

Abstract
Polycyclic diterpenes exhibit many important biological activities, but de novo synthetic access to these molecules is highly challenging due to their structural complexity. Semisynthetic access has also been limited by the lack of chemical tools for scaffold modifications. We report a chemoenzymatic platform to access highly oxidized diterpenes by a hybrid oxidative approach that strategically combines chemical and enzymatic oxidation methods. This approach allows for selective oxidations of previously inaccessible sites on the parent carbocycles and enables abiotic skeletal rearrangements to additional underlying architectures. We synthesized a total of nine complex natural products with rich oxygenation patterns and skeletal diversity in ten steps or less from ent-steviol.
One Sentence Summary:
Synthesis of nine complex diterpenes through combined chemical and enzymatic C–H oxidation methods.
The ent-kauranes, ent-atisanes and ent-trachylobanes (Figure 1A) are biosynthetically-related families of diterpene natural products with wide-ranging biological activities (1). These activities include inhibition of ion channels, signal transduction cascades and the inflammasome (2,3,4). The main structural difference between the three natural product families lies in the carbocyclic architecture of their C/D rings: ent-kauranes share a common [3.2.1] bicyclic ring system while ent-atisanes and ent-trachylobanes are characterized by the presence of a [2.2.2] bicycle and [3.2.1.0] tricycle, respectively. These three distinct ring systems are thought to arise from a common precursor (5), ent-copalyl pyrophosphate, through several Wagner–Meerwein shifts following initial formation of ent-pimarenyl cation (Figure S1). Oxidative tailoring and oxidation-enabled rearrangements could then take place on the minimally-oxidized carbocyclic skeleton, contributing to the enormous diversity found in the three natural product families. Despite recent discoveries on terpene synthases involved in the production of these families (6,7,8), most of the oxygenases that are responsible for the subsequent tailoring events have yet to be identified. Such limitations have rendered synthetic biology access to these privileged structures difficult.
Figure 1.
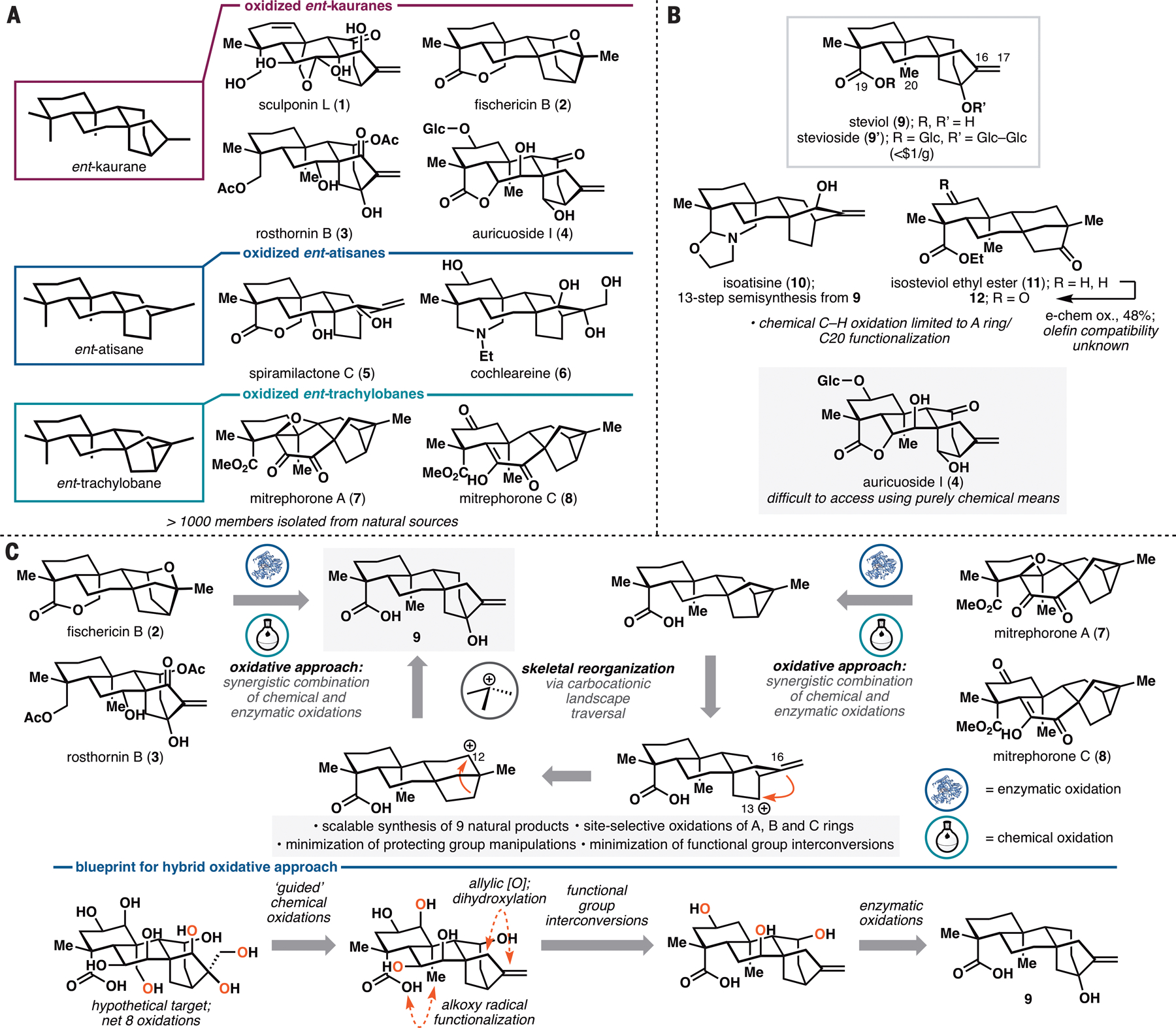
A. Selected examples of oxidized ent-kauranes, ent-atisanes and ent-trachylobanes. B. Limitations of purely chemical C–H oxidation approaches in steviol-based semisynthesis. C. Retrosynthetic analysis of oxidized ent-kauranes, ent-atisanes and ent-trachylobanes employing a hybrid oxidative approach that combines chemical and enzymatic C–H oxidations. Glc = β-D-glucopyranosyl.
Due to their intriguing architectures and promising biological activities, ent-kauranes, ent-atisanes and ent-trachylobanes have been the subject of many synthetic studies, more commonly through the use of de novo approaches (see refs. 9,10 for recent reviews; see Figure S2 for full graphical summary). Semisynthetic efforts, while not as popular, have also been pursued, including Mander’s pioneering synthesis of 6,7-seco-ent-kauranes from gibberellic acid (11) and recent syntheses (12,13) of atisane-type diterpene alkaloids (e.g. 10, Figure 1B) and neotripterifordin from stevioside. These examples suggest the possibility of developing a more systematic pursuit of highly oxidized ent-kauranes from stevioside, which at $0.65/g, represents an attractive starting point for synthesis. However, the realization of this concept has been hampered by the lack of useful methods for scaffold modification. As steviol (stevioside aglycone, 9) lacks any appropriate functional handles within its A, B or C ring, semisynthetic elaborations of the framework have mostly relied on the use of C19 functionality in Hofmann–Löffler–Freytag or Suarez hypoiodite reactions (12,13), which currently only allow modification of the C20 methyl group. Remote chemical oxidation of steviol has remained unexplored, likely due to the incompatibility of its C16–C17 exo olefin with C–H oxidation conditions. To the best of our knowledge, such oxidation has only been performed electrochemically (14) on isosteviol ethyl ester (11), an ent-beyerane compound lacking any olefin, producing the corresponding C2-keto product. An efficient, remote, and site-selective C–H functionalization toolkit that can act on ent-kaurane skeleton would overcome a major roadblock in converting steviol to ent-kauranes containing multiple oxidations on their A, B and/or C rings (e.g. auricuoside I, Figure 1B). Furthermore, oxidized ent-atisanes and ent-trachylobanes remain hard to access using semisynthetic approaches due to the need to convert the C/D ring of ent-kaurane to those of ent-atisane and ent-trachylobane in a facile manner (< five steps).
We have developed a chemoenzymatic synthetic strategy to access a wide array of oxidized ent-kauranes, ent-atisanes and ent-trachylobanes. A key feature of this strategy is the application of a hybrid oxidative approach featuring a combination of remote biocatalytic hydroxylations and ‘guided’ C–H oxidation methods (15) to access to a variety of oxidation patterns previously unattainable using purely chemical means. In prior applications of C–H functionalization in natural product synthesis, each of these oxidation strategies has been employed independently (16,17), and our approach illustrates the value in combining them in a synergistic fashion. We also designed a skeletal reorganization sequence towards ent-atisane and ent-trachylobane frameworks starting from steviol (Figure 1C), which can enter analogous oxidation series to afford highly decorated members of these families. A total of nine natural product targets were synthesized with high synthetic ideality, as well as redox- and step-economy, highlighting the enabling nature of our strategy.
Enzymatic Tool Development for Scaffold Modifications
The overarching premise of our synthetic strategy is to identify biocatalytic oxidation methods to address the methodology gap in the selective functionalization of the A, B, and C rings of minimally oxidized ent-kaurane, ent-atisane and ent-trachylobane skeletons. Each of the newly introduced hydroxyl groups is viewed as a gateway for further manipulations to access many members of these diterpene families through a combination of functional group interconversions and chemical C–H oxidation methods (Figure 1C). To access the ent-atisane and ent-trachylobane frameworks, we drew inspiration from the postulated biogenetic relationship between the two and the ent-kaurane framework. One hypothesis proposed that they arise from a common carbocation via divergent alkyl/H shifts (5). As such rearrangements could potentially be reversible in nature, we hypothesized that under suitable conditions, an ent-beyerane skeleton could be converted to an ent-atisane or ent-trachylobane product. Synthetic entry into this sequence could be achieved readily via the well-precedented conversion of the ent-kaurene stevioside to the ent-beyerane isosteviol (18). We postulated that further carbocation generation at C12 would trigger a Wagner–Meerwein rearrangement to an ent-atisane product, from which access to the ent-trachylobane framework could be realized via C–C bond formation between C13 and C16. Following such skeletal reorganization, an array of oxidative transformations on minimally-oxidized ent-atisane and ent-trachylobane skeleton would provide rapid access to targets such as spiramilactone C (5) and the mitrephorones (7, 8).
Successful execution of the aforementioned strategy would thus hinge on the identification of the appropriate enzymes for selective and practical oxidations of the A, B and C rings of steviol. Prior investigations in this area (Figure S3) have not resulted in the development of synthetically-useful methods. Recent characterization of the platensimycin biosynthesis pathway has revealed the presence of several dedicated ent-kaurane hydroxylases for selective C–H oxidations (19,20). Early in the pathway, a P450 monooxygenase, PtmO5, catalyzes a remote C–H hydroxylation at the C11 position of ent-kauranol, followed by an intramolecular cyclization to form the ether bridge. Next, two functionally-redundant α-ketoglutarate-dependent dioxygenases (Fe/αKGs), PtmO3 and PtmO6, hydroxylate the C7 carbon from the β face en route to the construction of the enone functionality of platensimycin. If these enzymes possess sufficient substrate promiscuity to accept steviol or ent-kaurenoic acid as substrate, and could do so with high reaction efficiency, they would comprise ideal biocatalysts for use in our synthetic campaign. PtmO3 and PtmO6 are highly homologous (99% identical). However, the latter was better overproduced upon expression in E. coli and was used exclusively here. Selective C7 hydroxylation of 9 and 13 could be observed with high total turnover numbers (Figure 2), suggesting this enzyme could be useful for preparative-scale B ring oxidation of ent-kauranes.
Figure 2.
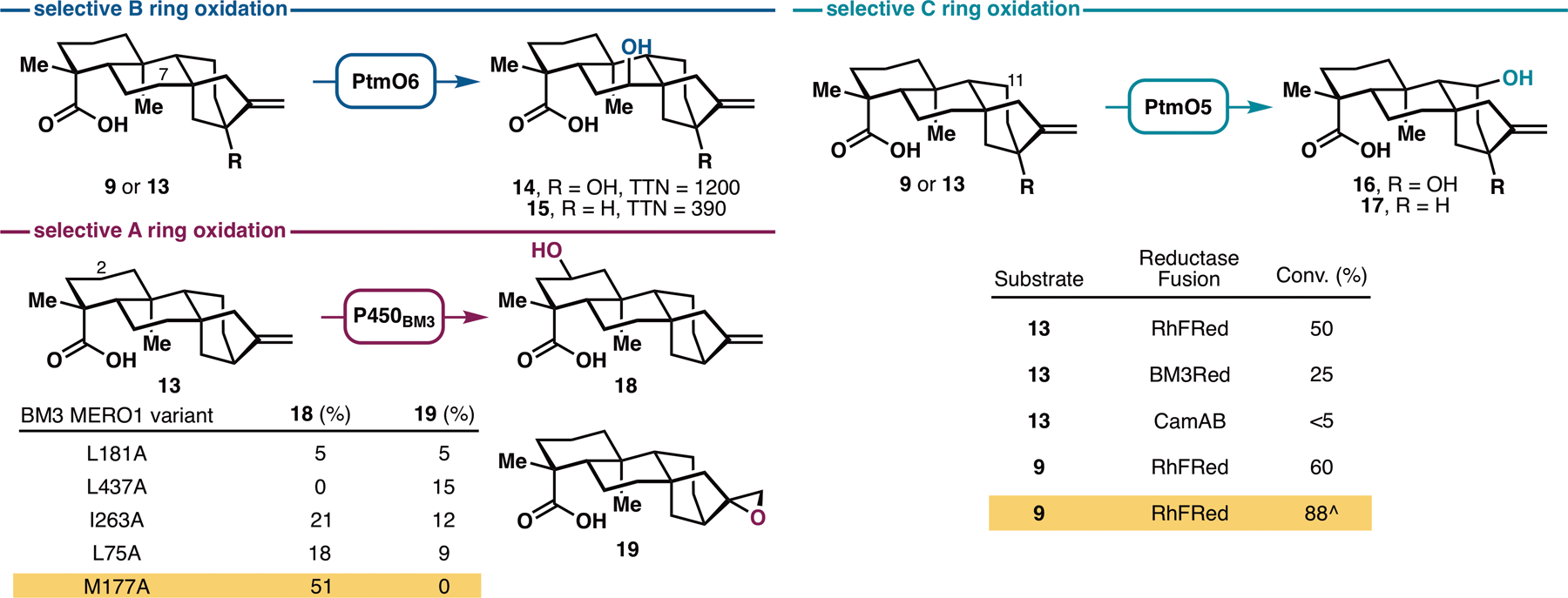
Discovery of three enzymes, PtmO6, PtmO5-RhFRed and BM3 MERO1 M177A for site-selective oxidations at C7, C11, and C2 respectively. See Supplementary Materials for plasmid construction (Figure S4) and reaction conditions. ^Performed by co-expressing PtmO5-RhFRed, Opt13 and GroES/EL in a single E. coli C41(DE3) strain.
Annotated as a class I P450, PtmO5 requires a separate reductase partner to support its function. We have previously employed the CamA/B reductase system for the functional characterization of PtmO5 (19) but found that this system gave very low reaction conversion in whole-cell and lysate reactions. This observation prompted us to examine alternative reductase partners. Given prior successes (21,22), artificial fusion with a reductase partner were deemed as a particularly viable solution. Among the chimeras tested, PtmO5-RhFRed, generated by linking PtmO5 with the reductase domain of P450RhF, provided the most promising outcome in the hydroxylation of 9 and 13. Further co-expression of PtmO5-RhFRed, GroES/EL chaperone and the phosphite dehydrogenase Opt13 for NADPH regeneration in a single E. coli C41(DE3) strain allowed selective C11 hydroxylation of 9 to be attained with 88% isolated yield on preparative scale.
Variants of P450BM3 have proven to be highly effective biocatalysts for selective oxidations of readily available terpene scaffolds (Figure S5, 23), including the A ring oxidation of decalin-containing terpenes (24,25,26,27). Furthermore, they have been shown to exhibit exceptional substrate promiscuity and excellent evolvability for new reactions. Based on these precedents, we postulated that some of these variants would be capable of performing similar oxidation on 9 or 13. To test this hypothesis, we conducted preliminary screening of P450BM3 alanine-scanning variants in our enzyme library (27) for the hydroxylation of 9 or 13. No hydroxylation activity could be observed with 9, but oxidized product(s) were formed from 13 with some of the variants tested. Variant BM3 MERO1 M177A in particular produced the C2-hydroxylated product 18 selectively without any over-oxidation or formation of C16–C17 epoxide side product. These results demonstrate the ready tunability of these oxidation biocatalysts to achieve selective reaction in the presence of other reactive functional group(s), a notable advantage over chemical oxidation methods. Importantly, this discovery identified a solution for the A ring oxidation problem.
Chemoenzymatic synthesis of oxidized ent-kauranes
Preliminary substrate scope examination of PtmO6, PtmO5-RhFRed and BM3 MERO1 M177A (Figure S6) suggested that they are able to accept other ent-kaurane and ent-atisane substrates bearing alternative functional group arrangements. This observation, in combination with the promiscuity of many other bacterial oxygenases (17,28) suggested that they could be useful for divergent synthesis. We initially targeted three ent-kauranes that would require the use of only remote B ring oxidation (Figure 3): mitrekaurenone (21), fujenoic acid (23), and pharboside aglycone (25). These three molecules contain different oxidation states and stereochemical configurations at C6 and C7, and their divergent synthesis would provide an ideal testbed for the synthetic versatility of biocatalytic oxidation with PtmO6. Steviol (9) was first converted to ent-kaurenoic acid (13) via a two-step protocol involving brominative displacement of the 3° alcohol and radical dehalogenation. The use of PtmO6 on 13 allowed selective installation of a 2° alcohol at C7, delivering the product as a single diastereomer with good conversion and yield. This transformation could be carried out routinely on gram scale using clarified lysate of E. coli cells expressing PtmO6. Conversion of alcohol 15 to the corresponding ketone (20) was accomplished by treatment with DMP. Introduction of the C6 α-OH of mitrekaurenone would require oxidation from the more hindered face of the skeleton. Thus, a strategy featuring an SN2-type displacement of a β-disposed leaving group at C6 by the C19 acid was pursued. α-oxidation with pyridinium tribromide was found to elicit simultaneous intramolecular lactonization by the C19 acid, thereby completing the synthesis of 21 in five steps.
Figure 3.
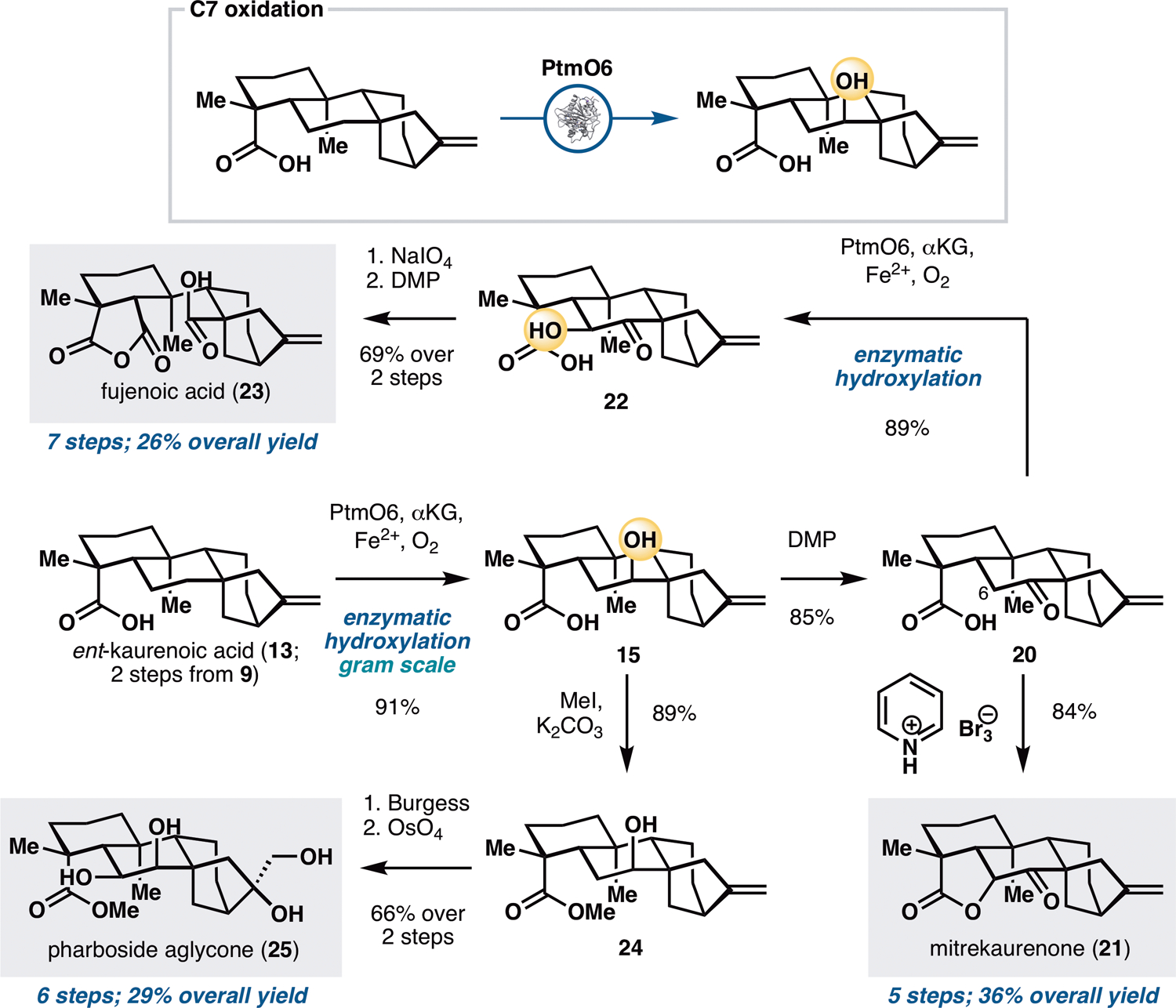
Application of PtmO6 in the chemoenzymatic total synthesis of mitrekaurenone (21), fujenoic acid (23) and pharboside aglycone (25). See Supplementary Materials for reaction conditions.
To access fujenoic acid (23), a net ten electron oxidation needed to be carried out on the B ring of the scaffold. Chemical methods for α-hydroxy ketone synthesis were initially attempted on 20 but we fortuitously found that PtmO6 is capable of installing the C6 alcohol with superior yield. Oxidative cleavage of the C6–C7 bond with NaIO4, followed by hemiketal oxidation with DMP furnished 23 in seven steps from 9. Pharboside aglycone (25), on the other hand, contains a β-disposed syn-diol motif at C6 and C7. While 22 could potentially be a viable synthetic intermediate, the conversion of its α-hydroxy ketone functionality to the desired diol motif would require a difficult reduction from the more hindered face. As an alternative, the 2° alcohol of methyl ester 24 was dehydrated to the corresponding olefin with Burgess reagent, thereby allowing the C6,C7 syn-diol motif to be introduced via dihydroxylation. The use of OsO4 and NMO simultaneously converted the two olefins to the corresponding syn-diol units and completed the synthesis of 25 in six steps from steviol.
Next, we sought to demonstrate the utility of our strategy in the preparation of ent-kauranes that contain oxidations on multiple rings (Figure 4), such as rosthornins B (3) and C (30), and fischericin B (2). These targets were chosen to highlight how multiple enzymatic hydroxylation reactions could be combined together or used strategically in combination with the concept of innate and guided C–H functionalization logic (15). Access to 3 and 30 from steviol would require hydroxylation at C7 and C11 with PtmO6 and PtmO5-RhFRed, reduction of the carboxylic acid at C19, and introduction of a carbonyl group at C15. To install the two alcohols at C7 and C11, a strategic decision had to be made in terms of the ordering of the two enzymatic hydroxylation steps. Performing C11 hydroxylation prior to C7 hydroxylation would necessitate nontrivial differentiation between the two alcohols for subsequent acetylation at C11 and stereochemical inversion at C7. Conversely, oxidation at C7 prior to that at C11 would allow the use of a carbonyl motif at C7 as a ‘masking’ group for the C7 α-OH and minimize potential chemoselectivity issues in subsequent manipulations. We found that ketone 26, accessed in two steps from 9 via PtmO6 hydroxylation and PDC oxidation, could undergo a regioselective hydroxylation at C11 with PtmO5-RhFRed, albeit with only moderate conversion. Using lysates of E. coli expressing PtmO5-RHFRed and Opt13, C11 hydroxylation of 26 could be carried out with 65% isolated yield. The use of Ac2O and DMAP allowed selective acetylation of the C11 alcohol without any undesired side reaction with the 3° alcohol at C13. At this stage, the C19 carboxylic acid needed to be reduced to the alcohol without concomitant removal of the acetate group at C11. This was achieved by first converting the acid to the corresponding acyl imidazole (28), followed by treatment with NaBH4, which also led to concomitant reduction of the C7 ketone to the α-disposed alcohol. Installation of the enone unit on the D ring via selective C15–OH oxidation with SeO2 and IBX completed the synthesis of rosthornin C (30) in seven steps overall. Finally, conversion of 30 to rosthornin B (3) could be effected via selective acetylation of the primary alcohol at C19.
Figure 4.
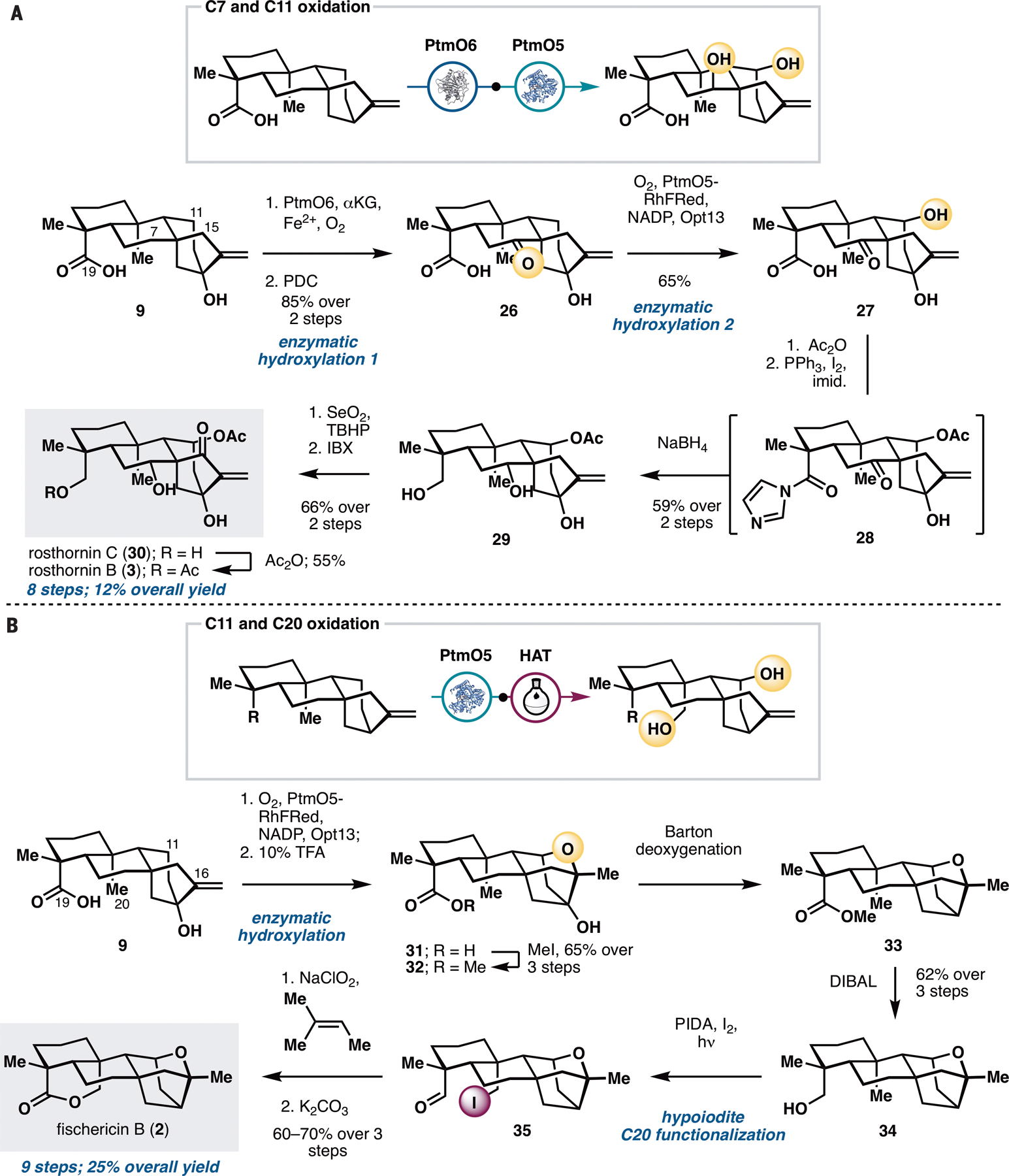
Application of PtmO5-RhFRed in the chemoenzymatic total synthesis of rosthornins B (3) and C (30) and fischericin B (2). See Supplementary Materials for reaction conditions.
Fischericin B (2) contains a caged ether motif that is reminiscent of platensimycin and a bridging lactone ring between C19 and C20. Synthesis of 2 would thus provide an opportunity to develop a hybrid strategy that combines enzymatic hydroxylation at C11 and alkoxy-radical-based C–H functionalization at C20. Our synthesis commenced with PtmO5-catalyzed C11 hydroxylation of 9. Using information gleaned from prior biosynthetic studies of platensimycin (19), the hydroxylated product could be treated with strong acidic conditions to construct the desired caged ether motif. In the presence of this motif, the C13 tertiary alcohol proved inert to conversion to the corresponding bromide for subsequent radical debromination. As a workaround, the free acid of 31 was first methylated and a Barton deoxygenation was performed on its C13 tertiary alcohol. The key hypoiodite–mediated C20 functionalization under Suarez conditions (13,29) cleanly delivered iodoaldehyde 35, which could be further oxidized and subjected to intramolecular ring closure to complete our synthesis of fischericin B (2) in just nine steps.
Oxidation-enabled skeletal rearrangement to ent-atisane and ent-trachylobane
Access to minimally oxidized ent-atisane and ent-trachylobane skeletons commenced from isosteviol (36), an ent-beyerane available in one step via acid-catalyzed degradation/rearrangement of stevioside (Figure 5A). Execution of our synthetic blueprint required the installation of a functional group at C12 that is suited for subsequent carbocation generation. We envisioned first C11 hydroxylation of 36 with PtmO5-RhFRed, followed by a C11 to C12 transposition of the resulting alcohol. Hydroxylation of isosteviol with PtmO5-RhFRed unexpectedly proceeded at its C12 carbon instead of C11, thereby obviating the need for any further functional group interconversions. Furthermore, this reaction proceeded with high conversion and yield, and could be carried out routinely on multi-gram scale in a single pass. The unexpected switch in regioselectivity could be rationalized by the difference in C ring conformation of ent-kaurane and ent-beyerane. PtmO5 oxidizes the axial C11 β-H of its native substrate. In contrast, the equivalent C11 β-H on 36 adopts an equatorial configuration, and is therefore inaccessible for abstraction by the active Fe(IV)-oxo species. Instead, C–H abstraction takes place at the adjacent axial C12 β-H. Treatment of 37 with TfOH initiated the intended Wagner–Meerwein rearrangement, delivering 38, which contains the requisite ent-atisane [2.2.2] C/D ring bicycle. Access to ent-trachylobane skeleton from 38 required the formation of a new C–C bond between C13 and C16. Examination of several different methods (30) to forge this bond led to the discovery of a reductive rearrangement in the presence of BF3•Et2O and Et3SiH, which afforded an ent-trachylobane product 40 from 39 in 61% yield after 3x recycling. We propose that this rearrangement proceeds via ionization of C13 alcohol, followed by formation of a non-classical carbocation and selective reductive quenching at C15. Overall, this synthetic sequence provides rapid and controlled access to ent-atisane and ent-trachylobane frameworks from isosteviol in two and four steps respectively and is made possible by the use of PtmO5-catalyzed hydroxylation of 36.
Figure 5.
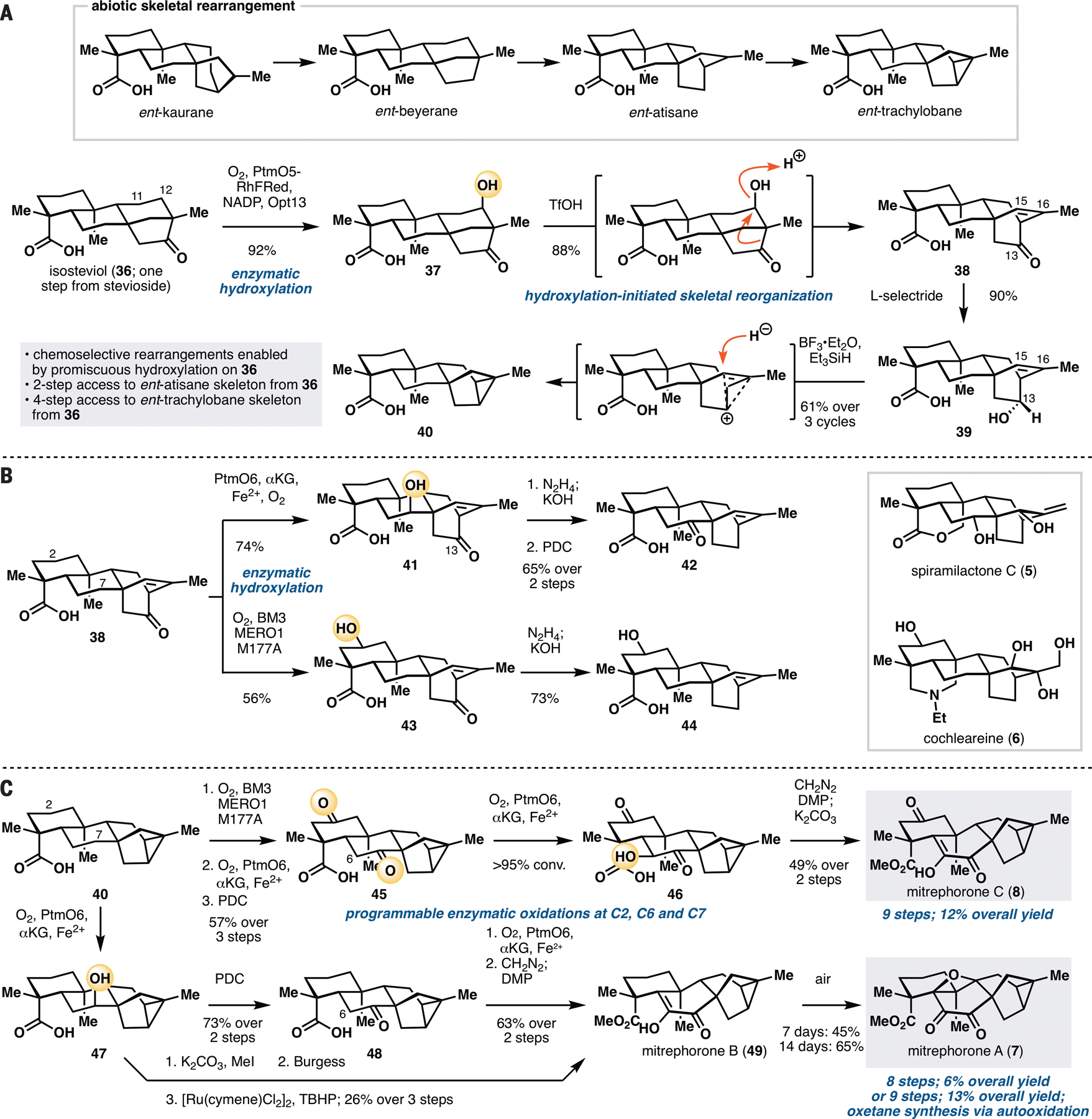
A. Conversion of isosteviol (36) to ent-atisane and ent-trachylobane products via site-selective C12 hydroxylation and carbocationic rearrangements. B. Further site-selective oxidations of 38 using PtmO6 and BM3 MERO1 M177A. C. Divergent chemoenzymatic total synthesis of the mitrephorones starting from 40. See Supplementary Materials for reaction conditions.
Preliminary investigation suggests that 38, bearing high structural resemblance to the native ent-atiserenoic acid precursor of plantensin (6,20), is a useful intermediate for accessing more oxidized ent-atisanes via enzymatic hydroxylation (Figure 5B). It is capable of undergoing oxidation at C7 with PtmO6 and is hydroxylated in a more efficient fashion than its C13-deoxy counterpart. Wolff–Kishner deoxygenation of C13 ketone and PDC oxidation of C7-OH afforded ketone 42, which represents a potential intermediate towards spiramilactone C (5). In an alternative sequence, 38 could first be oxidized at C2 with BM3 MERO1 M177A to acid 43 without any observable epoxidation of its C15–C16 olefin. Wolff–Kishner reduction delivered 44, which possesses the appropriate functionalities to be converted to the diterpenoid alkaloid cochleareine (6).
Chemoenzymatic synthesis of the mitrephorones
Intermediate 40 could be used to divergently prepare mitrephorones A, B and C (7, 49, 8, Figure 5C). In accord with its structural similarity to 9 or 13, C2 oxidation of 40 with BM3 MERO1 M177A proceeded very efficiently. At high enough enzyme-to-substrate ratio, this process also led to iterative oxidation to install a ketone moiety at C2. Enzymatic oxidation with PtmO6, followed by PDC oxidation, furnished diketone 45. As is the case with fujenoic acid (27), PtmO6 was capable of installing C6-OH on 45. Intermediate 46 proved to be unstable, requiring rapid methylation of the C19 acid and mild oxidation at C6. Thus, the synthesis of mitrephorone C (8) was completed by methylation of the C19 acid with CH2N2, and further oxidation at C6 with DMP, followed by keto-enol tautomerization.
Synthesis of mitrephorone B (49) from 40 commenced via C7 enzymatic oxidation with PtmO6. Initial attempts focused on the dehydration of the C7-OH to the corresponding olefin, followed by a ruthenium-catalyzed direct oxidation (31) to install the C6–C7 dione moiety. Unfortunately, this sequence was found to be low yielding and accompanied by significant amounts of side-product even at low conversion. It is also worth noting that this sequence provided no desired product at all when the C2 carbon exists in the ketone oxidation state (i.e., for the mitrephorone C series). As a workaround, 40 was oxidized to the corresponding ketone (48), enzymatically hydroxylated at C6 with PtmO6, and then methylated and oxidized with PDC. While this route led to one more step than the dehydration/oxidation sequence, it provided a superior overall yield. According to Magauer’s report (32), conversion of 49 to mitrephorone A (7) could be achieved through the use of electrochemical oxidation or White’s Fe(PDP) catalyst. We found serendipitously that 49 is capable of undergoing a slow autoxidation to form 7 (45% yield after 7 days, or 65% yield after 14 days). Such autoxidation did not take place with 8, suggesting that distal substituents are capable of modulating the reactivity of the diosphenol motif of 49. This discovery allowed us to divergently prepare the entire known family members of the mitrephorones from 40. The use of enzymatic oxidations with PtmO5-RhFRed, PtmO6 and BM3 MERO1 M177A proved to be highly enabling as all three members of the family could be prepared in less than ten steps, a marked improvement over previous routes to the mitrephorones (32,33).
Conclusion
Using this chemoenzymatic strategy, we prepared nine highly oxidized ent-kaurane and ent-trachylobane natural products in less than ten steps each. Central to this strategy is the use of three selective and scalable biocatalytic processes that are able to hydroxylate the A, B and C rings of the parent carbocyclic structures with site-selectivity and functional group compatibility unmatched by any known small-molecule reagents or catalysts. Leveraging the newly-introduced hydroxyl groups in a series of carbocationic rearrangements enables rapid traversal of the diterpene landscape spanning the ent-kaurane, ent-atisane and ent-trachylobane families. By virtue of the substrate promiscuity of the enzymes, the biocatalytic oxidations can also be carefully permuted and used in conjunction with chemical C–H oxidations in multi-step synthetic sequences for streamlined access to complex natural products with minimal functional group interconversions and protecting group manipulations (34). The marriage of chemical and enzymatic C–H oxidations, in particular, constitutes a powerful means to streamline access to highly oxidized terpenes and avoids circuitous oxidative transformations, which are at times necessary in the ‘two-phase’ strategy for terpene synthesis (35) due to the exclusive use of small-molecule reagents or catalysts. The strategy outlined here not only opens the door for rapid access to a wide array of ent-kauranes, ent-atisanes and ent-trachylobanes, but also provides a blueprint for combining emerging synthetic paradigms with biocatalysis in the preparation of privileged molecular scaffolds. Generalization of this hybrid oxidative approach to other complex terpene families can be readily envisioned by way of identifying and profiling new oxygenases for site-selective modifications of building blocks (36) or late-stage functionalization (37). Finally, continued advancement in enzyme engineering strategies (38) and the development of new-to-nature transformations (39) will further expand the pool of reactions available for use and ultimately encourage broader adoption of this strategy in multi-step synthesis.
Supplementary Material
Acknowledgments:
We thank Profs. Phil S. Baran, Ryan A. Shenvi, Thomas J. Maimone and Keary M. Engle for useful discussions.
Funding: This work is supported, in part, by the National Institutes of Health Grant GM134954 (B.S.), GM128895 (H.R.) and GM124461 (J.D.R.).
Footnotes
Competing interests: PtmO3, PtmO5, and PtmO6 are three gene products of the platensimycin and platencin biosynthetic gene cluster, and the platensimycin and platencin biosynthetic gene cluster is included in the United States Patent No 8,652,838, titled “Platensimycin biosynthetic gene cluster of Streptomyces platensis”, published on February 18, 2014.
Data and materials availability: All data is available in the main text or the supplementary materials.
References and Notes:
- 1.Liu M; Wang W-G; Sun H-D; Pu J-X Nat. Prod. Rep 2017, 34, 1090. [DOI] [PubMed] [Google Scholar]
- 2.Friese J; Gleitz J; Guster UT; Heubach JF; Matthiesen T; Wilffert B; Selve N Eur. J. Pharmacol 1997, 337, 165. [DOI] [PubMed] [Google Scholar]
- 3.Liao Y-J; Bai H-Y; Li Z-H; Zou J; Chen J-W; Zheng F; Zhang J-X; Mai S-J; Zeng M-S Sun H-D; Pu J-X; Xie D Cell Death Dis. 2014, 5, e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He H; Jiang H; Chen Y; Ye J; Wang A; Wang C; Liu Q; Liang G; Deng X; Jiang W; Zhou R Nat. Commun 2018, 9, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong YJ; Tantillo DJ J. Am. Chem. Soc 2010, 132, 5375. [DOI] [PubMed] [Google Scholar]
- 6.Smanski MJ; Yu Z; Casper J; Lin S; Peterson RM; Chen Y; Wendt-Pienkowski E; Rajski SR; Shen B Proc. Natl. Acad. Sci. USA 2011, 108, 13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson AJ; Hershey DM; Chestnut T; Xu M; Peters RJ Phytochemistry 2014, 103, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin B; Cui G; Guo J; Tang J; Duan L; Lin H; Shen Y; Zhang H; Huang L Plant Physiol. 2017, 174, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riehl PS; DePorre YC; Armaly AM; Groso EJ; Schindler CS Tetrahedron 2015, 71, 6629–6650. [Google Scholar]
- 10.Lazarski KE; Moritz BJ; Thomson RJ Angew. Chem. Int. Ed 2014, 53, 10588–10599. [DOI] [PubMed] [Google Scholar]
- 11.Kenny MJ; Mander LN; Sethi SP Tetrahedron Lett. 1986, 27, 3927. [Google Scholar]
- 12.Cherney EC; Lopchuk JM; Green JC; Baran PS J. Am. Chem. Soc 2014, 136, 12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi S; Shibukawa K; Hamada Y; Kuruma T; Kawabata A; Masuyama A J. Org. Chem 2018, 83, 1606. [DOI] [PubMed] [Google Scholar]
- 14.Kawamata Y; Yan M; Liu Z; Bao D-H; Chen J; Starr JT; Baran PS J. Am. Chem. Soc 2017, 139, 7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brückl T; Baxter RD; Ishihara Y; Baran PS Acc. Chem. Res 2012, 45, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung K; Condakes ML; Novaes LFT; Harwood SJ; Morikawa T; Yang Z; Maimone TJ J. Am. Chem. Soc 2019, 141, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowell AN; Demars MD II; Slocum ST; Yu F; Anand K; Chemler JA; Koravaki N; Priessnitz JK; Park SR; Koch AA; Schultz PJ; Sherman DH J. Am. Chem. Soc 2017, 139, 7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutt OE; Doan TL; Georg GI Org. Lett 2013, 15, 1602–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudolf JD; Dong L-B; Zhang X; Renata H; Shen B J. Am. Chem. Soc 2018, 140, 12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong L-B; Zhang X; Rudolf JD; Deng M-R; Kalkreuter E; Cepeda AJ; Renata H; Shen B J. Am. Chem. Soc 2019, 141, 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S; Podust LM; Sherman DH J. Am. Chem. Soc 2007, 129, 12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C; Shen F; Wang S; Wang Y; Liu J; Bai W-J; Wang X ACS Catal. 2018, 8, 5794. [Google Scholar]
- 23.Whitehouse CJC; Bell SG; Wong L-L Chem. Soc. Rev 2012, 41, 1218. [DOI] [PubMed] [Google Scholar]
- 24.Lewis JC; Mantovani SM; Fu Y; Snow CD; Komor RS; Wong CH; Arnold FH ChemBioChem 2010, 11, 2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kille S; Zilly FE; Acevedo JP; Reetz MT Nat. Chem 2011, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z; El Damaty S; Fasan R J. Am. Chem. Soc 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]
- 27.Li J; Li F; King-Smith E; Renata H Nat. Chem 2020, 12, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt JJ; Khatri Y; Brody SI; Zhu C; Pietraszkiewicz H; Valeriote FA; Sherman DH ACS Chem. Biol 2020, 15, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu W; Hjerrild P; Overgaard J; Poulsen TB Angew. Chem. Int. Ed 2016, 55, 8294. [DOI] [PubMed] [Google Scholar]
- 30.Kelly RB; Beckett BA; Eber J; Hung H-K; Zamecnik J Can. J. Chem 1975, 53, 143. [Google Scholar]
- 31.Kawamura S; Chu H; Felding J; Baran PS Nature 2016, 532, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wein LA; Wurst K; Angyal P; Weisheit L; Magauer T J. Am. Chem. Soc 2019, 141, 19589–19593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter MJR; Schneider M; Brandstätter M; Krautwald S; Carreira EM J. Am. Chem. Soc 2018, 140, 16704–16710. [DOI] [PubMed] [Google Scholar]
- 34.Gaich T; Baran PS J. Org. Chem 2010, 75, 4657. [DOI] [PubMed] [Google Scholar]
- 35.Kanda Y; Nakamura H; Umemiya S; Puthukanoori RK; Appala VRM; Gaddamanugu GK; Paraselli BR; Baran PS J. Am. Chem. Soc 2020, accepted. DOI: 10.1021/jacs.0c03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hernandez-Ortega A; Vinaixa M; Zebec Z; Takano E; Scrutton NS Sci. Rep 2018, 8, 14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong B; Luo T; Lei X ACS Cent. Sci 2020, 6, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galanie S; Entwistle D; Lalonde J Nat. Prod. Rep 2020, Advance Article. DOI: 10.1039/C9NP00071B. [DOI] [PubMed] [Google Scholar]
- 39.Chen K; Arnold FH Nat. Catal 2020, 3, 203–213. [Google Scholar]
- 40.Hellwell CA; Chandler PM; Poole A; Dennis ES; Peacock WJ Proc. Natl. Acad. Sci. USA 2001, 98, 2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nett RS; Montaranes M; Marcassa A; Lu X; Nagel TC; Charles P; Hedden P; Rojas MC; Peters RJ Nat. Chem. Biol 2017, 13, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi JA; Gomes DC; Lyra FH; dos Santos GF; Martins LR Molecules 2014, 19, 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernando H; Halpert JR; Davydov DR Arch. Biochem. Biophys 2008, 471, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munro AW Biochem. Soc. Trans 1993, 21, 216S. [DOI] [PubMed] [Google Scholar]
- 45.Munro AW; Lindsay JG; Coggins JR; Kelly SM; Price NC Biochim. Biophys. Acta 1996, 1296, 127–137. [DOI] [PubMed] [Google Scholar]
- 46.Jamakhandi AP; Jeffus BC; Dass VR; Miller GP Arch. Biochem. Biophys 2005, 439, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miles JS; Munro AW; Rospendowski BN; Smith WE; McKnight J; Thomson AJ Biochem. J 1992, 288, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fasan R ACS Catal. 2012, 2, 647–666. [Google Scholar]
- 49.Munro AW; Girvan HM; McLean KJ Biochim. Biophys. Acta 2007, 1770, 345–359. [DOI] [PubMed] [Google Scholar]
- 50.Sabbadin F; Hyde R; Robin A; Hilgarth E-M; Delenne M; Flitsch S; Turner N; Grogan G; Bruce NC ChemBioChem 2010, 11, 987–994. [DOI] [PubMed] [Google Scholar]
- 51.Sibbesen O; De Voss JJ; Ortiz de Montellano PR J. Biol. Chem 1996, 271, 22462–22469. [DOI] [PubMed] [Google Scholar]
- 52.Matos PM; Mahoney B; Chan Y; Day DP; Cabral MMW; Martins CHG; Santos RA; Bastos JK; Page PCB; Heleno VCG Molecules 2015, 20, 18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C; Lee D; Graf TN; Phifer SS; Nakanishi Y; Riswan S; Setyowati FM; Saribi AM; Soejarto DD; Farnsworth NR; Falkinham JO III; Kroll DJ; Kinghorn AD; Wani MC; Oberlies NH J. Nat. Prod 2009, 72, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fraga BM; Gonzalez P; Hernandez MG; Suarez S Tetrahedron 2005, 61, 5623. [Google Scholar]
- 55.Kim KH; Choi SU; Lee KR J. Nat. Prod 2009, 72, 1121. [DOI] [PubMed] [Google Scholar]
- 56.Gobu FR; Chen JJ; Zeng J; Wei WJ; Wang WF; Lin CJ; Gao K J. Nat. Prod 2017, 80, 2263. [DOI] [PubMed] [Google Scholar]
- 57.Xu YL; Li ZQ Acta Bot. Yunnanica 1988, 20, 97. [Google Scholar]
- 58.Xu YL; Ma YB Phytochemistry 1989, 28, 3235. [Google Scholar]
- 59.Li C et al. Org. Lett 2005, 7, 5709–5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.