

Summary
The beginning of the 21st century has been marked by three distinct waves of zoonotic coronavirus outbreaks into the human population. The COVID-19 (coronavirus disease 2019) pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and emerged as a global threat endangering the livelihoods of millions worldwide. Currently, and despite collaborative efforts, diverse therapeutic strategies from ongoing clinical trials are still debated. To address the need for such an immediate call of action, we leveraged the largest dataset of drug-induced transcriptomic perturbations, public SARS-CoV-2 transcriptomic datasets, and expression profiles from normal lung transcriptomes. Most importantly, our unbiased systems biology approach prioritized more than 50 repurposable drug candidates (e.g., corticosteroids, Janus kinase and Bruton kinase inhibitors). Further clinical investigation of these FDA-approved candidates as monotherapy or in combination with an antiviral regimen (e.g., remdesivir) could lead to promising outcomes in patients with COVID-19.
Subject Areas: Virology, Bioinformatics, Systems Biology
Graphical Abstract
Highlights
-
•
A large repertoire of drug repurposing across SARS-CoV-2 transcriptomic settings
-
•
Pathway enrichment revealed TNF-NF-κB signaling as a targetable hallmark for COVID-19
-
•
Drug enrichment identified BTK inhibitors as repurposable candidates for COVID-19
-
•
Identified drug candidates warrant investigation in models of SARS-CoV-2 infection
Virology; Bioinformatics; Systems Biology
Introduction
On December 31, 2019, the world held its breath as the first cases of pneumonia of unknown etiology detected in the Chinese city of Wuhan were brought to light by the WHO China Country Office. Ever since, this new pandemic caused by the single-strand positive RNA virus, SARS-CoV-2 spread quickly and caused a worldwide global panic. By May 22, 2020, the number of infected individuals has exponentially increased to 4,993,470 cases with the global death toll reaching 327,738 (Coronavirus Disease (COVID-19 May22), no date). This dramatic rise in cases owes itself on one hand to the nonspecific flu-like manifestations that afflict the vast majority of patients with COVID-19 and on the other to the heavy-travel profile characteristic of most populations in the current era of globalization. COVID-19 can clinically manifest on a spectrum ranging from mild non-specific flu-like symptoms to near-fatal acute respiratory distress syndrome (ARDS) leading to inflammation, pneumonia, acute lung injury (ALI), and sepsis especially in the elderly and persons with underlying comorbidities (Chen et al., 2020) (Wang et al., no date; Guan et al., 2020).
The current understanding of COVID-19 pathogenesis, etiology, and clinical features has been first extrapolated from knowledge gathered from other similar zoonotic infections associated with upper respiratory illness, such as the severe acute respiratory syndrome coronavirus (SARS-CoV) and the Middle East respiratory syndrome coronavirus (MERS-CoV) (Menter et al., 2020; Rota, 2003; Gu and Korteweg, 2007; Ng et al., 2016). It is now established that SARS-CoV-2 specifically recognizes the human angiotensin-converting enzyme 2 protein (ACE2) as its main target on the cell surface (Gheblawi et al., 2020; Hoffmann et al., 2020a). Following virus entry, the cell triggers the physiological response to the infection through several defense arms that are also thought to be implicated in the pathogenesis of the infection Thus, characterizing the downstream transcriptional hallmarks of COVID-19 is crucial to illuminating the mechanisms of this deadly respiratory infection and to guide potent therapeutic countermeasures. However, with the urgency of the current pandemic situation, developing new drugs capable of combating COVID-19 is deemed difficult. To that end, drug repurposing represents a rapid approach to expediting the process of finding a therapeutic candidate (Harrison, 2020; Gordon et al., 2020; Harrison, 2020). Cases of drug repurposing have been serendipitous rather than data-driven discoveries, and this extrapolates to COVID-19 research. The hydroxychloroquine-azithromycin combination has been proposed as a panacea for COVID-19 but has not been backed by a proven scientific rationale. Despite initial fanfare generated from favorable outcomes noted in an open-label, non-randomized clinical trial (Gautret et al., 2020), the azithromycin-hydroxychloroquine combination fell short of expectations in later trials with therapeutic inefficacy overshadowed by cardiovascular side effects associated with its use (Bessière et al., 2020; Geleris et al., 2020; Rosenberg et al., 2020; Tang et al., 2020). This setback highlights the dire need for an unbiased methodology to further identify drug-target associations that can be efficiently translated to the clinic.
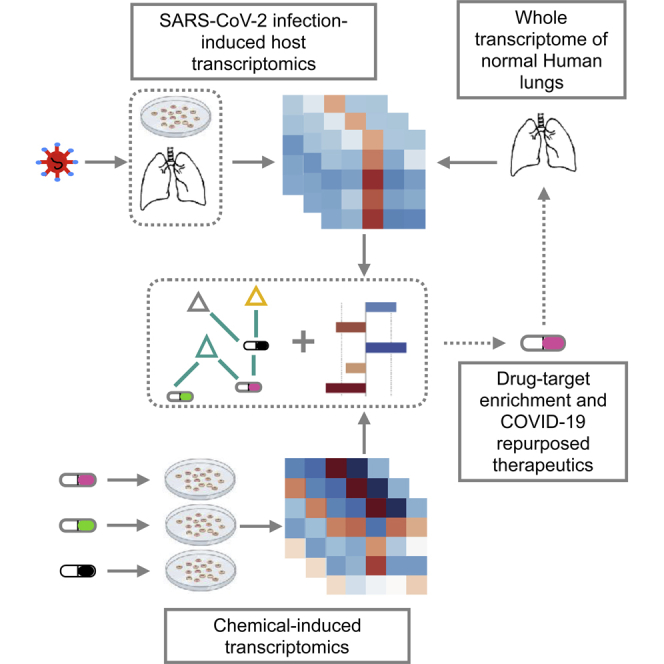
Motivated by both an unmatched global pandemic and a lack of plausible drug candidates to treat COVID-19, we have conducted a unique data-driven approach (Figure 1) that integrates three types of data: (1) a public resource of SARS-CoV-2 transcriptomics (Blanco-Melo et al., 2020), (2) drug-induced transcriptome profiles from the Library of Integrated Network-based Cellular Signatures (LINCS) Connectivity Map project (Subramanian et al., 2017), and (3) transcriptome-wide profiling in normal lung tissue from the Genotype-Tissue Expression (GTEx) project (GTEx Consortium, 2013). Briefly, our analyses highlighted several targetable hallmarks associated with the SARS-CoV-2 pathobiology and underscored plausible drug-target associations among which top-scoring hits have been acknowledged in ongoing COVID-19 clinical trials. Furthermore, our approach provides the largest set of pre-clinical candidates and US Food and Drug Administration (FDA)-approved drugs with known safety profiles to be further investigated for COVID-19 treatment. In comparison to other studies that have used the old version of Connectivity Map (Li et al., no date; Zhou et al., 2020) or the limited L1000 web interface (Duarte et al., no date), the novelty of our work is 2-fold: first, it provides the largest repertoire of drug repurposing candidates across available SARS-CoV-2, in vivo, and in vitro settings (R Shiny WebApp), and second, it has identified drug candidates that interfere with ACE2 co-expressed signatures in SARS-CoV-2-infected settings.
Figure 1.

Schematic Overview of the Computational Approach
Schematic overview of the computational approach. Molecular signatures induced by SARS-CoV-2, chemical-induced gene expression profiles, gene and drug set enrichment analyses, and drug-target databases have been integrated into the analytical framework to understand the molecular pathophysiology of SARS-CoV-2 and to identify novel therapeutic options.
Results
To fill the unmet need for effective COVID-19 treatments, we leveraged the LINCS—the largest body of drug-induced transcriptional perturbations profiles available to date, the healthy lung transcriptome from the GTEx project, and the recently published SARS-CoV-2-induced transcriptomic profiles from in vivo and in vitro settings (all settings are described in the method section and illustrated in Table 1).
Table 1.
Summary of Public Datasets Used in the Analytical Pipeline
| Group | Mock Controls | SARS-COV-2 | Treatment | Dataset | Duration | Setting |
|---|---|---|---|---|---|---|
| SARS2_A549_ACE2 | 3 | 3 | - | GSE147507 | 1 d | Lung adenocarcinoma, in vitro + ACE2 vector |
| SARS2_A549_ACE2_RUXO | - | 3 | 3 (500 nM pre-treatment) | GSE147507 | 1 d | Lung adenocarcinoma, in vitro |
| SARS2_Calu3 | 3 | 3 | - | GSE147507 | 1 d | Lung adenocarcinoma, in vitro |
| SARS2_LUNG | 2 (Healthy lung biopsies) | 2 (Technical rep, same deceased patient) | - | GSE147507 | 1 d | Ex vivo, patient biopsy |
| SARS2_NHBE | 3 | 3 | - | GSE147507 | 1 d | Normal lung cell |
| SARS2_BALF_WUHAN_1 | 3 (BALF, healthy) | 2 (Replicates BALF, COVID-19) | - | PMID: 32228226,PMID: 31836714 | - | In vivo, Bronchoalveolar lavage fluid |
| SARS2_BALF_WUHAN_2 | 3 (BALF, healthy) | 2 (Replicates BALF, COVID-19) | - | PMID: 32228226,PMID: 31836714 | - | In vivo, Bronchoalveolar lavage fluid |
| GTEX | 374 (Healthy postmortem) | - | - | https://gtexportal.org/home/ | - | Ex vivo, patient postmortem biopsy |
Hallmarks of SARS-CoV-2 Infection across In Vivo and In Vitro Settings
To address pharmacological gaps in SARS-CoV-2 research, it was of paramount importance to understand the molecular mechanisms underlying the SARS-CoV-2 infection. To this end, we have collected public SARS-CoV-2 transcriptome-wide datasets and normal lung transcriptomics from GTEx. Subsequently, we conducted a differential expression analysis to compare SARS-CoV-2 conditions with their respective control groups. Next, all genes from the different comparisons were merged by the shrunken log2 fold change values (no-cutoff) to yield a full matrix corresponding to 7,433 genes × 7 contrasts. These are referred to as SARS2_BALF_WUHAN1-2, SARS2_LUNG, SARS2_NHBE, SARS2_A549_ACE2, SARS2_Calu3, and SARS2_A549_ACE2_RUXO (ruxolitinib-treated cells).
For each of these seven contrasts, the fraction of significant up- and downregulated genes (|Log2FoldChange| > 1 and p-adjusted value < 0.05) and the corresponding overlap between contrasts is given in Figure S1. As for the transcriptomic data from the GTEx project (N = 374 samples), gene-wise Pearson correlation of the ACE2 gene and all other expressed genes is referred to as ACE2_GTEX (Pearson correlation ∈[-0.47, 0.59]), out of which 7,427 genes were shared with the seven SARS-CoV-2 contrasts and genes were ranked accordingly.
The ranked lists of genes across all SARS2 contrasts as well as genes ranked by their correlation coefficient from ACE2_GTEX were investigated at the pathway level using gene set enrichment analysis (GSEA). We assessed enrichment against a collection of hallmark gene sets and a curated set of genes associated with SARS_CoV infection from the literature (see Transparent Methods section). To assess the similarity between settings, we first computed all pairwise correlations of pathway enrichment scores from all GSEA analysis (Figure 2A). Interestingly, both SARS2_A549_ACE2_RUXO and ACE2_GTEX were strongly concordant (rho = 0.63, p < 0.01). This suggests that ruxolitinib treatment in ACE2-expressing A549 cells and ACE2 baseline expression in the lungs engage a similar set of molecular pathways. Indeed, similarly to ACE2_GTEX, ruxolitinib-induced pathways were highly anti-correlated with those from all the SARS2 settings (Figure 2A). Although SARS2_LUNG was correlated with bronchoalveolar lavage fluid (BALF) settings, this correlation was not significant (p value > 0.05, Figure 2A), suggesting a functional diversity in response to SARS-CoV-2.
Figure 2.

Hallmark Enrichment Analyses
SARS2 and ACE2_GTEX settings.
(A) Pairwise Spearman rank correlations from the Hallmark enrichment analyses. Red and blue colors represent positive and negative correlations, respectively, whereas blank cells show non-significant correlations at a given p value threshold of 0.05.
(B) Dot plot visualization of enriched terms in all SARS2 and ACE2_GTEX settings. Gene set enrichment analyses (GSEA) were performed against the Hallmark dataset from MsigDB and a collection of genes related to SARS-CoV infection from the literature (gene shot, 224 genes). The color of the dots represents the enrichment score, and the FDR value of significance was set to 0.05 for all sets. Size represents the total number of genes in the gene set.
(C) Heatmap depicting the expression levels of 169 genes linked to the innate immune system across all SARS2 settings. These genes were extracted from the leading edge of the GSEA and belong to the following hallmarks/sets: TNFA SIGNALING VIA NFKB, INTERFERON GAMMA RESPONSE, INTERFERON ALPHA RESPONSE, IL6-JAK-STAT3 SIGNALING, INFLAMMATORY RESPONSE, and the geneshot_SARS_CoV gene list. The graph illustrates the shrunken log2 fold change from all SARS2 contrasts. The sidebar plots show the number of settings for which the genes have a p-adjusted value of less than 0.05. The heatmap shows five distinct groups from k-mean clustering. A side heatmap depicts the correlation values for these same genes against ACE2 expression from GTEx (all genes have negative correlation values because all mentioned hallmarks were significantly downregulated in ACE2_GTEX).
We further investigated the enriched hallmarks/pathways in both the normal lung and SARS-CoV-2-infected in vivo and in vitro settings. By keeping all gene sets with a normalized enrichment score |NES| greater or equal to 1 and a p-adjusted value threshold of 5%, our analysis identified a highly conserved hallmark process, the TNF-α alpha (tumor necrosis factor alpha) signaling via nuclear factor (NF)-κB. This hallmark was significantly upregulated by the SARS-CoV-2 infection but downregulated in both ruxolitinib-treated and ACE2_GTEX groups (Figure 2B). Moreover, to investigate the pathogenic cellular response of SARS-CoV-2, we proceeded as follows: (1) we extracted the union of all leading-edge genes from the conserved hallmark and (2) we extracted leading-edge genes belonging to the following enriched hallmarks: INTERFERON GAMMA RESPONSE, INTERFERON ALPHA RESPONSE, INFLAMMATORY RESPONSE, IL6-JAK-STAT3 signaling, and the curated set of SARS-CoV-associated genes from the literature (geneshot_SARS_CoV). It is noteworthy to mention that enriched hallmarks from (2) were negatively scoring gene sets in ACE2_GTEX and positively enriched in all SARS2 settings but the WUHAN_BALF samples (Figure 2B). Across all settings, the resulting genes were assigned either the shrunken log2 fold change from their corresponding differential gene expression (DEG) analysis or the correlation value for ACE2_GTEX (Figure 2C). From this collection of relevant genes of innate immunity, we identified a set of conserved upregulated genes in SARS2 settings (group1, Figure 2C). These same upregulated genes were downregulated in ruxolitinib-treated cells and highly anti-correlated with reference to ACE2_GTEX. Most of these genes (e.g., CXCL10, CXCL11, OAS1, TNFSF10, MX1, DDX58, ISG15, IFIT2, IFIT3, EIF2AK2) are induced in response to interferons (IFNs). Notably, the induction of these genes was much more pronounced in SARS2_LUNG, SARS2_Calu3, and SARS2_A549_ACE2 (Figure 2C). For instance, the pro-inflammatory chemokine CXCL10/IP-10 had a 32-fold increase in SARS2_LUNG compared with a 3-fold increase in the WUHAN_BALF samples, and it was almost absent in NHBE-infected cells. Chemokine's expression in the postmortem lung sample (SARS2_LUNG) was similar to what is being observed in A549_ACE2 and Calu3 cells infected with high loads of virus (MOI:2). Although MOI of 2 was also used for NHBE cells, RNA sequencing profiling 24 h post-infection showed that these cells efficiently cleared the virus (no change observed for CXCL10-11). In contrast to groups 1 and 2 that were predominantly represented by IFN-induced chemokines and genes regulated by NF-κB in response to TNF-α, group 3 seems to represent a set of genes belonging to the IL6-JAK-STAT3 pathway (CSF1, IFNGR2, IL4R, IFNGR1, HMOX1, TYK2, TGFB1, TNFRSF1A) and inflammatory responses (PTGER4, IRS2, MAP2K3, AXL, C5AR1, PTAFR); these genes were antagonized by SARS-CoV-2 specifically in BALF samples and not the cell lines. Last, group 4 showed a similar pattern to group 1 and its gene members are also related to the IFN alpha/beta/gamma, and complement signaling (e.g., TRIM25, UBE2L6, STAT1, JAK2, IL1B, IL2RG, GBP2, PLAUR). For instance, STAT1, a known master regulator of IFN genes, has more than a 5-fold-induction in SARS2_LUNG, A549, and Calu3, but its expression was not tangible in the other in vivo settings or SARS2_NHBE. In summary, expression dynamics from the deceased patients are highly similar to those from SARS2_A549_ACE2 and SARS2_Calu3 but dissimilar to WUHAN_BALF and NHBE cells. The former elicits marked pro-inflammatory events, which could explain the development of severe cases of COVID-19 symptoms and lung injury as seen in the postmortem samples.
Identification of New COVID-19 Repurposing Opportunities through Mining L1000 Connectivity Map
In the previous section, we highlighted molecular vulnerabilities providing a rationale for targeting hyperinflammatory response in COVID-19. Although ruxolitinib provided a proof of concept that abrogating inflammation is an appealing therapeutic strategy, we conducted a full data-driven computational approach to identify potential drug candidates that, in addition to ruxolitinib, could perturb the pathological mechanism induced by the new SARS-CoV. To this end, we have interrogated the L1000, the largest repository of chemical-induced gene perturbation profiles. The curation and pre-filtering of the L1000 dataset used in this study are described in the Transparent Methods section. Briefly, we have assembled expression perturbations of 978 landmark genes across 4,487 chemical compounds from LINCS L1000. Of the 978 genes 727 (~75%) were shared with our SARS2 and ACE2_GTEX settings. To identify potential drug repurposing candidates, we used the cosine similarity distance between vectors of gene expression signatures. For each of the drug-induced expression profiles, a cosine score was calculated against all SARS2 and ACE2_GTEX settings (shinyApp table provided upon request). A positive cosine score corresponds to similar expression patterns (useful to identify drugs inducing similar modes of action/molecular mechanisms), whereas a negative score would suggest an opposite expression pattern (likely beneficial in the context of a disease, specifically COVID-19).
We first validated our approach on ruxolitinib signature in SARS-CoV2-infected cells (SARS2_A549_ACE2_RUXO). Our pipeline could accurately and unbiasedly identify ruxolitinib from L1000 as the second-best matching hit out of 4,487 chemical compounds (cosine score = 0.18, p value from 10,000 random permutations = 2 × 10−4).
To further ascertain that our approach can identify not only ruxolitinib but its drug target class, we performed a drug set enrichment analysis. In this setting, a drug set is a target gene/protein that is associated with at least three drugs. We first ranked drugs by the cosine value and ran the enrichment analysis against 295 target genes (see Transparent Method section for drug-target preparation and database). With respect to SARS2_A549_ACE2_RUXO, we could confirm that indeed JAK2 inhibitors were significantly overrepresented at the top of the ranked list of cosine scores (ES = 0.825, adjusted p value = 0.08) (Figure 3C).
Figure 3.

Drug Set Enrichment Analysis and Enriched Drug-Target Associations
(A and B) (A) Dot plot visualization of enriched drug-target associations in all SARS2 including the generated consensus cosine score. Drug set enrichment analyses were performed against 295 drug-target families. Only those with absolute enriched score ES ≤ −0.5 and an FDR <0.25 are shown. (B) ACE2_GTEX settings. Drug set enrichment analyses were performed against 295 drug-target families. Only those with absolute enriched score ES ≥ 0.5 and an FDR <0 .25 are shown.
(C and D) (C) GSEA plot of the cosine signature for Janus kinase 2 inhibitors in SARS2_A549_ACE2_RUXO drug set enrichment analysis and (D) GSEA plot of the cosine signature for Bruton kinase inhibitors in ACE2_GTEX drug set enrichment analysis.
(E) Heat plot showing all enriched drug-target families with putative dual activities: restoring both ACE2 co-expression partners in the lungs and pathway perturbations following SARS-CoV-2 infection.
Next, we investigated drug-target classes that display a negative enrichment score (their drug members likely reverse the phenotype) against a computed consensus cosine score, which is the median value of the cosine spanning 4,487 drugs across all SARS2 settings. We postulate that the consensus score would emphasize drug classes that target generic processes induced by SARS-CoV-2 across diverse settings and infectivity phases/severity levels. Furthermore, drug-target enrichment analyses were conducted separately for each of the SARS2 settings (excluding RUXO group) (Figure 3A).
Importantly, when considering an enrichment drug score (ES) less or equal to −0.5 and adjusted-p value < 0.25 (Figure 3A), our consensus signature exhibited the strongest enrichment for JAK2 inhibitors (filgotinib, baricitinib, fostamatinib), Bruton kinase-BTK inhibitors (GDC-0834, ibrutinib, dasatinib; Figure 3D), CACNA1C blockers (mibefradil, diltiazem, verapamil), HCRTR1 (orexin) antagonists (SB-334867/almorexant/suvorexant), PTGER2 agonists (carbacyclin, treprostinil), KCNA7 channel blockers (amiodarone, flecainide), ASIC3 channel blocker (amiloride, nafamostat, diclofenac), PDE5A inhibitors (milrinone, zaprinast, vardenafil, ibudilast, sildenafil), and ADRA1B antagonists (prazosin, tamsulosin, alfuzosin, doxazosin, mianserin, ritanserin, phentolamine, indoramin) (Figure 3A). When leveraging the drug enrichment analysis for each of SARS2 settings, we observed that SARS2_LUNG exhibited very few enriched drug classes, among these enriched classes were BRD4 inhibitors (BI-2536, colchicine) and NR1I2 agonists (hyperforin, mifepristone, nifedipine, clotrimazole, paclitaxel). Interestingly, corticosteroid agonists (methylprednisolone, triamcinolone, budesonide, mometasone, beclomethasone, dexamethasone, fluocinonide, betamethasone, fluorometholone) were enriched in WUHAN_BALF, consensus, and NHBE settings.
Given the important role of ACE2 in both SARS-CoV-2 infectivity and normal lung physiology, we next sought to identify drugs that could counteract the SARS-CoV-2 invasion by restoring the baseline co-expression network of ACE2 in the healthy lung.
In this particular analysis and similar to SARS2_A549_ACE2_RUXO, a positive enrichment score is the preferred outcome (Figure 3B). A drug-target class with an enrichment score equal or greater than 0.5 is considered a putative candidate to maintain a normal ACE2 expression, whereas a drug-target with a negative enrichment score is likely to be beneficial for the virus. The search for such modulators identified drug-targets highly enriched for inhibitors of cereblon protein (lenalidomide, pomalidomide, thalidomide; ES = 0.93, adjusted-p value = 0.05) and Bruton kinase BTK (fostamatinib, ibrutinib, dasatinib, GDC-0834; ES = 0.86, adjusted-p value = 0.023). Strikingly, ACE inhibitors (enalapril, fosinopril, captopril, perindopril) also showed positive connectivity with ACE2_GTEX (ES = 0.61, adjusted-p value = 0.22). This could be explained as ACE expression from GTEx was highly anti-correlated with ACE2 (rho = −0.47). Last, and similar to Bruton kinase inhibitors, we identified drug-target classes such as NR3C1 agonists (corticosteroids), PDE5A inhibitors (milrinone, zaprinast, vardenafil, ibudilast, sildenafil), ADRA2B antagonists (phenoxybenzamine, prazosin, bromocriptine, apomorphine, terguride, ARC-239, chlorpromazine, mirtazapine, phentolamine), HTR2A agonists (MK-212, pergolide, pindolol, terguride, donitriptan, quipazine, lorcaserin, aripiprazole), and KCNA10 channel blockers (verapamil, pimozide). These classes might have a dual effect by modulating the inflammatory response, protecting the lung, and restoring ACE2-induced downregulation by the virus (Figure 3E). We are aware that only a limited fraction of the drugs is assigned to a target class (1,030 of 4,487 drugs; 23%). As such, all results from our analyses will be made publicly available to accelerate the search for COVID-19 treatments.
Pathways/Molecular Hallmarks Associated with Potential Modulators of COVID-19
To understand the mechanism by which the candidate drugs block the molecular events underlying COVID-19, we focused on significant drug-target lists associated with the previously described consensus score. This involved 25 drug-target families and an ensemble of 106 drugs (Figure S2). Given the small number of genes from the L1000 dataset (727 of the 978 landmark genes are shared with our data), we opted out to choose GSEA but instead, we conducted a row-wise Welch's t test to identify the landmark genes that show significant changes between the 106 drugs and the SARS2 groups. This analysis yielded a set of 124 genes that displayed extreme values from the two-tailed t test (false discovery rate <5%, 73 genes with positive t-stat value, 51 genes with negative t-stat values) (Figure 4A).
Figure 4.

Core Pathway Enrichment from SARS2 Settings in Which Gene Members were Significantly Downregulated/Upregulated in Response to Top Candidate Drugs
(A) Welch's t test was performed between gene levels induced by 106 selected drugs from L1000 and SARS-CoV-2 expression settings. Heatmap illustrating the expression level of 124 genes that significantly separate the two groups (p-adjusted value from the t test of less than 0.05).
(B) Heat plot for core pathways in which gene members were significantly downregulated in response to L1000 drugs.
(C) Heat plot for core pathways in which gene members were significantly upregulated in response to L1000 drugs, in contrast to SARS2 settings (p value < 0.05).
When performing a hypergeometric test, separately for the negatively and positively modulated genes considering the set of significant genes, we showed that several downregulated processes were enriched for cytokine signaling, Jak-STAT signaling, complement activation, TNF-α effects on cytokine activity, lung fibrosis, and inflammatory response. Gene members include transcripts such as RALA, CD40, FRS2, CRKL, SOCS2, UBE2L6, STX4, SMAD3, CCL2, IFNAR1, SPRED2, PIK3CA, and IKBKB (Figure 4B). In contrast, upregulated processes were enriched for metabolic processes, mainly glucose metabolism, cell cycle, and mRNA processing. Such transcripts include CTD, LBR, PGM1, NUP85, PFKL, PLCB3, PSMD10, SLC5A6, CAT, NUP133, HK1, HSD17B10, ETFB, NUP62, ADH5, NUP88, NNT, RPN1, GNAI2, VAPB, and SACM1L (Figure 4C). Finally, although L1000 contains a limited set of genes and a lot of transcriptome-wide information is missing, we could confirm that the identified drugs interfere with the hallmarks of the SARS-CoV-2 infection described earlier. More importantly, most of these drugs are FDA approved and patients would benefit from them before reaching severe courses of COVID-19.
Discussion
In this study, we focus on (1) defining aspects of molecular pathogenesis with regard to SARS-CoV-2 and (2) validating potential therapeutic targets using integrative data-driven computational analyses. This was carried on large transcriptomic datasets encompassing both in vitro and in vivo SARS-CoV-2-infected samples as well as normal human lung biopsy samples to provide a global solid basis to validate our results.
GSEA was initially carried out on all of the aforementioned datasets to ascertain the molecular pathways involved in SARS-CoV-2 pathogenesis. TNF-α signaling via NF-κB was found to be a highly conserved hallmark process across all SARS-CoV-2 models. This aligns with published research on the pathogenesis of betacoronaviruses (SARS-CoV in MERS-CoV) (DeDiego et al., 2014; Zhang et al., 2007; Kanzawa et al., 2006; Kanzawa et al., 2006; de Wit et al., 2016; Siu et al., 2019). Our analysis provides further validation of the central role of TNF-α signaling via NF-κB in SARS-CoV-2 pathogenesis. It has been reported that serum levels of TNF-α along with many other pro-inflammatory cytokines and chemokines were higher in patients with SARS-CoV-2 in the intensive care unit (ICU) relative to their non-ICU counterparts (Siu et al., 2019; Huang et al., 2020). TNF-α plays an instrumental role in orchestrating the clinical outcomes seen in severe SARS-CoV and SARS-CoV-2 infections (reviewed in Tay et al., 2020). Besides its role in the ominous cytokine storm, TNF-α strongly induces hyaluronan synthase-2 (HAS2) in pulmonary epithelial and fibroblasts potentially leading to ARDS (Xu et al., 2020). Downregulation of ACE2 expression by SARS-CoV-1 S protein has been incriminated in the ALI characteristic of severe SARS-CoV and possibly SARS-CoV-2 (Kuba et al., 2005; Hoffmann et al., 2020a). In one study, it was shown that SARS-CoV S protein downregulates ACE2 by upregulating TNF-α production, which in turn acts in an autocrine fashion to induce the TNF-α-converting enzyme (TACE)-dependent shedding of the ACE2 ectodomain (Haga et al., 2008). Very recently, Feldman and colleagues (Robinson et al., 2020) discussed the potential of anti-TNF therapies as a treatment for COVID-19 based on data from the SECURE-IBD registry and the COVID-19 Global Rheumatology Alliance registry. Both studies showed that specific TNF blockade is associated with better outcome and fewer hospital admissions. However, limited reports on the use of anti-TNF in patients with COVID-19 warrants further trials and consideration.
Our analyses highlighted a strong transcriptional response and induction of a subset of pro-inflammatory cytokines and IFN-induced genes. Subsequently, the in vivo dataset also allowed us to dichotomize the lung biopsy and BALF samples into two degrees of clinical severity. Lung biopsy samples are likely to represent severe SARS-CoV-2 groups, as they were derived from SARS-CoV-2 deceased victims, whereas milder SARS-CoV-2 infection was represented by BALF samples. Stronger estrogen enrichment in BALF samples as opposed to the SARS-CoV-2 samples alludes to a possible protective role for estrogen in SARS-CoV-2 infection. This notion is further strengthened by epidemiologic data showing a preponderance of males in patients with severe SARS-CoV-2 infection (Channappanavar et al., 2017; Bi et al., 2020; Stelzig et al., 2020). Furthermore, estrogen (E2) was found to decrease ACE2 expression in vitro cell models and thus potentially regulate SARS-CoV-2 infection (Bi et al., 2020; Stelzig et al., 2020). IFN-related genes such as STAT1 and CXCL10 as well as inflammatory response and IL-6-JAK-STAT pathway genes were significantly upregulated in the lung biopsy samples and minimally expressed in BALF tissue samples. The latter result possibly stems from the impressive arsenal of anti-IFN molecular tricks that betacoronaviruses have evolved to evade the impending immune response. The SARS-CoV nsp1 protein has demonstrated the ability to interfere with IFN signaling pathways by decreasing the levels of phosphorylated STAT1 (Wathelet et al., 2007), whereas MERS-CoV membrane protein (M protein) and SARS-CoV ORF3b and -6 were found to prevent nuclear IRF3 translocation and subsequent IFN-stimulated genes expression (Kopecky-Bromberg et al., 2007; Yang et al., 2013). A testament to its pathogenic prowess, SARS-CoV-2 was found to trigger significantly lower levels of IFNs than SARS-CoV while boasting a superior infection and replication rate in the lungs (Chu et al., 2020). In case of progression into severe SARS-CoV-2, the immune milieu adopts the transcriptomic profile of the lung sample dataset. Unabated viral replication eventually induces the delayed release of IFN-α/β and the subsequent influx of inflammatory macrophages. Consequently, proinflammatory cytokines (TNF-α, IL-6, and IL-1β) are released to devastating effect. Inevitably, the aforementioned “cytokine storm” hinders viral clearance via T cell apoptosis and damages pulmonary microvascular and alveolar epithelial cell barriers via endothelial and epithelial cell apoptosis. The resultant vascular leakage and alveolar edema induce ARDS and lung damage (Ye et al., 2020). Interestingly and in stark contrast to the other in vitro SARS-CoV-2 datasets, the NHBE SARS-CoV-2 set failed to register any tangible hallmark-associated gene expression. This could be explained by the cell tropism of SARS-CoV-2 in the human lungs that favors type I pneumocytes, type II pneumocytes, and alveolar macrophages over bronchial epithelia (Chu et al., 2020). Alternatively, NHBE might be innately capable of clearing the virus. Elucidation of the exact cause might be addressed in future cellular studies.
To identify potential repurposing candidates, we mined the L1000 connectivity map for drug inducing SARS-CoV-2-relevant gene perturbations. The ranked list of drugs was then subjected to drug enrichment analysis targeting two aspects of SARS-CoV-2 treatment: the reversal of the inflammatory SARS-CoV-2 phenotype and the maintenance of normal ACE2 expression. The output of these aforementioned analyses yielded a panoply of repurposing candidates that conformed to either one of the mentioned aspects of treatment or both. These data achieve two major goals depending on the drug in question. These data provide novel therapeutic targets for SARS-CoV-2 treatment. Investigators are then encouraged to seek further validation, biological or otherwise, en route to clinical consideration. For output drugs that are currently being trialed or under trial consideration such as colchicine (NCT04375202), chlorpromazine (ReCoVery trial; NCT04366739), corticosteroids (NCT04381936), prazosin (PREVENT trial; NCT04365257), lenalidomide (GETAFE trial; NCT04361643), thalidomide (NCT04273581), ibrutinib (NCT04375397), and sildenafil (NCT04304313) among others, our results provide an added layer of validation to imbue the concerned investigator with added confidence moving forward. As the drug class with the highest enrichment in our analysis, currently trialed JAK2 inhibitors such as ruxolitinib (NCT04348071) and baricitinib (BARI-COVID trial; NCT04320277) present promising therapeutic repurposing targets for SARS-CoV-2 treatment. Blockade can target IL-6 signaling via JAK-STAT, a process that was enriched in our hallmark analysis and a powerful driver of immune dysregulation in SARS-CoV-2 vis-à-vis its lymphopenic effect (Chu et al., 2020; Giamarellos-Bourboulis et al., 2020) as well as the pro-inflammatory angiotensin 2-AT1R signaling via JAK-STAT.
Other enriched targets interfere with viral entry. During SARS-CoV and SARS-CoV-2 entry, cell surface-associated transmembrane protease serine 2 (TMPRSS2) cleaves the S protein to permit viral and cellular membrane fusion (Glowacka et al., 2011). Nafamostat (RACONA trial; NCT04352400) and to a lesser extent, camostat (NCT04353284), both enriched in our analysis, have been shown to prevent SARS-CoV-2 entry by inhibiting TMPRSS2 (Glowacka et al., 2011; Hoffmann et al., 2020a, 2020b). Clathrin-mediated endocytosis then ensues with regulation afforded by numb-associated kinases (NAK), such as AP2-associated protein kinase 1 (AAK1) and cyclin G-associated kinase (GAK) (Sorrell et al., 2016). In addition to the JAK-STAT blockade discussed above, the JAK inhibitor baricitinib might also disrupt viral entry via AAK1 and GAK inhibition (Richardson et al., 2020). Interestingly, chloroquine and hydroxychloroquine were not significantly enriched in our drug analysis. This pertinent negative finding is in line with the failure of hydroxychloroquine to demonstrate clinical efficacy. In effect, this finding lends credence to our data-driven analysis as it outperformed extrapolated biological validation of chloroquine and hydroxychloroquine, albeit on non-SARS-CoV-2 cell models.
Finally, and given the limited set of genes profiled in LINCS L1000, we investigated whether the observed drug mechanism of action is associated with molecular events triggered by SARS-CoV-2 and not a ubiquitous modulation of pathways. Notably, the enriched drug candidates downregulated proinflammatory processes such as complement activation, JAK-STAT pathways, and lung fibrosis. Conversely, metabolic processes such as transcription, glucose metabolism, and cell cycle processes were upregulated. The observed resumption of normal function attests to SARS-CoV-2's disruptive influence on cellular resources during infection. This is demonstrated by the nsp1 protein expressed by SARS-CoV during infection. SARS-CoV nsp1 achieves translational inhibition either via direct binding to the 40S subunit and cleavage of cellular mRNA or via inhibition of the 48S initiation complex formation and its conversion to the 80S initiation (Kamitani et al., 2006, 2009; Lokugamage et al., 2012). This results in the shutoff mechanism whereby viral RNA is preferentially transcribed and translated over host mRNA. Moreover, the massive metabolic suppression observed concur with recent findings from proteomic and metabolomic characterization from a severe case of COVID-19 (Shen et al., 2020).
In conclusion, through a rigorous data-driven computational analysis, this study validates aspects of a complex pathogenetic framework for SARS-CoV-2 infection; however, targeted biological validation is still a requisite for a comprehensive understanding of the pathogenesis. A significant number of therapeutic repurposing candidates, many of which are FDA-approved, safe, and cheap, are presented to the scientific community for further validation and clinical consideration. The different mechanisms of action of the various validated candidates serve to emphasize the myriad of pathways involved in the pathogenesis and the implicit complexity of the virus. With that in mind and as time is of the essence, the notion of combining drug candidates with different mechanisms of actions might provide a synergistic effect while simultaneously decreasing the risk of associated adverse drug reactions and expediting the process of testing each therapeutic candidate based on the severity of COVID-19 symptoms.
Limitations of the Study
The study does not provide experimental validation.
Resource Availability
Lead Contact
Nehme El-Hachem.
Materials Availability
All datasets reported in this study are publicly available.
Data and Code Availability
Processed data will be available upon request. A shiny app has been made available for public use.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors would like to thank all researchers who shared their data publicly and made this project possible. This work is partially supported by a Discovery Grant from the National Sciences and Engineering Research Council of Canada (RGPIN/06101-2014) and an operating grant from the Cancer Research Society (OG24054). M.R. holds a Fonds de la Recherche en Santé du Québec Junior II Award.
All authors declare no conflict of interest.
Author Contributions
N.E.-H. conceived the study, performed all bioinformatic analyses, and wrote the manuscript with the significant input of E.E, G.N., B.H.-K., M.K., and M.R. G.D., O.A., S.Z., G.M., J.-P.B., and R.S. provided clinical insights, immunology expertise, and contributed to the critical review of the manuscript.
Declaration of Interests
R.S. is founder of IntelliStem Technologies Inc., Toronto, ON, Canada. The other authors have no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101697.
Contributor Information
Nehme El-Hachem, Email: nehme.el-hachem@umontreal.ca.
Mazen Kurban, Email: mk104@aub.edu.lb.
Moutih Rafei, Email: moutih.rafei.1@umontreal.ca.
Supplemental Information
References
- Bessière F., Roccia H., Delinière A., Charrière R., Chevalier P., Argaud L., Cour M. Assessment of QT intervals in a case series of patients with coronavirus disease 2019 (COVID-19) infection treated with hydroxychloroquine alone or in combination with azithromycin in an intensive care unit. JAMA Cardiol. 2020;5:1067. doi: 10.1001/jamacardio.2020.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Q., Wu Y., Mei S., Ye C., Zou X., Zhang Z., Liu X., Wei L., Truelove S.A., Zhang T Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: a retrospective cohort study. Lancet Infect. Dis. 2020;20:911–919. doi: 10.1016/S1473-3099(20)30287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fett C., Mack M., Ten Eyck P.P., Meyerholz D.K., Perlman S. Sex-based differences in susceptibility to severe acute respiratory syndrome coronavirus infection. J. Immunol. 2017;198:4046–4053. doi: 10.4049/jimmunol.1601896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H., Chan J.F.-W., Wang Y., Yuen T.T.-T., Chai Y., Hou Y., Shuai H., Yang D., Hu B., Huang X. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020;71:1400. doi: 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronavirus Disease . 2020. COVID-19 May22.https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200522-covid-19-sitrep-123.pdf?sfvrsn=5ad1bc3_4 [Google Scholar]
- DeDiego M.L., Nieto-Torres J.L., Regla-Nava J.A., Jimenez-Guardeño J.M., Fernandez-Delgado R., Fett C., Castaño-Rodriguez C., Perlman S., Enjuanes L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014;88:913–924. doi: 10.1128/JVI.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte, R.R.R., Copertino Jr. D.C., Iñiguez L.P., Marston J.L., Nixon D.F., Powell T.R. (no date) ‘Repurposing FDA-approved drugs for COVID-19 using a data-driven approach. doi: 10.26434/chemrxiv.12148764. [DOI] [PMC free article] [PubMed]
- Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;56:105949. doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Geleris J., Sun Y., Platt J., Zucker J., Baldwin M., Hripcsak G., Labella A., Manson D.K., Kubin C., Barr R.G. Observational study of hydroxychloroquine in hospitalized patients with covid-19. N. Engl. J. Med. 2020;382:2411. doi: 10.1056/NEJMoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheblawi M., Wang K., Viveiros A., Nguyen Q., Zhong J.C., Turner A.J., Raizada M.K., Grant M.B., Oudit G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020;126:1456–1474. doi: 10.1161/CIRCRESAHA.120.317015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.-E., Katsaounou P. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Müller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.-J., Liang W.H., Zhao Y., Liang H.R., Chen Z.S., Li Y.M., Liu X.Q., Chen R.C., Tang C.L., Wang T. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur. Respir. J. 2020;55:2000547. doi: 10.1183/13993003.00547-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007;170:1136–1147. doi: 10.2353/ajpath.2007.061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga S., Yamamoto N., Nakai-Murakami C., Osawa Y., Tokunaga K., Sata T., Yamamoto N., Sasazuki T., Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. U S A. 2008;105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020;38:379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Schroeder S., Kleine-Weber H., Müller M.A., Drosten C., Pöhlmann S. Nafamostat mesylate blocks activation of SARS-CoV-2: new treatment option for COVID-19. Antimicrob. Agents Chemother. 2020;64:e00754-20. doi: 10.1128/AAC.00754-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. U S A. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa N., Nishigaki K., Hayashi T., Ishii Y., Furukawa S., Niiro A., Yasui F., Kohara M., Morita K., Matsushima K. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett. 2006;580:6807–6812. doi: 10.1016/j.febslet.2006.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martínez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Bai T., Yang L., Hou X. (no date) ‘Discovery of potential drugs for COVID-19 based on the connectivity map. doi: 10.21203/rs.2.24684/v1.
- Lokugamage K.G., Narayanan K., Huang C., Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012;86:13598–13608. doi: 10.1128/JVI.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menter T., Haslbauer J.D., Nienhold R., Savic S., Hopfer H., Deigendesch N., Frank S., Turek D., Willi N., Pargger H. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77:198. doi: 10.1111/his.14134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D.L., Al Hosani F., Keating M.K., Gerber S.I., Jones T.L., Metcalfe M.G., Tong S., Tao Y., Alami N.N., Haynes L.M. Clinicopathologic, immunohistochemical, and ultrastructural findings of a fatal case of Middle East respiratory syndrome coronavirus infection in the United Arab Emirates, April 2014. Am. J. Pathol. 2016;186:652–658. doi: 10.1016/j.ajpath.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson P., Griffin I., Tucker C., Smith D., Oechsle O., Phelan A., Rawling M., Savory E., Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–e31. doi: 10.1016/S0140-6736(20)30304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P.C. Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol. 2020 doi: 10.1016/S2665-9913(20)30309-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E.S., Dufort E.M., Udo T., Wilberschied L.A., Kumar J., Tesoriero J., Weinberg P., Kirkwood J., Muse A., DeHovitz J. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA. 2020;323:2493. doi: 10.1001/jama.2020.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Peñaranda S., Bankamp B., Maher K., Chen M.H. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., Yuen K.S., Castano-Rodriguez C., Ye Z.W., Yeung M.L., Fung S.Y., Yuan S., Chan C.P., Yuen K.Y., Enjuanes L., Jin D.Y. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell F.J., Szklarz M., Abdul Azeez K.R., Elkins J.M., Knapp S. Family-wide structural analysis of human numb-associated protein kinases. Structure. 2016;24:401–411. doi: 10.1016/j.str.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzig K.E., Canepa-Escaro F., Schiliro M., Berdnikovs S., Prakash Y.S., Chiarella S.E. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020;318:L1280. doi: 10.1152/ajplung.00153.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171 doi: 10.1016/j.cell.2017.10.049. 1437.e17–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Cao Z., Han M., Wang Z., Chen J., Sun W., Wu Y., Xiao W., Liu S., Chen E. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ. 2020;369:m1849. doi: 10.1136/bmj.m1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Zhou Y., Yang Z., Xia D., Geng S. (no date) ‘Clinical characteristics of patients with severe pneumonia caused by the 2019 novel coronavirus in Wuhan, China. doi: 10.1101/2020.03.02.20029306. [DOI] [PMC free article] [PubMed]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Zhang L., Geng H., Deng Y., Huang B., Guo Y., Zhao Z., Tan W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q., Wang B., Mao J. ‘The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19’. J. Infect. 2020;80:607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wu K., Wang D., Yue X., Song D., Zhu Y., Wu J. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-kappaB. Virology. 2007;365:324–335. doi: 10.1016/j.virol.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data will be available upon request. A shiny app has been made available for public use.