

Abstract
The use of ultra performance liquid chromatography coupled to data independent tandem mass spectrometry with traveling wave ion mobility for detection and structural identification of ether-linked glycerophosphoethanolamine is described. The experimental design generates 4D data (chromatographic retention time, precursor accurate mass, drift time with associated calculated collisional cross-section, and time-aligned accurate mass diagnostic product ions) for each ionization mode. Confident structure identification depends on satisfying 4D data confirmation in both positive and negative ion mode. Using this methodology, a number of ether-linked glycerophosphoethanolamine lipids are structurally elucidated from mouse brain lysosomes. It is further determined that several ether-linked glycerophosphoethanolamine structures are differentially abundant between lysosomes isolated from mouse cortex following traumatic brain injury as compared to that of sham animals. The combined effort of aligning multi-dimensional mass spectrometry data with a well-defined traumatic brain injury model lays the foundation for gaining mechanistic insight in the role lysosomal membrane damage plays in neuronal cell death following brain injury.
Keywords: data independent tandem mass spectrometry, ether lipids, glycerophosphoethanolamine, lipid structure, lysosomes, traveling wave ion mobility, traumatic brain injury, UPLC-HDMSE
1. Introduction
Glycerophospholipids (GPL) are essential molecular components of biological systems. GPL play an integral role in membrane structure, dynamics, and signaling.[1] GPL are comprised of a glycerol backbone with aliphatic chains at the sn-1 and sn-2 position and a phosphate ester-linked polar head group at the sn-3 position.[2] GPL are categorized into sub-classes by structural variation at the phosphate head group.[3] Within each GPL sub-class further structural variation is introduced at both the sn-1 and sn-2 position by varying the aliphatic chain linkage, length and degree of saturation.[3] The consequence of the high degree of structural variation results in 1000s of potential GPL structures present in a given eukaryotic cell.
In addition to the number of possible GPL structures, modulation of the amphipathic nature of GPL has great impact on their biophysical properties which in turn influences membrane integrity and function. For example, the relatively smaller size of the phosphoethanolamine head group compared to phosphocholine results in a more conical molecular geometry for PE.[4] As a consequence, the increased inclusion of PE into phosphatidylcholine (PC) bilayers imposes membrane curvature stress. The induced curvature stress has been implicated in influencing membrane budding, fission, and fusion.[5] Structural variation in the aliphatic chains can also greatly affect the biophysical properties of GPL.[6] Aliphatic chain length and degree of unsaturation influences membrane fluidity by affecting the compactness of plasma membranes.
A third structural feature of GPL is the sn-1 linkage to the glycerol backbone. The sn-1 aliphatic chain can be present as a fatty acyl, alkyl ether (1-O-alkyl ether; plasmanyl) or vinyl ether (1-O-alk-1’-enyl ether; plasmenyl).[7] Example structures are provided in Figure S1, Supporting Information, detailing acyl, alkyl ether, and vinyl ether sn-1 structural configurations. Ether-linked GPL are present in a variety of mammalian cell types and are predominantly found as plasmanyl-PC and plasmenyl-PE.[8] The ether-linked aliphatic chain influences the biophysical properties of the GPL affecting membrane function. Specifically, plasmenyl-PE have been associated with modulating membrane fluidity, regulating membrane protein activity, and serving as anti-oxidants to mitigate oxidative stress.[9] In addition, plasmenyl-PE typically house a polyunsaturated fatty acid (PUFA) at the sn-2 position. The enzymatic release of PUFAs via plasmalogen-specific phospholipase A2 (PLA2) plays an integral role in the production of secondary messenger molecules such as eicosanoids and prostaglandins.[10] Deficiency in plasmenyl-PE levels has been linked to peroxisomal, respiratory, inflammatory, and neurodegenerative disorders.[11–13]
Our previous data demonstrate that similar to other neurodegenerative diseases traumatic brain injury (TBI) is associated with lysosomal membrane permeabilization.[14,15] The subsequent events following TBI which lead to compromised lysosomes are not fully understood, yet recent data suggest that leaky lysosomal membrane leads to impaired autophagy and neuronal cell death.[16] We have recently reported that the direct link between activation of cytosolic phospholipase A2 (cPLA2) following TBI and damage to lysosomal membranes involved disruption of GPL composition.[16] Furthermore, a number of ether-linked PE were significantly decreased in cortical lysosomes at 1 h following experimental TBI in the mouse model. The detection and identification of plasmenyl-PE has been primarily achieved via the use of mass spectrometry.[7,17,18] Herein, we report the use of ultra performance liquid chromatography (UPLC) coupled to data independent tandem mass spectrometry with traveling wave ion mobility (HDMSE) for detection and structural identification of ether-linked PE from mouse brain lysosomes.
2. Experimental Section
2.1. Materials
LC-MS grade acetonitrile, methanol, water, isopropanol, formic acid, and ammonium formate were purchased from Fisher Scientific (Pittsburg, PA). HPLC grade tert-Butyl methyl ether (MTBE) was purchased from Sigma Aldrich (St. Louis, MO). Lipid standards were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). The lipid standards included Splash Lipidomix and a number of individual lipid standards.
2.2. Sample Preparation
Lysosomes were isolated from mouse brains that were subjected to a controlled cortical impact as described previously.[15] Refer to the Supporting Information for a detailed description of the animal model. Isolated lysosomes were stored at −80 °C. Total lipid extracts from the lysosome samples were prepared using a MTBE lipid extraction protocol[19] with slight modifications. Refer to the Supporting Information for a detailed description of the MTBE lipid extraction protocol used.
2.3. Ultra Performance Liquid Chromatography (UPLC) Data Independent Tandem Mass Spectrometry with Traveling Wave Ion Mobility (HDMSE)
UPLC was performed on a Waters ACQUITY UPLC system (Milford, MA). The separation was achieved using a C18 CSH (1.7 μm; 2.1 × 100 mm) column (Waters, Milford, MA). UPLC parameters were adopted with slight modifications.[20] Mobile phase A was 10 mm ammonium formate with 0.1% formic acid in water/acetonitrile (40:60, v/v) and mobile phase B was 10 mm ammonium formate with 0.1% formic acid in acetonitrile/isopropanol (10:90, v/v). The gradient was ramped from 40% to 43% B in 2 min, ramped to 50% in 0.1 min, ramped to 54% B in 9.9 min, ramped to 70% in 0.1 min, and ramped to 99% B in 5.9 min. The gradient was returned to initial conditions in 0.5 min and held for 1.9 min for column equilibration. The flow rate was 0.4 mL min−1. The column was maintained at 55 °C and the auto-sampler was kept at 5 °C. A 2 μL injection was used for all samples.
HDMSE experiments were performed with a traveling wave ion-mobility enabled hybrid quadrupole orthogonal acceleration time-of-flight mass spectrometer (SYNAPT G2-S, Waters Corporation, Wilmslow, United Kingdom). HDMSE parameters were adopted with slight modifications.[20,21] Data were acquired over the m/z range of 100–1800 for both positive and negative ion modes. The capillary voltage was 2.0 kV and sampling cone voltage was 30 V. Nitrogen at a flow of 650 L/h was used as the desolvation gas with a constant desolvation temperature of 400 °C. The source temperature was set at 125 °C. During acquisition, the first quadrupole was set to pass all precursor ions with the “trap” T-wave operated at 4 V (T-wave velocity of 315 m s−1). The precursor ions were transferred to the traveling wave ion mobility cell and subjected to a nitrogen flow of 80 mL min−1 (T-wave velocity of 600 m s−1, wave height of 40 V). As the ions exited the ion mobility cell they were transferred to the “transfer” T-wave and underwent two discrete and alternating scan functions (T-wave velocity of 190 m s−1). The first scan was set at low-collision energy (4 eV) and used to collect precursor ion spectra. The second scan was set at elevated-collision energy and ramped from 30–55 eV which was used for generation of product ion spectra. Argon gas was used for collision-induced dissociation (CID). Leucine Enkephalin (0.1 mg mL−1 in water-acetonitrile with 0.1% formic acid) at a flow rate of 7.5 μL min−1 was used as the lock-mass to ensure high mass accuracy data acquisition. The ion mobility drift times of lipid precursor ions were determined experimentally with nitrogen as the drift gas, ion mobility wave height of 40 V, and ion mobility wave velocity of 600 m s−1. The calculated drift times were converted to collisional cross section (CCS) values using previously published methods with polyalanine as the calibrant.[21] The CCS values are reported in Angstrom (Å2).
Data were acquired with MassLynx v4.1 (Waters Corporation, Wilmslow, United Kingdom). Data were analyzed with MassLynx, MSE Data Viewer v1.2 (Waters), DriftScope HDMS v2.7 (Waters), and Progenesis QI v2.2 (Nonlinear Dynamics, Newcastle, United Kingdom).
3. Results and Discussion
Ether PE identification from brain lysosomal total lipid extracts was achieved via the use of a combination of C18 reverse-phase chromatography, traveling wave ion mobility, and high-resolution data-independent tandem mass spectrometry in both positive and negative ion modes. This approach is referred to as UPLC-HDMSE and yields 4D data per ionization mode: chromatographic retention time, precursor ion accurate mass, precursor ion drift time (and calculated CCS), and chromatographic- and mobiligraphic-aligned, accurate mass product ions.
3.1. Chromatographic Separation
The C18 reverse phase chromatography effectively separated extracted lysosomal lipids by aliphatic chain hydrophobicity (Figure 1 (negative ion mode) and Figure S2, Supporting Information (positive ion mode)). The total ion chromatogram (TIC) displayed a variety of glycerolipids, glycerophospholipids, and sphingolipids of which the most abundant lipids were PC and PE. Here we focus on the identification of alkyl ether PE (notated as PE-O) and vinyl ether PE (notated as PE-P). An extracted ion chromatogram (EIC) for a series of diacyl PE and vinyl ether PE are highlighted in Figures 1 and Figure S2, Supporting Information.
Figure 1.
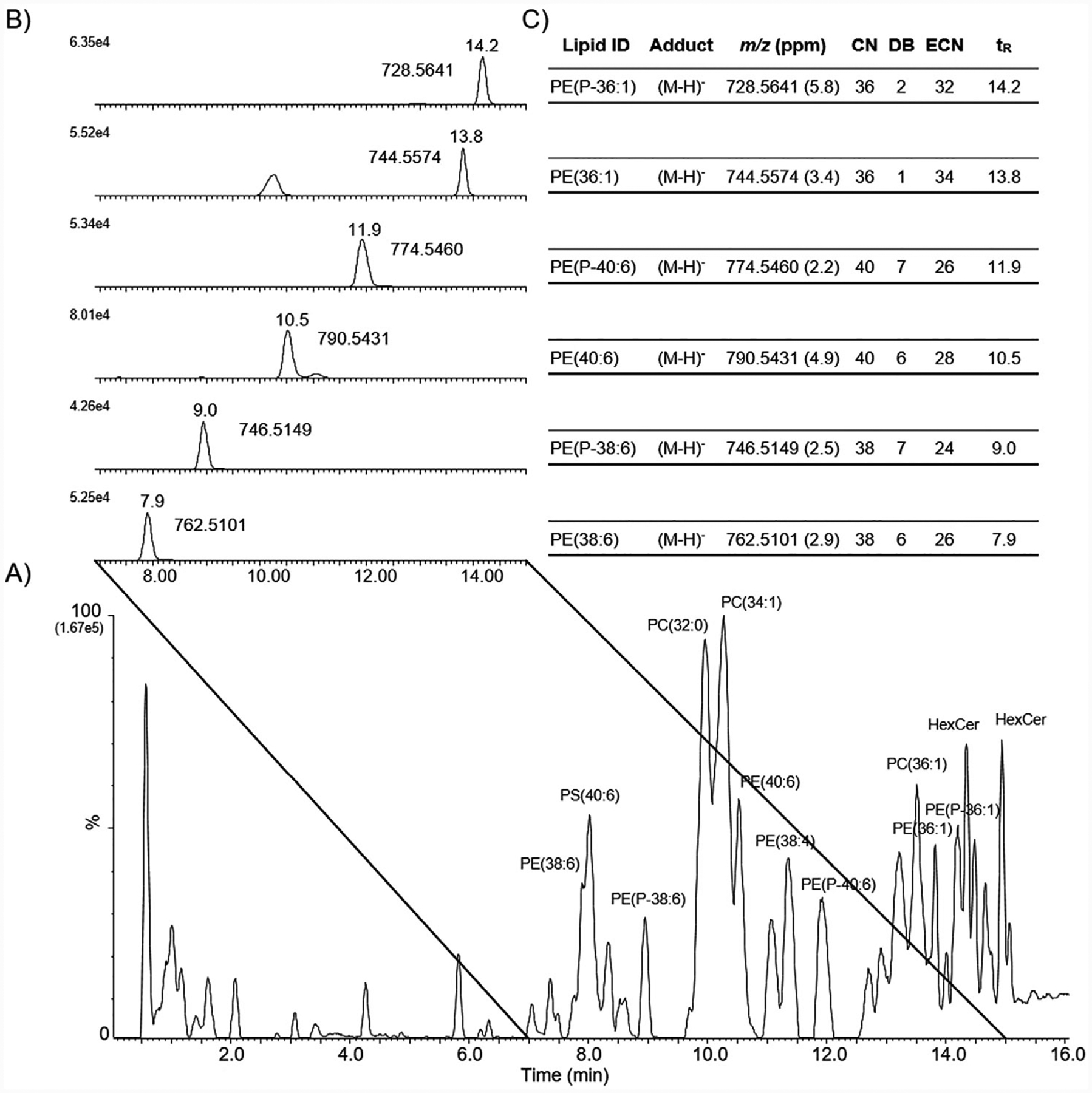
Chromatographic separation of lysosomal lipid extracts in negative ion mode. A) Reverse phase C18 UPLC, negative ion mode total ion chromatogram (TIC) of total lipid extract from mouse brain lysosomes. Several abundant peaks are labeled with putative lipid identification. Note, PC lipids were observed as deprotonated formate anions. B) Extracted ion chromatogram (EIC) of m/z values corresponding to identified PE and PE-P. Retention time and m/z value are listed next to the EIC peak. C) Table with lipid ID, adduct, m/z value with mass error, aliphatic chain number (CN), number of aliphatic double bonds (DB), equivalent carbon number (ECN), and retention time (tR) for each identified PE and PE-P from the EIC.
A useful measure of chromatographic retention for GPL is the calculation of the equivalent carbon number (ECN).[22] The ECN predicts for reverse-phase chromatography a direct relationship between ECN and retention time such that the lower the ECN the less chromatographic retention when comparing intra-class lipid species. The designation of intra-class lipid species refers to not only the lipid head group (e.g., PC or PE) but also the intra-class aliphatic chain configuration (e.g., diacyl PE or ether PE). For example, PE (38:6), PE (40:6), and PE (36:1) have ECN values of 26, 28, and 34 and chromatographic retention times of 7.9, 10.5, and 13.8 min, respectively. Conversely, PE(P-38:6), PE(P-40:6), and PE(P-36:0) have ECN values of 24, 26, and 32 and chromatographic retention times of 9.0, 11.9, and 14.2 min, respectively. Ether PE of similar ECN are more retained in reverse-phase chromatography than their corresponding diacyl PE. The difference (increase) in chromatographic retention for PE-P as compared to PE is explained by the polarity difference at the sn-1 chain containing an ether (for PE-P) or an ester (for PE). C18 chromatographic separation of diacyl and vinyl ether PE authentic standards provided verification for a number of PE structures and substantiated the aforementioned ECN GPL intra-class principle (Figure S3, Supporting Information). The difference in chromatographic retention between alkyl and vinyl ether PE is more subtle; here, ECN does not discriminate between the two.
3.2. HDMSE for Structure Identification for Vinyl Ether PE
Putative structure identification of vinyl ether PE (PE-P) was achieved via the use of HDMSE. The value of using HDMSE for confident lipid structure elucidation is demonstrated for PE(P-18:0/18:1). LIPIDMAPS[23] database searching with a 7.5 ppm threshold of the precursor ion at m/z 728.5641 (negative ion mode; tR = 14.2 min) and m/z 730.5730 (positive ion mode; tR = 14.2 min) indicated these m/z values corresponded to the following lipids: PC(O-33:2), PC(P-33:1), PE(O-36:2), and PE(P-36:1). The PC lipid candidates were eliminated as possible structures based on the absence of the diagnostic phosphocholine head-group at m/z 184.1 from the tandem mass spectrum and chromatographic retention of authentic standards.
Although chromatography greatly reduces lipid complexity in a temporal fashion it does not completely discriminate lipids of similar chemical composition. For example, the low-collision energy mass spectrum for the precursor at m/z 728.5641 (negative ion mode; tR = 14.2 min) clearly shows a number of other precursor ions that co-elute (Figure 2A). The corresponding elevated collision energy mass spectrum details product ions from not only the precursor ion at m/z 728.5641 but from all other precursor ions that co-elute at this retention time (Figure 2B). This results in a tandem mass spectrum populated by many product ions that originate from several possible precursor ions. The convoluted tandem mass spectrum makes assigning product ions to specific precursor ions challenging. The inclusion of a post-ionization gas-phase separation in the form of traveling wave ion mobility adds an additional separation that further reduces spectral complexity. The “cleaner,” less complex mass spectrum is a result of including ions that have a common drift time (drift time exclusion window was set to ±2% of reported drift time). The HDMSE spectra display precursor ions (low-collision energy spectrum) and product ions (elevated-collision energy spectrum) that are both chromatographically and mobiliographically aligned. The “cleaner,” less complex mass spectra yields a low- and elevated-collision energy scan for m/z 728.5641 that enables confident assignment of product ions (Figure 2C,D).
Figure 2.
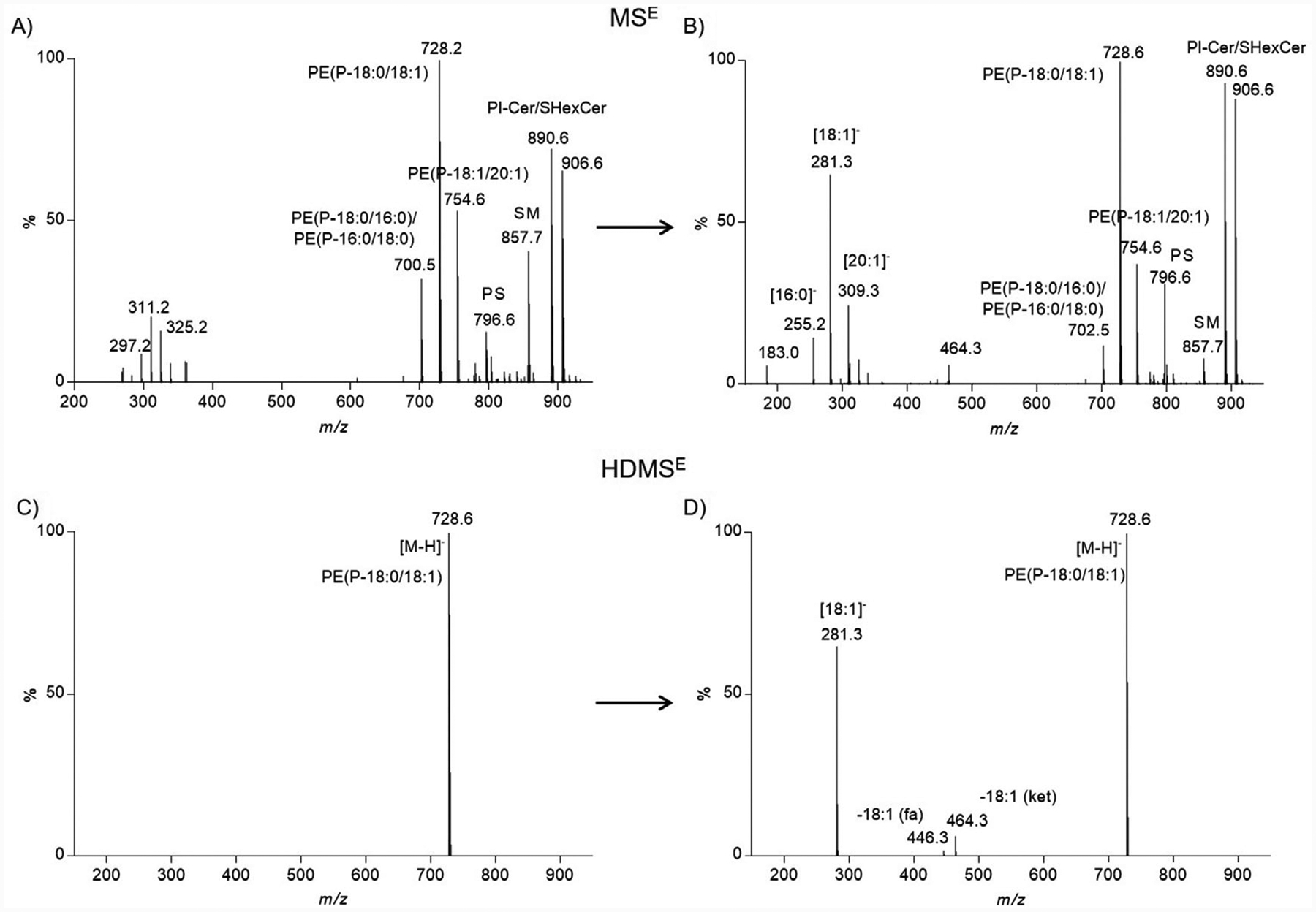
Spectral complexity of MSE vs. HDMSE. A) Negative ion mode mass spectrum corresponding to the low collision energy scan (MSE) for PE(P-36:1) (m/z value, 728.5641; tR, 14.2 min). Several other precursor ions co-eluted at this retention time and are present in the mass spectrum. B) Negative ion mode tandem mass spectrum corresponding to the elevated collision energy scan (MSE) for PE(P-36:1) (m/z value, 728.5641; tR, 14.2 min). Product ions observed could not confidently be assigned to specific precursor ions. C) Negative ion mode mass spectrum corresponding to the low collision energy scan (HDMSE) for PE(P-36:1) (m/z value, 728.5641; tR, 14.2 min; drift time bin threshold <2%). Only one precursor ion is observed with traveling wave ion mobility enabled. D) Negative ion mode tandem mass spectrum corresponding to the elevated collision energy scan (HDMSE) for PE(P-36:1) (m/z value, 728.5641; tR, 14.2 min; drift time bin threshold <2%). Product ions observed were assigned to a single precursor ion.
The negative ion mode HDMSE elevated-collision energy mass spectrum for m/z 728.5641 resulted in diagnostic product ions that corresponded to charge retention on the dissociated sn-2 acyl chain (m/z 281.3; [18:1]−) and the neutral loss of the sn-2 acyl chain as a free fatty acid (m/z 446.3; −18:1fa) or ketene (m/z 464.3; −18:1ket) (Figure S4, Supporting Information). CID of alkyl ether PE and vinyl ether PE precursor ions overwhelmingly fragment at the sn-2 position and not at both the sn-1 and sn-2 positions as is the case with diacyl PE (Figure S4A, Supporting Information).[7,18,24] The aforementioned product ions indicated there was an 18:1 acyl chain present at the sn-2 position. Thus, there are two possible lipid candidates PE(O-36:2) and PE(P-36:1) of which the following structures are possible: PE(O-18:1/18:1) and PE(P-18:0/18:1).
The positive ion mode HDMSE elevated-collision energy mass spectrum for m/z 730.5730 substantiated the structural identity (Figure 3). Positive ion mode CID of vinyl ether PE produces diagnostic product ions consistent with localizing the aliphatic chains to specific positions.[7,18] The sn-1 and sn-2 diagnostic ions were a result of 1) neutral loss of the sn-1 vinyl ether as an alkenyl alcohol (m/z 462.3; −(P-18:0)), 2) charge retention on a rearrangement reaction involving the sn-1 vinyl ether and the phosphoethanolamine (PEtN) head group (m/z 392.3; [P-18:0+PEtN]+), and 3) charge retention on the sn-2 acyl chain after neutral loss of the PEtN head group and neutral loss of the sn-1 vinyl ether as an alkene (m/z 339.3 ([18:1]+)). These product ions assigned the sn-1 position as a 18:0 vinyl ether and the sn-2 position as a 18:1 acyl giving a PE(P-18:0/18:1) lipid structure. The lipid structure assignment was confirmed by use of a PE(P-18:0/18:1) authentic standard (Figure S6, Supporting Information).
Figure 3.
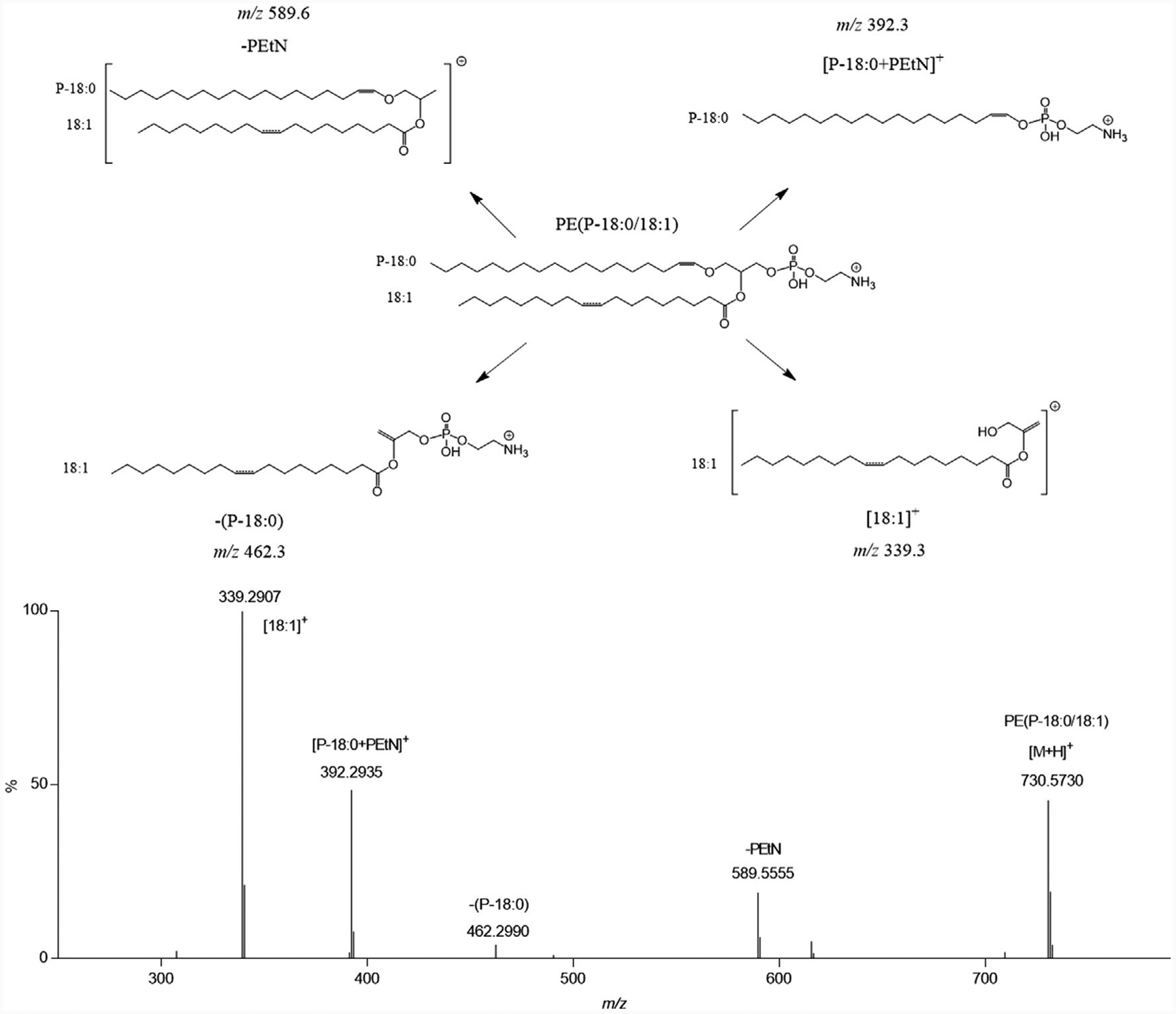
HDMSE-enabled structural assignment for a vinyl ether PE in positive ion mode. Positive ion mode tandem mass spectrum corresponding to the elevated collision energy scan (HDMSE, with traveling wave ion mobility) for PE(P-18:0/18:1) (m/z value, 730.5730; tR, 14.2 min; drift time bin threshold <2%). Product ions were assigned based on accurate mass measurements, authentic standard verification (Figure S6, Supporting Information), and literature precedent.[7] The inset structure shows likely product ion fragmentation. The double bond location on the sn-2 acyl chain is denoted as a dashed line indicating the double bond position was not determined.
3.3. HDMSE for Structure Identification for Alkyl Ether
The identification of alkyl ether PE (PE-O) followed a similar workflow as described for vinyl ether PE. An example of this workflow can be seen in the structure elucidation of the alkyl ether PE: PE(O-16:0/22:6) (Figure S7, Supporting Information). Refer to the Supporting Information for detailed description of this process.
3.4. Collisional Cross Section (CCS) Values for Alkyl Ether PE and Vinyl Ether PE
The experimentally derived traveling wave ion mobility drift time enable calculation of CCS values. CCS values have been reported to be highly reproducible and an added benefit for lipid identification.[21,25] The CCS values for the identified ether PE lipids are reported in Table 1. The CCS values were compared to in silico calculations using the on-line tool LipidCCS Predictor.[26] The calculated CCS values were within 2% of the predicted CCS values generated by LipidCCS predictor.
Table 1.
List of all ether PE structures that were identified in mouse brain lysosomes. Analysis of ether PE lipids by UPLC-HDMSE as described for purified cortical lysosomes from sham and TBI at 1 h after injury. The “O” and “P” in the lipid ID and lipid structure column refer to alkyl ether PE and vinyl ether PE, respectively. ppm = parts per million, tR = chromatographic retention time in minutes, CCS = collisional cross section (Å2), CN = aliphatic chain significance determined using a student t-test, carbon number, DB = number of double bonds in aliphatic chains, ECN = equivalent carbon number, (p > 0.05). All identified ether PE that reached significance were down- *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001. ns = non-significant regulated in TBI exposed lysosomes.
| Positive ion mode | Negative ion mode | Significance | Fold | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipid ID | m/z | ppm | tR | CCS | m/z | ppm | tR | CCS | CN | DB | ECN | Lipid structure | Sham/TBI | Change | |
| 1 | LPE(P-16:0) | 438.3003 | 5.5 | 1.7 | 209.2 | 436.2855 | 5 | 1.6 | 203 | 16 | 1 | 14 | LPE(P-16:0) | ns | ns |
| 2 | LPE(P-18:1) | 464.3166 | 6.5 | 1.7 | 213.2 | 462.3021 | 6.7 | 1.6 | 207.7 | 18 | 2 | 14 | LPE(P-18:1) | ns | ns |
| 3 | LPE(P-18:0) | 466.3294 | 0.4 | 2.5 | 215.7 | 464.3169 | 5 | 2.4 | 209.6 | 18 | 1 | 16 | LPE(P-18:0) | ns | ns |
| 4 | PE(P-38:6) | 748.5267 | 1.2 | 9 | 271.9 | 746.5186 | 7.5 | 8.9 | 265.5 | 38 | 7 | 24 | PE(P-16:0/22:6) | * (0.0059) | 1.5 |
| 5 | PE(P-36:4) | 724.5266 | 1.4 | 9.7 | 270.7 | 722.5163 | 4.6 | 9.7 | 261.4 | 36 | 5 | 26 | PE(P-16:0/20:4) | *** (0.0026) | 1.6 |
| 6 | PE(O-38:6) | 750.5418 | 1.9 | 9.4 | 274.5 | 748.53 | 1.9 | 9.4 | 266.7 | 38 | 6 | 26 | PE(O-16:0/22:6) | *** (0.0027) | 1.6 |
| 7 | PE(P-40:6) | 776.5583 | 0.8 | 12 | 279.6 | 774.546 | 2.2 | 11.9 | 271.3 | 40 | 7 | 26 | PE(P-18:0/22:6) | * (0.0106) | 1.1 |
| 8 | PE(P-38:4) | 752.5574 | 2 | 12.9 | 277.1 | 750.5453 | 1.3 | 12.9 | 267.3 | 38 | 5 | 28 | PE(P-18:0/20:4) | *** (0.005) | 1.1 |
| 9 | PE(P-42:6) | 804.5886 | 2 | 14 | 287.1 | 802.5741 | 1.9 | 14 | 276.9 | 42 | 7 | 28 | PE(P-20:0/22:6) | ** (0.0077) | 1.5 |
| 10 | PE(P-32:0) | 676.5275 | 0.1 | 12.4 | 265.8 | 674.5113 | 2.5 | 12.4 | 253.7 | 32 | 1 | 30 | PE(P-16:0/16:0) | ns | ns |
| 11 | PE(P-34:1) | 702.5437 | 0.7 | 12.7 | 269.3 | 700.5291 | 0.7 | 12.7 | 258.8 | 34 | 2 | 30 | PE(P-16:0/18:1)/PE(P-18:1/16:0) | * (0.0408) | 1.2 |
| 12 | PE(P-36:2) | 728.558 | 1.2 | 13 | 273.3 | 726.5468 | 3.4 | 13 | 262 | 36 | 3 | 30 | PE(P-18:1/18:1) | ns | ns |
| 13 | PE(P-38:3) | 754.5733 | 1.6 | 13.8 | 278.4 | 752.5599 | 0.1 | 13.8 | 268.6 | 38 | 4 | 30 | PE(P-18:0/20:3) | * (0.0328) | 1.3 |
| 14 | PE(P-40:4) | 780.5873 | 3.7 | 14 | 282.1 | 778.5802 | 5.9 | 14 | 273.5 | 40 | 5 | 30 | PE(P-18:0/22:4) | * (0.0147) | 1.2 |
| 15 | PE(P-34:0) | 704.5575 | 2 | 14.1 | 276.1 | 702.543 | 1.9 | 14.1 | 260.4 | 34 | 1 | 32 | PE(P-18:0/16:0)/PE(P-16:0/18:0) | ns | ns |
| 16 | PE(P-36:1) | 730.5741 | 0.5 | 14.2 | 278.6 | 728.558 | 2.6 | 14.2 | 264.8 | 36 | 2 | 32 | PE(P-18:0/18:1) | ns | ns |
| 17 | PE(P-38:2) | 756.5886 | 2.1 | 14.2 | 281 | 754.5784 | 3.7 | 14.2 | 270.9 | 38 | 3 | 32 | PE(P-18:1/20:1) | * (0.0337) | 1.3 |
| 18 | PE(O-40:4) | 782.6054 | 0.5 | 14.1 | 284.8 | 780.5909 | 0.4 | 14.1 | 274.1 | 40 | 4 | 32 | PE(O-20:0/20:4) | ns | ns |
| 19 | PE(P-38:1) | 758.6039 | 2.5 | 14.7 | 283.6 | 756.5948 | 4.8 | 14.7 | 271.2 | 38 | 2 | 34 | PE(P-18:0/20:1) | ns | ns |
| 20 | PE(P-40:2) | 784.6219 | 0.5 | 14.8 | 286.6 | 782.6074 | 0.6 | 14.7 | 276.1 | 40 | 3 | 34 | PE(P-18:1/22:1) | ns | ns |
| 21 | PE(P-38:0) | 760.62 | 2 | 15.3 | 285.5 | 758.6055 | 1.8 | 15.3 | 272.7 | 38 | 1 | 36 | PE(P-18:0/20:0)/PE(P-20:0/18:0) | ns | ns |
| 22 | PE(P-40:1) | 786.6386 | 1.9 | 15.2 | 287.2 | 784.6241 | 2 | 15.2 | 277.4 | 40 | 2 | 36 | PE(P-18:0/22:1) | ns | ns |
3.5. Identification of Alkyl Ether PE and Vinyl Ether PE from Brain Lysosomes
In total, there were 22 ether PE lipids that were identified via the UPLC-HDMSE platform (Table 1). Ether PE identification criteria were based on affirmative confirmation of ether PE structure in both positive and negative ion modes. Of these 22 ether PE, 20 of them were vinyl ether PE (PE-P) and two were alkyl ether PE (PE-O). For the PE-P lipids, three lyso PE-P lipids were identified and all contained a saturated fatty acid (SFA) or monounsaturated fatty acid (MUFA). There were 10 PE-P lipids that had either a SFA or MUFA at the sn-2 position. These acyl chains were of the following configurations: 16:0, 18:0, 18:1, 20:1, and 22:1. The remaining seven PE-P lipids had a PUFA at sn-2 position with the following configurations: 20:3, 20:4, 22:4, and 22:6. The two identified PE-O lipids contained either a 20:4 or 22:6 PUFA at the sn-2 position. Of note, the UPLC-HDMSE platform did not determine the position of the acyl chain double bond locations. For all ether PE lipids, the sn-1 alkyl or vinyl ether chain consisted of a 16:0, 16:1, 18:0, 18:1, or 20:0. To our knowledge, this is the first report detailing the identification of ether PE from brain lysosomal total lipid extracts.
With little reported literature as to the composition of lysosomal membranes, it is difficult to evaluate if the number and diversity of the identified ether PE is appropriate for brain lysosomes. Literature reports PE is a primary component in mammalian brain tissue and represents approximately 40% of total GPL.[27] Within this subgroup of total PE, ether PE (mainly PE-P) comprises approximately 50–80% of the total amount depending on the specific region of the brain.[12,28] The total PE content from the brain lysosomes was 35% of which ether PE abundance consisted of 38% of total PE (data not shown; data were derived from both the positive and negative ion mode TIC).
Our reported relative abundance of total PE and ether PE appear to be consistent with reported relative abundances for brain tissue (vide supra). What is less clear is if the number of identified ether PE are representative of brain lysosomes in general. One factor that could contribute to an under representation of reported ether PE was the stringent identification criteria. Although we were able to confidently identify ether PE structures, the identification process was dependent on high abundant lipids in order to effectively fragment precursor ions and observe them in both ionization polarities. We are currently investigating efforts to expand the depth of detection and identification of lysosomal derived ether PE. These efforts include sample preparation (e.g., use of acid hydrolysis post total lipid extraction), use of orthogonal chromatography techniques (e.g., HILIC), and the use of data-dependent inclusion lists for targeting suspected ether PE.
3.6. Differential Expression of Vinyl Ether PE from Brain Lysosomes after TBI in a Mouse Model
Brain lysosomes from cortices of mice exposed to TBI and from sham mice were comparatively analyzed to determine differential expression of ether PE lipids. Out of the 22 ether PE lipids identified, ten of these lipids were differentially expressed (all down regulated; p < 0.05) in mouse brain lysosomes 1 h after exposure to TBI (Table 1; Table S1, Supporting Information). The vast majority (7 of 10) of the significantly altered ether PE structures were vinyl ether and had a PUFA at the sn-2 position. Four of these vinyl ether PE are shown in Figure 4.
Figure 4.
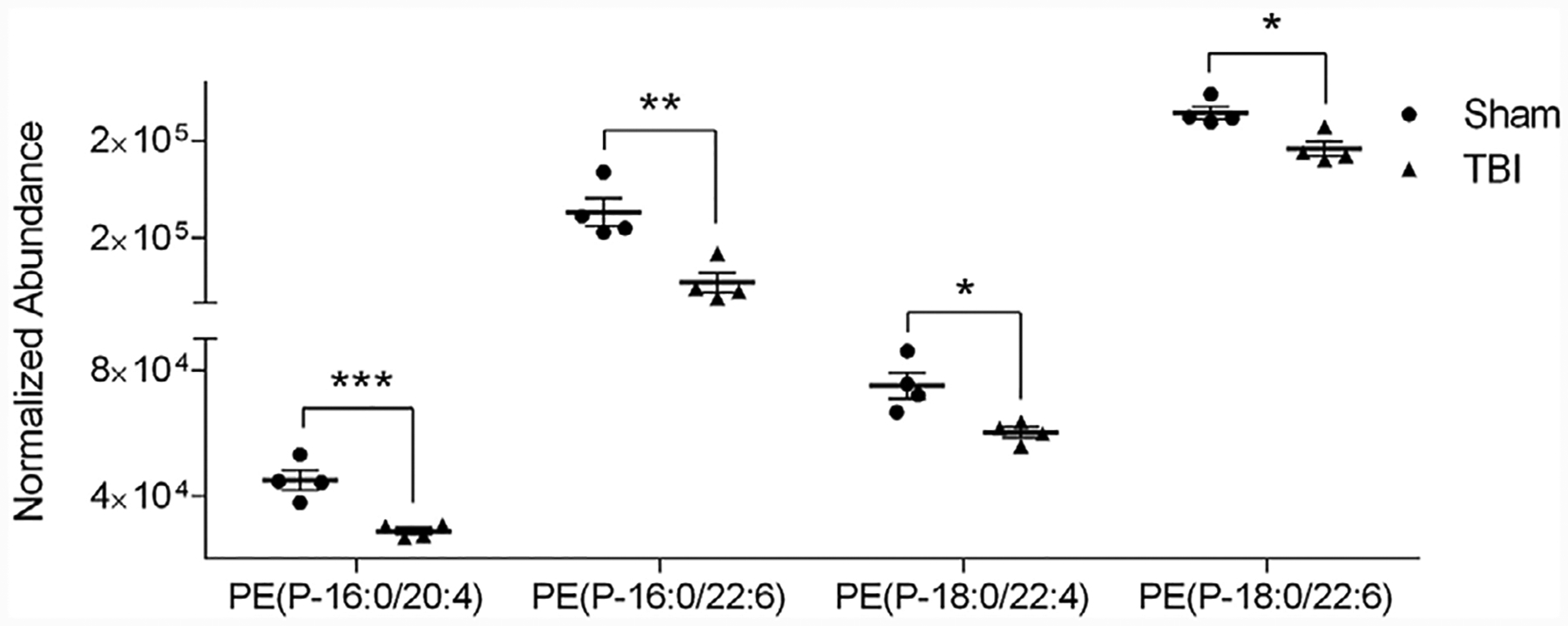
Vinyl ether PE structures that were differentially expressed in mouse brain lysosomes exposed to TBI. Analysis of ether PE lipids by UPLC-HDMSE as described for purified cortical lysosomes from sham and TBI at 1 h after injury. Abundance was plotted as mean SEM, N = 4, significance determined using a student t-test, *p < 0.05, ** ±p < 0.01, ***p < 0.005, ****p < 0.0001.
Reduced levels of vinyl ether PE, also referred to as plasmalogens, have been reported to be linked with several neurodegenerative disorders including Alzheimer disease, Down syndrome, and Parkinson’s disease.[12,13,29] The associated functional explanation for plasmalogen deficiency have been attributed to the vinyl ether PE’s structural attributes towards influencing membrane fluidity, oxidative potential, and acting as a reservoir for secondary messengers. The vinyl ether configuration at the sn-1 position effectively decreases membrane fluidity by elon-gating the sn-1 aliphatic chain.[30] This biophysical change alters membrane dynamics and has been associated with fusion and fission events.[31] In regards to the oxidative potential, it has been speculated that the vinyl ether bond is preferentially oxidized over acyl unsaturated fatty acids.[32] This would allow vinyl ether PE to buffer against oxidative damage. Last, plasmalogens have been noted to be enriched in PUFA, especially arachidonic acid and docosahexaenoic acid.[33] These biologically active lipid mediators can be released by enzymatic activity of PLA2 hydrolysis.[10]
We recently reported that TBI leads to lysosomal membrane permeabilization (LMP) mediated by activation of cPLA2.[16] The link between cPLA2 hydrolysis of membrane GPL and destabilization of lysosomal membranes may provide additional mechanistic insight toward understanding how cPLA2 mediated LMP is involved in inhibition of autophagy and neuronal cell death in neurotrauma and neurodegenerative diseases. The identification of a number of plasmalogen containing PUFA that are significantly attenuated following TBI in brain lysosomes is a novel finding and supports the evidence that lysosomal membrane structure and fluidity are impacted by neurotrama. Decreased lysosomal plasmalogen content could additionally impact the anti-oxidative buffering capacity of lysosomal membranes leading to increased susceptibility to oxidative stress. Furthermore, reduction of plasmalogens enriched with sn-2 containing PUFA could detrimentally impact eicosanoid metabolism thereby exacerbating deleterious cellular response following TBI.
4. Conclusions
Total lipid extracts of brain lysosomes from a traumatic brain injury mouse model were analyzed via UPLC-HDMSE. The UPLC-HDMSE platform generated 4D data that allowed for confident structure identification based on chromatographic retention, drift-time (and CCS), accurate mass precursor, and time & drift-aligned product ions. A number of alkyl ether PE and vinyl ether PE lipids were structurally identified. Furthermore, several vinyl ether PE were determined to be differentially abundant in lysosomal membranes from a mouse TBI model as compared to sham. On-going efforts are being pursued to further interrogate ether PE and the role ether PE play in the pathogenesis of neurodegeneration after traumatic brain injury.
Supplementary Material
Significance Statement.
More than 1.7 million new cases of traumatic brain injury (TBI) occur annually in the United States. The initial mechanical impact of TBI triggers a cascade of secondary biochemical events that lead to progressive neurodegeneration and chronic neuroinflammation. To date, there is no effective therapy available to restrict these changes and improve TBI outcomes. Correspondingly, there is a need to gain a greater understanding of the molecular mechanism(s) of injury progression following TBI. We recently identified disruption of the lysosomal membranes as one of the early molecular events after TBI and linked it to the impairment of autophagy and neuronal cell death. At present, little is known about the role of lipid structure and how changes in lipid composition contribute to lysosomal membrane disruption including lysosomal membrane permeabilization. To this end, we have developed an analytical platform that enables highly confident structure identification of ether glycerophosphoethanolamine (PE) from mouse cortical lysosomes. Furthermore, we demonstrate vinyl ether PE (plasmalogens) are differentially abundant in lysosomes isolated from TBI mouse brain. This finding provides evidence that certain glycerophospholipid structures, in particular vinyl ether PE, may be indicative of organelle-specific molecular damage after TBI that contributes to the pathogenesis of brain injury.
Acknowledgements
This project has been funded in part by the University of Maryland School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014) and by R01NS 091218 to M.M.L. and MSCRFE-2747 to C.S.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Jace W. Jones, University of Maryland, School of Pharmacy, Department of Pharmaceutical Sciences, Baltimore, MD 21201, USA
Chinmoy Sarkar, University of Maryland, School of Medicine, Department of Anesthesiology, Baltimore, MD 21201, USA.
Marta M. Lipinski, University of Maryland, School of Medicine, Department of Anesthesiology, Baltimore, MD 21201, USA
Maureen A. Kane, University of Maryland, School of Pharmacy, Department of Pharmaceutical Sciences, Baltimore, MD 21201, USA
References
- [1].Hishikawa D, Hashidate T, Shimizu T, Shindou H, J. Lipid Res 2014, 55, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Murphy RC, Axelsen PH, Mass Spectr. Rev 2011, 30, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH Jr., Murphy RC, Raetz CR, Russell DW, Subramaniam S, Nucl. Acids Res 2007, 35, D527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marsh D, Biophysical J. 2007, 93, 3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McMahon HT, Gallop JL, Nature 2005, 438, 590. [DOI] [PubMed] [Google Scholar]
- [6].Janmey PA, Kinnunen PK, Trends Cell Biol. 2006, 16, 538. [DOI] [PubMed] [Google Scholar]
- [7].Zemski Berry KA, Murphy RC, J. Am. Soc. Mass Spectr 2004, 15, 1499. [DOI] [PubMed] [Google Scholar]
- [8].Braverman NE, Moser AB, Biochimica et biophysica acta 2012, 1822, 1442. [DOI] [PubMed] [Google Scholar]
- [9].Sindelar PJ, Guan Z, Dallner G, Ernster L, Free Rad. Biol. Med 1999, 26, 318. [DOI] [PubMed] [Google Scholar]
- [10].Farooqui AA, Yang HC, Horrocks LA, Brain Res. Brain Res. Rev 1995, 21, 152. [DOI] [PubMed] [Google Scholar]
- [11].Rudiger M, von Baehr A, Haupt R, Wauer RR, Rustow B, Critic. Care Med 2000, 28, 1572. [DOI] [PubMed] [Google Scholar]
- [12].Han X, Holtzman DM, McKeel DW Jr., J. Neurochem 2001, 77, 1168. [DOI] [PubMed] [Google Scholar]
- [13].Fabelo N, Martin V, Santpere G, Marin R, Torrent L, Ferrer I, Diaz M, Molecular Medicine (Cambridge, Mass.) 2011, 17, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lipinski MM, Wu J, Faden AI, Sarkar C, Antioxidants Redox Signal. 2015, 23, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM, Autophagy 2014, 10, 2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sarkar C, Jones JW, Hegdekar N, Thayer JA, Kumar A, Faden AI, Kane MA, Lipinski MM, accepted Autophagy February 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Deeley JM, Thomas MC, Truscott RJ, Mitchell TW, Blanksby SJ, Anal. Chem 2009, 81, 1920; [DOI] [PubMed] [Google Scholar]; b) Ivanova PT, Milne SB, Brown HA, J. Lipid Res 2010, 51, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fhaner CJ, Liu S, Zhou X, Reid GE, Mass Spectr. 2013, 2, S0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hsu FF, Turk J, J. Am. Soc. Mass Spectr 2007, 18, 2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D, J. Lipid Res 2008, 49, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Damen CW, Isaac G, Langridge J, Hankemeier T, Vreeken RJ, J. Lipid Res 2014, 55, 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Paglia G, Angel P, Williams JP, Richardson K, Olivos HJ, Thompson JW, Menikarachchi L, Lai S, Walsh C, Moseley A, Plumb RS, Grant DF, Palsson BO, Langridge J, Geromanos S, Astarita G, Anal. Chem 2015, 87, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Jones JW, Carter CL, Li F, Yu J, Pierzchalski K, Jackson IL, Vujaskovic Z, Kane MA, Biomed. Chromat.: BMC 2017, 31; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lisa M, Cifkova E, Holcapek M, J. Chromat. A 2011, 1218, 5146. [DOI] [PubMed] [Google Scholar]
- [23].Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA, J Lipid Res. 2009, 50, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hsu FF, Turk J, J. Am. Soc. Mass Spectr 2000, 11, 892. [DOI] [PubMed] [Google Scholar]
- [25].a) Berry KA, Barkley RM, Berry JJ, Hankin JA, Hoyes E, Brown JM, Murphy RC, Anal. Chem 2017, 89, 916; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kliman M, May JC, McLean JA, Biochimica et biophysica acta 2011, 1811, 935; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kyle JE, Zhang X, Weitz KK, Monroe ME, Ibrahim YM, Moore RJ, Cha J, Sun X, Lovelace ES, Wagoner J, Polyak SJ, Metz TO, Dey SK, Smith RD, Burnum-Johnson KE, Baker ES, The Analyst 2016, 141, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou Z, Tu J, Xiong X, Shen X, Zhu ZJ, Anal. Chem 2017, 89, 9559. [DOI] [PubMed] [Google Scholar]
- [27].a) Guan Z, Soderberg M, Sindelar P, Prusiner SB, Kristensson K, Dallner G, J. Neurochem 1996, 66, 277; [DOI] [PubMed] [Google Scholar]; b) Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G, J. Biol. Chem 2012, 287, 2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rosenberger TA, Oki J, Purdon AD, Rapoport SI, Murphy EJ, J. Lipid Res 2002, 43, 59. [PubMed] [Google Scholar]
- [29].Murphy EJ, Schapiro MB, Rapoport SI, Shetty HU, Brain Res. 2000, 867, 9. [DOI] [PubMed] [Google Scholar]
- [30].Paltauf F, Chem. Phy. Lipids 1994, 74, 101. [DOI] [PubMed] [Google Scholar]
- [31].Han X, Gross RW, Biophysical J. 1992, 63, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Broniec A, Klosinski R, Pawlak A, Wrona-Krol M, Thompson D, Sarna T, Free Rad. Biol. Med 2011, 50, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gorgas K, Teigler A, Komljenovic D, Just WW, Biochimica et biophysica acta 2006, 1763, 1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.