

Abstract
Human immunodeficiency virus (HIV) is an attractive target for chimeric antigen receptor (CAR) therapy. CAR T cells have proved remarkably potent in targeted killing of cancer cells, and we surmised that CAR T cells could prove useful in eradicating HIV-infected cells. Toward this goal, we interrogate several neutralizing single-chain variable fragments (scFvs) that target different regions of the HIV envelope glycoprotein, gp120. We find here that CAR T cells with scFv from NIH45-46 antibody demonstrated the highest cytotoxicity. Although NIH45-46 CAR T cells are capable of eliminating antigen-expressing cells, we wanted to address HIV reactivation from ex vivo culture of HIV patient-derived CAR T cells. In order to capitalize on the HIV reactivation, we developed a conditionally replicating lentiviral vector (crLV). The crLV can hijack HIV machinery, forming a chimeric lentivirus (LV) instead of HIV and delivered to uninfected cells. We find that CAR T cells generated with crLVs have similar CAR-mediated functionality as traditional CARs. We also demonstrate crLVs’ capability of expanding CAR percentage and protecting CD4 CAR T cell in HIV donors. Collectively, we demonstrate here that the novel crLV NIH45-46 CAR can serve as a strategy to combat HIV, as well as overcome HIV reactivation in CD4+ CAR T cells.
Keywords: HIV, CAR T-cell, 45-46, Conditionally replicating vectors
Graphical Abstract
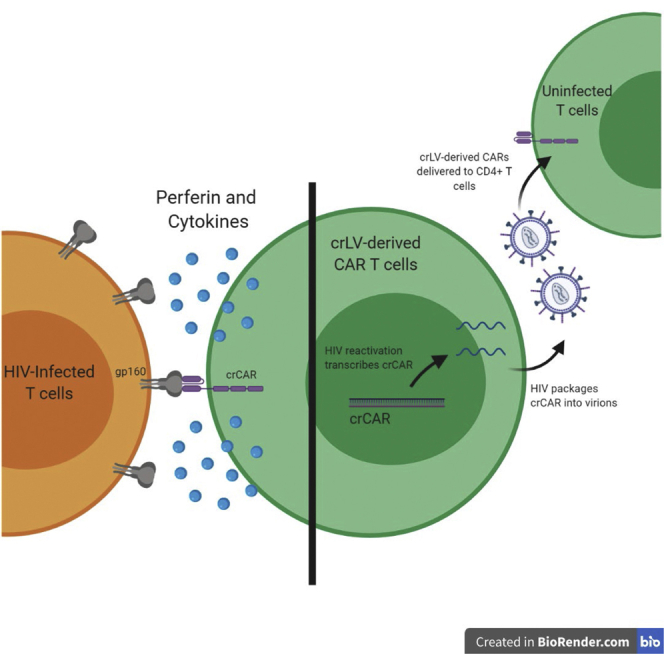
In this paper, Urak et al. develop an innovative means to generating anti-HIV chimeric antigen receptor (CAR) T cells. By utilizing conditionally replicating lentiviral vectors, they show that HIV-infected CD4+ T cells can be used to spread CAR to new T cells, ultimately demonstrating that HIV-infected T cells can be used to generate anti-HIV CAR.
Introduction
Recent clinical successes of T cell immunotherapy using chimeric antigen receptors (CARs) have demonstrated dramatic regression of B cell malignancies.1,2 Because of this efficacy against B cell malignancies, the field of T cell immunotherapy has expanded to a variety of other diseases, such as human immunodeficiency virus (HIV).3, 4, 5, 6 HIV is a worthwhile target for CAR therapy because extensive research in neutralizing antibody therapy has provided for an abundance of single-chain variable fragments (scFvs) to use in the design of CAR constructs. However, unlike B cell malignancies, CAR T cells against HIV have a variety of challenges that first need to be overcome in order for this therapy to be efficacious. One of the most predominant challenges in treating HIV with CAR T cells is the ability of HIV to infect and eliminate target CD4+ T cells.7 Recent studies have indicated that the CD4+ T cell population is a key component of CAR T cell persistence and efficacy,8 which suggests that maintaining or expanding this population may be associated with an improved therapy.
Many experimental approaches developed to date attempt to preserve the CD4 population by preventing HIV from infecting the T cells. Although adding antiretrovirals to the ex vivo culture during CAR production would suppress the reactivation, it also hinders the integration of CAR LV within the T cells,9,10 ultimately demonstrating the need to develop novel strategies for preserving the CD4 population. These strategies have included editing the T cells themselves, such as knocking out the CCR5 gene, which expressed a critical co-receptor for HIV infection,4 or by including fusion inhibitors in the CAR.3 Although these methods prevent HIV infection of T cells, they are useful only for donor-derived CAR T cell products. HIV patient-derived T cells will have virus integrated within the T cells, which can be reactivated and eliminate the CD4+ population during ex vivo culture.11
In order to capitalize on the issue of viral reactivation in the HIV patient-derived CAR T cell products, we propose developing conditionally replication lentivirus (crLV)-derived CAR that parasitizes HIV machinery to encapsulate itself within the virion,12,13 potentially converting other CD4+ T cells into HIV CARs. By parasitizing the virus, crLVs will add a negative selective pressure on HIV by acting as an interfering particle, while expanding the CAR to more CD4+ T cells.14 Based on this notion, we evaluated various scFvs from different neutralizing antibodies, designed a crLV-derived CAR, and tested the hypothesis that anti-HIV CAR T cells can be developed from virus-infected cells to target HIV-infected cells. We find here that the novel neutralizing antibody-derived scFv, NIH45-46, has a greater efficacy against gp120-expressing cell lines than other neutralizing antibodies tested, and crLV-derived CAR T cells demonstrate similar transduction, expansion, and efficacy to conventional LV-derived CAR T cells. We also find that in the presence of HIV, crLV-derived CARs are capable of mobilizing CAR to CD4+-expressing cells and protect CD4 in HIV patient-derived CAR T cells. These data suggest that crLV-derived CARs are a viable approach to expand CARs in HIV patient-derived T cell products and may prove a viable treatment for people living with HIV.
Results
NIH45-46 CAR T Cells Exhibit Greater Efficacy Than CARs Derived from Other Neutralizing Antibodies
There is a plethora of neutralizing antibodies that target the gp120 envelope of HIV,15 and scFvs were derived from broadly neutralizing antibodies that have been reported to have greater than 90% coverage over HIV strains.16, 17, 18, 19, 20 These broad neutralizing antibodies bind to distinct locations of the gp120: PGT121 and PGT128 bind to the V3 glycan, 3BC176 binds to the CD4/V3 loop, and NIH45-46 binds to the CD4 binding domain.16, 17, 18, 19, 20 These anti-GP120 scFvs were expressed on a second generation CAR, in which the IgG4 Fc linked with point mutations at L235E and N297Q to prevent macrophage CD16 and CD32 binding, CD4 transmembrane (TM) domain to anchor to the cell membrane, 4-1BB co-stimulator domain for persistence, and CD3ζ for cytotoxicity21,22 in frame with a truncated human epidermal growth factor receptor (huEGFRt), a marker for CAR expression23 (Figure 1A). To determine whether the CARs were functional, we performed an activation assay. T cells transduced with second generation LV-derived CARs were co-cultured for 24 h with HEK293 cells with or without gp160 expression and analyzed for CD137. The activation assay showed PGT121, PGT128, and NIH45-46, but not 3BC176, were all capable of activating upon gp160 antigen (Figure 1B). To determine which CAR would be most efficacious against a GP120 target, we co-cultured T cells transduced with second generation LV-derived CAR for 4 days with 8e5 cells, which are CEM cells that contain a defective provirus-expressing gp12024 and a stable GFP reporter (8e5.GFP; Figure S1B). A reduction in the number of GFP-positive cells is an indicator of anti-GP120 CAR-mediated cell death. The novel NIH45-46 CAR T cells demonstrated superior cytotoxicity in three donors (Figures 1C, 1D, and S1A).
Figure 1.

Determination of the Optimal scFv for Anti-HIV CAR Therapy
(A) Gene structure of second generation LV-derived NIH45-46 CAR containing 5′ and 3′ truncated LTRs, IgG4 linker (L) containing mutations L235E and N297Q, a CD4 transmembrane (TM) domain, 4-1BB costimulatory domain, and CD3ζ domain. A T2A skip separates a truncated EGFR (EGFRt) from the CAR. (B) Transduced T cells with various second-generation CAR constructs were co-cultured with GFP-expressing HEK.GP160s or HEKs for 24 h prior to analyzing CD137 expression by flow cytometry. (C and D) Donor 1 (C) and donor 2 (D) T cells were transduced with various second generation CAR constructs derived from neutralizing antibodies that were cultured with GFP-expressing 8e5 or CEM cells for 4 days prior to analyzing cell line survival by flow cytometry. Mock-transduced T cells were used as a control. All samples, except CEM control in donor 2, were run in duplicates.
A crLV-Derived CAR Vector Does Not Impact Lentiviral Production and Transduction
To generate a crLV-derived CAR, the CAR was inserted into the crLV vector that contained the long term repeat (LTR) promoter of HIV-1 and essential regulatory RNA motifs present in the gag, pol, and envelope regions, which include the Psi packaging element involved in genome packaging into the virion, the polypurine tract (PPT) needed for reverse transcription of the viral genome, and the Rev response element (RRE) required for nuclear export of viral RNA, respectively12 (Figure 2A). Based on previous studies, this crLV-derived CAR should be expressed and packaged into budding virions by both viral and host proteins specifically imbued in HIV-infected CD4+ T cell populations.14 However, previous studies have also shown that increasing vector size can have deleterious effects on LV packaging and reduced transgene expression.25 We evaluated whether extending the CAR construct by ∼2 kb of regulatory sequence would impact the generation of LV. Following LV production, CARs were generated and titered on Jurkat cells, a T cell line, and expression was confirmed by detecting truncated epidermal growth factor receptor (tEGFR) expression by flow cytometric analysis. Notably, no observed significant difference in LV packaging was observed from six separate LV productions (Figure S2A). Next, we determined to what extent the crLV-derived CAR impacted CAR expression in primary T cells. Following activation and lentiviral transduction of CD3-enriched T cells, both crLV- and LV-derived NIH45-46 CARs were comparably expressed (Figures S2C and S2D). Collectively, these data indicate that the incorporation of the conditionally replicating components to the NIH45-46 CAR does not appear to impact the production and expression of the LV required for CAR T cell production.
Figure 2.

Functionality of crLV-Derived NIH45-46 CAR T Cells
(A) Gene structure of second generation crLV-derived NIH45-46 CAR containing 5′ and 3′ full-length LTRs, non-coding regions for gag, pol, and envelope. (B) Transduced T cells with crLV- or LV-derived NIH45-46 were co-cultured with GFP-expressing HEK.GP160s or HEKs for 24 h prior to analyzing CD137 expression by flow cytometry. (C) crLV- and LV-derived NIH45-46 CAR T cells were cultured with GFP-expressing 8e5 or CEM cells for 4 days prior to analyzing cell line survival by flow cytometry. Mock-transduced T cells were used as a control. All samples were run in duplicates. (D) The crLV- and LV-derived NIH45-46 CAR T cells were co-cultured with NL4-3 infected or uninfected GFP-expressing CEM cells for 4 days prior to analyzing cell line survival by flow cytometry. Mock-transduced T cells were used as control. All samples were run in duplicates.
Conditionally Replicating Anti-HIV CAR T Cells Exhibit Cytotoxic Efficacy against gp120-Expressing Cells
Next, we determined whether the crLV-derived NIH45-46 CAR exhibited comparable efficacy to LV-derived NIH45-46 CAR. We first determined whether the crLV-derived CAR-expressing cell could activate upon recognition of gp120 antigen. Transduced CAR Jurkat cells or T cells were co-cultured with either HEK293 or gp160-expressing HEK293 cells and analyzed for CD69 or CD137 expression, respectively, markers of T cell activation.26 The crLV-derived NIH45-46 CAR Jurkats and primary T cell had comparable activation to the NIH45-46 CAR-treated cells (Figures S2B and 2B).
We next evaluated the cytotoxicity of crLV-derived NIH45-46 CAR T cells when compared with LV-derived NIH45-46 CAR T cells. CD3-enriched T cells from healthy donor PBMCs were transduced with the crLV and LV CARs, co-cultured for 4 days with either 8e5.GFP or CEM.GFP control cells, and analyzed for cytotoxicity by flow cytometry. All crLV-derived NIH45-46 CAR T cell donors (n = 3) had ∼60%–80% cytotoxicity against 8e5.GFP, which was comparable with LV-derived NIH45-46 CAR T cells (Figures 2C, S2E, and S2F). Neither CAR construct had efficacy against gp120-negative CEM.GFP cells. These data indicate that the crLV vector did not impact the functionality of the CAR T cells. Next, to determine whether crLV-derived NIH45-46 CAR cells eliminated virally infected cells, we used CEM.GFP cells infected with a X4-tropic virus, NL4-3, and cultured for 7 days, and verified GP120 expression through flow cytometry (Figure S2G). The crLV-derived and LV-derived NIH45-46 CARs were added at a 1:5 ratio of T cells to infected cells and incubated for 4 days. We observed that both crLV-derived and LV-derived NIH45-46 CARs had comparable cytotoxicity against infected CEM.GFP cells (Figure 2D). Collectively, these data demonstrate that the crLV-derived NIH45-46 CAR T cells are comparable with the conventional NIH45-46 CAR T cells in eliminating HIV-infected cells.
A Conditionally Replicating CAR Vector Can Be Mobilized to Uninfected Cells
To determine whether crLV-derived NIH45-46 CARs are capable of being mobilized and transferred to uninfected cells, we developed a mobilization assay (Figure S3A). In this assay, CEM cells are transduced with either crLV or conventional NIH45-46 CAR and infected with X4-tropic HIV (NL4-3 or LAI). CAR CEM.CCR5 cells were used for this assay because when we performed a cytotoxicity assay, they had no activity against 8e5 cells (Figure S2H). This is important because we wanted to avoid possible expansion or elimination of CAR+ cells upon transfer of supernatant. Cells were cultured for 7 days to allow for HIV production, and the supernatant was then transferred to uninfected CEM.GFP cells. The efficiency of crLV-derived CAR mobilization was determined by identifying CEM.CCR5 cells that were both GFP and CAR positive; these cells were analyzed for CAR expression at days 11 and 14 (4 and 7 days posttransfer of supernatant), respectively. We observed that crLV-derived NIH45-46 CAR, but not the LV NIH45-46 CAR, was capable of being mobilized and transferred to uninfected cells (Figures 3B and 3C). These data suggest that crLV-derived CAR can be mobilized in HIV-infected donors and delivered to uninfected CD4+ cells.
Figure 3.

Mobilization of crLV-Derived NIH45-46 CAR
(A) crLV vectors are capable of functioning only in cells that are infected with HIV (1) that is actively reverse transcribed (2), integrated (3), replicating (5), packaged (7), and released (8), whereby they will hijack the machinery to package the crLV-derived CAR into the virion. The crLV-derived CAR will use the viral machinery to be transcribed (6), packaged (7), and released into the virion (9). (B) Flow cytometric analysis of one of the NL4-3 mobilization assays. Cells were collected on days 4 and 7 after supernatant transfer and stained for EGFR. (C) Graphical analysis of mobilization of crLV-derived CAR using two different X4-tropic viruses at day 7. LAI data are the representation of two experiments, and NL4-3 is the representation of four experiments.
Conditionally Replicating CAR Vector Increases CAR Percentage and Protects CD4 T Cells in HIV Donor
To determine whether crLV-derived NIH45-46 CARs would increase CAR percentage during ex vivo production, we transduced Jurkats with crLV-derived NIH45-46 CAR at a low percentage (8%), then infected them with NL4-3. We observed, by day 8, the percentage of CAR increased to 35% positivity and the mean fluorescence intensity (MFI) had also increased (Figure S3B). To confirm this finding, we transduced HIV-donor T cells with crLV- or LV-derived NIH45-46 CAR. On day seven, following bead removal, we observed increased CAR percentage and MFI in CD4+ T cells from two donors (Figures 4A and S3C) that was not observed in CD8+ T cells (Figure 4B). We also observed a higher percentage of CD4+ T cells in crLV-derived NIH45-46 (43%) when compared with LV-derived NIH45-46 (23%) or mock transduced (24%), suggesting that the crLV-derived NIH45-46 CAR may offer some protection to the CD4+ T cells (Figure 4C). Finally, we investigated the activation capacity of crLV-derived CARs in HIV-infected donors. In one donor, we observed similar activation between crLV- and LV-derived NIH45-46 CAR T cells in two donors (Figures 4D and S3D).
Figure 4.

Flow Cytometric Analysis of crLV-Derived NIH45-46 CAR T Cells from HIV Donor
(A and B) CD4+ (A) and CD8+ (B) crLV- and LV- derived NIH45-46 CAR expression in HIV-donor T cells. (C) CD4+ and CD8+ percentage on day 7 in crLV- and LV-derived NIH45-46 CAR HIV-donor T cells. (D) Transduced T cells with crLV- or LV-derived NIH45-46 were co-cultured with GFP-expressing HEK.GP160s or HEKs for 24 h prior to analyzing CD137 expression by flow cytometry. This is a representative sample of technical replicates (n = 2).
Discussion
HIV reservoirs and viral escape pose an immense challenge when developing a cure strategy for HIV.7 Although broadly neutralizing antibodies have shown phenomenal neutralization of HIV clinically, long-term efficacy is lacking.18,27, 28, 29 Alternatively, using the scFvs of these neutralizing antibodies in CAR T cell therapies offers a potential therapeutic that can eliminate the latent reservoirs and possibly lead to a cure.7 However, developing an anti-HIV CAR T cell has several unique challenges.
CAR T cell therapy relies on effective targeting motifs and, in the case of HIV, there are a wide variety of neutralizing antibodies to develop into CARs.15 However, it is relatively unclear which antibody or ligand is most efficacious for anti-HIV CAR therapy. Hale et al.4 and Ali et al.30 developed multiple anti-gp120 CARs, and all had similar activation and cytotoxicity. Although we observed similar activation of PGT128, PGT121, and NIH45-46 (Figure 1B), we observed differences in CAR functionality across donors (Figures 1C, S1A, and S1B). Specifically, we observed only a partial response in the PGT128 scFv CAR that previously showed significant activity with multiple donors.4,30 This inconsistency could reflect differences in CAR architecture (hinge and TM domain) or the authors using high T cell-to-target ratios (CAR/target 3:1, 5:1, or 10:1), which may rescue the function of the weaker CAR constructs.31 Furthermore, we observed a significant cytotoxic effect from 3BC176 CAR on non-target cells, suggesting potential off-target toxicity in one of the donors (Figure S2D), which also helped us to delineate scFv-specific effects. Under our assays conditions, we show that the novel NIH45-46 CAR is more efficacious than the other three neutralizing antibodies scFVs against GP160 cells, resulting in potent elimination of target cells across multiple donors. Importantly, NIH45-46 CAR T cells may have broader efficacy against a variety of isolates of HIV-1 because the NIH45-46 antibody has been shown to have over 95% neutralization of HIV-1 isolates, although this remains to be determined for CAR T cells.16,20 Overall, these data support the development of NIH45-46 as a potent CAR for the elimination of HIV-infected cells; however, this is not the only challenge for developing anti-HIV CAR therapy.
We have observed that when expanding HIV-donor T cells ex vivo, HIV will become reactivated, which could have detrimental effects on the CD4 percentage, which will decrease over expansion (Figure S1C). The study presented here demonstrates a unique approach to addressing HIV infection by using a crLV CAR. The crLV-derived CARs had equivalent activation and cytotoxicity against gp120-expressing cell lines and HIV-infected cells but also have a secondary function. In the presence of HIV, crLV-derived CARs can be mobilized to uninfected CD4+ T cells (Figure 3A). HIV reactivation has the potential to expand the viral infection to other CD4+ T cells during ex vivo CAR T cell expansion; however, the crLV-derived CARs can also utilize this infection to expand the CAR.7,32 Indeed, we observed that in the presence of HIV, crLV-derived CARs mobilize to uninfected CD4+ cells. The expression of mobilized CAR steadily increases from ∼2% to ∼9% of the population by day 14. We also observed, when transducing T cells from HIV-infected donors, that the crLV-derived CAR, when compared with LV-derived CAR, will have a higher CAR percentage in CD4 T cells with a higher MFI. This phenomenon is not observed in the CD8 T cells, suggesting that there is crLV-derived CAR-mediated mobilization and expansion occurring. Furthermore, only crLV-derived CAR T cells, when compared with LV-derived CAR or mock-transduced T cells, had a higher CD4 percentage (Figure 4C), suggesting that crLV-derived CARs can protect CD4+ T cells ex vivo. Indeed, previous studies have found that conditionally replicating vectors add a level of protection to uninfected cells by diluting the amount of active free virus,12,14,33 although such a notion was not assessed here.
Overall, we find here that crLV-derived CAR constructs can functionally expand T cells into anti-HIV CARs. Earlier studies with conditionally replicating vectors have demonstrated mobilization of small interfering particles and RNAs.14,33 The observations presented here demonstrate not only the mobilization of a large transgene, but the spread of CAR function by the mobilization of the anti-HIV CAR. Such an observation is impacting because it suggests that a single lentiviral vector treatment strategy to cure HIV infection is theoretically feasible. In essence a single crLV-derived CAR, with a suicide gene, treatment in an HIV-infected individual could result in the expansion of the anti-HIV CAR and eventual CAR-mediated eradication of HIV-infected cells. Although a tantalizing prospect, to what extent crLV vectors can be used to parasitize HIV and spread anti-HIV CAR cells in vivo remains to be clinically tested, but may one day prove to be a safe, effective, single-treatment functional cure for HIV infection.
Materials and Methods
DNA Construct
The NIH45-46 s-generation LV-derived CAR construct (pLV-CAR) was provided by Dr. Forman’s Lab at City of Hope, which was generated by inserting a gBlock (IDT) (Table S1) into the NheI and BstEII sites using Gibson Assembly Master Mix (NEB). Generation of all other anti-gp120 broadly neutralizing antibodies (PGT128, PGT121, or 3BC176) was generated by ordering a gBlock (IDT) of the scFVs sequence (Table S1), and using the NEBuilder HiFi DNA Assembly Master Mix, we inserted the sequence into NheI and NotI sites of the second-generation LV plasmid (Figure 2A).21 The crLV CAR plasmid (pcrLV-CAR) was generated by PCR amplifying the CAR using primers (F primer: 5′-GGATCTGCGATCGCTCCGGTGC-3′, R primer 5′-CACTGAAGAGGCCGATCCCCAG-3′), and using the NEBuilder HiFi DNA Assembly Master Mix, we inserted the sequence into NcoI and BstEII digested crLV vector. Infectious molecular clones, pLAI and pNL4-3, were obtained from the NIH AIDS reagent program.34
Extracellular Staining and Flow Cytometry
For flow cytometry analysis, cells were resuspended in phosphate-buffered saline (PBS; Fisher Scientific) containing 2% fetal bovine serum (FBS). For extracellular staining, cells were washed twice prior to incubation with antibody for 30 min at 4°C in the dark. For GP120 staining, cells were incubated with the VRC01 antibody (NIH AIDS reagent program, 12033)35 for 30 min at 4°C in the dark, washed twice, and then incubated with Anti-Human-IgG APC antibody (Clone: IS11-3B2.2.3; Miltenyi). Other antibodies used in this study were CD3 (Clone: SK7; BD Biosciences), CD69 (Clone: L78; BD Biosciences), CD8 (SK1; BD Biosciences), CD4 (SK3; BD Biosciences), and EGFR (Clone: Ay13; BioLegend). Immediately before analyzing, 1/3 volume of 4′,6-diamidino-2-phenylindole (DAPI) was added to each sample for viability.
All samples were analyzed on the MACSQuant Analyzer 10 (Miltenyi). Greater than 100,000 events were collected and analyzed on FlowJo software (version 10; Tree Star). All experiments were gated for density using forward and side scatter, then single cells using area and height, and finally viability using DAPI before analysis.
Lentiviral and HIV Production
The LV- and crLV-derived CARs were generated by seeding 293T cells (20 × 106 cells/T225) 8 h before co-transfecting. 293T cells were co-transfected with a four-plasmid system: pLV-CAR or pcrLV-CAR construct (61 μg), pGagPol (40 μg), pRev (4 μg), and pVSV-g (8 μg) with 2.5M calcium phosphate and 2× Hank’s balanced salt solution as previously described.36 Following overnight incubation, media were replaced with DMEM with 2% FBS and 1:1,000 sodium butyrate. Cells were incubated for 72 h before supernatant was collected. Supernatant was centrifuged 2,000 rpm for 20 min and filtered through 0.45-μm filter to remove cell debris. The virus was precipitated by adding 40% (w/v) polyethylene glycol (PEG) solution and incubated at 4°C overnight. The precipitated virus was centrifuged at 3,000 rpm for 20 min, resuspended in FBS-free DMEM, and ultra-centrifuged at 24,500 rpm for 1.5 h. Isolated virus was resuspended in FBS-free DMEM (Corning) and frozen at −80°C for later use. LV titers, as determined by truncated EGFR (EGFRt) or GFP expression, were quantified by transducing Jurkats (0.1 × 106) with various dilutions of virus (1 μL crude virus or 90, 30, or 10 μL of 1:200 diluted virus in RPMI) and allowed to incubate for 7 days before flow cytometric analysis. The data were analyzed on FlowJo (v.10), and the titer was calculated using the formula (cells transduced × percent expression × 10/virus volume).37
X4-tropic HIV-1 subtype B (LAI and NL4-3) was generated by transfecting infectious molecular clone (20 μg) with Lipofectamine 2000 into 293T cells (4 × 106 cells/T175). Cells were incubated for 48–72 h before supernatant was collected. The supernatant was centrifuged at 1,800 rpm, filtered through 0.45-μm filter, and frozen at −80°C until later use (as described by McCutchan et al.38 and Robertson et al.39). The viral titer was calculated using a TZM-bl assay.40
Cell Lines
Jurkats (ATCC TIB-152), CEM (ATCC CRL-2265), and 8e5 (ATCC CRL-8993) cell lines were obtained from ATCC. CEM.NKR CCR5+ cells (CEM.CCR5), which were transduced to express CCR5, were obtained from the NIH AIDS reagent program.41 Cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS (Gemini). HEK293 and HEK293 cells that stably express gp160 (HEK293-GP160) were maintained in DMEM supplemented with 10% heat-inactivated FBS. HEK293 cells (ATCC CRL-11268) were obtained from ATCC, and HEK293-gp160 cells from the 92UG037.8 strain of HIV were kindly provided by Dr. Chen.42 Cell lines transduced to express GFP and Firefly luciferase (ffluc) were generated by transducing with lentiviral vector encoding for epHIV-7-EGFP-ffluc and then sorted using the BD FACSAria SORP cell sorter (BD Biosciences) for >98% purity.22
Generation of CAR T Cells
Healthy donor whole blood was obtained from the City of Hope blood donor center, and HIV-donor whole blood used for HIV reactivation study was obtained from ZenBio, which HIV donors received for mobilization assay obtained from Dr. Cardoso’s Lab at City of Hope. Blood was ficolled for peripheral blood mononuclear cells (PBMCs),22 and CD3+ cells were then isolated from PBMCs using the EasySep Human T Cell Isolation Kit according to manufacturer’s instructions (STEMCELL Technologies). Freshly isolated CD3+ cells were activated with Dynabeads human T-activator CD3/CD28 (Life Technologies) and transduced 24 h after with a LV encoding for either crLV- or LV-derived NIH45-46CAR:4-1BB:CD3ζ/huEGFRt at an MOI of 2 and expanded in the presence of 50 U/mL recombinant human interleukin-2 (rhIL-2; Novartis) and 0.5 ng/mL interleukin-15 (rhIL-15; Novartis), which was supplemented every 48–72 h during the course of ex vivo expansion for 14–21 days.22 Mock-transduced T cells (no LV) were used as control. HIV expression was determined by an Alliance HIV-1 p24 ELISA (Perkin Elmer).
Jurkat Activation Assay
Ten thousand crLV-derived and LV-derived CAR-expressing Jurkat cells were co-cultured with equivalent numbers of HEK293 or HEK293-gp160 cells in a round-bottom 96-well plate. The Jurkats were collected after 24 h, analyzed for CD69 expression by flow cytometry in the MACSQuant Analyzer 10 (Miltenyi), and quantified using FlowJo Software (v.10).
T Cell Activation Assay
Ten thousand crLV-derived and LV-derived CAR-expressing T cells were co-cultured with equivalent numbers of HEK293 or HEK293-gp160 cells in a flat-bottom 96-well plate. The T cells were collected after 24 h, analyzed for CD137 expression by flow cytometry in the MACSQuant Analyzer 10 (Miltenyi), and quantified using FlowJo Software (v.10).
T Cell Cytotoxicity Assay
Ex vivo expanded CD3+ crLV- or LV-derived CAR T cells (0.5 × 105) were co-cultured with CEM or 8e5 GFP+ cells (2 × 105) in a round-bottom 96-well plate. Cells were collected after 4 days, and cell lines were analyzed for GFP expression by flow cytometry on a MACSQuant Analyzer 10 (Miltenyi). For HIV-infected cells, CEM cells (10 × 105) were infected with NL4-3 MOI 0.01 for 6 h before washing with PBS and then expanded for 7 days before being cultured with ex vivo expanded CD3+ crLV- and LV-derived CAR T cells for 4 days. Cells were analyzed by averaging surviving tumor counts from mock-transduced technical replicate, then dividing each individual sample from the average to determine percent cytotoxicity.
Mobilization Assay
CEM.CCR5+ (5 × 105) cells were transduced with either crLV- and LV-derived CAR (MOI 2) and then enriched for EGFR using EasySep EGFR enrichment kit (STEMCELL Technologies). Enriched CAR CEM.CCR5+ cells (10 × 105) were infected with NL4-3 or LAI virus at a MOI of 0.1 and incubated for 18 h before a PBS wash. Cells were grown for 7 days before 500 μL of supernatant was collected and transferred to uninfected GFP-expressing CEM cells (5 × 105) in 1 mL DMEM/10% FBS in a 24-well plate (Corning). The cells were analyzed at days 11 and 14, days 4 and 7 after supernatant transfer, for EGFR expression by flow cytometry.
Jurkats were transduced at low percentage of crLV-derived NIH45-46 CAR, roughly 10%, and 1 × 106 infected with NL4-3 virus at MOI of 0.1. After 6 h, the cells were washed three times with PBS. Cells were cultured for 8 days before analyzing for EGFR expression by flow cytometry.
Statistics
Data are presented as mean and standard deviation. Statistical comparisons between groups were performed on GraphPad Prism (v.8.3.1) using the unpaired two-tailed t test to calculate p value.
Author Contributions
Conceptualization, R.Z.U., X.W., and K.V.M.; Methodology, R.Z.U. and L.L.; Investigation, R.Z.U., L.L., G.S., and C.S.; Writing – Original Draft: R.Z.U.; Writing – Review & Editing: T.S., X.W., and K.V.M.; Funding Acquisition, K.V.M.; Resources, X.W. and K.V.M; Supervision, T.S., X.W., and K.V.M.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Mental Health, USA (grant R01 113407-01 to K.V.M. and City of Hope).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.09.014.
Supplemental Information
References
- 1.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anthony-Gonda K., Bardhi A., Ray A., Flerin N., Li M., Chen W., Ochsenbauer C., Kappes J.C., Krueger W., Worden A. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 2019;11:eaav5685. doi: 10.1126/scitranslmed.aav5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hale M., Mesojednik T., Romano Ibarra G.S., Sahni J., Bernard A., Sommer K., Scharenberg A.M., Rawlings D.J., Wagner T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017;25:570–579. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazarzaei A., Vafaei M., Ghasemian A., Mirforughi S.A., Rajabi Vardanjani H., Alwan N.A.S. Memory and CAR-NK cell-based novel approaches for HIV vaccination and eradication. J. Cell. Physiol. 2019;234:14812–14817. doi: 10.1002/jcp.28280. [DOI] [PubMed] [Google Scholar]
- 6.Herzig E., Kim K.C., Packard T.A., Vardi N., Schwarzer R., Gramatica A., Deeks S.G., Williams S.R., Landgraf K., Killeen N. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell. 2019;179:880–894 e10. doi: 10.1016/j.cell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner T.A. Quarter Century of Anti-HIV CAR T Cells. Curr. HIV/AIDS Rep. 2018;15:147–154. doi: 10.1007/s11904-018-0388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D., Aguilar B., Starr R., Alizadeh D., Brito A., Sarkissian A., Ostberg J.R., Forman S.J., Brown C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3:e99048. doi: 10.1172/jci.insight.99048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman A.N., Singh P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018;75:2491–2507. doi: 10.1007/s00018-018-2772-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchand C., Johnson A.A., Semenova E., Pommier Y. Mechanisms and inhibition of HIV integration. Drug Discov. Today Dis. Mech. 2006;3:253–260. doi: 10.1016/j.ddmec.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agosto L.M., Henderson A.J. CD4+ T cell subsets and pathways to HIV latency. AIDS Res. Hum. Retroviruses. 2018;34:780–789. doi: 10.1089/aid.2018.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dropulić B., Hĕrmánková M., Pitha P.M. A conditionally replicating HIV-1 vector interferes with wild-type HIV-1 replication and spread. Proc. Natl. Acad. Sci. USA. 1996;93:11103–11108. doi: 10.1073/pnas.93.20.11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberger L.S., Schaffer D.V., Arkin A.P. Theoretical design of a gene therapy to prevent AIDS but not human immunodeficiency virus type 1 infection. J. Virol. 2003;77:10028–10036. doi: 10.1128/JVI.77.18.10028-10036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shrivastava S., Charlins P., Ackley A., Embree H., Dropulic B., Akkina R., Weinberg M.S., Morris K.V. Stable Transcriptional Repression and Parasitism of HIV-1. Mol. Ther. Nucleic Acids. 2018;12:12–18. doi: 10.1016/j.omtn.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dashti A., DeVico A.L., Lewis G.K., Sajadi M.M. Broadly Neutralizing Antibodies against HIV: Back to Blood. Trends Mol. Med. 2019;25:228–240. doi: 10.1016/j.molmed.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diskin R., Scheid J.F., Marcovecchio P.M., West A.P., Jr., Klein F., Gao H., Gnanapragasam P.N., Abadir A., Seaman M.S., Nussenzweig M.C., Bjorkman P.J. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334:1289–1293. doi: 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Julien J.-P., Sok D., Khayat R., Lee J.H., Doores K.J., Walker L.M., Ramos A., Diwanji D.C., Pejchal R., Cupo A. Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS Pathog. 2013;9:e1003342. doi: 10.1371/journal.ppat.1003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krumm S.A., Mohammed H., Le K.M., Crispin M., Wrin T., Poignard P., Burton D.R., Doores K.J. Mechanisms of escape from the PGT128 family of anti-HIV broadly neutralizing antibodies. Retrovirology. 2016;13:8. doi: 10.1186/s12977-016-0241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pancera M., Changela A., Kwong P.D. How HIV-1 entry mechanism and broadly neutralizing antibodies guide structure-based vaccine design. Curr. Opin. HIV AIDS. 2017;12:229–240. doi: 10.1097/COH.0000000000000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song R., Pace C., Seaman M.S., Fang Q., Sun M., Andrews C.D., Wu A., Padte N.N., Ho D.D. Distinct HIV-1 neutralization potency profiles of ibalizumab-based bispecific antibodies. J. Acquir. Immune Defic. Syndr. 2016;73:365–373. doi: 10.1097/QAI.0000000000001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonnalagadda M., Mardiros A., Urak R., Wang X., Hoffman L.J., Bernanke A., Chang W.C., Bretzlaff W., Starr R., Priceman S. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther. 2015;23:757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urak R., Walter M., Lim L., Wong C.W., Budde L.E., Thomas S., Forman S.J., Wang X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer. 2017;5:26. doi: 10.1186/s40425-017-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilburn K.M., Mwandumba H.C., Jambo K.C., Boliar S., Solouki S., Russell D.G., Gludish D.W. Heterogeneous loss of HIV transcription and proviral DNA from 8E5/LAV lymphoblastic leukemia cells revealed by RNA FISH:FLOW analyses. Retrovirology. 2016;13:55. doi: 10.1186/s12977-016-0289-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar M., Keller B., Makalou N., Sutton R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001;12:1893–1905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- 26.Chang Z.L., Lorenzini M.H., Chen X., Tran U., Bangayan N.J., Chen Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018;14:317–324. doi: 10.1038/nchembio.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein F., Halper-Stromberg A., Horwitz J.A., Gruell H., Scheid J.F., Bournazos S., Mouquet H., Spatz L.A., Diskin R., Abadir A. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492:118–122. doi: 10.1038/nature11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu L., Pegu A., Rao E., Doria-Rose N., Beninga J., McKee K., Lord D.M., Wei R.R., Deng G., Louder M. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358:85–90. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bardhi A., Wu Y., Chen W., Li W., Zhu Z., Zheng J.H., Wong H., Jeng E., Jones J., Ochsenbauer C. Potent in vivo NK cell-mediated elimination of HIV-1-infected cells mobilized by a gp120-bispecific and hexavalent broadly neutralizing fusion protein. J. Virol. 2017;91 doi: 10.1128/JVI.00937-17. e00937-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali A., Kitchen S.G., Chen I.S.Y., Ng H.L., Zack J.A., Yang O.O. HIV-1-Specific Chimeric Antigen Receptors Based on Broadly Neutralizing Antibodies. J. Virol. 2016;90:6999–7006. doi: 10.1128/JVI.00805-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolnikov A., Klamer G., Chitranjan A., Xu N., Shen S., Micklethwaite K.P., Lock R.B., O’Brien T. Identifying the Factors Modulating the Efficacy of CAR-T Cell Therapy. Blood. 2014;124:5813. [Google Scholar]
- 32.Tschumi B.O., Dumauthioz N., Marti B., Zhang L., Lanitis E., Irving M., Schneider P., Mach J.P., Coukos G., Romero P., Donda A. CART cells are prone to Fas- and DR5-mediated cell death. J. Immunother. Cancer. 2018;6:71. doi: 10.1186/s40425-018-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanner E.J., Jung S.-Y., Glazier J., Thompson C., Zhou Y., Martin B., Son H.-I., Riley J.L., Weinberger L.S. Discovery and Engineering of a Therapeutic Interfering Particle (TIP): a combination self-renewing antiviral. bioRxiv. 2019 doi: 10.1101/820456. [DOI] [Google Scholar]
- 34.Adachi A., Gendelman H.E., Koenig S., Folks T., Willey R., Rabson A., Martin M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X., Yang Z.Y., Li Y., Hogerkorp C.M., Schief W.R., Seaman M.S., Zhou T., Schmidt S.D., Wu L., Xu L. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cribbs A.P., Kennedy A., Gregory B., Brennan F.M. Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnol. 2013;13:98. doi: 10.1186/1472-6750-13-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B., Metharom P., Jullie H., Ellem K.A., Cleghorn G., West M.J., Wei M.Q. The significance of controlled conditions in lentiviral vector titration and in the use of multiplicity of infection (MOI) for predicting gene transfer events. Genet. Vaccines Ther. 2004;2:6. doi: 10.1186/1479-0556-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCutchan F.E., Salminen M.O., Carr J.K., Burke D.S. HIV-1 genetic diversity. AIDS. 1996;10(Suppl 3):S13–S20. [PubMed] [Google Scholar]
- 39.Robertson D.L., Anderson J.P., Bradac J.A., Carr J.K., Foley B., Funkhouser R.K., Gao F., Hahn B.H., Kalish M.L., Kuiken C. HIV-1 nomenclature proposal. Science. 2000;288:55–56. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 40.Johnson V., Byington R. Infectivity assay (virus yield assay) In: Aldovini A., Walker B.D., editors. Techniques in HIV research. Stockton Press; 1990. pp. 71–76. [Google Scholar]
- 41.Trkola A., Matthews J., Gordon C., Ketas T., Moore J.P. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J. Virol. 1999;73:8966–8974. doi: 10.1128/jvi.73.11.8966-8974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J., Kovacs J.M., Peng H., Rits-Volloch S., Lu J., Park D., Zablowsky E., Seaman M.S., Chen B. HIV-1 ENVELOPE. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science. 2015;349:191–195. doi: 10.1126/science.aaa9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.