencing to therapeutic decision making in lung cancer. Ann Oncol. 2015;26(12):2477-2482. doi: 10.1093/annonc/mdv475 [DOI] [PubMed] [Google Scholar]
- 19.Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ; Working Group of the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee . ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15(9):733-747. doi: 10.1038/gim.2013.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fizazi K, Greco FA, Pavlidis N, Daugaard G, Oien K, Pentheroudakis G; ESMO Guidelines Committee . Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl 5):v133-v138. doi: 10.1093/annonc/mdv305 [DOI] [PubMed] [Google Scholar]
- 21.Greco FA, Burris HA III, Litchy S, et al. Gemcitabine, carboplatin, and paclitaxel for patients with carcinoma of unknown primary site: a Minnie Pearl Cancer Research Network study. J Clin Oncol. 2002;20(6):1651-1656. doi: 10.1200/JCO.2002.20.6.1651 [DOI] [PubMed] [Google Scholar]
- 22.Greco FA, Rodriguez GI, Shaffer DW, et al. Carcinoma of unknown primary site: sequential treatment with paclitaxel/carboplatin/etoposide and gemcitabine/irinotecan: a Minnie Pearl Cancer Research Network phase II trial. Oncologist. 2004;9(6):644-652. doi: 10.1634/theoncologist.9-6-644 [DOI] [PubMed] [Google Scholar]
- 23.Hainsworth JD, Spigel DR, Clark BL, et al. Paclitaxel/carboplatin/etoposide versus gemcitabine/irinotecan in the first-line treatment of patients with carcinoma of unknown primary site: a randomized, phase III Sarah Cannon Oncology Research Consortium Trial. Cancer J. 2010;16(1):70-75. doi: 10.1097/PPO.0b013e3181c6aa89 [DOI] [PubMed] [Google Scholar]
- 24.Hainsworth JD, Daugaard G, Lesimple T, et al. Paclitaxel/carboplatin with or without belinostat as empiric first-line treatment for patients with carcinoma of unknown primary site: a randomized, phase 2 trial. Cancer. 2015;121(10):1654-1661. doi: 10.1002/cncr.29229 [DOI] [PubMed] [Google Scholar]
- 25.Ross JS, Wang K, Gay L, et al. Comprehensive genomic profiling of carcinoma of unknown primary site: new routes to targeted therapies. JAMA Oncol. 2015;1(1):40-49. doi: 10.1001/jamaoncol.2014.216 [DOI] [PubMed] [Google Scholar]
- 26.Greco FA, Lennington WJ, Spigel DR, Hainsworth JD. Molecular profiling diagnosis in unknown primary cancer: accuracy and ability to complement standard pathology. J Natl Cancer Inst. 2013;105(11):782-790. doi: 10.1093/jnci/djt099 [DOI] [PubMed] [Google Scholar]
- 27.Erlander MG, Ma XJ, Kesty NC, Bao L, Salunga R, Schnabel CA. Performance and clinical evaluation of the 92-gene real-time PCR assay for tumor classification. J Mol Diagn. 2011;13(5):493-503. doi: 10.1016/j.jmoldx.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meiri E, Mueller WC, Rosenwald S, et al. A second-generation microRNA-based assay for diagnosing tumor tissue origin. Oncologist. 2012;17(6):801-812. doi: 10.1634/theoncologist.2011-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varghese AM, Arora A, Capanu M, et al. Clinical and molecular characterization of patients with cancer of unknown primary in the modern era. Ann Oncol. 2017;28(12):3015-3021. doi: 10.1093/annonc/mdx545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400-409. doi: 10.1158/2159-8290.CD-16-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Román M, Baraibar I, López I, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer. 2018;17(1):33. doi: 10.1186/s12943-018-0789-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagasaka M, Li Y, Sukari A, Ou SI, Al-Hallak MN, Azmi AS. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev. 2020;84:101974. doi: 10.1016/j.ctrv.2020.101974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinicrope FA, Okamoto K, Kasi PM, Kawakami H. Molecular biomarkers in the personalized treatment of colorectal cancer. Clin Gastroenterol Hepatol. 2016;14(5):651-658. doi: 10.1016/j.cgh.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726-736. doi: 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haratani K, Hayashi H, Takahama T, et al. Clinical and immune profiling for cancer of unknown primary site. J Immunother Cancer. 2019;7(1):251. doi: 10.1186/s40425-019-0720-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
eMethods
eTable 1. Gene List for RNA Sequencing
eTable 2. The results of validation study about tumor classification
eTable 3. The example of prior probability for ERBB2 gene alteration
eTable4. Details of Poststudy Cancer Therapy
eTable5. Adverse Events of Any Grade in at Least 10% of Patients (n = 97)
eFigure. Patient Disposition