

Abstract
Malignant pleural mesothelioma (MPM) is an aggressive cancer, associated with poor prognosis. We assessed the feasibility of patient-derived cell cultures to serve as an ex vivo model of MPM. Patient-derived MPM cell cultures (n=16) exhibited stemness features and reflected intratumour and interpatient heterogeneity. A subset of the cells were subjected to high-throughput drug screening and coculture assays with cancer-specific cytotoxic T cells and showed diverse responses. Some of the biphasic MPM cells were capable of processing and presenting the neoantigen SSX-2 endogenously. In conclusion, patient-derived MPM cell cultures are a promising and faithful ex vivo model of MPM.
Keywords: mesothelioma, pleural disease
Introduction
Malignant pleural mesothelioma (MPM) is an aggressive malignancy, with increasing incidence in many countries.1–3 Patients often present with advanced stage malignancy and malignant pleural effusion (MPE).2 Current treatment modalities offer limited life span benefit; thus, MPM is associated with poor prognosis and bears high mortality rate.3
Due to the complexity and heterogeneity of MPM, a biomarker-driven approach to therapy is likely to be required to control tumour progression, fluid formation and improve patients’ quality of life and survival. Preclinical in-vitro MPM models are an invaluable tool to study the disease and develop novel treatments.4–7 We designed a study to assess the feasibility of patient-derived MPM cell cultures, to serve as a faithful translational ex vivo platform suitable for the development of biomarker-guided therapies.
Results
Patient-derived MPM cell cultures established from MPE specimens displayed morphological heterogeneity and features of cancer stemness
We generated a panel of 16 patient-derived MPM cell cultures originated from MPE specimens (online supplementary figures 1-2, online supplementary table 1). All cell cultures were in vitro morphologically stable, and displayed diverse growth rates and phenotypes, including cobblestone and spindle shapes (figure 1A–E). A blinded National Health Service Consultant Pathologist examined May-Grunwald Giemsa stained cytospin specimens of the MPM cell cultures and confirmed their malignant nature. They exhibited several cytological cancer hallmarks: atypical, prominent, pleomorphic, large and multiple nucleoli (figure 1A–E). MPM cell cultures were able to form tumour-spheroids exhibiting stemness potency and indicating the existence of a tumour stem cell subpopulation (figure 1F–M).
Figure 1.
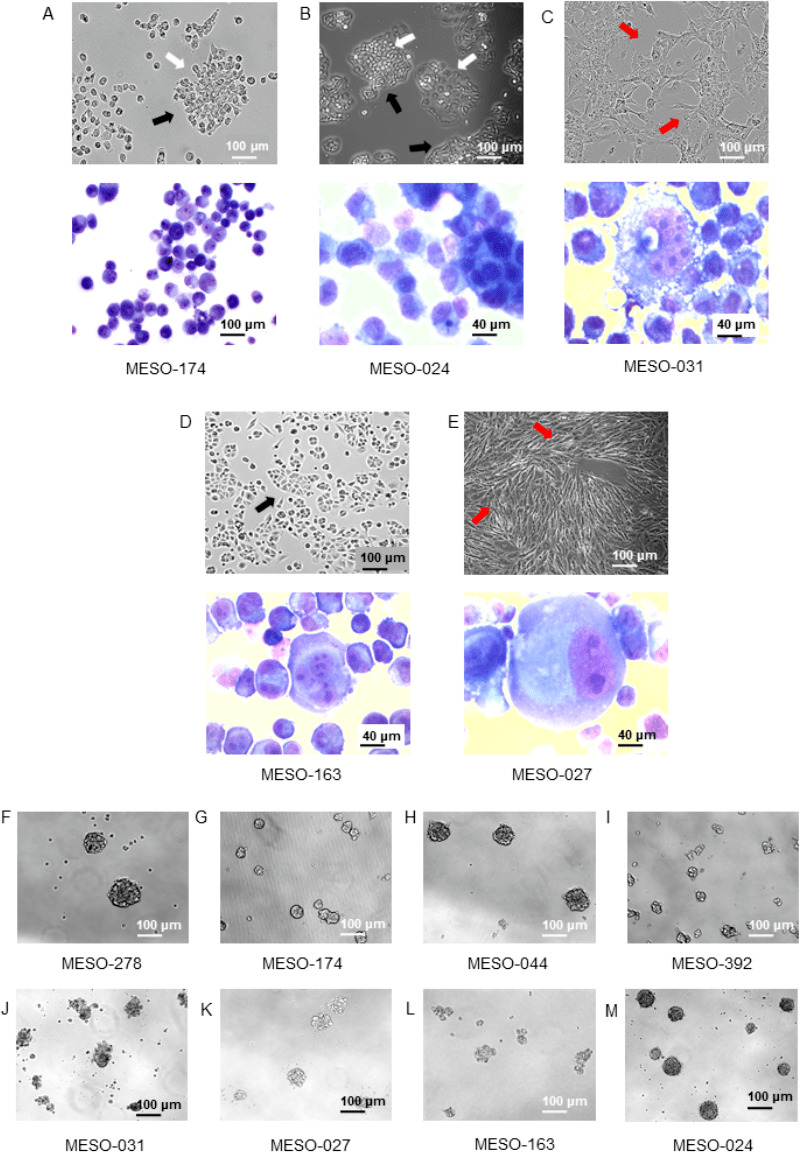
Patient-derived malignant pleural mesothelioma (MPM) cell cultures originated from malignant pleural effusion specimens are truly cancerous and showed tumour stemness properties. (A–E) Top: phase contrast images (10× magnification) of representative MPM cells in culture showing colony formation (white arrows), cobblestone (black arrows) and spindle (red arrows) shapes. Bottom: May Grunwald Giemsa-stained cytospin specimens of selected MPM cell cultures showing (A) pleomorphic and multiple nucleoli (10× magnification), (B) small atypical nucleoli and two-tone cytoplasm typical of mesothelial morphology (40× magnification), (C) atypical features with large nucleus and very large nucleoli (40× magnification), (D) bizarre nucleus with multiple nucleoli (40× magnification), (E) large and multiple nuclei (binucleate) and atypical and multiple nucleoli (40× magnification). (F–M) Phase contrast images of tumour-spheroids formed by MPM patient-derived cancer cell cultures (10× magnification). Patient-derived MPM cell cultures were able to form tumour-spheres highlighting tumour stemness properties and the existence of a cancer stem cell subpopulation.
thoraxjnl-2020-215027supp001.pdf (7MB, pdf)
Genomic profiling of the patient-derived MPM cell cultures exhibited interpatient variation
We subjected to whole-genome sequencing three of the MPM cell cultures to profile their mutational landscape (online supplementary figures 3-5 and online supplementary table 2) and determine the mutational status of the known MPM-associated tumour suppressor genes: TRP53, NF2, BAP1, LATS2, SETD2 and CDKN2A.8 The analysis revealed a diverse spectrum of mutational profiles (non-synonymous, stop codon gain and frameshift insertion mutations) for these genes highlighting the importance of personalised and targeted therapies in MPM (figure 2A–B). These findings are in line with previously published data and indicate a resemblance at the molecular level between the patient-derived cell cultures and MPM tumours.8
Figure 2.

Whole-genome sequencing and high-throughput drug screening of the malignant pleural mesothelioma (MPM) patient-derived cell cultures revealed heterogeneous mutational profiles and different responses to anticancer agents. The MPM cell cultures reflect ex vivo the interpatient heterogeneity. (A–B) To screen the interindividual mutational landscape variation and detect mutations caused by serial passaging, we subjected three of the cell cultures (two epithelioid: MESO-163, MESO-031 and one biphasic: MESO-174) to whole-genome sequencing at two different timepoints: an early passage, when the cells were seeded (passage 0 (P0)) and a late time point (passage 20 (P20)). (A) Graph showing the functional genomic region for each mutation that was detected at the early passage cell cultures for the MPM-related genes: TRP53, NF2, BAP1, LATS2, SETD2 and CDKN2A. Each colour represents a different genomic region (intergenic, UTR5, exonic, intronic, UTR3). (B) Graph displaying the exonic variant function for the mutations detected in the MPM-related genes TRP53, NF2, BAP1, LATS2, SETD2, CDKN2A. Each colour represents a different functional consequence of the point mutation (synonymous, non-synonymous, stop gain, frameshift insertion, non-frameshift insertion). (C–E) To investigate the potential value of the patient-derived MPM cell cultures as an ex vivo platform suitable to develop and assess novel treatment agents, a proof-of-concept high-throughput drug screening assay was performed with the three genome sequenced cell cultures (MESO-163, MESO-174, MESO-031). To examine the drug response of the cells, we used a library (n=316, online supplementary table 3) of antitumour agents approved for different primary malignancies. (C) Heatmap of unsupervised hierarchical clustering of the drug responses for the MPM cell cultures subjected to high-throughput drug screening. Drugs were added 24 hours post seeding the cells at three different concentrations (100 nM, 1 µM, 10 μM). Cell viability was measured 48 hours post treatment and was compared (z-score) to the combination of 10 μM/1.6 μM pemetrexed/cisplatin (positive control), the current clinical first-line chemotherapy. Each column represents an MPM cell line and each row a drug (10 μM). The colour scale represents cell viability z-scores. Bright red is for maximal viability (no drug response), while dark blue is for best drug response. (D) Heatmap of unsupervised hierarchical clustering of the drug responses for the top six agents (bortezomib, carfilzomib, dactinomycin, dinaciclib, omacetaxine mepesuccinate and idarubicin) and the combination pemetrexed/cisplatin (positive control). These drugs were more efficient than the positive control, 10 μM/1.6 μM pemetrexed/cisplatin at all concentrations (100 nM, 1 μM, 10 μM) for all the cell cultures. The heatmap is for the 10 μM concentration. The colour scale represents cell viability z-scores. Bright red is for maximal viability (no drug response), while dark blue is for best drug response. (E) Drug response curve of the patient-derived cell cultures MESO-163, MESO-031 and MESO-174 for the combination of pemetrexed and cisplatin, the current clinical first-line chemotherapy regimen for MPM. MESO-163 on the left shows the highest response among the three cell cultures, followed by MESO-031 and MESO-174. Interestingly, the clinical data correlate with the drug screening findings as patient MESO-163 responded to chemotherapy while patient MESO-031 did not respond.
High-throughput drug screening of patient-derived MPM cell cultures revealed heterogeneous response profiles
The three sequenced MPM cell cultures were subjected to a high-throughput drug screening assay (316 Food and Drug Administration (FDA) approved antitumour agents, online supplementary figure 6) and exhibited different response profiles, a finding that reflects the interpatient heterogeneity (figure 2C). We detected six drugs: bortezomib, carfilzomib, dactinomycin, dinaciclib, omacetaxine and idarubicin, which were consistently more cytotoxic compared with pemetrexed/cisplatin (positive control) the current clinical first-line chemotherapy regimen (figure 2D). Clusters of topoisomerase, mitotic (including microtubule stabilisers), histone deacetylase and mammalian target of rapamycin (mTOR) inhibitors were among the most effective drugs (online supplementary figure 7). We calculated the drug response curves to pemetrexed/cisplatin for the three MPM cell cultures. MESO-163 was more sensitive compared with MESO-031 and MESO-174 (figure 2E).
HLA-A2-restricted cancer-specific CD8+ T cells identified SSX2+ patient-derived MPM cell cultures
To evaluate the feasibility to use the MPM cell panel in T cell immunotherapy assays, we performed a series of coculture experiments with HLA-A2-restricted SSX2-specific cytotoxic CD8+ T cells (CTCs) and four HLA-A2+ MPM cell cultures (two epithelioid: MESO-044, MESO-278 and two biphasic: MESO-174, MESO-392; online supplementary figure 8).
As expected, the exposure of cancer-specific CTCs, to SSX2-peptide loaded MPM cells at varying peptide concentrations triggered the induction of cytotoxic immune responses (online supplementary figures 9-13). Interestingly, the two biphasic MPM cell cultures were able to induce a CTC immune response even without the SSX2-peptide pulse. In specific, we detected increased: CD107a expression, killing capacity and production of two antitumour cytokines: interferon γ and tumour necrosis factor α (figure 2A–C) by the CTCs. The biphasic cell cultures MESO-174 and MESO-392 exhibited statistically significantly higher levels of SSX2 gene expression compared with the epithelioid MESO-044 and MESO-278 (figure 3D). These findings suggest that the two biphasic MPM cell cultures were able to endogenously process and present the SSX-2 epitope peptide.
Figure 3.

Coculture of the biphasic malignant pleural mesothelioma (MPM) cell cultures: MESO-174 and MESO-392 with HLA-A2-restricted, SSX-2 specific, CD8+ T cells-induced T cell degranulation, killing capacity and production of the cytotoxic cytokines interferon γ (IFNγ) and tumour necrosis factor α (TNFα) without the peptide pulse. (A) Barplot presenting the percentage of CD107a+ T cells on coculture with the epithelioid: MESO-044, MESO-278 and the biphasic: MESO-174, MESO-392 MPM cell cultures, without the SSX2-peptide loading. The biphasic cell cultures induced production of CD107a. Data are summarised as mean±SEM. The table below shows the comparisons by one-way analysis of variance (ANOVA) with Tukey’s correction for multiple testing. Quadruplicate samples were used per cell culture/condition. (B) Graph presenting the killing capacity of the T cells on coculture with the epithelioid: MESO-044, MESO-278 and the biphasic: MESO-174, MESO-392 MPM cell cultures, without the peptide loading. The vertical axis shows the percentage of MPM cell death and the horizontal the MPM cell culture. Notably, the biphasic MPM cells triggered T cell cytotoxicity. Data are summarised as mean±SEM. The table below shows the comparisons by one-way ANOVA with Tukey’s correction for multiple testing. Quadruplicate samples were used per cell culture/condition. (C) Barplots of IFNγ (left) and TNFα (right) expression by the T cells on coculture with the epithelioid: MESO-044, MESO-278 and the biphasic: MESO-174, MESO-392 MPM cell cultures, without the peptide pulse. The biphasic MPM cell cultures induced the expression of the cytotoxic cytokines IFNγ and TNFα. Data are summarised as mean±SEM. The tables below show the comparisons by one-way ANOVA with Tukey’s correction for multiple testing. Quadruplicate samples were used per cell culture/condition. (D) SSX-2 mRNA expression by qPCR of the epithelioid cell cultures: MESO-044 and MESO-278 and the biphasic: MESO-174 and MESO-392 corrected to GAPDH and compared with MESO-044 expression. The two biphasic cell cultures displayed increased SSX2 expression levels compared with the epithelioid ones. These data combined suggest the existence of an SSX2+ subpopulation within the biphasic MPM cell cultures MESO-174 and MESO-392. Data are summarised as mean±SEM. The table on the right shows the comparisons by one-way ANOVA with Tukey’s correction for multiple testing. Triplicate samples were used per cell culture/condition.
Discussion
We report the establishment and validation of a panel of 16 patient-derived MPM cell cultures as an ex-vivo model of the disease. The cells were polyclonal, displayed tumour stemness features and demonstrated diverse morphologies, growth rates and mutational landscapes. Three and four MPM cell cultures were subjected to high-throughput drug screening and CTCs coculture assays and exhibited diverse responses. We detected a correlation between the response to pemetrexed/cisplatin and the observed clinical phenotype and discovered two biphasic cell cultures that harboured an SSX2+ subpopulation. Cell cultures from the same patient were established at different clinical course timepoints, which may provide the potential to study molecular alterations due to MPM progression.
Strengths and limitations
The patient-derived ex vivo MPM model offers the potential to serve as a faithful and practical tool to study MPM and accelerate research.4–7 The diverse profiles of MPM cell cultures reflect the interpatient variability. Single sequencing in combination with high-throughput drug screening of the MPM cell panel offer the potential to study treatment response mechanisms, identify neoantigens and importantly investigate the molecular pathways which are involved in tumour stem cell resistance to treatments and tumour relapse.9 The discovery of neoantigens, such as SSX2, is important as they are an attractive drug target due to specificity.10 The development and validation of tumour classification molecular signatures with regards to treatment response could improve patient clinical management. Coculture of tumour and cancer-specific CTCs has the potential to unveil their multifaceted crosstalk and develop novel immunotherapies.
The model described herein has limitations. The establishment of a cell culture is a relative lengthy procedure (1–2 months), the success rate (45%) of cell culture establishment requires improvement and the reasons for which some samples fail to generate a cell line require investigation. The drug screening hits need to be validated with further experiments that directly measure cell death.
Conclusion
In conclusion, patient-derived MPM cell cultures exhibit a remarkable resemblance to the clinical phenotype and features of MPM, which are suggestive of a faithful ex-vivo human model.
Acknowledgments
The authors would like to thank the Oxford Respiratory Trials Unit, the Target Discovery Institute and the Nuffield Department of Medicine, University of Oxford for the support to conduct the research project.
Footnotes
Contributors: NIK, NMR and IP conceived and designed the study. NIK, RA, MAH, XY and YP did laboratory assays and analysed the data. MM did cytological and histological analyses. NIK and MAH did gene expression assays and analysed the data. NIK, RA and SM analysed the whole-genome sequencing data. EB, RMM, RJH and VG collected clinical data. NIK and RA analysed clinical data. NIK, RA, SH and DE did the high-throughput drug screening and analysed the data. NIK, SJ, CV and MD provided resources and prepared the ethics protocols. GTS and TD provided materials and knowledge. NMR and IP supervised the study. All authors reviewed the manuscript.
Funding: Oxford Centre for Histopathology Research and the Oxford Radcliffe Biobank are supported by the NIHR Oxford Biomedical Research Centre (Molecular Diagnostics theme), the Oxford Cancer Centre (CRUK), the NIHR Thames Valley and South Midlands Clinical Research Network, individual stakeholders, and the University of Oxford’s Medical Sciences Division. NIK and NMR are supported by a National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) grant. TD and YP are supported by Medical Research Council, UK. This work was supported by an Oxfordshire Health Services Research Committee (OHSRC) Research Grant (ID: 1336) to NIK and a Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS, ID: 2018-I2M-2–002) to TD.
Competing interests: IP works for AstraZeneca in a non-related field.
Patient consent for publication: Not required.
Ethics approval: Oxford Radcliffe Biobank Tissue Access Committee approval reference number: 19/A107.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Leong SL, Zainudin R, Kazan-Allen L, et al. . Asbestos in Asia. Respirology 2015;20:548–55. 10.1111/resp.12517 [DOI] [PubMed] [Google Scholar]
- 2. Bibby AC, Tsim S, Kanellakis N, et al. . Malignant pleural mesothelioma: an update on investigation, diagnosis and treatment. Eur Respir Rev 2016;25:472–86. 10.1183/16000617.0063-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Woolhouse I, Bishop L, Darlison L, et al. . British thoracic Society guideline for the investigation and management of malignant pleural mesothelioma. Thorax 2018;73:i1–30. 10.1136/thoraxjnl-2017-211321 [DOI] [PubMed] [Google Scholar]
- 4. Kobayashi M, Takeuchi T, Ohtsuki Y. Establishment of three novel human malignant pleural mesothelioma cell lines: morphological and cytogenetical studies and EGFR mutation status. Anticancer Res 2008;28:197–208. [PubMed] [Google Scholar]
- 5. de Cupis A, Pirani P, Favoni RE. Establishment and preliminary characterization of human malignant mesothelioma cell lines. Monaldi Arch Chest Dis 1998;53:188–92. [PubMed] [Google Scholar]
- 6. Manning LS, Whitaker D, Murch AR, et al. . Establishment and characterization of five human malignant mesothelioma cell lines derived from pleural effusions. Int J Cancer 1991;47:285–90. 10.1002/ijc.2910470219 [DOI] [PubMed] [Google Scholar]
- 7. Versnel MA, Bouts MJ, Hoogsteden HC, et al. . Establishment of human malignant mesothelioma cell lines. Int J Cancer 1989;44:256–60. 10.1002/ijc.2910440212 [DOI] [PubMed] [Google Scholar]
- 8. Hmeljak J, Sanchez-Vega F, Hoadley KA, et al. . Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov 2018;8:1548–65. 10.1158/2159-8290.CD-18-0804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–84. 10.1038/nrc1590 [DOI] [PubMed] [Google Scholar]
- 10. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2020-215027supp001.pdf (7MB, pdf)