

Abstract
Background
Intracranial aneurysms (IAs) are dangerous because of their potential to rupture. We previously found significant RNA expression differences in circulating neutrophils between patients with and without unruptured IAs and trained machine learning models to predict presence of IA using 40 neutrophil transcriptomes. Here, we aim to develop a predictive model for unruptured IA using neutrophil transcriptomes from a larger population and more robust machine learning methods.
Methods
Neutrophil RNA extracted from the blood of 134 patients (55 with IA, 79 IA-free controls) was subjected to next-generation RNA sequencing. In a randomly-selected training cohort (n = 94), the Least Absolute Shrinkage and Selection Operator (LASSO) selected transcripts, from which we constructed prediction models via 4 well-established supervised machine-learning algorithms (K-Nearest Neighbors, Random Forest, and Support Vector Machines with Gaussian and cubic kernels). We tested the models in the remaining samples (n = 40) and assessed model performance by receiver-operating-characteristic (ROC) curves. Real-time quantitative polymerase chain reaction (RT-qPCR) of 9 IA-associated genes was used to verify gene expression in a subset of 49 neutrophil RNA samples. We also examined the potential influence of demographics and comorbidities on model prediction.
Results
Feature selection using LASSO in the training cohort identified 37 IA-associated transcripts. Models trained using these transcripts had a maximum accuracy of 90% in the testing cohort. The testing performance across all methods had an average area under ROC curve (AUC) = 0.97, an improvement over our previous models. The Random Forest model performed best across both training and testing cohorts. RT-qPCR confirmed expression differences in 7 of 9 genes tested. Gene ontology and IPA network analyses performed on the 37 model genes reflected dysregulated inflammation, cell signaling, and apoptosis processes. In our data, demographics and comorbidities did not affect model performance.
Conclusions
We improved upon our previous IA prediction models based on circulating neutrophil transcriptomes by increasing sample size and by implementing LASSO and more robust machine learning methods. Future studies are needed to validate these models in larger cohorts and further investigate effect of covariates.
Keywords: Intracranial aneurysm, Neutrophil, Transcriptomics, Machine learning, Inflammation, Prediction model
Background
Intracranial aneurysms (IAs) are present in up to 6% of the general population, but only about 10% show any symptoms prior to rupture [1]. The rupture of an IA is the leading cause of nontraumatic subarachnoid hemorrhage, which has a mortality rate up to 50% [2–4]. Clinical studies have shown that the incidence of future rupture can be decreased with elective endovascular or surgical treatment [5, 6]. However, because most IAs are asymptomatic, unruptured aneurysms are usually only detected incidentally on cerebral imaging performed for other medical reasons. Imaging by magnetic resonance imaging/angiography (MRI/MRA), computed tomography angiography (CTA), and digital subtraction angiography (DSA) are not routinely used for IA screening because they are expensive and carry the risk for serious complications due to their invasive nature (e.g., DSA) or exposure to radiation (e.g., DSA, CTA). Thus, a blood test to identify individuals with unruptured IAs could facilitate a paradigm shift to proactive IA management by enabling routine screening and preventive treatment.
We hypothesized that changes in circulating neutrophil gene expression are correlated with the presence of IA in the cerebral vasculature. Neutrophils, the most abundant leukocyte in human blood, are important sentinels for tissue damage and defense against microorganisms, and play a central role in the coordination of innate immunity and inflammation [7]. Recent evidence has suggested that neutrophils have a large degree of heterogeneity in both phenotype and function, playing a wide range of roles in homeostasis and disease-specific responses [8]. Indeed, in previous publications, our group has demonstrated different circulating neutrophil signatures between individuals with cystic fibrosis and idiopathic arthritis [7], suggesting that neutrophils have the ability to adapt their transcriptomes to specific biologic contexts.
Recent studies have begun to show that neutrophils may display disease-specific responses during IA pathophysiology. Investigations of resected aneurysms have found elevated levels of key proteins released from neutrophils, namely myeloperoxidase (MPO) and neutrophil gelatinase-associated lipocalin (NGAL) in the IA wall [9, 10]. These proteins have also been found to be elevated in plasma levels of blood from patients with IA [10, 11]. These proteins may play a key role in extracellular matrix (ECM) degradation and neutrophil activation [12, 13] during aneurysm natural history via their production of reactive oxygen species (ROS) and protection of MMP-9 degradation, respectively [9, 10]. Neutrophils can produce a wide range of cytokines, namely inflammatory and immunoregulatory cytokines, chemokines (necessary for monocyte infiltration), angiogenic and fibrogenic factors, and tumor necrosis factor (TNF) superfamily members [14]. Studies have shown that TNF is upregulated in human IA tissues [15], and from animal models of the disease, is essential for IA formation [16, 17]. Additionally, based on their reported role in progression and rupture of abdominal aortic aneurysms, neutrophils may also release neutrophil extracellular traps (NETs) in IA [18–21].
In a small, proof-of-concept study, we previously performed differential expression analysis in case-controlled cohorts (n = 11 IA, n = 11 control) and found an 82-gene signature that distinguishes IAs from controls [22]. Bioinformatics analyses broadly reflected peripheral neutrophil activation in patients with IA, as genes with elevated expression in the IA group were associated with leukocyte activation, cell activation, and defense response [22]. In a follow-up study, we next utilized machine learning to determine whether an algorithm could predict the presence of unruptured IA using differential gene expression [23]. In an unmatched cohort (n = 30), 26 highly-informative neutrophil transcripts (FDR < 0.05, abs[fold-change] ≥ 1.5) were used to construct a diagonal Linear Discriminant Analysis model, which predicted the presence of IA with an accuracy of 90% in a small independent cohort (n = 10) [23].
While these results were exciting, due to the small sample size, it was difficult to generalize our findings to a broader population. Therefore, in this study, we aimed at confirming these results in a larger cohort of patients. Importantly, this increased sample size would enable us to: (A) implement more advanced feature selection methods (in place of basic thresholding) and machine learning techniques (i.e. Random Forest) to improve prediction accuracy, and (B) examine potential effect of demographics and comorbidities on model prediction.
Methods
Study enrollment
This study was approved by University at Buffalo Institutional Review Board (study no. 030-474433). All methods followed the approved protocol. Written informed consent was obtained from all subjects prior to sample collection. Patients receiving cerebral DSA at Gates Vascular Institute, Buffalo, NY with and without IA diagnosis were enrolled in this study. Most indications for DSA included confirmation of noninvasive imaging results of presence of IAs or other cerebral vascular conditions, follow-up of non-invasive imaging for headache or visual disturbance, or follow-up of previously identified IAs. All patients who consented to participate in this study were over 18 years, English speaking, and had not previously been treated for IA. Patients with potentially altered immune systems were excluded, including, for example, patients who had recent invasive surgery, were receiving chemotherapy, had a fever (> 100°F), had received solid organ transplants, had autoimmune diseases, or were taking prednisone or other immunomodulating drugs.
Between December 2013 and September 2018, we collected 232 blood samples from cerebral DSA patients at Gates Vascular Institute (103 from patients with IA, and 129 from IA-free controls). Forty-three of these samples had been sequenced as a part of our previous studies [22, 23]. In all cases, IA diagnosis was confirmed by DSA images. Patient medical record data was also collected to study demographics and comorbidities.
Neutrophil isolation
During DSA, 16 mL of blood was drawn from the access catheter in the femoral artery and transferred into two 8 mL, citrated, cell preparation tubes (BD, Franklin Lakes, NJ). Neutrophils were isolated within 1 h of blood collection, as described elsewhere [7]. Briefly, cell preparation tubes were centrifuged at 1700×g for 25 min to separate erythrocytes and neutrophils from mononuclear cells and plasma in the peripheral blood samples via a Ficoll density gradient. Erythrocytes and neutrophils were collected into a 3 mL syringe. Following hypotonic lysis of red blood cells, neutrophils were isolated by centrifugation at 400×g for 10 min, disrupted and stored in TRIzol reagent (Life Technologies, Carlsbad, CA) at − 80 °C until further processing. Neutrophils isolated in this fashion are more than 98% CD66b+ by flow cytometry and contain no contaminating CD14+ monocytes [24].
RNA preparation
Neutrophil RNA was extracted as described previously [22] using TRIzol, according to the manufacturer’s instructions. Trace DNA was removed by DNase I (Life Technologies, Carlsbad, CA) treatment. RNA was purified using the RNeasy MinElute Cleanup Kit (Qiagen, Venlo, Limburg, Netherlands) and suspended in RNase-free water. The purity and concentration of RNA in each sample were measured by absorbance at 260 nm and 280 nm on a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA), and 200 ng to 400 ng of RNA was reserved for sequencing. Precise RNA concentration was measured via the Quant-iT RiboGreen Assay (Invitrogen, Carlsbad, CA) with a TBS-380 Fluorometer (Promega, Madison, WI), and the quality of the RNA samples was measured with an Agilent 2100 BioAnalyzer RNA 6000 Pico Chip (Agilent, Las Vegas, NV). RNA samples to be sequenced had acceptable purity (260/280 ratio of ~ 1.8 or greater, range: 1.76–2.12) and integrity (RIN of ~ 5 or greater, range: 4.5–9.1) prior to RNA sequencing.
RNA sequencing
For newly processed samples, the Illumina TruSeq Stranded Total RNA Gold Library Preparation Kit (Illumina, San Diego, CA) was used for library preparation. Samples were subjected to 50-cycle, single-read sequencing in a HiSeq2500 system (Illumina) and demultiplexed using Bcl2Fastq. To increase sample size, we combined reads from these new samples with reads from our previous samples [22, 23] that were sequenced in the same manner, but for which libraries were constructed using the Illumina TruSeq RNA library Prep Kit V2 (Illumina, San Diego, CA). For all data, per-cycle base-call (BCL) files generated by the Illumina HiSeq2500 were converted to per-read FASTQ files using bcl2fastq version 2.20.0.422 using default parameters. The quality of the sequencing was reviewed using FastQC version 0.11.5. Detection of potential contamination was done using FastQ Screen version 0.11.1. FastQC and FastQ Screen quality reports were summarized using MultiQC version 1.5. No adapter sequences were detected, so no trimming was performed. Genomic alignments were performed using HISAT2 version 2.1.0 using default parameters. NCBI reference GRCh38 was used for the reference genome and gene annotation set. Sequence alignments were compressed and sorted into binary alignment map (BAM) files using samtools version 1.3. Counting of mapped reads for genomic features was performed using Subread featureCounts version 1.6.2 using the parameters -s 2 –g gene_id –t exon –Q 60, the annotation file specified with—a was the NCBI GRCh38 reference from Illumina iGenomes. Aggregate quality control data (i.e. alignment statistics and feature assignment statistics) were again summarized using MultiQC.
Differential expression analysis and data exploration
Before implementing our machine learning pipeline, we performed differential expression analysis on the whole dataset to identify transcripts that were significantly differentially expressed in IA using Bioconductor package edgeR version 3.24.0. After estimating dispersion, edgeR identified differentially expressed genes by using a negative binomial distribution with generalized linear models and a quasi-likelihood F-test to identify differentially expressed genes [25, 26]. We incorporated the two sequencing batches into the design matrix to correct for any potential batch effects due to different library preparation kits. Genes with a counts sum > 0 across all samples were used as input. Multiple hypothesis testing correction was performed using Benjamini–Hochberg false discovery rate (FDR) correction [27]. Transcripts with an FDR-corrected p-value (q-value) < 0.05 were considered significantly differentially expressed. To explore how transcriptomes separated patients with and without IA on a broad scale, we performed hierarchical clustering, using the hclust package in R under default settings (complete linkage).
Next-generation sequencing is typically performed on high quality RNA (RIN > 7), when possible [28]. However, clinical samples, particularly human neutrophils that contain high levels of endonucleases as part of the host defense response to bacteria, rarely produce RNA without any degree of degradation [29]. Between IA and control groups in our study, there was no statistically significant difference in RIN (p = 0.18, Student’s t-test). Yet, given that some samples had low RIN and others had high RIN, we performed co-variate correlation analysis as shown in Xiong et al. [30] in order to determine if RNA quality could have affected the expression levels of differentially expressed transcripts. A correlation between gene expression and RIN was considered if it had both a Pearson correlation coefficient r > 0.80 and a p-value < 0.01.
Verification of expression differences by qPCR in a sub-cohort
To verify expression differences in differentially expressed genes, quantitative polymerase chain reaction (qPCR) was performed. Due to limitations in RNA quantity, qPCR was performed on 10 transcripts in a subset of 50 of the 134 samples (20 IA and 30 control). We followed the protocol described previously [22]. In brief, oligonucleotide primers were designed using Primer3 software (Primer3Web 0.4.0) and Primer BLAST (NCBI, Bethesda, MD) to have a 60 °C melting temperature, a length of 15–25 nucleotides, and a product of 50–250 base pairs (with at least one primer that spans an exon-exon junction), as well as an estimated efficiency > 0.8 (actual range: 0.82–1.1, average: 0.96, median: 1.0) [31]. Primer sequences, annealing temperatures, efficiencies, and product lengths are reported in Additional file 1: Table S1. For reverse transcription, first-strand cDNA was generated from total RNA using qScript cDNA Synthesis kit (Quantabio, Beverly, MA, USA) according to the manufacturer’s directions. qPCR was run with 5 ng of cDNA in 20 µL reactions in triplicate in Bio-Rad CFX Connect (Bio-Rad, Hercules, California) using the qScript One-Step SYBR Green Master Mix kit (Quantabio, Beverly, MA, USA) and gene-specific primers at a concentration of 0.02 μM each. The temperature profile consisted of an initial step of 95 °C for 1 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min, and then a final melting curve analysis from 60 to 95 °C over 20 min.
Gene-specific amplification was demonstrated by a single peak using the Bio-Rad dissociation melt curve. Samples were normalized based on HPRT1, GAPDH, and GPI (housekeeping genes [32–34]) expression, which were run in parallel reactions to the genes of interest. Additional file 2: Fig. S1 shows the stability in the expression (from RNA sequencing, Additional file 2: Fig. S1A) and Ct values (from qPCR, Additional file 2: Fig. S1B) of the housekeeping genes in the control and IA groups. All had similar coefficient of variation values, and there was no statistical significance in their variation between the two groups for sequencing or qPCR (all p-values > 0.01, F-test). Their Ct values were used to calculate gene-specific average fold-change between the two groups using the 2−ΔΔCt method [35]. These values were then averaged across all housekeeping genes. For comparison with qPCR data, RNA sequencing data from the same samples was used to calculate the average fold-change in gene expression between the IA and the control groups. This fold-change value (presented as an absolute fold-change in the positive or negative direction [for fold-change < 1]) was then compared to the same metric calculated by the 2−ΔΔCt method in qPCR data in order to determine if the absolute fold-change in expression was in the same direction and statistically different (Student’s t-test, significance at p-value < 0.05).
Feature selection for classification model development
To build predictive models for IA, we began with raw counts to eliminate bias and uncertainty associated with distribution modeling incorporated in edgeR. Raw counts were then normalized to transcript per million (TPM) values to facilitate comparison of expression between samples by normalizing by both gene length and sequencing depth. Then, we applied an abundance filtering by only selecting protein coding genes with average TPM > 1 across all samples, reducing the set of potential transcripts to 18,833. To account for the two sequencing batches in our study design (as done in edgeR analyses), we performed batch effect correction using ComBat under the default settings in R [36, 37]. Then, 70% of samples were randomly allocated to a training cohort (n = 39 IA and n = 55 control) and 30% to a testing cohort (n = 16 IA and n = 24 control), maintaining the proportion of IA and controls.
For feature selection in the training cohort, we performed a supervised feature selection by using the Hilbert–Schmidt Independence Criterion Least Absolute Shrinkage and Selection Operator (HSIC LASSO) method. HSIC LASSO was implemented in the ComBat corrected dataset to select features for the model. To visualize how those selected transcripts separated samples from patients with and without IAs, we performed principal component analysis (PCA) using the prcomp package under the default settings [38]. Co-variate correlation analysis following Xiong et al. [30] was performed again to determine if RNA quality (i.e. RIN) could have affected the expression levels of TPM-normalized transcripts selected by LASSO. A correlation between gene expression and RIN was considered if it had both a Pearson correlation coefficient r > 0.80 and a p-value < 0.01.
Model training
We used MATLAB Statistics and Machine Learning Toolbox (MathWorks, Natick, MA) to train 4 popular algorithms (K-Nearest Neighbor, Random Forest, Support Vector Machine with Gaussian and cubic kernels) on 2 different gene panels—the 37 transcripts identified by LASSO in this study and the 26 transcripts identified by filtering in our previous study. While these algorithms have been used in other disease classification applications [39–44], we implemented all 4 algorithms to determine which best suited our data. Specific parameters for each algorithm are as follows:
For K-Nearest Neighbors, we used a Euclidean metric and 10 neighbors (k). The resulting model classified test samples by calculating their distance to each training sample and the test sample labels were predicted by choosing the class that was most common among their 10 nearest neighbors.
For Random Forest, which constructs a multitude of decision trees in training and outputs the mode of the classes as the predicted class [45], we set the number of trees as 1000. The Random Forest was built by constructing a multitude of decision trees based on subsets of the training data generated by random sampling with replacement and the resulting model classified testing samples by the majority vote of the decision trees.
For Support Vector Machines, we used two different kernels [46], Gaussian and cubic. To separate a binary-labeled sample, the Support Vector Machine transforms them into a multidimensional space using the kernel, and then a hyper-plane, which maximizes the distance to samples of either class, is established. The resulting model classified testing samples by transforming them into a higher dimensional space with the corresponding kernel and making decisions based on their signed distance to the hyper-plane.
Model assessment
We estimated the performance of each model for the new LASSO-identified features as well as our previously-identified 26 features by a leave-one-out (LOO) cross-validation within the training cohort. Model predictions were compared to each patient’s clinical diagnosis from imaging, and the true positives, true negatives, false positives, and false negatives were tallied. We then calculated each model’s sensitivity, specificity, and accuracy, as described elsewhere [23]. Based on model predictions, we created receiver operating characteristic (ROC) curves and calculated the area under the ROC curve (AUC) to assess model performance. Additionally, to gauge predictive value of models, we determined positive predictive value (PPV) and negative predictive value (NPV). PPV and NPV were estimated using formulas based on Bayes’ theorem as previously described [23] with 5% aneurysm prevalence, which is within the range of IA prevalence reported in the literature (3.2–7% [47–50]). The classification models were then independently evaluated in the testing cohort (n = 40), and classification results were compared to clinical diagnoses to calculate the true sensitivity, specificity, and accuracy for each model. ROC curves were constructed and AUCs, along with PPV and NPV, were used to assess the performance of each classifier. This was performed for algorithms trained on LASSO-selected features and the previous 26 features.
Testing influence of clinical covariates on gene expression differences
While we randomly assigned samples to training or testing cohorts, this study was not cohort-controlled and used a large, heterogeneous population. Consequently, it is possible that factors other than IA status, such as demographics or comorbidities, could be affecting differential expression and model performance. To determine if patient characteristics influenced model performance, we first performed a chi-square test to determine if there were different rates in the aneurysm and control populations. We examined gender, hypertension, heart disease, stroke, high cholesterol, cancer, diabetes, arthritis, asthma, smoking status, and age. Additionally, we performed covariate matching in the MatchIt program in R to create 6 subclasses under default settings with similar distribution of covariates (age [60 and under vs over 60], sex, smoking status [non-smoker vs current smoker], hypertension, heart disease, stroke history, high cholesterol, cancer, diabetes, arthritis, asthma, and IA family history) for aneurysm and control populations [51, 52]. To create subclasses, we used a distance measure determined by a logistic regression model to estimate the propensity score. We then examined misclassification rate in each of the subclasses to determine if any group with a specific “covariate profile” was associated with greater misclassification.
Bioinformatics
Gene ontology enrichment analysis was performed using Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GORILLA) [53]. A background list of neutrophil expression from 3 healthy individuals (average fragments per kilobase million > 0.5) was used to compute hypergeometric statistics and assign significance to GO terms [7]. GO functions and processes with a p-value < 0.001 were reported. Ingenuity Pathway Analysis (IPA) software (Qiagen Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) was used to investigate networks associated with the differentially expressed genes identified by edgeR (q < 0.05, fold-change > 2) and those selected by LASSO during feature selection. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Knowledge Base and overlaid onto a molecular network derived from information accumulated in the Knowledge Base. Gene networks were algorithmically generated based on their “connectivity” derived from known interactions between the products of these genes. Networks were considered significant if their p-scores were ≥ 15. Network score is calculated as p-score = −log10(p-value), so a score of 15 corresponds to a p-value of 1E-15 [54].
Results
Study population
We obtained and analyzed an additional 91 samples from individuals undergoing cerebral DSA that met data and RNA quality criteria. Combined with the 43 samples we previously analyzed, our total dataset was 134 neutrophil transcriptomes—55 from patients with IA and 79 from control patients. The characteristics of study population are presented in Table 1; detailed aneurysm characteristics in Additional file 3: Table S2. Overall, the 134 samples had an average 260/280 ratio of 2.04 (median: 2.05) and an average RIN of 6.7 (median: 6.7), as shown in the quality data reported in Additional file 4: Table S3. Patients with IA had 73 aneurysms (as 12 individuals had multiple IAs), which ranged in size from 1 to 19 mm measured by largest diameter on 2D images.
Table 1.
Clinical characteristics of training and testing cohorts
| Training Cohort | Testing Cohort | |||
|---|---|---|---|---|
| Control (n = 55) | Aneurysm (n = 39) | Control (n = 24) | Aneurysm (n = 16) |
|
| Age (Mean ± SE) | 62 ± 2.0 | 61 ± 1.7 | 59 ± 2.9 | 57 ± 3.3 |
| Age [Median (Q1/Q3)] | 66 (54/72) | 60 (54/68) | 59 (54/68) | 58.5 (49.25/63.25) |
| Sex (% of patients) | ||||
| Female | 56.36% | 69.23% | 50% | 75% |
| Smoker (% of patients) | ||||
| Yes | 10.91% | 26.64% | 20.83% | 43.75% |
| Comorbidities (% of patients) | ||||
| Hypertension | 61.82% | 53.85% | 54.17% | 50% |
| Heart disease | 30.91% | 23.08% | 25% | 18.75% |
| High cholesterol | 52.73% | 48.72% | 62.50% | 50% |
| Stroke history | 12.73% | 10.26% | 25% | 0% |
| Diabetes | 29.09% | 17.95% | 8.33% | 31.25% |
| Arthritis | 16.36% | 30.77% | 16.67% | 18.75% |
Clinical characteristics of the randomly-created training and testing cohorts. With the exception of age, these factors were quantified as binary data points. The clinical factors were retrieved from the patients’ medical records via the latest “Patient Medical History” form administered prior to imaging
Differential RNA expression in neutrophils from patients with IA vs. controls
RNA sequencing data were used to identify differentially expressed neutrophil transcripts between IA and control groups. Overall, our sequencing experiments had an average of 55.06 million reads per sample and a 95% read mapping rate (or % aligned), as reported in Additional file 4: Table S3. The scatter plot in Fig. 1a shows neutrophil expression differences between IA patients and controls in terms of average fold-change in expression and significance level. Differential expression analysis in edgeR identified 65 transcripts that were significantly differentially expressed (q < 0.05, fold-change > 2) (red and blue points in Fig. 1b). Twenty-three genes showed lower expression in the IA group, and 42 showed higher expression. For all differentially expressed transcripts, correlation analysis demonstrated that RIN was not a significant co-variate; the maximum absolute Pearson correlation coefficient was 0.21, or a coefficient of determinization of R2 = 0.043 (range in R2: 2.76E-6 to 0.043), and none had p-value < 0.01 (Additional file 5: Fig. S2A). Using all transcriptome data, we performed supervised hierarchical clustering to determine if gene expression in general could also discriminate patients with IAs from controls. On the dendrogram in Fig. 1c, samples from IA and control groups are separated. The dendrogram shows 7 clusters of primarily IA or control samples (highlighted sections). Overall, hierarchical clustering congregated 73% of the samples with their respective groups.
Fig. 1.

RNAseq data from whole dataset (n = 134). a The scatter plot demonstrates the dispersion in expression between the IA and control groups. b The volcano plot produced following edgeR analysis demonstrates that there are 65 differentially expressed genes. Red points are increased in IA group and blue points are decreased in IA group. c Clustering performed on all transcriptome data demonstrates several distinct clusters of IA and control samples. Overall, 73% of samples were assigned to the correct group
Bioinformatics results
To gain biological insight into the observed neutrophil RNA expression differences between IA and control groups, we performed bioinformatics analyses using gene set enrichment analysis and physiological pathway modeling. We used GORILLA to analyze the ontologies associated with the edgeR genes with increased and decreased expression in IA as compared to background of healthy individuals. Genes with increased expression in IA were associated with cell migration, cell motility, T cell migration, and lymphocyte migration processes. On the other hand, genes with decreased expression in IA had functions related to sodium channel activity, ion channel activity, and gated channel activity, as well as signaling and regulation of membrane potential processes. A full list of ontologies associated with edgeR genes is reported in Additional file 6: Table S4. IPA gene network analysis identified 3 significant networks with p-scores of 21, 21, and 15, respectively (Fig. 2). The first network was associated with cell morphology, cell-to-cell signaling and interaction, nervous system development and function, with hubs around IRS1, GRIK1, GRIN2A, and L-glutamic acid. The second network is associated with connective tissue development and function, dermatological diseases and conditions, organismal injury and abnormalities, with hubs of ELAVL1, CCND1, and FMOD. Finally, the last network was enriched for cell death and survival, connective tissue disorders, and inflammatory disease functions, reflected by a predominant hub being TNF. Associated molecules and diseases/functions for each network are listed in Additional file 7: Table S5.
Fig. 2.

Networks derived from IPA of the 65 differentially expressed transcripts (q < 0.05, fold-change > 2). Transcripts with increased expression in IA are red; transcripts with lower expression in IA are green; fold-change is represented by intensity. a This network (p-score = 21) has related functions of cell-to-cell signaling and interaction, nervous system development and function, and cell morphology. b This network (p-score = 21) associated with dermatological diseases and conditions, organismal injury and abnormalities, and connective tissue development and function. c This network (p-score = 15) has ties to cell death and survival, connective tissue disorders, and inflammatory disease
Verification of expression differences by RT-qPCR
We confirmed expression differences of 9 prominent IA-associated transcripts (C1QL1, GPR15, HES4, PVRL2, CD163, CYP1B1, CDH2, ZBTB16, PTGDS) using RT-qPCR (qPCR was attempted on PDE9A, but results were not included due to low efficiency of the primer pairs; efficiency < 0.50). These genes were selected because they were prominently differentially expressed transcripts, i.e. were in the models we trained, were highly abundant in at least one cohort, or were significantly differentially expressed. This confirmation was performed in a subset of 49 patient samples, as one IA sample did not provide sufficient data for analysis across all genes and so that data is not included here. Figure 3 demonstrates that the expression differences between patients with and without IA were of the same direction and of similar magnitudes when calculated by both RNA sequencing and RT-qPCR, with the exception of CDH2 and ZBTB16. There was a statistical difference between qPCR and RNA sequencing for C1QL1 and ZBTB16 (both p-values < 0.034). Only ZBTB16 had both a significant and different direction in fold-change than that reported by RNA sequencing.
Fig. 3.
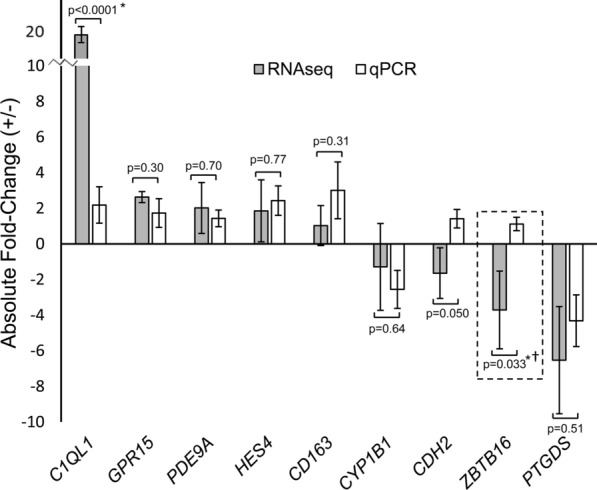
Verification of RNA-Sequencing data for 9 transcripts by qPCR. A total of 49 of the sequenced samples were analyzed by RT-qPCR, as the other samples did not have enough RNA for the additional reactions. Seven of the 9 transcripts in samples in a subset of patients had the same direction of expression difference on qPCR. There was a statistically significant difference in fold-change in expression (indicated by *) between RNAseq and qPCR for C1QL1 and ZBTB16. Only ZBTB16 had both a significant and different fold-change direction (indicated by †) than that calculated with RNA sequencing data. (Negative fold-change values calculated by negative inverse of fold-change, error bars = standard error.)
Selected transcripts and model training
Feature selection using LASSO identified 37 IA-associated transcripts with significant expression in the training cohort, which were used to create models with 4 machine learning algorithms. Table 2 reports gene-specific accuracy, sensitivity, and specificity of the 37 model genes. For all transcripts selected by LASSO, correlation analysis also demonstrated that RIN was not a significant co-variate; the maximum absolute Pearson correlation coefficient was < 0.08 at 0.54 or R2 = 0.29 (range in R2: 4.36E−6 to 0.29), although 11 genes had a p-value < 0.01 (Additional file 5: Fig. S2B).
Table 2.
The 37 transcripts selected for classification model training
| Gene | Gene ID | Accession # | Training Cohort | Testing Cohort | ||||
|---|---|---|---|---|---|---|---|---|
| Acc | Sen | Spe | Acc | Sen | Spe | |||
| AC011380.1 | – | AC011380 | 0.56 | 0.64 | 0.51 | 0.58 | 0.69 | 0.50 |
| C1QL1† | 10882 | NM_006688.5 | 0.80 | 0.56 | 0.96 | 0.93 | 0.88 | 0.96 |
| CCDC42B | 387885 | NM_001144872.2 | 0.59 | 0.05 | 0.96 | 0.65 | 0.19 | 0.96 |
| CEP295NL† | 100653515 | NM_001243541.1 | 0.71 | 0.59 | 0.80 | 0.80 | 0.56 | 0.96 |
| CERS4† | 79603 | NM_024552.3 | 0.79 | 0.72 | 0.84 | 0.85 | 0.88 | 0.83 |
| CLP1 | 10978 | NM_006831.3 | 0.57 | 0.00 | 0.98 | 0.58 | 0.00 | 0.96 |
| DCUN1D1 | 54165 | NM_020640.3 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| EIF4EBP3 | 8637 | NM_003732.3 | 0.47 | 0.26 | 0.62 | 0.68 | 0.69 | 0.67 |
| FLT1 | 2321 | NM_002019.4 | 0.52 | 0.00 | 0.89 | 0.50 | 0.00 | 0.83 |
| GBGT1† | 26301 | NM_021996.6 | 0.71 | 0.74 | 0.69 | 0.70 | 0.88 | 0.58 |
| GPR15† | 2838 | NM_005290 | 0.79 | 0.79 | 0.78 | 0.93 | 1.00 | 0.88 |
| GPR157 | 80045 | NM_024980.5 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| GTF2B | 2959 | NM_001514.6 | 0.55 | 0.03 | 0.93 | 0.55 | 0.00 | 0.92 |
| HBB | 3043 | NM_000518.5 | 0.79 | 1.00 | 0.64 | 0.58 | 0.94 | 0.33 |
| HIST1H4E | 8367 | NM_003545.3 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| HIST2H2AB | 317,72 | NM_175065.2 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| ISY1 | 57461 | NM_001199469.1 | 0.60 | 0.05 | 0.98 | 0.65 | 0.13 | 1.00 |
| KIAA1324 | 57535 | NM_020775.5 | 0.59 | 0.05 | 0.96 | 0.55 | 0.06 | 0.88 |
| KIAA1614 | 57710 | NM_020950.2 | 0.63 | 0.15 | 0.96 | 0.63 | 0.06 | 1.00 |
| LOC100129697 | 100129697 | NM_001290330.2 | 0.65 | 0.26 | 0.93 | 0.58 | 0.06 | 0.92 |
| LOC105377284 | 105377284 | XR_938891.2 | 0.59 | 0.05 | 0.96 | 0.65 | 0.13 | 1.00 |
| LRRN3† | 54674 | NM_001099658.2 | 0.78 | 0.79 | 0.76 | 0.85 | 1.00 | 0.75 |
| MFSD6L | 162387 | NM_152599.3 | 0.67 | 0.59 | 0.73 | 0.65 | 0.50 | 0.75 |
| MORC3 | 23515 | NM_015358.3 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| MTRNR2L1 | 100462977 | NM_001190452.1 | 0.57 | 0.03 | 0.96 | 0.60 | 0.00 | 1.00 |
| NECAB1 | 64168 | NM_022351.5 | 0.62 | 0.15 | 0.95 | 0.58 | 0.06 | 0.92 |
| NEIL3 | 55247 | NM_018248.3 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| PDCD10 | 11235 | NM_007217.4 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
| PGM5 | 5239 | NM_021965.4 | 0.55 | 0.00 | 0.95 | 0.50 | 0.00 | 0.83 |
| RFFL | 117584 | NR_037713.1 | 0.57 | 0.00 | 0.98 | 0.58 | 0.00 | 0.96 |
| SDCBP2 | 27111 | NM_080489.5 | 0.71 | 0.33 | 0.98 | 0.65 | 0.31 | 0.88 |
| SMIM8 | 57150 | NM_001042493.3 | 0.57 | 0.03 | 0.96 | 0.50 | 0.00 | 0.83 |
| SYP | 6855 | NM_003179.2 | 0.59 | 0.05 | 0.96 | 0.63 | 0.13 | 0.96 |
| TGS1† | 96764 | NM_024831.7 | 0.80 | 0.79 | 0.80 | 0.78 | 0.94 | 0.67 |
| TMC4 | 147798 | NM_001145303.2 | 0.64 | 0.49 | 0.75 | 0.63 | 0.44 | 0.75 |
| USF1 | 7391 | NM_007122.5 | 0.59 | 0.03 | 0.98 | 0.60 | 0.06 | 0.96 |
| UTY | 7404 | NM_182660.1 | 0.57 | 0.00 | 0.98 | 0.60 | 0.00 | 1.00 |
Acc accuracy, Sen sensitivity, Spe specificity
We show the per-transcript performance in the training and testing dataset. Transcripts with high accuracy (> 0.70) in both training and testing cohorts are denoted by †
Figure 4 demonstrates PCA (a and d), performance metrics (b and e), and ROC curves (c and f) for the training of four types of machine learning models that utilize either the new 37-transcript panel or our original 26-gene panel. The PCA in Fig. 4a illustrates these transcripts’ ability to clearly separate aneurysm samples from control in training cohort. Compared to the PCA using the 26 previously identified genes in Fig. 4d, it is visually evident that the transcripts identified by LASSO were able to better separate IA and control groups.
Fig. 4.

Models’ performance in the training dataset. a PCA using the 37 selected transcripts demonstrated clear separation between samples from patients with IA and those from controls. b Estimation of model performance during LOO C-V in the training cohort demonstrated that models performed with an accuracy of 0.85–0.91. Considering a 5% prevalence of IA, PPV ranged from 0.33–1 and NPV ranged from 0.98 to 0.99. c ROC analysis showed that all models had AUCs ≥ 0.95. d PCA using the 26 previously-identified transcripts demonstrated inferior separation between IA and control cases. e Estimation of model performance during LOO C-V in the training cohort demonstrated that models performed with an accuracy of 0.71–0.80. Considering a 5% prevalence, PPV and NPV ranged from 0.13–0.41 and 0.97–0.98, respectively. f ROC analysis also showed subpar performance compared to newly identified transcripts (AUC range 0.71–0.92). (AUC = area under the ROC curve, C-V = cross validation, cSVM = cubic support vector machines, gSVM = Gaussian support vector machines, KNN = k-nearest neighbors, LOO = leave-one-out, NPV = negative predictive value, PCA = principal component analysis, PPV = positive predictive value, RF = random forests, ROC = receiver operator characteristic)
Sensitivity, specificity, accuracy, NPV, and PPV estimated by LOO cross-validation in the training cohort are reported in Fig. 4b for models using the new 37-transcript panel. Each classification method achieved high performance, with accuracies that ranged from 0.85 to 0.91. Evaluation by ROC curve analysis showed a range in AUCs from 0.95 to 0.98 (Fig. 4c) across all methods. All models had high NPV of approximately 1 (0.98–1). Random Forest outperformed K-Nearest Neighbor and both Support Vector Machine algorithms, with a sensitivity of 0.87, specificity of 0.95, accuracy of 0.92, AUC of 0.98, NPV of 0.99, and PPV of 0.46. Figure 4e reports performances of the 4 classification models trained with the 26 previously-identified genes. Sensitivity, specificity, accuracy, NPV, and PPV were estimated by LOO cross-validation in the training cohort. AUCs ranged from 0.71 to 0.92, as shown in Fig. 4f. Overall performance in the training cohort was superior using the transcripts selected by LASSO; all metrics (accuracy, sensitivity, specificity, AUC, 5% PPV, 5% NPV) when averaged across the 4 models were greater using the new 37-gene panel.
Predictive models of IA have high performance and high NPV in testing
Figure 5 demonstrates PCA (a and d), performance metrics (b and e), and ROC curves (c and f) for the testing of the machine learning models, which utilize either the new 37-transcript panel or our original 26-gene panel. PCA performed on the testing data (Fig. 5a) shows that the 37 transcripts could discriminate patients with IAs from controls. The separation between classes was more obvious using the 37 newly-identified transcripts than the 26 previously-identified transcripts (Fig. 5d). Using the 37 features selected by LASSO, the models predicted aneurysm status in the testing cohort with accuracies ranging from 0.83 to 0.90 (Fig. 5b). The ROC analysis in Fig. 5c shows that model AUCs ranged from 0.95 to 0.99. In the testing cohort, the Random Forest model again performed well, with a sensitivity of 1.0, specificity of 0.75, accuracy of 0.85, and AUC of 0.99. The performance of the previously identified 26-gene panel (Fig. 5e, f) was similar to that of the 37-gene panel in the testing cohort with accuracies ranging from 0.83 to 0.93 and AUCs of 0.84–0.97. While average accuracy for the 4 models was the same (86%) using the 37-gene panel identified by LASSO and the 26-gene panel previously identified, the models using the 37 LASSO features had greater average AUC (0.97 vs 0.91).
Fig. 5.

Models’ performance in the testing dataset. a PCA using the 37 selected transcripts in this independent dataset also demonstrated strong separation between samples from patients with IA and from controls. b Assessment of true model performance showed that models performed with an accuracy of 0.83–0.90. In this dataset all models had a sensitivity of 1. At 5% IA prevalence, the PPV ranged from 0.15 to 0.24 and NPV was 1 for all models. c ROC analysis showed that all models again had AUCs ≥ 0.95. d PCA using the 26 previously-identified transcripts demonstrated mediocre separation between IA and control cases. e Estimation of model performance in the testing cohort demonstrated that models performed with an accuracy of 0.83–0.93. Considering a 5% prevalence, PPV and NPV ranged from 0.15–0.52 and 0.99–1, respectively. f ROC analysis also showed inferior performance compared to newly identified transcripts (AUC range 0.84–0.97). (AUC = area under the ROC curve, C-V = cross validation, cSVM = cubic support vector machines, gSVM = Gaussian support vector machines, KNN = k-nearest neighbors, LOO = leave-one-out, NPV = negative predictive value, PCA = principal component analysis, PPV = positive predictive value, RF = random forests, ROC = receiver operator characteristic)
Bioinformatics of LASSO-selected transcripts
To investigate how the biological underpinnings of IA specifically influence the genes selected by LASSO, we performed bioinformatics analyses using only the 37 new panel genes (n = 23 with decreased expression in IA group, n = 14 with increased expression in IA group). Model genes with decreased expression in the IA group were associated with negative regulation of execution phase of apoptosis, negative regulation of endothelial cell proliferation, and regulation of execution phase of apoptosis (Additional file 8: Table S6). However, model genes with increased expression in the IA group did not return any significant functions or processes. Two networks using all 37 genes produced by IPA had significant p-scores (equal to 47 and 25). The first network showed hubs around TNF and MMP3 and was associated with cancer, cellular movement, and connective tissue disorders. The second had hubs around HBB and MAPK and was associated with cell cycle, cellular assembly and organization, DNA replication, recombination, and repair. We note TNF was incorporated in networks generated using both edgeR and LASSO gene sets. See Fig. 6 and Additional file 9: Table S7 for details on these networks, including associated molecules and top diseases and functions.
Fig. 6.

Networks derived from IPA of the 37 genes identified by LASSO. Transcripts with increased expression in IA are red; transcripts with lower expression in IA are green; fold-change is represented by intensity. a This network (p-score = 47) affiliated with cancer, cellular movement, and connective tissue disorders. b This network (p-score = 25) has associated functions of cell cycle, cellular assembly and organization, DNA replication, recombination, and repair
Presence of clinical covariates and effect on model performance
Table 3 shows the rates of demographics and comorbidities in aneurysm and control populations. Only smoking was significantly higher in the IA population (p = 0.017), which can be expected as it is a well-known risk factor for IA and IA rupture [55]. We created 5 subclasses using MatchIt as there were too few samples in the 6th subclass. Misclassification by the 37-gene prediction model for each subclass ranged from 8 to 19%, indicating no one subclass could be driving misclassification.
Table 3.
Clinical characteristic differences in entire population
| Control (n = 79) |
Aneurysm (n = 55) |
Chi-square test | |
|---|---|---|---|
| Age (Mean ± SE) | 61 ± 1.7 | 60 ± 1.5 |
(age 60 cutoff) 0.243 |
| Age [Median (Q1/Q3)] | 65 (54/72) | 60 (54/67) | |
| Sex (% of patients) | |||
| Female | 54.43% | 70.91% | 0.054 |
| Smoking (% of patients) | |||
| Current | 13.92% | 30.91% | 0.017† |
| Comorbidities (% of patients) | |||
| Arthritis | 16.46% | 27.27% | 0.130 |
| Asthma | 7.59% | 18.18% | 0.063 |
| Cancer | 11.39% | 9.09% | 0.668 |
| Diabetes | 22.78% | 21.82% | 0.895 |
| Heart disease | 29.11% | 21.82% | 0.344 |
| High cholesterol | 55.70% | 49.09% | 0.451 |
| Hypertension | 59.49% | 52.73% | 0.437 |
| IA family history | 7.69% | 12.73% | 0.336 |
| Stroke history | 18.99% | 9.09% | 0.114 |
None of the reported covariates were significantly different in either group (chi-square test < 0.05) except for smoking†
Discussion
More robust machine learning strategy improves biomarker performance
In this study, we implemented a new machine learning strategy for IA biomarker discovery, which consisted of a larger dataset (94 training, 40 testing), LASSO for feature selection, and more robust algorithms, K-Nearest Neighbor, Random Forest, and Support Vector Machine with cubic and Gaussian kernels. Our larger dataset and LASSO feature selection led to a new panel of 37 genes to use in IA predictive models. Two genes of these 37 genes, C1QL1 and TGS1, were also in our previously-discovered 26-gene panel. The new learning algorithms trained using the 37 genes all performed very well in the testing cohort with accuracies of 0.83–0.90 and AUCs of 0.95–0.99, a marked increase over our previous algorithms. Interestingly, all 4 new models had an NPV of 1, indicating that in the testing dataset there were no false negatives. This may be important for future applications of these biomarkers as a prescreen, since false negatives would be particularly deleterious.
To examine how the increased sample size and improved algorithms affected model performance, we retrained the previous 26-gene panel using the new algorithms in the current, larger dataset. The performance of the retrained models in the testing set (n = 40) using the 26-gene panel improved from our previous study with accuracies ranging from 0.83 to 0.93 and AUCs of 0.84–0.97. Despite this increase in performance using the new algorithms, models using the previously identified 26 genes still fell short of those using the newly identified 37 genes; the average testing AUC using 26 genes was 0.91 compared to 0.97 when using 37 genes. This suggests that the 37 features identified by LASSO are more reliable for IA prediction than the 26 selected by filtering in our last study.
Improved IA prediction could be attributed to our increased sample size, which afforded several advantages. First, it allowed us to use LASSO to identify features instead of simple filtering methods. Thresholding filters, as we had previously used, consider each gene independently, which can neglect groups of genes that function together in pathophysiologic mechanisms and could be useful as a biomarker. Filtering methods can also select highly correlated, redundant genes that increase the number of features required to make accurate predictions. HSIC LASSO, a nonlinear feature selection method, overcomes these issues and identifies combinations of non-redundant genes with strong dependence on disease status. Implementing LASSO in the training dataset identified 37 unique IA-associated genes, two of which (C1QL1, TGS1) had also been identified as part of the 26-gene panel in our last expression profiling study [23]. The identification of non-redundant features may be one reason why the biomarkers created in this study outperform our past efforts, as some of the 26 features (with the exception of C1QL1 and TGS1) may have ultimately been uninformative for classification.
Secondly, a larger sample size also enabled us to leverage more complex machine learning models, namely Support Vector Machine and Random Forests which perform better in larger datasets [56]. In our previous effort we did implement Support Vector Machine, but only achieved a testing accuracy of 0.70, possibly because the training dataset contained only 30 patients [23]. In this larger study we were able to achieve an accuracy of 0.85 for Support Vector Machine (Gaussian kernel). Nevertheless, we found that in our data Random Forest consistently performed the best, with a testing accuracy of 0.85 and AUC of 0.99. Both Random Forests and K-Nearest Neighbors are weighted neighbors schemes. However, the K-Nearest Neighbors algorithm may have had poorer performance because this classifier simply uses the training data for prediction instead of learning a discriminative rule. The performance of the K-Nearest Neighbors classifier is reliant on the quality of the training data, which in the case of transcriptomes derived from human samples may be noisy. However, this problem is well-solved in Random Forest. Through the random sampling process, Random Forest handles outliers by binning them. Also, by averaging the decision trees, the Random Forest method provides a low bias and moderate variance model, which improves the generalizability of the output model. In other words, Random Forest not only attains a good performance in the training data but also performs well in unknown (testing) data. And while Support Vector Machine performed well here, Random Forest likely surpassed Support Vector Machine by avoiding overfitting and achieving better predictive power.
We note that increasing sample size may have introduced more variability in our data due to a larger, heterogeneous population that was not cohort-controlled. For example, in our entire population we found smoking was significantly higher in patients with IA (χ = 0.017), which may be because smoking is a well-known risk factor for IA formation and rupture [57–59]. Indeed two genes in our model, LRRN3 and GPR15, are among the top differentially expressed genes in blood between current and never smokers according to a meta-analysis by Huan et al. [60] Their presence in our predictive model may be because of the higher proportion of smokers in IA group or because these genes are capturing biological mechanisms related to smoking that are important in IA pathogenesis, such as endothelial dysfunction [61–63]. Still, when we performed covariate analysis using MatchIt to create subgroups with similar distributions of covariates between IA and control groups, we found that no one subgroup had significantly higher misclassification rates. For instance, 61% of all subjects in “Subclass 5” were smokers, and this subgroup had a misclassification rate of 13%. Yet, “Subclass 1”, which had 0% smokers, had a misclassification rate of 14%. These results suggest that our prediction models may not be affected greatly by covariate imbalance, albeit testing this in even larger cohorts will be needed to confirm these results.
Complex role of circulating neutrophils in intracranial aneurysm
Inflammation is widely-recognized to play a central role in the pathophysiology of IA [64–66]. It is commonly thought that in IA neutrophils are recruited to the sac, where they infiltrate the wall and coordinate the inflammatory responses [64, 67, 68]. In this study, gene ontology enrichment analysis showed that genes with higher expression in IA identified by edgeR in the entire dataset were related to cell migration and lymphocyte migration ontologies. These processes, which were also observed in neutrophils from patients with IAs in our previous studies [22, 23], increase upon peripheral activation and prompt inflammatory cell migration and infiltration of diseased tissue [64, 69]. IPA analysis mirrored these results, showing 3 significant networks, 2 of which were involved in activation-related processes: cell-to-cell signaling and interaction, and inflammatory disease function. Interestingly, one of the largest nodes of gene connectivity in all the networks was TNF, a proinflammatory cytokine with many functions including regulation of cell proliferation and apoptosis. TNF has been shown to have a mechanistic role in IA formation in animal models, [70] and an increased presence in human IA tissue compared to superficial temporal artery control tissue [71]. In this network, TNF has a predicted connection to DEFA1, which was significantly elevated in neutrophils of IA patients. Higher levels of this cytotoxic defensin protein that is contained within neutrophil granules have been reported in IA tissue, suggesting that production of this protein may occur peripherally before neutrophils enter the IA wall [72]. Here the molecules CCR4 (which is a receptor of MIP-1, RANTES, CCL17, and MCP-1) [73, 74] and CCR6 (which is a receptor of MIP-3α) [75] were also related to the TNF node. We suspect that these receptors, which play a role in dendritic and T cell migration and recruitment during inflammation, [76] may coordinate inflammatory cell migration once expressed in aneurysm tissue.
We also observed the dysregulation of inflammation and a potential role of TNF in our bioinformatics analyses of model genes selected by LASSO in the training dataset. TNF was a hub of connectivity in networks created using the LASSO genes. In these networks, we observed an indirect relationship between TNF and the complement system (i.e. C1QTNF1), which is also associated with C1QL1 (one of 37 model genes). This may be because complement activation plays a critical role in the inflammatory response, [77] has been implicated in IA wall degradation and rupture, [78] and involves proteins that are increased in human IA tissue (including CFB, CFH, C1Q, and C3AR1 [79]). We suspect that the complement alternative pathway may be one mechanism through which neutrophils become activated as it can amplify activation through a positive feedback mechanism [80]. In addition to complement members, the TNF node was also related to CD44, a cell surface glycoprotein critical to neutrophil recruitment during inflammation. Because neutrophils interact with CD44, PSGL-1, and E-selectin ligand 1 as they roll along activated endothelial cells, this result may reflect neutrophils transmigrating into inflamed endothelium [81]. Our data shows TNF may also interact with the transcription factor TP53, a node with connections to numerous molecules, many of which have decreased expression. TP53 plays a variety of roles in inflammation, such as acting on the NF- κB pathway [82]. NF- κB is a key transcription factor in the pathogenesis of IA, as it controls the inflammatory responses in the vessel wall [83]. The activation of NF-κB leads to the upregulation of MCP-1 and VCAM that function to recruit monocytes to the IA lumen, where they become macrophages and secrete MMP-2 and MMP-9 to degrade the extracellular matrix [84]. Overall, our bioinformatics analyses of genes selected by LASSO, while not overlapping greatly with the differentially expressed genes selected by edgeR in the entire dataset (with the exception of C1QL1, GRP15), show that the biology of neutrophil activation and inflammation responses are captured by the IA prediction model gene panel.
In addition to neutrophil activation and heightened inflammatory signaling, we observed other aberrant neutrophil functions not specifically characterized in IA, including our previous studies [22, 23]. In genes identified in the whole dataset by edgeR, gene ontology enrichment analysis showed that the differentially expressed genes with decreased expression in IA had functions related to sodium channel activity, ion channel activity, and gated channel activity, as well as signaling and regulation of membrane potential processes (ASIC2, GRIK3, SCN5A). GRIK3, glutamate receptor 7, is particularly interesting as glutamate is a chemotactic factor for neutrophils after injury or infection [85]. Glutamate binding to its receptors can trigger release of cytokines and MMPs and can activate immune responses, all critical processes in IA [86, 87]. Future studies are needed to better understand how these channel activities impact IA pathogenesis.
New ontologies were also captured using the genes identified by LASSO in the training dataset. Using the LASSO genes with lower expression in IA, we found dysregulation of apoptosis as gene ontology enrichment analysis reported both negative regulation of execution phase of apoptosis and regulation of execution phase of apoptosis. These were associated with RFFL, which has been shown to be related to TNF signaling [88]. Ontologies were also related to MTRNR2L1 (MTRNR2-Like 1) that may function similarly to Humanin (MTRNR2), which is protective against cell death by suppressing several apoptotic pathways. Several studies have shown that, in this way, Humanin may be a neuroprotective factor that can influence Alzheimer’s disease and other angiopathy-associated neurodegenerative diseases [89, 90]. Perhaps in our study, dysregulated MTRNR2L1 expression (as well as TP53, which also induces apoptosis) [91, 92] may be responsible for increasing the lifespan of neutrophils, which would provide further evidence of neutrophil activation in IA. These results are echoed in the blood profiling study of IA published by Jin et al. [93]. They reported hsa-miR-21, an upregulated miRNA in IA serum, induces apoptosis by extracellular signals, potentially triggering more apoptotic reactions to facilitate the medial thinning and destructive remodeling, a hallmark of IA pathogenesis [94–97]. Overall, we suspect that capturing neutrophil activation and inflammation responses involved in IA is the reason why the 37-gene panel was able to detect IA.
Limitations
In this study, we increased sample size from our previous study by adding 94 samples to 40 samples we previously analyzed. However, these two batches used different versions of the Illumina kit for library preparation, which necessitated the implementation of batch effect correction that could potentially have introduced bias or skewed our dataset [98]. Secondly, RNA sequencing was performed on samples with a wide range of RIN values. While we demonstrated that RIN was not a significant co-variant of differential expression, in the future methods such as DegNorm developed by Xiong et al. [30] could be implemented to correct for expression variation due to differences in RNA degradation and potentially yield more accurate results. Thirdly, all samples were recruited from patients receiving cerebral imaging at a single center, which may introduce selection bias. Future studies are needed to validate our predictive models using broader patient populations from multiple centers. Fourthly, inflammatory or vascular diseases other than IA could affect model prediction. Larger studies with multiple control groups of individuals with other vascular and inflammatory conditions are needed to refine our model. Lastly, more rigorous qPCR performed specifically on model genes with more efficient probes (primers) will be needed to translate this gene panel into an assay that can show linearity in output and reproducibility over technical replicates.
Conclusions
We improved IA predictive model performance from circulating neutrophil transcripts by using LASSO for feature selection and powerful machine learning techniques in a large dataset. The Random Forest algorithm performed the best with a testing AUC of 0.99. Bioinformatics using all 134 samples implicated inflammation through TNF and neutrophil activation as key processes in IA. IPA networks using the 37 LASSO-selected genes also reflected these increased inflammatory and signaling pathways. Comorbidities and demographics did not significantly affect IA prediction. Future studies are needed to validate these predictive models.
Supplementary information
Additional file 1: Table S1. Primers used for qPCR.
Additional file 2: Figure S1. Stability of housekeeping genes in RNA sequencing and qPCR.
Additional file 3: Table S2. Characteristics of 73 aneurysms in all patients with intracranial aneurysms.
Additional file 4: Table S3. Batch Assignment, RNA QC, and sequencing QC for the testing and training cohorts.
Additional file 5: Figure S2. Co-variate correlation analysis between RNA quality (RIN) and differentially expressed genes.
Additional file 6: Table S4. GORILLA enriched ontologies for edgeR genes.
Additional file 7: Table S5. Transcripts and functions for the significant networks constructed by Ingenuity Pathway Analysis (IPA) using genes identified by edgeR.
Additional file 8: Table S6. GORILLA enriched ontologies for LASSO genes.
Additional file 9: Table S7. Transcripts and functions for the significant networks constructed by Ingenuity Pathway Analysis (IPA) using genes identified by LASSO.
Acknowledgements
We thank the patients who participated in this study, Jonathan Bard MA and Brandon Marzullo MS for RNA sequencing data analysis assistance, and Jennifer L. Gay CCRP for study protocol management. This work was performed in part at the New York State Center of Excellence in Bioinformatics and Life Sciences’ Genomics and Bioinformatics Core.
Abbreviations
- AUC
Area under ROC curve
- BAM
Binary alignment map
- BCL
Per-cycle base-call
- CTA
Computed tomography angiography
- DSA
Digital subtraction angiography
- ECM
Extracellular matrix
- FDR
False discovery rate
- GO
Gene ontology
- GORILLA
Gene Ontology enRIchment anaLysis and visuaLizAtion
- HSIC
Hilbert–Schmidt independence criterion
- IA
Intracranial aneurysm
- IPA
Ingenuity pathway analysis
- LASSO
Least absolute shrinkage and selection operator
- LOO
Leave one-out
- MPO
Myeloperoxidase
- MRA
Magnetic resonance angiography
- MRI
Magnetic resonance imaging
- NGAL
Neutrophil gelatinase-associated lipocalin
- NPV
Negative predictive value
- PCA
Principal component analysis
- PPV
Positive predictive value
- qPCR
Quantitative polymerase chain reaction
- RIN
RNA integrity
- ROC
Receiver-operating-characteristic
- ROS
Reactive oxygen species
- RT-qPCR
Real-time quantitative polymerase chain reaction
- TPM
Transcript per million
Authors’ contributions
Study conception and design: KEP, VMT, JNJ, JK, HM. Acquisition of data: KEP, VMT, MW, AJ, LC, KVS, EIL, AHS. Analysis and interpretation of data: KEP, VMT, LL, LC, KJ, JNJ, YS, JK, HM. Drafting of manuscript: KEP, VMT, LL, JNJ, YS, JK, HM. Critical revision: KEP, VMT, LL, MW, AJ, LC, KJ, JNJ, YS, KVS, EIL, AHS, KJ, HM. All authors read and approved the final manuscript.
Funding
This work was funded by National Science Foundation Award No. 1746694—through Neurovascular Diagnostics, Inc., the Brain Aneurysm Foundation, the New York State Center for Advanced Technology in Big Data and Health Sciences, and the Cummings Foundation.
Availability of data and materials
The datasets used in the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the University at Buffalo Health Sciences Institutional Review Board (study no. 030–474433). All methods were carried out in accordance with approved protocol and informed consent was obtained from all subjects.
Consent for publication
No specific patient information or images are given in the manuscript.
Competing interests
KEP, LL, MW, AJ, LC, KJ, YS, JK—None. VMT— Principal investigator: National Science Foundation Award No. 1746694, and the Brain Aneurysm Foundation grant, Center for Advanced Technology grant, and Cummings Foundation grant mentioned above. Co-founder: Neurovascular Diagnostics, Inc. JNJ—Principal Investigator: NIH Grant R01-AR-060604. KVS—Consulting and teaching for Canon Medical Systems Corporation, Penumbra Inc., Medtronic, and Jacobs Institute. Co-Founder: Neurovascular Diagnostics, Inc. EIL—Intratech Medical Ltd., NeXtGen Biologics. Principal investigator: Medtronic US SWIFT PRIME Trials. Honoraria–Medtronic. Consultant–Pulsar Vascular. Advisory Board-Stryker, NeXtGen Biologics, MEDX. Cognition Medical. Other financial support—Abbott Vascular for carotid training sessions. AHS—Financial Interest/Investor/Stock Options/Ownership: Amnis Therapeutics, Apama Medi- cal, BlinkTBI, Inc, Buffalo Technology Partners, Inc., Cardinal Health, Cerebrotech Medical Systems, Inc, Claret Medical, Cognition Medical, Endostream Medical, Ltd, Imperative Care, International Medical Distribution Partners, Rebound Therapeutics Corp., Silk Road Medical, StimMed, Synchron, Three Rivers Medi- cal, Inc., Viseon Spine, Inc. Consultant/Advisory Board: Amnis Therapeutics, Boston Scientific, Canon Medical Systems USA, Inc., Cerebrotech Medical Systems, Inc., Cerenovus, Claret Medical, Corindus, Inc., Endostream Medical, Ltd, Guidepoint Global Consulting, Imperative Care, Integra, Medtronic, Micro- Vention, Northwest University—DSMB Chair for HEAT Trial, Penumbra, Rapid Medical, Rebound Therapeutics Corp., Silk Road Medical, StimMed, Stryker, Three Rivers Medical, Inc., VasSol, W.L. Gore & Associates. National PI/Steering Committees: Cerenovus LARGE Trial and ARISE II Trial, Medtronic SWIFT PRIME and SWIFT DIRECT Trials, MicroVention FRED Trial & CONFIDENCE Study, MUSC POSITIVE Trial, Penumbra 3D Separator Trial, COMPASS Trial, INVEST Trial. HM— Principal investigator: NIH Grants R01-NS-091075 and R01-NS-064592. Grant support: Canon Medical Systems. Co-founder: Neurovascular Diagnostics, Inc.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kerry E. Poppenberg and Vincent M. Tutino contributed equally to this work
Contributor Information
Kerry E. Poppenberg, Email: kerrypop@buffalo.edu
Vincent M. Tutino, Email: vincentt@buffalo.edu
Lu Li, Email: lli59@buffalo.edu.
Muhammad Waqas, Email: mwaqas@ubns.com.
Armond June, Email: armondju@buffalo.edu.
Lee Chaves, Email: leechave@buffalo.edu.
Kaiyu Jiang, Email: kaiyujia@buffalo.edu.
James N. Jarvis, Email: jamesjar@buffalo.edu
Yijun Sun, Email: yijunsun@buffalo.edu.
Kenneth V. Snyder, Email: ksnyder@ubns.com
Elad I. Levy, Email: elevy@ubns.com
Adnan H. Siddiqui, Email: asiddiqui@ubns.com
John Kolega, Email: kolega@buffalo.edu.
Hui Meng, Email: huimeng@buffalo.edu.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12967-020-02550-2.
References
- 1.Vega C, Kwoon JV, Lavine SD. Intracranial aneurysms: Current evidence and clinical practice. Am Fam Physician. 2002;66:601–608. [PubMed] [Google Scholar]
- 2.Olafsson E, Hauser WA, Gudmundsson G. A population-based study of prognosis of ruptured cerebral aneurysm: mortality and recurrence of subarachnoid hemorrhage. Neurology. 1997;48:1191–1195. doi: 10.1212/WNL.48.5.1191. [DOI] [PubMed] [Google Scholar]
- 3.Hop Jeannette W, Rinkel Gabriel JE, Algra A, van Gijn J. Case-fatality rates and functional outcome after subarachnoid hemorrhage: a systematic review. Stroke. 1997;38:660–664. doi: 10.1161/01.str.28.3.660. [DOI] [PubMed] [Google Scholar]
- 4.Nieuwkamp DJ, Setz LE, Algra A, Linn FHH, de Rooij NK, Rinkel GJE. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol. 2009;8:635–642. doi: 10.1016/S1474-4422(09)70126-7. [DOI] [PubMed] [Google Scholar]
- 5.Greving JP, Rinkel GJ, Buskens E, Algra A. Cost-effectiveness of preventive treatment of intracranial aneurysms: new data and uncertainties. Neurology. 2009;73:258–265. doi: 10.1212/01.wnl.0b013e3181a2a4ea. [DOI] [PubMed] [Google Scholar]
- 6.Juvela S. Treatment options of unruptured intracranial aneurysms. Stroke. 2004;35:372–374. doi: 10.1161/01.STR.0000115299.02909.68. [DOI] [PubMed] [Google Scholar]
- 7.Jiang K, Sun X, Chen Y, Shen Y, Jarvis JN. Rna sequencing from human neutrophils reveals distinct transcriptional differences associated with chronic inflammatory states. BMC Med Genomics. 2015;8:55. doi: 10.1186/s12920-015-0128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. 2018;9:113. doi: 10.3389/fphys.2018.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gounis MJ, Vedantham S, Weaver JP, Puri AS, Brooks CS, Wakhloo AK, et al. Myeloperoxidase in human intracranial aneurysms: preliminary evidence. Stroke. 2014;45:1474–1477. doi: 10.1161/STROKEAHA.114.004956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serra R, Volpentesta G, Gallelli L, Grande R, Buffone G, Lavano A, et al. Metalloproteinase-9 and neutrophil gelatinase-associated lipocalin plasma and tissue levels evaluation in middle cerebral artery aneurysms. British J Neurosurg. 2014;1–5. [DOI] [PubMed]
- 11.Chu Y, Wilson K, Gu H, Wegman-Points L, Dooley SA, Pierce GL, et al. Myeloperoxidase is increased in human cerebral aneurysms and increases formation and rupture of cerebral aneurysms in mice. Stroke. 2015;46:1651–1656. doi: 10.1161/STROKEAHA.114.008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stapleton PP, Redmond HP, Bouchier-Hayes DJ. Myeloperoxidase (mpo) may mediate neutrophil adherence to the endothelium through upregulation of cd11b expression–an effect downregulated by taurine. Adv Exp Med Biol. 1998;442:183. doi: 10.1007/978-1-4899-0117-0_24. [DOI] [PubMed] [Google Scholar]
- 13.Leopold JA. The central role of neutrophil gelatinase-associated lipocalin in cardiovascular fibrosis. Hypertension. 2015;66:20–22. doi: 10.1161/HYPERTENSIONAHA.115.05479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: Facts beyond expression. Front Immunol. 2014;5:508. doi: 10.3389/fimmu.2014.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayaraman T, Paget A, Shin YS, Li X, Mayer J, Chaudhry H, et al. Tnf-alpha-mediated inflammation in cerebral aneurysms: A potential link to growth and rupture. Vasc Health Risk Manag. 2008;4:805–817. doi: 10.2147/VHRM.S2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of tnf-alpha-tnfr1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun. 2014;2:34. doi: 10.1186/2051-5960-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Critical role of tnf-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation. 2014;11:77. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korai M, Furukawa H, Kamio Y, Mitsui K, Nagahiro S, Hashimoto T. Abstract 197: roles of neutrophil extracellular trap in the rupture of intracranial aneurysm. Stroke. 2017;48:A197–A197. [Google Scholar]
- 19.Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, et al. Novel role of il (interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2018;38:843–853. doi: 10.1161/ATVBAHA.117.309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinosa M, Su G, Salmon MD, Lu G, Cullen JM, Fashandi AZ, et al. Resolvin d1 decreases abdominal aortic aneurysm formation by inhibiting netosis in a mouse model. J Vasc Surg. 2018;68:93S–103S. doi: 10.1016/j.jvs.2018.05.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan H, Zhou H-F, Akk A, Hu Y, Springer Luke E, Ennis Terri L, et al. Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler Thromb Vasc Biol. 2016;36:1660–1669. doi: 10.1161/ATVBAHA.116.307786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tutino VM, Poppenberg KE, Jiang K, Jarvis JN, Sun Y, Sonig A, et al. Circulating neutrophil transcriptome may reveal intracranial aneurysm signature. PLoS ONE. 2018;13:e0191407. doi: 10.1371/journal.pone.0191407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tutino VM, Poppenberg KE, Li L, Shallwani H, Jiang K, Jarvis JN, et al. Biomarkers from circulating neutrophil transcriptomes have potential to detect unruptured intracranial aneurysms. J Transl Med. 2018;16:373–373. doi: 10.1186/s12967-018-1749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarvis JN, Dozmorov I, Jiang K, Frank MB, Szodoray P, Alex P, et al. Novel approaches to gene expression analysis of active polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther. 2004;6:R15–r32. doi: 10.1186/ar1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor rna-seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc: Ser B (Methodol) 1995;57:289–300. [Google Scholar]
- 28.Shen Y, Li R, Tian F, Chen Z, Lu N, Bai Y, et al. Impact of rna integrity and blood sample storage conditions on the gene expression analysis. Onco Targets Ther. 2018;11:3573–3581. doi: 10.2147/OTT.S158868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anzai N, Kawabata H, Hirama T, Masutani H, Ueda Y, Yoshida Y, et al. Types of nuclear endonuclease activity capable of inducing internucleosomal DNA fragmentation are completely different between human cd34+ cells and their granulocytic descendants. Blood. 1995;86:917–923. doi: 10.1182/blood.V86.3.917.917. [DOI] [PubMed] [Google Scholar]
- 30.Xiong B, Yang Y, Fineis FR, Wang JP. Degnorm: Normalization of generalized transcript degradation improves accuracy in rna-seq analysis. Genome Biol. 2019;20:75. doi: 10.1186/s13059-019-1682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallona I, Weiss J, Egea-Cortines M. Pcrefficiency: A web tool for pcr amplification efficiency prediction. BMC Bioinformatics. 2011;12:404. doi: 10.1186/1471-2105-12-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, et al. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2005;85:154–159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- 33.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Scarlet D, Ertl R, Aurich C, Steinborn R. The orthology clause in the next generation sequencing era: Novel reference genes identified by rna-seq in humans improve normalization of neonatal equine ovary rt-qpcr data. PLoS ONE. 2015;10:e0142122. doi: 10.1371/journal.pone.0142122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics. 2006;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 38.Hothorn T, Everitt BS. A handbook of statistical analyses using r. Boca Raton: Chapman and Hall/CRC; 2014. [Google Scholar]
- 39.Jabbar MA, Deekshatulu BL, Chandra P. Classification of heart disease using k- nearest neighbor and genetic algorithm. Procedia Technol. 2013;10:85–94. doi: 10.1016/j.protcy.2013.12.340. [DOI] [Google Scholar]
- 40.Chen H-L, Huang C-C, Yu X-G, Xu X, Sun X, Wang G, et al. An efficient diagnosis system for detection of parkinson’s disease using fuzzy k-nearest neighbor approach. Expert Syst Appl. 2013;40:263–271. doi: 10.1016/j.eswa.2012.07.014. [DOI] [Google Scholar]
- 41.Moore PJ, Lyons TJ, Gallacher J, for the Alzheimer’s Disease Neuroimaging I Random forest prediction of alzheimer’s disease using pairwise selection from time series data. PLoS ONE. 2019;14:e0211558. doi: 10.1371/journal.pone.0211558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebedev AV, Westman E, Van Westen GJP, Kramberger MG, Lundervold A, Aarsland D, et al. Random forest ensembles for detection and prediction of alzheimer's disease with a good between-cohort robustness. NeuroImage Clin. 2014;6:115–125. doi: 10.1016/j.nicl.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee Y, Lee C-K. Classification of multiple cancer types by multicategory support vector machines using gene expression data. Bioinformatics. 2003;19:1132–1139. doi: 10.1093/bioinformatics/btg102. [DOI] [PubMed] [Google Scholar]
- 44.Yu W, Liu T, Valdez R, Gwinn M, Khoury MJ. Application of support vector machine modeling for prediction of common diseases: The case of diabetes and pre-diabetes. BMC Med Inform Decis Mak. 2010;10:16–16. doi: 10.1186/1472-6947-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liaw A, Wiener M. Classification and regression by randomforest. R News. 2002;2:18–22. [Google Scholar]
- 46.Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 47.Harada K, Fukuyama K, Shirouzu T, Ichinose M, Fujimura H, Kakumoto K, et al. Prevalence of unruptured intracranial aneurysms in healthy asymptomatic Japanese adults: differences in gender and age. Acta Neurochirurgica. 2013;155(11):2037–2043. doi: 10.1007/s00701-013-1841-7. [DOI] [PubMed] [Google Scholar]
- 48.Li MH, Chen SW, Li YD, Chen YC, Cheng YS, Hu DJ, et al. Prevalence of unruptured cerebral aneurysms in chinese adults aged 35 to 75 years. Ann Intern Med. 2013;159(8):514–521. doi: 10.7326/0003-4819-159-8-201310150-00004. [DOI] [PubMed] [Google Scholar]
- 49.Rinkel GJE. Intracranial aneurysm screening: Indications and advice for practice. Lancet Neurol12. 2005;4(2):122–8. doi: 10.1016/S1474-4422(05)00993-2. [DOI] [PubMed] [Google Scholar]
- 50.Vlak MHM, Algra A, Brandenburg R, Rinkel GJE. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 2011;10(7):626–636. doi: 10.1016/S1474-4422(11)70109-0. [DOI] [PubMed] [Google Scholar]
- 51.Ho DE, Imai K, King G, Stuart EA. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit Anal. 2007;15:199–236. doi: 10.1093/pan/mpl013. [DOI] [Google Scholar]
- 52.Ho D, Imai K, King G, Stuart EA. Matchit: Nonparametric preprocessing for parametric causal inference. J Stat Softw. 2011;42:28. doi: 10.18637/jss.v042.i08. [DOI] [Google Scholar]
- 53.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. Gorilla: A tool for discovery and visualization of enriched go terms in ranked gene lists. BMC Bioinformatics. 2009;10:48–48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gopurappilly R, Bhonde R. Transcriptional profiling and functional network analyses of islet-like clusters (ilcs) generated from pancreatic stem cells in vitro. Genomics. 2015;105:211–219. doi: 10.1016/j.ygeno.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Müller Tomm B, Vik A, Romundstad Pål R, Sandvei MS. Risk factors for unruptured intracranial aneurysms and subarachnoid hemorrhage in a prospective population-based study. Stroke. 2019;50:2952–2955. doi: 10.1161/STROKEAHA.119.025951. [DOI] [PubMed] [Google Scholar]
- 56.Kim S-Y. Effects of sample size on robustness and prediction accuracy of a prognostic gene signature. BMC Bioinformatics. 2009;10:147–147. doi: 10.1186/1471-2105-10-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woo D, Khoury J, Haverbusch MM, Sekar P, Flaherty ML, Kleindorfer DO, et al. Smoking and family history and risk of aneurysmal subarachnoid hemorrhage. Neurology. 2009;72:69–72. doi: 10.1212/01.wnl.0000338567.90260.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Juvela S, Poussa K, Porras M. Factors affecting formation and growth of intracranial aneurysms. Stroke. 2001;32:485–491. doi: 10.1161/01.STR.32.2.485. [DOI] [PubMed] [Google Scholar]
- 59.Juvela S, Hillbom M, Numminen H, Koskinen P. Cigarette smoking and alcohol consumption as risk factors for aneurysmal subarachnoid hemorrhage. Stroke. 1993;24:639–646. doi: 10.1161/01.STR.24.5.639. [DOI] [PubMed] [Google Scholar]
- 60.Huan T, Joehanes R, Schurmann C, Schramm K, Pilling LC, Peters MJ, et al. A whole-blood transcriptome meta-analysis identifies gene expression signatures of cigarette smoking. Hum Mol Genet. 2016;25:4611–4623. doi: 10.1093/hmg/ddw288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamio Y, Miyamoto T, Kimura T, Mitsui K, Furukawa H, Zhang D, et al. Roles of nicotine in the development of intracranial aneurysm rupture. Stroke. 2018;49:2445–2452. doi: 10.1161/STROKEAHA.118.021706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Can A, Castro VM, Ozdemir YH, Dagen S, Yu S, Dligach D, et al. Association of intracranial aneurysm rupture with smoking duration, intensity, and cessation. Neurology. 2017;89:1408–1415. doi: 10.1212/WNL.0000000000004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chalouhi N, Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, et al. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediators Inflamm. 2012;2012:271582. doi: 10.1155/2012/271582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab. 2012;32:1659–1676. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery. 1999;45:1137–1147. doi: 10.1097/00006123-199911000-00024. [DOI] [PubMed] [Google Scholar]
- 66.Turkmani AH, Edwards NJ, Chen PR. The role of inflammation in cerebral aneurysms. Neuroimmunol Neuroinflamm. 2015;2(2):2015. [Google Scholar]
- 67.Strong M, Amenta P, Dumont A, Medel R. The role of leukocytes in the formation and rupture of intracranial aneurysms. Neuro Immunol Inflamm. 2015;2:107–114. [Google Scholar]
- 68.Chalouhi N, Points L, Pierce GL, Ballas Z, Jabbour P, Hasan D. Localized increase of chemokines in the lumen of human cerebral aneurysms. Stroke. 2013;44:2594–2597. doi: 10.1161/STROKEAHA.113.002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sawyer DM, Pace LA, Pascale CL, Kutchin AC, O'Neill BE, Starke RM, et al. Lymphocytes influence intracranial aneurysm formation and rupture: Role of extracellular matrix remodeling and phenotypic modulation of vascular smooth muscle cells. J Neuroinflamm. 2016;13:185–185. doi: 10.1186/s12974-016-0654-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Critical role of tnf-α in cerebral aneurysm formation and progression to rupture. J Neuroinflamm. 2014;11:77–77. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jayaraman T, Berenstein V, Li XG, Mayer J, Silane M, Shin YS, et al. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery. 2005;57:558–563. doi: 10.1227/01.NEU.0000170439.89041.D6. [DOI] [PubMed] [Google Scholar]
- 72.Wang C, Qu B, Wang Z, Ju J, Wang Y, Wang Z, et al. Proteomic identification of differentially expressed proteins in vascular wall of patients with ruptured intracranial aneurysms. Atherosclerosis. 2015;238:201–206. doi: 10.1016/j.atherosclerosis.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 73.Sun W, Li WJ, Wei FQ, Wong TS, Lei WB, Zhu XL, et al. Blockade of mcp-1/ccr4 signaling-induced recruitment of activated regulatory cells evokes an antitumor immune response in head and neck squamous cell carcinoma. Oncotarget. 2016;7:37714–37727. doi: 10.18632/oncotarget.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshie O, Matsushima K. Ccr4 and its ligands: From bench to bedside. Int Immunol. 2015;27:11–20. doi: 10.1093/intimm/dxu079. [DOI] [PubMed] [Google Scholar]
- 75.Aziz N, Detels R, Chang LC, Butch AW. Macrophage inflammatory protein-3 alpha (mip-3α)/ccl20 in hiv-1-infected individuals. J AIDS Clin Res. 2016;7:587. doi: 10.4172/2155-6113.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. Ccr6 regulates the migration of inflammatory and regulatory t cells. J Immunol. 2008;181:8391–8401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343:227–235. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tulamo R, Frösen J, Junnikkala S, Paetau A, Pitkäniemi J, Kangasniemi M, et al. Complement activation associates with saccularcerebral artery aneurysm wall degeneration and rupture. Neurosurgery. 2006;59:1069–1077. doi: 10.1227/01.NEU.0000245598.84698.26. [DOI] [PubMed] [Google Scholar]
- 79.Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, et al. Genomics of human intracranial aneurysm wall. Stroke. 2009;40:1252. doi: 10.1161/STROKEAHA.108.532036. [DOI] [PubMed] [Google Scholar]
- 80.Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 81.Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages psgl-1 and cd44 through a common signaling pathway to induce integrin alphalbeta2-mediated slow leukocyte rolling. Blood. 2010;116:485–494. doi: 10.1182/blood-2009-12-259556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooks T, Harris CC, Oren M. Caught in the cross fire: P53 in inflammation. Carcinogenesis. 2014;35:1680–1690. doi: 10.1093/carcin/bgu134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pawlowska E, Szczepanska J, Wisniewski K, Tokarz P, Jaskólski DJ, Blasiak J. Nf-κb-mediated inflammation in the pathogenesis of intracranial aneurysm and subarachnoid hemorrhage. Does autophagy play a role? Int J Mol Sci. 2018;19:1245. doi: 10.3390/ijms19041245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, et al. Nf-kappab is a key mediator of cerebral aneurysm formation. Circulation. 2007;116:2830–2840. doi: 10.1161/CIRCULATIONAHA.107.728303. [DOI] [PubMed] [Google Scholar]
- 85.Gupta R, Palchaudhuri S, Chattopadhyay D. Glutamate induces neutrophil cell migration by activating class i metabotropic glutamate receptors. Amino Acids. 2013;44:757–767. doi: 10.1007/s00726-012-1400-1. [DOI] [PubMed] [Google Scholar]
- 86.Flood S, Parri R, Williams A, Duance V, Mason D. Modulation of interleukin-6 and matrix metalloproteinase 2 expression in human fibroblast-like synoviocytes by functional ionotropic glutamate receptors. Arthritis Rheum. 2007;56:2523–2534. doi: 10.1002/art.22829. [DOI] [PubMed] [Google Scholar]
- 87.Lin Y-J, Chang J-S, Liu X, Tsang H, Lin T-H, Liao C-C, et al. Genetic variants of glutamate receptor gene family in taiwanese kawasaki disease children with coronary artery aneurysms. Cell Biosci. 2014;4:67–67. doi: 10.1186/2045-3701-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liao W, Xiao Q, Tchikov V, Fujita K-I, Yang W, Wincovitch S, et al. Carp-2 is an endosome-associated ubiquitin ligase for rip and regulates tnf-induced nf-kappab activation. Curr Biol. 2008;18:641–649. doi: 10.1016/j.cub.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yen K, Wan J, Mehta HH, Miller B, Christensen A, Levine ME, et al. Humanin prevents age-related cognitive decline in mice and is associated with improved cognitive age in humans. Sci Rep. 2018;8:14212. doi: 10.1038/s41598-018-32616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zapała B, Staszel T, Kieć-Wilk B, Polus A, Knapp A, Wybrańska I, et al. Humanin and its derivatives as peptides with potential antiapoptotic and confirmed neuroprotective activities. Przegl Lek. 2011;68:372–377. [PubMed] [Google Scholar]
- 91.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 93.Jin H, Li C, Ge H, Jiang Y, Li Y. Circulating microrna: A novel potential biomarker for early diagnosis of intracranial aneurysm rupture a case control study. J Transl Med. 2013;11:296. doi: 10.1186/1479-5876-11-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Metaxa E, Tremmel M, Natarajan Sabareesh K, Xiang J, Paluch Rocco A, Mandelbaum M, et al. Characterization of critical hemodynamics contributing to aneurysmal remodeling at the basilar terminus in a rabbit model. Stroke. 2010;41:1774–1782. doi: 10.1161/STROKEAHA.110.585992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, et al. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol. 2012;123:773–786. doi: 10.1007/s00401-011-0939-3. [DOI] [PubMed] [Google Scholar]
- 96.Kolega J, Gao L, Mandelbaum M, Mocco J, Siddiqui AH, Natarajan SK, et al. Cellular and molecular responses of the basilar terminus to hemodynamics during intracranial aneurysm initiation in a rabbit model. J Vasc Res. 2011;48:429–442. doi: 10.1159/000324840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meng H, Metaxa E, Gao L, Liaw N, Natarajan SK, Swartz DD, et al. Progressive aneurysm development following hemodynamic insult. J Neurosurg. 2011;114:1095. doi: 10.3171/2010.9.JNS10368. [DOI] [PubMed] [Google Scholar]
- 98.Nygaard V, Rødland EA, Hovig E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics. 2015;17:29–39. doi: 10.1093/biostatistics/kxv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Primers used for qPCR.
Additional file 2: Figure S1. Stability of housekeeping genes in RNA sequencing and qPCR.
Additional file 3: Table S2. Characteristics of 73 aneurysms in all patients with intracranial aneurysms.
Additional file 4: Table S3. Batch Assignment, RNA QC, and sequencing QC for the testing and training cohorts.
Additional file 5: Figure S2. Co-variate correlation analysis between RNA quality (RIN) and differentially expressed genes.
Additional file 6: Table S4. GORILLA enriched ontologies for edgeR genes.
Additional file 7: Table S5. Transcripts and functions for the significant networks constructed by Ingenuity Pathway Analysis (IPA) using genes identified by edgeR.
Additional file 8: Table S6. GORILLA enriched ontologies for LASSO genes.
Additional file 9: Table S7. Transcripts and functions for the significant networks constructed by Ingenuity Pathway Analysis (IPA) using genes identified by LASSO.
Data Availability Statement
The datasets used in the current study are available from the corresponding author on reasonable request.