

Abstract
We investigate permeation of three blocked dipeptides with different side chain polarity across a phospholipid membrane and their behavior at the water–membrane interface by way of molecular dynamics simulations. Hydrophilic serine-serine dipeptide is found to desorb from the interface to aqueous phase, whereas hydrophobic phenylalanine–leucine and amphiphilic serine-leucine tend to accumulate at the interface with a free energy minimum of –3 kcal/mol. All three dipeptides exhibit free energy barriers to permeation across the membrane located at the center of the bilayer. The height of the barrier is strongly sequence dependent and increases with the dipeptide polarity. It is equal to 3.5, 6.4, and 10.0 kcal/mol for phenylalanine-leucine, serine-leucine, and serine-serine, respectively. The corresponding permeability coefficients are equal to 4.6 × 10−3, 4.5 × 10−5, and 8.7 × 10−8 cm/s. The apparent insensitivity of membrane permeability to hydrophobicity of dipeptides, found in some experiments, is attributed to neglecting corrections for unstirred water layers near membrane surface, which are significant for hydrophobic species. Different hydrophobicity of the dipeptides also influences their conformations and orientations, both at the interface and inside the membrane. In particular, penetration of hydrophilic serine-serine dipeptide causes the formation of water-filled defects in the bilayer. These results are relevant to the delivery of peptide-based therapeutic agents.
Graphical Abstract
INTRODUCTION
Peptides have diverse biological functions, acting as hormones, neurotransmitters, growth factors, ion channel ligands, cytokines, toxins, and antimicrobials.1–4 This makes them potentially powerful therapeutic agents in treating pain, neurological, metabolic and cardiovascular diseases, diabetes, and cancer.1,5–9 This is because they combine a number of features desired in drugs: selectivity, efficiency, and safety.10 Often, however, naturally occurring peptides cannot be readily used as therapeutics because they are degraded by proteases and poorly permeate cell membranes.2,5,11,12 For example, bioavailability of neurological peptide drugs and opioids in the brain10,13 is limited due to the presence of the blood–brain barrier.14 A variety of strategies are used to improve metabolic durability of drugs and their ability to penetrate cellular membranes, which include derivatization, incorporation of noncanonical amino acids, and synthesis of peptide analogues. Another highly promising strategy is the application of liposomes as vehicles for drug delivery. Liposomal delivery has been shown to stabilize therapeutic compounds, improve their uptake in cells and tissues, reduce toxicity, and improve targeting to the desired sites.12,15,16
Whether the goal is enhancing the passive diffusion of peptide drugs to increase their uptake at the target site or retaining them inside liposomes for improved bioavailability, membrane permeability to these drugs is of critical importance. A common predictor of permeability used in pharmacokinetics is lipophilicity, most frequently measured as the water–octanol partition coefficient.17 Although this measure has been relatively successful and has led to predictive models for peptides18,19 and other drugs,20–22 it is recognized that their accuracy is highly dependent on the peptides studied.17 For this reason, molecular-level understanding of permeation properties of peptides and their derivatives, and in particular their dependence on sequence, is of considerable relevance to drug delivery.
Dipeptides and their permeation through membranes are of interest in pharmacology and related fields not only as the smallest units for sequence dependence studies but also in their own rights.23 For example, β-Ala-Tyr24 is an antimicrobial agent, L-glutamine-L-tryptophan is an anticancer drug,25 histidine dipeptide is used for cell stress control,26 and Tyr-Ala dipeptide is used as antiaging medication.27 The neuropeptide N-acetylaspartylglutamate (NAAG) is the third most common neurotransmitter in the mammalian nervous system behind glutamate and GABA.28 Peptides that are analogues of NAAG are considered potential, effective inhibitors of NAAG peptidase. These inhibitors have been shown to have neuroprotective effects in the stroke model and are considered in treatment of peripheral neuropathies resulting from trauma, diabetes, or chemotherapy.28–30
Dipeptides also play an increasingly important role in optimizing cell culture media for manufacturing biopharmaceuticals. Since the 20 natural amino acids differ widely in their aqueous solubility and lipophilicity, liquid media often provide imbalanced amino acid feed to the cells leading to retarded cell growth and decreased efficiency of extracting pharmaceuticals. Recently, it has been shown that properly chosen dipeptides can have markedly improved solubility in both aqueous media and membranes and can reach their targets in vivo faster. Then, they are hydrolyzed to their parent amino acids at the target.31 This is, for example, the case for tyrosine or cysteine in dipeptides with glycine and alanine.32 In general, the knowledge of permeation properties of dipeptides can lead to substantial improvements in efficiency of producing biopharmaceuticals from cell cultures.
A different perspective on the significance of dipeptides follows from a study of Adamala and Szostak.33 They demonstrated that hydrophobic dipeptides, made inside fatty acid vesicles from activated blocked amino acids in a reaction of peptide bond formation catalyzed by another dipeptide, promote vesicle growth through intake of fatty acid micelles present in the surrounding aqueous medium. The process involves permeation of dipeptides across the membrane that forms the vesicles, and it represents an example of coupling between the growth and division of protocellular compartments and metabolism encapsulated inside them, a necessary condition for supporting evolutionary processes at the origin of life.
Although permeation of single amino acids or side chain analogues through membranes has been a subject of extensive experimental34–41 and simulation40–42 investigations, permeation of dipeptides has been studied experimentally less frequently,35,43 and no computational studies of this process have been published so far. Dipeptides are markedly more complex than amino acids, as their two side chains can have different polarity. This will influence their interactions with both polar head groups and nonpolar alkyl tails of membrane lipids. Thus, it is expected that permeation of dipeptides and their interactions with the water–membrane interface will be sequence dependent. To understand this dependence, we investigate three dipeptides blocked with acetyl group at the N-methylamide at the N-terminal and C-terminal. These are the hydrophobic phenylalanine-leucine dipeptide, amphiphilic serine-leucine, and hydrophilic serine-serine. All three have been studied in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane with molecular dynamics (MD) simulations. Here, we first discuss interaction, conformation, and orientation of the dipeptides at the water–membrane interface, followed by calculations of their free energy profiles across membrane and permeability of the POPC bilayer to these dipeptides.
METHODS
Molecular Dynamics Simulations.
The package NAMD44 was used to carry out all-atom MD simulations. The simulated system consisted of a dipeptide in a planar membrane–water lamellae system, which included 142 POPC lipid molecules and 8019 water molecules. The size of the simulation cell was ∼69.6 × 69.6 × 85.1 Å with periodic boundary conditions applied along x, y, and z directions. The x and y dimensions of the cell were kept fixed to ensure that the surface area per lipid headgroup was equal to the value of 68.3 Å2 measured in the recent X-ray scattering experiments.45 The pressure was kept at 1 atm along the z-direction, perpendicular to the membrane surface. A constant temperature of 303 K was maintained using the Langevin friction force with the damping coefficient at 5 ps−1. In all simulations, a time step of 2 fs was used. The particle mesh Ewald scheme was applied to calculate long-ranged electrostatic interactions, with the grid 72 × 72 × 90. The cutoff of 13.5 Å was used for van der Waals interactions.
The updated version of CHARMM potentials for phospholipids46–48 and the TIP3P model49 of water were used to describe interatomic interactions in the system. The three selected dipeptides—phenylalanine-leucine, serine-leucine, and serine-serine—are all blocked at the N- and C-terminal with acetyl group and N-methylamide, and are referred to as Ace-Phe-Leu-NMe, Ace-Ser-Leu-NMe, and Ace-Ser-Ser-NMe, respectively. The recently optimized CHARMM force field for proteins50 was applied to the dipeptides. The CMAP term51 for the backbone torsion angle was applied to the N- and C-termini.
To test the potential functions, benchmark MD simulations were carried out for the Ace-Phe-Leu-NMe dipeptide in water and in decane, which forms a nonpolar environment similar to the lipid tail region. The simulations in water were 250 ns in length, with constant pressure of 1 atm and temperature of 298 K in a simulation box of 38.8 × 38.8 × 38.8 Å. The dipeptide backbone was mainly distributed between three states: polyproline II (PII), β, and αR (see the Ramachandran map plotted in Figure S1). The extended PII and β states were populated more frequently than the coiled αR state: 65% vs 23% for Phe and 63% vs 28% for Leu. In addition, there were small contributions (total ∼10%) from other states. The observed dominance of the PII and β states is in agreement with the recent NMR and circular dichroism (CD) spectroscopic studies on blocked Ace-X1-X2-NH2 peptides in water.52 This reflects the sought-after improvement in the updated CHARMM force field for proteins50 compared to the old one,46,51 which systematically overestimated the αR state.50 The high population of the PII state shown for a number of short peptides in previous spectroscopic and theoretical studies (see references in a recent review53) is the cornerstone for the PII hypothesis initially put forward by Tiffany and Krimm54,55 to understand the structure of unfolded proteins.53,56
For the dipeptide in decane, a trajectory 200 ns in length was obtained at T = 310 K and constant pressure of 1 atm. The dimensions of the simulation box were 57.9 × 57.9 × 57.9 Å. The conformation of the backbone was found predominantly in the extended β state (the populations of β, PII, and αR were 72%, 17%, and 10%, respectively, for Phe and 75%, 14%, and 10% for Leu; see also the Ramachandran map plotted in the upper panel of Figure S2). These preferences are similar to those calculated for alanine dipeptide in the gas phase.57 For comparison, population of the β state in aqueous solution is only 16% for Phe and 30% for Leu. A similar dependence of the backbone conformation on solvent polarity was previously observed in experimental studies on different tripeptides containing alanine.58 In these studies, it was found that β-sheet conformation dominated in DMSO, whereas conformational distribution in water was markedly broadened. Similar backbone conformations were found for the dipeptide at the POPC membrane center (see the Ramachandran map plotted in the lower panel of Figure S2). The interconversion between the different conformation states was rather fast, occurring on the nanosecond time scale.
Free Energy Calculations.
The adaptive biasing force (ABF) method59,60 was used to calculate the free energy profile, A(z), for the dipeptide traversing the POPC membrane along the reaction coordinate z, defined as the z-component of the distance between the centers of mass of the dipeptide and the lipid bilayer. In the ABF method, the uniform sampling of the reaction coordinates is achieved by subtracting averaged thermodynamics force from the instantaneous force along the chosen reaction coordinates in an adaptive manner.59,60 To calculate the free energy profile, A(z), the range of −26 Å < z < 0 Å was divided into four strata (“windows”), corresponding to the following ranges of z: [−26 Å, –18 Å], [−18 Å, –12 Å], [−12 Å, –6 Å], and [−6 Å, 0 Å]). This covers the region from bulk water to the membrane center. ABF simulations of 200 ns in length were conducted in each window. To keep the dipeptide in each window, harmonic restraints were applied at both edges of the window. To improve statistics for the slow dynamics involving molecular rotation of Ace-Ser-Leu-NMe and Ace-Ser-Ser-NMe near the membrane center, additional ABF simulations of 70–80 ns in length were carried out in the range −3 Å < z < −1 Å and additional 150–250 ns equilibrium MD trajectories were obtained for the range −1 Å < z < 1 Å.
RESULTS AND DISCUSSION
1. Adsorption at Membrane Interface.
To study interfacial behavior of the selected dipeptides, each of them was placed and equilibrated at the water–membrane interface. This was followed by unconstrained MD simulations of 100 ns in length. During these simulations, Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe dipeptide remained at the interface, whereas Ace-Ser-Ser-NMe desorbed after 45 ns and diffused to the aqueous phase. This behavior is in agreement with the general trend, according to which molecules that contain both polar and nonpolar groups, but not polar species, are attracted to interfaces between water and nonpolar phases.61
Plotted in Figure 1 are the density profiles of water, POPC lipid, and the two dipeptides that remained absorbed at the interface. As can be seen in the figure, the hydrophobic side chains of Phe and Leu in Ace-Phe-Leu-NMe are buried in the nonpolar lipid tail region, while the backbone O and N atoms are closer to the headgroup region and water. Similarly, the hydrophobic side chain of Leu in Ace-Ser-Leu-NMe prefers lipid tails, whereas the polar side chain of Ser resides mostly in the more hydrophilic environment formed by lipid head groups and water. Coordination numbers, ⟨CN⟩, defined as the average number of oxygen atoms in water and POPC head groups that are nearest neighbors of a selected atom in the solute molecule (within the first peak of the radial distribution function), support this conclusion. For the hydroxyl oxygen atom (OG) on the serine side chain, the calculated ⟨CN⟩ is 2.3, of which 1.8 is contributed by water molecules and the remaining 0.5 comes from head groups. This confirms that serine is largely exposed to the polar environment. In contrast, the value of ⟨CN⟩ is substantially smaller for backbone O and N atoms and varies between 1 and 1.4. The coordination numbers for both the dipeptides are listed in Table S1.
Figure 1.

Density profiles of Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe at the water–membrane interface. For comparison, the profiles for Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe are plotted on the left and the right side of the membrane interface, respectively, even though simulations for these dipeptides were carried out independently. For Ace-Phe-Leu-NMe, the densities of the Phe side chain, Leu side chain, backbone N, and O of atoms are plotted in solid black, dashed black, solid red, and dashed red, respectively. For Ace-Ser-Leu-NMe the densities of the Leu side chain, Ser side chain, backbone N, and O atoms are plotted in solid green, dashed green, solid purple, and dashed purple, respectively, and the densities are scaled up by a factor of 50 for better visibility. The densities of water, POPC tail atoms, and POPC headgroup atoms are plotted in brown, solid blue, and dashed blue, respectively.
Orientation of different residues with respect to the water–membrane interface can be probed through examining the angle θ between the vector RCα–Cβ pointing from atom Cα to Cβ and the Z-axis. The average value of this angle for each residue in the dipeptides is plotted in Figure S3. In Ace-Ser-Leu-NMe, Ser and Leu side chains point in the opposite directions relative to the membrane interface, with the former directed toward the water phase (θ > 90°) and the latter directed toward the lipid tail region (θ < 90°). In Ace-Phe-Leu-NMe, which contains only hydrophobic side chains, both Phe and Leu point toward the tail region. Preferences of hydrophobic and hydrophilic residues for phases of different polarity also influence conformational preferences of the dipeptides. Because of its amphipathic character, Ace-Ser-Leu-NMe favors a more extended structure than Ace-Phe-Leu-NMe. Its most populated conformation is the extended β–β state (∼22%), which places the side chains on the opposite sides of the backbone (the angle between RCα–Cβ of Ser and Leu peaks at 140°). For Ace-Phe-Leu-NMe, in contrast, the population of β–β is only 2%, and the most favored backbone conformation is a less extended β–αR state (67%). The interconversion between the conformation states is fast, as it happens at the nanosecond time scale. Preferences for amphiphatic conformations at aqueous interfaces have been observed in computer simulations of several other peptides.61 For example, folding of a peptide consisting of leucine and glutamine to an amphiphatic α-helix at the water–decane interface proceeds through a series of amphiphatic intermediates.62
2. Free Energy Profiles of the Dipeptides across the Water–Membrane System.
Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe.
The free energy profiles, A(z), for Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe, shown in Figure 2, exhibit interfacial minima of ∼−3 kcal/mol at the water–membrane interface and a free energy barrier at the center of the bilayer. This barrier with respect to the aqueous phase is equal to 6.5 kcal/mol for Ace-Ser-Leu-NMe, which consists of a hydrophilic and a hydrophobic amino acid, but is only 3.5 kcal/mol for Ace-Phe-Leu-NMe, which contains only hydrophobic side chains that partition preferentially to the nonpolar core of the membrane. The existence of the interfacial minima indicates that dipeptides have amphiphatic character even if both side chains are strongly hydrophobic. This is due to the polar character of the peptide backbone.
Figure 2.

Free energy profiles of Ace-Phe-Leu-NMe (solid black curve), Ace-Ser-Leu-NMe (dashed red curve), and Ace-Ser-Ser-NMe (dotted-dashed blue curve) as a function of the distance of the center mass of the dipeptide from the center mass of the POPC membrane (z = 0), along z. The water–membrane interface is at approximately −15 Å, and the water phase is on the left side of the interface.
Although the free energy profiles are qualitatively similar for both dipeptides, orientations of these two molecules during their transfer across the membrane differ. To track these orientations, we define angle θ between the normal of the membrane to the water–membrane interface and a unit vector, r, pointing from Cα of the first residue (Phe or Ser for Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe, respectively) to Cα of the second residue, Leu (for schematic representation of θ see Figure S4a). If θ is close to 0, the dipeptide is approximately aligned with the normal. If the reaction coordinate, z, is negative, the first residue is closer to water than the second residue. The opposite is true for positive z. If θ is close to 90°, then the dipeptide is approximately parallel to the interface. The two-dimensional probability distributions, P(θ,z), are shown in Figure S4b,c. At the interface, both dipeptides prefer orientations parallel to the membrane. In both cases, this allows for adopting the maximally amphiphatic arrangement. In the membrane, P(θ,z) for Ace-Phe-Leu-NMe is quite broad, indicating that the peptide rotates freely. In contrast, Ace-Ser-Leu-NMe progressively aligns itself with the interface normal, such that polar serine remains close to the water phase and hydrophilic lipid head groups. Only when the dipeptide moves sufficiently deep into the membrane that retaining contacts with the polar environment is no longer possible, P(θ,z) becomes much more uniform.
At the center of the bilayer, P(θ,z) has to be symmetric with respect to z due to the symmetry of the system. If this is not the case, the calculated A(z) will be inaccurate. We tested this behavior for Ace-Ser-Leu-NMe in an additional MD simulation of 300 ns in length in which the dipeptide was constrained in the center of the membrane (z = 0 Å) but was free to rotate. As it can be seen in Figure 3, the dipeptide adopts orientation in the whole range of θ, 0° < θ 180°, in the course of the simulation. The autocorrelation function of the unit vector r for molecular orientation, f(t)〈r̂(t)·r̂(0)〉 plotted in the inset in Figure 3, decays exponentially. The decay time, approximately equal to 7 ns, is sufficiently short to allow adequate sampling of all orientations of the dipeptide at the time scale of our simulations.
Figure 3.

Orientation angle θ for Ace-Ser-Leu-NMe molecule constrained to the center of the POPC membrane as a function of time. Inset: the autocorrelation funtion of the unit vector r characterizing the orientation of the peptide as a function of time.
Single, blocked hydrophobic amino acids exhibit free energy profiles across water–membrane interfaces similar to dipeptides. Probably the best studied is the derivative of tryptophan, N-acetyltryptophanamide (Ace-Trp-NH2).40,41,63 Both MD simulations of this system yielded an interfacial minimum and a barrier in the center of the bilayer, but the corresponding free energies were only in a qualitative agreement with each other. A(z) was also calculated for Ace-Tyr-NH2 and Ace-Phe-NH2,41 the analogues of Ace-Trp-NH2 containing tyrosine and phenylalanine instead of tryptophan. In order to have the direct comparison with these calculations, we determined the free energy profile across the water–membrane interface for Ace-Phe-NMe, a compound that is similar to the dipeptides considered in this study. This profile is shown in Figure S5. The agreement with the earlier simulations is very good. The interfacial minimum and the free energy barrier obtained by Lee et al.41 are equal to −4.3 and 2.5 kcal/mol, respectively, whereas our calculations yielded −3.5 and 2.5 kcal/mol for the same quantities. Similarly good agreement with earlier calculations,40,42 carried out with the aid of OPLS,64 Berger,65 and SPC water66 force fields, was obtained for A(z) of 3-methylindole, the side chain of tryptophan. This compound is interfacially active but, in contrast to blocked amino acids and dipeptides, partitions preferentially to the hydrophobic interior of the membrane (see Figure S5). The free energy profiles for Ace-Phe-NMe and Ace-Phe-Leu-NMe are quite similar. The interfacial minima are nearly the same and the barrier for Ace-Phe-Leu-NMe is 1 kcal/mol larger than the barrier for Ace-Phe-NMe. The reason for this increase is that the dipeptide contains more polar backbone atoms than the blocked amino acid.
Ace-Ser-Ser-NMe.
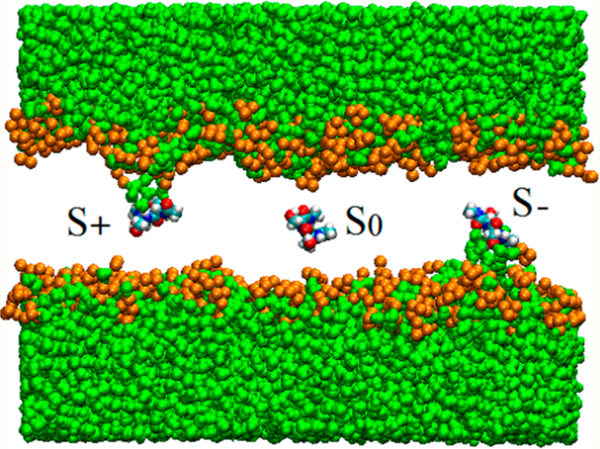
Determining A(z) for Ace-Ser-Ser-NMe is markedly more complicated than determining A(z) for the other two dipeptides studied here. The polar Ace-Ser-Ser-NMe does not have an interfacial free energy minimum. As it moves inside the membrane, water molecules and some neighboring lipid headgroups follow, creating defects in the membrane termed “water fingers”. They were first identified by Benjamin in MD simulations in which Cl− was transferred from water to a hydrophobic liquid phase.67 They were also observed in simulations of ion transfer across a water–membrane interface.68 Subsequently, it was found that water fingers could be induced by other ions,69–71 amino acids,40,42,72 and acid molecules73 permeating phospholipid membranes. For Ace-Ser-Ser-NMe, the defects, shown in Figure 4, are present almost always when the dipeptide is in the membrane near the interface. Even near the center of the membrane, they persist for hundreds of nanoseconds, before the system returns to a defect-free configuration in which the dipetide is detached from both water and lipid headgroup (Figure 4c). Symmetry dictates that a water finger should appear from the opposite side of the membrane as the dipeptide crosses the z = 0 plane. More generally, the peptide at a given value of z in the membrane can be considered as existing in an equilibrium between three states: S− (Figure 4a) and S+ (Figure 4b), characterized by water fingers connected to the aqueous phases at negative and positive values of z, respectively, and S0 (Figure 4c), which is defect-free. The populations of these states are functions of z. Near the center of the bilayer, all three states are expected to be appreciably populated. For z = 0, populations of S− and S+ should be equal by symmetry. When z decreases, population of S− is expected to increase while the populations of S+ and S0 decrease. When z increases, S+ becomes dominant, while S− and S0 progressively vanish.
Figure 4.

Snapshot of Ace-Ser-Ser-NMe at positions near the center of the bilayer. Panels a, b, and c represent respectively states S−, S+, and S0. Water finger defects are clearly seen in (a) and (b). Green: water; gold: head groups of POPC lipids (oxygen atoms). Phospholipid tails were removed for clarity.
Considering that transitions between the S−, S+, and S0 states are rare, obtaining an equilibrated free energy profile near the center of the bilayer and assigning reliable error estimates would require a long MD trajectory. For this reason, we took an alternative approach. We carried out simulations approximately 400 ns in length in which the dipeptide was constrained at z = –0.1, −0.2, −0.3, −0.4, −0.5, −0.6, −0.7, or −0.8 Å from the center of the bilayer and calculated the force, ⟨fz⟩, acting on the dipeptide along the z direction averaged over a time interval of 5 ns. Averaging was carried out to suppress large fluctuations of the instantaneous force. The time dependence of ⟨fz⟩ for simulations at the three smallest values of z is shown in Figure 5. The behavior of ⟨fz⟩ for the remaining five trajectories is similar. Independently of the value of z, the average force exhibits three plateaus at –1.43, 1.43, and 0 kcal/(mol Å), which correspond respectively to S−, S+, and S0 state. The constant values of ⟨fz⟩ near the center of the bilayer mean that the free energy in this region changes linearly with z in the S− and S+ states and remains constant in S0. Results of a separate MD simulation of 600 ns in length, in which the dipeptide, constrained to the range −2.0 Å < z < 2.0 Å, was trapped in the S− state confirm that the average force near the center of the bilayer is approximately constant. In this simulation, the population of the dipeptide as a function of its position along z changes exponentially. This dependence can be described very well by a function exp(–βfz) with f equal to 1.37 kcal/(mol Å), in a very good agreement with the estimate of the average force obtained from the z-constrained simulations.
Figure 5.

Force acting on Ace-Ser-Ser-NMe as a function of time. The force is averaged over 5 ns intervals. Black, red, and blue curves are for the trajectories in which the dipeptide was fixed at z = −0.1, −0.2, and −0.3 Å, respectively.
If we abbreviate the average force in S+ as f, then the probabilities of observing the dipeptide in state S− and S+ as functions of z are proportional to exp(−βfz) and exp(βfz), respectively. For a given f, the ratio of these probabilities is uniquely defined for each z and is equal to exp(−2βfz). No adjustable parameters are needed to determine this ratio. The probability of observing the dipeptide in state S0 can be written as depending on exp(−βΔA0), where ΔA0 is the free energy difference between S0 and S− or S+ at z = 0. The free energies of the latter two states have to be equal by symmetry at this value of z. To ensure that the probabilities of S−, S+, and S0 add to one for each z, they are normalized by their sum at this value of z, exp(−βfz) + exp(βfz) + exp(−βΔA0) = 2 cosh(βfz) + exp(−βΔA0). The only unknown parameter needed to calculate the probabilities of interest is ΔA0. The value of this parameter can be determined from the constrained simulations at different values of z, as described above, with the aid of a Bayesian approach, as described in detail in the Supporting Information. The most likely value of ΔA0 is calculated as 0.05 kcal/mol, which indicates that all three states are nearly equally populated at the center of the bilayer. From the Bayesian method, one can obtain not only the most likely value of ΔA0 but also the full probability distribution of ΔA0. This distribution is shown in the Supporting Information (Figure S6). The standard error in ΔA0 estimated from the distribution is equal to 0.56 kcal/mol.
As z increases away from the center of the membrane, the probability of state S+ also increases at the expense of S− and S0. As z decreases, the probability of S− increases at the expense of S+ and S0. At the largest values of z for which f was determined to be constant, the probabilities of S− and S0 are sufficiently small that they can be neglected without an appreciable loss of accuracy. Then, the free energy profile near the center of the bilayer, integrated over the averaged forces from the states S−, S+, and S0, can be seamlessly connected with the profile at larger values of |z|, calculated by way of ABF. In these calculations, the dipeptide was in the S− state, connected to the aqueous phase by a water finger. The full free energy profile, shown in Figure 2, exhibits the barrier at the center of the bilayer equal to 10 kcal/mol. This barrier is substantially higher than the barrier for the other two dipeptides. This is due to the hydrophilic character of both amino acid side chains in Ace-Ser-Ser-NMe.
3. Permeability of Membranes to Dipeptides.
A common approach74 to calculating membrane permeability, Pm, of a solute on the basis of computer simulations is to integrate the diffusion equation in the steady state approximation.75,76 This yields
| (1) |
where Dz(z) is the position-dependent diffusion coefficient along z, z1, and z2 are the boundaries between the bulk water phases and the water–membrane interfaces on both sides of the membrane, kB is the Boltzmann constant, and T is temperature. Applying the 1-D diffusion model is justified because rotational dynamics of Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe inside the POPC membrane is markedly faster than the simulation time. As can be seen in Figure 3 and Figure S4, different orientations of the dipeptides are frequently sampled during the simulations. Thus, orientational degrees of freedom do not have to be explicitly included but instead can be folded into the 1-D potential of mean force, A(z). Even for Ace-Ser-Ser-NMe the interconversion between the S0, S+, and S− states takes place at the time scale of several hundred nanoseconds, which is still much faster than the time needed to traverse the free energy barrier of 10 kcal/mol separating water phases on both sides of the membrane. In this case, however, interconversion between different orientations of the dipeptide with respect to the membrane normal is retarded by the presence of water fingers. This is why special care had to be taken to obtain the equilibrated potential of mean force.
Besides A(z), shown in Figure 2, calculating permeability from eq 1 requires the knowledge of the diffusion coefficient as a function of z. Its values, calculated from the autocorrelation function of random force77 acting on each dipeptide, are listed in Table S2. Dz(z) inside the POPC membrane, equal to (0.2–0.6) × 10−6 cm2/s, is approximately an order of magnitude lower than the diffusion constant in bulk water, calculated from the Einstein relation to be 3 × 10−6 cm2/s. A similar decrease was found for a number of other solutes permeating membranes.78,79 Permeabilty values calculated from eq 1 are 8.7 × 10−8, 4.5 × 10−5, and 4.6 × 10−3 cm/s for Ace-Ser-Ser-NMe, Ace-Ser-Leu-NMe, and Ace-Phe-Leu-NMe, respectively. From these values it clearly follows that permeability is strongly dependent on residue type. Permeability to Ace-Phe-Leu-NMe, which contains two hydrophobic side chains, is 4 orders of magnitude faster than permeability to polar Ace-Ser-Ser-NMe. This result is in agreement with the conventional view, first put forward by Meyer80 and Overton,81 according to which permeability increases with hydrophobicity of the permeant measured by way of its solubility in nonpolar liquids.
So far, no measurements of permeability of pure bilayers to dipeptides have been reported. There are, however, measurements of permeability to blocked single amino acids, in several instances accompanied by complementary computer simulations. Chakrabarti et al.36,82 found that lysine methyl ester permeated egg phosphatidylcholine membranes quite rapidly, as Pm was equal to 10−2 cm/s. Cardenas et al.40 studied permeability of 1,2-dioleoyl-sn-glicero-3-phosphocholine (DOPC) membrane to blocked tryptophan, Ace-Trp-NH2, by way of the parallel artificial membrane permeation assay (PAMPA). Their measurements yielded Pm equal to 10−7 cm/s. A similar value was measured for Ace-Phe-NH2 and Ace-Tyr-NH2 by Lee et al.,41 also by way of PAMPA. Transport of several blocked or zwitterionic amino acids and dipeptides across a more complicated Caco-2 cell membrane was measured by way of PAMPA38,43 and other methods.38 In all cases, membrane permeabilities fell in a narrow range of 10−6–10−7 cm/s. No correlation was found between the observed permeabilities and the lipophilicity of the peptides assessed via their water/octanol partition coefficients.38 This is at variance with the Meyer–Overton rule and measured83–85 or calculated86,87 permeabilities of small molecules. One explanation is that a major contribution to permeation of peptides through a membrane comes from the free energy needed to break their hydrogen bonds with water at the water–membrane interface.35,38
An alternative explanation of the apparent insensitivity of membrane permeability to hydrophobicity of peptides is related to the contribution to a measured effective permeability of an unstirred water layer adjacent to a membrane. To extract the true membrane permeability, this contribution has to be correctly accounted for.88–90 This is the case especially for hydrophobic molecules for which diffusion through the unstirred water layer may become the rate limit step in the permeation process. It has been shown that the high intrinsic membrane permeability for a number of lipophilic drug molecules, such as imipramine or ibuprofen, is recovered in PAMPA experiments only after applying corrections that remove the contribution from the unstirred water layer.90,91 Similarly, an unexpectedly slow apparent permeability of Caco-2 cell membranes to lipophilic drugs, such as testosterone and desipramine, was ascribed to contributions from an unstirred water layer.38 For the hydrophobic Ace-Phe-Leu-NMe, the permeability, Pu, of unstirred water layer was estimated at 10−6–10−5 cm/s. To arrive at this estimate, Pu was calculated as D/2L88–90 with the diffusion constant D in water equal to 3 × 10−6 cm2/s and the thickness L of the unstirred layer taken as a few hundred to a thousand micrometers, as previously assumed in experimental studies that used different methods to measure membrane permeability,88,89 including PAMPA.90 Similar values of Pu were obtained for other dipeptides and are close to previously measured permeabilities. These values are markedly lower than the Pm calculated for Ace-Phe-Leu-NMe, which means that Pu is the dominant contribution to the apparent membrane permeability to this dipeptide. In contrast, the contribution of unstirred water layer to permeation of hydrophilic Ace-Ser-Ser-NMe is negligible, since the calculated Pm is approximately 2 orders of magnitude smaller than Pu.
Enhancement of the permeability of peptides due to hydrophobic moieties provides an efficient mechanism of fast delivery for drug compounds across the membrane. One example is dipeptide derivatives of 5-aminolevulinic acid (5ala) that have been found useful in photodynamic therapy92 and in treatment of several types of cancer.93–95 The hydrophilic nature of 5ala impairs its ability to cross cell membranes limiting its efficacy. However, N-acetyl-terminated methyl esters of dipeptides containing 5ala and leucine or phenylalanine do not suffer from this disadvantage and therefore are more effective than 5ala alone.96 Conjugates of hydrophobic dipeptides with small molecule drugs often exhibit improved bioavailability, compared to the drugs alone, especially in oral formulations, because their transport across human intestines is mediated by the oligopeptide transporter, hPEPT1, which recognizes all dipeptides and tripeptides.22 The dipeptides exhibit sufficient hydrophobicity to penetrate cell membranes, so that the prodrug can reach its target. Specifically, it has been shown that the conjugate of Val-Ala and an antiviral drug AZT combines good chemical stability, lipophilicity, and affinity for hPEPT1 to have reduced cytotoxicity and improved antiretroviral index relative to AZT.97
CONCLUSIONS
Free energy profiles of dipeptides permeating POPC membranes and the corresponding permeability coefficients strongly depend on polarity of the amino acid side chains. As hydrophobicity increases, the free energy barrier inside the membrane decreases and the permeability coefficient increases. Dipeptides with hydrophobic or mixed, hydrophobic/hydrophilic side chains accumulate preferentially at the water–membrane interface, whereas hydrophilic dipeptides desorb from the interface. The tendency to match polarity of the side chains and the environment influences conformation and orientation of dipeptides both at the interface and in the membrane. In the case of hydrophilic dipeptides, it may induce local membrane deformations that promote access of water to polar side chains.
The ability to calculate permeation properties of dipeptides and other small peptides might provide an efficient, cost-effective way to assist in designing peptide-based drugs, including those containing noncanonical amino acids and peptide derivatives that would be charaterized by good ability to penetrate cell walls and, consequently, improved bioavailability. Similar calculations can also help in designing drug–dipeptide conjugates with desired permeation properties. Finally, the knowledge of permeability coefficients for dipeptides with different sequences might be useful for optimizing delivery of a balanced spectrum of amino acids to cell culture media for manufacturing biopharmaceuticals.
Interfacial activity of some dipeptides appears to be significant for fusion events. This has been already demonstrated for fusion of vesicles and micelles made of fatty acids and discussed in the context of the origin of life. It would be of considerable interest to explore whether small, hydrophobic peptides are also capable of promoting vesicle fusion.
In general, calculations of sequence-dependent permeation properties of dipeptides might be quite useful for a number of problems in cell biology and pharmacology. However, this would require not only high quality computer simulations but also calibrating calculated results against reliable measurement of permeability coefficients properly corrected for such artifacts as unstirred water layer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NASA Exobiology Program. All simulations were performed at the NASA Advanced Supercomputing (NAS) Division and on the Anton computer at the Pittsburgh Supercomputer Center.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.jpcb.7b08238.
Ramachandran map for the backbone conformation of Ace-Phe-Leu-NMe in water and decane, side chain orientation distribution Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe at water–membrane surface, 2D molecular orientation plot of Ace-Phe-Leu-NMe and Ace-Ser-Leu-NMe from water to the center of membrane, free energy profile for Ace-Phe-NMe and 3-methylindole across POPC membrane, and normalized posterior probability distribution P(ΔA0|M,S) in Bayesian analysis; tables of coordination number and position dependent diffusion coefficient for dipeptide (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Otvos L Jr.; Wade JD Current challenges in peptide-based drug discovery. Front. Chem 2014, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Uhlig T; Kyprianou T; Martinelli FG; Oppici CA; Heiligers D; Hills D; Calvo XR; Verhaert P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPa Open Proteomics 2014, 4, 58–69. [Google Scholar]
- (3).Hancock RE; Sahl HG Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol 2006, 24, 1551–1557. [DOI] [PubMed] [Google Scholar]
- (4).Epand RM; Vogel HJ Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta, Biomembr 1999, 1462, 11–28. [DOI] [PubMed] [Google Scholar]
- (5).Fosgerau K; Hoffmann T. Peptide therapeutics: Current status and future directions. Drug Discovery Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- (6).Kaspar AA; Reichert JM Future directions for peptide therapeutics development. Drug Discovery Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]
- (7).Craik DJ; Fairlie DP; Liras S; Price D. The future of peptide-based drugs. Chem. Biol. Drug Des 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- (8).Yamada A; Sasada T; Noguchi M; Itoh K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013, 104, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dietrich U; Durr R; Koch J. Peptides as drugs: From screening to application. Curr. Pharm. Biotechnol 2013, 14, 501–512. [DOI] [PubMed] [Google Scholar]
- (10).Egleton RD; Davis TP Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx 2005, 2, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lopez-Otin C; Matrisian LM Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [DOI] [PubMed] [Google Scholar]
- (12).Sercombe L; Veerati T; Moheimani F; Wu SY; Sood AK; Hua S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol 2015, 6, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Aldrich JV; McLaughlin JP Opioid peptides: Potential for drug development. Drug Discovery Today: Technol. 2012, 9, e23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hawkins BT; Davis TP The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev 2005, 57, 173–185. [DOI] [PubMed] [Google Scholar]
- (15).Swaminathan J; Ehrhardt C. Liposomal delivery of proteins and peptides. Expert Opin. Drug Delivery 2012, 9, 1489–1503. [DOI] [PubMed] [Google Scholar]
- (16).Li P; Nielsen HM; llertz A. Oral delivery of peptides and proteins using lipid-based drug delivery systems. Expert Opin. Drug Delivery 2012, 9, 1289–1304. [DOI] [PubMed] [Google Scholar]
- (17).Banks WA; Kastin AJ Peptides and the blood-brain barrier: Lipophilicity as a predictor of permeability. Brain Res. Bull 1985, 15, 287–292. [DOI] [PubMed] [Google Scholar]
- (18).Buchwald P; Bodor N. Octanol-water partition: Searching for predictive models. Curr. Med. Chem 1998, 5, 353–380. [PubMed] [Google Scholar]
- (19).Buchwald P; Bodor N. Octanol-water partition of non-zwitterionic peptides: Predictive power of a molecular size-based model. Proteins: Struct., Funct., Genet 1998, 30, 86–99. [DOI] [PubMed] [Google Scholar]
- (20).Egan WJ; Merz KM; Baldwin JJ Prediction of drug absorption using multivariate statistics. J. Med. Chem 2000, 43, 3867–3877. [DOI] [PubMed] [Google Scholar]
- (21).Van De Waterbeemd H; Smith DA; Beaumont K; Walker DK. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem 2001, 44, 1313–1333. [DOI] [PubMed] [Google Scholar]
- (22).Mannhold R; Poda GI; Ostermann C; Tetko IV Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J. Pharm. Sci 2009, 98, 861–893. [DOI] [PubMed] [Google Scholar]
- (23).Sala-Rabanal M; Loo DD; Hirayama BA; Turk E; Wright EM Molecular interactions between dipeptides, drugs and the human intestinal H+–oligopeptide cotransporter hPEPT1. J. Physiol 2006, 574, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kyrikou I; Benetis NP; Chatzigeorgiou P; Zervou M; Viras K; Poulos C; Mavromoustakos T. Interactions of the dipeptide paralysin beta-Ala-Tyr and the aminoacid Glu with phospholipid bilayers. Biochim. Biophys. Acta, Biomembr 2008, 1778, 113–124. [DOI] [PubMed] [Google Scholar]
- (25).Deplanque G; Madhusudan S; Jones PH; Wellmann S; Christodoulos K; Talbot DC; Ganesan TS; Blann A; Harris AL Phase II trial of the antiangiogenic agent IM862 in metastatic renal cell carcinoma. Br. J. Cancer 2004, 91, 1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Song BC; Joo N-S; Aldini G; Yeum K-J Biological functions of histidine-dipeptides and metabolic syndrome. Nutr. Res. Pract 2014, 8, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang Z; Zhao Y; Wang X; Lin R; Zhang Y; Ma H; Guo Y; Xu L; Zhao B. The novel dipeptide Tyr-Ala (TA) significantly enhances the lifespan and healthspan of caenorhabditis elegans. Food Funct. 2016, 7, 1975–1984. [DOI] [PubMed] [Google Scholar]
- (28).Neale JH; Olszewski RT; Zuo D; Janczura KJ; Profaci CP; Lavin KM; Madore JC; Bzdega T. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J. Neurochem 2011, 118, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang W; Slusher B; Murakawa Y; Wozniak KM; Tsukamoto T; Jackson PF; Sima AA GCPII (NAALADase) inhibition prevents long-term diabetic neuropathy in type 1 diabetic BB/Wor rats. J. Neurol. Sci 2002, 194, 21–28. [DOI] [PubMed] [Google Scholar]
- (30).Carozzi VA; Chiorazzi A; Canta A; Lapidus RG; Slusher BS; Wozniak KM; Cavaletti G. Glutamate carboxypeptidase inhibition reduces the severity of chemotherapy-induced peripheral neurotoxicity in rat. Neurotoxic. Res 2010, 17, 380–391. [DOI] [PubMed] [Google Scholar]
- (31).Grayson I; Kessler C. Modern applications of amino acids and dipeptides. Chim. Oggi-Chem. Today 2015, 33, 46–51. [Google Scholar]
- (32).Kang S; Mullen J; Miranda LP; Deshpande R. Utilization of tyrosine- and histidine-containing dipeptides to enhance productivity and culture viability. Biotechnol. Bioeng 2012, 109, 2286–2294. [DOI] [PubMed] [Google Scholar]
- (33).Adamala K; Szostak JW Competition between model protocells driven by an encapsulated catalyst. Nat. Chem 2013, 5, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bean RC; Shepherd WC; Chan H. Permeability of lipid bilayer membranes to organic solutes. J. Gen. Physiol 1968, 52, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Conradi RA; Hilgers AR; Ho NF; Burton PS The influence of peptide structure on transport across Caco-2 cells. Pharm. Res 1991, 8, 1453–1460. [DOI] [PubMed] [Google Scholar]
- (36).Chakrabarti AC; Clark-Lewis I; Harrigan PR; Cullis PR Uptake of basic amino acids and peptides into liposomes in response to transmembrane pH gradients. Biophys. J 1992, 61, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chakrabarti AC; Deamer DW Permeability of lipid bilayers to amino acids and phosphate. Biochim. Biophys. Acta, Biomembr 1992, 1111, 171–177. [DOI] [PubMed] [Google Scholar]
- (38).Fujikawa M; Ano R; Nakao K; Shimizu R; Akamatsu M. Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: Application to prediction of Caco-2 cell permeability. Bioorg. Med. Chem 2005, 13, 4721–4732. [DOI] [PubMed] [Google Scholar]
- (39).Cheng C-Y; Goor OJGM; Han S. Quantitative analysis of molecular transport across liposomal bilayer by J-mediated 13C Overhauser dynamic nuclear polarization. Anal. Chem 2012, 84, 8936–8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cardenas AE; Jas GS; DeLeon KY; Hegefeld WA; Kuczera K; Elber R. Unassisted transport of N-acetyl-L tryptophanamide through membrane: Experiment and simulation of kinetics. J. Phys. Chem. B 2012, 116, 2739–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lee BL; Kuczera K; Middaugh CR; Jas GS Permeation of the three aromatic dipeptides through lipid bilayers: Experimental and computational study. J. Chem. Phys 2016, 144, 245103. [DOI] [PubMed] [Google Scholar]
- (42).MacCallum JL; Bennett WFD; Tieleman DP Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J 2008, 94, 3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ano R; Kimura Y; Urakami M; Shima M; Matsuno R; Ueno T; Akamatsu M. Relationship between structure and permeability of dipeptide derivatives containing tryptophan and related compounds across human intestinal epithelial (Caco-2) cells. Bioorg. Med. Chem 2004, 12, 249–255. [DOI] [PubMed] [Google Scholar]
- (44).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kucerka N; Tristram-Nagle S; Nagle JF Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol 2006, 208, 193–202. [DOI] [PubMed] [Google Scholar]
- (46).MacKerell AD Jr.; Bashford D; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (47).Feller S; MacKerell AD Jr. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar]
- (48).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (50).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).MacKerell AD Jr.; Feig M; Brooks CL III Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc 2004, 126, 698–699. [DOI] [PubMed] [Google Scholar]
- (52).Oh K-I; Lee K-K; Park E-K; Jung Y; Hwang G-S; Cho M. A comprehensive library of blocked dipeptides reveals intrinsic backbone conformational propensities of unfolded proteins. Proteins: Struct., Funct., Genet 2012, 80, 977–990. [DOI] [PubMed] [Google Scholar]
- (53).Adzhubei AA; Sternberg MJE; Makarov AA Polyproline-II helix in proteins: Structure and function. J. Mol. Biol 2013, 425, 2100–2132. [DOI] [PubMed] [Google Scholar]
- (54).Tiffany ML; Krimm S. Circular dichroism of the “random” polypeptide chain. Biopolymers 1969, 8, 347–359. [Google Scholar]
- (55).Krimm S; Tiffany ML The circular dichroism spectrum and structure of unordered polypeptides and proteins. Isr. J. Chem 1974, 12, 189–200. [Google Scholar]
- (56).Shi Z; Woody RW; Kallenbach NR Is polyproline II a major backbone conformation in unfolded proteins? Adv. Protein Chem 2002, 62, 163–240. [DOI] [PubMed] [Google Scholar]
- (57).Han WG; Jalkanen KJ; Elstner M; Suhai S. Theoretical study of aqueous N-acetyl-L-alanine N’-methylamide: Structures and Raman, VCD, and ROA spectra. J. Phys. Chem. B 1998, 102, 2587–2602. [Google Scholar]
- (58).Eker F; Cao X; Nafie L; Huang Q; Schweitzer-Stenner R. The structure of alanine based tripeptides in water and dimethyl sulfoxide probed by vibrational spectroscopy. J. Phys. Chem. B 2003, 107, 358–365. [Google Scholar]
- (59).Darve E; Pohorille A. Calculating free energies using average force. J. Chem. Phys 2001, 115, 9169. [Google Scholar]
- (60).Darve E; Rodríguez-Gomez D; Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys 2008, 128, 144120. [DOI] [PubMed] [Google Scholar]
- (61).Pratt LR; Pohorille A. Hydrophobic effects and modeling of biophysical aqueous solution interfaces. Chem. Rev 2002, 102, 2671–2692. [DOI] [PubMed] [Google Scholar]
- (62).Chipot C; Pohorille A. Conformational equilibria of terminally blocked single amino acids at the water-hexane interface: a molecular dynamics study. J. Phys. Chem. B 1998, 102, 281–290. [DOI] [PubMed] [Google Scholar]
- (63).Cardenas AE; Shrestha R; Webb LJ; Elber R. Membrane permeation of a peptide: It is better to be positive. J. Phys. Chem. B 2015, 119, 6412–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jorgensen WL; Tiradorives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- (65).Berger O; Edholm O; Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J 1997, 72, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Berendsen HJC; Grigera JR; Straatsma TP The missing term in effective pair potentials. J. Phys. Chem 1987, 91, 6269–6271. [Google Scholar]
- (67).Benjamin I. Mechanism and dynamics of ion transfer across a liquid-liquid interface. Science 1993, 261, 1558–1560. [DOI] [PubMed] [Google Scholar]
- (68).Wilson MA; Pohorille A. Mechanism of unassisted ion transport across membrane bilayers. J. Am. Chem. Soc 1996, 118, 6580–6587. [DOI] [PubMed] [Google Scholar]
- (69).Tepper HL; Voth GA Mechanisms of passive ion permeation through lipid bilayers: Insights from simulations. J. Phys. Chem. B 2006, 110, 21327–21337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Khavrutskii IV; Gorfe AA; Lu B; McCammon JA Free energy for the permeation of Na+ and Cl- ions and their ion-pair through a zwitterionic dimyristoyl phosphatidylcholine lipid bilayer by umbrella integration with harmonic Fourier beads. J. Am. Chem. Soc 2009, 131, 1706–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Vorobyov I; Olson TE; Kim JH; Koeppe RE II; Andersen OS; Allen TW Ion-induced defect permeation of lipid membranes. Biophys. J 2014, 106, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Bonhenry D; Tarek M; Dehez F. Effects of phospholipid composition on the transfer of a small cationic peptide across a model biological membrane. J. Chem. Theory Comput 2013, 9, 5675–5684. [DOI] [PubMed] [Google Scholar]
- (73).Bemporad D; Luttmann C; Essex JW Behaviour of small solutes and large drugs in a lipid bilayer from computer simulations. Biochim. Biophys. Acta, Biomembr 2005, 1718, 1–21. [DOI] [PubMed] [Google Scholar]
- (74).Wijmans JG; Baker RW The solution-diffusion model: A review. J. Membr. Sci 1995, 107, 1–21. [Google Scholar]
- (75).Diamond JM; Katz Y. Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water. J. Membr. Biol 1974, 17, 121–154. [DOI] [PubMed] [Google Scholar]
- (76).Marrink S; Berendsen H. Simulation of water transport through a lipid membrane. J. Phys. Chem 1994, 98, 4155–4168. [Google Scholar]
- (77).Kubo R. The fluctuation-dissipation theorem. Rep. Prog. Phys 1966, 29, 255–284. [Google Scholar]
- (78).Orsi M; Essex JW Permeability of drugs and hormones through a lipid bilayer: Insights from dual-resolution molecular dynamics. Soft Matter 2010, 6, 3797–3808. [Google Scholar]
- (79).Wei C; Pohorille A. Permeation of nucleosides through lipid bilayers. J. Phys. Chem. B 2011, 115, 3681–3688. [DOI] [PubMed] [Google Scholar]
- (80).Meyer H. (1899) Zur theorie der alkoholnarkose. Naunyn-Schmiedeberg’s Arch. Pharmacol 1899, 42, 109–118. [Google Scholar]
- (81).Overton E Studien über die Narkose zugleich ein Beitrag zur allgemeinen Pharmakologie; Gustav Fischer: Jena, Switzerland, 1901. [Google Scholar]
- (82).Chakrabarti AC; Deamer DW Permeability of lipid bilayers to amino acids and phosphate. Biochim. Biophys. Acta, Biomembr 1992, 1111, 171–177. [DOI] [PubMed] [Google Scholar]
- (83).Finkelstein A. Water and nonelectrolyte permeability of lipid bilayer membranes. J. Gen. Physiol 1976, 68, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Orbach E; Finkelstein A. The nonelectrolyte permeability of planar lipid bilayer membranes. J. Gen. Physiol 1980, 75, 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Walter A; Gutknecht J. Monocarboxylic acid permeation through lipid bilayer membranes. J. Membr. Biol 1984, 77, 255–264. [DOI] [PubMed] [Google Scholar]
- (86).Bemporad D; Essex JW; Luttmann C. Permeation of small molecules through a lipid bilayer: A computer simulation study. J. Phys. Chem. B 2004, 108, 4875–4884. [Google Scholar]
- (87).Wei C; Pohorille A. Permeation of membranes by ribose and its diastereomers. J. Am. Chem. Soc 2009, 131, 10237–10245. [DOI] [PubMed] [Google Scholar]
- (88).Walter A; Hastings D; Gutknecht J. Weak acid permeability through lipid bilayer membranes. J. Gen. Physiol 1982, 79, 917–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Barry PH; Diamond JM Effects of unstirred layers on membrane phenomena. Physiol. Rev 1984, 64, 763–872. [DOI] [PubMed] [Google Scholar]
- (90).Avdeef A Absorption and Drug Development: Solubility, Permeability, and Charge State; John Wiley & Sons Inc.: Hoboken, NJ, 2003; Chapter 7. [Google Scholar]
- (91).Nielsen PE; Avdeef A. PAMPA–a drug absorption in vitro model 8. Apparent filter porosity and the unstirred water layer. Eur. J. Pharm. Sci 2004, 22, 33–41. [DOI] [PubMed] [Google Scholar]
- (92).Brown SB; Brown EA; Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004, 5, 497–508. [DOI] [PubMed] [Google Scholar]
- (93).Stummer W; Pichlmeier U; Meinel T; Wiestler OD; Zanella F; Reulen HJ Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [DOI] [PubMed] [Google Scholar]
- (94).Casas A; Batlle A. Aminolevulinic acid derivatives and liposome delivery as strategies for improving 5-aminolevulinic acid-mediated photodynamic therapy. Curr. Med. Chem. 2006, 13, 1157–1168. [DOI] [PubMed] [Google Scholar]
- (95).Harris F; Pierpoint L. Photodynamic therapy based on 5-aminolevulinic acid and its use as an antimicrobial agent. Med. Res. Rev 2012, 32, 1292–1327. [DOI] [PubMed] [Google Scholar]
- (96).Di Venosa G; Vallecorsa P; Giuntini F; Mamone L; Batlle A; Vanzuli S; Juarranz A; MacRobert AJ; Eggleston IM; Casas A. The use of dipeptide derivatives of 5-aminolaevulinic acid promotes their entry to tumor cells and improves tumor selectivity of photodynamic therapy. Mol. Cancer Ther 2015, 14, 440–451. [DOI] [PubMed] [Google Scholar]
- (97).Santos C; Morais J; Gouveia L; De Clercq E; Pannecouque C; Nielsen CU; Steffansen B; Moreira R; Gomes P. Dipeptide derivatives of AZT: Synthesis, chemical stability, activation in human plasma, hPEPT1 affinity, and antiviral activity. ChemMedChem. 2008, 3, 970–978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.