

SUMMARY
Accurately profiling systemic immune responses to cancer initiation and progression is necessary for understanding tumor surveillance and, ultimately, improving therapy. Here, we describe the SYLARAS software tool (systemic lymphoid architecture response assessment) and a dataset collected with SYLARAS that describes the frequencies of immune cells in primary and secondary lymphoid organs and in the tumor microenvironment of mice engrafted with a standard syngeneic glioblastoma (GBM) model. The data resource involves profiles of 5 lymphoid tissues in 48 mice and shows that GBM causes wide-spread changes in the local and systemic immune architecture. We use SYLARAS to identify a subset of CD45R/B220+ CD8+ T cells that is depleted from circulation but accumulates in the tumor mass and confirm this finding using multiplexed immunofluorescence microscopy. SYLARAS is freely available for download at (https://github.com/gjbaker/sylaras). A record of this paper’s transparent peer review process is included in the Supplemental Information.
Graphical Abstract
In Brief
Localized tumors such as glioblastoma alter the composition of the immune system in peripheral organs including the spleen, lymph nodes, bone marrow, and thymus. SYLARAS enables efficient, systematic analysis of immune system architecture across many organs and samples to reveal subtle, recurrent changes on a background of between-sample biological variability.
INTRODUCTION
Glioblastoma (GBM) is an aggressive and incurable brain tumor characterized by high intrinsic and adaptive resistance to immunotherapy (Jackson et al., 2019). Like many solid cancers, it dampens the effector function of tumor-resident immune cells by producing anti-inflammatory cytokines and catabolites (Maxwell et al., 1992; Huettner et al., 1997; Crane et al., 2014; Wainwright et al., 2012; Zhou et al., 2015), lectins (Baker et al., 2014, 2016), and immune checkpoint molecules (Wainwright et al., 2014; Bloch et al., 2013). Interest in using immunotherapy to treat GBM is driven by evidence of dramatic tumor regression in some orthotopic immunocompetent murine models (Reardon et al., 2016) and encouraging but sporadic responses to immune checkpoint inhibitors (ICIs) in human patients (Cloughesy et al., 2019; Schalper et al., 2019; Zhao et al., 2019; Ito et al., 2019). However, the success of ICI therapy for GBM and other tumors of the central nervous system likely depends on a more complete description of immune cell interactions within and across lymphoid tissues in response to tumor growth, the cell and molecular repertoires necessary for efficacious ICI therapy, and biomarkers predictive of ICI response. In this paper, we tackle the first of these challenges.
The immune system comprises a complex network of specialized cells that communicate with each other and traffic to distinct tissues to confer resistance to foreign and self-antigens. Key primary and secondary lymphoid tissues include the blood, bone marrow, lymph nodes, spleen, and thymus each of which plays complementary roles in the priming and maintenance of robust anti-tumor immunity.
Despite this, cancer immunology has focused primarily on tumor-infiltrating immune cells and their behavior within the tumor microenvironment (TME). Recent results from animal models of cancer show that effective immunotherapy depends on the peripheral immune system (Spitzer et al., 2017), although the effect of cancer on immunological events taking place across the peripheral immune system remains unclear. This is due in part to lack of effective tools for processing, analyzing, and visualizing large sets of immuno-profiling data characterizing multiple lymphoid organs across time and disease status.
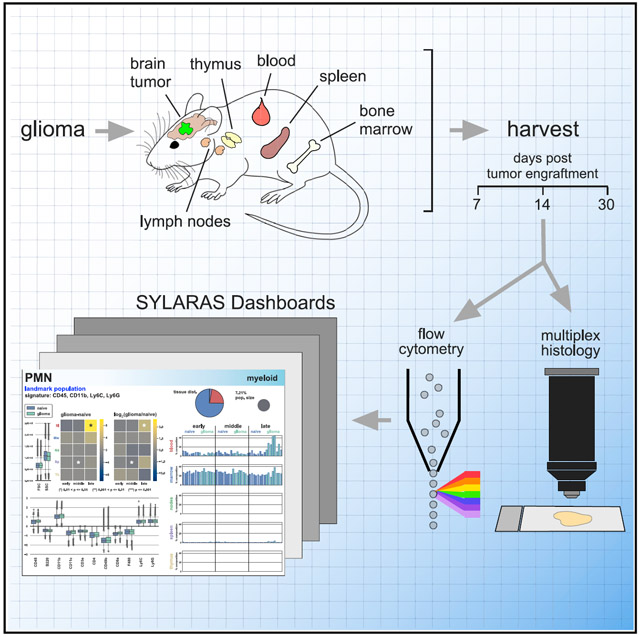
Here, we describe SYLARAS (systemic lymphoid architecture response assessment), a tool for studying systemic immune responses. SYLARAS combines multiplex immunophenotyping with software for transforming complex single-cell datasets into a visual compendium of time and tissue-dependent changes in immune cell frequencies and the relationships between these frequencies. We focus on perturbations imposed by GBM, but our approach is applicable to other cancers, infectious or autoimmune disease, vaccines, immunotherapy, etc. Typically, SYLARAS is deployed in three stages. In the first stage, longitudinal immunophenotyping data are collected from multiple mouse lymphoid organs of test and control subjects using an approach such as multiplex flow cytometry. In the second stage, raw flow cytometry standard (FCS) files are spectrally compensated, filtered for viable cells and then stratified into distinct immune cell classes via graphical user interface (GUI)-assisted manual gating or clustering algorithms such as PhenoGraph (Levine et al., 2015) and FlowSOM (Van Gassen et al., 2015). In a final stage, data-rich graphical dashboards are generated, one per immune cell type as means of summarizing basal immune status and response to perturbation.
We demonstrate the use of SYLARAS by studying the impact of intracranially engrafted GL261 mouse glioma on peripheral immune cell composition in five major lymphoid organs using conventional flow cytometry. Although highly multiplexed methods for immune system profiling such as mass cytometry (CyTOF) are capable of measuring many more features per cell, the method is relatively expensive. Flow cytometry retains an advantage when large numbers of samples must be analyzed: multiplexed flow cytometry is rapid, robust, relatively inexpensive, and widely available. However, the SYLARAS approach is compatible with any method for acquiring multiplexed single-cell data including cytometry by time-of-flight (CyTOF) (Spitzer and Nolan, 2016) and cellular barcoding strategies (Bodenmiller et al., 2012). The resource described in this paper comprises, (1) a well-validated 12-channel flow cytometry panel able to distinguish major murine immune cell types, (2) immuno-profiles for ~1 × 108 cells from 240-tissues in control and GBM-bearing mice, and (3) Python-based SYLARAS software for the programmatic identification of tumor-induced changes in systemic immune composition.
As one illustration of the utility of this data, we identify a change in immune architecture caused by GBM that involves a specialized subset of CD8+ T lymphocytes characterized by expression of the CD45R/B220 isoform of the CD45 protein tyrosine phosphatase type C receptor (Ptprc; henceforth B220+ CD8T cells). The previously described ability of B220+ CD8T cells to attenuate immune response to self-antigens (Marvel and Mayer, 1988; Takeuchi et al., 1989; James and Kwok, 2007; Yu et al., 2018) suggests that they may have immunosuppressive activity in GBM. We find that these cells are depleted from the circulation of tumor-bearing animals and infiltrate the TME of mouse gliomas. Relative to conventional CD8+ T cells, murine B220+ CD8T cells express different genes, are morphologically distinct, and localize to different regions of the brain tumor mass, suggesting that they represent a functionally distinct lymphocyte population. Experimental protocols, datasets, and source code for the SYLARAS project are freely available via Synapse (https://www.synapse.org/#!Synapse:syn21038562/wiki/597169), GitHub (https://github.com/gjbaker/sylaras), and links at the SYLARAS project website (https://www.sylaras.org).
RESULTS
A Visual Compendium of the Peripheral Immune Response to GBM
To collect a dataset on the effects of GBM on the peripheral immune system, we harvested blood, bone marrow, deep and superficial cervical lymph nodes, spleen, and thymus from 48 age-matched, immunocompetent C57BL6/J mice engrafted with syngeneic GL261 glioma cells (National Cancer Institute Tumor Repository) or vehicle alone at three time points post-engraftment (7, 14, and 30 days; n = 8 mice/group/time point). GL261 cells are widely used as a mouse model of a glioma responsive to combination therapy against the immune checkpoint proteins PD-1 and CTLA-4 (Reardon et al., 2016). GL261 glioma was chemically induced using methylcholanthrene in 1970 and represents one of a few syngeneic implantable models of GBM (Oh et al., 2014). The reproducibly in growth rate of GL261 tumors from one animal to the next is an advantage as compared with spontaneous tumors generated in situ, especially for large-scale multidimensional screens. Brains were also collected at the time of euthanasia for multiplexed tissue imaging by t-CyCIF (Lin et al., 2015, 2018), a method in which high-plex images are assembled from iterative rounds of conventional four-color immunofluorescence. Stereotactic injection of 3 × 104 GL261 cells into the brain striatum generated rapidly growing tumors with a median survival of 36 days (Figure S1A). Immune tissues were disaggregated and immunolabeled with an optimized panel of 11 fluorophore-conjugated antibodies and then analyzed by flow cytometry (Figures 1, S1B-S1E, and S2; Table S1). Average cell viability was 98.6% (range: 90.0%−99.7%) across the ~108 cell dataset (Figure S3A). Immune cell abundance varied across the five lymphoid organs. To correct for these differences, a weighted random sample (WRS) of 1 × 107 cells was drawn from the full dataset based on the cellularity of each tissue. This normalized the number of cells per tissue sample to an average of ~ 4 × 104 (Figure S3B).
Figure 1. Immunoprofiling GBM-Bearing Mice by 12-Color Flow Cytometry.

(1) GBM cells (or vehicle control) were stereotactically engrafted into the brain striata of age-matched mice.
(2) Lymphoid tissues were harvested from 8 replicate mice of GBM injected or control mice 7, 14, or 30 days after tumor engraftment.
(3) Tissues were disaggregated and plated in a 96-well V-bottom plate. Plate locations for wells corresponding to unstained splenocytes (UNS), fixable viability dye (FVD), CD45 isotype control (ISO), single-color compensation controls (target names), and PMT calibration beads run before and after data acquisition (PRE, POST) are indicated.
(4) Cells were immunolabeled with 11 fluorophore-conjugated antibodies and then stained with FVD.
(5) Single-cell data were acquired by high-throughput flow cytometry.
(6) Raw data were spectrally deconvolved and viable single-cells were selected for analysis.
(7) Preprocessed data underwent a histogram gating procedure (as described in Figure 3) prior to computational analysis using SYLARAS software. See also Figures S1-S3; Table S1.
To provide an overview of the 240-tissue dataset, we used SYLARAS to generate graphical dashboards capturing the statistics of specific immune cell subsets (Figures 2 and S4). Dashboards specified the cell-type alias (e.g., PMN for polymorphonuclear neutrophils), lineage (myeloid versus lymphoid), immunomarker signature (e.g., CD45+ CD11b+, LyC+, and Ly6G+), distribution among 5 lymphoid organs, and fractional contribution to all cells in the dataset (Figures 2A-2E). Quantitative information was also provided on light scattering as measured by flow cytometry, immunomarker expression profiles (Figures 2F and 2G), mean differences and fold-changes between age-matched control and tumor-bearing mice across three time points in tumor progression (Figures 2H and 2I), as well as cell-type frequency in each tissue for every animal in the study (Figure 2J). The modularity of the SYLARAS algorithm allows dashboard features to be changed with minor revisions to the source code.
Figure 2. SYLARAS Dashboard Summarizing a GBM-Induced Immune System Perturbation.

(A–C) Example SYLARAS dashboard for polymorphonuclear (PMN) immune cells displaying 9 cell-type-specific attributes: (A) brief alias, (B) lineage, and (C) immunomarker signature indicating whether the immunophenotype corresponds to 1 of 14 major “landmark populations” (see text for details).
(D) Distribution of cells across 5 lymphoid tissues color-coded as in (H), (I), and (J).
(E) Percentage of this cell type relative to all immune cells (in the random sample subjected to detailed analysis).
(F) Forward and side scatter (FSC/SSC).
(G) Logicle-transformed, background-subtracted immunomarker signal intensity.
(H) Time and tissue-specific difference in mean cell frequency (percentage) between GBM-burdened and mock-engrafted animals; asterisks denote one of three levels of statistical significance as indicated (FDR-corrected, two-tailed independent Student’s t tests, n = 8 mice/group).
(I) Time and tissue-specific log2 fold-change in mean cell frequency (percentage) between GBM-burdened and mock-engrafted animals; asterisks are as in (H). (J) Percentage of each lymphoid tissue composed of PMN cells across the study’s 48 mice. See also Figure S4.
Manual and Automated Approaches to Cell Subset Identification with SYLARAS
Manual gating of cytometry data is based on prior knowledge about patterns of CD (cluster of differentiation) antigen expression and is usually performed manually using software programs such as FlowJo. However, the 2,640 fluorescence intensity versus cell count histograms comprising our dataset (48 mice × 11 immunomarkers × 5 tissues) required the implementation of a more efficient approach. We therefore represented each histogram on a Logicle scale (Parks et al., 2006) (a generalization of hyperbolic sine functions widely used to display compensated flow cytometry data) and formatted them as a scrolling HTML table viewable in a web browser. This facilitated comparison between tissues, time points, and replicates and enabled rapid identification and recording of gate values between positive and negative signal intensities (Figure 3A). Gate values were then subtracted from intensity distributions to center the gate at zero and make non-specific fluorescence negative-valued (Figure 3B). Gating the data in this way, we were able to process the entire dataset in <2 h. Peak finding algorithms and Gaussian mixture models (Melnykov and Maitra, 2010) were evaluated as a means to automatically set gates but provided no discernable advantage over manual curation when the time for human review was included. Following the gating procedure, SYLARAS was used to binarize intensities according to mathematical sign, resulting in an M-dimensional Boolean immunophenotype (e.g., CD1+, CD2−, CD3+… CDM+) for each cell where M is the number of immunomarkers used in the analysis (Figure 3C).
Figure 3. The SYLARAS Approach to Immune Cell Subset Identification.

(A) Illustration of an iterative procedure in which intensity versus cell number histograms were gated to distinguish positive staining from background autofluorescence. Example histograms are shown (CD1, green; CD2, blue). Black outlines superimposed on each histogram represent the signal intensity distribution of unlabeled (control) splenocytes which SYLARAS uses as a fiducial reference for background signal intensities. Gate values were entered into a .txt (or .xls) file preformatted with sample metadata and used to update the histograms with a vertical line (dashed red line) at the curated gate for review and further refinement if needed.
(B) Bivariate scatter plot showing the same signal intensity data as shown in (A) following Logicle-transformation and subtraction of respective gate values from cell signal intensities. The two-way intensity distribution demonstrates the binning of cells among 22 = 4 possible immunophenotype quadrants.
(C) Extension of quadrant gating for two-dimensional data to octant gating for three-dimensional data where cells are binned among 23 = 8 possible immunophenotype octants. This procedure is extended to the total number of antibody markers in the data.
Of the 211 (2,048) Boolean immunophenotypes that can be specified with 11 markers, 604 were represented by ≥1 cell, and 30 were populated by >1% of cells in one or more of the 240 tissues analyzed. Together these 30 immunophenotypes accounted for 97% of viable immune cells and were the focus of further analysis. The 30 immunophenotypes were divided into 14 landmark immune cell subsets based on marker expression (e.g., CD4+ or CD8+ T cells, etc.; Figure 4A, inner wedges). Landmark populations were further divided into subclasses based on expression of additional immunomarkers. For example, CD8+ T cells characterized as CD45+, CD3ϵ+, and CD8α+, which also expressed Ly6C, correspond to Ly6C+ CD8+ memory T cells in mice (Walunas et al., 1995) (Figure 4A, outer wedges). Relative to the total number of cells, the abundance of cells in the 30 immunophenotypes ranged from ~30% for B cells to ~0.01% for dendritic cells (DCs) (Figure 4B). In agreement with the established light scattering properties of different immune cell subsets, forward and side scatter (FSC and SSC) were lowest among cells classified as adaptive lymphocytes (e.g., CD4+ and CD8+ T cells), intermediate in non-polymorphonuclear myeloid cells and innate lymphocytes (e.g., monocytes, macrophages, DCs, and NK cells), and highest among granulocytes (e.g., neutrophils) (Figure 4C). The exceptionally high SSC of a subset of CD45+, CD11b+, F4/80+ cells allowed them to be classified as eosinophils (as opposed to macrophages).
Figure 4. Mouse Immune Cell Subset Identification via Knowledge-Based and Data-Driven Methods.

(A) Sunburst plot mapping immunophenotypes to known cell lineages. Fourteen landmark immune cell populations are indicated by the inner wedges of the plot and are based on a large body of literature on immune cell types. Outer wedges represent refinements on the landmark populations made possible with additional markers. Immunomarkers expressed by each cell type are specified.
(B) Binary heatmap showing the immunomarker signature of each cell type together with their fractional contribution to the WRS (left of heatmap) and a literature reference (right of heatmap; PMID, PubMed identifier).
(C) FSC and SSC distributions for cells from control (blue boxplots) and GBM-burdened mice (green boxplots) annotated to each cell type and shown in ascending rank order from left-to-right according to the median value of the control data. Box plot elements: horizontal line, median; box limits, first to third quartile (Q1 to Q3); whiskers, from Q1-1.5 × interquartile range (IQR) to Q3 + 1.5 × IQR; diamond points, outliers.
(D) The fraction of data accounted for by each of 30 PhenoGraph clusters (k = 20, Euclidean distance; left of heatmap). Heatmap shows mean immunomarker signal intensity of cells constituting each cluster. Background signal intensities are set to zero to highlight foreground antibody signal. Cell-type calls made based on prior knowledge and accounting for >50 of cells in each cluster are shown (right of heatmap).
(E) The fraction of data accounted for by each of 30 FlowSOM clusters (nClus = 30; left of heatmap). Heatmap shows mean immunomarker signal intensity of cells constituting each cluster. Background signal intensities are set to zero to highlight foreground antibody signal. Cell-type calls made based on prior knowledge and accounting for >50 of cells in each cluster are shown (right of heatmap). (F) Logicle-transformed signal intensity distributions of PhenoGraph (top) and FlowSOM (bottom) cluster pairs enriched for B cells demonstrating population splitting based on non-specific signal intensities. Signal intensities above zero indicate foreground antibody signal while negative values indicate non-specific fluorescence as determined by histogram gating. Arrowheads denote immunomarker channels exhibiting considerable difference in non-specific signal intensity between clusters. Box plot elements are as in (C). See also Figure S5.
Unsupervised clustering is a common way to identify immune cell types in large datasets since it circumvents the need for gating (Weber and Robinson, 2016). We integrated results from two widely used clustering algorithms into the SYLARAS pipeline: PhenoGraph (Levine et al., 2015), which is based on nearest-neighbor clustering, and FlowSOM (Van Gassen et al., 2015), which uses a minimum spanning tree algorithm. Because of the computational costs of PhenoGraph, random sampling was used to reduce the dataset a further 5-fold. To facilitate comparison between clustering methods and immune cell classification by gating, the number of nearest neighbors (k) in PhenoGraph and number of metaclusters (nClus) in FlowSOM were adjusted so that both algorithms generated 30 clusters: the same number of cell subsets identified manually. Unexpectedly, no clusters generated by either method were comprised entirely of cells identified by manual gating as a single immunophenotype. The best agreement was obtained for 19 PhenoGraph clusters and 21 FlowSOM clusters in which >50% of clustering cells were assigned a single immunophenotype (Figures 4D, 4E, and S5). For an additional 8 PhenoGraph and 7 FlowSOM clusters, cell populations were made up of two to three immunophenotypes, while the remaining clusters had no obvious correspondence with a known immune cell subset.
We asked why the results of automated clustering differed from those generated by manual gating. In some cases, it was clear that cells of the same manually assigned immunophenotype were split into multiple clusters based only on differences in intensity values of background autofluorescence as determined by human inspection. PhenoGraph clusters 1 and 7 and FlowSOM clusters 1 and 6 were good examples of this phenomenon; cells in these clusters expressed similar levels of CD45 and B220 (an immunomarker of B cells) but differed in background fluorescence in channels used for AF647, BV605, PE-CF594, and BV711 (corresponding to CD11c, CD4, CD8α, and Ly6G respectively) (Figures 4F and S5). These background intensities were at least 10–100-fold lower than CD11c, CD4, CD8α, and Ly6G staining on other cells in the dataset (e.g., CD4 on T cells). By manual gating, low signal intensities are simply assigned a value of zero; in contrast, automated clustering interprets small absolute differences in background as arising from on-target antibody binding and uses them to assign cluster membership. We speculate that, with a large number of relatively low dimensional flow cytometry histograms, manual gating—which in essence is a supervised method of identifying subgroups—may generally be superior to unsupervised clustering.
Impact of GBM on Peripheral Immune Cell Frequency and Network-Level Architecture
The composition of five lymphoid organs with respect to the 30 immunophenotypes identified by manual gating was as expected: the spleen, cervical lymph nodes, and blood were primarily made up of B cells, CD4+ T cells, and CD8+ T cells, the bone marrow was primarily composed of PMN cells, B cells, and monocytes/macrophages, and the thymus was predominantly composed of double-positive T (DPT) cells and immature single-positive T (ISPT) cells (Figures 5A and 5B). When the data were analyzed by principle component analysis (PCA), we found that the first two principle components (PCs) explained >60% of dataset variation—a good performance for PCA. The scores plot for these 2 PCs revealed the presence of five primary clusters separated by tissue type (Figure 5C). As expected, spleen, lymph nodes, and blood exhibited substantial overlap since the cellular composition of these secondary lymphoid tissues are more similar then that of primary lymphoid organs (i.e., bone marrow and thymus). GBM-engrafted mouse 3 at t = 30-days was an outlier (Figure 5C). This mouse had more DCs, macrophages, and Ly6C− PMN cells in its blood and bone marrow relative to any other GBM-burdened animal at the 30-day time point (Figure 5D).
Figure 5. Breakdown of Mouse Lymphoid Tissue Composition in 30 Cellular Immunophenotypes.

(A) Individual (bars) and cumulative (stairs) percentage of 5 lymphoid tissues accounted for by successively scarce cell types.
(B) Distribution of cell types among lymphoid tissues (left y axis) shown in ascending rank order from left-to-right according to their information entropy (dashed black line, right y axis). DPT and ISPT cells had the lowest entropy due to their near-exclusive restriction to the thymus, while mature CD4+ T and CD8+ T cells had the highest entropy due to their broad distribution across multiple tissues.
(C) Scores plot of the first 2 PCs of a PCA performed on a 240-row × 30-column data table of the percentage of each tissue sample (rows) accounted for by each cell type (columns). Circles highlight samples from GBM-burdened mouse 3 at t = 30 days.
(D) Cluster maps of the log2 ratio of the tissue-specific frequency of each cell type in late-stage GBM-burdened mice relative to the median value for the group of 8 mice. Cell types most enriched per tissue in mouse three, which was an outlier with respect to all animals analyzed, are enumerated.
To identify time and tissue-dependent differences in immunophenotypes between healthy and disease-burdened animals we performed 450 Student’s t tests (30 immunophenotypes × 5 tissues × 3 time points). After adjusting for multiple hypothesis testing using the false discovery rate (FDR) method, 25 statistically significant differences were identified (q value <0.05; Figure 6A), most of which were observable at t = 30-days. However, increases in the abundance of double-negative T (DNT) cells in the spleen and lymph nodes of tumor-bearing mice relative to controls were observed as early as 7 days post-engraftment. This likely reflects the immune system’s initial response to tumor cell inoculation as opposed to tumor burden per se and is consistent with the role played by DNT cells in the acute response to inflammation (D’Acquisto and Crompton, 2011).
Figure 6. GBM Perturbs Peripheral Immune Cell Subset Frequency and Correlation Structure.

(A) Mean log2 fold-change in time and tissue-specific cell-type frequency between GBM-bearing and mock-engrafted mice. Asterisks denote one of three levels of statistical significance (FDR-corrected, two-tailed independent Student’s t test, n = 8 mice/group).
(B) Mean percent change in time and tissue-specific cell-type frequencies between GBM-bearing and mock-engrafted mice. Only statistically significant cell subsets are shown (q ≤ 0.05, FDR-corrected, two-tailed independent Student’s t test, n = 8 mice/group).
(C) Agglomerative hierarchical clustering of Spearman’s rank-order correlation coefficients computed across replicates of control (left) and GBM-burdened (right) mice (n = 24 mice/group). Members of GBM-burdened cluster 3 are enumerated in order from top-to-bottom.
Tumor-associated decreases in immune cell frequency were observed for circulating B cells, splenic lymphoid tissue inducer (LTi) cells (Lane et al., 2005), splenic biphenotypic B-macrophages (Borrello and Phipps, 1999), CD45+ Lin− immune progenitor cells in the lymph nodes and spleen, and Ly6C− PMN cells in the bone marrow. Tumor-associated increases were also observed. CD3ϵ+ post-selection thymocytes accumulated in the bone marrow of tumor-bearing mice. This result was consistent with the work of Chongsathidkiet et al. showing that GBM-induced T cell lymphopenia is caused in part by sequestration of T cells in the bone marrow (Chongsathidkiet et al., 2018). Ly6C+ plasma cells (Wrammert et al., 2002) also increased in number in the cervical lymph nodes, suggestive of an active humoral immune response and consistent with the known ability of brain-derived antigens to traffic to the cervical lymph nodes and elicit plasma cell responses (Cserr et al., 1992). Myeloid cells also changed in response to tumor burden: for example, increases in circulating CD11b+ Ly6C+ Ly6G+ PMN cells, consistent with granulocytic myeloid derived suppressor cells (MDSCs). MDSCs are known to accumulate in the blood of GBM-burdened mice and humans (Kamran et al., 2017) (Raychaudhuri et al., 2015) and were 18% more abundant on average in diseased animals at t = 30 days (Figure 6B). Ly6C−-patrolling monocytes, eosinophils, and many other myeloid populations also increased in some tissues while decreasing in others. All of these changes are detailed in Supplemental Information and summarized in the data available through SYLARAS dashboards (Figure S4).
Variation in the frequencies of immune cells in different tissues among mice of the same treatment group suggested a way to investigate immune system homeostasis. Between-subject biological variation (BSBV) (Sebastián-Gámbaro et al., 1997) is a potential confounder in some statistical tests due to increasing data variance and correspondingly weaker p values; however, correlated variation (as determined by correlation analysis) is suggestive of functional interaction between cell types. The effect of GBM on correlated variation between peripheral immune cells was investigated by computing Spearman’s rank-order correlation independently on the tissue-specific immunophenotypes of healthy and diseased animals. Cell populations having a frequency <0.1 % were excluded from the analysis to minimize noise, leaving 76 tissue-specific immunophenotypes for which correlations were computed. Agglomerative hierarchical clustering on the resulting correlation matrices revealed 8 clusters per treatment group (as determined by silhouette analysis; Figure 6C).
Cell-cell correlations for GBM-burdened and control mice were substantially different. For example, GBM-burdened mice exhibited a cluster (cluster 3) significantly enriched for DNT cells in the spleen, thymus, and draining lymph nodes (p = 0.0003, one-tailed binomial test) and PMN cells in the blood and spleen (p = 0.04, one-tailed binomial test) that was not present in control data, suggesting that GBM induces coordinated changes in these two immune cell subsets across multiple tissues. The results of Coffelt et al. (Coffelt et al., 2015) support this finding; they show that IL-17A-producing γδ T cells—which likely correspond to DNT cells in our study—induce a systemic inflammatory cascade leading to the accumulation of circulating PMN neutrophils in a mouse model of breast cancer. More generally, we propose that correlation analysis can be used to probe the self-regulatory architecture of the peripheral immune system and its perturbation by disease. Our data show that peripheral immune architecture is broadly altered by GBM.
B220+ CD8T Cells Are Depleted in the Blood of GBM-Bearing Mice and Accumulate in the Tumor Microenvironment
Among the statistically significant differences in immune cell frequency that were detected between healthy and tumor-bearing animals, two cell populations stood out as exhibiting monotonic changes with increasing statistical significance across time: splenic NK cells and circulating B220+ CD8T cells. Both populations decreased from t = 14 days (q = 0.017 for NK cells; q = 0.026 for B220+ CD8T cells) to t = 30 days (q = 0.0009 for NK cells; q = 0.002 for B220+ CD8T cells) relative to age-matched control mice. NK cells have previously been described in GBM immunology (Baker et al., 2014, 2015, 2016), but the disease has not previously been shown to involve B220+ CD8+ T lymphocytes, cells known to attenuate immune responses to self-antigens in mice and humans (Marvel and Mayer, 1988; Takeuchi et al., 1989; James and Kwok, 2007).
B220, also known as CD45R, is an isoform of the Ptprc gene expressed by murine B cells during all developmental stages from pro-B cells through mature B cells. Unlike the uniformly low B220 expression by CD4+ T cells and high expression by B cells—the canonical B220-expressing immune cell type—B220 expression by CD8+ T cells spanned several orders of magnitude; ~5%–10% of CD8+ T cells expressed B220at levels similar to those of B cells (Figure 7A). Three lines of evidence suggested that these B220hi CD8+ T cells represent a distinct cell subset from conventional CD8+ T cells. First, whereas the abundance of circulating B220+ CD8T cells decreased with advancing disease, CD8+ T cells were unaffected (Figure 7B). Second, B220+ CD8T cells were remarkably rare in the thymus but more abundant in the bone marrow and spleen given their fractional contribution to total CD8+ T cell counts (Figure 7C). Third, although the frequency of splenic B220+ CD8T and CD8+ T cells was strongly correlated in most mice (R2 between 0.6 and 0.9), this correlation was lost in late-stage, disease-bearing animals (R2 = 0.01), suggesting a differential response to the presence of GBM (Figure 7D).
Figure 7. B220+ CD8T Cells Are Diminished in the Blood but Found in the Tumor Microenvironment of Mice Bearing GL261 Glioma.

(A) B220 (x axis) versus CD8α (y axis) scatter plot showing signal intensity distributions for four different immunophenotypes. The broad distribution in CD8α-positive staining is interpreted as reflecting the presence of two overlapping cell populations.
(B) Mean log2 fold-change in time and tissue-specific frequency of the CD8T (left) and B220+ CD8T (right) immunophenotypes (*: 0.01 < q <= 0.05; **: 0.001 < q <= 0.01). Bl, blood; Mw, marrow; Nds, lymph nodes; Sp, spleen; Th, thymus.
(C) Pie charts showing differences in the tissue distribution of CD8T (top) and B220+ CD8T (bottom) cells. Mw, marrow; Th, thymus; Bl, blood; Sp, spleen; Nds, lymph nodes.
(D) Regression analysis comparing the frequencies of CD8T (x axis) and B220+ CD8T (y axis) cells in the spleens of control (left column) and GBM-bearing (right column) mice at 3 time points in tumor progression (rows). Data points represent individual mice. Transparency around the regression line denotes the 95% confidence interval for the regression. R2 = coefficient of determination.
(E) Line plot showing the number of cells corresponding to the seven most abundant immunophenotypes (example images shown) in the GL261 brain tumor microenvironment 36 days after engraftment.
(F) Examples of CD8α single-positive (top) and B220/CD8α double-positive (bottom) lymphocytes identified in the same tumor microenvironment as the cells shown in (E). X and Y tile coordinates are shown above each example to allow for cross-referencing with Figure S7A showing the complete tumor microenvironment at 40×.
(G) Box plot distributions of CD8α signal intensity for the CD8T (left) and B220+ CD8T (right) immunophenotypes in the blood of the 48 mice used in this study (p < 1 × 10−4, two-tailed independent Student’s t test, n = 48 mice). Box plot elements: horizontal line, median; box limits, first to third quartile (Q1 to Q3); whiskers, from Q1-1.5 × interquartile range (IQR) to Q3 + 1.5 × IQR; diamond points, outliers.
(H) Spatial correlation between immunophenotypes within the brain tumor microenvironment. The low degree of correlation between CD8α single-positive and B220/CD8α double-positive cells is highlighted by the red box.
(I) Kernel density estimates of the spatial localization of CD8α single-positive (blue contours) and B220/CD8α double-positive (red contours) cells superimposed on a dot plot diagram of the x and y coordinates of vimentin+ Lin− GBM cells in the same tumor microenvironment. See also Figures S6 and S7 and Table S2.
We postulated that the observed reduction in circulating B220+ CD8T cells in tumor-bearing mice might be a consequence of their extravasation from the circulation into the brain TME. We therefore characterized tumor-infiltrating immune cells using cycles of multiplex immunofluorescence microscopy (t-CyCIF, see STAR Methods for details). An antibody panel functionally similar to the one used for flow cytometry was optimized and validated (Figure S6; Table S2) and then used to acquire 12-channel images of the late-stage brain TME. 168 fields of view (tiles) were collected at 40× magnification, registered and then stitched together to form a composite image from which protein expression data were extracted from ~9 × 104 singe cells comprising the tumor mass following image segmentation (Figures S7A and S7B). Using the immune cell identification features in SYLARAS, we identified multiple tumor-infiltrating immunophenotypes, including B220+ CD8T cells (Figures 7E and S7C-S7E). Relative to lymphocytes single-positive for CD8, the double-positive cells had a distinctive morphology: they were more eccentric, their plasma membranes were smoother, and their CD8α staining was more intense (Figures 7F, S7C, and S7D). Their higher CD8α staining was consistent with data on peripheral B220+ CD8T cells, for which median CD8α staining was ~1.3-fold higher than other circulating CD8+ T cells (Figure 7G). Tumor-infiltrating single-positive and double-positive lymphocytes also occupied different regions within the tumor mass: the former were more frequent at the tumor borders in areas of low tumor cell density, whereas double-positive cells were more evenly distributed in the center of the tumor (Figures 7H and 7I).
DISCUSSION
In this paper, we describe a software tool for efficient analysis of the systemic immune system (SYLARAS) as well as a dataset collected using the tool describing the impact of GBM on immune cells in multiple immune organs using a well-established syngeneic mouse model. Each of ~108 cells from five primary and secondary immune organs was assayed for 11 CD antigens indicative of cell type and differentiation status as well as cell viability, FSC, and SSC. An 11-plex Boolean immunophenotype can specify 2,048 possible cell states, but we found that only 30 such states were populated by >1% of cells in any single tissue sample; across the entire dataset, the frequency of these immunophenotypes ranged from 0.01% for CD45+, CD11c+, CD11b+, Ly6C+, and F4/80+ dendritic cells to 32% for CD45+, B220+ B cells. In aggregate, the cells described in our dataset comprised >97% of viable CD45+ cells. We observed substantial variation (BSBV) in these frequencies, even among age-matched and genetically identical animals, and patterns of correlation and anti-correlation computed from this variability provides insight into the overall architecture of the immune system. One GBM-dependent change in immune cell frequencies we observed involves an atypical subpopulation of CD8+ T cells expressing the CD45R/B220 splice variant of the phosphotyrosine phosphatase CD45 (James and Kwok, 2007; Yu et al., 2018). Across animals, conventional splenic CD8+ T and B220+ CD8+ T cells are highly correlated in abundance except in late-stage GBM-bearing animals in which they are depleted from the circulation. Multiplex tissue imaging shows that depletion is likely to be a consequence of their accumulation in the GBM TME. Based on these and other data, we conclude that immune cell frequencies in the spleen, thymus, blood, cervical lymph nodes, and bone marrow are broadly altered by GBM, in some cases by recruitment to the tumor TME. Thus, localized disease imposes a broad, systems-level perturbation of the immune system.
SYLARAS software presents information on immune cell types in the form of dashboards, one per immune cell type. These dashboards summarize data on frequency, marker status, abundance in different tissue etc. as well as significant differences associated with disease progression. Dashboard attributes can be substituted with other types of information by simple changes in the SYLARAS code, making the software modular and compatible with flow cytometry, CyTOF (Spitzer and Nolan, 2016), and a set of emerging image-based single-cell technologies (Lin et al., 2015, 2018; Goltsev et al., 2018; Gut et al., 2018; Schubert et al., 2006). We found that the scripts in SYLARAS were necessary for processing our GBM immunoprofiling data consisting of nearly 100M cells across more than 200 tissue samples. Data on this scale could not easily be analyzed using GUI-based tools for flow cytometry data analysis such as FlowJo. We integrated results from two tools for unsupervised data clustering into the SYLARAS workflow, PhenoGraph (Levine et al., 2015), and FlowSOM (Van Gassen et al., 2015) but found that manual gating of fluorescence histograms was superior in performance and also rapid: using SYLARAS, it was possible to gate nearly 3,000 histograms in less than 2 h without the need for automated peak finding or Gaussian mixture models.
We were surprised to find that neither FlowSOM nor PhenoGraph yielded clusters similar in marker distribution to those generated by manual gating; in both cases, the majority of clusters contained multiple cell types and some clusters corresponded to cell types that have not previously been described. This appears to arise because unsupervised clustering is sensitive to small variation in non-specific or background fluorescence that is easily recognized as an experimental artifact by humans. In the case of high-dimensional data such as single-cell RNA sequencing (RNA-seq), unsupervised clustering is efficient, eliminates bias, and enables identification of new cell types (Zhang et al., 2019). However, in the case of antibody-based immunoprofiling, prior knowledge is already imposed at the point of antibody selection, particularly because CD antigens are often considered as dichotomous variables either present or absent on immune cells. Under these circumstances, gating appears to be more robust to fluctuations in background signal intensity and peak shape as compared with clustering methods, particularly when the number of samples is high, and fluctuations in autofluorescence intensity accumulate. We do not doubt that an unsupervised workflow could be developed to recapitulate the results of human gating, but manual gating in SYLARAS is simple, reliable, and should be effective with datasets substantially larger than the one presented here. We hope that the data resource on peripheral immune responses to GBM described here can serve as a foundation for more extensive characterization of systemic immunity in disease and therapy.
Limitations of Study
The analysis in this paper is limited to a single implantable GL261 mouse model (Oh et al., 2014). It is likely that spontaneous tumors having greater genetic diversity would be more representative of brain cancer in humans. However, analysis of such de novo tumor models would substantially complicate the type of analysis presented here, since more animals would be required for each genetic subtype. A limitation of syngeneic models is that orthotopic tumor implantation, which involves injecting cancer cells into a native tissue environment using a fine-gauged needle, can generate an inflammatory response. We attempt to account for this by including a control cohort of age-matched C57BL/6J mice injected with vehicle alone. An alternative and potentially better strategy, particularly as a control for samples collected soon after inoculation, would involve injection of primary glial cells as a control.
As compared with single-cell RNA-seq multiplexed flow cytometry measures relatively few parameters; unlike multiplex imaging (Lin et al., 2015, 2018; Goltsev et al., 2018; Gut et al., 2018; Schubert et al., 2006) flow cytometry does not provide spatial information. However, flow cytometry is robust, inexpensive and reproducible, and well-suited to the study of immune cell types. In future studies we aim to combine imaging more fully with flow cytometry on genetically engineered mouse models (GEMMs) and further study interactions between peripheral and tumor-resident immune systems.
Key Changes Prompted by Reviewer Comments
In response to reviewer’s concerns we have added a description of limitations of our study and have removed preliminary RNA-seq and imaging data suggesting that cells similar to B220+ CD8T murine cells can also be found in human GBM. These data will be expanded and described in a future publication. We have also modified the description of SYLARAS to describe how it might be used with barcoding and other multiplexing strategies. For context, the complete transparent peer review record is included within the Supplemental Information.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter K. Sorger (peter_sorger@hms.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Single-cell data files, SYLARAS dashboards, antigen expression boxplots, and t-CyCIF data generated in this study have been deposited to the Sage Synapse data repository (https://www.synapse.org/#!Synapse:syn21038562/wiki/597169) and are located in directories under the following synapse IDs: syn21038618 (FCS files), syn22314373 (combined CSV file), syn22263977 (antigen expression boxplots), syn22249852 (SYLARAS dashboards), syn22249837 (t-CyCIF data). SYLARAS source code is written in the Python programming language and available for academic re-use under an MIT license agreement at Github (https://github.com/gjbaker/sylaras).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Twelve (12)-week-old female C57BL/6J mice (syngeneic to the GL261 mouse glioma cell line) were purchased commercially from the Jackson Laboratory (Stock Number: 000664, Bar Harbor, ME). Mice were randomly assigned to experimental groups and housed under standard conditions. Animal procedures were documented in a protocol (IS00000178) pre-approved by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School. Mice were housed five per microisolator cage adhering to the guidelines outlined by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Harvard Center for Comparative Medicine (HCCM).
Glioma Cells
GL261 (Glioma 261; RRID: CVCL_Y003) mouse glioma cells were obtained from the Developmental Therapeutics Program (DTP), Division of Cancer Treatment and Diagnosis (DCTD) Tumor Repository through a material transfer agreement with the Biological Testing Branch (BTB) of the National Cancer Institute (NCI). The cells were determined by the Section of Comparative Medicine at Yale University’s School of Medicine to be free of common viruses and mycoplasma bacteria (MPV, LCMV, TMEV, SENDAI, MVM, MHV, ECTRO, REO, MYCO) on December 11, 2015. The cells were cultured in T75 or T175 tissue culture flasks under humidified conditions in 95% air/5% CO2 at 37°C. Culture medium consisted of Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 0.3 mg/mL L-glutamine, 50 U/mL penicillin, and 50 mg/mL streptomycin. Cells were passaged every 3-5 days depending on initial seeding density.
METHOD DETAILS
Study Design
To screen the GL261 glioma model by SYLARAS, we randomized a cohort of age-matched C57BL/6J mice (N=48) to one of two treatment groups. One group of mice were engrafted with GL261 glioma cells suspended in cell culture media; the other served as an experimental control and were injected with vehicle alone to account for neuroinflammation caused by tumor implantation and physiological changes in immune system architecture over time. Each of the two cohorts were divided into three subgroups to be euthanized at different time points in tumor progression (n=8 mice/treatment-specific subgroup). Time points were 7-, 14-, and 30-days post tumor engraftment.
Stereotactic Brain Tumor Engraftment
Details on the stereotactic engraftment of glioma cells into the mouse brain can be found here: (Baker et al., 2015). Briefly, mice were anesthetized with an intraperitoneal injection of 75 mg/kg ketamine and 0.5 mg/kg dexmedetomidine followed by a subcutaneous injection of 5 mg/kg carprofen and were found non-responsive to toe and tail pinch prior to proceeding with the procedure. Fur above the cranium was trimmed using surgical clippers and the cranium was prepped with povidone-iodine and scrubbed with 70% isopropyl alcohol. Petrolatum ophthalmic ointment was applied to the eyes to prevent drying before the mouse skull was secured in the stereotactic frame. Using a scalpel, a 1 cm midline incision from the frontal bone to the occipital bone was made. The skin above the cranium was retracted with Colibri retractors and a hole was drilled into the cranium at the stereotactic coordinates: +0.5 mm AP, +2.5 mm ML. A bolus of 3x104 GL261 cells suspended in FBS-free DMEM was then injected 3.0 mm below the surface of the brain using a microliter syringe and a 33G needle. The scalp was sutured with three 3-0 nylon monofilament sutures. Post-operative mice were given 0.1 mg/kg of buprenorphine hydrochloride subcutaneously and 2.5 mg/kg atipamezole hydrochloride intramuscularly for pain and anesthesia reversal and allowed to recover in fresh cages with access to food and water.
General Reagents
ddH20; RPMI-1640 (Corning, Cat. No. 10-040-CV); L-glutamine (Gibco, Cat. No. 25030-081); penicillin-streptomycin (10,000 U/mL) (ThermoFisher Scientific, Cat. No. 15140-163); heat-inactivated (HI) fetal bovine serum (FBS) (Gibco, Cat. No. 16140-071); Dulbecco’s phosphate buffered saline (DPBS) w/o CaCl2, MgCl2 (Corning, Cat. No. 21-040-CV); 5% (w/v) sodium azide (NaN3) (BDH, Cat. No. BDH7465-2); ethylenediaminetetraacetic acid (EDTA) disodium salt dehydrate (C10H14N2Na2O8·2H2O) (Sigma Aldrich, Cat. No. ED2SS); 15 mL polypropylene conical tubes (Falcon, Cat. No. 352097); 50 mL polypropylene conical tubes (Falcon, Cat. No. 352098); 5 mL polystyrene serological pipettes (Corning, Cat. No. 4050); 10 mL polystyrene serological pipettes (Corning, Cat. No. 4100); micropipettes (1000 μL, 200 μL, 20 μL, 10 μL) (Gilson); research plus 12-channel pipette (50-300 μl), (Eppendorf, Cat. No. 3122000060); 0.1-10 μl TipOne natural pipet tips (USA Scientific, Cat. No. 1111-3200); 1.0-20 μl TipOne natural pipet tips (USA Scientific, Cat. No. 1120-1810); 1-200 μl TipOne natural pipet tips (USA Scientific, Cat. No. 1111-1200); 101-1,000 μl TipOne natural pipet tips (USA Scientific, Cat. No. 1111-2820); 40 μm nylon mesh cell strainers (Falcon, Cat. No. 352340); 2 L polyethylene Dewar flask (Nalgene, Cat. No. 4150-2000); sterile cryogenic storage vials (Sigma-Aldrich, Cat. No. V7634); mini vortexer 120V (VWR, Cat. No. 58816-121); polypropylene general-purpose test tube racks (Nalgene, Cat. No. 5930-0020); 96-well reversible microcentrifuge tube rack (Bio Plas, Cat. No. 0091); S1 pipet filler (ThermoFisher Scientific, Cat. No. 9531); 9 L TruCool rectangular ethylene-vinyl acetate foam ice pans (BioCision, Cat. No. BCS-112); 1.5 mL microcentrifuge tubes (USA Scientific, Cat. No. 1615-5500); gel loading tips (Costar, Cat. No. 4853); 60 mm x 15 mm polystyrene tissue culture dishes (Falcon, Cat. No. 353002); 0.4% Trypan Blue solution (Gibco, Cat. No. 15250061)
Reagents Germane to Mouse Euthanasia, Perfusion, and Tissue Processing
ketamine hydrochloride injection (VEDCO, NDC: 50989-996-06); xylazine hydrochloride injection (AKORN INC, NDC: 59399-111-50); 0.9% sodium chloride (NaCl) injection, USP (Hospira, NDC 0409-4888-10); 1 mL Norm-Ject® sterile Luer-slip syringes (Henke Sass Wolf, Cat. No. 4010.200V0); PrecisionGlide needles - 26G x ½ (0.45 mm x 13 mm) (BD, Cat. No. 305111); sodium chloride (NaCl) (Sigma Aldrich, Cat. No. S9888); calcium chloride (CaCl2•2H2O) (Sigma Aldrich, Cat. No. C8106); sodium phosphate monobasic (NaH2PO4•2H2O) (Sigma Aldrich, Cat. No. 71505); D-glucose (C6H12O6) (Sigma Aldrich, Cat. No. G8270); sodium bicarbonate (NaHCO3) (Sigma Aldrich, Cat. No. S5761); potassium chloride (KCl) (Sigma Aldrich, Cat. No. P9333); heparin sodium salt from porcine intestinal mucosa (Sigma-Aldrich, Cat. No. H4784); extruded polystyrene foam block (2); Halsted-mosquito hemostat (2) (Fine Science Tools, Cat. No. 13008-12); fine scissors—martensitic stainless steel (2) (Fine ScienceTools, Cat. No. 14094-11); Friedman rongeur (Fine Science Tools, Cat. No. 16000-14); Littauer bone cutters (Fine Science Tools, Cat. No. 16152-12); cover-glass forceps (Fine Science Tools, Cat. No. 11073-10); Dumont #5 forceps (2) (Fine Science Tools, Cat. No. 11252-40); Graefe forceps (2) (Fine Science Tools, Cat. No. 11051-10); Masterflex L/S digital pump system with easy-load II pump head, 600 RPM, 115/230V (Cole-Parmer, Cat. No. EW-77921-75); 20 G x 1 1/2" aluminum hub blunt needles (Kendall, Cat. No. 8881202363); razor blades (VWR, Cat. No. 55411-050); frosted microscope slides (Fisher Scientific, Cat. No. 12-550-343); 3 mL Luer-Lok® syringes (BD, Cat. No. 309657); PrecisionGlide needles - 23G x 1 (0.6 mm x 25 mm) (BD, Cat. No. 305145); Falcon 3 mL polyethylene transfer pipets (Corning, Cat. No. 357524)
Reagents Germane to Immunolabeling
Brilliant Stain Buffer (BSB) (BD Biosciences, Cat. No. 563794); TruStain FcX anti-mouse CD16/32 antibody (BioLegend, Cat. No. 101320); fixable viability dye, eFluor 455UV (eBioscience, Cat. No. 65-0868-14); Brilliant Ultraviolet 737-conjugated anti-mouse CD11b, clone: M1/70, isotype: rat DA/HA IgG2b, κ (BD Biosciences, Cat. No. 564443); V500-conjugated anti-mouse CD45, clone: 30-F11, isotype: rat LOU/M IgG2b, κ (BD Biosciences, Cat. No. 561487); Brilliant Violet 605-conjugated anti-mouse CD4, clone: RM4-5, isotype: rat IgG2a, κ (BioLegend, Cat. No. 100548); Brilliant Violet 711-conjugated anti-mouse Ly6G, clone: 1A8, isotype: rat IgG2a, κ (BioLegend, Cat. No. 127643); Alexa Fluor 488-conjugated anti-mouse CD3ϵ, clone: 145-2C11, isotype: Armenian hamster IgG (eBioscience, Cat. No. 53-0031-82); PE/Cy7-conjugated anti-mouse CD49b, clone: HMα2, isotype: Armenian hamster IgG (BioLegend, Cat. No. 103518); PE-conjugated anti-mouse F4/80, clone: BM8, isotype: rat IgG2a, κ (BioLegend, Cat. No. 123110); PE-CF594-conjugated anti-mouse CD8α, clone: 53-6.7, isotype: rat LOU/M IgG2a, κ (BD Biosciences, Cat. No. 562283); PerCP/Cy5.5-conjugated anti-mouse/human CD45R/B220, clone: RA3-6B2, isotype: rat IgG2a, κ (BioLegend, Cat. No. 103236); Alexa Fluor 647-conjugated anti-mouse CD11c, clone: N418, isotype: Armenian hamster IgG (BioLegend, Cat. No. 117312); APC/Cy7-conjugated anti-mouse Ly6C, clone: HK1.4, isotype: rat IgG2c, κ (BioLegend, Cat. No. 128026); V500-conjugated rat IgG2b, κ isotype control antibody, clone: A95-1, isotype: rat LOU/M IgG2b, κ (BD Biosciences, Cat. No. 560784); fixation/permeabilization solution kit (BD Biosciences, Cat. No. 554714); 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (ThermoFisher Scientific, Cat. No. D1306); 96-well V-bottom, non-treated, polystyrene microplate (Costar, Cat No. 3897); 12-well, V-bottom reagent reservoir (Argos Technologies, Cat. No. B3135); Microseal ‘F’ foil seal (Bio-Rad; Cat. No. MSF1001)
Reagents Germane to Flow Cytometry
Sphero rainbow fluorescent particles (3.0-3.4 μm) (BD Biosciences, Cat. No. 556291); FACSDiva CS&T research beads (BD Biosciences, Cat. No. 655051)
Preparation of Reagents
Supplemented RPMI-1640
A 500 mL bottle of RPMI-1640 with L-glutamine was supplemented to achieve the following reagent concentrations: 100 U/mL penicillin, 100 U/mL streptomycin, 10% HI-FBS, 0.05% sodium azide, and 0.1% (w/v) EDTA.
Heparinized Tyrode’s Solution
A 1 L glass screw-cap storage bottle containing a stirring bar was filled with ddH2O and stirred continuously. The following reagents were added: 8.0 g sodium chloride, 0.264 g calcium chloride, 0.05 g sodium phosphate monobasic, 1.0 g D-glucose, 1.0 g sodium bicarbonate, 0.2 g potassium chloride, 100 U of heparin sodium. Stored at 4°C.
Ammonium-Chloride-Potassium (ACK) Lysis Buffer (1X)
A 1 L glass screw-cap storage bottle containing a stirring bar was filled with ddH20 and stirred continuously. The following reagents were added: 8.29 g of ammonium chloride, 1.0 g of potassium bicarbonate, 37.2 mg of sodium EDTA. pH adjusted to 7.4. Stored at room temperature.
Flow Buffer
1X DPBS containing 0.5% HI-FBS (100-200 mL). Stored at 4°C.
Flow Buffer + Azide
Flow Buffer containing 0.05% (w/v) sodium azide (100-200 mL). Stored at 4°C.
EDTA Solution
1X DPBS containing 10% (w/v) disodium (100 mL). Ultrasonicated and stored at 4°C.
Fc Block
Flow Buffer containing 15 μg/mL mouse monoclonal anti-CD16/32 antibodies (see Table S1 for preparation details).
Fixable Viability Dye
eFluor 455UV fixable viability dye diluted 1.5:1,000 with 1X DPBS (see Table S1 for preparation details).
Major Equipment and Parameter Settings
Flow Cytometer
BD LSR II Special Order Research Product (SORP) flow cytometer with BD High Throughput Sampler (HTS) — laser lines and wattages were as follows: 488 nm (20 mw, run at 20 mw); 405 nm (50 mw, run at 50 mw); 594 nm (200 mw, run at 125 mw); 355 nm laser (20 mw, run at 20 mw).
Plate Washer
BioTek EL406 automated microplate washer/dispenser — instrument configurations were as follows: plate type = 96-well, W-aspirate, vacuum filtration = false, travel rate = 1 (4.1 & 1.0 mm/sec), delay = 0 msec, z-offset = 55 steps (6.99 mm above carrier), x-offset = 0 steps (center of well), y-offset = 0 steps (center of well), secondary aspirate = no.
Centrifuge and Rotor
Beckman Coulter Avanti J-26XP centrifuge equipped with a Beckman Coulter JS-5.3 anodized aluminum swinging-bucket rotor
Ultrasonic Bath
Bransonic CPXH ultrasonic cleaning bath (model 3800)
Antibody Titration
Preparation of Splenocytes
To identify optimal immunolabeling concentrations for 11 mouse immune cell lineage antibodies, the spleens of two 12-week-old female C57BL/6J mice were harvested according to the procedure described in the “Lymphoid Tissue Harvesting and Processing” section of the STAR Methods. Spleens were gently macerated using opposing frosted sides of two glass microscope slides, then rinsed thoroughly into a single 60 x 15 mm polystyrene Petri dish on ice containing 4 mL of supplemented RPMI-1640. Splenocytes were transferred from the Petri dish to a 15 mL conical tube and centrifuged at 350 x g (max RCF) for 10 minutes at 4°C. The cell supernatant was discarded and the pellet was resuspended with 8 mL of 1X ACK lysis buffer. The tube was incubated on ice for 5 minutes before 6 mL of Flow Buffer + Azide was added to inhibit further lysis. The cell suspension was passed through a 40 μm nylon mesh into a fresh 15 mL conical tube, centrifuged at 350 x g (max RCF) for 10 minutes at 4°C to pellet the cells, and resuspended with 2 mL of Flow Buffer + Azide. Cell counting was performed using a hemocytometer and Trypan Blue viability dye; splenocyte concentration was adjusted accordingly with additional Flow Buffer + Azide to achieve 1x107 cells/mL.
Immunolabeling
Using a multichannel pipette, 200 μL of the 1x107 cells/mL splenocyte suspension was transferred into 11 concentric columns of a 96-well V-bottom microplate on ice (8 rows per column) to test the following 2-fold serial dilution series for each antibody: 24, 12, 6, 3, 1.5, 0.75, 0.375, 0.1875 μg/mL. The plate was next centrifuged at 100 x g (max RCF) for 3 minutes at 4°C. One-hundred and fifty (150) μL of cell supernatant were aspirated from each well using a BioTek EL406 automated microplate washer/dispenser and resuspended with 100 μL of 15 μg/mL Fc Block (see Table S1 for preparation details). Splenocytes were allowed to incubate on ice for 5 minutes, then centrifuged at 100 x g (max RCF) for 3 minutes at 4°C. One-hundred (100) μL were aspirated from each well and resuspended with 100 μL of antibodies diluted in BSB. Splenocytes were allowed to immunolabel on ice for 15 minutes in the dark. The plate was centrifuged at 100 x g (max RCF) for 3 minutes at 4°C. Two-hundred (200) μL of supernatant were aspirated from each well and resuspended with 100 μL of Flow Buffer + Azide as a wash step. The cells were again centrifuged at 100 x g (max RCF) for 3 minutes at 4°C and washed for a second time using 200 μL Flow Buffer + Azide. Immunolabeled cells were incubated in a solution of 1 μg/mL DAPI for 5 minutes prior to flow cytometric analysis.
Data Acquisition
Immunolabeled splenocytes were analyzed by flow cytometry using a BD LSR II SORP flow cytometer equipped with a BD High Throughput Sampler (HTS) for the automated acquisition of data from 96-well plates. The following gating strategy was used: (FSC-A vs. SSC-A) → (SSC-H vs. SSC-W) → (FSC-H vs. FSC-W) → (DAPI-A vs. FSC-A) → (CDx vs. count). Ten-thousand (10,000) viable singlets were analyzed per well. A staining index (SI) was calculated for each antibody at each immunolabeling concentration. Concentrations yielding the maximum SI for each antibody were chosen for use in our SYLARAS screen of the GL261 mouse glioma model.
Lymphoid Tissue Harvesting and Processing
Preparation of Reagents
The following disposable reagents were gathered before each of the study’s three time points: (80) 0.5 mL microcentrifuge tubes, labeled by tissue type, treatment condition, and biological replicate; (64) 60 x 15 mm polystyrene Petri dishes on ice, labeled by tissue type (excluding blood), treatment condition, and biological replicate; (16) 15 mL conical tubes for blood collection, labeled by treatment condition and biological replicate; (16) 3 mL syringes equipped with 23G needles, for bone marrow aspiration, labeled by treatment condition and biological replicate; (16) 1 mL tuberculin syringes equipped with 26G needles for transcardial blood draws, labeled by treatment condition, biological replicate; and a single 1 mL tuberculin syringe equipped with a 26G needle for anesthesia administration.
One-hundred (100) μL of a 10% EDTA solution were added to each 15 mL conical tube. Four (4) mL of supplemented RPMI-1640 were added to each Petri dish with the exception of those labeled bone marrow, which received 2 mL of media. The other 2 mL were deposited into the respectively-labeled 3 mL syringes for bone marrow aspiration. One-hundred ninety-eight (198) μL of Flow Buffer were added to each microcentrifuge tube. Fifty (50) μL of a 10% EDTA solution were added to each 1 mL tuberculin syringe for drawing blood. Conical tubes, Petri dishes, blood drawing syringes, etc. were kept on ice or stored at 4°C throughout the tissue harvesting and processing procedure.
Mouse Anesthesia
At each time point, 8 GBM-bearing mice and 8 age-matched, mock-engrafted controls were terminally anesthetized in series using 150 mg/kg of ketamine hydrochloride and 20 mg/kg xylazine hydrochloride diluted in sterile 0.9% NaCl per mouse delivered intraperitoneally using a 1 mL tuberculin syringe (See Table S1 for preparation details). Once non-responsive to both toe and tail pinch, each mouse was pinned ventral-side-up to an extruded polystyrene foam block by their front and hind paws using four 26G needles (one per paw). The abdomen was sprayed with 70% ethanol to disinfect the incision site and obstruct fur from entering the dissection cavity.
Tissue Excision
Five (5) lymphoid organs of each mouse were harvested in the following order: blood, thymus, spleen, deep and superficial cervical lymph nodes, bone marrow.
Blood: A “Y” incision was made from the abdomen to the rib cage exposing the heart. Blood was aspirated from the right ventricle into a 1 mL tuberculin syringe. Syringe needles were removed prior to expelling blood from the tuberculin syringe into a respectively-labeled 15 mL conical tube. Mice underwent transcardial perfusion with heparinized and oxygenated (95% O2/5% CO2) Tyrode’s solution at a rate of 4.0 mL/minute for at least 2 minutes in a laminar flow hood to achieve complete exsanguination. More detailed methods on transcardial perfusion of mice can be found here: (Baker et al., 2015).
Thymus: Post-exsanguination, mice were returned to the extruded polystyrene foam block for thymus excision with small dissection scissors and fine-tipped, bent forceps. Care was taken to avoid contaminating adipose tissue and mediastinal lymph nodes. Thymi were stored in respectively-labeled 60 x 15 mm polystyrene Petri dishes on ice.
Spleen: Spleens were excised using small dissection scissors and fine-tipped, bent forceps and stored in respectively-labeled 60 x 15 mm polystyrene Petri dishes on ice.
Superficial/deep cervical lymph nodes: Under a dissection microscope, lymph nodes were removed with small dissection scissors and fine-tipped, bent forceps and stored in respectively labeled 60 x 15 mm polystyrene Petri dishes on ice.
Bone marrow: The right hind limb was removed using bone cutters and trimmed to isolate the femur and tibia. Proximal and distal epiphyses of each bone were removed using a single-edged razor blade. Bone marrow was flushed into respectively-labeled 60 x 15 mm polystyrene Petri dishes on ice using 3 mL syringes containing 2 mL of supplemented RPMI-1640.
Tissue Processing
Spleens, lymph nodes, and thymi were gently macerated using opposing frosted sides of two glass microscope slides, then rinsed thoroughly into respectively-labeled 60 x 15 mm polystyrene Petri dishes on ice containing 4 mL of supplemented RPMI-1640. For cervical lymph nodes, plastic Pasteur pipettes were used to transfer the nodes onto the frosted side of a single microscope slide for maceration. Five (5) mL of ice-cold 1X DPBS were added to each Petri dish and cell suspensions were filtered through clean 40 μm nylon meshes into respectively-labeled 15 mL conical tubes using 10 mL serological pipettes. Tubes were centrifuged at 400 xg (max RCF) for 10 minutes at 4°C before cell pellets were resuspended in 4 mL of a 1X ACK lysis buffer using 5 mL serological pipettes. Tubes were incubated on ice for 5 minutes, centrifuged at 400 x g (max RCF) for 10 minutes at 4°C, and resuspended with varying amounts of Flow Buffer + Azide. The following tissue-specific volumes were used for initial resuspension: 1000 μL (thymus), 1000 μL (spleen), 200 μL (bone marrow), 100 μL (combined deep and superficial cervical lymph nodes).
Blood samples underwent RBC lysis for 5 minutes on ice by adding 10 mL of 1X ACK lysis buffer to each 15 mL conical tube (or until blood changed from dark burgundy to bright red). The samples were then centrifuged at 400 x g (max RCF) for 10 minutes at 4°C and resuspended with 200 μL of Flow Buffer + Azide.
Cell Counting
Using a P20 micropipette equipped with a gel loading tip, 2 μL of each tissue sample were added to respectively-labeled 0.5 mL microcentrifuge tubes containing 198 μL of Flow Buffer. Ten (10) μL of the resulting 1:100 dilutions were further diluted 1:1 with 0.4% Trypan Blue. Ten (10) μL of these dilutions were used for cell counting with a hemocytometer. Cell yields varied by tissue type: ~3x105 – 1x106 (blood), ~3x107 – 8x107 (spleens), ~6x107 – 8x107 (thymi), ~6x106 – 1.5x107 (deep/superficial cervical lymph nodes), ~9x106 – 1.5x107 (bone marrow). Counts were recorded in an Excel spreadsheet formatted to compute the necessary dilutions to achieve a concentration of 2x107 cells/mL across all samples (Table S1). Blood samples typically contained less than 2x106 cells and were not diluted further.
Immunolabeling
Reagent Preparation
The following disposable reagents were gathered at each time point in the study: (22) 1.5 mL microcentrifuge tubes, labeled in duplicate with antibody names (including the CD45 isotype control) and organized into two duplicate rows of a microcentrifuge rack stored on ice; (1) 12-well, V-bottom reagent reservoir labeled with antibody names (including the CD45 isotype control); (3) 15 mL conical tubes, one labeled “FVD”, one labeled “Fc Block”, and one labeled “Cocktail”; (1) 96-well, V-bottom microplate.
Antibody Dilution
Antibodies (11 immune lineage markers plus CD45 isotype control) were diluted 1:10 with BSB in respectively-labeled microcentrifuge tubes on ice (see Table S1 for preparation details). A fraction of each working dilution was used in preparing single-color compensation controls. The remaining working dilutions (excluding the CD45 isotype control) were combined into the 15 mL conical tube labeled “Cocktail”, which served as the antibody master mix for multiplex immunolabeling. Antibody stocks and dilutions were kept on ice in the dark.
Labeling
Using a multichannel pipette, (100) μL of each lymphoid tissue sample (1x106 cells) were added to respective wells of a 96-well V-bottom microplate according to a predefined plate layout (Figure 1). To obtain enough CD49b+ cells for fluorescence compensation of the CD49b flow cytometer channel, 100 μL of each 200 μL white blood cell suspension was deposited into a common CD49b single-color compensation control well (G9); the other 100 μL were deposited into the respectively-labeled experimental well. The plate was then centrifuged at 100 x g (max RCF) for 6 minutes at 4°C. Fifty (50) μL of cell supernatant were aspirated from each well using a BioTek EL406 automated microplate washer/dispenser such that a residual 50 μL remained in each well. One hundred (100) μL of 15 μg/mL Fc Block (see Table S1 for preparation details) were added to each well of the 96-well plate using a multichannel pipette, mixed by pipetting, and incubated on ice for 5minutes. The plate was centrifuged at 100 x g (max RCF) for 5 minutes at 4°C and 50 μL of cell supernatant were aspirated from each well. Each of 80 experimental tissue samples received 100 μL of the master antibody mix from the 15 mL conical tube labeled “Cocktail”. One-hundred (100) μL of each single-color compensation control were added to its respective well; 100 μL of CD45 isotype antibodies were added to the “ISO” control well, and 100 μL of stock BSB were added to the wells labeled “UNS” and “FVD”.
Cells were incubated for 15 minutes on ice in the dark before 100 μL of 1X DPBS were added using a multichannel pipette and pipetted thoroughly to mix. The plate was then centrifuged at 100 x g (max RCF) for 5 minutes at 4°C. One-hundred ninety-one (191) μL of cell supernatant were aspired from each well and resuspended with 200 μL of 1X DPBS using a multichannel pipette followed by mixing. The plate was again centrifuged at 100 x g (max RCF) for 5 minutes at 4°C. Two hundred (200) μL of cell supernatant were removed from each well.
Viability Staining
With the exception of the well labeled “UNS”, 100 μL of a 1.5:1,000 dilution of fixable viability dye from the 15 mL conical tube labeled “FVD” (see Table S1 for preparation details) were added to each well of the 96-well V-bottom microplate. This achieved a final staining concentration of 1:1,1000. Cells were then incubated on ice in the dark for 30 minutes. One-hundred (100) μL of 1X DPBS were added to each well and pipetted to wash. The plate was centrifuged at 100 x g (max RCF) for 5 minutes at 4°C. Two-hundred (200) μL of cell supernatant were aspirated from each well and resuspended with 200 μL of 1X DPBS. The plate was again centrifuged at 100 x g (max RCF) for 5 minutes at 4°C followed by the aspiration of 200 μL of cell supernatant.
Cell Fixation and Short-term Storage
One-hundred (100) μL of BD fixation/permeabilization solution were added to each well of the 96-well plate and pipetted to prevent cell crosslinking. The plate was then incubated on ice in the dark for 20 minutes. One-hundred (100) μL of Flow Buffer were added to the wells and pipetted to mix before the plate was centrifuged at 100 x g (max RCF) for 5 minutes at 4°C. Two-hundred (200) μL of cell supernatant were aspired from each well and resuspended with 200 μL of Flow Buffer. The prepared microplate plate was covered with a Microseal ‘F’ foil seal to prevent sample dehydration, wrapped in aluminum foil to block light, then stored at 4°C before flow cytometric analysis.
Cytometer Setup and PMT Calibration
PMT voltages on a BD LSR II SORP flow cytometer were calibrated such that signal intensities corresponding to viable unlabeled splenocytes (well E10) were on scale and to the left of center in each detection channel. To prevent downstream compensation values from exceeding 100%, optical spillover of single-color compensation controls into off-target detection channels was checked to ensure that peak signal intensities occurred in their respective detection channel. Sphero Rainbow Fluorescent Particles (i.e. SP beads) were run at the outset of our study to predefine tolerability ranges for laser intensity, stability, and alignment, and to prevent run-to-run variation so that fluctuations in laser emission power between data acquisition cycles could be accounted for with calibration. SP beads were gated using (FSC-A vs. SSC-A) and visualized as biexponential histograms in each detection channel.
Data Acquisition
Cytometer setup & tracking was performed using FACSDiva CS&T research beads to optimize and standardize instrument performance across data acquisition cycles. During each acquisition, the 96-well V-bottom microplate was placed in a BD High Throughput Sampler (HTS) affixed to a BD LSR II SORP flow cytometer. The HTS system was programmed to collect events from all wells of the plate in a predefined order. Because fluidic anomalies can impact laser delay stability, wells corresponding to single-positive (SP) beads were run first to check that PMT voltages remained within previously defined tolerability ranges. Optical controls were then run in the following order: UNS, FVD, ISO, single-color compensation controls, experimental samples, SP beads (to check that PMT voltages were stable over the data acquisition period). Normal C57BL/6J splenocytes were used for all compensation controls except the CD49b single-positive control, which consisted of pooled WBCs from all samples to achieve enough CD49b+ cells for fluorescence compensation. The following acquisition gating strategy was used: (FSC-A vs. SSC-A) → (SSC-H vs. SSC-W) → (FSC-H vs. FSC-W) → (BUV395-A vs. FSC-A) → (CDx vs. count). Histograms were plotted on biexponential scale. Antibodies were detected using the combinations of laser lines, band pass filters, and long pass filters shown in (Figure S1D). Raw data were exported as FCS3.0 files.
Fluorescence Compensation
Raw flow cytometry data were spectrally deconvolved using commercial software (FlowJo). Optical controls were imported into the compensation group of a FlowJo workspace. Single-color compensation controls plus cells from the FVD optical-control well (E10) were gated for viable singlets according to the following strategy: (FSC-A vs. SSC-A) → (SSC-H vs. SSC-W) → (FSC-H vs. FSC-W) → (BUV395-A vs. FSC-A). Data in the last plot of the gating strategy (BUV395-A vs. FSC-A) were viewed in contour at the 2% level. Viable singlets were then backgated to FSC-A vs. SSC-A to isolate subsets of viable singlets suitable for fluorescence compensation of each detection channel.
Unimodal CD45 histograms made it difficult to compensate this channel. This was overcome by combining the data from the CD45 isotype control well (E11) with that of the CD45 single-color compensation control well (F9) using FlowJo’s concatenate feature. This resulted in a bimodal distribution which was saved as a FCS3.0 file to the compensation group of the FlowJo workspace and compensated in an otherwise typical fashion. Data corresponding to wells E11 and F9 were no longer needed and deleted from the workspace.
Compensation control data were visualized as histograms in their respective detection channels. Histograms were gated at the interface of the first and second signal intensity peaks using FlowJo’s bisector tool. Using FlowJo’s compensation tool, data corresponding to the left and right subsets were dragged into the “negative” and “positive” software interface fields. After performing this process on each of the 11 immunomarkes plus the FVD control, a new group was created in the workspace and given the name “Cocktail”. Data corresponding to the 80 experimental tissue samples were imported into this group. The compensation matrix was then applied to the “Cocktail” group. The resulting compensated experimental data were gated according to the same strategy as optical control samples to achieve compensated, viable singlets which were exported as new FCS3.0 files.
Gating Strategy
Data from compensated viable singlets were visualized as a set of 2,640 histograms (80 experimental tissue samples displayed in 11 immunomarker channels at 3 time points in tumor progression). Histograms were plotted as scalable vector graphics (SVGs) on Logicle scale (Parks et al., 2006) and displayed as a scrolling HTML table. Kernel density estimates (KDEs) generated for each detection channel using data from well E10 (unlabeled control splenocytes) at each time point were superimposed over their corresponding experimental histograms to determine the point at which antibody signal intensities superseded cell-intrinsic autofluorescence. Points at the interface between signal and noise were manually curated and recorded in a .TXT file.
CD45 signal intensity distributions were unimodal with no discernable local minima. Thus, for each combination of time point and tissue, a common CD45 gate was curated by pooling the corresponding samples, computing Q25 – [1.5 * [Q75 - Q25]] then rounding to the nearest multiple of 5 (where Q25 and Q75 were the first and third quartiles of the Logicle-transformed data, respectively). The .TXT file containing gate points was used to update the HTML table with vertical lines at the curated gate points for confirmation or refinement.
Compensated viable singlets and the curated gate points were Logicle transformed (Parks et al., 2006). The transformed gate points were subtracted from each of the transformed signal intensity values comprising their corresponding distributions. This caused the gate points to become zero and autofluorescence signal intensities to become negative-valued. A spurious population of CD49b+ granulocytes were identified in blood samples. As this was a suspected artifact caused by the interaction between residual CD49b+ platelets and Ly6G+ neutrophils in blood samples (Barnard et al., 2005), we only considering CD49b immunomarker status when the immunophenotype of an immune cell was otherwise consistent with that of an NKcell (e.g. CD45+, CD49b+, CD11b+). The Logicle-transformed and background-subtracted data were saved as individual FCS3.0 files (https://www.synapse.org/#!Synapse:syn21038618) and a combined CSV table (https://www.synapse.org/#!Synapse:syn22314373) which served as input into the SYLARAS software tool.
t-CyCIF
5μm-thick coronal sections of GL261-bearing mouse brain sections were cut from formalin-fixed, paraffin-embedded (FFPE) tissue blocks. Tissues were mounted on glass microscope slides and iteratively immunolabeled through multiple rounds of 4-color immunofluorescence (i.e. Hoechst, AF488, AF555, AF657) using the t-CyCIF method (Lin et al., 2018). Briefly, tissue sections underwent nuclear counterstaining for 30 minutes at room temperature through incubation with Hoechst dye (10 mg/mL stock) diluted 1:5,000 in 1X PBS. Slides were washed thoroughly in 1X PBS then incubated in Odyssey Blocking Buffer at room temperature for 1 hour to reduce non-specific antibody binding. Autofluorescence background was imaged before each immunolabeling cycle to increase signal-to-noise ratios in the final images. Antibodies were used against the following targets: Ly6C, CD8α, CD68, CD45R/B220, CD4, CD49b, Ly6G, Foxp3, CD11b, Ki67, Vimentin (See Table S2 for antibody details). Immunolabeling was performed at 4°C overnight in opaque and humidified chambers. Tissues were washed thoroughly in 1X PBS and temporarily coverslipped in 1X PBS containing 10% glycerol before imaging with a CyteFinder slide-scanning fluorescence microscope (RareCyte, Seattle, WA, USA) using a 40X (0.6NA) objective and 2x2 binning. After each imaging session, slides were submersed in Coplin jars containing 1X PBS so that the coverslip would spontaneously come away from the microscope slide. Antibody fluorophores were quenched after each round of imaging by incubating the tissues in a 3% H2O2 solution diluted with 1X PBS and containing 20 mM NaOH for 2 hours at room temperature in the presence of intense fluorescent light.
One-hundred sixty-eight (168) 400x300μm imaging fields were acquired at each imaging cycle. Imaging fields across cycles were aligned and registered on Hoechst signal intensity using ImageJ’s Multistack Registration Plugin. Registered images were segmented on Hoechst-stained nuclei using ImageJ’s Analyze Particles function. The segmentation mask of each cell was dilated by three pixels to capture antibody signal within the cell cytoplasm and at the plasma membrane. Median immunomarker signal intensities of ~9x104 cells from within the brain tumor microenvironment were computed and analyzed with SYLARAS software.
Software
FACSDiva
version 8.0
FlowJo
version 10.3.0
Python
version 3.6.1
PhenoGraph
version 1.5.2
FlowSOM
version 1.14.1
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using documented and validated statistical functions in the SciPy.stats library for statistical computing (https://docs.scipy.org/doc/scipy/reference/stats.html). Specific tests and sample sizes are indicated at their point of reference either in the main text or related figure legends. Hypothesis tests were two-tailed and performed using independent observations except for the binomial tests for cluster enrichment associated with data shown in Figure 6C. A statistical significance threshold was chosen for all tests at an FDR-adjusted p-value (q-value) of less than 0.05. Spearman’s rank-order tests for correlation were used under the assumption of monotonically-related ordinal data. Coefficients of determination (R2) are reported for all regression analyses. Agglomerative hierarchical clustering was performed using the unweighted pair group method with arithmetic mean (UPGMA) linkage algorithm and Euclidean distance metric. Tissue-weighted random sampling of flow cytometry data was performed by assigning a weight to each cell in the data according to the following formula: 1/(ω x Ni), where ω is the number of unique tissue types and Ni is the number of events associated with the ith tissue (e.g. blood, marrow, nodes, spleen, thymus).
ADDITIONAL RESOURCES
Additional information related to SYLARAS: www.sylaras.org.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD45 (clone: 30-F11) | BD Biosciences | Cat#561487; RRID: AB_10697046 |
| Rat monoclonal anti-CD11b (clone: M1/70) | BD Biosciences | Cat#564443; RRID: AB_2738811 |
| Hamster monoclonal anti-CD3ϵ (clone: 145-2C11) | eBioscience | Cat#53-0031-82; RRID: AB_469889 |
| Rat monoclonal anti-CD4 (clone: RM4-5) | BioLegend | Cat#100548; RRID: AB_2563054 |
| Rat monoclonal anti-CD8α (clone: 53-6.7) | BD Biosciences | Cat#562283; RRID: AB_11152075 |
| Hamster monoclonal anti-CD49b (clone: HMα2) | BioLegend | Cat#103518; RRID: AB_2566103 |
| Rat monoclonal anti-F4/80 (clone: BM8) | BioLegend | Cat#123110; RRID: AB_893486 |
| Rat monoclonal anti-Ly6G (clone: 1A8) | BioLegend | Cat#127643; RRID: AB_2565971 |
| Rat monoclonal anti-Ly6C (clone: HK1.4) | BioLegend | Cat#128026; RRID: AB_10640120 |
| Rat monoclonal anti-CD45R/B220 (clone: RA3-6B2) | BioLegend | Cat#103236; RRID: AB_893354 |
| Hamster monoclonal anti-CD11c (clone: N418) | BioLegend | Cat#117312; RRID: AB_389328 |
| Rat monoclonal anti-CD16/32 (clone: 93) | BioLegend | Cat#101320; RRID: AB_1574975 |
| Rat monoclonal IgG2b, κ isotype (clone: A95-1) | BD Biosciences | Cat#560784; RRID: AB_1937321 |
| Biological Samples | ||
| Formalin-fixed, paraffin-embedded, GL261 glioma-bearing mouse brain tissue sections (36-days post tumor engraftment) | This paper | N/A |
| Deposited Data | ||
| Flow Cytometry Standard (FCS) files | This paper | https://www.synapse.org/#!Synapse:syn21038618 |
| Combined CSV table | This paper | https://www.synapse.org/#!Synapse:syn22314373 |
| SYLARAS Dashboards | This paper | https://www.synapse.org/#!Synapse:syn22249852 |
| Antigen expression by different mouse immune cell subsets identified via prior knowledge and unsupervised clustering algorithms | This paper | https://www.synapse.org/#!Synapse:syn22263977 |
| t-CyCIF data (mouse lymph node, GL261 glioma-36dpi) | This paper | https://www.synapse.org/#!Synapse:syn22249837 |
| Experimental Models: Cell Lines | ||
| Mouse: GL261 glioma cells | Developmental Therapeutics Program (DTP), Division of Cancer Treatment and Diagnosis (DCTD) Tumor Repository, National Cancer Institute; https://dtp.cancer.gov/organization/btb/tumor_repositories.htm | RRID: CVCL_Y003 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Software and Algorithms | ||
| SYLARAS software | This paper | https://github.com/gjbaker/sylaras |
| FACSDiva (version 8.0) | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software | |
| FlowJo (version 10.3.0) | https://www.flowjo.com/ | |
| Python (version 3.6.1) | https://www.python.org/ | |
| R (version 3.5.3) | https://www.r-project.org/ | |
| PhenoGraph (version 1.5.2) | Levine et al., 2015 | https://github.com/jacoblevine/PhenoGraph |
| FlowSOM (version 1.14.1) | Van Gassen et al., 2015 | https://bioconductor.org/packages/release/bioc/html/FlowSOM.html |
| Other | ||
| SYLARAS website | This paper | https://www.sylaras.org/ |
Highlights.
Tumors broadly alter peripheral immune architecture in a syngeneic mouse model
SYLARAS and flow cytometry facilitate exploration of multiorgan immune phenotypes
Manual gating outperforms clustering in assigning immune cell types
B220+ CD8T cells are one type depleted from circulation and recruited to tumors
ACKNOWLEDGMENTS
This work was supported by American Cancer Society Postdoctoral fellowship PF-16-197-01-LIB to G.J.B., by NIH/NCI grants P50-GM107618 and U54-CA225088 to P.K.S., and by the Harvard Ludwig Center. We gratefully acknowledge G. Berriz and A. Sokolov for their computational support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2020.08.001.
DECLARATION OF INTERESTS
P.K.S. is a member of the SAB or board of directors of Glencoe Software, Applied Biomath, and RareCyte and has equity in these companies; he is also on the SAB of NanoString. In the last 5 years, the Sorger lab has received research funding from Merck. Sorger declares that none of these relationships are directly or indirectly related to the content of this manuscript.
REFERENCES
- Baker GJ, Castro MG, and Lowenstein PR (2015). Isolation and flow cytometric analysis of glioma-infiltrating peripheral blood mononuclear cells. J. Vis. Exp 105, 53676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GJ, Chockley P, Yadav VN, Doherty R, Ritt M, Sivaramakrishnan S, Castro MG, and Lowenstein PR (2014). Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer Res. 74, 5079–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GJ, Chockley P, Zamler D, Castro MG, and Lowenstein PR (2016). Natural killer cells require monocytic Gr-1(+)/CD11b(+) myeloid cells to eradicate orthotopically engrafted glioma cells. Oncoimmunology 5, e1163461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard MR, Linden MD, Frelinger AL, Li Y, Fox ML, Furman MI, and Michelson AD (2005). Effects of platelet binding on whole blood flow cytometry assays of monocyte and neutrophil procoagulant activity. J. Thromb. Haemost 3, 2563–2570. [DOI] [PubMed] [Google Scholar]
- Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, and Parsa AT (2013). Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res 19, 3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, Simonds EF, Bendall SC, Sachs K, Krutzik PO, and Nolan GP (2012). Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol 30, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MA, and Phipps RP (1999). Fibroblast-secreted macrophage colony-stimulating factor is responsible for generation of biphenotypic B/macrophage cells from a subset of mouse B lymphocytes. J. Immunol 168, 3605–3611. [PubMed] [Google Scholar]
- Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, et al. (2018). Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med 24, 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intra-tumoral and systemic immune responses in recurrent glioblastoma. Nat. Med 25, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, and de Visser KE (2015). IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, Fong L, Campbell MJ, Cooper S, Oakes SA, et al. (2014). Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. USA 111, 12823–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr HF, Harling-Berg CJ, and Knopf PM (1992). Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 2, 269–276. [DOI] [PubMed] [Google Scholar]
- D’Acquisto F, and Crompton T (2011). CD3+CD4-CD8− (double negative) T cells: saviours or villains of the immune response? Biochem. Pharmacol 82, 333–340. [DOI] [PubMed] [Google Scholar]
- Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, and Nolan GP (2018). Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut G, Herrmann MD, and Pelkmans L (2018). Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042. [DOI] [PubMed] [Google Scholar]
- Huettner C, Czub S, Kerkau S, Roggendorf W, and Tonn JC (1997). Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 17, 3217–3224. [PubMed] [Google Scholar]
- Ito H, Nakashima H, and Chiocca EA (2019). Molecular responses to immune checkpoint blockade in glioblastoma. Nat. Med 25, 359–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CM, Choi J, and Lim M (2019). Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol 20, 1100–1109. [DOI] [PubMed] [Google Scholar]
- James EA, and Kwok WW (2007). CD8+ suppressor-mediated regulation of human CD4+ T cell responses to glutamic acid decarboxylase 65. Eur. J. Immunol 37, 78–86. [DOI] [PubMed] [Google Scholar]
- Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, Raja N, Shah D, Lowenstein PR, and Castro MG (2017). Immunosuppressive myeloid cells’ blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol. Ther 25, 232–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane PJL, Gaspal FMC, and Kim MY (2005). Two sides of a cellular coin: CD4(+)CD3− cells regulate memory responses and lymph-node organization. Nat. Rev. Immunol 5, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el-A.D., Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Fallahi-Sichani M, and Sorger PK (2015). Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvel J, and Mayer A (1988). CD45R gives immunofluorescence and transduces signals on mouse T cells. Eur. J. Immunol 18, 825–828. [DOI] [PubMed] [Google Scholar]
- Maxwell M, Galanopoulos T, Neville-Golden J, and Antoniades HN (1992). Effect of the expression of transforming growth factor-beta 2 in primary human glioblastomas on immunosuppression and loss of immune surveillance. J. Neurosurg 76, 799–804. [DOI] [PubMed] [Google Scholar]
- Melnykov V, and Maitra R (2010). Finite mixture models and model-based clustering. Statist. Surv 4, 80–116. [Google Scholar]
- Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, and Parsa AT (2014). Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DR, Roederer M, and Moore WA (2006). A new “logicle” display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry A 69, 541–551. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, and Vogelbaum MA (2015). Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neurooncol 122, 293–301. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. (2016). Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res 4, 124–135. [DOI] [PubMed] [Google Scholar]
- Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea C, López-Diaz de Cerio A, Tejada S, et al. (2019). Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med 25, 470–476. [DOI] [PubMed] [Google Scholar]
- Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Böckelmann R, Malykh Y, Gollnick H, Friedenberger M, Bode M, and Dress AWM (2006). Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol 24, 1270–1278. [DOI] [PubMed] [Google Scholar]
- Sebastián-Gámbaro MA, Lirón-Hernández FJ, and Fuentes-Arderiu X (1997). Intra- and inter-individual biological variability data bank. Eur. J. Clin. Chem. Clin. Biochem 35, 845–852. [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic immunity is required for effective cancer immunotherapy. Cell 168, 487–502.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, and Nolan GP (2016). Mass cytometry: single cells, many features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Rudd CE, Tanaka S, Rothstein DM, Schlossman SF, and Morimoto C (1989). Functional characterization of the CD45R (2H4) molecule on CD8 (T8) cells in the autologous mixed lymphocyte reaction system. Eur. J. Immunol 19, 747–755. [DOI] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, Tobias AL, Han Y, and Lesniak MS (2012). IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res 18, 6110–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al. (2014). Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res 20, 5290–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Bruce DS, Dustin L, Loh DY, and Bluestone JA (1995). Ly-6C is a marker of memory CD8+ T cells. J. Immunol 155, 1873–1883. [PubMed] [Google Scholar]
- Weber LM, and Robinson MD (2016). Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry A 89, 1084–1096. [DOI] [PubMed] [Google Scholar]
- Wrammert J, Källberg E, Agace WW, and Leanderson T (2002). Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur. J. Immunol 32, 97–103. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ma X, Gong R, Zhu J, Wei L, and Yao J (2018). Recent advances in CD8+ regulatory T cell research. Oncol. Lett 15, 8187–8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B, et al. (2019). Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 16, 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med 25, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. (2015). Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol 17, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell data files, SYLARAS dashboards, antigen expression boxplots, and t-CyCIF data generated in this study have been deposited to the Sage Synapse data repository (https://www.synapse.org/#!Synapse:syn21038562/wiki/597169) and are located in directories under the following synapse IDs: syn21038618 (FCS files), syn22314373 (combined CSV file), syn22263977 (antigen expression boxplots), syn22249852 (SYLARAS dashboards), syn22249837 (t-CyCIF data). SYLARAS source code is written in the Python programming language and available for academic re-use under an MIT license agreement at Github (https://github.com/gjbaker/sylaras).