

Abstract
Objective
Metabolic syndrome (MetS) is a cluster of concurrent risk factors for cardiovascular disease and type 2 diabetes. This post hoc analysis explored key efficacy and safety endpoints in patients with psoriatic arthritis (PsA) and MetS treated with tofacitinib.
Methods
Tofacitinib 5 and 10 mg twice daily and placebo data were pooled from two Phase 3 studies (OPAL Broaden [12 months; ClinicalTrials.gov identifier NCT01877668]; OPAL Beyond [6 months; ClinicalTrials.gov identifier NCT01882439]); patients received one background conventional synthetic disease‐modifying antirheumatic drug. Patients were stratified by baseline presence/absence of MetS. Efficacy and safety were reported to month 3 (tofacitinib and placebo) and 6 (tofacitinib only). Efficacy outcomes included: American College of Rheumatology (ACR)20/50/70, Health Assessment Questionnaire‐Disability Index (HAQ‐DI) response, Psoriasis Area Severity Index (PASI)75 response, and enthesitis/dactylitis resolution rates; and changes from baseline (Δ) in C‐reactive protein, HAQ‐DI, Patient’s/Physician’s Global Assessment of Arthritis, and patient‐reported outcomes. Safety outcomes included treatment‐emergent all‐causality adverse events (AEs), Δ in lipid/hepatic values, and liver parameter increases.
Results
Of 710 patients, 41.4% (n = 294) had baseline MetS. All efficacy outcomes improved with both tofacitinib doses versus placebo, to month 3; tofacitinib efficacy was consistent to month 6, regardless of MetS status. MetS did not appear to affect the incidence of AEs or Δ in lipid/hepatic values with tofacitinib up to month 3 or 6. Arterial thromboembolism and myocardial infarction (adjudicated major adverse cardiovascular events) were each reported once in tofacitinib‐treated patients with MetS.
Conclusion
Regardless of baseline MetS status, tofacitinib showed greater efficacy versus placebo in patients with active PsA. The tofacitinib safety profile appeared similar in patients with versus without MetS.
Introduction
Psoriatic arthritis (PsA) is a chronic, immune‐mediated inflammatory musculoskeletal disorder associated with a high prevalence of metabolic syndrome (MetS) (1), a cluster of concurrent risk factors that can lead to higher incidence of cardiovascular (CV) disease and type 2 diabetes (2, 3, 4). According to a 2009 international consensus statement, abnormal findings in at least three of five components (central obesity, elevated triglycerides, reduced high‐density lipoprotein [HDL]‐cholesterol, elevated blood pressure, and elevated fasting glucose) constitute MetS (2). As MetS and its components comprise CV risk factors, these patients should be identified clinically and managed accordingly.
MetS is known to be associated with insulin resistance and chronic systemic inflammation (5). Central obesity, the most prevalent manifestation of MetS, may play a role in both, with excess adipose tissue resulting in higher levels of hormones and proinflammatory cytokines such as leptin, interleukin (IL)‐6, and tumor necrosis factor (TNF)‐α (5, 6).
Elevated levels of leptin, IL‐6, TNF‐α, and other proinflammatory cytokines are also associated with inflammatory diseases, such as PsA (7, 8); the prevalence of MetS is significantly higher in patients with PsA than in patients with noninflammatory musculoskeletal disease (9), and the presence of MetS was associated with more severe PsA (1).
Of note, patients with both PsA and MetS are less likely to achieve minimal disease activity when treated with TNF‐α inhibitors (TNFi), compared with TNFi‐treated patients with PsA but without MetS (10). Furthermore, in a meta‐analysis, obesity was associated with a decreased response to TNFi in patients with PsA (11).
Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of PsA. The efficacy and safety of tofacitinib 5 and 10 mg twice daily (BID) were demonstrated in two Phase 3 studies of up to 6 or 12 months’ duration in patients with PsA who had an inadequate response (IR) to csDMARDs or TNFi (12, 13), and in a long‐term extension study (NCT01976364) (14). The impact of MetS on tofacitinib efficacy has not previously been assessed; however, a higher body mass index (BMI; ≥ 35 kg/m2) is associated with reduced tofacitinib response in patients with PsA (15).
The objective of this post hoc analysis was to compare key efficacy and safety endpoints in patients with PsA who were treated with tofacitinib in combination with a background csDMARD in the Phase 3 studies according to the presence or absence of comorbid MetS.
METHODS
Study design
This post hoc analysis included pooled data from two Phase 3 studies, the primary results of which have been reported elsewhere (12, 13). OPAL Broaden (NCT01877668) and OPAL Beyond (NCT01882439) were randomized, double‐blind, placebo‐controlled studies that enrolled patients aged 18 years or older who were diagnosed with PsA (≥ 6 months; based on the ClASsification criteria for Psoriatic ARthritis [CASPAR] [24]), with active arthritis (≥ 3 swollen joints and ≥ 3 tender/painful joints on motion) at screening and baseline, and active plaque psoriasis at screening.
OPAL Broaden was a 12‐month study that enrolled patients who had an IR to one or more csDMARD and had not previously received a TNFi (TNFi‐naïve) (12). OPAL Beyond was a 6‐month study that enrolled patients who had an IR to one or more TNFi (TNFi‐IR) (13). Patients were randomized to receive tofacitinib 5 mg BID, tofacitinib 10 mg BID, adalimumab 40 mg by subcutaneous injection every other week (OPAL Broaden only), or placebo advancing to tofacitinib 5 or 10 mg BID at month 3. All patients in both studies continued on a stable dose of a single csDMARD.
Post hoc analysis
This post hoc analysis included all patients who received one or more dose of tofacitinib or placebo in OPAL Broaden and OPAL Beyond. For patients who were randomized to and received tofacitinib, data were pooled from both studies up to month 6. Patients who received placebo and switched to tofacitinib at month 3 were not included in analyses up to month 6. For patients who received placebo, data were pooled up to month 3 (end of placebo‐controlled period). Patients who received adalimumab in OPAL Broaden were excluded from this analysis, as the objective of this analysis was to compare tofacitinib and placebo in data pooled from both studies, and adalimumab was not administered in OPAL Beyond. The studies were also analyzed separately to assess the tofacitinib efficacy in patients with PsA by prior TNFi experience (TNFi‐naïve [OPAL Broaden] vs TNFi‐IR [OPAL Beyond]) and MetS at baseline.
Efficacy endpoints at months 3 and 6, and safety endpoints to months 3 and 6, were analyzed according to the presence or absence of MetS at baseline. MetS was defined per previously standardized criteria (2).
Efficacy analysis
Efficacy assessments included the primary endpoints from OPAL Broaden and OPAL Beyond: the proportion of patients achieving an American College of Rheumatology (ACR)20 response (≥ 20% improvement from baseline in ACR components) (16) at month 3, and change from baseline to month 3 in Health Assessment Questionnaire‐Disability Index (HAQ‐DI; range: 0‐3; higher scores indicate greater disability).
Further binary efficacy endpoints at months 3 and 6 include achievement of ACR20 (at month 6), ACR50, and ACR70 (≥ 50% and ≥ 70% improvements from baseline in ACR components, respectively) responses, achievement of HAQ‐DI minimally clinically important response (reduction from baseline of ≥ 0.35 points) (17) in patients with baseline HAQ‐DI ≥ 0.35, ≥ 75% reduction from baseline Psoriasis Area and Severity Index (PASI75) response in patients with plaque psoriasis affecting ≥ 3% body surface area at baseline and a baseline PASI score > 0, enthesitis resolution (using Leeds Enthesitis Index [LEI]) in patients with baseline LEI > 0, and dactylitis resolution (using Dactylitis Severity Score [DSS]) in patients with baseline DSS > 0.
Additional continuous endpoints included change from baseline in HAQ‐DI at month 6, and changes from baseline at months 3 and 6 in Physician’s Global Assessment of Arthritis (PGA; measured by Visual Analog Scale [VAS], 0‐100 mm), swollen joint count (SJC; 66 joints), tender/painful joint count (TJC; 68 joints), C‐reactive protein (CRP; further stratification by baseline CRP levels ≤ 2.87 or > 2.87 mg/L), and patient‐reported outcomes (PROs), including Patient’s Global Assessment of Arthritis (PtGA; VAS, 0‐100 mm), Patient’s Assessment of Arthritis Pain (‘Pain’; VAS, 0‐100 mm), Functional Assessment of Chronic Illness Therapy‐Fatigue (FACIT‐F) total score, Short Form‐36 Health Survey Acute Version 2 (SF‐36v2; reporting component and norm‐based domain scores) (18), and EuroQol 5‐Dimensions 3‐Level (EQ‐5D‐3L; reporting total and dimension scores) and EQ‐VAS (mm; your own health state today) (19).
Statistical analysis of efficacy endpoints
For binary endpoints using pooled data from two studies, treatment differences (95% confidence interval [CI]) were analyzed using large sample approximation of the difference in binomial proportions using a Cochran‐Mantel‐Haenszel approach adjusting for study. Response rates (standard error [SE]) were reported by treatment group and baseline MetS status, and treatment differences (95% CI) were calculated for tofacitinib doses versus placebo at month 3. A missing response was considered a nonresponse to treatment.
Continuous endpoints, except CRP, using pooled data from two studies, were analyzed using a mixed model for repeated measures, with fixed effects of treatment, visit, study, treatment‐by‐visit, baseline MetS, treatment‐by‐baseline MetS, visit‐by‐baseline MetS, treatment‐by‐visit‐by‐baseline MetS interactions, geographical region, and baseline value. Missing values were not imputed in the model. A common unstructured covariance matrix was used. Least squares (LS) mean changes from baseline were calculated at months 3 and 6 using two separate models. Results at month 3 were based on a model that included tofacitinib 5 mg BID, tofacitinib 10 mg BID, and placebo treatment groups; as the placebo group was only included up to month 3 (end of placebo‐controlled period), the results at month 6 were based on a separate model that included only tofacitinib 5 mg BID and tofacitinib 10 mg BID treatment groups. Treatment differences (95% CI) were calculated for tofacitinib doses versus placebo at month 3, by baseline MetS.
For analyses of efficacy endpoints performed by study (prior TNFi experience), similar methods were used as for pooled data, but without adjustment to study.
CRP data were pooled from the two studies. Mean (SE) change from baseline was calculated descriptively at months 3 and 6, by treatment group, stratified by baseline MetS status and CRP cutoff (≤ 2.87 vs > 2.87 mg/L, where 2.87 mg/L is the upper limit of normal range).
Interaction analysis
To evaluate whether treatment effects were different between patients with and without baseline MetS, the interaction effects of baseline MetS status and treatment group on ACR20/50 and PASI75 response, and on changes from baseline in HAQ‐DI and CRP, were assessed at month 3. ACR20/50 and PASI75 responses were assessed using a multiple logistic regression model, and changes from baseline in HAQ‐DI and CRP were assessed using an analysis of covariance model. Both models used fixed effects of treatment, baseline MetS, treatment‐by‐baseline MetS, study, geographical region, age, and PsA disease duration. For assessments of interaction effects for ACR20/50 and PASI75, a missing response was considered a nonresponse; for HAQ‐DI and CRP, patients with missing change from baseline values were excluded from the analysis. A value of P ≤ 0.05, based on Wald test of type 3 analysis for interaction effect, was considered statistically significant.
Safety analysis
Treatment‐emergent all‐causality adverse events (AEs) were reported up to months 3 and 6 by Medical Dictionary for Regulatory Activities (MedDRA) Preferred Term. AEs of potential interest in patients with MetS, including fasting blood glucose increase, hyperglycemia, diabetes, hypertension, and hepatic steatosis were analyzed descriptively. Major adverse cardiovascular events (MACE) were adjudicated by an external, independent committee. The following laboratory parameters were assessed at months 3 and 6: percent change from baseline in fasting lipid values (low‐density lipoprotein [LDL]‐cholesterol, HDL‐cholesterol, total cholesterol, and triglycerides), change from baseline in hepatic laboratory values (total bilirubin, aspartate aminotransferase [AST], alanine aminotransferase [ALT], and gamma glutamyl transferase [GT]); and incidence of liver parameters as multiples of the upper limit of normal (ULN) (for total bilirubin, AST, ALT, and gamma GT). Change from baseline in hepatic values and incidence of liver parameters were assessed without regard for baseline abnormality.
RESULTS
Patients
Of the 710 patients in this pooled analysis, 294 (41.4%) had baseline MetS (120 from OPAL Broaden; 174 from OPAL Beyond).
Demographics and baseline characteristics are summarized in Table 1. Across all treatment groups in both the pooled analysis and individual studies, patients with baseline MetS tended to be older with higher mean BMI versus patients without MetS. A greater proportion of patients with baseline MetS had abdominal obesity, a CRP level > 2.87 mg/L, and were taking lipid‐lowering medications at baseline, versus patients without baseline MetS. Baseline PRO values appeared similar regardless of baseline MetS status (Table 1 and Online Supplementary Table 1). Baseline demographics, disease characteristics, and PRO values were generally similar among patients in each study, regardless of prior TNFi experience (Online Supplementary Table 2).
Table 1.
Demographics and baseline disease characteristics by treatment group and baseline MetS status; pooled data from OPAL Broaden and OPAL Beyond
|
Tofacitinib 5 mg BID a (N = 238) |
Tofacitinib 10 mg BID a (N = 236) |
Placebo a (N = 236) |
||||
|---|---|---|---|---|---|---|
|
With baseline MetS (N = 99) |
Without baseline MetS (N = 139) |
With baseline MetS (N = 101) |
Without baseline MetS (N = 135) |
With baseline MetS (N = 94) |
Without baseline MetS (N = 142) |
|
| OPAL Broaden b , N | 38 | 69 | 36 | 68 | 46 | 59 |
| OPAL Beyond c , N | 61 | 70 | 65 | 67 | 48 | 83 |
| Demographics | ||||||
| Age (y), mean (SD) | 53.0 (11.0) | 46.9 (12.8) | 53.2 (10.4) | 46.5 (11.9) | 53.2 (11.4) | 45.2 (12.2) |
| Female, n (%) | 51 (51.5) | 70 (50.4) | 59 (58.4) | 77 (57.0) | 48 (51.1) | 88 (62.0) |
| Race, n (%) | ||||||
| White | 95 (96.0) | 131 (94.2) | 98 (97.0) | 123 (91.1) | 90 (95.7) | 132 (93.0) |
| Other | 4 (4.0) | 8 (5.8) | 3 (3.0) | 12 (8.9) | 4 (4.3) | 10 (7.0) |
| BMI (kg/m2), mean (SD) | 33.3 (6.1) | 27.4 (5.4) | 33.4 (5.7) | 27.8 (5.5) | 32.9 (4.8) | 26.8 (4.8) |
| Abdominal obesity d , n (%) | 88 (88.9) | 63 (45.3) | 93 (92.1) | 54 (40.0) | 86 (91.5) | 54 (38.0) |
| Patients taking concomitant lipid‐lowering medications at baseline e , n (%) | 25 (25.3) | 4 (2.9) | 32 (31.7) | 7 (5.2) | 21 (22.3) | 5 (3.5) |
| Disease characteristics | ||||||
| Psoriatic arthritis duration (y), mean (SD) | 9.2 (7.9) | 8.2 (7.9) | 8.2 (6.5) | 6.9 (6.6) | 7.2 (6.3) | 8.7 (8.2) |
| SJC (66), mean (SD) | 13.6 (11.9) | 11.6 (8.9) | 14.2 (11.6) | 10.9 (8.0) | 11.8 (9.4) | 10.4 (8.5) |
| TJC (68), mean (SD) | 21.3 (13.4) | 20.0 (12.4) | 26.9 (17.5) | 20.5 (13.9) | 22.9 (14.9) | 18.3 (14.2) |
| CRP (mg/L) | ||||||
| > 2.87 f , n (%) | 69 (69.7) | 84 (60.4) | 68 (67.3) | 80 (59.3) | 62 (66.0) | 81 (57.0) |
| Mean (SD) | 11.1 (18.5) | 13.1 (21.9) | 10.1 (16.3) | 13.4 (25.2) | 11.9 (18.4) | 11.0 (21.4) |
| Median (range) | 5.2 (0.2‐126.0) | 5.8 (0.2‐115.0) | 5.0 (0.4‐100.0) | 5.0 (0.2‐163.0) | 5.3 (0.3‐100.0) | 4.0 (0.2‐164.0) |
| PASI, mean (SD) g | 9.5 (7.5) | 8.7 (8.1) | 10.6 (8.5) | 9.8 (7.4) | 12.0 (11.7) | 9.3 (8.5) |
| LEI, mean (SD) h | 2.9 (1.6) | 2.7 (1.5) | 3.5 (1.7) | 3.0 (1.7) | 2.7 (1.5) | 2.8 (1.5) |
| DSS, mean (SD) i | 8.6 (8.7) | 8.3 (9.3) | 10.3 (9.9) | 8.0 (6.4) | 7.9 (6.0) | 8.6 (8.0) |
| HAQ‐DI, mean (SD) | 1.3 (0.7) | 1.2 (0.6) | 1.4 (0.6) | 1.2 (0.6) | 1.3 (0.7) | 1.1 (0.7) |
| PtGA (mm), mean (SD) | 54.9 (22.8) | 57.1 (22.4) | 58.3 (21.2) | 54.9 (23.6) | 57.5 (23.7) | 53.2 (22.9) |
| PGA (mm), mean (SD) | 55.7 (20.0) | 52.8 (20.3) | 55.8 (20.1) | 55.3 (18.0) | 54.4 (19.0) | 53.3 (21.0) |
| Pain (mm), mean (SD) | 55.1 (22.7) | 56.7 (24.1) | 59.9 (19.0) | 55.3 (24.0) | 56.0 (24.7) | 52.9 (24.3) |
| FACIT‐F total score, mean (SD) | 26.0 (11.4) | 27.6 (11.5) | 26.2 (10.6) | 27.6 (10.3) | 27.8 (10.6) | 28.0 (10.8) |
| SF‐36v2 PCS, mean (SD) | 33.7 (7.8) | 34.8 (8.5) | 32.0 (8.7) | 35.1 (8.4) | 34.2 (8.4) | 36.0 (8.8) |
| SF‐36v2 MCS, mean (SD) | 41.1 (12.7) | 39.6 (11.1) | 40.3 (12.0) | 39.9 (12.3) | 40.2 (11.9) | 40.1 (11.7) |
| EQ‐VAS (mm), mean (SD) | 52.8 (21.7) | 50.8 (22.2) | 48.9 (21.6) | 51.7 (20.3) | 53.2 (22.8) | 51.6 (19.5) |
Abbreviations: BID, twice daily; BMI, body mass index; BSA, body surface area; CRP, C‐reactive protein; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; DSS, Dactylitis Severity Score; EQ, EuroQol; FACIT‐F, Functional Assessment of Chronic Illness Therapy‐Fatigue; HAQ‐DI, Health Assessment Questionnaire‐Disability Index; LEI, Leeds Enthesitis Index; MCS, Mental Component Summary; MetS, metabolic syndrome; PASI, Psoriasis Area and Severity Index; PCS, Physical Component Summary; PGA, Physician’s Global Assessment of Arthritis; PtGA, Patient’s Global Assessment of Arthritis; SD, standard deviation; SF‐36v2, Short Form‐36 Health Survey Acute Version 2; SJC, swollen joint count (out of 66 joints); TJC, tender/painful joint count (out of 68 joints); TNFi, tumor necrosis factor inhibitor; VAS, Visual Analog Scale.
All patients received a stable dose of one background csDMARD.
All patients had not previously received a TNFi, except one patient randomized to placebo who received etanercept prior to enrollment.
All patients had an inadequate response to ≥ 1 TNFi.
Based on elevated waist circumference; waist circumference thresholds are population‐ and country‐specific (2).
Baseline concomitant lipid‐lowering medications were defined as drugs taken at baseline (Day 1).
Upper limit of normal.
PASI was assessed only in patients with baseline BSA ≥ 3% and PASI > 0.
Baseline LEI was summarized only for patients with baseline LEI > 0.
Baseline DSS was summarized only for patients with baseline DSS > 0.
Efficacy
Regardless of baseline MetS status, at month 3, the proportions of patients achieving ACR20/50/70, PASI75, and HAQ‐DI response, as well as enthesitis and dactylitis resolution, were greater in patients receiving tofacitinib 5 or 10 mg BID versus placebo (Figure 1). This was supported by an analysis of the treatment differences between tofacitinib and placebo at month 3 (Online Supplementary Figure 1), showing that response rates tended to be in favor of both tofacitinib doses versus placebo. Response rates were generally similar in patients with versus without baseline MetS at months 3 and 6 (Figure 1); exceptions were HAQ‐DI response with tofacitinib 10 mg BID at both time points, and dactylitis resolution at month 6 with both tofacitinib doses, which were lower in patients with versus without baseline MetS. Among tofacitinib‐treated patients, response rates were consistent up to month 6. Analyses of the interaction effects between treatment groups and baseline MetS status at month 3 demonstrated no significant interactions for ACR20 (P = 0.23), ACR50 (P = 0.93), and PASI75 (P = 0.92) response rates.
Figure 1.
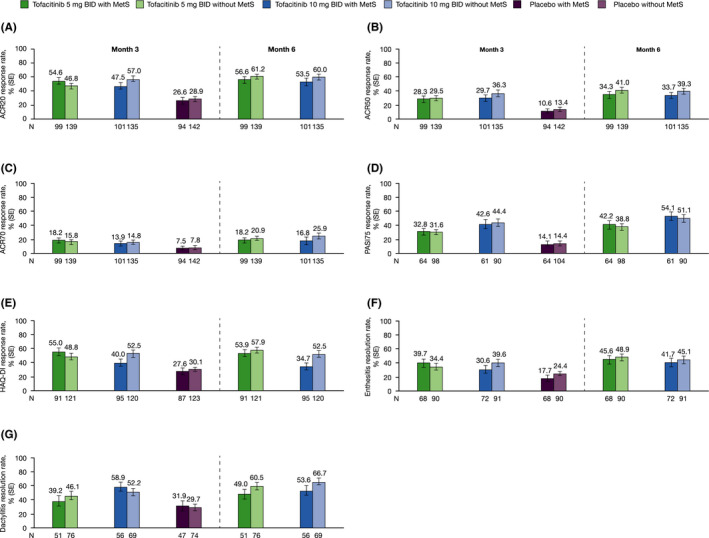
Response rate (SE) for ACR20 (A), ACR50 (B), ACR70 (C), PASI75a (D), and HAQ‐DI responseb (E), enthesitis resolutionc (F) and dactylitis resolutiond (G) rates at months 3 and 6, by treatment group and baseline MetS status; pooled data from OPAL Broaden and OPAL Beyond. aPASI was assessed only in patients with baseline BSA ≥ 3% and PASI > 0. bHAQ‐DI response is defined as a decrease ≥ 0.35 among patients with baseline HAQ‐DI score ≥ 0.35. cEnthesitis resolution was assessed only in patients with baseline LEI > 0. dDactylitis resolution was assessed only in patients with baseline DSS > 0. A patient with a missing value was considered a nonresponder. Abbreviations: ACR, American College of Rheumatology; BID, twice daily; BSA, body surface area; DSS, Dactylitis Severity Score; HAQ‐DI, Health Assessment Questionnaire‐Disability Index; LEI, Leeds Enthesitis Index; MetS, metabolic syndrome; N, number of patients with nonmissing baseline MetS; PASI, Psoriasis Area and Severity Index; SE, standard error.
When prior TNFi experience was taken into account, response rates across all outcomes were generally similar within each treatment group at months 3 and 6, irrespective of baseline MetS, and there were no clear trends between TNFi‐naïve (OPAL Broaden) and TNFi‐IR (OPAL Beyond) patients (Online Supplementary Figure 2); however, some small differences were observed. Compared with the pooled data and TNFi‐IR patients, numerically higher response rates were observed in TNFi‐naïve patients for ACR20 and ACR50 responses at months 3 and 6 with tofacitinib 10 mg BID, and for PASI75 response at month 3 with tofacitinib 5 mg BID and at month 6 for both tofacitinib doses. Enthesitis and dactylitis resolution rates appeared numerically more favorable in TNFi‐IR patients receiving tofacitinib 5 mg BID at month 3 compared with the pooled data and TNFi‐naïve patients.
Regardless of baseline MetS status, LS mean improvements from baseline in disease‐related measures and PROs were greater with both tofacitinib doses versus placebo at month 3; improvements observed with tofacitinib were consistent to month 6 (Figure 2A–E; Online Supplementary Figure 3), and treatment differences at month 3 were in favor of either tofacitinib dose versus placebo (Online Supplementary Figure 4). In patients with elevated CRP levels at baseline, mean reductions from baseline in CRP were similar in tofacitinib‐treated patients irrespective of baseline MetS status; except at month 3 in the tofacitinib 10 mg BID group, when patients with MetS experienced less pronounced reductions in CRP versus those without MetS (Figure 2F). In patients with baseline CRP ≤ 2.87 mg/L who received either dose of tofacitinib, CRP levels increased at month 3 in patients with baseline MetS, and remained stable in patients without MetS.
Figure 2.
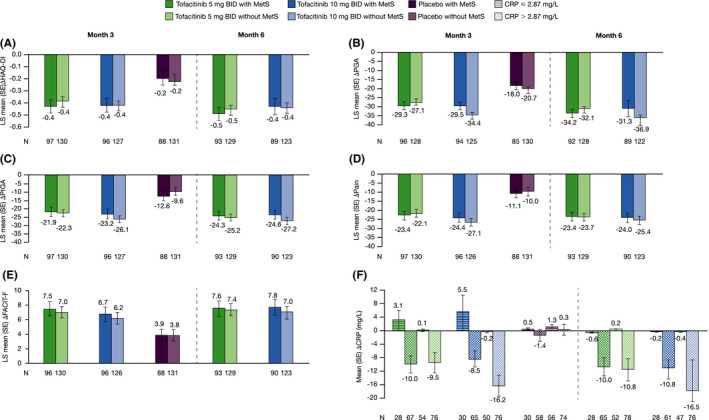
LS mean (SE) change from baseline in HAQ‐DI (A), PGA (VAS, mm) (B), PtGA (VAS, mm) (C), Pain (VAS, mm) (D), and FACIT‐F total score (E) at months 3 and 6 by treatment group and baseline MetS status, and mean (SE) change from baseline in CRP (F), at months 3 and 6, by treatment group, baseline MetS status, and baseline CRP ≤ 2.87 mg/L and > 2.87 mg/L; pooled data from OPAL Broaden and OPAL Beyond. Each of the endpoints (except CRP) were analyzed using a mixed model for repeated measures, with fixed effects of treatment, visit, study, treatment‐by‐visit, baseline MetS, treatment‐by‐baseline MetS, visit‐by‐baseline MetS, treatment‐by‐visit‐by‐baseline MetS interactions, geographical region, and baseline value. Missing values were not imputed. A common unstructured covariance matrix was used. LS mean (SE) changes from baseline were calculated at months 3 and 6 using two separate models. Results at month 3 were based on a model that included tofacitinib 5 mg BID, tofacitinib 10 mg BID, and placebo treatment groups; as the placebo treatment group was only included up to month 3 (end of placebo‐controlled period), the results at month 6 were based on a separate model that included only tofacitinib 5 mg BID and tofacitinib 10 mg BID treatment groups.
Arithmetic mean (SE) was calculated for change from baseline in CRP by treatment group, baseline MetS status, and CRP cutoff. Abbreviations: Δ, change from baseline; BID, twice daily; CRP, C‐reactive protein; FACIT‐F, Functional Assessment of Chronic Illness Therapy‐Fatigue; HAQ‐DI, Health Assessment Questionnaire‐Disability Index; LS mean, least squares mean; MetS, metabolic syndrome; N, number of patients evaluable at months 3 and 6; PtGA, Patient’s Global Assessment of Arthritis; PGA, Physician’s Global Assessment of Arthritis; SE, standard error; VAS, Visual Analog Scale.
Interaction analyses of baseline MetS status and treatment group also demonstrated no significant interaction on change from baseline in HAQ‐DI (P = 0.89) and CRP (P = 0.18) at month 3.
LS mean improvements from baseline in LEI, DSS, HAQ‐DI, PGA, PtGA, Pain, SJC, TJC, and CRP with tofacitinib were generally comparable at months 3 and 6, regardless of prior TNFi experience and baseline MetS (Online Supplementary Figures 5 and 6).
LS mean improvements from baseline in SF‐36v2 (especially bodily pain), EQ‐VAS, and EQ‐5D‐3L pain/discomfort score were greater with both tofacitinib doses versus placebo, both in patients with and without baseline MetS (Online Supplementary Figures 7 and 8).
Safety
The proportions of patients with treatment‐emergent AEs, serious AEs, and AEs leading to discontinuations were generally similar between patients with and without MetS receiving tofacitinib 5 mg BID, tofacitinib 10 mg BID, or placebo up to months 3 and 6 (Table 2).
Table 2.
Treatment‐emergent adverse events up to months 3 and 6; pooled data from OPAL Broaden and OPAL Beyond
|
Tofacitinib 5 mg BID a (N = 238) |
Tofacitinib 10 mg BID a (N = 236) |
(N = 236) |
||||
|---|---|---|---|---|---|---|
| n (%) |
With baseline MetS (N = 99) |
Without baseline MetS (N = 139) |
With baseline MetS (N = 101) |
Without baseline MetS (N = 135) |
With baseline MetS (N = 94) |
Without baseline MetS (N = 142) |
| Adverse events | ||||||
| Up to month 3 | 55 (55.6) | 59 (42.4) | 43 (42.6) | 74 (54.8) | 36 (38.3) | 59 (41.5) |
| Up to month 6 | 68 (68.7) | 86 (61.9) | 67 (66.3) | 92 (68.1) | NA | NA |
| Serious adverse events | ||||||
| Up to month 3 | 2 (2.0) | 2 (1.4) | 2 (2.0) | 2 (1.5) | 3 (3.2) | 1 (0.7) |
| Up to month 6 | 4 (4.0) | 6 (4.3) | 5 (5.0) | 6 (4.4) | NA | NA |
| Adverse events leading to discontinuation | ||||||
| Up to month 3 | 2 (2.0) | 3 (2.2) | 5 (5.0) | 5 (3.7) | 3 (3.2) | 3 (2.1) |
| Up to month 6 | 3 (3.0) | 7 (5.0) | 8 (7.9) | 6 (4.4) | NA | NA |
Abbreviations: BID, twice daily; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; MetS, metabolic syndrome; N, number of patients analyzed; n, number of patients with the adverse event of interest; NA, not applicable.
All patients received a stable dose of one background csDMARD.
Patients in the placebo group were advanced to tofacitinib 5 or 10 mg BID after 3 months of placebo treatment.
The most common AEs reported in ≥ 5% of patients (Online Supplementary Table 3) up to month 3 were generally similar across treatment groups: nasopharyngitis, upper respiratory tract infection, headache, and diarrhea. Additional commonly reported AEs up to month 6 were: bronchitis, lower respiratory tract infection, urinary tract infection, and hypertension. No clear trends in AEs were observed based on baseline MetS status. There also did not appear to be a clear trend in AEs potentially associated with MetS (Table 3). One patient with baseline MetS who received tofacitinib 5 mg BID up to month 3, experienced an arterial thromboembolic event (day 45; the MedDRA Preferred Term was amaurosis fugax; this was adjudicated as a transient ischemic attack); the treatment was stopped temporarily, and the AE was resolved. One patient with baseline MetS who received tofacitinib 5 mg BID up to month 6 (last day of tofacitinib treatment was day 169) experienced an acute myocardial infarction on day 245, which was adjudicated as a MACE.
Table 3.
AEs of potential interest in the context of MetS up to months 3 and 6, by treatment group and baseline MetS status; pooled data from OPAL Broaden and OPAL Beyond
| Up to Month 3 | Up to Month 6 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Tofacitinib 5 mg BID a (N = 238) |
Tofacitinib 10 mg BID a (N = 236) |
(N = 236) |
Tofacitinib 5 mg BID a (N = 238) |
Tofacitinib 10 mg BID a (N = 236) |
||||||
| n (%) |
With baseline MetS (N = 99) |
Without baseline MetS (N = 139) |
With baseline MetS (N = 101) |
Without baseline MetS (N = 135) |
With baseline MetS (N = 94) |
Without baseline MetS (N = 142) |
With baseline MetS (N = 99) |
Without baseline MetS (N = 139) |
With baseline MetS (N = 101) |
Without baseline MetS (N = 135) |
| Blood glucose increase | 0 | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 | 0 |
| Diabetes | 0 | 1 (0.7) | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 | 0 |
| Hyperglycemia | 1 (1.0) | 0 | 1 (1.0) | 0 | 0 | 0 | 1 (1.0) | 0 | 1 (1.0) | 0 |
| Hypertension | 2 (2.0) | 2 (1.4) | 4 (4.0) | 1 (0.7) | 1 (1.1) | 2 (1.4) | 5 (5.1) | 4 (2.9) | 4 (4.0) | 3 (2.2) |
| Hepatic steatosis | 0 | 0 | 1 (1.0) | 0 | 0 | 0 | 1 (1.0) | 0 | 1 (1.0) | 1 (0.7) |
| ATE | 1 (1.0) | 0 | 0 | 0 | 0 | 0 | 1 (1.0) | 0 | 0 | 0 |
| MI | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) c | 0 | 0 | 0 |
Abbreviations: AE, adverse event; ATE, arterial thromboembolism; BID, twice daily; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; MACE, major adverse cardiovascular event; MetS, metabolic syndrome; MI, myocardial infarction; N, number of patients evaluable for AEs; n, number of patients with AEs.
All patients received a stable dose of one background csDMARD.
Patients in the placebo group were advanced to tofacitinib 5 or 10 mg BID after 3 months of placebo treatment.
Adjudicated as a MACE.
At baseline, LDL‐cholesterol, total cholesterol, and triglyceride levels were higher, and HDL‐cholesterol levels were lower, in patients with baseline MetS versus without MetS (Table 4), as expected. Regardless of baseline MetS status, mean percent changes from baseline in fasting lipid values were greater with either tofacitinib dose versus placebo up to month 3, and results were consistent to month 6; tofacitinib 10 mg BID‐treated patients without baseline MetS experienced a substantially greater change in triglyceride levels versus other patient groups (Table 4). Between baseline and month 3, eight patients with baseline MetS began lipid‐lowering medications (tofacitinib 5 mg BID, n = 1 [1.0%]; tofacitinib 10 mg BID, n = 5 [5.0%]; placebo, n = 2 [2.1%]); two patients without baseline MetS began lipid‐lowering medications (tofacitinib 5 mg BID, n = 1 [0.7%]; tofacitinib 10 mg BID, n = 1 [0.7%]).
Table 4.
Baseline and percentage change from baseline in fasting lipid values at month 3 and month 6, by treatment group and baseline MetS status; pooled data from OPAL Broaden and OPAL Beyond
| Tofacitinib 5 mg BID a | Tofacitinib 10 mg BID a | Placebo a , b | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
With baseline MetS (N = 99) |
Without baseline MetS (N = 139) |
With baseline MetS (N = 101) |
Without baseline MetS (N = 135) |
With baseline MetS (N = 94) |
Without baseline MetS (N = 142) |
|||||||||||
| N1 |
Mean (SD) Median [IQR] |
N1 |
Mean (SD) Median [IQR] |
N1 |
Mean (SD) Median [IQR] |
N1 |
Mean (SD) Median [IQR] |
N1 |
Mean (SD) Median [IQR] |
N1 |
Mean (SD) Median [IQR] |
|||||
| LDL‐cholesterol | ||||||||||||||||
| Baseline (mg/dL) | 90 |
120.3 (35.0) 115.8 [94.2‐142.9] |
132 |
114.3 (30.1) 113.7 [93.8‐134.5] |
90 |
124.4 (37.6) 120.1 [100.0‐152.0] |
119 |
116.5 (36.7) 110.0 [90.0‐133.2] |
87 |
120.8 (36.4) 117.0 [95.0‐146.7] |
135 |
110.5 (30.8) 110.0 [87‐127.8] |
||||
|
% change from baseline to month 3 |
83 |
10.6 (23.0) 10.7 [−5.3 to 20.8] |
125 |
8.2 (18.4) 6.8 [−2.2 to 19.6] |
87 |
11.9 (25.8) 6.1 [−2.0 to 21.2] |
114 |
16.0 (25.9) 11.5 [−1.4 to 30.6] |
79 |
4.5 (17.7) 0.9 [−6.9 to 14.3] |
126 |
4.2 (20.0) 1.7 [−8.3 to 11.5] |
||||
| % change from baseline to month 6 | 86 |
10.0 (24.2) 10.3 [−4.3 to 22.6] |
124 |
9.6 (19.1) 4.6 [−2.9 to 20.7] |
79 |
12.8 (22.6) 11.8 [−4.0 to 29.5] |
117 |
15.4 (24.5) 11.7 [0.3 to 31.3] |
NA | NA | NA | NA | ||||
| HDL‐cholesterol | ||||||||||||||||
| Baseline (mg/dL) | 92 |
48.7 (15.3) 44.4 [38.5‐56.9] |
132 |
61.0 (16.7) 58.2 [28.6‐70.3] |
93 |
48.7 (15.7) 45.6 [38.6‐56.4] |
119 |
62.8 (21.1) 57.5 [47.1‐72.2] |
88 |
47.9 (13.3) 46.2 [39.6‐55.0] |
136 |
61.0 (18.0) 58.3 [48.8‐70.7] |
||||
|
% change from baseline to month 3 |
89 |
11.1 (19.7) 10.2 [−2.2 to 19.5] |
125 |
8.6 (20.6) 5.3 [−3.6 to 17.5] |
90 |
16.4 (18.9) 14.6 [5.8 to 25.4] |
116 |
12.6 (18.8) 11.6 [0.4 to 24.2] |
84 |
0.4 (15.2) 0.7 [−9.3 to 8.4] |
127 |
−1.3 (16.5) −1.6 [−8.6 to 9.4] |
||||
|
% change from baseline to month 6 |
93 |
10.0 (23.3) 9.8 [−2.9 to 16.5] |
124 |
8.2 (19.2) 7.5 [−4.1 to 18.0] |
83 |
16.3 (20.0) 14.1 [3.3 to 27.7] |
117 |
12.3 (20.8) 11.1 [−1.8 to 23.3] |
NA | NA | NA | NA | ||||
| Total cholesterol | ||||||||||||||||
| Baseline (mg/dL) | 92 |
206.5 (44.2) 206.8 [175.6‐234.8] |
132 |
197.3 (35.4) 197.8 [171.1‐220.1] |
94 |
209.8 (45.6) 207.0 [178.4‐239.8] |
120 |
199.0 (46.0) 192.3 [163.3‐227.8] |
88 |
203.4 (43.8) 200.2 [177.0‐235.8] |
136 |
193.2 (38.8) 193.2 [164.5‐214.7] |
||||
|
% change from baseline to month 3 |
90 |
9.6 (14.6) 8.7 [1.4‐15.6] |
125 |
7.4 (13.9) 7.1 [−0.7 to 14.0] |
91 |
10.2 (18.8) 11.0 [2.2‐18.9] |
117 |
13.9 (18.3) 11.5 [2.3‐22.6] |
84 |
2.5 (13.0) 0.8 [−4.9 to 7.1] |
127 |
1.7 (13.8) 1.1 [−7.0 to 8.2] |
||||
|
% change from baseline to month 6 |
93 |
8.4 (16.2) 7.3 [−1.2 to 17.6] |
124 |
7.7 (12.8) 6.7 [−2.5 to 16.0] |
84 |
14.3 (17.3) 12.9 [2.1‐24.8] |
117 |
14.2 (17.4) 12.1 [2.7‐23.9] |
NA | NA | NA | NA | ||||
| Triglycerides | ||||||||||||||||
| Baseline (mg/dL) | 92 |
193.4 (107.8) 179.1 [127.9‐222.1] |
132 |
110.0 (42.9) 100.9 [83.2‐131.5] |
93 |
202.7 (216.0) 162.0 [121.3‐210.6] |
119 |
97.3 (37.7) 95.6 [67.0‐119.5] |
88 |
177.7 (78.8) 169.0 [125.2‐216.8] |
136 |
107.7 (55.6) 93.8 [73.9‐117.3] |
||||
|
% change from baseline to month 3 |
90 |
9.2 (44.8) −1.0 [−21.1 to 27.6] |
125 |
9.4 (36.5) 4.5 [−15.0 to 27.7] |
90 |
7.6 (43.1) 3.8 [−20.2 to 25.6] |
116 |
24.1 (63.0) 8.6 [−11.1 to 49.0] |
84 |
7.4 (48.6) −4.0 [−21.4 to 22.6] |
127 |
11.4 (43.6) 1.4 [−15.7 to 38.0] |
||||
|
% change from baseline to month 6 |
93 |
6.1 (38.4) 2.1 [−19.0 to 28.1] |
124 |
6.6 (34.9) 1.7 [−18.6 to 26.1] |
83 |
14.9 (48.7) 8.3 [−19.8 to 34.9] |
117 |
25.3 (59.4) 17.9 [−17.2 to 46.4] |
NA | NA | NA | NA | ||||
Abbreviations: BID, twice daily; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; HDL, high‐density lipoprotein; IQR, interquartile range; LDL, low‐density lipoprotein; MetS, metabolic syndrome; N, number of patients treated without regard to baseline abnormality; N1, number of patients analyzed; NA, not assessed; SD, standard deviation.
All patients received a stable dose of one background csDMARD.
Patients in the placebo group were advanced to tofacitinib 5 or 10 mg BID after 3 months of placebo treatment.
At baseline, total bilirubin (for tofacitinib 5 mg BID only), AST, ALT, and gamma GT values were generally slightly higher in patients with versus without baseline MetS (Online Supplementary Table 4). In tofacitinib‐treated patients, AST and ALT levels slightly increased from baseline, and to a greater extent in those with MetS versus those without MetS (Online Supplementary Table 4). No clear pattern in the proportions of patients with liver parameters above ULN thresholds was observed between those with or without MetS (Online Supplementary Table 5).
DISCUSSION
This analysis compared the efficacy and safety of tofacitinib in patients with PsA, stratified by baseline MetS status. Regardless of baseline MetS status, tofacitinib 5 or 10 mg BID tended to show greater efficacy than placebo at month 3 in patients with active PsA, across all endpoints (ACR20/50/70, PASI75, HAQ‐DI response, enthesitis and dactylitis resolution, and changes from baseline in HAQ‐DI, PGA, SJC, TJC, CRP [in patients with baseline CRP > 2.87 mg/L], and additional PROs). These improvements were consistent to month 6 with tofacitinib. Additionally, baseline MetS status did not appear to affect tofacitinib safety up to month 3 or 6, as assessed by AEs, serious AEs, discontinuations due to AEs, and changes in lipid or liver parameters.
The efficacy of tofacitinib generally appeared unaffected by baseline MetS status at months 3 and 6, although some differences were noted in HAQ‐DI response at both time points with tofacitinib 10 mg BID, and dactylitis resolution with both tofacitinib doses at month 6. In patients with baseline CRP > 2.87 mg/L, CRP decrease from baseline with tofacitinib 10 mg BID appeared lower at month 3 in patients with baseline MetS versus those without baseline MetS; however, the reason for this remains unclear. The reduction in CRP levels observed with tofacitinib may relate to its downstream effect on IL‐6, which influences CRP levels (5). Although BMI has previously been reported to be positively correlated with CRP levels in PsA patients (20, 21), in this analysis, baseline CRP levels appeared similar in patients with and without baseline MetS, despite patients with baseline MetS having a higher baseline BMI. Additionally, at month 3, there was no interaction between treatment group, baseline MetS status, and efficacy endpoints, suggesting that baseline MetS status does not affect the treatment effects of tofacitinib doses differentially in PsA patients. Previous observations showing diminished responses to TNFi in patients with MetS or obesity (10, 11) were not observed here with tofacitinib.
Limited data are available assessing the effect of MetS on PsA treatment efficacy. A previous study of TNFi treatment in patients with PsA and MetS determined that those with MetS were less likely to achieve minimal disease activity (10). Higher BMI has also previously been associated with reduced treatment response in patients with PsA receiving tofacitinib (15) and TNFi (11). In contrast, the current analysis demonstrated that, with the exception of the endpoints discussed above, tofacitinib efficacy across clinical outcomes and PROs appeared similar in patients regardless of baseline MetS status and were also generally consistent with those seen in the OPAL Broaden and OPAL Beyond patient populations (12, 13). Tofacitinib efficacy generally did not appear to be affected by prior TNFi experience, although for some outcomes (ACR20, ACR50, and PASI75 response rates), the tofacitinib 10 mg BID dose appeared to result in numerically better responses in TNFi‐naïve versus TNFi‐IR patients, regardless of baseline MetS status; however, the variations may be due to the low patient numbers. The discrepancy in sample size between patients with versus without MetS in OPAL Broaden, and in some outcomes in OPAL Beyond, may also have impacted response rates. The elevated levels of TNF‐α and IL‐6 in patients with MetS may attenuate the efficacy of TNFi (10) to a greater extent than that of tofacitinib, which acts on the JAK‐STAT pathway and downregulates the levels of multiple cytokines involved in PsA (eg, IL‐2, IL‐6, IL‐17, IL‐22, and IL‐23) (7, 22, 23, 24, 25).
The safety profile of tofacitinib 5 and 10 mg BID was generally similar in patients with or without baseline MetS. Additional safety outcomes potentially important in the context of MetS (eg, fasting blood glucose levels, diabetes, and hypertension) were similar between treatment groups, and no new safety risks were identified, based on baseline MetS status (12, 13). One event each of arterial thromboembolism and myocardial infarction occurred in tofacitinib‐treated patients with baseline MetS. AEs of special interest (ie, herpes zoster, serious infections, opportunistic infections, gastrointestinal perforations, interstitial lung disease, and malignancies) have been reported previously (12, 13) and were not assessed in this analysis due to the low number of patients with these events in the index studies, which limits meaningful interpretation when analyzed further by baseline MetS subgroup. In tofacitinib‐treated patients, HDL‐cholesterol, LDL‐cholesterol, and triglyceride elevations at month 3 were similar to the index studies regardless of baseline MetS status. A pooled analysis of the OPAL Broaden and OPAL Beyond studies in patients with active PsA observed dose‐dependent increases in lipid levels of 10% to 15% following treatment with tofacitinib 5 and 10 mg BID at month 3, with no appreciable changes at month 6 (26). Relevant prescribing information recommends that lipid parameters are assessed between 4 and 8 weeks (US prescribing information) or 8 weeks (European Union summary of product characteristics) after tofacitinib initiation, and that patients are managed according to clinical guidelines (27, 28). The above pooled analysis also found that across OPAL Broaden, OPAL Beyond, and a long‐term extension study (median duration of tofacitinib exposure 594.0 days; range 1‐1,196 days), tofacitinib did not affect hypertension rates, CRP levels decreased, and the incidence of MACE was low (26). However, MACE have a long latency, and an ongoing randomized comparator study in rheumatoid arthritis is specifically investigating the risk of MACE with tofacitinib (ClinicalTrials.gov identifier NCT02092467). At month 3, slightly greater numerical increases from baseline in AST and ALT were generally observed in patients with baseline MetS versus those without baseline MetS. Given that MetS is a strong predictor of nonalcoholic fatty liver disease (29), there may also be a potential hepatic impact in this patient population, but observed increases were generally small and remained stable, and were not clinically relevant up to month 6. Routine monitoring of laboratory parameters, including liver enzymes, is recommended in patients treated with tofacitinib (28).
This post hoc analysis has limitations, and results should be interpreted with caution. Statistical analysis was estimation in nature, rather than hypothesis testing, with small patient numbers limiting potential conclusions, including comparisons between tofacitinib dose groups; furthermore, treatment differences between tofacitinib versus placebo, rather than patients with versus without MetS were assessed. Additionally, no universal consensus has been established for the diagnosis of MetS (30, 31); the criteria applied here were based upon a consensus statement from organizations that include the International Diabetes Federation and the National Heart, Lung, and Blood Institute (2). Therefore, these criteria may not fully capture the concepts of MetS impact on tofacitinib efficacy and safety in patients with PsA and MetS. Finally, it is also not known whether the lipid‐lowering medications initiated after baseline were prescribed due to tofacitinib therapy (as increases in lipid values after tofacitinib initiation have previously been observed) (32) or baseline MetS status.
In patients with active PsA, greater efficacy improvements were observed in patients who received tofacitinib 5 or 10 mg BID versus placebo, regardless of baseline MetS status. Additionally, MetS did not appear to affect the tofacitinib safety profile. Tofacitinib provides a treatment option for patients with active PsA, regardless of comorbid MetS. Future analyses of observational registry and/or clinical data will further inform the potential impact of MetS on the efficacy and safety of tofacitinib in patients with PsA.
ACKNOWLEDGMENTS
Medical writing support, under the guidance of the authors, was provided by Mark Bennett, PhD, and Christina Viegelmann, PhD, CMC Connect, McCann Health Medical Communications and was funded by Pfizer Inc, New York, NY, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461‐464).
AUTHOR CONTRIBUTIONS
All authors contributed to the interpretation of the data, critically revised each draft of the manuscript for intellectual content, provided final approval of the version submitted for publication, and accept accountability for the accuracy and integrity of the work.
Study conception and design
Ritchlin, Giles, Ogdie, Young, Wang, Wu, Woolcott, Stockert.
Acquisition of data
Helliwell, Wang, Wu.
Analysis and interpretation of data
Ritchlin, Giles, Ogdie, Gomez‐Reino, Helliwell, Young, Wang, Wu, Romero, Woolcott, Stockert.
Supporting information
Supplementary Material
This study was sponsored by Pfizer Inc.
Dr. Ritchlin has received grant/research support from AbbVie, Amgen, and UCB, and is a consultant for AbbVie, Amgen, Celgene, Janssen, Lilly, Novartis, Pfizer Inc, Sun, and UCB. Dr. Giles has received grant/research support from Pfizer Inc, and is a consultant for AbbVie, Bristol‐Myers Squibb, Genentech, UCB (less than $10,000 each), and Lilly (more than $10,000). Dr. Ogdie has received grant/research support from Novartis and Pfizer Inc to Penn and Amgen to Forward/The National Data Bank for Rheumatic Diseases (more than $10,000 each), and has served as a consultant for AbbVie, Amgen, Celgene, Corrona, Janssen, Lilly, Novartis, Pfizer Inc, Takeda, and UCB; her husband has received royalties from Novartis related to a patent (more than $10,000). Dr. Gomez‐Reino has received grant/research support from AbbVie, MSD, Pfizer Inc, and Roche; is a consultant for Pfizer Inc; and has participated in speakers’ bureaus for AbbVie, Biogen, Bristol‐Myers Squibb, Janssen, MSD, Pfizer Inc, and Roche. Dr. Helliwell has received research grants from AbbVie, Janssen, and Pfizer Inc; has received honoraria from AbbVie, Amgen, Gilead, Janssen, Pfizer Inc, and UCB; and has conducted other work for AbbVie, Janssen, and UCB. Drs. Young, Wang, Wu, Woolcott, and Ms. Stockert are employees and shareholders of Pfizer Inc. Dr. Romero was an employee and shareholder of Pfizer Inc at the time of this analysis. No other disclosures relevant to this article were reported.
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices 1) for indications that have been approved in the US and/or EU, or 2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1. Haroon M, Gallagher P, Heffernan E, FitzGerald O. High prevalence of metabolic syndrome and of insulin resistance in psoriatic arthritis is associated with the severity of underlying disease. J Rheumatol 2014;41:1357–65. [DOI] [PubMed] [Google Scholar]
- 2. Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009;120:1640–5. [DOI] [PubMed] [Google Scholar]
- 3. Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, et al. The metabolic syndrome and cardiovascular risk: a systematic review and meta‐analysis. J Am Coll Cardiol 2010;56:1113–32. [DOI] [PubMed] [Google Scholar]
- 4. Meigs JB, Wilson PWF, Fox CS, Vasan RS, Nathan DM, Sullivan LM, et al. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J Clin Endocrinol Metab 2006;91:2906–12. [DOI] [PubMed] [Google Scholar]
- 5. Wisse BE. The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity. J Am Soc Nephrol 2004;15:2792–800. [DOI] [PubMed] [Google Scholar]
- 6. Després J‐P, Lemieux I. Abdominal obesity and metabolic syndrome. Nature 2006;444:881–7. [DOI] [PubMed] [Google Scholar]
- 7. Coates LC, FitzGerald O, Helliwell PS, Paul C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: is all inflammation the same? . Semin Arthritis Rheum 2016;46:291–304. [DOI] [PubMed] [Google Scholar]
- 8. Versini M, Jeandel P‐Y, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev 2014;13:981–1000. [DOI] [PubMed] [Google Scholar]
- 9. Haroon M, Rafiq Chaudhry AB, Fitzgerald O. Higher prevalence of metabolic syndrome in patients with psoriatic arthritis: a comparison with a control group of noninflammatory rheumatologic conditions. J Rheumatol 2016;43:463–4. [DOI] [PubMed] [Google Scholar]
- 10. Costa L, Caso F, Ramonda R, Del Puente A, Cantarini L, Darda MA, et al. Metabolic syndrome and its relationship with the achievement of minimal disease activity state in psoriatic arthritis patients: an observational study. Immunol Res 2015;61:147–53. [DOI] [PubMed] [Google Scholar]
- 11. Singh S, Facciorusso A, Singh AG, Vande Casteele N, Zarrinpar A, Prokop LJ, et al. Obesity and response to anti‐tumor necrosis factor‐α agents in patients with select immune‐mediated inflammatory diseases: a systematic review and meta‐analysis. PLoS One 2018;13:e0195123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila‐Zapata F, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 2017;377:1537–50. [DOI] [PubMed] [Google Scholar]
- 13. Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med 2017;377:1525–36. [DOI] [PubMed] [Google Scholar]
- 14. Nash P, Coates LC, Kivitz AJ, Mease PJ, Gladman DD, Covarrubias‐Cobos JA, et al. SAT0293 Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, up to 36 months in patients with active psoriatic arthritis: data from the third interim analysis of OPAL Balance, an open‐label long‐term extension study [abstract]. Ann Rheum Dis 2018;77 (Suppl 2) http://scientific.sparx‐ip.net/archiveeular/?searchfor=nash&view=1&c=a&item=2018SAT0293. [Google Scholar]
- 15. Ritchlin C, Ogdie A, Giles J, Gomez‐Reino J, Helliwell P, Stockert L, et al. Impact of baseline body mass index on the efficacy and safety of tofacitinib in patients with psoriatic arthritis [abstract]. Arthritis Rheumatol 2019;71 (Suppl 10) URL: https://acrabstracts.org/abstract/impact‐of‐baseline‐body‐mass‐index‐on‐the‐efficacy‐and‐safety‐of‐tofacitinib‐in‐patients‐with‐psoriatic‐arthritis/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 17. Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient‐rated importance and satisfaction. J Rheumatol 2011;38:2461–5. [DOI] [PubMed] [Google Scholar]
- 18. Ware JE, Kosinski M Jr, Bjorner JB, Turner‐Bowker DM, Gandek B, Maruish ME. User’s manual for the SF‐36v2® Health Survey. 2nd ed Lincoln (RI): QualityMetric Incorporated; 2007. [Google Scholar]
- 19. EuroQoL Group . EuroQol–a new facility for the measurement of health‐related quality of life. Health Policy 1990;16:199–208. [DOI] [PubMed] [Google Scholar]
- 20. Klingberg E, Bilberg A, Björkman S, Hedberg M, Jacobsson L, Forsblad‐d'Elia H, et al. Weight loss improves disease activity in patients with psoriatic arthritis and obesity: an interventional study. Arthritis Res Ther 2019;21:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strober B, Teller C, Yamauchi P, Miller JL, Hooper M, Yang Y‐C, et al. Effects of etanercept on C‐reactive protein levels in psoriasis and psoriatic arthritis. Br J Dermatol 2008;159:322–30. [DOI] [PubMed] [Google Scholar]
- 22. Migita K, Miyashita T, Izumi Y, Koga T, Komori A, Maeda Y, et al. Inhibitory effects of the JAK inhibitor CP690,550 on human CD4(+) T lymphocyte cytokine production. BMC Immunol 2011;12:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meyer DM, Jesson MI, Li X, Elrick MM, Funckes‐Shippy CL, Warner JD, et al. Anti‐inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP‐690,550, in rat adjuvant‐induced arthritis. J Inflamm (Lond) 2010;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther 2019;21:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raychaudhuri SK, Abria C, Raychaudhuri SP. Regulatory role of the JAK STAT kinase signalling system on the IL‐23/IL‐17 cytokine axis in psoriatic arthritis. Ann Rheum Dis 2017;76:e36. [DOI] [PubMed] [Google Scholar]
- 26. Gladman DD, Charles‐Schoeman C, McInnes IB, Veale DJ, Thiers B, Nurmohamed M, et al. Changes in lipid levels and incidence of cardiovascular events following tofacitinib treatment in patients with psoriatic arthritis: a pooled analysis across phase III and long‐term extension studies. Arthritis Care Res (Hoboken) 2019;71:1387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. European Medicines Agency . Xeljanz (tofacitinib): summary of product characteristics. 2020. URL: https://www.ema.europa.eu/en/documents/product‐information/xeljanz‐epar‐product‐information_en.pdf.
- 28. US Food and Drug Administration . XELJANZ® (tofacitinib): highlights of prescribing information. 2019. URL: https://labeling.pfizer.com/ShowLabeling.aspx?id=959.
- 29. Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med 2005;143:722–8. [DOI] [PubMed] [Google Scholar]
- 30. Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med 2011;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Neill S, O'Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev 2015;16:1–12. [DOI] [PubMed] [Google Scholar]
- 32. Charles‐Schoeman C, Wicker P, Gonzalez‐Gay MA, Boy M, Zuckerman A, Soma K, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum 2016;46:261–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices 1) for indications that have been approved in the US and/or EU, or 2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.