

Abstract
Mesenchymal stem cells (MSCs) have been widely used for regenerative therapy. In most current clinical applications, MSCs are delivered by injection but face significant issues with cell viability and penetration into the target tissue due to a limited migration capacity. Some therapies have attempted to improve MSC stability by their encapsulation within biomaterials; however, these treatments still require an enormous number of cells to achieve therapeutic efficacy due to low efficiency. Additionally, while local injection allows for targeted delivery, injections with conventional syringes are highly invasive. Due to the challenges associated with stem cell delivery, a local and minimally invasive approach with high efficiency and improved cell viability is highly desired. In this study, we present a detachable hybrid microneedle depot (d-HMND) for cell delivery. Our system consists of an array of microneedles with an outer poly(lactic-co-glycolic) acid (PLGA) shell and an internal gelatin methacryloyl (GelMA)-MSC mixture (GMM). The GMM was characterized and optimized for cell viability and mechanical strength of the d-HMND required to penetrate mouse skin tissue was also determined. MSC viability and function within the d-HMND was characterized in vitro and the regenerative efficacy of the d-HMND was demonstrated in vivo using a mouse skin wound model.
Keywords: microneedle, mesenchymal stem cell, regenerative therapy, biodegradable polymer, GelMA hydrogel
Graphical Abstract
1. Introduction
The application of stem cell biology to tissue regeneration has undergone a remarkable evolution and has generated great interest due to its potent ability to make, repair, and maintain tissues and organs after injury [1]. Stem cells can not only differentiate into different functional cells which can promote the overall process of regeneration, but also can regenerate damaged tissue through the secretion of functional growth factors that stimulate and promote tissue regeneration by angiogenesis, remodeling, cellular recruitment and immune modulation [2]. Among several types of stem cells, mesenchymal stem cells (MSCs) have demonstrated the most clinical promise for treating tissue damage because of their wide tissue distribution, ease of isolation, compatibility with ex vivo culture and their immunomodulatory abilities [3]. Thus, MSCs have been intensively studied in preclinical and clinical studies for regenerative applications and treating inflammation caused by cardiovascular disease [4], myocardial infarction [5], brain and spinal cord injury [6], bone and cartilage injuries [7] and liver fibrosis [8].
Historically, MSCs have been administrated by injection into blood vessels [9]. Bone marrow-derived MSCs could be used to treat cardiac damage by catheter-based trans-endocardial injections [10]. Chronically infarcted myocardium has been regenerated via long-term engraftment and trilineage differentiation of MSCs. Multiple MSC transplantations were made safe and effective by intrathecal injection [11]. Other delivery methods include intravenous and intranasal injection of MSCs for brain injuries [6a, 6b], intra-articular injection of autologous MSCs for knee osteoarthritis [7], and intrasplenic injection of exosomes secreted by MSCs for liver injuries [12]. However, achieving a therapeutic response requires the delivery of a massive number of stem cells to a specific site with high precision. Therefore, clinical translation of MSC therapeutics is difficult to achieve due to low engraftment efficiency. As a result, significant effort has been devoted to increasing the efficiency and stability of MSC delivery by combining metallic [13], polymeric [14], and hydrogel-based microparticles [8b]. These approaches enhance the reparative potential of conventional MSCs and promote proper cellular function which improves cryopreservation and lyophilization stability. Although advanced local transplantation or injection of MSCs shows some regenerative potential, these methods still require excessive cell production and in situ injection to the target tissue may cause adverse effects or further damage to the tissue due to the usage of hypodermic needles [15]. Despite these risks, direct injection of MSCs into the lesion, particularly for myocardial infarction (MI), have been introduced and are the leading clinical practice [16]. These procedures are invasive and have potential side effects in addition to leading to the development of scar tissue.
Various attempts have also been made to improve localization of cells once delivered. MSCs have been embedded in scaffolds consisting of a wide range of biomaterials. For example, the entrapment of MSCs in thermally expandable hydrogel patches has been demonstrated to promote cell adhesion and spreading activity [17]. In addition, a polysaccharide-incorporated silk fibroin, chitosan, and hyaluronic acid-hybrid patch was designed to enhance the proliferation and cardiomyogenic differentiation of MSCs [18]. This hybrid patch with embedded MSCs was implanted into a rat MI model [19]. Furthermore, a commercial product, CardioCel®, made from processed bovine pericardium has been manufactured and also embedded with MSCs. Although CardioCel® showed good therapeutic efficacy for cardiovascular cell therapy [20], dynamic tissue barriers prevent minimally invasive, effective implementation of these therapies. The inability to target these cells to tissues of interest with high efficiency and engraftment has inhibited widespread clinical adoption. Additionally, these MSC-embedded scaffolds usually exhibit low strength of adhesion due to poor cohesive properties which also decreases the cell migration efficiency into injury sites and reduces tissue remodeling efficacy.
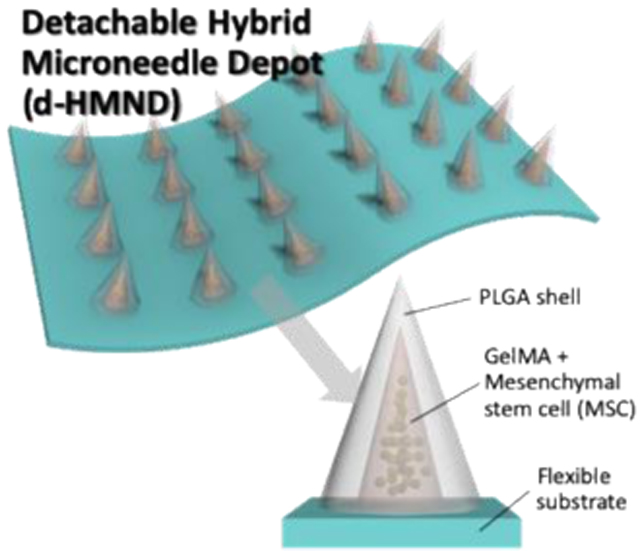
To address these challenges, straightforward delivery with minimal tissue damage is required for successful therapeutic application beyond conventional intrastromal/venous injections and patches. Microneedles (MNs) have been shown to be an effective drug delivery vehicle while also minimizing the dose required through localization. Furthermore, recent MN research for vascular [21] or ocular tissues [22] have demonstrated their spatial precision for drug delivery while overcoming the complex and dynamic barriers of the body including multilayered vascular structures or tear turnover and eye blinking. However, conventional molding processes for the fabrication of MNs are not compatible with maintaining cell viability; MNs are generally dried to achieve the rigidity needed for tissue penetration, but live cells cannot survive this dry environment. Therefore, we engineered a “detachable hybrid microneedle depot (d-HMND)” on a flexible patch for delivering MSCs to a variety of tissues and organs for tissue regeneration (Figure 1 A&B). The d-HMND is made up of gelatin methacryloyl (GelMA) and poly(lactic-co-glycolic)acid 50/50 (PLGA50/50), both of which are biocompatible and biodegradable. We hypothesize that encapsulating the MSCs in a GelMA matrix will improve cell viability after delivery by providing a biologically relevant extracellular matrix (ECM) containing peptides for cell adhesion and matrix degradation and remodeling. In addition, the PLGA shell protects the GelMA-MSC mixture (GMM) before insertion into the targeted tissue. The solid PLGA shell of the d-HMND facilitates facile insertion of the needles into the wound bed with minimal collateral tissue damage. While the MSCs are directly delivered to the target tissue, they are encased by the PLGA shell. The degradation of the PLGA (> 2 weeks) ensures that the MSCs do not migrate into the surrounding tissue but this manner facilitates healing through secreted markers to aid wound regeneration. To do this, the d-HMND is designed to be separated after application so that no foreign materials remain at the site of the wound long-term. In this study, we designed and fabricated the d-HMND, optimized the fabrication methods, and characterized GelMA mechanical properties for the enhancement of MSC viability. Finally, we tested the regenerative efficacy of the d-HMND using a mouse skin wound model.
Figure 1.

Graphical abstract of this study indicating (A) an assembled d-HMND consisting of an array of MNs with PLGA shells, filled with a GelMA-MSC mixture, and fixed to a flexible substrate. (B) The working mechanism of the MNs delivered by the d-HMND after application to target tissue. (C) Sequential fabrication of a d-HMND. i. Preparing GelMA + MSC mixture (GMM). ii. Multiple casting steps for PLGA shell fabrication iii. Loading GMM into the PLGA shells. iv. Assembly d-HMND using double-sided scotch tape.
2. Results and Discussion
2.1. Characterization of GelMA-MSC mixture for cell viability
We first synthesized GelMA to compose the needle-filling scaffold housing the MSCs within the PLGA MN shell. GelMA was prepared according to previously reported protocols [23]. MSCs (passages 3~7) from ATCC were used for the experiments. To load the GMM into the shell of the d-HMND, 10 % (w/v) freeze dried GelMA and 0.5% (w/v) photoinitiator were dissolved into MSC culture medium. The isolated MSCs were then mixed with the GelMA prepolymer solution at a cell density of 107 cells/mL.
MSC viability is highly dependent on the surrounding environment cues including mechanical properties [24] and material patterns [25]. In order to optimize cellular behavior to maximize regenerative capacity, we first manipulated the mechanical properties of GelMA by varying the crosslinking time between 2 and 5 minutes. Thus, to analyze the mechanical properties of the gel, uniform disks (2 mm height × 8 mm diameter) of GMM were crosslinked with a UV light intensity of 14 mW/cm2. This intensity was selected as various cell types, including fibroblasts and liver cells, have been incorporated into GelMA using a similar intensity[26]. We observed that with this light intensity the required amount of time to crosslink the gel disks did not significantly impact the cell viability. MSC viability exceeded 90% up to 6 minutes of crosslinking (Figure S1). After crosslinking with different exposure time, the compressive moduli of GMM disks were measured using a universal testing system (Figure S2). As shown in Figure 2A (black bar), the stiffness of the GMM disk can be tuned between 10 and 50 kPa by increasing crosslinking time. To evaluate the influence of stiffness on the viability of encapsulated MSCs, an array of GMM disks with different stiffnesses were made and stored in cell media for 24 h. Subsequently, the viability of MSCs within the GMM disks was analyzed using live/dead cell assay. We found that the cell viability was over 90% when MSCs were encapsulated in materials with a stiffness between 25 and 35 kPa and a crosslinking time around 3 to 4 min (Figure 2A). Both overly soft and stiff environments (top images of Figure 2B) led to poor cell viability, indicating that there is an ideal range of stiffnesses that maximize MSC survival. We confirmed cell viability across the samples with over 90% cell viability (samples crosslinked for 3, 3.5, and 4 min) compared to the two worst cases (samples crosslinked for 2 and 5 min) using a Cell Counting Kit-8 (CCK-8) assay (Figure S3). The average CCK-8 signals of 3, 3.5, and 4 min samples were higher than that of 2- and 5-min samples. In addition, the 4-min samples were statistically significant compared to both the 2- and 5-min samples. The high viability demonstrated by the 4-min crosslinking condition indicates that it is the best condition for making the GMM.
Figure 2.

(A) MSC viability within 3D GMM structure with respect to different compressive moduli of GelMA tuned by crosslinking time after 1 day. (B) Representative images of MSC viability from live & dead assay. Top images showing poor viability in GelMA that is too soft (2 min crosslinking) and too stiff (5 min crosslinking). Bottom images showing excellent viability of MSCs in GelMA with compressive moduli around 20~30 kPa. (scale bar = 100 μm)
GelMA is one of the biomaterials that has been vigorously studied and used for bioengineering and biomedical applications [27]. Since GelMA has the ability to interact with cells, it is advantageous for three-dimensional (3D) cell culture by acting as ECM [28]. A variety of manufacturing methods have been used to create GelMA scaffolds for cell growth including layer-by-layer stacking [29], micromolding [30] or patterning [31], and 3D printing [32]. GelMA, in each individual study, has been characterized and optimized for the specific cell types and tissues used. In this study, one of the main parameters to be optimized was the stiffness of the GelMA hydrogel surrounding the MSCs. We confirmed that the culture medium used in the fabrication of GelMA solutions facilitated the supply of nutrition to the MSCs in a closed system. Finally, we demonstrated that a compressive modulus of approximately 30 kPa maximized MSC viability in this system.
2.2. Fabrication of d-HMND
A female polydimethylsiloxane (PDMS) MN array, with MNs 700 μm in length, 1850 μm center-to-center spacing, and 1.5 aspect ratio, was molded and treated with O2 plasma. To form the PLGA shell, a mixture of PLGA and dimethyl sulfoxide (DMSO) was cast into the mold (Figure 1C). The O2 plasma treatment renders the surface hydrophilic, allowing the PLGA solution to fill the MN cavities and better adhere to the walls of the mold. Excess PLGA was removed by doctor blading, ensuring consistency amongst the MNs, and the DMSO was evaporated to solidify the shell. To achieve the desired PLGA shell characteristics and uniformity, we repeated the casting process, casting additional PLGA solution over the previously casted shells.
To investigate the formation of the PLGA shell, the PLGA solution was spiked with rhodamine B (RB), a visualizing agent, and imaged using confocal microscopy after the first and second molding. Each successive casting step deposited additional material to form the MN shell (Figure 3A). At each confocal cross-section (i-iii, Figure 3A), the PLGA shell lined the wall. After a single casting step, the thickness of the PLGA shell at the tip (i) was greater than that of the middle layer (iii). However, after two casting steps the overall thickness of the PLGA shell from the tip (i) to the top layer (vi) was distributed uniformly to mimic the profile of the wall (iv-vi, Figure 3A). Next, as described in Figure 1C, we filled the PLGA shell with GMM, removed the excess material to yield a flat surface, and crosslinked the GMM within the shell. To better release the MNs from the mold, the PDMS mold was manually stretched to form gaps at the interface between the PLGA shell and PDMS surface. Then, commercial double-sided tape was attached to the top of the PDMS mold and peeled off to yield the final d-HMND (Figure 3B).
Figure 3.

(A) Confocal microscopic images of PLGA shell after the 1st and 2nd molding processes. Left two images are axial cross-sections and the right images are top-viewed cross-sections along the length of the needle (i~vi). (Scale bars = 200 μm) (B) Magnified image of the d-HMND loaded with GMM inside the red-dyed PLGA shell. (scale bar = 3 mm)
2.3. Mechanical property and tissue insertion of d-HMND
Once the manufacturing process was complete, we evaluated material stability over time. Indeed, the moisture from the GMM inside the shell can partially degrade the PLGA and diminish the structural integrity of the MN. For this reason, we investigated the effect of moisture from the GMM on the mechanical strength of the MN by tuning the crosslinking time. Increased crosslinking time leads to additional curing which results in less moisture released from the GMM, and therefore reduces the degradation of the MN shell. We prepared five different MNs: d-HMND without GMM (only PLGA shell) and d-HMND with GMM crosslinked for 1, 2, 3 and 4 min, respectively). Then, the mechanical strength of the d-HMNDs were measured after 24 h using a custom force measurement system. A metal pillar with a flat surface gently pressed down a d-HMND fixed on top of a load-cell at a controlled speed of 1.8 μm/s. The reaction force was recorded in real time over the displacement of the metal pillar. The first peak in the force-response curve indicates the yield strength of a d-HMND (inset of Figure 4A). The mechanical strength of the d-HMND decreased with the reduce degree of crosslinking. The d-HMNDs crosslinked for 1 and 2 min showed a statistically significant decrease in mechanical integrity relative to the exclusively PLGA shell (control group). Conversely, there was no statistically significant change in failure force when comparing d-HMNDs crosslinked for 3 and 4 min. Further study showed that average failure force was higher for samples crosslinked for 4 min as opposed to 3 min. Additionally, the properties of the d-HMND cured for 4 min closely matched the rigidity of MN composed only of PLGA shell (Figure 4B). Beyond the mechanical properties of the MN, we also observed that MSC viability was greater than 90% at crosslinking times between 3 and 4 min (Figure 2A). Therefore, in order to preserve the mechanical strength and cell viability, the GMM curing time was selected to be 4 min.
Figure 4.

(A) Compression test of d-HMNDs containing variably cross-linked GMMs. The inset shows the definition of failure force on graphed on the y-axis. (B) Mechanical behavior of d-HMNDs against compressive deformation. (C) Detachability rate of d-HMND with respect to different application times from 0.5 min to 2 min. (D) An image of cryosectioned mouse skin after applying the d-HMND (red one, we used RB as a visualizing agent). The inset is a top-view image after d-HMND application. (Scale bars = 500 μm, 3mm(inset)). *p < 0.01, compared with PLGA shell only group. All data are presented as the mean ± SD.
After optimizing the fabrication, we tested the insertion ability of the d-HMND using ex vivo mouse skin. The ex vivo tissue was harvested and used within 4 h. Once again, to visualize the d-HMND within the tissue, RB was used. After patching, the treated tissue was frozen and cryosectioned. Immediately before the d-HMND was applied to the skin, the surface of the tissue was wetted with saline solution. After applying the MN to the tissue, the PLGA shells were expected to slightly degrade via hydrolysis and the dismantlement of the d-HMND would proceed. Therefore, we also optimized the application time of the MNs to balance successful MN detachment from the flexible substrate and the minimization of patching time. After studying an array of different contact times, only those in excess of 1 min resulted in the detachment and proper placement of greater than 90% of MNs (Figure 4C). We found that the MNs were successfully transferred from the flexible substrate to the target tissue and the d-HMND penetrated deep enough into the tissue to facilitate localized MSC delivery (Figure 4D).
2.4. MSCs in d-HMND
Despite effective manufacturing protocols and ideal mechanical properties for tissue penetration, the application of regenerative therapies using the d-HMND cannot be realized without viable, functional MSCs encapsulated in the d-HMND. To demonstrate the ability of our system for the delivery of live cells, we investigated the viability of MSCs in the GMM loaded in the PLGA shell. Although the PLGA shell initially inhibits the diffusion of nutrients to sustain the cells, the internal GMM is based on nutrient-rich media that can to sustain the MSCs for a certain period of time. We prepared d-HMNDs and incubated them for up to 48 h in an incubator at 36.5 °C. The needles were deconstructed, and the flexible substrate was removed to assay MSC viability at 1, 6, 12, 24, 36 and 48 h time-points after d-HMND fabrication. We divided the structure into three equal length segments (top, middle, and bottom), as shown in Figure 5, to better observe regional MSC viability. The results were also averaged to provide a representative viability for the entire structure. As hypothesized, cell viability remained above 90% up to 24 h after production; however, later time points showed a sharp decrease with approximately 10% viability after 48 h. This experiment indicated that the d-HMND should be used earlier than 24 h after fabrication. Future animal experiments used d-HMND within 12~24 h after production.
Figure 5.

(A) Representative images of MSC viability at time points of 1, 24 and 48 h. (Scale bars =100 μm). (B) Time-dependent MSC viability. (C) Quantification of VEGF concentration secreted by MSCs cultured over 7 days in two GMMs - one made and used immediately (black), the other made and stored for 24 h (red). (D) Accumulated VEGF concentrations from (C). (E) Stemness of MSCs in GMMs. All data are presented as the mean ± SD.
Even though the cell viability is maintained above 90% for 24 h within the PLGA shell, it is also essential that the MSCs function normally to enhance their regenerative effect. To verify this behavior, we investigated the paracrine signaling of MSCs. To study the secretion of pro-angiogenic molecules, we selected vascular endothelial growth factor (VEGF) as a representative biomarker promoting angiogenesis, as it is a well-known mitogen for endothelial cells [33]. VEGF was quantified by assaying the conditioned media from MSCs cultured in two different disks of GMM (2 mm height × 8 mm diameter). One sample was stored in cell media immediately after GMM production (0 h group), and the other was stored in cell media after being stored for 24 h covered with a PDMS mold and PLGA film after GMM production (24 h group). We then collected the conditioned media at days 1, 4 and 7 and performed an enzyme-linked immunospecific assay (ELISA). MSCs in both samples secreted VEGF throughout the study showing the highest secretion between days 1 and 4 (Figure 5C). Interestingly, we found no statistically significant difference in VEGF secretion between the two groups at all time points. This indicates that the MSCs in the d-HMND can maintain both their viability and functionality for up to 24 h post-fabrication. Both groups also demonstrated stable VEGF secretion over one week (Figure 5D). Maintaining the natural function of the MSCs is also important for further in vivo tests. We assayed the stemness of the MSCs upon loading and after 24 h (Figure 5E and S4) to ensure the processing did not change their phenotype. We used 3 positive antibodies (CD90, CD44, CD29) and 2 negative antibodies (CD45, CD34) and found that both time points had similar stemness with respect to all both positive and negative antibodies.
2.5. Therapeutic effect of d-HMND
The excisional wound model is one of the most commonly employed mouse models for studying general regeneration processes [34]. A full skin thickness excisional wound extending through the panniculus carnosus was created on the dorsum to make injury group (no treatment). Then MSC injection, d-HMND with and without MSCs (d-HMND group and d-HMND w/o MSC group, respectively) groups were prepared. The MSC injection group was intradermally injected with 0.1 mL of phosphate-buffered saline (PBS) seeded with 106 MSCs near the wound site. Relative to the number of cells delivered by our 8×8 d-HMND array in this study, 200 times more cells were injected into the wound. As for the d-HMND used in the in vivo study, the volume of each microneedle was approximately 7×10−3 μL and contained 70 cells, assuming the density of the prepolymer solution with MSCs is 1 g/mL. Resultingly, the 8×8 d-HMND array delivered approximately 4500 cells in total. The d-HMND group was studied by comparing with the d-HMND w/o MSC group as a control. Wounds were photographed at days 7 and 14, and wound closure was determined based on wound size relative to the original wound dimensions (Figure 6A&B). Wound healing rates were calculated at 7 and 14 days after injury. The rates were 74.2±9.5% and 11.4±0.5%, respectively, in the injury group; 71.7±6.3% and 11.5±3.6% in the MSC injection group; 67.0±9.8% and 10.9±4.0% in the d-HMND w/o MSC group; and 63.4±4.4% and 7.3±4.5% in the d-HMND group. Our findings suggested that the wound healing rate was slowed at both time points with treatment from the d-HMND. After 7 days, d-HMND treated wound was recovered the most with a statistical significance (Figure 6C). Even though there was no statistically significant difference in wound area amongst the various groups, the d-HMND treated group showed the minimal wound area showing that the untreated injury group was found to have the slowest wound contraction over the entire period (Figure S5). Resultingly, the wound contraction was most rapid in animals treated with the d-HMND treated group. Additionally, mice that did not receive treatment did not show hair regeneration around the wound, whereas in all treated groups (MSC, d-HMND w/o MSC and d-HMND) hair regrew as the wound closed. Interestingly, animals treated with the d-HMND group showed increased hair regeneration, even compared to other treated groups.
Figure 6.

(A) Images of wound healing in different experimental groups: untreated (control), MSC-injected, d-HMND without MSCs, and d-HMND with MSCs. (Scale bars = 10 mm). (B) Wound area of each group after 1 week. (C) Representative histologic images of wound bed treated by each method at days 7 and 14. (Scale bars = 1 mm). (D) Re-epithelialization after 2 weeks. (E) MET length after 2 weeks. (F) Representative immunofluorescence images of the wound edge treated by each method each day. (Scale bars = 200 μm). (G) Quantitative analysis of CD 31 positive area. *p < 0.05 and **p < 0.01, compared with Injury group. #p < 0.05 and ##p < 0.01, compared with MSC injection group. †p < 0.05 and ††p < 0.01, compared with d-HMND w/o MSC group. All data are presented as the mean ± SD.
The wound healing effect of the d-HMND was further analyzed through histological evaluation. Low-magnification histological analysis revealed that treatment with the d-HMND improved connectivity (tissue migration) in the wound bed compared to other groups. After 14 days, inflammation and granulation tissue persisted in the untreated group, whereas granulation tissue maturation was observed in all treated groups (MSC, d-HMND w/o MSC and d-HMND). In particular, the d-HMND group showed evidence of transition to the remodeling phase of wound healing, indicated by a decrease in the thickness of the fibrotic tissue and an increase in the number of hair follicles around the wound. Re-epithelialization of the wound bed was measured in cross-sectioned tissue at day 7. The d-HMND group demonstrated significantly more re-epithelialization (48.1±12.9%, n=5) than the other groups (injury; 21.0±2.9%, MSC injection; 35.6±8.7% and d-HMND w/o MSC; 35.5±5.7%). Furthermore, the d-HMND group enhanced keratinocyte migration towards the center of the wound leading to increased migrating epidermal tongue (MET) length relative to the other groups (Figure 6E). Finally, we verified angiogenesis in vivo using CD31 immunofluorescence staining to the observe the effect of increased VEGF release by the MSCs in the d-HMND. Interestingly, on day 7, the percentage of CD31 positive (red) area was found to be greater in the treatment groups (MSC injection, d-HMND w, w/o MSC) than in the injury only group (Figure 6F). In particular, CD31 positive area was highest in the d-HMND group, corroborating the results of the other wound healing metrics (re-epithelialization %, MET length). The d-HMND significantly increased CD31 area % by 802.0 ± 69.1% compared to the other groups (injury; 100.0 ± 48.3%, MSC injection; 573.3 ± 142.7%, and d-HMND w / o MSC; 600.8 ± 135.7%) (Figure 6G). Based upon the various tissue analyses conducted, our in vivo experiments validate the findings of our in vitro experiments and confirm that the d-HMND can be used to improve skin wound healing through the delivery of MSCs.
Furthermore, the d-HMND w/o MSC group (d-HMND without MSCs) elevated wound closure rates and improved re-epithelialization. Recent studies have reported that microneedle application alone produces a variety of biological responses. According to El-Domyati et al, microneedling stimulates the dermal collagen layer, increasing the expression of collagen Type I, III, VII, and tropoelastin, allowing the collagen layer to remain more dense [35]. In addition, microneedling has been reported to aid in the migration of fibroblasts, keratinocytes, and blood vessels during healing process in chronic skin wound conditions [15]. Another study also revealed that microneedling induced the upregulation of tissue remodeling and wound healing related genes (COL3A1, COL8A1, TIMP3), epithelial proliferation and differentiation markers (KRT13, IGF1), and immune cell recruitment gene (CCL11) [36]. Considering the beneficial effects of microneedling without added therapeutics, our in vivo study suggests that the HMND w/o MSCs may also have stimulated the wound healing process.
3. Conclusions
In this study, we have developed a novel device for enhancing wound healing called the d-HMND. It is the first device to use a MN array to facilitate localized MSC delivery with a minimal dose of cells. A biodegradable PLGA shell was fabricated by a two-step molding process using plasma surface modification. GMM with a 30 kPa compressive modulus was characterized and optimized for MSC viability. GMM was loaded into the PLGA shells and the d-HMNDs were transferred to commercial double-sided tape. The array of MNs showed excellent mechanical integrity with strength sufficient to penetrate the target tissue. The d-HMND were able to be separated from the substrate after application to the target tissue. This delivery mechanism ensures that the substrate does not elicit a foreign body response as the needles composed of PLGA, MSCs, and a biodegradable GelMA scaffold have all been shown to be biocompatible. The use of cell media for preparing pre-polymer solution to make the GMM ensured that the nutrient supply was sufficient to maintain cell viability above 90% for 24 h after d-HMND production. Furthermore, the functionality of the delivered MSCs was observed through their secretion of VEGF in closed system (no nutrient supply from outside). Thus, MSCs loaded in GMM and surrounded by PDMS and PLGA for 24 h, showed VEGF secretion profiles similar to that of cultured MSCs. Lastly, we used a full-thickness skin excisional wound mouse model to test the in vivo therapeutic efficacy of the d-HMND. Animals treated with the d-HMND showed elevated wound closure rates, improved re-epithelialization, and increased CD31-positive microvasculature compared to controls. The d-HMND is an innovative cell therapy delivery technology that shows great promise for improved treatment of skin wounds using MSCs.
4. Experimental Section
Synthesis of GelMA:
A 10 % (w/v) gelatin solution was made by dissolving gelatin powder from porcine skin (G1890, Sigma Aldrich, USA) in 100 mL PBS at 50 °C for 1 h and then 8 mL of methacrylic anhydride (276685, Sigma Aldrich, USA) was added using a burette to the gelatin solution and stirred at 50 °C for 2 h before an additional 100 mL of PBS was added. Subsequently, the solution was dialyzed for 5 days at 40 °C using dialysis bag (888–11530, Spectrum Chemical Mfg. Corp., USA) to remove impurities. The purified solution was filtered by a vacuum filtration cup with 0.22 μm pores and frozen to −80 °C and subsequently lyophilized. The frozen GelMA was lyophilized for 3 days. The prepolymer solution of the GMM was prepared by dissolving 10% (w/v) lyophilized GelMA and 0.5% (w/v) photoinitiator (2-Hydroxy40-(2-hydroxyethoxy)-2-methylpropiophenone, 410896–10G, Sigma Aldrich, USA) in cell culture media.
MSC Culture:
Human MSCs from bone marrow were purchased from ATCC (Cat. No. PCS-500–012) and cultured in Dulbecco’s Modified Eagles Medium-low glucose (Sigma Aldrich, USA) supplemented with 10% fetal bovine serum (Sigma Aldrich, USA) and 1% penicillin streptomycin (Invitrogen, USA). Cells were passaged or collected at approximately 80% confluency. When passaging, cells were rinsed 3 times with PBS and trypsin-EDTA (Gibco, USA) solution was added. When the cells were detached from the flask, an equal volume of cell culture medium was added to quench the trypsin. Dissociated cells were transferred to centrifuge tube for further use. Passage 3~5 MSCs were used in the experiments.
Fabrication of d-HMND:
To obtain the MN structures, lithography, multiple layer deposition, and wet etching processes were used. To alter the size and shape of the MNs, the size of the square micropattern (1,750 X 1,750 μm2) on the photomask was manipulated. At first, we deposited oxide and a nitride layers on a 4-inch wafer by furnace and low-pressure chemical vapor deposition, in sequence. An SU-8 layer was then spread by spin coating and exposed to UV light through a lithographic film photomask bearing an array of squares. After photoresist ashing, the exposed Si3N4 and SiO2 layers were removed by reactive ion etching to leave a micropattern of square islands. The patterned wafer was dipped in 29 % KOH solution at 79 °C and washed in a bubbling water bath. Finally, an octagonal cone-shaped, 700 μm length male MN array was fabricated with a 1.5 aspect ratio. Subsequently, a polydimethylsiloxane (PDMS) mold containing an array of MN cavities was prepared by casting the PDMS over the male MNs. To make the PLGA shell of the MN, PLGA50/50 (P2191, Sigma Aldrich, USA) solution (PLGA:DMSO = 1:3, w/w) was cast into the mold multiple times. Prior to casting, the surface of the PDMS mold was modified by O2 plasma to make the surface hydrophilic and allow the PLGA solution to fill the cavity by adhering to the walls of the PDMS cavity. PLGA was cast over the mold and evaporated under vacuum to solidify the PLGA shell and remove bubbles. The same process was repeated to thicken and reinforce the PLGA shell. Once the shell was formed, GMM was cast over the mold containing the PLGA shells and covered by the double-sided tape to yield the assembled d-HMND.
Mechanical testing of GelMA and d-HMND and MSC viability test:
Compressive tests to measure the stiffness of the hydrogels were conducted using a universal testing machine (Instron 5524, Instron, USA) with a 10 N load cell. To make the samples for mechanical testing, a standardized gel structure crosslinked with different UV exposure times was used to vary the mechanical properties. 14 mW/cm2 UV light was used. The compressive modulus of gels crosslinked for between 2 and 5 min was found. In parallel, MSC viability was investigated in the gels with varying stiffness. Cell viability was evaluated with a LIVE/DEAD Viability/Cytotoxicity Kit. A solution consisting of 0.05% calcein and 0.2% ethidium homodimer-1 in PBS was added to each well and incubated for 30 mins. After three washes with PBS the samples were imaged with a fluorescent microscope to find the optimal GelMA property. Because the d-HMND is a closed system that does not provide additional nutrients once manufactured, we feared that MSC viability would decrease rapidly with time. We prepared several d-HMNDs and quantify cell viability at different time points up to 3 days. In addition, a CCK-8 assay (Sigma Aldrich, USA) was performed to verify MSC viability within the GMM. GMM disks crosslinked with different exposure times ranging from 2 to 5 min were put in a 48-well plate and cultured in DMEM with 10% FBS for 24 h. 2D control groups were prepared for the CCK-8 assay as well. We made GelMA substrates crosslinked for 4 min and cultured the same number of MSCs in the wells as in the GMM (105 cells). Using a plate reader (BIOTEK Fluorescent plate reader, Synergy HTX multimode reader), CCK-8 signal was measured.
ELISA assay and evaluation of stemness of MSC:
The human VEGF sandwich assay ELISA (Sigma Aldrich, USA) was performed based on the supplier’s instructions. Briefly, 100 μl of each standard and sample were pipetted into wells coated with capture antibodies and placed in an incubator for 2.5 h. In succession, 100 μl of detection antibody were added to each well and left for 1 h, then 100 μl of streptavidin for 45 min, 100 μl of substrate solution for 20 min, and 50 μl of stop solution to halt the reactions. In between each step, the wells were washed four times with wash buffer. Using a plate reader, absorbance of 450 nm light was used to assay each well. The stemness of the MSCs was evaluated using anti-CD90-FITC, anti-CD44-FITC, anti-CD29-FITC, anti-CD34-FITC, and anti-CD45-FITC antibodies (Sigma Aldrich, USA). GMM disks were put in a 24-well plate and were washed with PBS 3 times, each time 10 min. Then, cells were fixed with 4% formaldehyde for 20 min at room temperature followed by 3 washes, each time for 10 min. After washing, the cells were permeabilized with 0.1% Triton X-100 in PBS solution for 5 min and washed 3 times with PBS. The samples were then treated with 2% BSA solution to block non-specific binding. Next, the cells were incubated for 30 min at 4 °C with the specific antibody diluted in 2% BSA. The cells were washed with PBS 3 times after antibody conjugation and the cell nuclei were stained with DAPI for 15 min. Finally, cells were observed with a fluorescent microscope and signal intensity (integrated density) was measured by ImageJ.
In vivo therapeutic test:
All animal experiments were approved by the UCLA Animal Research Committee (UCLA ARC #2018–003-01E). All animals were treated in compliance with the National Research Council criteria as outlined in the “Guide for the Care of Laboratory Animals” prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health, USA. Forty 7-week-old, C57BL/6J male mice (average weight: 20 grams) were purchased from Jackson Laboratory (Sacramento, CA, USA). Full-thickness excisional wounds were made on anesthetized mice (gas anesthesia, 1.5% isoflurane in 100% O2). A circular incision was made on the middle line of the dorsal skin using a 10-mm biopsy punch (Miltex, York, PA, USA). Full-thickness skin tissue, including the epidermis, dermis, subcutis, and muscularis, was separated by the blunt tip of a Metzenbaum scissors, and cut off along the incision. Mice were sacrificed for histology 7 and 14 days post-operation.
A total of 40 mice were used in the study and randomly divided into four groups. For the untreated injury group (n=5, control), 100 μL of PBS was applied onto the wound bed. Mice in the MSC injection group (n=5) were intradermally injected with 100 μL of PBS containing 107 cells per mL at four injection sites around the wound. Another group of mice had d-HMNDs without MSCs applied (n=5), 8×8 d-HMND arrays without MSCs were applied directly to the wound bed. Similarly, the d-HMND group (n=5) had 8×8 d-HMND arrays containing MSCs also applied directly to the wound bed.
Histology and wound healing evaluation:
Digital photographs of each wound was taken at days 0, 7, and 14. Wound closure time was defined as when the wound bed was re-epithelialized and completely filled with new tissue. Wound area was defined by tracing the margin of wound and measured by ImageJ program (National Institute of Health, USA). The investigators who measured the wound were blinded. Wound healing rate was calculated as follows: (Areaoriginal wound-Arearemaining wound)/Areaoriginal wound × 100. Skin specimens taken from the wound and the surrounding unwounded area were collected and fixed in 10% neutral buffered formalin (Leica Biosystems, IL, USA). This was followed by a general procedure for histological analysis and embedded in paraffin. 4-μm sections were processed with routine hematoxylin (Leica Biosystems) and eosin (Sigma Aldrich) (HE) stain. Histology images were acquired on a Nikon inverted microscope. Quantitative data, such as re-epithelialization % and MET length, was measured using the AmScope image analysis software (AmScope, Irvine, CA, USA). The re-epithelialization ratio (%) was measured in the HE-stained sections on day 7 (n=5/group). The width of the wound and distance covered by newly formed epithelium was measured and the re-epithelialization percentage was calculated by the following formula: % re-epithelialization = (distance covered by the epithelium/width of wound bed) × 100%. All histology results in this study were expressed as mean ± SD and statistical analysis was performed using GraphPad Instat (Graphpad software Inc., La Jolla, CA, USA). The statistical significance of differences was assessed by one-way ANOVA and Bonferroni post-hoc paired comparisons tests. P-values less than 0.05 between experimental groups were considered statistically significant.
Immunofluorescent staining:
A series of tissue sections were deparaffinized and processed with heat-induced antigen retrieval using citrated buffer according to the protocol. After 30 minutes in 0.3% Triton PBST for permeabilization, the antigen was further blocked using goat serum for 30 minutes. The sections were incubated with rabbit polyclonal CD31 primary antibody (1:200; Abcam, Cambridge, UK). After incubation, the sections were washed twice with PBST and incubated with secondary antibodies (1:1000, ThermoFisher, Alexa 555) at room temperature for 60 min and counterstained with DAPI for 5 min. The fluorescent images were imaged using a Nikon Eclipse Ti-S Inverted Phase Contrast Fluorescent Microscope.
Supplementary Material
Acknowledgements
The authors also acknowledge funding from the National Institutes of Health (EB021857, AR066193, AR057837, CA214411, HL137193, EB024403, EB023052, and EB022403).
Footnotes
The authors have no competing interests.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
KangJu Lee, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Yumeng Xue, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Junmin Lee, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Han-Jun Kim, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Yaowen Liu, College of Food Science, Sichuan Agricultural University, Yaan, 625014, China.
Peyton Tebon, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Einollah Sarikhani, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Wujin Sun, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Shiming Zhang, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Reihaneh Haghniaz, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Betül Çelebi-Saltik, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Stem Cell Sciences, Graduate School of Health Sciences, Hacettepe University, Sihhiye, 06100, Ankara, Turkey.
Xingwu Zhou, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Serge Ostrovidov, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Radiological Sciences, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Samad Ahadian, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA.
Nureddin Ashammakhi, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Radiological Sciences, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Mehmet R. Dokmeci, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA Department of Radiological Sciences, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Ali Khademhosseini, Department of Bioengineering and Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Radiological Sciences, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
References
- [1].a) Asahara T, Kawamoto A, American Journal of Physiology-Cell Physiology 2004, 287, C572; [DOI] [PubMed] [Google Scholar]; b) Murry CE, Field LJ, Menasché P, Circulation 2005, 112, 3174; [DOI] [PubMed] [Google Scholar]; c) Lindvall O, Kokaia Z, Martinez-Serrano A, Nature medicine 2004, 10, S42; [DOI] [PubMed] [Google Scholar]; d) Bonner-Weir S, Weir GC, Nature biotechnology 2005, 23, 857. [DOI] [PubMed] [Google Scholar]
- [2].a) Phinney DG, Prockop DJ, Stem cells 2007, 25, 2896; [DOI] [PubMed] [Google Scholar]; b) Stappenbeck TS, Miyoshi H, Science 2009, 324, 1666; [DOI] [PubMed] [Google Scholar]; c) Bajada S, Mazakova I, Richardson JB, Ashammakhi N, Journal of tissue engineering and regenerative medicine 2008, 2, 169. [DOI] [PubMed] [Google Scholar]
- [3].a) Shi Y, Hu G, Su J, Li W, Chen Q, Shou P, Xu C, Chen X, Huang Y, Zhu Z, Cell research 2010, 20, 510; [DOI] [PubMed] [Google Scholar]; b) Nasef A, Ashammakhi N, Fouillard L, 2008. [DOI] [PubMed] [Google Scholar]
- [4].Ranganath SH, Levy O, Inamdar MS, Karp JM, Cell stem cell 2012, 10, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miyahara Y, Nagaya N, Kataoka M, Yanagawa B, Tanaka K, Hao H, Ishino K, Ishida H, Shimizu T, Kangawa K, Nature medicine 2006, 12, 459. [DOI] [PubMed] [Google Scholar]
- [6].a) Donega V, Nijboer CH, Van Velthoven CT, Youssef SA, De Bruin A, Van Bel F, Kavelaars A, Heijnen CJ, Pediatric research 2015, 78, 520; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen M, Li X, Zhang X, He X, Lai L, Liu Y, Zhu G, Li W, Li H, Fang Q, Journal of neuroinflammation 2015, 12, 61; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ashammakhi N, Kim H-J, Ehsanipour A, Bierman RD, Kaarela O, Xue C, Khademhosseini A, Seidlits SK, Tissue Engineering Part B: Reviews 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koh Y-G, Jo S-B, Kwon O-R, Suh D-S, Lee S-W, Park S-H, Choi Y-J, Arthroscopy: The Journal of Arthroscopic & Related Surgery 2013, 29, 748. [DOI] [PubMed] [Google Scholar]
- [8].a) Mohamadnejad M, Namiri M, Bagheri M, Hashemi SM, Ghanaati H, Mehrjardi NZ, Ashtiani SK, Malekzadeh R, Baharvand H, World journal of gastroenterology: WJG 2007, 13, 3359; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meier RP, Mahou R, Morel P, Meyer J, Montanari E, Muller YD, Christofilopoulos P, Wandrey C, Gonelle-Gispert C, Bühler LH, Journal of hepatology 2015, 62, 634. [DOI] [PubMed] [Google Scholar]
- [9].Hare JM, Fishman JE, Gerstenblith G, Velazquez DLD, Zambrano JP, Suncion VY, Tracy M, Ghersin E, Johnston PV, Brinker JA, Jama 2012, 308, 2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Quevedo HC, Hatzistergos KE, Oskouei BN, Feigenbaum GS, Rodriguez JE, Valdes D, Pattany PM, Zambrano JP, Hu Q, McNiece I, Proceedings of the National Academy of Sciences 2009, 106, 14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jarocha D, Milczarek O, Wedrychowicz A, Kwiatkowski S, Majka M, Cell transplantation 2015, 24, 661. [DOI] [PubMed] [Google Scholar]
- [12].Tan CY, Lai RC, Wong W, Dan YY, Lim S-K, Ho HK, Stem cell research & therapy 2014, 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Han J, Kim B, Shin J-Y, Ryu S, Noh M, Woo J, Park J-S, Lee Y, Lee N, Hyeon T, ACS nano 2015, 9, 2805. [DOI] [PubMed] [Google Scholar]
- [14].Lorenz MR, Holzapfel V, Musyanovych A, Nothelfer K, Walther P, Frank H, Landfester K, Schrezenmeier H, Mailänder V, Biomaterials 2006, 27, 2820. [DOI] [PubMed] [Google Scholar]
- [15].Liebl H, Kloth LC, Journal of the American College of Clinical Wound Specialists 2012, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo L, Tang J, Nishi K, Yan C, Dinh P-U, Cores J, Kudo T, Zhang J, Li T-S, Cheng K, Circulation research 2017, 120, 1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jun I, Lee YB, Choi YS, Engler AJ, Park H, Shin H, Biomaterials 2015, 54, 44. [DOI] [PubMed] [Google Scholar]
- [18].Yang M-C, Wang S-S, Chou N-K, Chi N-H, Huang Y-Y, Chang Y-L, Shieh M-J, Chung T-W, Biomaterials 2009, 30, 3757. [DOI] [PubMed] [Google Scholar]
- [19].Chi N-H, Yang M-C, Chung T-W, Chen J-Y, Chou N-K, Wang S-S, Biomaterials 2012, 33, 5541. [DOI] [PubMed] [Google Scholar]
- [20].Vashi AV, White JF, McLean KM, Neethling WM, Rhodes DI, Ramshaw JA, Werkmeister JA, Journal of Biomedical Materials Research Part A 2015, 103, 1999. [DOI] [PubMed] [Google Scholar]
- [21].a) Lee KJ, Park SH, Lee JY, Joo HC, Jang EH, Youn Y-N, Ryu W, Journal of controlled release 2014, 192, 174; [DOI] [PubMed] [Google Scholar]; b) Lee J, Kim D-H, Lee KJ, Seo IH, Park SH, Jang EH, Park Y, Youn Y-N, Ryu W, Journal of Controlled Release 2017, 268, 237; [DOI] [PubMed] [Google Scholar]; c) Lee K, Park SH, Lee J, Ryu S, Joo C, Ryu W, Pharmaceutics 2019, 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Song HB, Lee KJ, Seo IH, Lee JY, Lee S-M, Kim JH, Kim JH, Ryu W, Journal of Controlled Release 2015, 209, 272; [DOI] [PubMed] [Google Scholar]; b) Lee K, Song HB, Cho W, Kim JH, Kim JH, Ryu W, Acta biomaterialia 2018, 80, 48. [DOI] [PubMed] [Google Scholar]
- [23].Luo Z, Sun W, Fang J, Lee K, Li S, Gu Z, Dokmeci MR, Khademhosseini A, Advanced healthcare materials 2019, 8, 1801054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Justin RT, Engler AJ, PloS one 2011, 6, e15978; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pek YS, Wan AC, Ying JY, Biomaterials 2010, 31, 385; [DOI] [PubMed] [Google Scholar]; c) Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H.-p., Lippens E, Duda GN, Nature materials 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Dumas V, Guignandon A, Vico L, Mauclair C, Zapata X, Linossier MT, Bouleftour W, Granier J, Peyroche S, Dumas J-C, Biomedical Materials 2015, 10, 055002; [DOI] [PubMed] [Google Scholar]; b) Yang Y, Wang X, Huang T-C, Hu X, Kawazoe N, Tsai W-B, Yang Y, Chen G, Journal of Materials Chemistry B 2018, 6, 5424. [DOI] [PubMed] [Google Scholar]
- [26].a) Suurmond CAE, Lasli S, van den Dolder FW, Ung A, Kim HJ, Bandaru P, Lee K, Cho HJ, Ahadian S, Ashammakhi N, Advanced healthcare materials 2019, 8, 1901379; [DOI] [PubMed] [Google Scholar]; b) Bertassoni LE, Cardoso JC, Manoharan V, Cristino AL, Bhise NS, Araujo WA, Zorlutuna P, Vrana NE, Ghaemmaghami AM, Dokmeci MR, Biofabrication 2014, 6, 024105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Loessner D, Meinert C, Kaemmerer E, Martine LC, Yue K, Levett PA, Klein TJ, Melchels FP, Khademhosseini A, Hutmacher DW, Nature protocols 2016, 11, 727. [DOI] [PubMed] [Google Scholar]
- [29].Gauvin R, Chen Y-C, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A, Biomaterials 2012, 33, 3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hosseini V, Ahadian S, Ostrovidov S, Camci-Unal G, Chen S, Kaji H, Ramalingam M, Khademhosseini A, Tissue Engineering Part A 2012, 18, 2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nikkhah M, Eshak N, Zorlutuna P, Annabi N, Castello M, Kim K, Dolatshahi-Pirouz A, Edalat F, Bae H, Yang Y, Biomaterials 2012, 33, 9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Miri AK, Nieto D, Iglesias L, Goodarzi Hosseinabadi H, Maharjan S, Ruiz-Esparza GU, Khoshakhlagh P, Manbachi A, Dokmeci MR, Chen S, Advanced Materials 2018, 30, 1800242; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ying GL, Jiang N, Maharjan S, Yin YX, Chai RR, Cao X, Yang JZ, Miri AK, Hassan S, Zhang YS, Advanced Materials 2018, 30, 1805460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Zisa D, Shabbir A, Suzuki G, Lee T, Biochemical and biophysical research communications 2009, 390, 834; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Song S-Y, Chung H-M, Sung J-H, Expert opinion on biological therapy 2010, 10, 1529. [DOI] [PubMed] [Google Scholar]
- [34].a) Isakson M, De Blacam C, Whelan D, McArdle A, Clover A, Stem cells international 2015, 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dorsett-Martin WA, Wound Repair and Regeneration 2004, 12, 591. [DOI] [PubMed] [Google Scholar]
- [35].El-Domyati M, Barakat M, Awad S, Medhat W, El-Fakahany H, Farag H, International journal of dermatology 2015, 54, 1361. [DOI] [PubMed] [Google Scholar]
- [36].Schmitt L, Marquardt Y, Amann P, Heise R, Huth L, Wagner-Schiffler S, Huth S, Baron J-M, PloS one 2018, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.