

Abstract
Daratumumab (Dara), a multiple myeloma (MM) therapy, is an antibody against the surface receptor CD38, which is expressed not only on plasma cells but also on NK cells and monocytes. Correlative data have highlighted the immune-modulatory role of Dara, despite the paradoxical observation that Dara regimens decrease the frequency of total NK cells. Here we show that, despite this reduction, NK cells play a pivotal role in Dara anti-MM activity. CD38 on NK cells is essential for Dara-induced immune modulation, and its expression is restricted to NK cells with effector function. We also show that Dara induces rapid CD38 protein degradation associated with NK cell activation, leaving an activated CD38-negative NK cell population. CD38+ NK cell targeting by Dara also promotes monocyte activation, inducing an increase in T cell costimulatory molecules (CD86/80) and enhancing anti-MM phagocytosis activity ex-vivo and in vivo. In support of Dara’s immunomodulating role, we show that MM patients that discontinued Dara therapy because of progression maintain targetable unmutated surface CD38 expression on their MM cells, but retain effector cells with impaired cellular immune function. In summary, we report that CD38+ NK cells may be an unexplored therapeutic target for priming the immune system of MM patients.
Introduction
Daratumumab (Dara) is a humanized IgG1 (ĸ subclass) antibody against the highly expressed plasma cell (PC) receptor CD38.1–5 It has been approved by the Food and Drug Administration for the treatment of relapsed and newly diagnosed multiple myeloma (MM).1, 3–8 The main anti-MM effect of Dara has thus far been attributed to its ability to target the MM cells by inducing immune activation cell killing,9 but unfortunately, despite its significant efficacy, relapse or resistance remains an issue. In support of its function as an ectoenzyme, we and others recently reported that a fraction of the entire CD38 molecule is actively internalized when cancer cells, including MM cells, are treated with CD38-specific antibodies.10, 11 The correlation between CD38 surface levels on the MM cells and response to Dara treatment remains controversial.12, 13 Whereas some researchers reported a significant downregulation of CD38 expression on the surface of the MM-PCs in patients progressing under Dara treatment,12 others have instead shown that detection of CD38 on these cells was hindered by competitive binding of Dara, which resulted in a “false” MM CD38-negative population.14 A restoration of CD38 expression on the cancer cells six months after Dara discontinuation has been also described.12 Despite the importance of CD38 expression on the myeloma cells, correlative studies have highlighted that MM patients who participated in Dara monotherapy trials (SIRIUS and GEN501) show significant lower levels of total NK cells but an increase in a CD38(−), activated NK cell population (CD69+), associated with an increase in CD8+ T-cell activation after two months of treatment.15 Although a recent published study has highlighted the possible effect of Dara in killing CD38+ NK cells with subsequent expansion of a more active CD38(−) NK cell population,16 the expansion of this population has not been observed in patients, and CD38 signaling has mainly been implicated in NK cell and Th1 activation.17–20 By using patient samples in conjunction with in vivo and ex vivo testing, we report that Dara binding to CD38 in NK cells induces its internalization and concomitant activation of a CD38+ NK cell population, a step we believe is essential in inducing immune activation against cancer cells. We also report that patients resistant to a Dara-containing treatment regimen retain CD38 surface expression on their myeloma cells but with impaired Dara-induced effector function.
Materials and Methods
Results
Dara-induced MM cell killing through CD38+/CD16+NK cells
Confirming previously published data with single-agent Dara,15 our data show that relapsed patients responding to Dara-containing combinations display a significantly lower total NK cell frequency in their peripheral blood compared to Dara-untreated patients (RRMM) (Fig.1A). Despite the reduction of the frequency of this population, these cells could still play a role in Dara anti-MM activity. The effect of NK cells was tested in NSG mice engrafted with CD38+ MM cells (MM.1S Gfp/Luc+). Two weeks after MM cell injection, mice with comparative tumor burden were randomly separated into four different groups (n=6 mice for group) and co-injected with the following: 1x106 healthy donor-derived peripheral blood mononuclear cells (PBMCs) plus Dara (group 1) or a non-MM specific humanized IgG1 (κ subclass) control antibody trastuzumab (Trast, group 2); or PBMCs depleted of the NK population [PBMC NK(−)] plus Dara (group 3) or trastuzumab (group 4) (Fig.1B). Each treatment was repeated once a week for three weeks. Mice treated with PBMCs+Dara have a significantly longer survival compared to that in mice treated with PBMCs+Trast (p=0.001). Mice treated with PBMC NK(−) + Dara had significantly shorter survival compared to the mice injected with PBMCs+Dara (p=0.015) (Fig.1C). We then investigated whether CD38 surface expression on immune effectors was essential for Dara-induced cell killing. PBMCs (effectors, E) were pretreated with Dara (ED), washed, and incubated with the target (T) cells (ED:T). Our data show that only CD38+ effector cells induced MM killing upon Dara treatment (Fig.1D, Sup Fig.1A) when pretreated with Dara (ED:T), whereas the PB CD38-negative fraction [PB-CD38(−)], were unable to induce MM killing under the same conditions (Fig.1D, Sup Fig.1A). To avoid the possibility that the PB-CD38(−) fraction was inducing less MM killing because the presence of a reduced number of NK cells19, we purified this population. Consistent with the importance of CD38 expression on the surface of immune effector cells, the same effect was also observed in purified total NK cells at different effector-target ratios (Fig.1E) (Sup.Fig.1 B,C). Conversely, CD38(−) NK cells obtained from the same donor did not induce killing in the same experimental conditions (Fig.1E). Since CD16 expression is critical for NK cell activation against cancer cells by antibody dependent cellular cytotoxicity (ADCC), we investigated whether surface CD16 levels may differ in CD38+ and CD38(−) NK cell populations. Our data show that CD38(−) NK cells have almost 80% less surface CD16 surface expression (MFI 16) compared to that on CD38+NK cells (MFI 70) (Fig.1F). Mass cytometry analysis also confirmed that in both healthy and patient donors, CD38 distribution in CD56+NK cells is mainly restricted to the CD16+ population (Sup. Fig.1D). We then assessed whether Dara could directly engage CD16 on the surface of NK cells even in absence of CD38+ target cells. We observed surface CD16 down-modulation when NK cells were treated overnight with Dara (Fig.1G), and the same result was observed in cells obtained from patients actively under Dara treatment (Fig.1H) (p=0.008). Interestingly, when the Dara-treated NK cells were washed twice to eliminate the excess of antibody and incubated with the target (CD38+ MM cells, ED:T), an antibody binding the same epitope, CD38-Mono-PE, could not detect CD38 on the surface of MM cells, in contrast to detection following control human IgG (C-IgG) but similarly to when MM cells were directly exposed to Dara (ED:TD) (Sup. Fig.1E), clearly showing that when the Fc of Dara is first bound to CD16, the Fab of Dara can efficiently recognize CD38 on the surface of MM cells. Blocking the FcR only in the effector cells (E-FcBlock-D:T) almost completely reverted Dara induced MM cell killing (Sup. Fig.1F).
Fig. 1. CD38+ NK cells are essential for Dara-induced killing of MM cells.
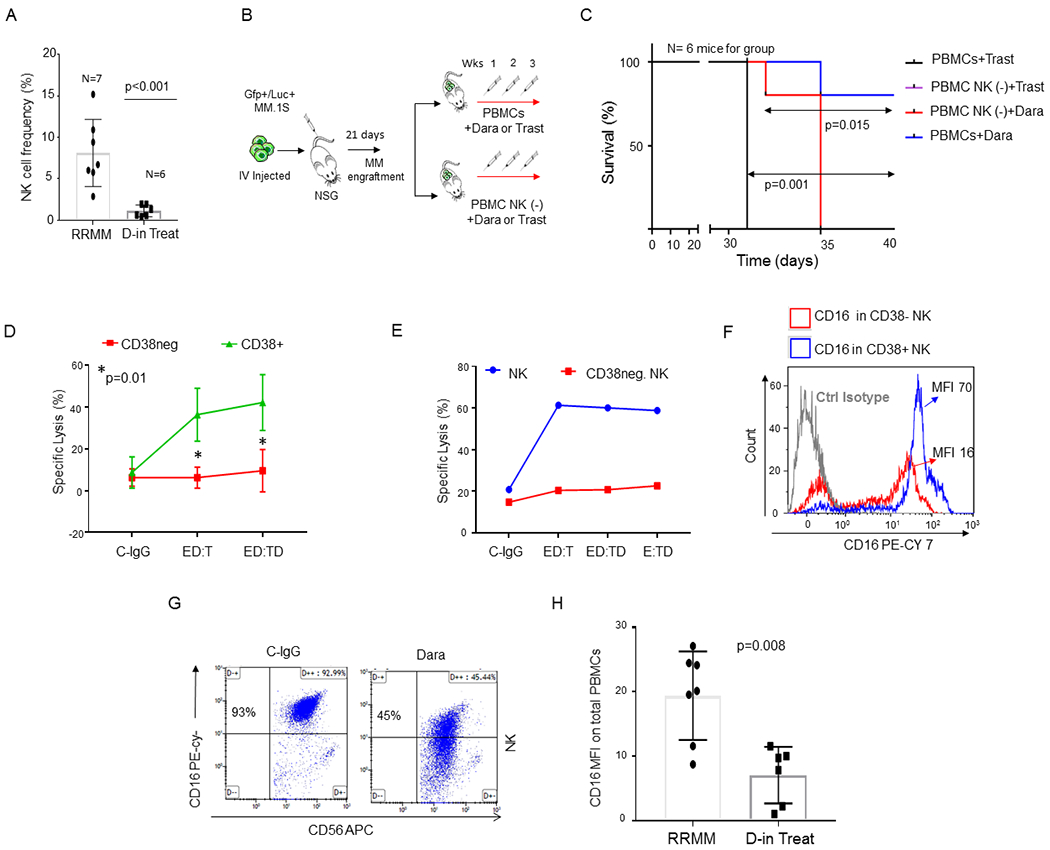
A) Bar graphs showing NK cell reduction in the PBMCs isolated from refractory MM patients (RRMM) and refractory MM patients actively on Dara treatment (D-in Treat) (p<0.001). Mann-Whitney-Wilcoxon test was performed; B) Schematic illustration of the animal treatment schedule. 5x106 MM.1S GFP+/Luc+ cells were injected intravenously (IV) into NSG mice. On day 13, after engraftment reached approximately ≥2x106 photons/sec/cm2/sr, mice were randomly distributed into 4 experimental groups and injected three times once a week by intravenous injection with 1x106 PBMCs or PBMCs depleted of total NK cells [PBMCs NK(−)] pretreated for 30 min with 125 μg Dara or trastuzumab (Trast), and co-injected; C) Survival curves of the 4 different mice groups described in B. Mice treated with PBMCs+Trast and treated with NK cell–depleted PBMCs [PBMC NK (−)+Trast] simultaneously reached IACUC guidelines for euthanasia and are reported with overlapping lines (black/purple); D) Line graph reporting the percentage of dead cells as 7-AAD-positive among target cells (MM.1S GFP+ cells) of at least n=3 HDs repeated in triplicate; E) Killing assay in terms of specific lysis (%) by total primary NK cells or CD38(−) NK cells (E) and incubated overnight with C-IgG and Dara, washed, and co-cultured with Gfp+ MM.1S cells (T) for 4hrs. The same HD donor was used for the comparison analysis; F) Overlay histogram showing that CD16 levels are almost 80% less on the surface of CD38(−) NK cells compared to that in the CD38+ NK cell population in the blood of the same HD. Two HDs were analyzed by flow analysis. CyTOF analysis was also performed in 1 independent HD and 1 MM patient (see Sup. Fig. 1D); G) Representative flow cytometry of CD16 surface expression of NK cells after an overnight treatment with control IgG (C-IgG) or Dara; H) Bar graph showing CD16 levels in non-refractory MM patients actively on Dara treatment (D-in Treat) compared to that in untreated RRMM patients (p=0.008).
Dara induces CD38+ NK cell activation
Since CD38+ effector cells appear crucial for the engagement of the Dara-induced anti-MM immune response but previously published data have shown that Dara-treated patients are left with an activated CD69+CD38(−) NK cell population,15 we decided to explore this apparent discordancy ex vivo. We observed CD69 to be significantly upregulated in NK cells when PBMCs were treated overnight with Dara (Sup.Fig.2A). CD69 upregulation in NK cells was also observed in the CD38+ PBMC fraction, but not in the PBMC CD38(−) population (Sup.Fig.2A). To assess whether Dara could have a direct activating effect on NK cells, we performed the same experiment using CD38+ and CD38(−) NK cell populations (Sup.Fig.2B). We observed a significant increase in CD69 expression in the CD38+ and in the NK cell population after overnight treatment with Dara, which was not observed in the CD38(−) fraction (p<0.001) (Fig.2A,B). Because previous data have shown that CD38 and CD16 are functionally dependent to induce mechanisms of NK cell activation21 and that Dara could induce fratricide of NK cells by its concomitant binding (bridging) to the Fc receptor and their surface CD38,16 we investigated whether targeting CD38 using only the Dara Fab or Fc fragments (Sup.Fig.2C) would still induce NK cell activation. One hour of incubation with the Dara Fab fragment cannot induce NK cell activation (CD69+ cells: 51% from Dara versus 15% from Fab) and degranulation (CD107a+ cells: 24% in Dara versus 0.66% from Fab) (Fig.2C). However, the addition of the purified Dara Fc fragments 30 min after the full saturation of CD38 on NK cells by Fab (Fig. 2D) completely restored the activation observed using the intact antibody (CD69+: Dara 51% versus Fab+Fc 54%; CD107a+: Dara 24% versus Fab+Fc 17%), excluding a possible mechanism of ADCC between NK cells in which both receptors were occupied but not physically connected (Fig.2D). NK cell activation was not observed when trastuzumab was used in the same experimental condition, but elevated CD69 and CD107a was again observed when Dara was added (Fig.2D). Western blot analysis revealed that up to 48hrs of treatment with Dara Fab did not induce CD38 down-modulation, but the addition of an intact Dara Fc again decreased CD38 levels comparable to those in Dara-treated NK cells (Fig.2E). Interestingly, we did not observe CD38 mRNA down modulation in NK cells upon incubation with Dara, neither at 24 or 48 hours of treatment (Sup. Fig. 2D), supporting that ex vivo Dara treatment does not select a CD38(−) NK cell population, which in patients retains lower CD38 mRNA levels compared to that in CD38+ NK cells (Sup. Fig.2E). Since previous data have shown that modulation of CD38 signaling pathways induces increases in IFN-γ and GM-CSF levels in NK cells,21 we investigated whether Dara could also modulate the release of these cytokines. We found an increase in IFN-γ and GM-CSF production in NK cells treated at different time points (1-16 hrs) at different Dara concentrations (1–100 μg/ml) (Fig. 2F,G). As expected, IFN-γ and GM-CSF increases were associated with upregulation of the NK cell degranulation marker (CD107a) after 1 hr. of Dara treatment (Sup.Fig.2F, p=0.02).
Fig. 2. Dara induces direct CD38+ NK cell activation.
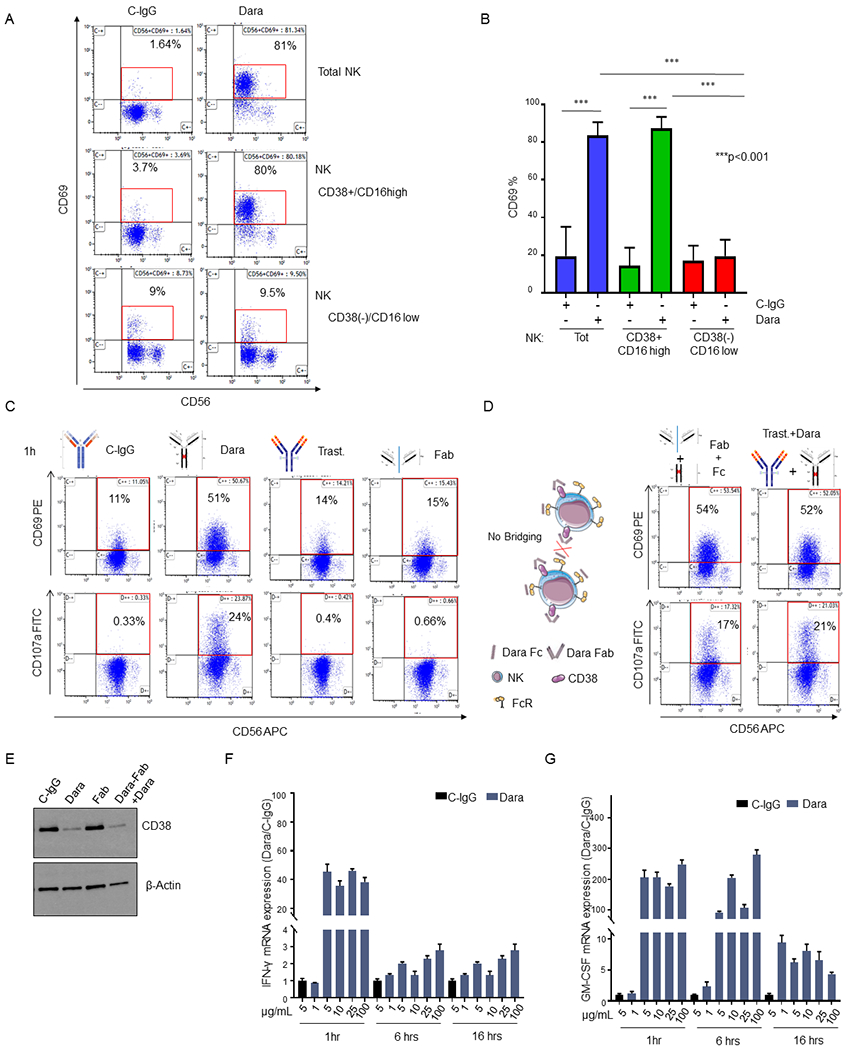
A) Representative flow analysis of CD69 on total NK, CD38+NK and CD38(−) NK cell fractions isolated from a healthy donor upon overnight Dara treatment; B) Bar graph representing the median ±SD of CD69 expression in total, CD38(−) and CD38+ NK cells treated with Dara or C-IgG (p<0.001). The experiment was repeated using three healthy donors in triplicate; C,D) Flow cytometry analysis of CD69 (top panels) and CD107a (bottom panels) in total NK cells treated for 1hr with C-IgG (C), Dara (C), Trast (C), Dara Fab (Fab) (C), Dara Fab + Dara Fc (Fab+Fc) (D), or Trast+Dara (D) at 10 μg/mL for 1 hr using the same HD (n=1) for each treatment and internal controls; D) Graphical Illustration (left panel) showing the fully saturated CD38 on NK cells after the addition of the purified Dara Fc fragment for 30 min, which followed the addition of Dara Fab, excluding a possible bridging between NK cells; E) Western Blot analysis showing CD38 protein levels in NK cells treated with either C-IgG (10 μg/mL), Dara (10 μg/mL), Dara-Fab or Dara-Fab+Dara for 48 hrs. The experiment was run in duplicate; F, G) IFN-γ and GM-CSF mRNA expression levels in NK cells under Dara treatment at different concentrations (from 1 μg/ml to 100 μg/ml) and different time points (1-16 hrs) compared to C-IgG; GADPH mRNA was used for normalization.
Dara induces CD38 protein degradation in NK cells
Flow cytometry analysis using anti-CD38-Multi-FITC, a non-competitive antibody, shows that CD38 on the surface of NK cells is strongly downregulated upon Dara treatment, from 70% to 40% after 1hr and to 6% after overnight treatment, an effect not observed with C-IgG (Fig.3A). Despite this surface downregulation, the total CD38 protein content upon 1 hour of incubation was unaffected (Fig.3B). Regardless of differences in CD38 levels, a decrease in CD38 protein was observed as soon as 6hrs in all donors tested (n=13; median age 56), in contrast to CD38 levels in C-IgG–treated NK cells (p=0.008) (Fig.3B,C). We then investigated whether Dara binding on the CD38+ NK cells could cause Dara/CD38 complex internalization and subsequent degradation. We used our recently published Dara conjugated to Alexa-Fluor 647 (DARA-AF-647).22 We first blocked DARA-AF-647 active NK cell internalization using ice and assessed maximum surface signal after 120 min of incubation; minimum surface signal in the same cells was alternatively assessed after surface acid wash (a.w.). NK cells were then incubated with DARA-AF-647 at 37°C at different time points (15–120 minutes), and the internalized fluorescence left after acid wash was compared to the minimum and maximum surface signals (Sup. Fig. 3A,B). We observed a rapid DARA-AF-647 internalization in CD38+ NK cells, a signal that reached a maximum internalization level at 60 min and remained elevated at 120 min after incubation. This result is aligned with the stability in total CD38 protein levels in the first few hours of treatment (Fig.3B). Our data show that Dara treatment induces an increase in intracellular Ca2+ mobilization in NK cells, with a 40% increase in magnitude after 30 seconds of Dara addition (Sup. Fig. 3C). Since we previously published that CD38 can be internalized though the auto-phagosome apparatus, we assessed whether CD38 protein down-modulation in NK cells could be linked to lysosomal degradation. Dara treatment induces a decrease of the phagosome marker microtubule-associated protein light chain 3 (LC3), and increase in GLUT4, supporting an increase in glucose uptake induced by autophagy and lysosomal degradation (Fig.3D). We also observed that primary NK cells treated with the autophagy inducer rapamycin (10, 50 nM) for 6 hours in the presence of either Dara or C-IgG show CD38 protein downregulation, an effect that was further potentiated by the addition of Dara (Fig.3E). We then investigated whether CD38 protein degradation after Dara binding could be reversed. When NK cells were treated overnight with the autophagy inhibitor wortmannin (3, 50 nM), we observed a significant rescue of CD38 protein degradation upon Dara binding compared to the control (Fig.3F, G). Consistent with these data, CD38 surface expression was also partially rescued by the addition of wortmannin (Fig.3H).
Fig. 3. Dara induces CD38 protein reduction in NK cells and Ca2+ mobilization.
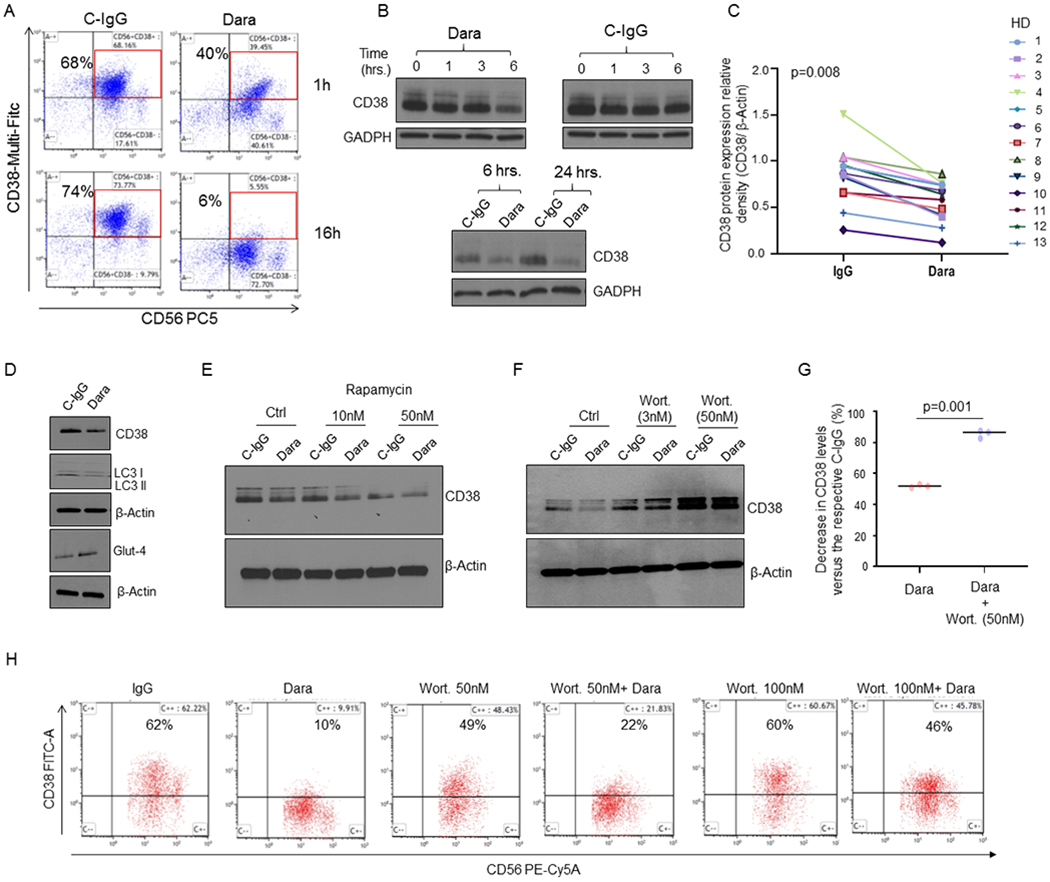
A) CD38 surface expression on NK cells at 1hr and 16hrs as detected by flow cytometry, showing down-regulation upon Dara treatment at 1hr and 16hrs compared to C-IgG; B) Western Blot analysis of CD38 protein level expression in NK cells treated over time with Dara (10 μg/mL) or IgG (10 μg/mL). C) Thirteen tumor-free donors (HD) were analyzed, and CD38 levels were assessed by densitometry analysis using β-actin as internal housekeeping. The Shapiro-Wilk test suggests that the samples are generated from a Gaussian distribution (α=0.05). Welch t-test statistic = 2.5963 with p=0.008; D) Western Blot analysis showing CD38 levels in primary NK cells treated for 6 hrs with Dara (10 μg/ml) or C-IgG, showing LC3II protein downregulation and GLUT4 upregulation; The experiment was performed in at least n=3 independent donors; E) Western Blot analysis showing CD38 levels in primary NK cells treated for 16 hours with rapamycin (10-50nM) as indicated in the presence of either C-IgG or Dara; F,G) Western Blot analysis showing CD38 levels in primary NK cells treated for 6 hours with wortmannin (Wort.) as indicated in the presence of either C-IgG or Dara. The experiment was repeated in biological triplicate, and CD38 relative density levels (CD38/β-actin) were normalized to C-IgG–treated NK cells for each HD donor, the differences reported (G); H) Flow cytometry analysis showing that Wort. treatment can partially rescue CD38 surface expression on CD56+CD3(−) NK cells after 6 hrs of Dara treatment.
Dara-treated CD38+ NK cells induce monocyte activation
Because IFN-γ and GM-CSF release are essential not only for NK cells to directly induce target cell cytolysis but also to indirectly activate the innate immune response, leading to monocyte activation and polarization towards the tumor site (Fig.4A),23 we investigated whether patients responding to Dara had an effective increase in monocytic-derived macrophages in their bone marrow. We collected BM aspirates from 8 relapsed MM patients who became resistant to Dara-based treatment prior to starting a subsequent therapy with a minimum of 1% CD138+ MM-PCs among total BM cells (Dara-resistant). We compared them (Dara-resistant, n=8) with 8 Dara-naive and 2 Dara-exposed (>1 year prior to the analysis) extensively pretreated patients (RRMM n=10) that were progressing on a Dara-free regimen. We also compared with Dara responding (n=5) patients, including 1 patient with a partial response, 2 with a very good partial response, and 2 with a complete response. Our data showed a significant increase in the frequency of the BM CD14+ population in Dara-responding patients compared with either RRMM or Dara-resistant (p<0.001) (Fig.4B). To assess the role of NK cells here, we tested whether Dara in an ex-vivo model could induce monocyte expansion and polarization. We observed upregulation of costimulatory T cell surface antigens (CD80/CD86) on the surface of the monocytes (CD14+) in PBMCs treated with Dara for 24 hrs (Fig.4C). We then investigated whether the effect of Dara on NK cells may be responsible for monocyte activation (CD80/CD86). CD14+ cells co-cultured with Dara-treated NK cells significantly upregulated CD80/86 (Fig.4D–E) (median average 36%), an effect not observed with C-IgG–treated NK cells (median average 19%, p<0.01, using three different healthy donors). CD80/86 upregulation was not detected in the absence of CD38+NK cells when CD14+ cells were directly exposed to Dara or C-IgG (Fig.4F). However, an increase in the CD80/86-positive population was observed with the addition of 50 ng/mL human recombinant (hr-) IFN-γ for 24 hrs (Fig.4F). Phagocytosis assays showed that CD14+ cells incubated with the conditioned media of Dara-treated NK cells for 72hrs display an almost 4-fold increase in CD38+ MM cell death over that from C-IgG–treated NK cells (Fig.4G). As expected, the same phagocytosis effect was observed when isolated CD14+ cells were cultured with hr-IFN-γ at the same time-point (Fig.4G).
Fig. 4. NK cell activation by Dara is essential for monocyte activation and polarization.
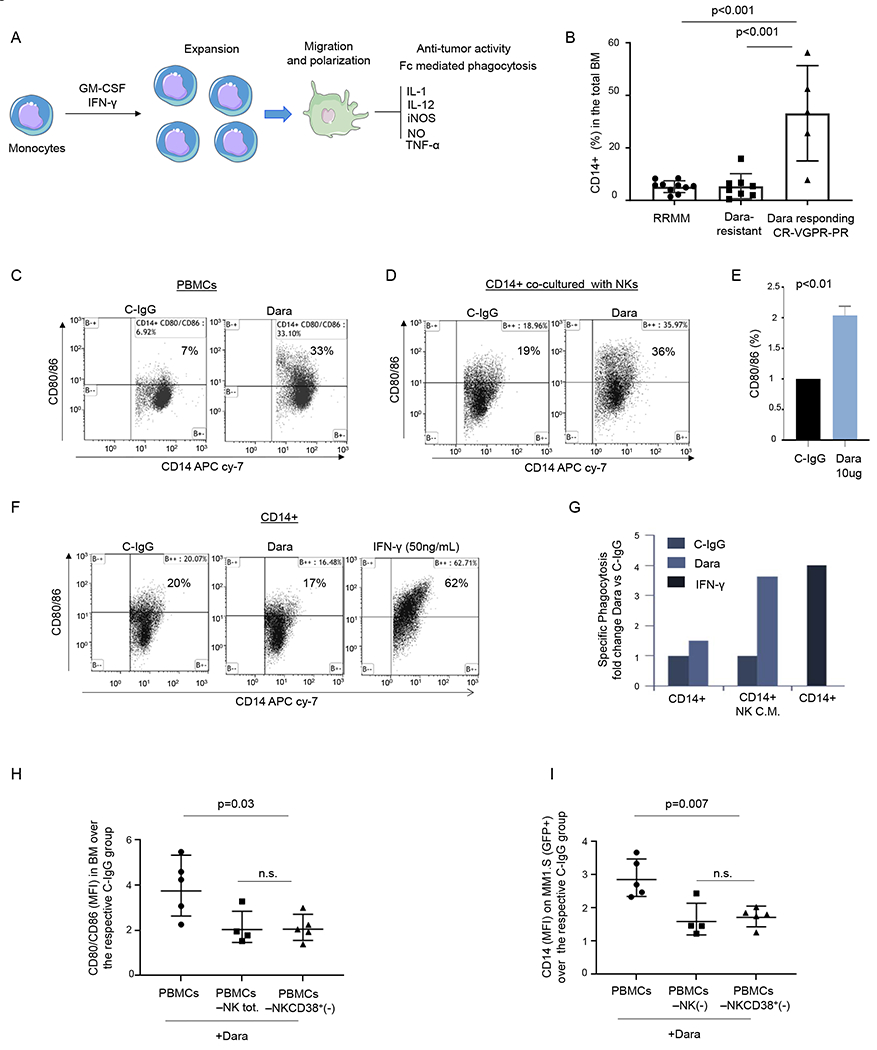
A) Illustration showing monocyte activation and polarization to M1 monocytic-derived macrophages with anti-tumor activity; B) Percent of CD14+ cells in the total BM in RRMM, Dara-resistant, and patients actively responding to Dara (Dara responding); C) Representative flow cytometric analysis of CD80/86 in PBMCs (n=2 HD) and treated overnight with Dara (10 μg/ml), showing up-regulation of CD80/86 antigens in contrast to that from C-IgG; D) Representative flow cytometric analysis of CD80/86 in monocytes isolated from a healthy donor co-cultured with CD38+NK cells (isolated from the same healthy donor) treated overnight with C-IgG or Dara (10 μg/ml); The experiment was repeated in n=3 HDs. E) Bar graph showing CD80/86 upregulation in CD14+ cells co-cultured with Dara-treated NK cells versus C-IgG–treated cells. The experiment was repeated using 3 HDs and the average ±SD is reported; p values were calculated using t test, unpaired (tails = 2); F) Flow cytometry analysis of CD80/86 in CD14+ cells treated overnight with C-IgG, Dara or IFN-γ (50 ng/ml); The experiment was repeated in n=3 HDs. G) Phagocytosis assay was performed on the CD14+ population isolated from a healthy donor (n=1), treated for 72hrs with conditioned media (CM) collected from NK cells treated overnight with C-IgG (10 μg/ml) or Dara (10 μg/ml), and compared to the CD14+ isolated population treated for 72hrs with IFN-γ (50 ng/ml) at the same timepoint; H-I) Dot plot graphs showing the CD80/86+/CD14+ population MFI) and the GFP+/CD14+ population (MFI), as assessed by flow cytometry, isolated from the BM of mice injected with total human PBMCs, PBMC NK(−), and PBMC depleted of CD38+ NK cells[PBMC NKCD38+(−)]. Data reported in H) and I) represent the mean ± SD; p values were calculated using multi comparison one-way ANOVA.
The effect of NK cells on monocyte activation upon Dara treatment was also tested in NSG mice engrafted with CD38+ MM cells (MM.1S Gfp/Luc+) (Fig.4H–I). We found a significant upregulation of the CD80/86 marker on the surface of human CD14+ (hCD14+) cells isolated from the BM of the mice treated with PBMCs+Dara compared to the BM levels found in mice injected with Dara-treated PBMCs that were depleted of total or CD38+ NK cells (Fig.4H). CD80/86 upregulation in Dara-treated PBMCs was associated with increased recruitment of hCD14+ cells in the mouse BM engrafted with MM.1S Gfp+ cells, as evidenced by an increase of the CD14+/Gfp+ double-positive population, compared to that in mice treated with PBMCs depleted of either total (PBMCs-NKtot) or CD38+ NK cells (PBMCs-NK/CD38+) (Fig.4I).
Dara resistant patients maintain CD38+ MM cells but retain effector cells with impaired immune function
Having observed that changes in CD38 surface levels in NK cells are associated with a Dara-induced immune response, we decided to investigate whether changes in CD38 expression on the MM cells could be involved in mechanisms of resistance. To analyze whether CD38 was still present on the surface of CD138+ MM plasma cells (MM-PCs) from Dara-resistant patients, we used a non-competitive anti-human CD38 antibody (CD38-Multi-FITC), which binds surface CD38 in the presence of Dara (Sup. Fig.4A,B), versus a CD38 monoclonal antibody (CD38-Mono PE) that shares with Dara the same targeted CD38 epitope (clone IB6) (Sup.Fig.4C,D), as confirmed in cell lines and primary samples (Sup.Fig.4A–D). We did not find significant differences in CD38 surface molecules (levels expressed as mean fluorescence intensity [MFI]) or in percent of BM CD38+ MM cells with respect to Dara-resistant and RRMM (Fig.5A–C). Immunohistochemistry analysis shows CD38 expression in the CD138+ MM-PCs in the BM biopsies of two independent Dara-resistant patients in whom marrow was collected before Dara treatment. A second marrow was collected for each patient, 1 patient at the time of biochemical progression, and 1 patient at the time of clinical progression (Sup. Fig.4E,F). Next, we studied whether CD38 on the surface of MM cells from Dara-resistant patients was still recognizable by Dara. Ex-vivo Dara treatment of Dara-resistant MM cells show no CD38+ signal upon staining with a Dara-competitive fluorescent antibody (-PE) (Fig.5D,F, Sup.Fig.4G). In contrast, surface CD38 was detected when the non-competitive antibody was used (Fig.5D,F and Sup.Fig.4H). We were unable to identify any missense mutations in 3 out of 5 of the MM-PCs isolated from Dara-resistant patients, which were used for ex-vivo Dara binding experiments (data not shown), and by using the MMRF IA9 released data set, we found no CD38 mutational or mRNA downregulation in the MM cells isolated from 3 patients who were analyzed before and after progressing on Dara-based therapies. Although the data from our cross-sectional study (RRMM versus Dara-resistant) show that Dara-resistant patients maintain targetable CD38 expression on their cancer cells, it could not be excluded that Dara-resistant patients progressed because of lowered CD38 surface molecules on their cancer cells. We therefore investigated whether Dara-induced MM cell killing could be instead proportional to the MM CD38 surface level expression. We generated five Gfp+/Luc+ MM cell lines having various CD38 surface expression levels (MM.1S, RPMI-8226, KMS11, JJN3 and U266) (Sup. Fig. 5A). We found no significant differences in Dara-induced MM cell killing among these cells regardless of amount of expression of surface CD38 (MM.1S [75%+], RPMI-8226 [98%], KMS-11 [11%+], JJN3 [9%+]) (Sup. Fig. 5B). Conversely, specific Dara-induced MM cell killing was not observed in a CD38 surface-negative MM cell line (U266) (Sup. Fig. 5B) and in acute myeloid leukemia cell line HL60 with deletion of CD38 (CD38 CRISPR knockout) compared to the parental cell line (Sup. Fig. 5C,D).24 We thus assumed that mechanisms of progression could not be related to the complete or even partial loss of CD38 target on MM-cells. Hence, we investigated whether lack of killing of CD38+ MM cells could instead come from changes in the microenvironment of MM patients. A flow cytometry-based killing assay showed that effector cells obtained from Dara-resistant patients (n=6) incubated with Dara were unable to induce killing of CD38+ MM cells (MM.1S), an effect that was instead observed when effector cells isolated from RRMM patients (n=4) were used (p<0.001) (Fig.5G), as was also observed in Dara-responding patients as previously published25. Mass cytometry analysis (CyTOF) of the PBMCs of Dara-resistant patients (n=3) that responded to the Dara regiment for more than 18 months (18–22 cycles) and then progressed showed as expected complete absence of the total NK cell population (data not shown) and functional impairment in the CD8+ immune reactive T cell population, as shown by a decrease in HLA-DR and CD127 expression, in contrast to Dara-responding patients (n=3) that were still in complete remission at the time of the analysis (Fig.5H) and who also displayed an immune signature that fully resembled the one observed in single agent Dara-responding patients, as recently published (Fig.5H).15 Increased expression of the immature monocytic population (CD14+), as assessed by an increase in CD33 and decrease in CD16 expression, was also observed in all Dara-resistant patients compared to that in the responding patients (Fig. 5H).
Fig. 5. Dara-resistant patients retain surface CD38 on MM cells.
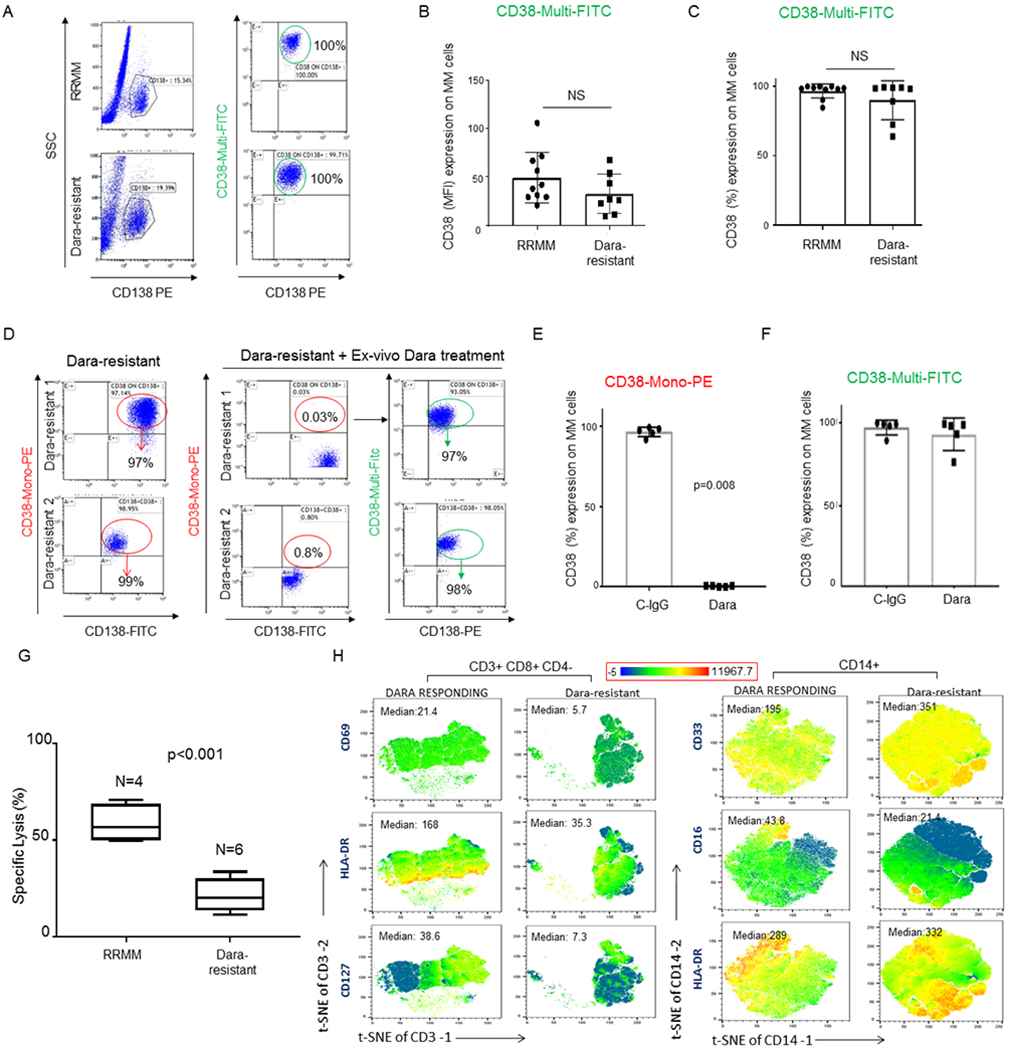
A) Representative flow analysis of CD38 expression showing no significant modulation of CD38 expression (%) on CD138+ MM-cells obtained from Dara-resistant (n=8) compared to RRMM patients (n=10); B,C) Bar graphs showing CD38 surface expression by MFI (B) and % (C) among Dara-resistant and RRMM patients. The Mann-Whitney-Wilcoxon test was performed; D) Representative flow analysis of CD38 on CD138+ MM cells of two Dara-resistant patients where BMCs were treated ex vivo with Dara for 1 hr, showing lack of CD38 in MM cells treated with Dara and stained with CD38-Mono PE, and presence of CD38 when the same cells were stained with CD38-Multi FITC; E,F) Bar graphs showing CD38 surface levels in CD138+ MM cells of 5 Dara-resistant patients after ex-vivo Dara incubation and stained with CD38-Mono PE (E) and CD38-Multi FITC antibodies (F); G) Bar graph showing killing induction (%) assessed by flow based assay in a set of RRMM patients (n=4) and Dara-resistant patients (n=6) treated with Dara ex vivo. Percentage of dead cells was calculated by gating in 7-AAD positive cells among target cells (MM1.S GFP+ cells); H) tSNE heatmap statistic plot obtained by CyTOF analysis showing CD69, HLA-DR and CD127 expression (Median) in the CD8+ cell subpopulation (gated in total CD3+ cells) of PBMCs isolated from Dara-resistant and Dara-responding patients and tSNE heatmap statistic plot obtained by CyTOF analysis showing CD33 and CD16 expression (Median) of PBMCs isolated from a representative Dara-resistant and Dara-responding patients.
Discussion
The role of NK cells in the mechanism of action of Dara has been debated; here we show that the therapeutic targeting of CD38+NK cells may play a pivotal role in initiating a Th1-mediated immune response,26, 27 which can be an essential component in mounting a powerful anti-CD38 immune response against myeloma cells as recently reported by Atanackovicet al.28 In agreement with Wang et al.,16 we demonstrate that Dara induces NK cell degranulation and IFN-γ release of only the CD16+CD38+ NK cell population. We also observed a considerable increase in CD69 expression, GM-CSF production, and Ca2+ mobilization in NK cells treated with Dara, suggesting that Dara induces substantial NK cell activation, potentially explaining the significant drop in NK cell frequency in Dara-treated patients.15 In line with the role of CD38 in immune activation, we observe that the immunosuppressant autophagosome inducer rapamycin downregulates CD38 protein expression in NK cells, whereas the autophagosome/PI3K inhibitor wortmannin significantly reverses this effect, supporting the idea that Dara is not selecting a CD38(−) NK population but instead is inducing CD38 degradation thereby activating NK cells. In agreement with previous data, which have shown a pivotal role of Dara in inducing macrophage-mediated phagocytosis;29, 30our results demonstrate that NK cell activation by Dara induces monocyte activation and polarization, increasing their anti-myeloma activity. Upregulation of CD80/CD86 on macrophages with Dara treatment via NK cell activation plays a pivotal role not only for macrophage polarization but also as the initial step of T cell activation and expansion31 through its binding to the T cell receptor CD28 as recently observed in Dara treated patients.15, 32 Although we did not perform longitudinal analyses of RRMM and Dara-resistant patients, we believe this type of cross-sectional cohort analysis reflects the immunoparesis in Dara-resistant patients and paves the way to investigate Dara immune mechanisms of acquired resistance in longitudinal clinical studies. We are currently conducting an investigator initiated trial of single-agent Dara as consolidation and maintenance therapy after autologous stem cell transplantation (NCT#03346135), with longitudinal correlative studies in to test whether changes in the immune microenvironment can interfere with response to Dara-based therapies. Our data indicate that Dara-resistant patients retain targetable unmutated CD38 in almost all myeloma cells, but these cells are surrounded by impaired effector cells. In summary, we propose that Dara induces NK cell activation and degranulation, an effect that can reduce their number but also leads to increased expression of CD80/CD86 T cell costimulatory molecules on monocytes, induces monocyte activation against MM cells and stimulates T cell expansion through CD28 binding (Fig.6). We report that CD38+NK cells prime the immune system of myeloma patients and may play an important therapeutic role for antibody-treated cancer patients in general. Our findings highlight that Dara-progressing patients display comparatively lower CD16 expression on their effector cells, a phenomenon that we observed both at the protein (Fig.5H) and RNA levels (data not shown). Although further research is required before clinical recommendations could be made, the data lead us to speculate that the use of other MM targeting antibodies that have a mechanism of ADCC mediated by surface CD16 on effector cells, including the anti-CD38 antibody isatuximab and the anti-CS1 antibody elotuzumab, would be unlikely to preserve all effectiveness when used after Dara-based therapy. To date, there have been no reports of patients salvaged by isatuximab or elotuzumab after failing Dara.33 Since anti-CD38 bispecific T cell engager antibodies, anti-CD38 antibodies with toxic payloads, and CAR-T cells against CD38 are now under study, the presence of CD38 on MM cells in Dara refractory patients and a decreased presence in the marrow microenvironment can boost the rationale for novel CD38-targeted therapeutic interventions, which may be more effective than unconjugated antibody therapies in Dara progressing patients.
Fig.6.
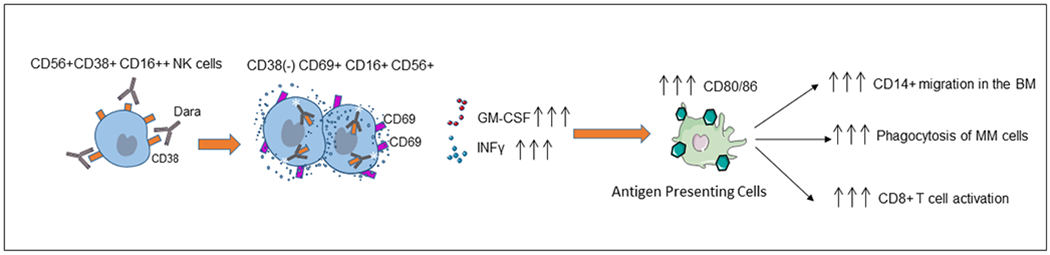
Graphical representation of the proposed mechanism of action of Dara, showing that anti-CD38 targeting induces NK cell activation and degranulation and leads to increased expression of CD80/CD86 T cell costimulatory molecules on monocytes, which induces monocyte polarization and activation and stimulates T cell expansion through CD28 binding, further associated with an immune cascade targeting CD38+ MM cells.
Supplementary Material
Acknowledgments:
We thank Dori Triplet, Evelyn Flores, Elizabeth Hartman and Debbie Flood for administrative support. We also thank Suzan King and Steve Allen for their support. Research was in part supported by the National Institute of Health under grant number NIH-2-R01-CA201382(FP, CCH) and in part under the NIH-2-R01-CA238429-01 (FP, JS, XW) and in part by the Steven Gordon and Briskin Family Innovation Grant. Research reported in this publication included work performed at the Liquid Tissue Bank, Analytical Cytometry, and Integrative Genomics and Bioinformatics shared resource cores supported by the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests: AK is a consultant for Celgene and Janssen, serves on the speakers’ bureau for SutroBioPharma, zPredicta, Celgene, Amgen, and Takeda, and has stock ownership in Celgene. CCH has received research grants from Takeda & Oncolytics Biotech; research and personal grants from Janssen, BMS, Sanofi, Nektar, and Karyopharm; and personal grants from Imbrium Pharmaceuticals and Oncopeptides, all outside the submitted work.
Footnotes
Supplementary information is available at Leukemia’s website.
References
- 1.Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. The New England journal of medicine. 2016; 375: 1319–31. [DOI] [PubMed] [Google Scholar]
- 2.Plesner T, Arkenau HT, Gimsing P, Krejcik J, Lemech C, Minnema MC, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood. 2016; 128: 1821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016; 375: 754–66. [DOI] [PubMed] [Google Scholar]
- 4.Rajan AM and Kumar S. New investigational drugs with single-agent activity in multiple myeloma. Blood cancer journal. 2016; 6: e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. The New England journal of medicine. 2015; 373: 1207–19. [DOI] [PubMed] [Google Scholar]
- 6.Plesner T, Arkenau HT, Gimsing P, Krejcik J, Lemech C, Minnema MC, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. The New England journal of medicine. 2018; 378: 518–28. [DOI] [PubMed] [Google Scholar]
- 8.Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. The New England journal of medicine. 2019; 380: 2104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phipps C, Chen Y, Gopalakrishnan S and Tan D. Daratumumab and its potential in the treatment of multiple myeloma: overview of the preclinical and clinical development. Therapeutic advances in hematology. 2015; 6: 120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghose J, Terrazas C, Viola D, Caserta E, Krishnan A, Hofmeister CC, et al. Daratumumab Impairs Myeloma Cell Adhesion Mediated Drug Resistance through CD38 Internalization. Blood. 2016; 128: abstract#4479. [Google Scholar]
- 11.Funaro A, Reinis M, Trubiani O, Santi S, Di Primio R and Malavasi F. CD38 functions are regulated through an internalization step. J Immunol. 1998; 160: 2238–47. [PubMed] [Google Scholar]
- 12.Pick M, Vainstein V, Goldschmidt N, Lavie D, Libster D, Gural A, et al. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur J Haematol. 2018; 100: 494–501. [DOI] [PubMed] [Google Scholar]
- 13.Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015; 29: 2039–49. [DOI] [PubMed] [Google Scholar]
- 14.Oberle A, Brandt A, Alawi M, Langebrake C, Janjetovic S, Wolschke C, et al. Long-term CD38 saturation by daratumumab interferes with diagnostic myeloma cell detection. Haematologica. 2017; 102: e368–e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams HC 3rd, Stevenaert F, Krejcik J, Van der Borght K, Smets T, Bald J, et al. High-Parameter Mass Cytometry Evaluation of Relapsed/Refractory Multiple Myeloma Patients Treated with Daratumumab Demonstrates Immune Modulation as a Novel Mechanism of Action. Cytometry A 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin Cancer Res. 2018; 24: 4006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lischke T, Heesch K, Schumacher V, Schneider M, Haag F, Koch-Nolte F, et al. CD38 controls the innate immune response against Listeria monocytogenes. Infect Immun. 2013; 81: 4091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rah SY, Kwak JY, Chung YJ and Kim UH. ADP-ribose/TRPM2-mediated Ca2+ signaling is essential for cytolytic degranulation and antitumor activity of natural killer cells. Sci Rep. 2015; 5: 9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallone R, Funaro A, Zubiaur M, Baj G, Ausiello CM, Tacchetti C, et al. Signaling through CD38 induces NK cell activation. Int Immunol. 2001; 13: 397–409. [DOI] [PubMed] [Google Scholar]
- 20.Postigo J, Iglesias M, Cerezo-Wallis D, Rosal-Vela A, Garcia-Rodriguez S, Zubiaur M, et al. Mice deficient in CD38 develop an attenuated form of collagen type II-induced arthritis. PLoS One. 2012; 7: e33534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. 2002; 99: 2490–8. [DOI] [PubMed] [Google Scholar]
- 22.Ghose J, Viola D, Terrazas C, Caserta E, Troadec E, Khalife J, et al. Daratumumab induces CD38 internalization and impairs myeloma cell adhesion. Oncoimmunology. 2018; 7: e1486948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R, Jaw JJ, Stutzman NC, Zou Z and Sun PD. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. 2012; 91: 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald RJ, Shrimp JH, Jiang H, Zhang L, Lin H and Yen A. Probing the requirement for CD38 in retinoic acid-induced HL-60 cell differentiation with a small molecule dimerizer and genetic knockout. Scientific reports. 2017; 7: 17406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I, et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood advances. 2017; 1: 2105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T, et al. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood. 2006; 107: 2392–9. [DOI] [PubMed] [Google Scholar]
- 27.Frasca L, Nasso M, Spensieri F, Fedele G, Palazzo R, Malavasi F, et al. IFN-gamma arms human dendritic cells to perform multiple effector functions. J Immunol. 2008; 180: 1471–81. [DOI] [PubMed] [Google Scholar]
- 28.Atanackovic D, Yousef S, Shorter C, Tantravahi SK, Steinbach M, Iglesias F, et al. In vivo vaccination effect in multiple myeloma patients treated with the monoclonal antibody isatuximab. Leukemia. 2019. [DOI] [PubMed] [Google Scholar]
- 29.Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs. 2015; 7: 311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fatehchand K, McMichael EL, Reader BF, Fang H, Santhanam R, Gautam S, et al. Interferon-gamma Promotes Antibody-mediated Fratricide of Acute Myeloid Leukemia Cells. J Biol Chem. 2016; 291: 25656–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiel M, Wolfs MJ, Bauer S, Wenning AS, Burckhart T, Schwarz EC, et al. Efficiency of T-cell costimulation by CD80 and CD86 cross-linking correlates with calcium entry. Immunology. 2010; 129: 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Usmani SZ, Khan I, Chiu C, Foureau D, Druhan LJ, Rigby K, et al. Deep sustained response to daratumumab monotherapy associated with T-cell expansion in triple refractory myeloma. Experimental hematology & oncology. 2018; 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoylman E, Brown A, Perissinotti AJ, Marini BL, Pianko M, Ye JC, et al. Optimal sequence of daratumumab and elotuzumab in relapsed and refractory multiple myeloma. Leukemia & lymphoma. 2020; 61: 691–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.