

Abstract
Synaptic plasticity requires a tight control of mRNA levels in dendrites. Consistently, RNA translation and degradation pathways have been recently linked to neurodevelopmental and neuropsychiatric diseases, suggesting a role for RNA regulation in synaptic plasticity and cognition. While the local translation of specific mRNAs has been implicated in synaptic plasticity, the tightly-controlled mechanisms that regulate local quantity of specific mRNAs remains poorly understood. Despite being the only RNA regulatory pathway that is associated with multiple mental illnesses, the Nonsense-Mediated mRNA Decay (NMD) pathway presents an unexplored regulatory mechanism for synaptic function and plasticity. Here, we show that neuron-specific disruption of UPF2, an NMD component, in adulthood attenuates learning, memory, spine density, synaptic plasticity (L-LTP), and potentiates perseverative/repetitive behavior. We report that the NMD pathway operates within dendrites to regulate GLUR1 surface levels. Specifically, UPF2 modulates the internalization of GLUR1 and promotes its local synthesis in dendrites. We identified PRKAG3 as a mechanistic substrate for NMD that contributes to the UPF2-mediated regulation of GLUR1 by limiting total GLUR1 levels. These data establish that UPF2 regulates synaptic plasticity, cognition, and local protein synthesis in dendrites, providing fundamental insight into the neuron-specific function of NMD within the brain.
Introduction
NMD is a process that regulates protein expression through degradation of specific mRNAs [1-3]. NMD was first identified as an mRNA surveillance pathway that degrades mRNAs undergoing premature termination of translation [4-6]. In most mRNAs, all exon-junction complexes (EJCs), a group of proteins bound to mRNA after splicing, are upstream of the termination codon. While Up-Frameshift (UPF) proteins UPF2 and UPF3B are part of the EJC, UPF1 is only recruited to mRNA if an EJC is a downstream of a termination codon, making this codon perceived as premature, and the mRNA considered nonsense. This initiates NMD and ultimately leads to mRNA degradation [7, 8]. Recently, it has been shown that NMD also targets physiological mRNAs that naturally have NMD-inducing features but code for functional proteins [9-12]. For instance, the mRNA of the synaptic plasticity protein ARC is a known target of NMD due to intron splicing in its 3’ UTR [13]. ARC is highly expressed in dendrites [14-17] and is required for synaptic plasticity and memory consolidation [18-21]. Similar to Arc mRNA, various mRNAs with intron splicing in 3’ UTR are localized to dendrites [22] and have been implicated in synaptic plasticity and memory [19, 23-26]. However, the extent to which NMD is physiologically relevant to synaptic plasticity is not clear.
Despite its putative link to many mental disorders including autism and schizophrenia [27-32], whether NMD is relevant to the synaptic pathology and behavioral deficits of these diseases remains unclear. It has been reported that NMD maintains synapse architecture and synaptic vesicle cycle efficacy in drosophila [33]. NMD has also been implicated in epileptogenesis [34]. In addition, a recent study showed that Upf3b-null mice exhibit deficits in fear-conditioned learning, supporting a role for NMD in regulating synaptic plasticity and cognition [35]. However, because NMD is ubiquitous and required for neuronal differentiation [36], disruption of this pathway during development hinders determining the direct role of NMD in these processes. Therefore, understanding the precise, cell-type specific role of NMD in developing and adult brain is likely to provide novel insights into key mechanisms critical for synaptic plasticity and behavior.
Synaptic plasticity takes several forms, including the modification of synaptic strength and spine structure. Synaptic transmission can be influenced by activity, becoming either enhanced, through long-term potentiation (LTP), or depressed, through long-term depression (LTD) [37]. Many cognitive disorders are associated with alterations in spine density and synaptic plasticity [38, 39]. Both LTP and LTD has been associated with local protein synthesis in dendrites [21, 40]. Deficits in mRNA regulation can result in ectopic protein synthesis and lead to cognitive disabilities [41-47], having downstream effects on synaptic plasticity and cognition. For example, exaggerated mRNA translation leads to alterations in spine density, LTP and LTD [44, 47-49] suggesting that a tight control of protein synthesis is required at many levels within dendrites [42]. A great amount of prior research has addressed the pathways that regulate translational derepression in dendrites. However, the mechanisms that control mRNA quantity during synaptic function remain unknown.
Here we show that both synaptic plasticity and cognitive function is physiologically regulated by neuronal NMD. Conditional deletion of the NMD gene Upf2 in adult glutamatergic neurons in vivo disrupts learning and memory and potentiates perseverative/repetitive behavior. Deletion of Upf2 also results in altered spine density and LTP in the adult hippocampus. This phenotype is accompanied by a significant reduction in total GLUR1 levels in conditional knockout mice. We show that UPF2 regulates GLUR1 surface levels through mechanisms that influences GLUR1 internalization and nascent protein synthesis. Selective disruption of UPF2 protein from cultured hippocampal neurons reduces both total and surface expression of GLUR1 protein without affecting GluR1 mRNA levels. This phenotype is recapitulated when NMD is disrupted in dendrites via the targeted-inhibition of UPF3B local synthesis. Disruption of UPF2 increases internalization, due to elevated ARC levels, and decreases synthesis, due to elevated PRKAG3 levels, of GLUR1 protein in dendrites. Normalizing the levels of Arc and Prkag3 mRNAs, together, completely corrects surface GLUR1 levels in UPF2-deficient dendrites. These data demonstrate a role for NMD in regulating cognitive function, the dendritic proteome, and synaptic plasticity, as well as potentially other essential dendritic functions.
Results
UPF2 is required for learning and memory and suppresses perseverative behavior.
We first sought to determine the requirement for neuronal NMD in cognitive function and synaptic plasticity. To do this, we genetically disrupted NMD in postmitotic glutamatergic neurons using a conditional knockout mouse of Upf2 (Upf2fl/fl) [50] [11, 50, 51]. We temporally controlled UPF2 disruption by crossing the Upf2fl/fl line with a mouse line that expresses tamoxifen-inducible Cre under the αCaMKII promoter (αCaMKII:CreERT2) [52]. In all behavioral experiments, we induced Cre in 2-month old conditional-knockout (Upf2fl/fl;αCaMKII:CreERT2. CKO) mice as well as in their control littermates (Upf2wt/wt;αCaMKII:CreERT2; CTRL) by tamoxifen. Loss of UPF2 protein in excitatory neurons was successfully achieved by 200mg/kg of tamoxifen administered every other day for 5 doses (Figure S1a). No difference in locomotor activity, anxiety-related behavior, or sociability phenotypes, were detected between Upf2 CKO mice and controls (Figure S1b-e).
We next examined hippocampus-dependent learning and memory in Upf2 CKO mice. On the Y-maze, Upf2 CKO mice showed no preference for the novel arm relative to the other arms, and displayed significantly worse novel arm preference than the control group (Figure 1a), suggesting that Upf2 CKO mice have disrupted short-term spatial memory. As an additional measure of hippocampal function, we tested Upf2 CKO mice for contextual fear memory. In this test, Upf2 CKO mice froze for significantly less time than controls by the last tone-shock pairing during conditioning, and this conditioning deficit resulted in worse contextual fear memory in Upf2 CKO mice relative to control mice 24 hr later (Figure 1b-c). In the Morris water maze, Upf2 CKO mice displayed a 1) robust attenuation of learning behavior, 2) increased latencies to enter the hidden platform quadrant during the probe trial, and 3) decreased exploration of this quadrant during probe testing (Figure 1d-f). These data collectively suggest that Upf2 CKO mice exhibit attenuated learning behavior, which produces worse performance on common assays of hippocampus-dependent memory function.
Figure 1. UPF2 is required for learning, memory, spine density, and LTP.
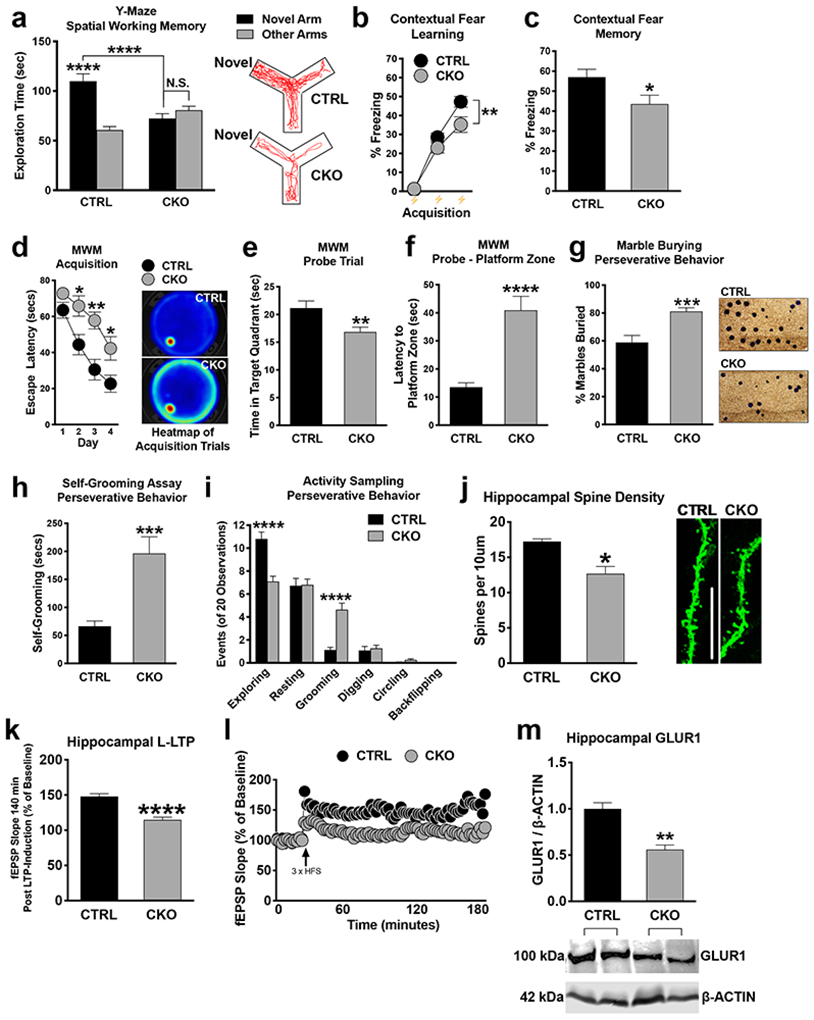
a-c, Impaired short-term spatial and contextual fear memory in CKO mice.
Quantitations for relative arm preference on the Y-Maze (n=24 CTRL and 22 CKO mice), freezing during contextual fear conditioning (n = 22 per group, b) and freezing during context re-exposure (n = 22 per group, c).
d-f, CKO mice exhibit disrupted hippocampus-dependent spatial acquisition.
In the MWM, CKO mice exhibited significantly delayed escape latencies on days 2, 3, and 4 compared to CTRL mice (n=18 for each group) (d). During the probe trial, CKO spent significantly less time exploring the target quadrant and took significantly longer to enter the platform zone compared to CTRL mice (n=18 for each group; f).
g-i, Increased repetitive and compulsive-like behavior in CKO mice.
CKO mice buried significantly more marbles (n=25 per group) as well as exhibited increased grooming relative to CTRL mice without evidence of further compulsive-like traits (i).
j-l, Disrupted hippocampal spine density and synaptic plasticity in CKO mice.
Spine quantification (n=3 mice per group; 4-10 independent apical dendrite; j) and LTP measurements (n=12 slices, 7 CTRL mice: n=14 slices, 8 CKO mice) (k and l) in CTRL and CKO mice.
m, Disrupted GLUR1 expression in the CKO hippocampus.
Western blot of total hippocampal GLUR1 revealed significantly decreased GLUR1 expression in CKO relative to CTRL mice (n= 3 CTRL and 5 CKO mice).
Data are represented as mean ± SEM; *p < 0.05, **p < 0.01, ***p <0.001 and ****p < 0.0001. N.S. represents “Not Significant”. Scale bar: 5 μm.
Because the NMD pathway has been previously linked to autism and schizophrenia [27-32], where stereotypies are common to both disorders, we also tested for perseverative/repetitive behaviors [47, 53-56]. In our marble burying assay, Upf2 CKO mice buried significantly more marbles than control mice (Figure 1g; 81.14% vs 58.86% of marbles). In an activity-sampling test session, where grooming behavior was continuously monitored for 20 min, CKO mice groomed for longer and displayed a greater frequency of grooming bouts compared to the control group (Figure 1h-i). Together, these data suggest that NMD is required for proper learning and memory, and its disruption potentiates perseverative/repetitive behaviors.
UPF2 ensures proper spine density and L-LTP in hippocampus.
To study spine density in Upf2 CKO mice, we injected AAV-αCaMKII-eGFP virus into the dorsal CA1 field 7d prior to perfusion. We observed significantly reduced CA1 spine density in Upf2 CKO hippocampal dendrites compared to control dendrites in vivo (Figure 1j). We next assessed both LTD, early phase LTP (E-LTP), and late phase LTP (L-LTP), in these mice. LTD was not different in UPF2-deficient slices compared to control slices (Figure S1f-g), Similarly, we found that E-LTP, which is thought not to require new gene transcription or mRNA translation, was unaltered in UPF2-deficient slices. However, we found that L-LTP, which requires new gene transcription, translation, and AMPA-receptor increase, was significantly decreased in UPF2-deficient slices compared to control slices (Figure 1k-l). These data suggest that NMD is required for proper LTP and spine density but not for LTD in hippocampus.
UPF2 positively regulates GLUR1 surface expression.
We next sought to identify mechanisms through which NMD regulates synaptic plasticity and cognitive function. It was previously shown that disruption of the EJC, a critical component for targeting mRNAs to the NMD pathway, causes an increase in the levels of AMPA receptor GLUR1 in hippocampal dendrites in vitro [13]. However, EJC proteins have multiple other functions, including mRNA transport and translation [57]. For example, genetic screens in drosophila showed that knockdown of EJC core components caused skipping of several exons in pre-mRNAs, which contain long introns [58]. Therefore, disruption of EJC might lead to differential phenotypes that are divergent from what occurs when the NMD machinery is discretely targeted. Intriguingly, contrary to what is observed in studies of disrupted EJC proteins, we found that UPF2 deficiency led to a decrease in total GLUR1 levels in vivo (Figure 1m). Additionally, this data is congruent with disrupted hippocampal LTP in Upf2 CKO mice given that LTP can be represented by the synaptic insertion of AMPA receptors [59]. To explore this phenotype in vitro, we infected hippocampal neurons with Upf2-shRNA:GFP lentivirus to interrupt endogenous NMD activity at 7 days in vitro (DIV7) and examined GLUR1 expression on DIV21. Using 2 unique Upf2-shRNA lentiviruses (Figure S2), we found that total levels of GLUR1 were decreased both in cell bodies and dendrites of hippocampal neurons upon knockdown of Upf2 (Figure S3a-c).
We next asked whether UPF2 regulates GLUR1 surface expression in neurons. An essential component of GLUR1 signaling involves the dynamic insertion and removal of the GLUR1 receptor to the synaptic surface [60]. Similar to total GLUR1 levels, UPF2 deficiency led to decreased surface levels of GLUR1 (Figure 2; see also Figure S3 and S4). This suggests that NMD positively regulates GLUR1 surface expression. While GluR1 mRNA is not a canonical target of NMD, it is known that some mRNAs that do not carry identifiable NMD-inducing features can also be degraded by NMD [61]. In addition, it has been suggested that GluR1 transcription is regulated by ARC [62]. Consistent with Arc mRNA being an established target of NMD [11, 13, 63-67] (see also Figure S5), the levels of both Arc mRNA and ARC protein are increased in hippocampal neurons upon knockdown of Upf2 (Figure S5). We, therefore, examined GluR1 mRNA levels in UPF2-deficient neurons. Overall GluR1 mRNA levels were unaltered following knockdown of Upf2 in hippocampal neurons, indicating that the reduction in total and surface GLUR1 protein does not arise from changes in mRNA availability. These data thus suggest that NMD modulates GLUR1 signaling via an indirect restraining mechanism that negatively regulates the synthesis, surface localization or surface turnover of GLUR1 in dendrites.
Figure 2. UPF2 is required for surface expression of GLUR1 on dendrites.
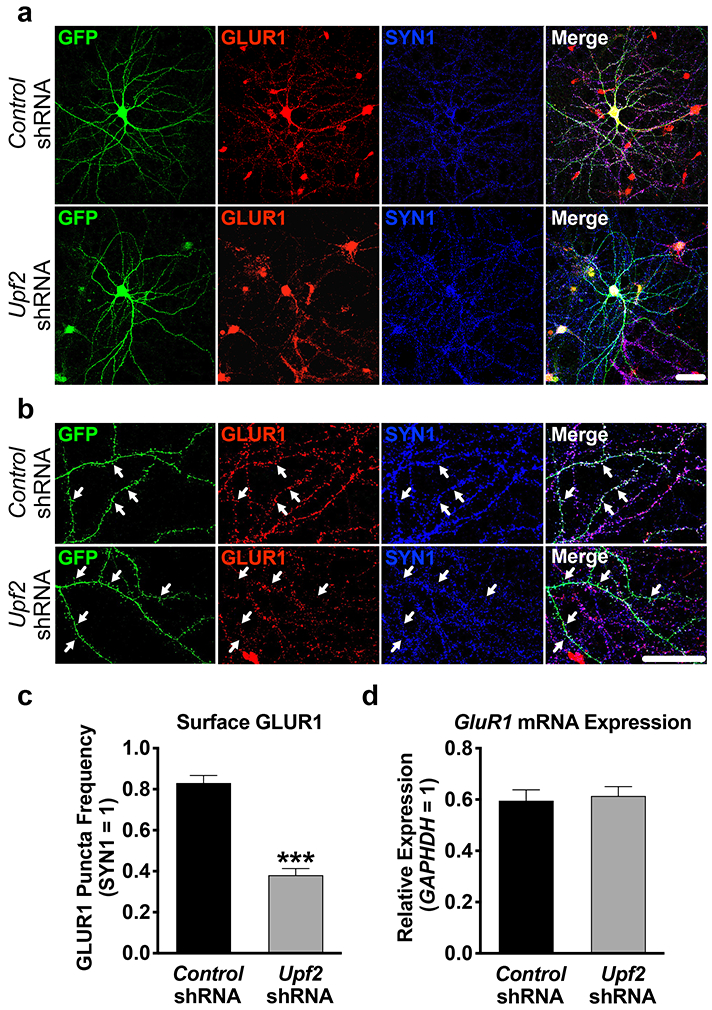
a-b, Hippocampal neurons were infected at DIV7 with control- or Upf2-shRNA lentivirus and fixed at DIV21. Surface GLUR1 expression was detected using an anti-N-terminus-GluR1 antibody (see Methods). Arrows indicate positive and negative GLUR1 signal at spines.
c, UPF2 positively regulates surface GLUR1 levels.
Quantifications of surface GLUR1 frequency in dendrites (n=3/group; 10 neurons-24 dendrites/group). Knockdown of Upf2 did not change SYN1 density (see Figure S4 for PSD-95 staining) indicating unaltered synaptic potential.
d, Knockdown of Upf2 does not alter GluR1 mRNA levels.
qRT-PCR of GluR1 mRNA levels in control- and Upf2-shRNA infected neurons (n=3/group; see Figure S7 for infection efficiency).
Data are represented as mean ± (SEM); ***p < 0.001. Scale bar: 20 μm.
UPF2 negatively regulates GLUR1 internalization in dendrites.
We next sought to determine how UPF2 regulates surface expression of GLUR1 in dendrites. It is possible that the reduction in total GLUR1 levels might account for decreased surface levels of this receptor. However, locally synthesized ARC is also known to cause GLUR1 endocytosis, leading to a decrease in the surface levels of this receptor [21, 68]. To determine if GLUR1 endocytosis is altered after disruption of NMD, we measured the basal internalization rate of GLUR1 upon knockdown of Upf2 in hippocampal dendrites using a previously established protocol [69]. We observed significantly increased GLUR1 basal internalization in UPF2-deficient dendrites (Figure 3a-b). However, ARC-mediated internalization of GLUR1 only influences surface but not total levels of GLUR1 [70], indicating that UPF2 must regulate total GLUR1 levels via other, yet defined, NMD targets.
Figure 3. UPF2 regulates GLUR1 internalization and local synthesis in dendrites.
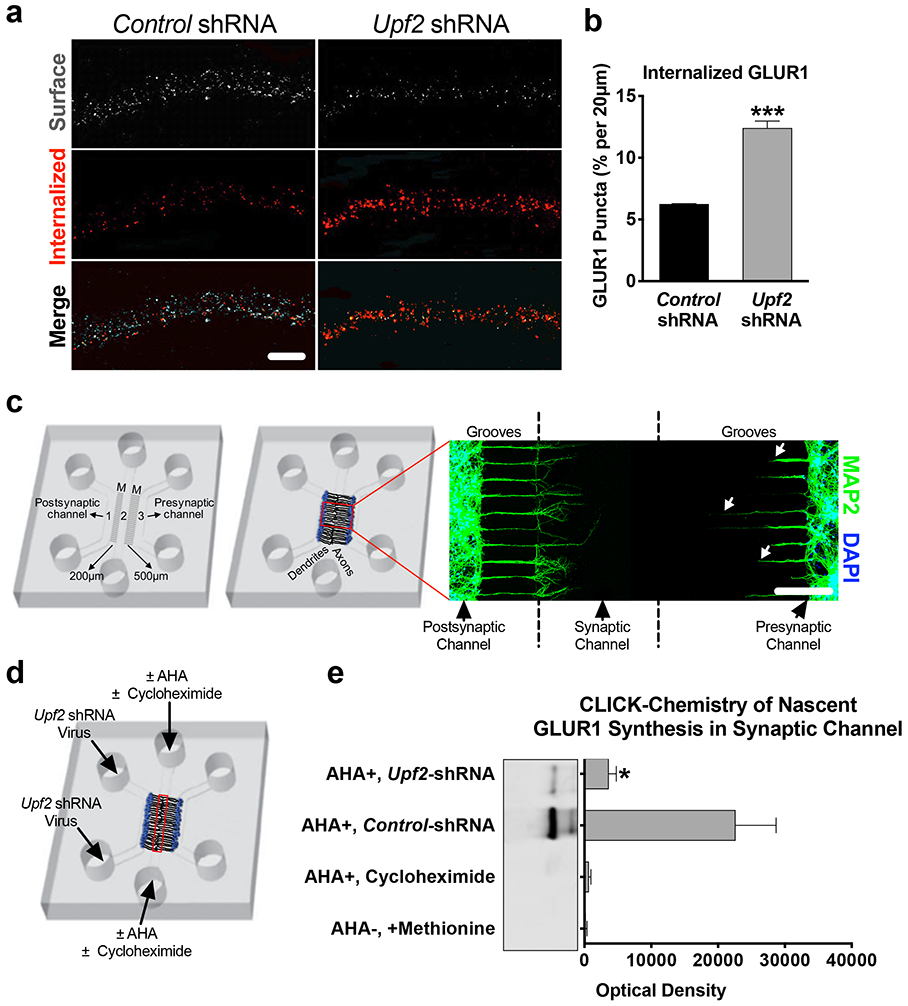
a-b, Increased rates of GLUR1 internalization in UPF2-deficient dendrites.
Basal internalization rates (for 10 minutes) of GLUR1 receptors was achieved by live-staining neurons using anti-N-terminus GLUR1 antibody at DIV 21 (see Methods) (n=3 per control-shRNA [13 neurons, 33 dendrites] and per Upf2-shRNA [20 neurons, 53 dendrites]).
c, Custom tripartite microfluidic device for compartmentalized synapse access.
In this customized tripartite chamber, the different microgroove (M) lengths between channels created a postsynaptic neuron channel, a presynaptic neuron channel, and a synaptic channel, which enabled selective treatment of these compartments (see Methods). By DIV8, the synaptic channel is exclusively populated with dendrites (MAP2) emerging from the postsynaptic channel. Arrows highlight that dendrites from the presynaptic channel do not reach the synaptic channel.
d-e, Decreased local synthesis of GLUR1 in UPF2-deficient dendrites.
We cultured E16 hippocampal neurons in tripartite chambers and applied control- or Upf2-shRNA virus only to postsynaptic neurons at DIV7. The media was exchanged to methionine-free media containing the methionine analog azidohomoalanine (AHA, 25 μM). After harvesting material from the synaptic channels (red box), the proteins were separated by SDS-PAGE, and nascent GLUR1 was detected with an anti-C-terminus antibody (see also Methods).
Data are represented as mean ± SEM; *p < 0.05 and ***p < 0.001. Scale bar: a 20 μm, c 200 μm.
UPF2 positively regulates local synthesis of GLUR1 in dendrites.
The total levels of dendritic GLUR1 are determined by the amount of GLUR1 protein trafficked to dendrites from cell bodies as well as a balance of local protein synthesis [71] and local protein degradation [72, 73] in dendrites. Thus, we also investigated the possibility of enhanced GLUR1 proteolysis or decreased GLUR1 synthesis in UPF2-deficient dendrites. To do this, we used a custom-fabricated tripartite microfluidic device, which, unlike traditional microfluidic chambers with two channels [74], contains a third “synaptic” channel (Figure 3c and Figure S6-7). This tripartite compartmentalization creates physical and fluidic isolation of presynaptic neurons, postsynaptic neurons, and their synapsing projections, enabling us to selectively isolate, treat and harvest dendrites in subsequent experiments.
When AMPA receptors are internalized, endocytosed receptors undergo lysosome- or proteasome-mediated degradation [73, 75-78]. To study local degradation of GLUR1, we cultured E16 hippocampal neurons in tripartite chambers and applied control- or Upf2-shRNA viruses only to postsynaptic cell channels at DIV7. This resulted in disruption of NMD only in postsynaptic cell bodies and dendrites but not in presynaptic cells (Figure S7). At DIV21, we inhibited both proteosomal and lysosomal degradation by treating the synaptic channels with MG132 and leupeptin. Co-inhibition of lysosome- and proteasome-mediated degradation did not restore GLUR1 levels in UPF2-deficient dendrites (Figure S8). Thus, degradation of GLUR1 receptors at the protein-level does not contribute to the reduction in surface GLUR1 observed in UPF2-deficient dendrites.
We next asked whether local synthesis of the GLUR1 receptor is altered in UPF2-deficient dendrites. To measure the dendritic synthesis of GLUR1, we used a CLICK-chemistry based approach and labeled locally synthesized nascent proteins at synapses (Figure 3c-e). We cultured E16 hippocampal neurons in tripartite chambers and targeted NMD only in postsynaptic cells by applying the Upf2-shRNA virus into the postsynaptic cell channel at DIV7. At DIV21, we exchanged the growth medium in the synaptic channels to methionine-free medium containing the methionine analog azidohomoalanine (AHA) [79] to label all newly synthesized proteins at synapses (Figure 3d). The amount of GLUR1 protein that bound the streptavidin in the UPF2-deficient synapses was significantly lower compared to the amount of GLUR1 that bound the streptavidin in the control synapses (Figure 3e). This data suggests that dendritic synthesis of GLUR1 is reduced upon disruption of NMD.
Local degradation of Arc and Prkag3 mRNAs by NMD in dendrites.
We next sought to determine whether local NMD contributes to the regulation of GLUR1 protein synthesis in dendrites. Similar to Arc mRNA, the mRNA of PRKAG3 protein, the non-catalytic subunit of AMP-activated protein kinase (AMPK), is also derived from removal of a 3’UTR intron [13] making the Prkag3 mRNA a potential substrate for NMD. Indeed, the half-life and mRNA/pre-mRNA ratio of Prkag3 is increased upon loss of UPF2 in hippocampal neurons (Figure S5). Interestingly, AMPK has been suggested to negatively regulate GLUR1 protein levels in dendrites in a translation dependent manner [25]. Intriguingly, both Arc and Prkag3 mRNAs are localized to dendrites, suggesting that they might be degraded by NMD in these compartments.
We found that the core proteins of the NMD machinery are localized to dendrites (Figure 4a), suggesting that local NMD might contribute to regulation of GLUR1 levels in these compartments. We first examined Arc and Prkag3 mRNA levels in UPF2-deficient synaptic regions. To do this, at DIV7, we selectively infected postsynaptic neurons with Upf2-shRNA lentivirus and isolated RNA from synaptic channels at DIV21. qRT-PCR showed an increase in the levels of both Arc and Prkag3 mRNAs in UPF2-deficient synaptic channels compared to control synaptic channels (Figure 4b). NMD is initiated when UPF1 recognizes NMD-inducing features in mRNAs during translation [80, 81]. Thus, inhibition of translation hinders NMD and leads to the accumulation of NMD-target mRNAs [7]. To confirm whether Arc and Prkag3 undergo translation-dependent degradation in synaptic regions, we cultured E16 neurons in tripartite chambers and, at DIV21, applied translation inhibitor cycloheximide (CHX) selectively to synaptic channels. qRT-PCR on harvested synaptic-channel material revealed that both Arc and Prkag3 mRNAs accumulated in these compartments upon inhibition of translation (Figure 4c).
Figure 4. Establishing local regulation of GLUR1 by NMD in dendrites.
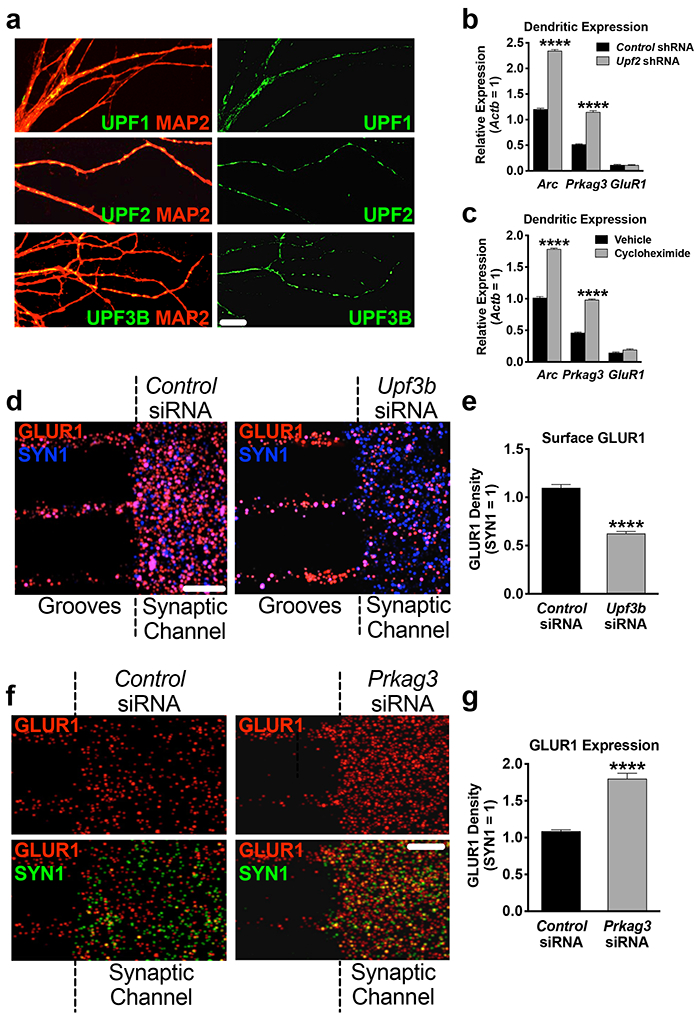
a, The NMD machinery is localized to dendrites.
UPF1, UPF2 and UPF3B immunostainings in hippocampal dendrites at DIV7.
b, Arc and Prkag3 mRNAs are increased in UPF2-deficient dendrites.
Synaptic material was harvested from tripartite chambers containing postsynaptic neurons infected with either control- or Upf2-shRNA virus (n=3) and qRT-PCR for Arc and Prkag3 mRNAs was performed.
c, Arc and Prkag3 mRNAs are subjected to translation-dependent degradation in dendrites. Selective treatment of synaptic channels with the translation inhibitor CHX (10 μM) at DIV21 for 3 hr and qRT-PCR for Arc and Prkag3 mRNAs (n=3).
d, NMD locally regulates surface expression of GLUR1 in dendrites.
Repetitive treatment of synaptic channels with non-overlapping Upf3b-siRNAs (10 nM) for 7 days led to complete loss of UPF3B protein (Figure S9d) and a >40% decrease in surface GLUR1 compared to control cultures (10 neurons, 10 dendrites per group).
e, Locally synthesized PRKAG3 negatively regulates GLUR1 levels in dendrites. Quantifications of total GLUR1 levels following non-overlapping Prkag3 siRNA treatment of synaptic channels for 7d (n=3 per control-siRNA [6 neurons, 9 dendrites] and per Prkag3-siRNA [8 neurons, 9 dendrites]).
Data are represented as mean ± SEM; ****p < 0.0001. Scale bar: 30 μm.
We next asked whether NMD contributes to the regulation of GLUR1 surface levels locally in dendrites. To address this, we inhibited the local synthesis of UPF3B, a protein specific to the NMD pathway [82, 83], selectively in synaptic channels. The local translation of Upf3b mRNA is a source for UPF3B protein in dendrites (Figure S9a-b). We cultured E16 neurons in cell-body channels and, starting at DIV14, repeatedly treated synaptic channels with siRNAs against Upf3b mRNA for seven days. This resulted in complete loss of UPF3B protein in synaptic channels (Figure S9d). Similar to the knockdown of Upf2 in postsynaptic cells, the surface frequency of GLUR1 was reduced upon selective loss of UPF3B protein in synaptic channels (Figure 4d).
Taken together, these experiments establish the local requirement for NMD in synaptic regions and that the local regulation of GLUR1 by NMD might, in addition to degrading Arc, also involve Prkag3 degradation.
PRKAG3 negatively regulates GLUR1 expression in dendrites.
We next examined whether PRKAG3 protein locally influences GLUR1 expression in synaptic channels. To do this, we cultured E16 neurons in cell-body channels and, starting at DIV14, repeatedly treated synaptic channels with siRNAs against the Prkag3 mRNA for seven days (Figure S9e). Selective loss of PRKAG3 in synaptic channels significantly increased the total levels of GLUR1 (Figure 4e).
Because the synaptic channels of tripartite chambers contain both dendrites and axons, it is possible that GLUR1 phenotypes may arise also from axonal changes and therefore not selectively from dendrites. To clarify this, we examined the proteins of the NMD machinery and targets of NMD in mature axons. Neither the components of the NMD machinery, nor Arc or Prkag3 mRNAs, were present in isolated hippocampal axons at DIV21 (Figure S10). In fact, the axonal transcriptome significantly shrinks as axons mature [84] and does not include any known NMD targets [85, 86]. These data indicate that alterations in GLUR1 expression upon selective disruption of either UPF3B or PRKAG3 proteins in synaptic channels therefore solely originate from a local effect within dendrites.
Normalization of Prkag3 and Arc mRNA levels rescues GLUR1 surface expression and spine density in UPF2-deficient dendrites.
We functionally tested a mechanistic role of locally synthesized ARC and PRKAG3 in the NMD-mediated regulation of surface GLUR1 expression. To do this, we systematically modulated the elevated levels of Arc and Prkag3 mRNAs in UPF2-deficient dendrites by siRNA transfection of synaptic channels. We first determined the concentration of siRNA required to correct Arc and Prkag3 levels in UPF2-deficient dendrites (Figure S11). Using optimized concentrations, we next modulated the levels of Arc and Prkag3 mRNAs in synaptic channels containing UPF2-deficient dendrites (Figure 5a). Compared to synaptic channels with intact NMD, cultures infected with Upf2-shRNA lentivirus had significantly lower GLUR1 density as expected. Restoring expression levels of Arc or Prkag3 mRNAs in synaptic channels led to a significant but incomplete recovery of surface GLUR1 in these compartments for each target (Figure 5b-c). This is consistent with observations that targeting Arc only rescues the internalization of GLUR1, while normalization of Prkag3 only rescues total GLUR1 levels but not the increased rates of internalization (Figure S12). Thus, we reinstated both Arc and Prkag3 mRNA levels together, which led to a complete recovery of surface GLUR1 density in UPF2-deficient dendrites (Figure 5b-c). To test this mechanism in vivo, we next sought to rescue spine density, which is dependent upon a 3D environment and not readily testable in vitro. To normalize Arc and Prkag3 levels in vivo, we infected CA1 hippocampal neurons of Upf2 CKO mice with lentiviruses expressing shRNAs against Arc and Prkag3. We used shRNA sequences that sufficiently modulated the increased levels of ARC and PRKAG3 proteins in UPF2-deficient neurons (Figure S13). The simultaneous modulation of Arc and Prkag3 in Upf2 CKO hippocampal neurons increased spine density in vivo to levels consistent with CTRL mice (Figure 5f). Together, these data indicate a functional role for UPF2-mediated modulation of ARC and PRKAG3 levels within dendrites, which together function to regulate GLUR1 signaling and synaptic plasticity.
Figure 5. UPF2 mechanistically regulates surface GLUR1 levels and spine density via degradation of Arc and Prkag3 in dendrites.
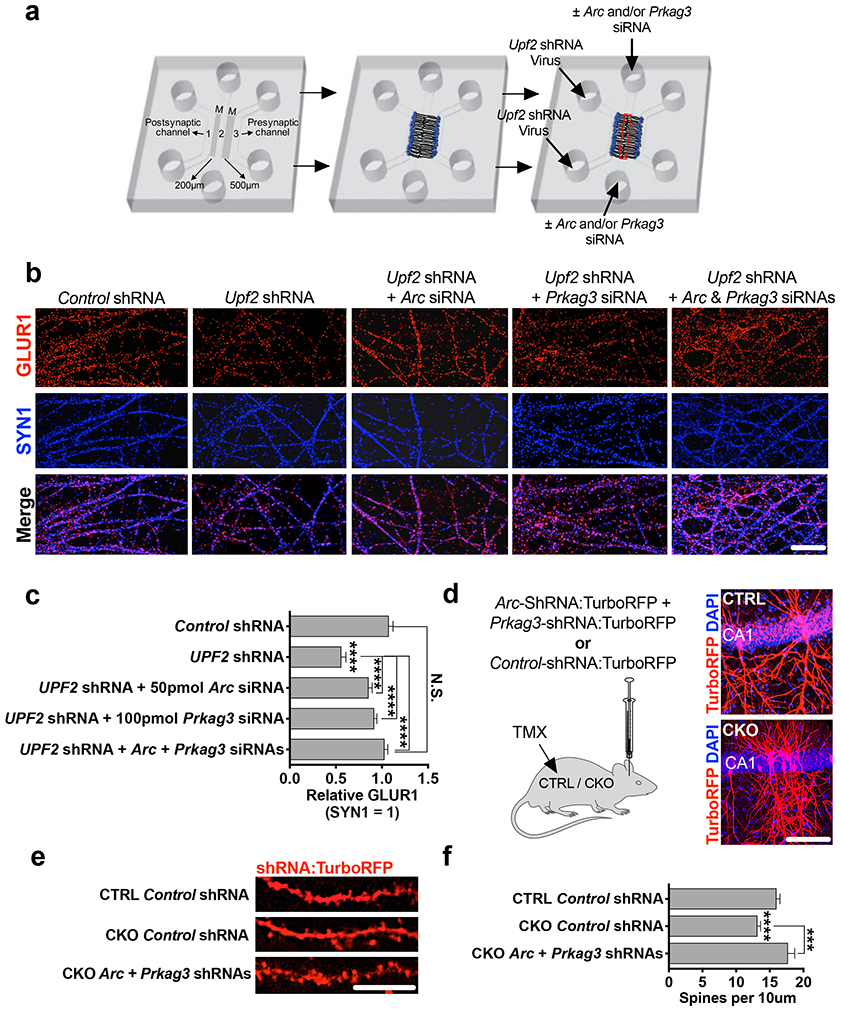
a-c, Modulating Arc and Prkag3 levels in UPF2-deficient dendrites rescues surface GLUR1. Schematic of the experimental design (a; see Figure S11 for knockdown validation) and immunostainings for surface GLUR1 in dendrites upon treatment of UPF2-deficient dendrites with Arc and Prkag3 siRNAs (b). siRNA transfection of synaptic channels started at DIV14 and lasted for a week. Quantifications of surface GLUR1 density following normalization of each target (n=3 per control-siRNA [10 neurons, 10 dendrites], Arc-siRNA [10 neurons, 10 dendrites] and Prkag3-siRNA [10 neurons, 10 dendrites]) (c).
d-f, Modulating Arc and Prkag3 levels in vivo rescues hippocampal spine density. Mice were stereotaxically injected with mCMV>Arc-shRNA:TurboRFP and mCMV>Prkag3-shRNA:TurboRFP lentiviruses to the dorsal CA1 region of the hippocampus (see Figure S13 for shRNA knockdown validation) (d). Representative images (e) and quantifications (f) of spine density from virus-injected mice (n=3 mice per group [n=14 CTRL + Control shRNA dendrite segments, n=22 CKO + Control shRNA dendrite segments, and n=16 CKO + Arc+Prkag3 shRNA dendrite segments]).
Data are represented as mean ± SEM; ****p < 0.0001, N.S. represents “Not Significant”. Scale bar: b 75 μm, d 100 μm, e 5 μm.
Discussion
Our study identifies neuronal NMD as a mechanism that contributes to the regulation of synaptic plasticity and cognitive function. Specifically, we report that NMD influences synaptic plasticity as well as learning and memory. In addition, our data suggests that local NMD participates in GLUR1 regulation by limiting ARC and PRKAG3 protein expression in dendrites.
UPF2 regulates synaptic plasticity and cognitive function.
Our data indicate that proper synaptic plasticity and cognitive function requires UPF2. Our data show that UPF2-deficient hippocampal slices show decreased L-LTP. In addition, spine density in the hippocampus of conditional Upf2 mutant mice is reduced relative to control mice. Mice also show attenuated learning and memory upon conditional ablation of Upf2 in excitatory neurons. Memory deficits were readily detectable in UPF2-deficient mice on three independent hippocampus-dependent behavioral assays. In addition to attenuated learning and memory, loss of UPF2 in excitatory neurons resulted in perseverative behavior in mice. Together, these data implicate a role for NMD in both synaptic plasticity and behavioral endophenotypes common to a spectrum of neurodevelopmental diseases including autism and schizophrenia.
Our data also provide comprehensive insight into the neuron-specific and spatiotemporal requirement for UPF2 in plasticity and behavior. Recently, a Upf3b null mutant mouse line was shown to have fear-conditioning defects but, unlike our observations in Upf2 CKO mice, no spatial learning deficit [35]. There may be several reasons for this. For instance, NMD is ubiquitous and required for neuronal differentiation [36, 87]. Consistent with this, global interruption of NMD by ablation of Upf3b during brain development alters neuronal maturation and is thereby likely to contribute to the behavioral phenotypes observed in the Upf3b null mouse line. In addition, while UPF2 and UPF3 proteins are known to be specific to the NMD machinery, it has been suggested that NMD might have different branches in which NMD exerts its function independent of UPF2 or UPF3B [83, 88, 89]. Therefore, studies of conditional Upf2 deletion provide a platform to dissect not only the temporal and cell-type specific functions of the NMD machinery, but also branch-specific functions of NMD in brain development and behavior.
UPF2-mediated GLUR1 regulation and its contribution to behavior.
Our data also demonstrate that UPF2 positively regulates GLUR1 signaling in hippocampus. We report that loss of UPF2 protein in adult hippocampal neurons caused a concomitant decrease in total and surface levels of this receptor. Our CLICK-chemistry experiments suggest that UPF2 positively regulates GLUR1 protein synthesis, whereby GLUR1 synthesis in dendrites is repressed in the absence of UPF2 protein leading to reduced local GLUR1 protein. Similarly, we confirm that the mRNA of the AMPK γ subunit (PRKAG3) is a substrate of NMD (Figure 4, S5, and S12). Our data suggests that disruption of PRKAG3 within dendrites increases GLUR1 levels (Figure 4). Activated AMPK is known to interrupt translation by either inhibiting mTOR kinase phosphorylation or the translation elongation factor 2 [90-95]. Consistent with this, upon knockdown of Upf2 we observed an increase in phosphorylated mTOR, which was rescued when PRKAG3 levels were modulated in these neurons (Figure S14). This modulation also restored total GLUR1 levels in UPF2-deficient dendrites. These data suggest that AMPK represses GLUR1 synthesis, and that NMD limits the amount of the AMPK γ subunit thereby serving as a balance in regulating total levels of the GLUR1 receptor. In addition, loss of UPF2 results in a higher GLUR1 internalization rate. ARC protein induces GLUR1 endocytosis, leading to a decrease in the surface levels of this receptor [21, 68]. Indeed, modulation of ARC levels in UPF2-deficient neurons rescues the GLUR1 internalization phenotype without altering total GLUR1 levels (Figure S12). Therefore, neither ARC modulation nor PRKAG3 modulation alone is capable of restoring the surface expression of GLUR1 upon disruption of UPF2. However, concomitant modulation of both ARC and PRKAG3 protein levels together results in a complete rescue of surface GLUR1 phenotype in UPF2-deficient dendrites (Figure 5). Thus, our data suggests that UPF2-dependent NMD regulates local synthesis and basal rates of GLUR1 internalization by degrading Prkag3 and Arc mRNAs, respectively.
Modulation of AMPA-receptor integrity plays a crucial role in synaptic strength. Strengthening of synapses by LTP or weakening of synaptic strength by LTD can be represented by the synaptic insertion of AMPA receptors [59]. Consistent with this, alterations in GLUR1 levels are linked to impairments in LTP as well as abnormalities in memory [53-55]. Our data explicates that hippocampal L-LTP, which is thought to underlie memory consolidation, is decreased in conditional Upf2 mutant mice. Activation of glutamate receptors facilitates LTP either by through the enlargement of pre-existing spines or promoting the formation of new spines. Intriguingly, AMPK has been suggested to have negative effects on LTP induction [96]. Therefore, a reduction in spine density is consistent with a decrease in L-LTP in conditional Upf2 mutants. This phenotype is accompanied by behavioral defects, including a lack of short-term spatial memory in the Y-maze, defective associative fear learning and memory, and attenuated spatial learning and memory on the Morris water maze. Each of these cognitive phenomenon requires activity-dependent synaptic strengthening and/or GLUR1-initiated signaling [53, 97, 98]. Reductions in GLUR1 expression were shown to cause spatial memory dissociations in mice [99]. Contextual conditioning requires not just increased levels of GLUR1, but specifically surface-localized GLUR1, for task performance [25]. NMD-dependent control of GLUR1 signaling is therefore likely to contribute to the plasticity and behavioral phenotypes Upf2 CKO mice exhibit. However, we cannot exclude the possibility that NMD regulates other receptors or intracellular signaling pathways that might also contribute to LTP, learning and memory. Induction of either mGluR- or NMDAR-LTD is thought to be associated with reduced spine density. In our electrophysiology experiments we only measured mGluR-based LTD due to our focus on GLUR1, which was not significantly altered in Upf2 cKO mice. However, we did observe a decrease in spine density, which is often associated with LTD. There are examples that spine density is decreased without an accompanying alteration in mGluR-LTD [100]. Nonetheless, we also cannot rule out alterations in other forms of LTD, such as NMDAR-derived LTD. Although NMDARs are more static than AMPA receptors in mature neurons, subunits of NMDARs are also locally translated in dendrites [101] suggesting that NMD and other dendritically-confined phenomenon may regulate NMDAR dynamics.
Local NMD as a regulator of the dendritic proteome.
We report that NMD can occur locally in dendrites. Arc mRNA was shown to be locally degraded in hippocampus [102]. However, the involvement of NMD in this, as well as more broadly hippocampal function, has not been demonstrated. Our data show that the NMD machinery is localized to dendrites, and the levels of the NMD targets Arc and Prkag3 are elevated upon selective inhibition of translation in these compartments (Figure 4c). Local inhibition of dendritic UPF3B synthesis results in reduced surface density of GLUR1, suggesting that the UPF3B-dependent branch of NMD locally influences GLUR1 levels within dendrites.
Our data demonstrate that local translation is regulated by mechanisms that control mRNA degradation in dendrites. NMD is likely to have more targets than Arc and Prkag3 in dendrites. In addition to its canonical targets, NMD may also degrade mRNAs that do not carry identifiable NMD-inducing features [61]. However, the mechanisms by which NMD recognizes its atypical targets remain unclear. This raises the possibility that NMD might be regulating various physiological mRNAs in dendrites that cannot be predicted based on sequence information. Therefore, we cannot exclude the possibility that atypical targets of NMD may also contribute to the NMD-mediated GLUR1 regulation at synapses as well as other synaptic events.
Conclusion.
Our experiments provide fundamental support for the idea that NMD modulates dendritic mRNA availability and therefore synaptic plasticity, learning, and memory. Through a number of locally-confined experiments in microfluidic devices and behavioral/LTP assays in neuron-restricted UPF2 conditional knockout mice, we define a specific role for the UPF2-dependent NMD pathway in dendrites and synaptic function (see schematic model in Figure S15). Therefore, our paper provides novel insights into basic mechanisms that regulate synaptic function that are also likely to play a role in NMD-implicated diseases such as autism and schizophrenia.
Methods
Mice and constructs
Hippocampal neuronal cultures were prepared from C57BL/6J mouse embryos (Charles River Laboratories). Upf2-shRNA 1 virus is on a piLenti-shRNA-GFP backbone carrying one shRNA against the Upf2 mRNA (AGGCGTATTCTGCACTCTAAAGGCGAGCT). Upf2-shRNA 2 virus is on a piLenti-shRNA-GFP backbone carrying four shRNAs against the Upf2 mRNA (TGAAAGACTATGTTATTTGTTGTATGATA). In experiments with Upf2 deletion in excitatory neurons, a Upf2 conditional knockout mouse line, which carries flanked loxP sites in the second exon of this gene, [1] was crossed with αCaMKII:CreERT2 line [2], and Upf2wt/wt;αCaMKII:CreERT2(CTRL) or Upf2fl/fl;αCaMKII:CreERT2 (CKO) adults were used. To induce Cre, mice were treated with 200 mg tamoxifen/kg body weight every other day for five days. To outline dendrites and spines, an AAV-CaMKIIa-eGFP virus (Addgene; 50469-AAV5) was injected into the adult hippocampus of CTRL and CKO mice. All animal experiments were completed with the approval of Weill Cornell Medical College ethical committees and per Research Animal Resource Center (RARC) guidelines.
Hippocampal neuronal cultures
For culturing hippocampal neurons, we followed a previously described protocol with some modifications [3]. We typically isolated hippocampal neurons from embryos at embryonic day 16 (E16) and plated 1×105 neurons per coverslip (diameter: 12 mm) pre-coated with poly D-lysine (PDL) in Neurobasal medium containing 2% B27, 1 mM sodium pyruvate, 2 mM Glutamax, 30% D-glucose and penicillin (100 units/ml) / streptomycin (100 μg/ml).
Compartmentalized culture in microfluidic devices.
Tripartite microfluidic device was designed and manufactured by the Jeon lab. Microfluidic chambers were prepared in house as previously described [4]. To isolate synaptic regions or to selectively treat the synaptic regions as well as post- or presynaptic cells with either pharmacological agents or sh/siRNAs, hippocampal neurons were cultured in tripartite microfluidic devices. The polymethyldimethylsiloxane (PDMS) devices were fabricated as previously described [4]. These devices have different channels separated by a physical barrier with embedded microgrooves (10 μm wide, 3 μm high). Unlike traditional microfluidic chambers with two channels [5], the tripartite PDMS chamber contains three channels (Figure 3). To access a high number of dendritic synapses, a ‘synaptic’ channel was placed 200 μm from the edge of one channel and 500 μm away from the other. Since the average length of dendrites extending into the microgrooves is less than 300 μm at 21 days in culture (DIV21), the closer “postsynaptic cell channel” allows a large number of dendrites to project from this channel to the new “synaptic” channel. On the other hand, the “presynaptic cell channel” that is located 500 μm away from the new channel ensures that only axons from this channel can extend into the synaptic channel. Fluidic isolation is established across the microgrooves, making it possible to isolate synaptic regions or to selectively treat the synaptic regions as well as post- or presynaptic cells. Reagents applied to synaptic channels were as follows: Cycloheximide (10 μM, Calbiochem), MG132 (10 μM, Sigma), and Leupeptin (10 μM, Sigma). siRNAs were applied to synaptic channels at different concentrations using 10% NeuroPORTER (Sigma) following manufacturer’s instructions.
For microfluidic cultures, we reversibly affixed the tripartite chambers to PDL-coated coverslips (Assistant; 50x24mm; two per coverslip) and then place them in 10 cm dishes. For culturing hippocampal neurons, we followed a previously described protocol with some modifications [3]. We typically isolated hippocampal neurons from embryos at embryonic day 16 and plate 1x105 neurons per coverslip (diameter: 12mm) pre-coated with poly D-lysine (PDL) in Neurobasal medium containing 2% B27, 1 mM sodium pyruvate, 2 mM Glutamax, 30% D-glucose and penicillin (100 units/mL) /streptomycin (100 μg/mL). We plated 8x104 neurons in cell-body channels and maintain these cultures in neuronal medium for up to 21 days. For immunostaining, we fixed neurons inside microfluidic chambers and continued with the staining procedure upon careful removal of chambers.
Immunohistochemistry
Hippocampal neuronal cultures were fixed in 4% paraformaldehyde (PFA)/PBS for 15 min, which was followed by 3 x 10 min washes in PBS. In most experiments, standard immunohistochemistry was performed as previously described [4]. Briefly, samples were incubated overnight in PBS containing 1% Triton X-100, 10% normal goat serum and the relevant dilution of primary antibodies. The following morning, samples were washed with PBS (3 x 10 min) to remove any residual primary antibody. Secondary antibodies were incubated in PBS containing 1% Triton X-100 and 10% normal goat serum for 2 hr. Samples were subsequently washed with PBS (3 x 20 min) and mounted (Vectashield, Vector Laboratories). Primary commercial antibodies included PRKAG3 (rabbit; Thermofisher, PA5-13797), ARC (rabbit; Synaptic Systems, 156002), MAP2 (mouse; Abcam, ab11267), SYNAPSIN 1 (SYN1) (rabbit; Abcam, ab64581), UPF3B (rabbit; Sigma, HPA001882), GFP (chick; Thermofisher, A10262), RFP (rabbit; Thermofisher, R10367), mTOR (mouse; Thermofisher, AHO1232), phospho-mTOR-Ser2448 (rabbit; Cell Signaling #2971), SQSTM1/p62 (mouse; ABCAM, ab56416), NEUN (rabbit; ABCAM, ab177487, GAP-43 (Rabbit; SANTA CRUZ, sc-10786). Two commercial GLUR1 antibodies were used: mouse GLUR1 antibody (Abcam, ab174785) for stainings with SYNAPSIN and rabbit GLUR1 antibody (Calbiochem, pc246) for the internalization assay. Anti-UPF1 and anti-UPF2 antibodies (both rabbit) were a gift from Jens Lykke-Andersen. Secondary antibodies were Alexa 488, 546, and 647 conjugated (Molecular Probes, 1:2000). Immunostaining for PRKAG3 was performed by using a peroxidase-based signal amplification system (Tyramid Signal Amplification, TSA, PerkinElmer) according to manufacturer’s instructions. TSA enables much higher dilution of primary antibodies than standard protocols. Higher dilution of primary antibodies reduced nonspecific interactions and improved the specificity of staining.
qRT-PCR, mRNA half-life and siRNA constructs
RNA was isolated from the synaptic channels of microfluidic devices by perfusion with TRIzol reagent (Invitrogen), with RNA being subsequently extracted from lysates following the manufacturer’s instructions. Purified RNA was incubated with DNase I (RNase-Free DNase Set, Qiagen) over a mini kit column (RNAeasy, Qiagen) to ensure DNA digestion. SuperScript III First-Strand Synthesis SuperMix (Invitrogen) was used to amplify cDNAs in a 20 μl reaction using 1 μg of RNA. Quantitative Real-Time PCR (qRT-PCR) reactions were performed as previously described [4] using the iQ SYBR Green Supermix (Bio-Rad) and an Eppendorf Mastercycler EP Realplex Thermocycler. For each reaction 10 ng cDNA was used. Relative expression levels were either normalized to Gapdh or Beta-Actin. To determine the estimated half-life of Arc and Prkag3 mRNAs, a final concentration of 10 μg/ml actinomycin D was added to control and UPF2-deficient neurons. The RNA was harvested at 0, 1 hr and 2 hr time points, and normalized against baseline measurements at 0 hr. The cDNAs were prepared and qRT-PCR was performed as described above.
Primers used for qRT-PCR:
Arc Primers:
(FW) 5’ GGGTGAGCTGAAGCCACAAAT 3’
(REV) 5’ GAGCTGAGCTCTGCTCTTCTT 3’
Arc Pre-mRNA Primers:
(FW) 5’ GGGTGAGCTGAAGCCACAAAT 3’
(REV) 5’ CTCCACCCTTGCAACTAATTT 3’
Prkag3 primers:
(FW) 5’ CAGGTCTACATGCACTTCATGC 3’
(REV) 5’ AAAGCTCTGCTTCTTGCTGTCC 3’
Prkag3 Pre-mRNA Primers:
(FW) 5’ TGGCCACCAGCTCAGAAAGA 3’
(REV) 5’ GTCAAGCTCTGCTCTGGAATT 3’
GluR1 primers:
(FW) 5’AGAGAAGAGGAGGAGAGCAG 3’
(REV) 5’ CTATCTGGATATTGTTGGGGA 3’
Gapdh primers:
(FW) 5’ ACATGGTCTACATGTTCC 3’
(REV) 5’ CAGATCCACAACGGAATAC 3’
Beta-Actin primers:
(FW) 5’ AGTGTGACGTTGACATCCGT 3’
(REV) 5’ TGCTAGGAGCCAGAGCAGTA 3’
Dicer-substrate siRNAs were acquired from Integrated DNA Technologies, and were predesigned using their proprietary algorithm, which integrates design principles from 21mer siRNA theory with updated 27mer criteria. This design process ensures siRNA target sites do not match known single nucleotide polymorphisms or exons that are the product of alternative splicing, with all sequences having been pre-screened using Smith-Waterman analysis to reduce off-target binding.
Arc siRNA Cocktail
Duplex 1:
5’ CUGAUGGCUAUGACUAUACCGUUAG 3’
3’ UCGACUACCGAUACUGAUAUGGCAAUC 5’
Duplex 2:
5’ CUACAUGGACUGAACAUCAAGAAGC 3’
3’ ACGAUGUACCUGACUUGUAGUUCUUCG 5’
Prkag3 siRNA
5’ GUCAAGAGAUGGACUUCUUAGAACA 3’
3’ GAGUCAAGAGATGGACUUCUUAGAACA 5’
Upf3b siRNA Cocktail
Duplex 1:
5’ AAAUUGAAGCCAAAAAUCGAGAATT 3’
3’ CCUUUAACUUCGGUUUUUAGCUCUUAA 5’
Duplex 2:
5’ GGGUCAAGAAUAUCAUGCUAUAGTA 3’
3’ UUCCCAGUUCUUAUAGUACGAUAUCAU 5’
Fluorescent in-situ hybridization (FISH) for Upf3b mRNA
For the detection of Upf3b mRNA, a mix of five non-overlapping antisense probes were used. Each oligonucleotide was 49 base pairs in length. FISH was visualized using the Tyramide In Situ System (Perkin Elmer) as previously described [4]. Briefly, samples were pretreated with hybridization buffer (50% formaldehyde, 5 x SSC, 0.1% tween, 100 ug/ml tRNA [Roche], 50 ug/ml heparin [Sigma]) for 2 hr at 65°C. Riboprobes were (100 ng/μl) were next heated to 97°C for 7 min and left to cool on ice. Hybridization was performed overnight at 65°C, and was followed by a series of washes at room temperature in hybridization buffer (no tRNA or heparin, 2 x 30 min), hybridization buffer/PBT (0.1% tween in PBS, 2 x 30 min), and lastly just PBT (4 x 30 min). Samples were next blocked for 1 hr (Roche Blocking Solution) and incubated with anti-DIG-POD (Roche) 1:800 in PBT overnight at 4°C. The following morning, samples were washed in PBT (3 x 20 min). Detection was visualized with tyramide-488 1:50 in supplied amplification buffer from the TSA-Plus in situ kit (Perkin Elmer).
Oligonucleotides used for antisense Upf3b probes
1- GGAAGAAAAAGATCATAGGCCTAAGGAGAAACGAGTGACCCTGTTTACG
2- GTCAAGAATATCATGCTATAGTAGAATTTGCACCATTTCAAAAAGCTGC
3- AAGAAAAGAGAGAAGAAAGGAGGAGACGGGAAATAGAGAGGAAAAGGCA
4- AAGAAAGAGCCAGTGGGCACAGTTATACTCTGCCCAGGCGTTCTGATGT
5- GGCACTTCGAGATAAAGGAAAGAAGAGTGAGAATACAGAATCAATATGC
Oligonucleotides used for sense Upf3b probes
1- ATAGGAGTATACATACGGTCTCGTATATAGTTGAAATTTTTGGTTCTTC
2- GCTTCTAATGGAGGGTGAAACTGGTTCCTTGTCGAAGTCCTTGTAGAAG
3- CCTCTCAATACGGTGTCTGTTACTCTTTTACTGTAGATGAGGTCTCTGT
4- CTTAAGGTCTTTCCCTCTTTTATTCCCTACTTGGTTTCTAATTCAACGA
5- CTAATGTCTCTGTCCCTGGCCCTGATACTCTCTCTAGTCCTCGCGTACT
Oligonucleotides used for antisense Gapdh probes
1- GGCTGCCATTTGCAGTGGCAAAGTGGAGATTGTTGCCATCAACGACCCC
2- TTGTCATCAACGGGAAGCCCATCACCATCTTCCAGGAGCGAGACCCCAC
3- GGTGGAGCCAAAAGGGTCATCATCTCCGCCCCTTCTGCCGATGCCCCCA
4- ACCCAGAAGACTGTGGATGGCCCCTCTGGAAAGCTGTGGCGTGATGGCC
5- AAGTATGATGACATCAAGAAGGTGGTGAAGCAGGCATCTGAGGGCCCAC
Oligonucleotides used for sense Gapdh probes
1- CAGATGTACAAGGTCATACTGAGGTGAGTGCCGTTTAAGTTGCCGTGTC
2- GCCACGACTCATACAGCACCTCAGATGACCACAGAAGTGGTGGTACCTC
3- CCGGTTCCAGTAGGTACTGTTGAAACCGTAACACCTTCCCGAGTACTGG
4- GTACCGGAAGGCACAAGGATGGGGGTTACACAGGCAGCACCTAGACTGC
5- GTAACGAGAGTTACTGTTGAAACAGTTCGAGTAAAGGACCATACTGTTA
GLUR1 internalization assay
GLUR1 internalization experiments were carried-out and quantified as previously described [6, 7]. Briefly, we incubated live DIV21 hippocampal cultures infected with control or Upf2 shRNAs at 37°C for 5 min with GLUR1 antibody (Calbiochem, PC246) to allow labeling of surface GLUR1. After washing, we incubated neurons at 37°C for 10 min to allow for basal internalization. We then fixed neurons with 4% PFA in PBS and blocked in a detergent-free blocking solution for 1 hr. Following blocking, we incubated neurons with AlexaFluor546 secondary antibody for 1 hr to label the surface population. To label the pre-labeled internalized fraction, we post-fixed neurons with 100% methanol at −20°C for 1 min and stained with Alexa-647 secondary antibody. We calculated the percent internalization by dividing the integrated far-red intensity (internalized GLUR1) by the total (red + far red) intensity between control and UPF2-deficient neurons.
Click chemistry and SDS-page
To measure the dendritic translation of GluR1, we used a click chemistry based approach and labeled locally synthesized GLUR1 at synaptic regions. At DIV21, we exchanged the growth media in the synaptic channels to methionine-free medium containing the methionine-analog azidohomoalanine (AHA; 25 μM) for 6 hr. AHA is incorporated into all newly synthesized proteins. After harvesting the material from the synaptic channels in RIPA, we incubated the lysate with DBCO-biotin [8] (30 μM; Click Chemistry Tools LLC) for 1 hr at ambient temperature. DBCO reacts with AHA [9], functionalizing newly synthesized proteins with biotin. We purified biotin-containing proteins with Dynabeads (150 μl per reaction; Dynabeads M-280 Streptavidin-Thermo Fisher Scientific). Dynabeads were separated over a magnetic rack and subsequently washed 2 times in PBS containing 0.1% Bovine Serum Albumin (BSA). Proteins were dissociated and denatured from beads by boiling Dynabeads for 5 min in 0.1% SDS. Lysates were subsequently analyzed for protein concentration via BCA Protein Assay (Thermofisher). Proteins were separated via SDS-PAGE, as previously described [10]. Briefly, protein aliquots were mixed with an equal volume of loading buffer, denatured at 95°C and loaded onto 8% gels before being subject to SDS-PAGE. Proteins were subsequently transferred to nitrocellulose membrane, incubated in 5% BSA diluted in TBST for 1 hr and then incubated with primary anti-C-terminus GLUR1 antibody overnight (Abcam, ab31232). Membranes were washed in TBST (3 x 5 min), incubated for 1 hr in IRDye 800CW goat anti-rabbit secondary (Licor) diluted in 5% low-fat milk diluted in TBST, and bands resolved on a Licor Odyssey CLx imager.
RNA Co-immunoprecipitation and RT-PCR
DIV7 hippocampal neurons were harvested in RNase-free PBS and quickly frozen in liquid nitrogen. The frozen cells were resuspended in 500 μl of B70 (70 mM KAc, 1 mM NaF, 5 mM MgOAc, 0.1% Triton X-100, 10% glycerol, 50 mM HEPES-KOH, pH 7.4, RNase-free) supplemented with fresh protease and phosphatase inhibitors and homogenized using a motor pestle. After the cell debris was removed by centrifugation, the supernatant was used for RNA co-immunoprecipitation following a published protocol (4). Briefly, the supernatant was divided into two equal samples, which were precleared on the Dynabeads Protein A (Invitrogen). Ten microliters of aliquot was transferred from each sample and saved as input samples. UPF1 antibody and IgG control were added to the remaining precleared supernatant samples, respectively, and incubated for 1 hr on a rotating wheel. The extract-antibody mixes were then added to the Dynabeads Protein A, which was preblocked with B70 containing 10 mg/ml BSA, 0.1 mg/ml yeast total RNA, and 0.1 mg/ml heparin, and incubated for 1 hr on a wheel. After extensive washing with B70, the immune-complexes on beads were directly used to synthesize the first strand cDNA by the SuperScript III CellsDirect cDNA Synthesis System (Invitrogen). Then 2 μl each of the first strand cDNA was used for a 25 μl PCR reaction. The aforementioned primers for Arc, Prkag3 and Gapdh were used for RT-PCR. The PCR conditions were 95°C, 30 s; 57°C, 30 s; 72°C, 30 s; 35 cycles.
Electrophysiology
The electrophysiology experiments were performed as previously described (114). All experiments were performed on transvers hippocampal slices (400 μm) from 3-month old (LTP studies) or 6-week-old (LTD studies) Upf2fl/fl;αCaMKII:CreERT2 mice and their control littermates (Upf2wt/wt;αCaMKII:CreERT2) following induction of Cre expression by tamoxifen (Sigma) at 2-months (for LTP) and 1-month (for LTD) of age. Given that Tamoxifen is fat soluble, we prepared our tamoxifen solution by dissolving the relevant quantity of drug into sunflower oil with 1:10 ETOH added. Tamoxifen solutions were incubated at 37°C with rocking for 6-8 hr. Tamoxifen was administered via oral gavage at 200 mg tamoxifen/kg body weight every other day for five total doses. This tamoxifen regimen led to successful ablation of UPF2 protein in the adult brain (Figure S1a). Slice preparation and aCSF composition were performed as described previously [11, 12]. Briefly, slices were isolated and transferred to recording chambers (preheated to 32°C), where they were superfused with oxygenated aCSF. We recorded field excitatory postsynaptic potentials (fEPSPs) from the stratum radiatum of CA1 using microelectrodes filled with artificial cerebrospinal fluid (ACSF) (resistance 1-4 MΩ). A bipolar Teflon-coated platinum electrode was placed in the stratum radiatum of CA3 to activate Schaffer collateral/commissural afferents at 0.05 Hz. Slices recovered in the recording chamber at least 1 hr before recordings began. In all experiments, basal field excitatory postsynaptic potentials (fEPSPs) were stable for at least 20 min before the start of each experiment. LTP was induced with either one (E-LTP) or three 1 sec 100-Hz high-frequency stimulation (HFS) trains, with an intertrain interval of 60 sec. To induce mGluR-LTD, slices were incubated with DHPG (100 μM) for 10 min. After induction of either E-LTP or mGluR-LTD, we collected fEPSPs for an additional 60 min (180 min for L-LTP). Slope values were compared from the conditional Upf2 mutant mice and their control littermates.
Stereotaxic injection of virus and hippocampal spine density analysis
To measure hippocampal spine density, the spine density of CA1 hippocampal neurons in control (CTRL: Upf2wt/wt;αCaMKII:CreERT2) and Conditional Knockout (CKO: Upf2fl/fl;αCaMKII:CreERT2) mice was examined post-labelling with GFP-expressing virus. To target the dorsal CA1 field, the stereotaxic coordinates AP: −2, ML: 1.6, DV: 1.5-.25 were used. 1 μl of AAV-CaMKIIa-eGFP (Addgene, #50469-AAV5) virus was injected bilaterally using a 10 μl nanofil syringe (World Precision Instruments) fitted with a 33-gauge beveled needle. Post-surgery, wounds were closed with tissue adhesive (Vetbond, 3M). For in vivo rescue experiments, equal volumes (50:50) of mCMV>Arc-shRNA:TurboRFP (Dharmacon, #V3SM7598-08EG11838) and mCMV>Prkag3-shRNA:TurboRFP (Dharmacon, #V3SM7598-08EG241113) lentivirus were pooled into a master-mix before injection into the dorsal CA1 field as described above. For all spine experiments, mice were transcardially perfused 7 days later with 4% PFA (without PBS flush), brains removed, and sectioned at 200 μm. Hippocampal dendrites were imaged on an Olympus FluoView-FV1000 confocal microscope. To identify spines, we measured maximal spine head width (WH), neck width (WN), and length (L) of spines using ImageJ. Spines were identified as Stubby (L ≤ 1 μm), Mushroom (1 < L ≤ 3 μm; WH ≥ 2 × WN), Long Thin (1 < L ≤ 3 μm; WH < 2 × WN), and Filopodia (3 < L ≤ 5 μm).
Arc-shRNA Lentiviral Sequence:
TATTCTTCAGAGCCACCCA
Prkag3-shRNA Lentiviral Sequence:
AGGCGCTGAGGGCATCGAT
Behavioral assays
To study the role of NMD in cognitive function, we induced Cre in 2-month old Upf2 CKO mice as well as in their control littermates by tamoxifen. This experimental design ensured that phenotypes are not caused by an acute effect of tamoxifen, or a non-specific effect of Cre at cryptic LoxP sites. A 2-week washout period followed tamoxifen dosing before behavior began, at approximately 3-months of age. The Y-maze [13, 14], fear conditioning [14, 15], Morris water maze [16, 17], marble burying [18], grooming (12), locomotor hyperactivity/open field [19], elevated-plus maze [19, 20] and three-chamber social interaction assay [21] were completed and analyzed as previously described, except where outlined below. Briefly, the Y-Maze comprised exposing mice to 2 of 3 arms of the maze for 10 min as a training trial. The third, blocked, arm of the maze was designated as the novel arm, and was pseudorandomized between mice. Following a 1 hr delay, mice were returned to the maze but were free to explore all 3 arms for 5 min. The time spent exploring the novel arm, relative to the other familiar arms, was quantified as our index of short-term spatial memory. To examine associative memory, we utilized a simple 2-day fear conditioned memory protocol. On day 1, mice were exposed to a 6 min conditioning trial that involved 3 tone-shock pairings. A 30-sec tone duration and 30 sec ISI was utilized, while all shock stimuli comprised a 0.7mA scrambled foot-shock that co-terminated with tone presentation. Learning was evaluated by plotting % freezing to each successive tone presentation as a time-course. On day 2, mice were returned to their conditioning context for 6 min, with % freezing over the test session being quantified as contextual fear memory. All experiments were completed in Colbourn fear chambers. The Morris water maze was used to test more sophisticated learning and longer-term spatial memory. Learning was measured by training mice to find a hidden platform (four trials per day, 75 sec/trial) over 4 consecutive days of testing. Trials on each day of testing were averaged, and plotted as a time-course, with gradually decreasing escape latencies being used as our index of spatial learning behavior. To assess spatial memory, we conducted a probe trial 24 hr after the last day of training on day 5. In this trial, the hidden escape platform was removed from the pool. Latency to enter the hidden platform zone, and time spent exploring the target quadrant, were quantified as measurements of spatial memory. Reversal learning was not evaluated as Upf2 CKO mice did not display adequate learning behavior, thus making it impossible to distinguish a general learning deficit from a reversal learning deficit even with addition of extra training trials. Assays were performed in order of least stressful (locomotor hyperactivity/open field) to most stressful (Morris water maze). Mice were bred and tested across 4-5 small cohorts, ensuring that all phenotypes were independent of idiosyncratic effects or nuisance variability that may contaminate any single day of testing. Where experiments were not scored using automated systems (e.g. marble burying), two experimenters scored task performance and in these cases at least one scorer was blinded.
Data analysis
Where total protein levels were measured via immunostaining, Image J was used to measure total fluorescence. In these analyses, a defined area containing dendrites were identified and the integrated fluorescent density was measured in each region. Background fluorescence was measured by selecting a region adjacent to dendrites, and was subtracted from positive fluorescent signal-measurements to derive corrected total fluorescence. Depending on the test, behavior was analyzed using a between-groups or mixed-model ANOVA with genotype and sex added as main effects. For tests that involved multiple observations, data were corrected using Tukey’s method. Consistent with prior research [10, 14, 22], as no significant interaction involving the main effect of sex was observed on any given behavioral assay – explicating no modulatory effect of sex on genotype-dependent effects – data from mice were pooled to increase power. No specific exclusion criteria were applied to the datasets, excepting the removal of outliers (defined as values falling outside of ± 2 S.D.). Significance was set at p=0.05 per Fisher’s tables, and datasets did not exhibit gross features requiring transformation to ensure test assumptions. Sampling was conducted to levels adequate to detect large effect sizes depending upon observed power, or to levels consistent with prior technical and/or experimental publications that predetermined these parameters. Both animal and cellular experiments were pseudorandomly selected for treatment allocations. Data analysis was completed using the Graphpad Prism software package.
Supplementary Material
Acknowledgements
We thank J. Lykke-Andersen for generously providing the anti-UPF1 and anti-UPF2 antibodies, M.E. Ross for helpful comments and suggestions and M. Toth for helpful suggestions regarding behavioral assays. This work was supported by a NHMRC CJ Martin Biomedical Fellowship awarded to M.N., KoreaNRF-2015R1A2A1A09005662 to N.L.J., NIH grants NS034007 and NS047384 to E.K, and NIH R01 MH114888 and Leon Levy Foundation Grants to D.C.
Footnotes
Conflict of Interests
The authors have no known conflict of interests to report.
References
- 1.Doma MK and Parker R, RNA quality control in eukaryotes. Cell, 2007. 131(4): p. 660–668. [DOI] [PubMed] [Google Scholar]
- 2.Hodgkin J, et al. , A new kind of informational suppression in the nematode caenorhabditis elegans. Genetics, 1989. 123(2): p. 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leeds P, et al. , Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol, 1992. 12(5): p. 2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lejeune F and Maquat LE, Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol, 2005. 17(3): p. 309–15. [DOI] [PubMed] [Google Scholar]
- 5.Li S and Wilkinson MF, Nonsense surveillance in lymphocytes? Immunity, 1998. 8(2): p. 135–41. [DOI] [PubMed] [Google Scholar]
- 6.Maquat LE, et al. , Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell, 1981. 27(3 Pt 2): p. 543–53. [DOI] [PubMed] [Google Scholar]
- 7.Carter MS, et al. , A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem, 1995. 270(48): p. 28995–9003. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, et al. , At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol, 1998. 18(9): p. 5272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karam R, et al. , Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta, 2013. 1829(6–7): p. 624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell JT, et al. , Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet, 2004. 36(10): p. 1073–8. [DOI] [PubMed] [Google Scholar]
- 11.Colak D, et al. , Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell, 2013. 153(6): p. 1252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rebbapragada I and Lykke-Andersen J, Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol, 2009. 21(3): p. 394–402. [DOI] [PubMed] [Google Scholar]
- 13.Giorgi C, et al. , The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell, 2007. 130(1): p. 179–91. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto T, et al. , Arc interacts with microtubules/microtubule-associated protein 2 and attenuates microtubule-associated protein 2 immunoreactivity in the dendrites. J Neurosci Res, 2004. 76(1): p. 51–63. [DOI] [PubMed] [Google Scholar]
- 15.Husi H, et al. , Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci, 2000. 3(7): p. 661–9. [DOI] [PubMed] [Google Scholar]
- 16.Moga DE, et al. , Activity-regulated cytoskeletal-associatedprotein is localized to recently activated excitatory synapses. Neuroscience, 2004. 125(1): p. 7–11. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez JJ, et al. , Long-term potentiation in the rat dentate gyrus is associated with enhanced Arc/Arg3.1 protein expression in spines, dendrites and glia. Eur J Neurosci, 2005. 21(9): p. 2384–96. [DOI] [PubMed] [Google Scholar]
- 18.Guzowski JF, et al. , Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci, 2000. 20(11): p. 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plath N, et al. , Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron, 2006. 52(3): p. 437–44. [DOI] [PubMed] [Google Scholar]
- 20.Smith-Hicks C, et al. , SRF binding to SRE 6.9 in the Arc promoter is essential for LTD in cultured Purkinje cells. Nat Neurosci, 2010. 13(9): p. 1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waung MW, et al. , Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron, 2008. 59(1): p. 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cajigas IJ, et al. , The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron, 2012. 74(3): p. 453–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown TC, et al. , NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron, 2005. 45(1): p. 81–94. [DOI] [PubMed] [Google Scholar]
- 24.Fu AK, et al. , APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci, 2011. 14(2): p. 181–9. [DOI] [PubMed] [Google Scholar]
- 25.Han Y, et al. , AMPK Signaling in the Dorsal Hippocampus Negatively Regulates Contextual Fear Memory Formation. Neuropsychopharmacology, 2016. 41(7): p. 1849–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd JD and Bear MF, New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci, 2011. 14(3): p. 279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Addington AM, et al. , A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry, 2011. 16(3): p. 238–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laumonnier F, et al. , Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry, 2010. 15(7): p. 767–76. [DOI] [PubMed] [Google Scholar]
- 29.Lynch SA, et al. , Broadening the phenotype associated with mutations in UPF3B: two further cases with renal dysplasia and variable developmental delay. Eur J Med Genet, 2012. 55(8-9): p. 476–9. [DOI] [PubMed] [Google Scholar]
- 30.Tarpey PS, et al. , Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet, 2007. 39(9): p. 1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen LS, et al. , Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet, 2013. 22(9): p. 1816–25. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, et al. , Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin Genet, 2013. 83(6): p. 560–4. [DOI] [PubMed] [Google Scholar]
- 33.Long AA, et al. , The nonsense-mediated decay pathway maintains synapse architecture and synaptic vesicle cycle efficacy. J Cell Sci, 2010. 123(19): p. 3303–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mooney CM, et al. , RNA sequencing of synaptic and cytoplasmic Upf1-bound transcripts supports contribution of nonsense-mediated decay to epileptogenesis. Scientific Reports, 2017. 7: p. 41517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L, et al. , A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol Psychiatry, 2018. 23(8): p. 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karam R and Wilkinson M, A conserved microRNA/NMD regulatory circuit controls gene expression. RNA Biol, 2012. 9(1): p. 22–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malenka RC and Bear MF, LTP and LTD: an embarrassment of riches. Neuron, 2004. 44(1): p. 5–21. [DOI] [PubMed] [Google Scholar]
- 38.Bhakar AL, Dolen G, and Bear MF, The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci, 2012. 35: p. 417–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crabtree GW and Gogos JA, Synaptic plasticity, neural circuits, and the emerging role of altered short-term information processing in schizophrenia. Front Synaptic Neurosci, 2014. 6: p. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Messaoudi E, et al. , Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci, 2007. 27(39): p. 10445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bassell GJ and Kelic S, Binding proteins for mRNA localization and local translation, and their dysfunction in genetic neurological disease. Curr Opin Neurobiol, 2004. 14(5): p. 574–81. [DOI] [PubMed] [Google Scholar]
- 42.Buffington SA, Huang W, and Costa-Mattioli M, Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci, 2014. 37: p. 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darnell JC and Klann E, The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci, 2013. 16(11): p. 1530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gkogkas CG, et al. , Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature, 2013. 493(7432): p. 371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelleher RJ 3rd and Bear MF, The autistic neuron: troubled translation? Cell, 2008. 135(3): p. 401–6. [DOI] [PubMed] [Google Scholar]
- 46.Liu-Yesucevitz L, et al. , Local RNA translation at the synapse and in disease. J Neurosci, 2011. 31(45): p. 16086–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santini E, et al. , Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature, 2013. 493(7432): p. 411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber KM, et al. , Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J Neurosci, 2015. 35(41): p. 13836–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidorov MS, Auerbach BD, and Bear MF, Fragile X mental retardation protein and synaptic plasticity. Mol Brain, 2013. 6: p. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weischenfeldt J, et al. , NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev, 2008. 22(10): p. 1381–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng S, et al. , PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci, 2012. 15(3): p. 381–8, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madisen L, et al. , A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci, 2010. 13(1): p. 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kessels HW and Malinow R, Synaptic AMPA receptor plasticity and behavior. Neuron, 2009. 61(3): p. 340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korb E and Finkbeiner S, Arc in synaptic plasticity: from gene to behavior. Trends Neurosci, 2011. 34(11): p. 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Penzes P, et al. , Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci, 2011. 14(3): p. 285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang G, et al. , Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron, 2014. 83(5): p. 1131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Hir H, Sauliere J, and Wang Z, The exon junction complex as a node of post-transcriptional networks. Nat Rev Mol Cell Biol, 2016. 17(1): p. 41–54. [DOI] [PubMed] [Google Scholar]
- 58.Ashton-Beaucage D, et al. , The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell, 2010. 143(2): p. 251–262. [DOI] [PubMed] [Google Scholar]
- 59.Fleming JJ and England PM, AMPA receptors and synaptic plasticity: a chemist's perspective. Nat Chem Biol, 2010. 6(2): p. 89–97. [DOI] [PubMed] [Google Scholar]
- 60.Anggono V and Huganir RL, Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol, 2012. 22(3): p. 461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He F and Jacobson A, Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu Rev Genet, 2015. 49: p. 339–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korb E, et al. , Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nat Neurosci, 2013. 16(7): p. 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soulé J, et al. , Balancing Arc Synthesis, mRNA Decay, and Proteasomal Degradation MAXIMAL PROTEIN EXPRESSION TRIGGERED BY RAPID EYE MOVEMENT SLEEP-LIKE BURSTS OF MUSCARINIC CHOLINERGIC RECEPTOR STIMULATION. J Biol Chem, 2012. 287(26): p. 22354–22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farris S, et al. , Selective localization of arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J Neurosci, 2014. 34(13): p. 4481–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ninomiya K, Ohno M, and Kataoka N, Dendritic transport element of human arc mRNA confers RNA degradation activity in a translation-dependent manner. Genes Cells, 2016. 21(11): p. 1263–1269. [DOI] [PubMed] [Google Scholar]
- 66.Paolantoni C, et al. , Arc 3’UTR splicing leads to dual and antagonistic effects in fine-tuning Arc expression upon BDNF signaling. Front Mol Neurosci, 2018. 11: p. 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steward O, et al. , Delayed degradation and impaired dendritic delivery of intron-lacking EGFP-Arc/Arg3. 1 mRNA in EGFP-Arc transgenic mice. Front Mol Neurosci, 2018. 10: p. 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chowdhury S, et al. , Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron, 2006. 52(3): p. 445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aoto J, et al. , Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell, 2013. 154(1): p. 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rial Verde EM, et al. , Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron, 2006. 52(3): p. 461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ju W, et al. , Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci, 2004. 7(3): p. 244–53. [DOI] [PubMed] [Google Scholar]
- 72.Lin A, et al. , Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem, 2011. 119(1): p. 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwarz LA, Hall BJ, and Patrick GN, Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci, 2010. 30(49): p. 16718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taylor AM, et al. , A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods, 2005. 2(8): p. 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ehlers MD, Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron, 2000. 28(2): p. 511–25. [DOI] [PubMed] [Google Scholar]
- 76.Patrick GN, et al. , Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol, 2003. 13(23): p. 2073–81. [DOI] [PubMed] [Google Scholar]
- 77.Widagdo J, et al. , Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang D, et al. , Na,K-ATPase activity regulates AMPA receptor turnover through proteasome-mediated proteolysis. J Neurosci, 2009. 29(14): p. 4498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dieterich DC, et al. , Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl Acad Sci U S A, 2006. 103(25): p. 9482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ishigaki Y, et al. , Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell, 2001. 106(5): p. 607–17. [DOI] [PubMed] [Google Scholar]
- 81.Singh G, et al. , Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol Cell, 2007. 27(5): p. 780–92. [DOI] [PubMed] [Google Scholar]
- 82.Chamieh H, et al. , NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol, 2008. 15(1): p. 85–93. [DOI] [PubMed] [Google Scholar]
- 83.Huang L, et al. , RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell, 201143(6): p. 950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deglincerti A and Jaffrey SR, Insights into the roles of local translation from the axonal transcriptome. Open Biol, 2012. 2(6): p. 120079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gumy LF, et al. , Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA, 2011. 17(1): p. 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor AM, et al. , Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci, 2009. 29(15): p. 4697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jolly LA, et al. , The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet, 2013. 22(23): p. 4673–4687. [DOI] [PubMed] [Google Scholar]
- 88.Chan WK, et al. , An alternative branch of the nonsense-mediated decay pathway. EMBO J, 2007. 26(7): p. 1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karam R, et al. , The unfolded protein response is shaped by the NMD pathway. EMBO Rep, 2015: p. e201439696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gwinn DM, et al. , AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell, 2008. 30(2): p. 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolster DR, et al. , AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem, 2002. 277(27): p. 23977–23980. [DOI] [PubMed] [Google Scholar]
- 92.Inoki K, Zhu T, and Guan K-L, TSC2 mediates cellular energy response to control cell growth and survival. Cell, 2003. 115(5): p. 577–590. [DOI] [PubMed] [Google Scholar]
- 93.Horman S, et al. , Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol, 2002. 12(16): p. 1419–1423. [DOI] [PubMed] [Google Scholar]
- 94.Kimura N, et al. , A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells, 2003. 8(1): p. 65–79. [DOI] [PubMed] [Google Scholar]
- 95.Sarbassov DD, Ali SM, and Sabatini DM, Growing roles for the mTOR pathway. Curr Opin Cell Biol, 2005. 17(6): p. 596–603. [DOI] [PubMed] [Google Scholar]
- 96.Ma T, et al. , Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. J Neurosci, 2014. 34(36): p. 12230–12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsuo N, Reijmers L, and Mayford M, Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science, 2008. 319(5866): p. 1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Whitlock JR, et al. , Learning induces long-term potentiation in the hippocampus. Science, 2006. 313(5790): p. 1093–1097. [DOI] [PubMed] [Google Scholar]
- 99.Reisel D, et al. , Spatial memory dissociations in mice lacking GluR1. Nat Neurosci, 2002. 5(9): p. 868–73. [DOI] [PubMed] [Google Scholar]
- 100.Moretti P, et al. , Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci, 2006. 26(1): p. 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swanger SA, et al. , Dendritic GluN2A synthesis mediates activity-induced NMDA receptor insertion. J Neurosci, 2013. 33(20): p. 8898–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Farris S, et al. , Selective localization of arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J Neurosci, 2014. 34(13): p. 4481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Methods
- 1.Weischenfeldt J, et al. , NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev, 2008. 22(10): p. 1381–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madisen L, et al. , A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci, 2010. 13(1): p. 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y, et al. , Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature, 2006. 442(7104): p. 814–7. [DOI] [PubMed] [Google Scholar]
- 4.Colak D, et al. , Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell, 2013. 153(6): p. 1252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor AM, et al. , A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods, 2005. 2(8): p. 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoto J, et al. , Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell, 2013. 154(1): p. 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin JW, et al. , Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci, 2000. 3: p. 1282–1290. [DOI] [PubMed] [Google Scholar]
- 8.Ning X, et al. , Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew Chem Int Ed Engl, 2008. 47(12): p. 2253–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agard NJ, Prescher JA, and Bertozzi CR, A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc, 2004. 126(46): p. 15046–7. [DOI] [PubMed] [Google Scholar]
- 10.Notaras M, et al. , The BDNF Val66Met polymorphism regulates glucocorticoid-induced corticohippocampal remodeling and behavioral despair. Transl Psychiatry, 2017: p. e1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trinh MA, et al. , The eIF2a kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn Mem, 2014. 21: p. 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma A, et al. , Dysregulation of mTOR signaling in fragile x syndrome. J Neurosci, 2010. 30(2): p. 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarnyai Z, et al. , Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci U S A, 2000. 97(26): p. 14731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notaras M, et al. , BDNF Val66Met genotype determines hippocampus-dependent behavior via sensitivity to glucocorticoid signaling. Mol Psychiatry, 2016. 21(6): p. 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen ZY, et al. , Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science, 2006. 314(5796): p. 140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumont M, et al. , Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J, 2009. 23(8): p. 2459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santini E, et al. , Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature, 2013. 493(7432): p. 411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angoa-Perez M, et al. , Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J Vis Exp, 2013. 82: p. 50978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell E, et al. , Behavioural traits propagate across generations via segregated iterative-somatic and gametic epigentic mechanisms. Nat Commun, 2016. 7: p. 11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walf AA and Frye CA, The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protocol, 2007. 2: p. 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moy SS, et al. , Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav, 2004. 3(5): p. 287–302. [DOI] [PubMed] [Google Scholar]
- 22.Notaras M, et al. , BDNF Val66Met genotype interacts with a history of simulated stress exposure to regulate sensorimotor gating and startle reactivity. Scz Bull, 2017. 43(3): p. 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.