

Summary
Understanding the antibody response is critical to developing vaccine and antibody-based therapies and has inspired the recent development of new methods to isolate antibodies. Methods to define the antibody-antigen interactions that determine specificity or allow escape have not kept pace. We developed Phage-DMS, a method that combines two powerful approaches—immunoprecipitation of phage peptide libraries and deep mutational scanning (DMS)—to enable high-throughput fine mapping of antibody epitopes. As an example, we designed sequences encoding all possible amino acid variants of HIV Envelope to create phage libraries. Using Phage-DMS, we identified sites of escape predicted using other approaches for four well-characterized HIV monoclonal antibodies with known linear epitopes. In some cases, the results of Phage-DMS refined the epitope beyond what was determined in previous studies. This method has the potential to rapidly and comprehensively screen many antibodies in a single experiment to define sites essential for binding interactions.
Subject Areas: Virology, Genomic Library
Graphical Abstract
Highlights
-
•
A high-throughput, comprehensive method to identify antibody epitopes is needed
-
•
Phage-DMS combines phage display technology and deep mutational scanning
-
•
Phage-DMS identified single mutations that lead to escape from HIV Env antibody binding
-
•
Effect of mutations in Phage-DMS correlate with results using a parallel approach
Virology; Genomic Library
Introduction
Antibodies are useful research tools, potential therapeutic molecules, and the end goal of many vaccines. Understanding the precise amino acids necessary for binding of antibody to its protein target can provide insights into pathways of escape and improve antigen design for vaccines. Defining these interactions can also enhance our knowledge of antibody function. Modern improvements in the isolation and cloning of monoclonal antibodies (mAbs) have resulted in a dramatic rise in the number of novel antibodies that can be produced, but current methods to map the epitopes of these antibodies cannot presently keep pace. Thus, there is a need for a rapid screening tool to finely map the epitopes of many antibodies in a high-throughput manner.
Structural studies of antibody-antigen complexes are the gold standard for defining key amino acids that directly interact with an antibody but typically are laborious and require large amounts of antibody. Recently, a method to display libraries of peptides on phage and probe for antibody binding via immunoprecipitation and deep sequencing has been described (Mohan et al., 2018). This method has been used to map the epitopes of novel HIV-specific mAbs (Doepker et al., 2020; Finton et al., 2013, 2014; Williams et al., 2019), characterize the human virome (Xu et al., 2015), and discover autoantigens (Larman et al., 2011). Phage libraries offer several advantages over peptide arrays and other mapping methods, namely, that phage libraries are easy to generate and store, are relatively low cost, and can be used to rapidly screen for peptide-antibody binding with very small amounts of antibody or plasma. However, although these overlapping peptide libraries are useful for identifying an epitope region, they are limited in their ability to pinpoint individual residues critical for antibody binding.
Several methods exist to more precisely map the specific residues that define an antibody epitope, including the amino acids that disrupt binding and lead to immune escape. Alanine scanning gives single amino acid resolution of antibody epitopes, but it does not provide a complete picture of the potential effect of all possible amino acid mutations at a site. A more comprehensive way to understand the consequences of mutations within the epitope site is to use deep mutational scanning (DMS), which is a technique where each residue of a protein or peptide can be mutated to every possible variant (Fowler and Fields, 2014). The resulting library of variants is then used in a functional screen that simultaneously detects the impact of each mutation through deep sequencing. We have previously developed methods employing viral DMS libraries to map the epitopes of HIV-specific antibodies using neutralization as a functional screen (Dingens et al., 2017). Although this approach detects viral escape from neutralization, it is not designed for use with antibodies that bind the viral antigen but mediate their effects through non-neutralizing functions. Therefore, creating a method of mapping antibody epitopes that measures binding agnostic of antibody function is needed.
We have built upon previous studies employing phage display in combination with DMS (Ernst et al., 2010; Fowler et al., 2010; Starita et al., 2013; Zinkus-Boltz et al., 2019), and here we describe Phage-DMS, a new method that allows high-throughput and high-resolution mapping of antibody epitopes that are proximal in primary sequence with only a single round of immunoprecipitation. Using Phage-DMS, we identified the epitope of four well-characterized HIV mAbs, confirming sites of escape predicted using other approaches as well as finding novel epitope sites. Thus, Phage-DMS represents a new tool for the comprehensive mapping of antibody-antigen interactions.
Results
Generation and Characterization of gp41/V3 HIV Envelope Phage-DMS Libraries
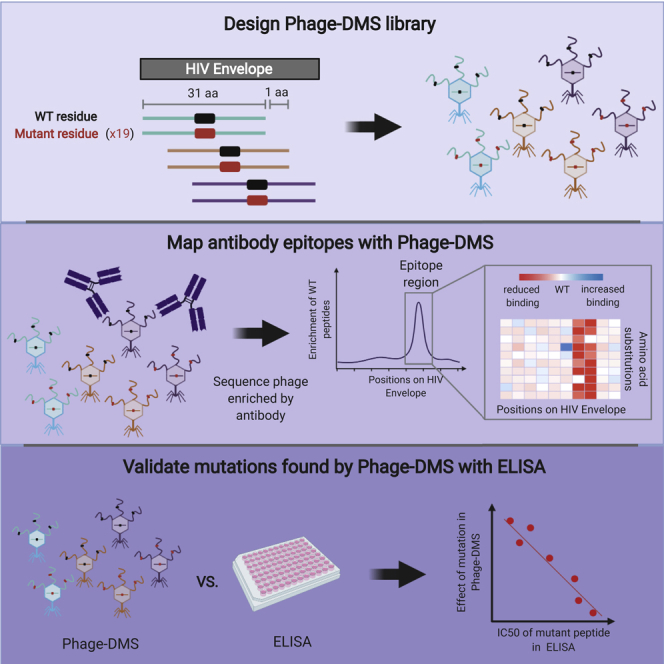
A schematic summarizing Phage-DMS is depicted in Figure 1. In brief, we computationally designed sequences that varied at the central position so that this residue contained every possible amino acid corresponding to either the wild-type residue or a mutant residue. These sequences were cloned into a phage display vector, and then this phage display DMS library was incubated with the antibodies of interest. The resulting phage-antibody complexes were immunoprecipitated, and the enriched phage were then subjected to deep sequencing to identify sequences specifically enriched or depleted in the presence of the antibody as compared with the initial phage display library.
Figure 1.

Schematic of Phage-DMS Library Design and Experimental Approach
(A) To build a Phage-DMS library, sequences are computationally designed to tile across an entire open reading frame of interest, with the central position varying to contain either the wild-type residue (shown in black) or a mutant residue (shown in red).
(B) Sequences are synthesized by releasable DNA microarray and then cloned into a T7 phage display vector.
(C and D) (C) The resulting phage display library is incubated with antibody, and (D) phage-antibody complexes are immunoprecipitated with magnetic beads.
(E) Sequences from enriched phage are PCR amplified and pooled samples are deeply sequenced.
(F) Finally, computational analysis is performed to determine the relative effect of single mutations on the binding of antibody to antigen. Figure made using BioRender.com.
A Phage-DMS library encoding peptides from two immunodominant regions of HIV Env—the V3 region of gp120 and the gp41 ectodomain—was generated in the background of envelope sequences from two common HIV-1 circulating clades (Hemelaar et al., 2019): clade A (two variants: BG505, BF520) and clade C (ZA1197) (Table S1). A total of 12,160 sequences were included. Duplicate libraries were made starting from the synthesized oligonucleotide array. Fifty-two percent of sequences from gp41/V3 library 1 and 37% of sequences from gp41/V3 library 2 matched the computationally generated sequences, based on sequencing individual plaques (Figure S1A). The other plaque sequences contained either indels, point mutations, frameshifts, or multiple errors.
The representation of all expected unique sequences was determined by deep sequencing, performed at a depth of 82- and 45-fold coverage for gp41/V3 library 1 and 2, respectively. A high degree of all unique sequences (91.6% and 87.9%, respectively) was counted at least once in each library (Figures S1B and S1C). There were some regions across gp41 and V3 that were not represented in the libraries, notably sequences corresponding to the fusion peptide domain of gp41. There were also differences in the representation of unique sequences by HIV strain, and we observed that peptides corresponding to BF520 Env were missing more than peptides from other strains. About half (51% and 50%, respectively) of all sequences detected were counted between 10 and 100 times within gp41/V3 library 1 and 2, indicating a modest degree of uniformity (Figure S1D).
Mapping the Epitope of gp41- and V3-Targeting Antibodies with Phage-DMS
To test the ability of our gp41/V3 Phage-DMS replicate libraries to define an antibody epitope, we performed experiments with Env-specific mAbs that have been previously characterized in detail and are known to bind to linear epitopes: gp41-specific mAbs 240D (Xu et al., 1991) and F240 (Cavacini et al., 1998); V3-specific mAbs 447-52D (Gorny et al., 1992) and 257D (Gorny et al., 1991).
Gp41-Specific mAbs
Deep sequencing of the phage bound by each of the gp41-specific antibodies showed enrichment of wild-type sequences corresponding to the expected epitope region of gp41, the immunodominant C-C loop (Figures 2A and 2B). For both mAbs, peptides in the background of all three Env strains showed enrichment within the same region and to a similar degree. There was a high degree of correlation between technical replicates and good correlation between biological replicates, demonstrating the reproducibility of the Phage-DMS approach (Figure S2).
Figure 2.

Enrichment and Scaled Differential Selection Results from Phage-DMS for gp41-Specific mAbs with gp41/V3 Libraries
(A and B) Line plot showing fold enrichment of wild-type peptides in the background of each HIV Env strain for (A) mAb 240D and (B) mAb F240.
(C and D) The color corresponding to each wild-type sequence is shown in the upper right. The x axis shows the amino acid position within HIV Env based on HXB2 reference sequence. (C and D) Heatmap showing the relative effect, as compared with wild-type BG505 Env, of each mutation on the binding to (C) mAb 240D and (D) mAb F240, within a selected region of gp41 shown with HXB2 numbering. Wild-type residues are marked by a black dot. Amino acids are grouped based on their properties as indicated to the left. All data shown are the average of two biological replicates.
See Quantification and Statistical Analysis for fold enrichment and scaled differential selection calculations.
To visualize the contribution of each amino acid to the epitope of the antibody, at each site we calculated the differential selection of each mutant-containing peptide as compared with the wild-type-containing peptide, scaled by the strength of enrichment of the wild-type peptide centered at that site, a metric hereafter termed “scaled differential selection.” Plotting the scaled differential selection for 240D and F240 in the context of the clade A Env protein (BG505) showed strong negative selection at positions C598 and C604 that form the disulfide C-C loop (Figures 2C and 2D). Mutations to either cysteine residue resulted in very low scaled differential selection values, with no amino acids showing higher preferred binding as compared with wild-type, as expected for these C-C loop specific antibodies.
For the 240D antibody, there was also selection against mutations to IWG (aa 595–597; Figure 2C), which was previously identified as the core epitope of 240D (Xu et al., 1991). However, mutations to 602 were also negatively selected as compared with wild-type, indicating that this site is also important for the epitope. There was enrichment above wild-type for most mutations to position L592, as well as single amino acids that are preferred above wild-type. For example, the variant N at position 599 and P at position 601 were preferred above wild-type, illustrating the potential of this method to identify a sequence that presents a more optimal epitope sequence than the wild-type viral sequence.
For the F240 antibody, nearly all mutations in C-C loop residues 597 through 604, with the exception of 601, resulted in depletion in Phage-DMS, suggesting they are part of the epitope (Figure 2D), consistent with structurally defined interactions for F240 (Gohain et al., 2016). Additionally, some mutations in regions flanking the predicted epitope were positively enriched in Phage-DMS, for example, sites 592 and 596.
V3-Specific mAbs
Phage-DMS with the V3-specific mAbs 447-52D and 257D enriched for wild-type peptides corresponding to the expected epitope region of the V3 loop (Gorny et al., 1992, 1993) (Figures 3A and 3B), with good correlation between replicates (Figure S3). For 447-52D, there was binding only to wild-type peptides from one of the clade A Env strains (BG505) and the clade C Env strain (ZA1197) but not the other clade A Env strain (BF520) (Figure 3A). This antibody has been reported to bind best to V3 sequences encoding the GPGR motif, but all the Env sequences in this library contain the less preferred GPGQ motif (Burke et al., 2009). Sequences outside of this core region differ between the strains that bind (IRIGPGQ) and the strain that does not bind (VHLGPGQ), suggesting these differences may influence binding to 447-52D in the context of the GPGQ motif. There was a similar result with 257D where only BG505 and ZA1197 V3 peptides within the expected epitope region were enriched, again indicating that naturally occurring sequence differences at the tip of the V3 loop may affect binding (Figure 3B).
Figure 3.

Enrichment and Scaled Differential Selection Results from Phage-DMS for V3-Specific mAbs with gp41/V3 Libraries
(A and B) Line plot showing fold enrichment of wild-type peptides in the background of each HIV Env strain for (A) mAb 447-52D and (B) mAb 257D.
(C and D) The color corresponding to each wild-type sequence is shown in the upper right. The x axis shows the amino acid position within HIV Env based on HXB2 reference sequence. (C and D) Heatmap showing the relative effect, as compared with wild-type BG505 Env, of each mutation on the binding to (C) mAb 447-52D and (D) mAb 257D, within the entire V3 region shown with HXB2 numbering. Wild-type residues are marked by a black dot. Amino acids are grouped based on their properties as indicated to the left. All data shown are the average of two biological replicates.
See Quantification and Statistical Analysis for fold enrichment and scaled differential selection calculations.
The amino acid positions that showed the strongest negative selection with 447-52D in the context of BG505 were in the GPGQ core epitope (Figure 3C). The exception to this is at position 315, where R was the most strongly preferred amino acid. Additionally, variation at sites 304–309 outside of this motif was negatively selected, suggesting these amino acids contribute to the epitope in this context. In particular, mutations to 304 demonstrated consistent negative selection, which has not been reported.
Most mutations to residues spanning 304 to 316 showed strong negative selection with 257D in the context of BG505 (Figure 3D). There were also a number of mutations in this region that demonstrated binding above the wild-type residue, including the R at position 315, consistent with the reported preference of 257D for the GPGR motif (Patel et al., 2008). H at position 308 was also strongly enriched above the wild-type sequence, which agrees with previous studies (Gorny et al., 1993; Patel et al., 2008). Interestingly, there were several other amino acids at position 308 (e.g., F, Y, W) that were more enriched than the wild-type R.
Generation of gp120 HIV Envelope Phage-DMS Libraries and Validation of V3 Epitopes
To test whether we would find similar results with a different Phage-DMS library, a second replicate set of Phage-DMS libraries was constructed, this time containing all peptides spanning the gp120 domain of Env. Given that 9%–13% of sequences were missing in the first library, we implemented two changes that were designed to minimize bias against sequences with higher GC content. First, we optimized the GC content of the sequences to be as uniform as possible, with an average GC content of 47%. Second, we implemented subcycling of the annealing and elongation steps between higher and lower temperatures during PCR because this was previously shown to significantly improve amplification of short template pools (Guido et al., 2016). A total of 28,840 sequences were generated in the background of a clade A (BG505), clade B (B41), and clade C (DU422) Env and cloned into duplicate phage libraries. Sequencing of individual plaques revealed that 47% of all sequences from gp120 library 1 and 41% of all sequences from gp120 library 2 were correct (Figure S4A).
We determined the representation of sequences within each library by deep sequencing at a depth of 71- and 65-fold coverage, respectively. Almost all (96.4% and 96.5%, respectively) unique sequences were present in gp120 library 1 and 2 (Figures S4B and S4C). About 66.7% of sequences were counted between 10 and 100 times for gp120 Library 1 and 68.1% for gp120 library 2, demonstrating improved uniformity as compared with the gp41/V3 libraries, where approximately half were represented at this frequency (Figure S4D). Our experiments also indicated that subcycling alone improved amplification (Figure S4E).
Because the gp120 Phage-DMS libraries we generated contained V3 sequences, we used the same two V3-specific mAbs as above, which enabled the comparison of results between the gp120 libraries and the gp41/V3 libraries. Technical and biological replicates performed with the V3 mAbs and the gp120 Phage-DMS libraries had good correlation (Figure S5). When measuring enrichment of wild-type sequences for 447-52D, we observed only strong enrichment of wild-type peptides from the clade B Env strain, which contains the preferred GPGR motif (aa 312–315) (Figure 4A). 447-52D did not enrich for V3 peptides from the two Env strains that encode GPGQ. For 257D, enrichment of wild-type peptides showed binding of this antibody was highest for peptides from the clade B strain, but there was also weak binding to peptides from the clade A strain (Figure 4B). There was no binding of 257D to peptides from the clade C strain.
Figure 4.

Enrichment and Scaled Differential Selection Results from Phage-DMS for V3-Specific mAbs with gp120 Libraries
(A and B) Line plot showing fold enrichment of wild-type peptides in the background of each HIV Env strain for (A) mAb 447-52D and (B) mAb 257D.
(C and D) The color corresponding to each wild-type sequence is shown in the upper right. The x axis shows the amino acid position within HIV Env based on HXB2 reference sequence. (C and D) Heatmap showing the relative effect, as compared with wild-type B41 Env, of each mutation on the binding to (C) mAb 447-52D and (D) mAb 257D, within a selected region of V3 shown with HXB2 numbering. Wild-type residues are marked by a black dot. Amino acids are grouped based on their properties as indicated to the left. All data shown are the average of two biological replicates.
See Quantification and Statistical Analysis for fold enrichment and scaled differential selection calculations.
Mutations to the residues 312–315 at the GPGR tip resulted in strong negative selection in Phage-DMS experiments with 447-52D (Figure 4C). We additionally saw moderate negative selection at residues upstream of the GPGR, and in particular all mutations to site R304 and H308 resulted in negative selection. For 257D, strongest negative selection was observed for mutations at sites 305, 307, and 312 as compared with wild-type (Figure 4D), which largely aligned with a previous study indicating that K-I--GP are the key residues for 257D binding (aa 305–313) (Patel et al., 2008). The exception to this was P313, where we did not observe strong negative selection in the peptide context of this library, although we did with the gp120 library. We additionally saw possibly weak contribution of sites on the N-terminal side of the V3 loop (sites 317 and 318) to the epitope.
The observed epitope for both V3 antibodies with the gp120 Phage-DMS libraries was, in general, consistent with previous results with the gp41/V3 libraries. For 447-52D, negative selection at sites 312–315 agreed with previous findings from the gp41/V3 libraries, although the relative strength of negative selection differed between the libraries. For 257D, fewer sites within the epitope region were sensitive to mutation in the context of clade B Env in the gp120 library when compared with results with the gp41/V3 library in the background of clade A Env (BG505). For example, we did not observe that mutations to P313 reduced binding, as we did with the gp41/V3 library, indicating its contribution to the epitope may depend on the context.
Validation of Phage-DMS Results Using Mutant Peptides in a Competition ELISA
To confirm that mutations identified by Phage-DMS disrupt binding to the V3- and gp41-specific antibodies, we performed competition enzyme-linked immunosorbent assays (ELISAs) with wild-type and mutant V3 or gp41 peptides (see Table S2). In this assay, peptides that bind to the antibody block activity against a coated antigen (gp120 or gp41); peptides with mutations in key epitope residues will no longer bind the antibody resulting in an increased IC50 value as compared with a wild-type peptide, and peptides that have improved binding to antibody will result in a decreased IC50 value as compared with a wild-type peptide.
The wild-type gp41 peptide competed for binding of both antibodies to gp41 protein (Figures S6A and S6B). The Phage-DMS results indicated strong preference for the cysteines that form the loop structure for both antibodies, and indeed a gp41 peptide with a mutation in one of these cysteines (C598D) no longer competes for binding to the antibodies at any concentration tested (Figure 5A). Site L602 was also identified as part of the epitope based on a strong depletion of peptides containing mutations to that site. For both 240D and F240, a peptide with a mutation at this site exhibited a 103- and 22-fold increase in IC50, respectively. The IC50 value of peptide S599K was similar to the wild-type peptide for 240D but was not detectable for F240, suggesting that the mutant peptide had lost is ability to bind F240, consistent with the differences observed for these two antibodies by Phage-DMS. A negative control gp41-binding antibody (5F3) did not show reduced activity in the presence of any gp41 peptides (Figure S6C).
Figure 5.

Comparison of Effects of Mutations on Peptide Binding in Competition ELISAs and Phage-DMS
(A and B) Bar plot showing the IC50 values of wild-type and mutant peptides in a competition ELISA for (A) gp41-specific mAbs with wells coated with MN gp41 protein or (B) V3-specific mAbs with wells coated with SF162 gp120 protein. Antibodies were pre-incubated with each peptide before addition to the wells. Peptides for which no inhibition of antibody binding could be detected at any concentration tested are indicated with an infinity symbol. Results for three replicate experiments are shown, with the mean ± SEM. Statistical significance was determined by an unpaired t test.
(C and D) Correlation between results of IC50 values from competition peptide ELISAs and scaled differential selection values from Phage-DMS for (C) gp41-specific mAbs and (D) V3-specific mAbs. Pearson's correlation shown at the top.
We confirmed that the wild-type V3 peptide competed for binding with both antibodies to gp120 protein (Figures S6D and S6E). With both 447-52D and 257D, the R315W peptide containing a mutation within the important GPGR motif resulted in a 72- and 5-fold increase in IC50, respectively (Figure 5B). Interestingly, the peptide containing a mutation at site 304, which was identified by Phage-DMS as being part of the epitope for mAb 447-52D but not found by any previous studies, demonstrated a 21-fold increase in IC50 as compared with the wild-type peptide. Sites 317 and 318 were identified by Phage-DMS as possibly weakly contributing to the epitope, and peptides containing mutations in those sites had a small but statistically significant difference in IC50 from the WT. Peptides with mutations in sites that were not identified by Phage-DMS as being part of the epitope, such as K305T with 447-52D and R304E with 257D, did not yield IC50 values that were significantly different from the wild-type peptide in competition ELISAs. VRC01 was used as a negative control gp120-binding antibody and did not show reduced binding in the presence of any V3 peptides (Figure S6F).
In order to examine whether the sites predicted to disrupt binding by Phage-DMS correlate with the binding results with corresponding peptides by ELISA, we compared IC50 values obtained for each WT and mutant peptide and differential selection values as measured by Phage-DMS. Differential selection measured for 240D and F240 in the context of the gp41/V3 library and for 447-52D and 257D in the context of the gp120 library each correlated well with the IC50 values measured by competition peptide ELISA (Figures 5C and 5D), with R2 values of 0.74 and 0.85, respectively (p = 0.013 and p = 4.2 × 10−6).
Discussion
The process of mapping the epitope of antibodies tends to be labor intensive with current methods and often does not provide a detailed understanding of the epitope. To address this, we have combined phage display technology and deep mutational scanning to create an approach called Phage-DMS that is capable of providing a comprehensive view of an antibody epitope in a high-throughput manner. As proof of concept, we created Phage-DMS libraries that display wild-type and mutant peptides derived from HIV Env strains, and we used these libraries to define the individual amino acids critical for binding to HIV-specific antibodies. The effects of specific mutations identified in this way were validated using the more traditional approach of peptide binding by ELISA. The binding measurements were highly correlated with Phage-DMS scaled differential selection, showing that Phage-DMS measurements capture mutational effects in a relatively quantitative way.
In addition to accurately identifying epitope residues that have been characterized by low-throughput mutagenesis and gold standard structural methods, Phage-DMS also refined and expanded upon the findings from previous studies. For example, mutations to R304 consistently resulted in decreased binding to 447-52D as compared with the wild-type sequence, yet this site had not been described to comprise the epitope in previous studies using overlapping hexapeptides (Gorny et al., 1992) or X-ray crystallography (Burke et al., 2009; Stanfield et al., 2004). Although neither crystal structure showed electron density at site R304, the prediction from Phage-DMS was validated by competition peptide ELISA, suggesting that Phage-DMS captures information other than physical proximity between antigen and antibody. Conversely, there were instances where previous studies had defined sites on Env with close proximity and hydrogen bonding to the antibody by X-ray crystallography, such as residues 595–609 for F240 (Gohain et al., 2016), but we did not observe strong negative selection taking place at sites 595, 596, or 605–609. Other studies have demonstrated that side-chain proximity does not necessarily predict the contribution of a site to the formation of a protein-protein complex (Dall'Acqua et al., 1998; Dingens et al., 2019; Falkowska et al., 2012; Jin et al., 1992; Kelley and O'Connell, 1993; Li et al., 2011; Muller et al., 1998; Pons et al., 1999), and this case with F240 further illustrates how analysis by Phage-DMS can yield information on the critical binding interactions of an antibody distinct from the identity of interfacing sites obtained by structural studies.
Identification of an antibody's epitope by Phage-DMS was reproducible, as demonstrated by the consistent results obtained by testing the same antibodies with two independently generated libraries. However, we did observe differences in the results with libraries built with different viral strains, highlighting the sensitivity of this assay to viral context. This is not surprising given that the strains included in the library differed by as much as 28% in their envelope sequences and thus the amino acid introduced by DMS will be influenced by these other sequences within the epitope. An example of this is 447-52D, where one library had a viral strain with a more optimal core epitope (B41 in the gp120 library), but the other library (gp41/V3) did not. In this case, other sites sensitive to mutation were revealed in the gp41/V3 library, presumably because none of the wild-type sequences contained the optimal core motif. 257D is another potential example of the importance of strain context and/or assay in determining fine antibody epitopes. We observed that P313 contributed to binding with 257D in the gp41/V3 library but not with the gp120 library. P313 had previously been identified as a key part of the epitope of 257D in a study using biopanning with random phage-displayed peptides (Patel et al., 2008) but was not recognized as contributing to binding in a different study that used overlapping hexapeptides from the clade B strain, MN (Gorny et al., 1993). The variable importance of site P313 could be related to HIV strain and/or to differences between methods or assay sensitivity.
Phage-DMS has several advantages over other mapping approaches. The high titer and renewable nature of phage libraries make them ideal for large-scale and high-throughput experiments. Although other groups have combined yeast display and deep mutational scanning to create a powerful tool for defining protein interactions (Kowalsky et al., 2015; Van Blarcom et al., 2015), these methods do have some limitations compared with Phage-DMS for these purposes. One major difference in throughput is because yeast display requires sorting by flow cytometry, which is time consuming and expensive, limiting the number of antibodies that can be tested, as previously noted (Van Blarcom et al., 2015). By contrast, Phage-DMS experiments can be conducted in a 96-well plate format with an independent test of an antibody in each well. In addition, there is often a requirement for long-read sequencing for yeast display approaches, which increases costs and complexity of analysis compared with Phage-DMS, which relies on short sequence reads. Yeast also have lower transformation efficiency and typically yield 107 to 109 transformants per microgram of DNA (Sheehan and Marasco, 2015). This creates a lower threshold for the number of variants that can be tested in a library as compared with phage, which can yield ∼1011 transformants, more than 100-fold higher than yeast. Therefore, with Phage-DMS, antibodies can be easily tested against multiple background sequences in parallel, which is especially important when studying antibodies against a pathogen as diverse as HIV. Other mutation-based approaches often involve creating and testing mutants in the background of a single Env strain. Phage-DMS can identify the binding sites for neutralizing antibodies, like 447-52D and 257D, and non-neutralizing antibodies, like 240D and F240, whereas many other methods usually rely on a functional readout, such as the ability to escape neutralization. Moreover, unlike methods that involve growth of the virus in culture, phage will display peptides encoding mutations irrespective of their effect on replication fitness, resulting in a more complete picture of the potential effect of all variants on binding. Given its versatility, Phage-DMS is not limited to mapping antibody epitopes and could theoretically be used to map binding sites between any two proteins of interest, similar to other studies that have utilized DMS to dissect protein-protein interactions using phage (Ernst et al., 2010; Fowler et al., 2010; Starita et al., 2013; Zinkus-Boltz et al., 2019).
There are some limitations of Phage-DMS. For one, post-translational modifications such as glycosylation are not present on the peptides displayed by the phage. Additionally, owing to the length of peptides displayed in these libraries, Phage-DMS in its current form is limited to mapping epitopes that depend on proximal amino acids and thus is not suited for complex conformational epitopes. To confirm these limitations, we tested known glycan-dependent (BF520.1, QA013.2) and discontinuous epitope (PGT145, 50-69) HIV Env-specific mAbs in Phage-DMS experiments and, as expected, did not observe enrichment of any peptide sequences (data not shown). However, our data and findings by others who have generated phage display libraries suggest that peptides can fold and present native-like conformations to some extent (Larman et al., 2011). For example, Phage-DMS showed a strong preference for cysteines that form a predicted loop structure within the epitope region of the gp41-specific antibodies.
Phage-DMS can be applied not just toward identifying epitope sites and potential pathways of antibody escape but also to improving the binding of antibody to antigen. We observed cases where mutations, such as F317T with antibody 447-52D, improved binding above wild-type levels by both Phage-DMS and competition peptide ELISA, demonstrating that Phage-DMS results could optimize antigen targeting and aid in rational vaccine design. Overall, we have demonstrated that Phage-DMS can map sites essential for antibody binding and could potentially accelerate the characterization of mAbs and development of vaccines.
Limitations of the Study
Because the Phage-DMS libraries described here display short linear peptides of 31 amino acids in length, generally this method is not optimal for identifying the full epitope of antibodies that rely on conformational presentation of the antigen. The peptides displayed in a Phage-DMS library will not undergo post-translational modification, and therefore, antibodies that rely on binding to glycans and other similar moieties cannot be defined using this method.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Julie Overbaugh (joverbau@fredhutch.org).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The accession number for the sequencing data reported in this paper is SRA: PRJNA630833. Aligned sequencing data and all code used to perform analyses and generate plots with Phage-DMS data can be found at https://github.com/meghangarrett/Phage-DMS.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank T. Gobillot and A. Dingens for helpful discussions, C. Sather and the Fred Hutch Genomics Core for assistance with sequencing, and L. Stamatatos for generous use of laboratory equipment. This work was supported by NIH R01 AI138709 and AI120961. M.E.G. was supported in part by T32 AI083203.
Author Contributions
J.O. conceived the project; M.E.G. and J.O. led the design of the study; M.E.G. and H.L.I. contributed to experimental design and performed experiments; M.E.G., K.H.D.C., R.B., and J.D.B. participated in computational and data analyses. M.E.G. and J.O. wrote the paper with input from all authors.
Declaration of Interests
M.E.G. and J.O. are inventors on a patent application on Phage-DMS.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101622.
Supplemental Information
References
- Burke V., Williams C., Sukumaran M., Kim S.S., Li H., Wang X.H., Gorny M.K., Zolla-Pazner S., Kong X.P. Structural basis of the cross-reactivity of genetically related human anti-HIV-1 mAbs: implications for design of V3-based immunogens. Structure. 2009;17:1538–1546. doi: 10.1016/j.str.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavacini L.A., Emes C.L., Wisnewski A.V., Power J., Lewis G., Montefiori D., Posner M.R. Functional and molecular characterization of human monoclonal antibody reactive with the immunodominant region of HIV type 1 glycoprotein 41. AIDS Res. Hum. Retroviruses. 1998;14:1271–1280. doi: 10.1089/aid.1998.14.1271. [DOI] [PubMed] [Google Scholar]
- Dall'Acqua W., Goldman E.R., Lin W., Teng C., Tsuchiya D., Li H., Ysern X., Braden B.C., Li Y., Smith-Gill S.J. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–7991. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- Dingens A.S., Arenz D., Weight H., Overbaugh J., Bloom J.D. An antigenic atlas of HIV-1 escape from broadly neutralizing antibodies distinguishes functional and structural epitopes. Immunity. 2019;50:520–532.e523. doi: 10.1016/j.immuni.2018.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingens A.S., Haddox H.K., Overbaugh J., Bloom J.D. Comprehensive mapping of HIV-1 escape from a broadly neutralizing antibody. Cell Host Microbe. 2017;21:777–787.e774. doi: 10.1016/j.chom.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doepker L.E., Simonich C.A., Ralph D., Shipley M.M., Garrett M., Gobillot T., Vigdorovich V., Sather D.N., Nduati R., Matsen F.A.t. Diversity and function of maternal HIV-1-specific antibodies at the time of vertical transmission. J. Virol. 2020;94 doi: 10.1128/JVI.01594-19. e01594–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A., Gfeller D., Kan Z., Seshagiri S., Kim P.M., Bader G.D., Sidhu S.S. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol. Biosyst. 2010;6:1782–1790. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- Falkowska E., Ramos A., Feng Y., Zhou T., Moquin S., Walker L.M., Wu X., Seaman M.S., Wrin T., Kwong P.D. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J. Virol. 2012;86:4394–4403. doi: 10.1128/JVI.06973-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finton K.A., Friend D., Jaffe J., Gewe M., Holmes M.A., Larman H.B., Stuart A., Larimore K., Greenberg P.D., Elledge S.J. Ontogeny of recognition specificity and functionality for the broadly neutralizing anti-HIV antibody 4E10. PLoS Pathog. 2014;10:e1004403. doi: 10.1371/journal.ppat.1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finton K.A., Larimore K., Larman H.B., Friend D., Correnti C., Rupert P.B., Elledge S.J., Greenberg P.D., Strong R.K. Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog. 2013;9:e1003639. doi: 10.1371/journal.ppat.1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D.M., Araya C.L., Fleishman S.J., Kellogg E.H., Stephany J.J., Baker D., Fields S. High-resolution mapping of protein sequence-function relationships. Nat. Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohain N., Tolbert W.D., Orlandi C., Richard J., Ding S., Chen X., Bonsor D.A., Sundberg E.J., Lu W., Ray K. Molecular basis for epitope recognition by non-neutralizing anti-gp41 antibody F240. Sci. Rep. 2016;6:36685. doi: 10.1038/srep36685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny M.K., Conley A.J., Karwowska S., Buchbinder A., Xu J.Y., Emini E.A., Koenig S., Zolla-Pazner S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 1992;66:7538–7542. doi: 10.1128/jvi.66.12.7538-7542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny M.K., Xu J.Y., Gianakakos V., Karwowska S., Williams C., Sheppard H.W., Hanson C.V., Zolla-Pazner S. Production of site-selected neutralizing human monoclonal antibodies against the third variable domain of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. U S A. 1991;88:3238–3242. doi: 10.1073/pnas.88.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny M.K., Xu J.Y., Karwowska S., Buchbinder A., Zolla-Pazner S. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J. Immunol. 1993;150:635–643. [PubMed] [Google Scholar]
- Guido N., Starostina E., Leake D., Saaem I. Improved PCR amplification of broad spectrum GC DNA templates. PLoS One. 2016;11:e0156478. doi: 10.1371/journal.pone.0156478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemelaar J., Elangovan R., Yun J., Dickson-Tetteh L., Fleminger I., Kirtley S., Williams B., Gouws-Williams E., Ghys P.D. Global and regional molecular epidemiology of HIV-1, 1990-2015: a systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2019;19:143–155. doi: 10.1016/S1473-3099(18)30647-9. [DOI] [PubMed] [Google Scholar]
- Jin L., Fendly B.M., Wells J.A. High resolution functional analysis of antibody-antigen interactions. J. Mol. Biol. 1992;226:851–865. doi: 10.1016/0022-2836(92)90636-x. [DOI] [PubMed] [Google Scholar]
- Kelley R.F., O'Connell M.P. Thermodynamic analysis of an antibody functional epitope. Biochemistry. 1993;32:6828–6835. doi: 10.1021/bi00078a005. [DOI] [PubMed] [Google Scholar]
- Kowalsky C.A., Faber M.S., Nath A., Dann H.E., Kelly V.W., Liu L., Shanker P., Wagner E.K., Maynard J.A., Chan C. Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J. Biol. Chem. 2015;290:26457–26470. doi: 10.1074/jbc.M115.676635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larman H.B., Zhao Z., Laserson U., Li M.Z., Ciccia A., Gakidis M.A., Church G.M., Kesari S., Leproust E.M., Solimini N.L. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., O'Dell S., Walker L.M., Wu X., Guenaga J., Feng Y., Schmidt S.D., McKee K., Louder M.K., Ledgerwood J.E. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J. Virol. 2011;85:8954–8967. doi: 10.1128/JVI.00754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan D., Wansley D.L., Sie B.M., Noon M.S., Baer A.N., Laserson U., Larman H.B. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 2018;13:1958–1978. doi: 10.1038/s41596-018-0025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller Y.A., Chen Y., Christinger H.W., Li B., Cunningham B.C., Lowman H.B., de Vos A.M. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998;6:1153–1167. doi: 10.1016/s0969-2126(98)00116-6. [DOI] [PubMed] [Google Scholar]
- Patel M.B., Hoffman N.G., Swanstrom R. Subtype-specific conformational differences within the V3 region of subtype B and subtype C human immunodeficiency virus type 1 Env proteins. J. Virol. 2008;82:903–916. doi: 10.1128/JVI.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons J., Rajpal A., Kirsch J.F. Energetic analysis of an antigen/antibody interface: alanine scanning mutagenesis and double mutant cycles on the HyHEL-10/lysozyme interaction. Protein Sci. 1999;8:958–968. doi: 10.1110/ps.8.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan J., Marasco W.A. Phage and yeast display. Microbiol. Spectr. 2015;3 doi: 10.1128/microbiolspec.AID-0028-2014. AID-0028-2014. [DOI] [PubMed] [Google Scholar]
- Stanfield R.L., Gorny M.K., Williams C., Zolla-Pazner S., Wilson I.A. Structural rationale for the broad neutralization of HIV-1 by human monoclonal antibody 447-52D. Structure. 2004;12:193–204. doi: 10.1016/j.str.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Starita L.M., Pruneda J.N., Lo R.S., Fowler D.M., Kim H.J., Hiatt J.B., Shendure J., Brzovic P.S., Fields S., Klevit R.E. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc. Natl. Acad. Sci. U S A. 2013;110:E1263–E1272. doi: 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blarcom T., Rossi A., Foletti D., Sundar P., Pitts S., Bee C., Melton Witt J., Melton Z., Hasa-Moreno A., Shaughnessy L. Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J. Mol. Biol. 2015;427:1513–1534. doi: 10.1016/j.jmb.2014.09.020. [DOI] [PubMed] [Google Scholar]
- Williams K.L., Stumpf M., Naiman N.E., Ding S., Garrett M., Gobillot T., Vezina D., Dusenbury K., Ramadoss N.S., Basom R. Identification of HIV gp41-specific antibodies that mediate killing of infected cells. PLoS Pathog. 2019;15:e1007572. doi: 10.1371/journal.ppat.1007572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.J., Kula T., Xu Q., Li M.Z., Vernon S.D., Ndung'u T., Ruxrungtham K., Sanchez J., Brander C., Chung R.T. Comprehensive serological profiling of human populations using a synthetic human virome. Science. 2015;348:aaa0698. doi: 10.1126/science.aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.Y., Gorny M.K., Palker T., Karwowska S., Zolla-Pazner S. Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J. Virol. 1991;65:4832–4838. doi: 10.1128/jvi.65.9.4832-4838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkus-Boltz J., DeValk C., Dickinson B.C. A phage-assisted continuous selection approach for deep mutational scanning of protein-protein interactions. ACS Chem. Biol. 2019;14:2757–2767. doi: 10.1021/acschembio.9b00669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is SRA: PRJNA630833. Aligned sequencing data and all code used to perform analyses and generate plots with Phage-DMS data can be found at https://github.com/meghangarrett/Phage-DMS.