

Abstract
Background:
No treatment exists for the most common dominantly inherited ataxia Machado-Joseph disease, or spinocerebellar ataxia type 3 (SCA3). Successful evaluation of candidate therapeutics will be facilitated by validated noninvasive biomarkers of disease pathology recapitulated by animal models.
Objective:
We sought to identify shared in vivo neurochemical signatures in two mouse models of SCA3 that reflect the human disease pathology.
Methods:
Cerebellar neurochemical concentrations in homozygous YACMJD84.2 (Q84/Q84) and hemizygous CMVMJD135 (Q135) mice were measured by in vivo magnetic resonance spectroscopy at 9.4 tesla. To validate the neurochemical biomarkers, levels of neurofilament medium (NFL, indicator of neuroaxonal integrity) and myelin basic protein (MBP, indicator of myelination) were measured in cerebellar lysates from a subset of mice and patients with SCA3. Finally, NFL and MBP levels were measured in cerebellar extracts of Q84/Q84 mice upon silencing of the mutant ATXN3 gene.
Results:
Both Q84/Q84 and Q135 mice displayed lower N-acetylaspartate than wild-type littermates, indicating neuroaxonal loss/dysfunction, and lower myo-inositol and total choline, indicating disturbances in phospholipid membrane metabolism and demyelination. Cerebellar NFL and MBP levels were accordingly lower in both models, as well as in the cerebellar cortex of patients with SCA3 than controls. Importantly, N-acetylaspartate and total choline correlated with NFL and MPB, respectively, in Q135 mice. Long-term sustained RNAi-mediated reduction of ATXN3 levels increased NFL and MBP in Q84/Q84 cerebella.
Conclusions:
N-acetylaspartate, myo-inositol and total choline levels in the cerebellum are candidate biomarkers of neuroaxonal and oligodendrocyte pathology in SCA3, aspects of pathology that are reversible by RNAi therapy.
Keywords: Machado-Joseph disease, ataxin-3, polyglutamine, demyelination, magnetic resonance spectroscopy
Introduction
With increasing human lifespan, numerous age-related neurodegenerative diseases of diverse etiologies are becoming more prevalent. Nine such disorders are caused by an expanded CAG triplet repeat in genes that encode an abnormally longer polyglutamine (polyQ) tract in the respective disease proteins1, 2. Since the identification of these disease genes in the 1990s, increased understanding of the molecular basis of polyQ disorders led to several potentially efficacious therapies that were tested in animal models. Nucleotide-based reduction of CAG-containing transcripts using nucleic acids has proven to be a particularly compelling therapeutic strategy for polyQ diseases in pre-clinical studies3–7, and clinical trials are underway for Huntington disease using antisense oligonucleotides (ASOs)8. Other gene and pharmacological therapeutic approaches for polyQ disorders are also in the pipeline9–11. Readiness for these upcoming clinical trials depends on the availability of disease biomarkers that allow noninvasive monitoring of aspects of cerebral disease pathology to assess therapeutic efficacy.
Spinocerebellar ataxia type 3 (SCA3) or Machado-Joseph disease (MJD) is a polyQ disease caused by an expanded CAG repeat in the ATXN3 gene12 that leads to selective dysfunction and loss of neurons of specific nuclei in the cerebellum, brainstem, midbrain, spinal cord, and peripheral nerves9–11, 13–21. The disease manifests in patients with SCA3 as ataxia and a variable degree of other manifestations, including extrapyramidal signs, neuropathy, ophthalmoplegia, and muscle atrophy22–26.
SCA3 is one of the polyQ diseases for which clinical trials are forthcoming. ASOs27–30, other RNA interference molecules31, 32, and pharmacological agents such as the selective serotonin reuptake inhibitor, citalopram33–35, showed preclinical efficacy to mitigate molecular, pathological, and phenotypic SCA3-like signs in transgenic mice.
The success of upcoming human trials of these agents will be facilitated by the availability of validated noninvasive biomarkers of cerebral and cerebellar pathology. To this end, a number of studies have demonstrated an ability to detect macro- and micro-structural and neurochemical abnormalities noninvasively by magnetic resonance imaging14, 36–38 (MRI) and spectroscopy39–41 (MRS) in SCA3. While macrostructural disease-associated changes detectable by conventional MRI clearly mark tissue loss, other MR metrics may allow noninvasive monitoring of aspects of pathology that precede cell loss and atrophy, and hence may be reversible with treatments. However, such MR metrics need to be validated by comparing them to tissue pathology.
Here, using MRS at 9.4 tesla (T), we sought to identify cerebellar neurochemical signatures in two mouse models of SCA3 that are extensively used in SCA3 preclinical trials: homozygous YACMJD84.2 (Q84/Q84)31, 42 and hemizygous CMVMJD135 (Q135)43 mice. We assessed two models to identify the common neurochemical abnormalities that may provide signatures of SCA3 pathology. To validate identified potential neurochemical biomarkers and determine which aspects of pathology they reflect, we evaluated cerebellar demyelination and neuroaxonal pathology in the same mice, as well as brain tissue from patients with SCA3. Finally, we determined whether RNAi-mediated silencing of the SCA3 gene reverses cerebellar demyelination and axonopathy in the Q84/Q84 model, since evidence of reversibility would suggest noninvasive myelin and neuronal markers could reflect therapeutic efficacy in future trials.
Materials and Methods
Experimental design
The study was designed to assess MRS neurochemical profiles of two transgenic models frequently used in preclinical trials for SCA3– homozygous YACMJD84.2 (Q84/Q84)42 and hemizygous CMVMJD135 (Q135)43 mice. All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals (Protocol PRO00003836) and by the University of Minnesota Institutional Animal Care and Use Committee (Protocol 1207A17510). Groups of Q84/Q84 (N=14 (7F/7M), 9–17 month-old) and their littermate nontransgenic (wt) mice (N=11 (7F/4M), 11–18 month-old), and Q135 (N=9 (3F/6M), 8–16 month-old) and their wt littermates (N=8 (5F/3M), 10–16 month-old) in the C57Bl6/J background (Supplementary Table 1) were scanned. Mice were housed in cages of maximum five animals and maintained in a standard 12-hours light/dark cycle with food and water ad libitum. Peripheral blood was collected by tail vein snip for a subgroup of Q84/Q84 mice (N=5) and littermate controls (N=4) to analyze liver profiles. Mice were shipped from the University of Michigan to the University of Minnesota for MR scanning, 2–4 weeks prior to scans. After MR scanning, mice were deeply anesthetized with sodium pentobarbital (100mg/kg i.p.), sacrificed by transcardial perfusion with pH 7.4 phosphate buffered saline and tissues were sent back to the University of Michigan for post-mortem analyses for demyelination (myelin basic protein (MBP), Luxol fast blue), axonopathy (neurofilament medium (NFL)), assessment of molecular layer thickness and Purkinje cell count.
To investigate if the pathological features identified in SCA3 mice reflect the human condition we obtained deidentified frozen and fixed samples from the cerebellar cortex of autopsied brains from molecularly confirmed SCA3 (N=6) and control individuals (N=5) from the University of Michigan Brain Bank. (See Supplementary Table 2 for demographic information).
To verify if RNAi-mediated depletion of human mutant ATXN3 rescues cerebellar molecular phenotypes in SCA3 mice, we used protein lysates from cerebella of end-stage Q84/Q84 mice (10–20 month old) that, at 6–8 weeks-of-age, were injected in the deep cerebellum nuclei with an adeno-associated virus (AAV2/1) encoding an artificial microRNA targeting mutant ATXN3 (miRATXN3) (N=10) or with vehicle (N=10)31.
Mouse genotyping and (CAG)n size assessment
Genotyping was performed by PCR using DNA isolated from tail biopsy at weaning as previously described42, 43. (CAG)n size in Q84/Q84 and Q135 mice (Supplementary Table 1), and in SCA3 and control individuals (Supplementary Table 2) was determined at Laragen Inc. by gene fragment analysis.
In vivo measurements
Magnetic resonance (MR) protocol
Procedures for anesthesia and MR scanning were identical to our prior studies44, 45. Mice were induced with 3–4% isoflurane and were maintained anesthetized with 1.5–2% isoflurane during scanning. Body temperature was maintained at 36–37°C and respiration rate at ~70–100 breaths per minute. The scanning time for each animal was approximately 50 minutes. MR scans were performed using a quadrature surface RF coil and a 9.4 T/31 cm magnet (Magnex Scientific, Abingdon, UK) interfaced to an Agilent console (Agilent, Inc., Palo Alto, CA, USA). A cerebellar volume-of-interest (VOI, 5–7μl, placed based on anatomical landmarks to avoid inclusion of cerebrospinal fluid (CSF), Fig. 1A, 2A) was selected on coronal and sagittal multi-slice images obtained with a rapid acquisition with relaxation enhancement (RARE) sequence46. First- and second-order shims were adjusted using FASTMAP47. Localized 1H MR spectra were acquired with a localization by adiabatic selective refocusing (LASER) sequence (echo time TE=15ms, repetition time TR=5s, 256 averages)48, as described previously44, 45. Spectra were saved as single scans, which were frequency and phase corrected49. Frequency drifts were corrected in the time domain after peak picking in frequency domain (the tCr peak at 3ppm) by using the first transient as reference. Similarly, phase correction was done in the time domain by calculating the phase difference between the reference (first) transient and other transients. Transients that showed evidence for motion (substantial deviations in frequency, linewidth, signal-to-noise ratio (SNR), loss of water suppression efficiency relative to other transients) were excluded prior to averaging49. In the 23 spectra obtained from the Q84/Q84 cohort (mice that were included in the MRS analysis), 1–2 shots (out of 256) were excluded in 3 spectra, 45 shots were excluded in one spectrum and the acquisition had to be stopped due to gasping in one mouse, generating 110 usable shots. The remaining 18 spectra had all 256 shots. In the 14 spectra obtained from the Q135 cohort (mice that were included in the MRS analysis), 5 shots were excluded in one spectrum and 93 shots were excluded in another spectrum. The remaining 12 spectra had all 256 shots. Finally, the averaged spectra were corrected for eddy current effects using the unsuppressed water signal acquired from the same VOI50.
Figure 1: Cerebellar neurochemical levels are altered in homozygous Q84/Q84 mice.
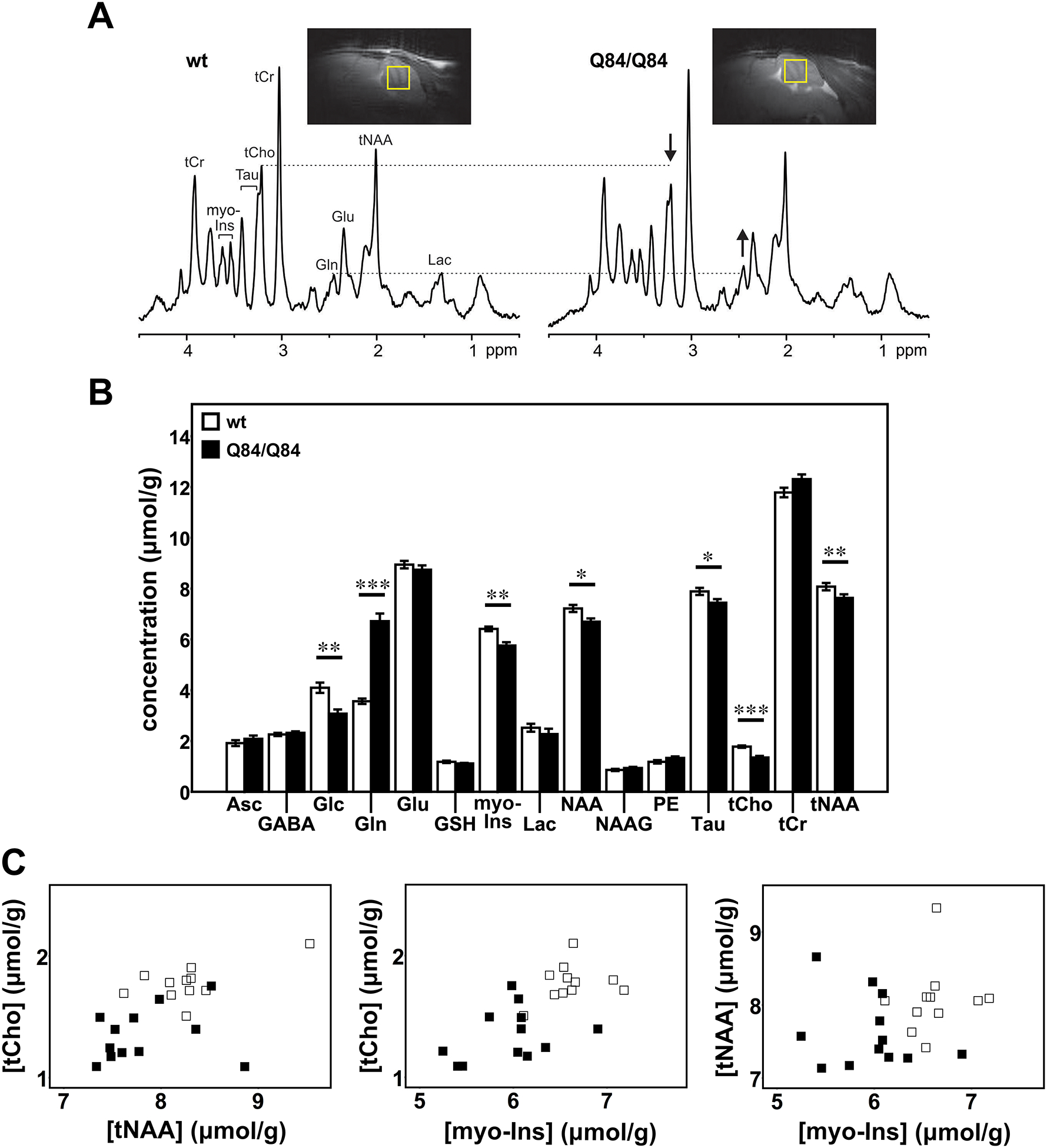
A) Representative localized proton MR spectra and midsagittal T2-weighted images of a Q84/Q84 and a wt littermate mouse. The most prominent neurochemical abnormalities, namely lower tCho and higher Gln in Q84/Q84 compared to wt mice, are denoted in the spectra by arrows. B) Cerebellar neurochemical profiles of Q84/Q84 mice (N=12, black bars) and wt littermates (N=11, white bars). Bars represent average neurochemical concentration ± SEM. Comparison between mouse genotypes was performed using Student’s t-test and statistical significance is indicated as: *P<0.05, **P<0.01, and ***P<0.001. C) Separation of Q84/Q84 mice (black squares) from controls (white squares) by plotting two altered metabolites against each other.
Figure 2: Cerebellar neurochemical levels are altered in hemizygous Q135 mice.
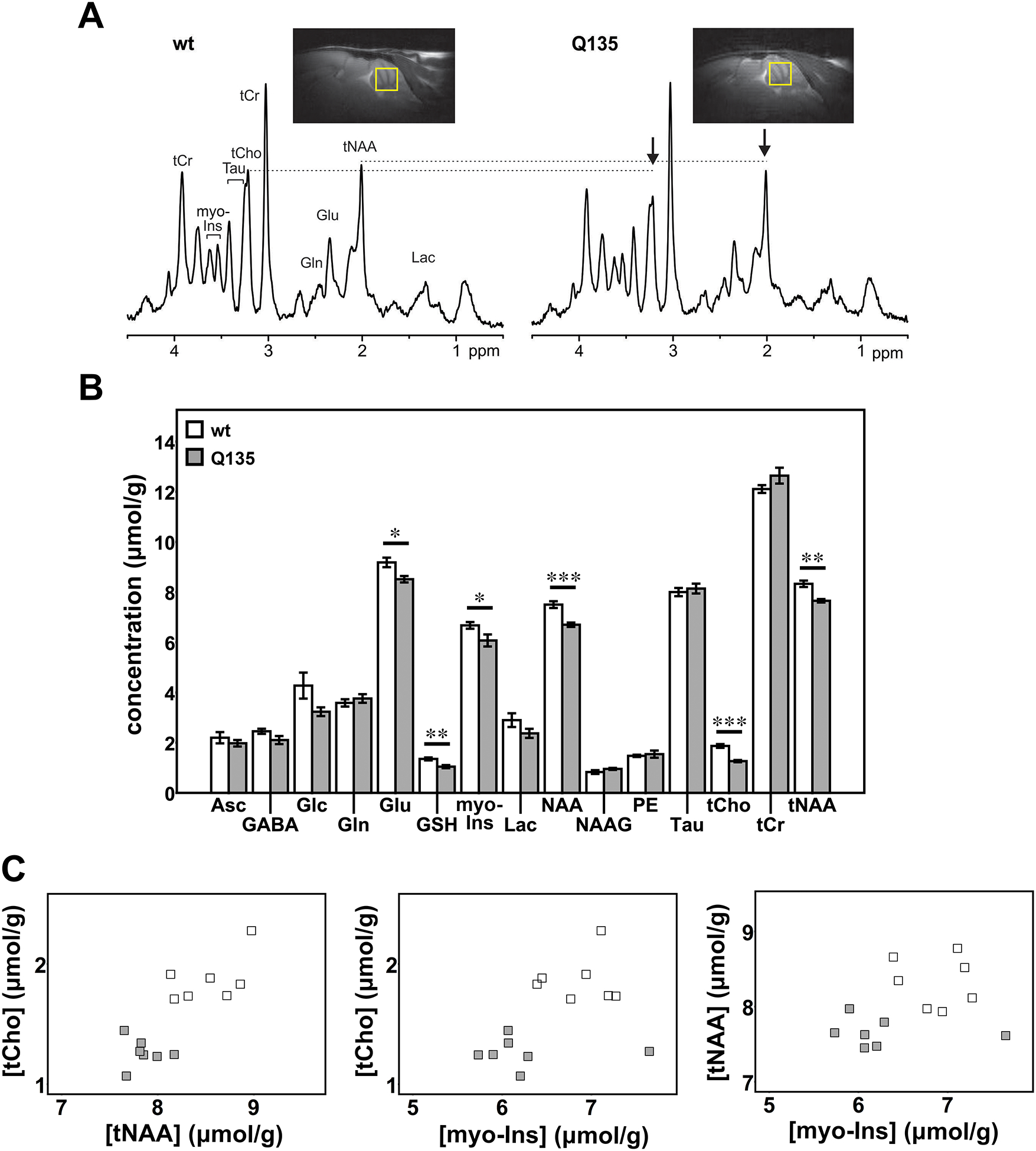
A) Representative localized proton MR spectra and midsagittal T2-weighted images of a Q135 and a wt littermate mouse. The most prominent neurochemical abnormalities, namely lower tCho and tNAA in Q135 compared to wt mice, are denoted in the spectra by arrows. B) Cerebellar neurochemical profiles of Q135 mice (N=7, grey bars) and wt littermates (N=7, white bars). Bars represent average chemical concentration ± SEM. Comparison between mouse genotypes was performed using Student’s t-test and statistical significance is indicated as: *P<0.05, **P<0.01, and ***P<0.001. C) Separation of Q135 mice (grey squares) from controls (white squares) by plotting two altered metabolites against each other.
Metabolite quantification
Metabolites were quantified using LCModel51 relative to unsuppressed water spectra acquired from the same VOI, as described before44, 45. Reliable concentrations were selected based on Cramér-Rao lower bounds (CRLB) criteria (quantification with CRLB≤20% in the majority of the spectra). Alanine, aspartate, glycine and scyllo-inositol were excluded from final analysis based on these criteria. If the correlation between two metabolites was consistently high (correlation coefficient<−0.5), their sum was reported52. Strong negative correlation was found between creatine and phosphocreatine and between glycerophosphocholine + phosphocholine, therefore total creatine (tCr) and total choline (tCho) were reported. Based on these reliability criteria,14 concentrations were evaluated: ascorbate (Asc), γ-aminobutyric acid (GABA), glucose (Glc), glutamine (Gln), glutamate (Glu), glutathione (GSH), myo-inositol (myo-Ins), lactate (Lac), N-acetylaspartate (NAA), N-acetylaspartylglutamate (NAAG), phosphoethanolamine (PE), taurine (Tau), tCr and tCho.
Liver profile
Peripheral blood collected from mouse tail vein snip was immediately centrifuged at maximum speed and serum was collected and stored at −80°C. Serum samples were thawed, centrifuged and supernatants were loaded in VetScan® Mammalian Liver Profile reagent rotors (Abaxis, Inc.), which in turn were read in a VetScan Chemistry Analyzer providing quantitative determinations of albumin (ALB), alkaline phosphatase (ALP), alanine aminotransferase (ALT), bile acids (BA), blood urea nitrogen (BUN), total cholesterol (CHOL), gamma glutamyl transferase (GGT).
Post-mortem measurements
Western blot
Extracts of soluble proteins in RIPA buffer from mouse cerebella or human cerebellar cortex were obtained using an established protocol31. Lysates from soluble protein fractions were resolved on 10% SDS-PAGE gels, and corresponding PVDF membranes were incubated overnight at 4°C with primary antibodies: rat anti-MBP (1:1000; MCA409S, AbDSerotec), mouse anti-160kD NFL (1:1000; ab7794, Abcam), rabbit anti-α-tubulin (1:5000; 2144S, Cell Signaling), mouse anti-GAPDH (1:10000; MAB374, Millipore), and rabbit anti-MJD (1:20000). Bound primary antibodies were visualized by peroxidase-conjugated anti-rat, anti-mouse or anti-rabbit secondary antibodies (1:10000; Jackson Immuno Research Laboratories) followed by treatment with ECL-plus reagent (PerkinElmer) and exposure to autoradiography films. Band intensity was quantified by densitometry using ImageJ.
Luxol fast blue staining
Five-μm paraffin sections of cerebellar lobules from aged-matched patients with SCA3 and unaffected individuals were simultaneously deparaffinized in xylene and stained with Luxol fast blue. Briefly, sections were dewaxed in xylene, washed in absolute ethanol and 95% ethanol, stained overnight in 0.1% Luxol fast blue in acidic 95% ethanol, washed in 95% ethanol, differentiated in 0.05% lithium carbonate solution for 30 seconds, rinsed in distilled water and washed in 70% and 90% ethanol, washed in absolute ethanol, fixed in xylene and mounted in DPX mountant. Slides were imaged in a bright-field Olympus BX51 microscope.
Immunofluorescence staining
Left mouse brain hemispheres were fixed overnight in 4% paraformaldehyde, embedded in 30% sucrose/phosphate-buffered saline, and sectioned on a sledge microtome (SM200R; Leica Biosystems). Triple immunofluorescent labelling of free-floating 40-μm sagittal sections was conducted as previously31, using rat anti-MBP (1:50), mouse anti-NFL (1:500), rabbit anti-ATXN3 (anti-MJD 1:2000), secondary mouse Alexa Fluor 488, rat Alexa Fluor 568 antibodies, and rabbit Alexa Fluor 647 (Invitrogen), and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI). Sections were mounted with Prolong Gold Antifade Reagent (Invitrogen) and imaged in an Olympus IX71 microscope. Measurements of molecular layer thickness were made as previously described53. Briefly, using images from the triple immunostaining, two measurements were made at the depth of the primary fissure and two others at 100 μm from there. Purkinje neuron cell counts were obtained as the total number of neurons in the region of the primary fissure folium.
Liver Hematoxylin & Eosin (H&E) histology
Livers from four Q84/Q84 mice and one littermate control used in the MRS studies were fixed in 10% formalin, paraffin embedded, and four-μm paraffin sections were stained with H&E and observed in a bright filed microscope.
Statistical Analysis
Characteristics of transgenic and control groups were compared using the two-tailed, unpaired Student’s t-test for age and spectral quality metrics (linewidth and SNR obtained from LCModel) and chi-square test for sex. Levels of neurochemicals and proteins were compared between the transgenic and control groups using Student’s t-test as all distributions were normal and showed homogeneous variances. Linear regression analyses were performed to evaluate the relationship between neurochemical concentrations and protein levels using Pearson correlation. A P<0.05 was considered statistically significant in all analyses. P-values were not corrected across the multiple metabolites due to the exploratory nature of our analysis and to avoid inflation of type II errors. Data were analyzed using IBM SPSS Statistics 22 software.
Results
Homozygous Q84/Q84 and hemizygous Q135 transgenic mice show shared neurochemical alterations in the cerebellum as measured by in vivo MRS
Q84/Q84 and Q135 SCA3 transgenic mice show early onset of SCA3-like phenotypes starting around 6 weeks-of-age31, 43 and therefore are often used in preclinical trials31, 35, 43, 53–56. We investigated common neurochemical biomarkers in these two models that could serve as outcome measures in preclinical trials.
Because no in vivo MRS studies were previously conducted in SCA3 mouse models, we obtained MR spectra at ultra-high field from the cerebellar vermis of symptomatic homozygous Q84/Q84, hemizygous Q135 and their littermate wt controls at an age when Purkinje cell dysfunction and pathology was previously shown in related transgenic mouse models35, 53. Two Q84/Q84 mice, two Q135 mice and one Q135 wt control were found to have an abnormal high Gln profile that was previously described in the brains of mice derived from the C57BL/6 strain57, 58. The abnormal profile consisted of 2–3 fold higher Gln, 35–60% lower myo-Ins and 10–35% lower Tau relative to other wt or SCA3 mice in the same group. Because this profile results from portosystemic shunting in the liver57, which causes alterations in gene expression in many organs, these 5 mice were excluded from further analysis.
SCA3 transgenic mice and their wt controls included in the MRS analyses were matched for age (Q84/Q84 13.4±2.1 (mean±SD) vs. wt 13.7±2.0 months, P=0.71; Q135 14.7±3.0 vs. wt 15.1±2.3 months, P=0.77) and sex (Q84/Q84 7M/5F vs. wt 4M/7F, P=0.41; Q135 4M/3F vs. wt 2M/5F, P=0.59). In addition, spectral quality was comparable in the SCA3 vs. wt groups (Linewidths Q84/Q84 10.0±2.2 (mean±SD) vs. wt 9.7±1.5 Hz, P=0.67; Q135 12.6±4.1 vs. wt 10.3±1.5 Hz, P=0.19; SNR Q84/Q84 30.6±7.6 vs. wt 31.7±4.0, P=0.67; Q135 28.7±3.5 vs. wt 27.4±3.2, P=0.49). Finally, structural images did not indicate substantial atrophy in SCA3 vs. wt mice (Figures 1A and 2A) and the VOI did not contain CSF. Therefore, group differences observed in the neurochemical profiles could not be due to group differences in age, sex, atrophy or spectral quality.
Neurochemical profiles of Q84/Q84 mice (N=12), with predominant CAG repeat size ranging from 69 to 74 triplets (Supplementary Table 1), showed significantly higher Gln and lower levels of total NAA (tNAA), NAA, Glc, myo-Ins, Tau, and tCho compared to wt littermates (N=11) (Figure 1A,B). Because sex matching was imperfect between groups and the severity of the phenotype may be sex-dependent in SCA3 mice, we further investigated the neurochemical profiles of male and female mice only. We observed the same neurochemical trends in male and female mice analyzed separately, except for the myo-Ins difference that appeared to be primarily driven by female mice (Supplementary Figure 1). This was consistent with a lack of sex-differences of the motor phenotype in Q84/Q84 mice31. To investigate if the Gln levels in Q84/Q84 mice that were almost two times higher than their wt controls were due to liver dysfunction, we analyzed serum liver profiles and liver pathology in a subgroup of the mice included in the MRS analysis. These analyses revealed typical liver histology with no signs of portosystemic shunt in four Q84/Q84 mice (data not shown), and normal levels of liver analytes in mice of both genotypes (Supplementary Table 3) except in one wt mouse that showed bile acids >250 μmol/L, but normal liver H/E histology (data not shown), thereby excluding liver dysfunction as the cause of the detected high levels of Gln in Q84/Q84 transgenic mice.
Cerebellar neurometabolite profiles of Q135 mice (N=7), with CAG repeat length ranging from 125 and 129 trinucleotides (Supplementary Table 1), revealed significantly lower levels of tNAA, NAA, Glu, myo-Ins, GSH, and tCho compared to controls (N=7) (Figure 2A,B). Similar to the Q84/Q84 mice, the same neurochemical trends were observed in male and female Q135 mice analyzed separately (Supplementary Figure 2). Therefore, lower NAA and tNAA, tCho and myo-Ins represent the common neurochemical abnormalities in both SCA3 models (Supplementary Figure 3). In addition, groups of Q84/Q84 and Q135 mice were separated from wt controls with little or no overlap by plotting concentrations of these metabolites against each other (Figure 1C,2C).
SCA3 transgenic mice show depletion of myelin basic protein (MBP) and neurofilament medium (NFL) in cerebellar extracts and slices and thinning of the molecular layer
Because the three neurochemicals that allowed clear separation of both SCA3 transgenic mouse models from their respective controls suggest disturbances in the metabolism of membrane and myelin phospholipids59, 60 (myo-Ins and Cho) and neuroaxonal pathology61 (NAA), and were all decreased in cerebella of SCA3 mice, we investigated whether these mice show cerebellar demyelination and axonal atrophy (Figure 3).
Figure 3: Cerebella of SCA3 transgenic mice show decreased levels of myelin basic protein (MBP) and neurofilament medium (NFL) that correlate with concentrations of tCho and tNAA, respectively, in Q135 mice.
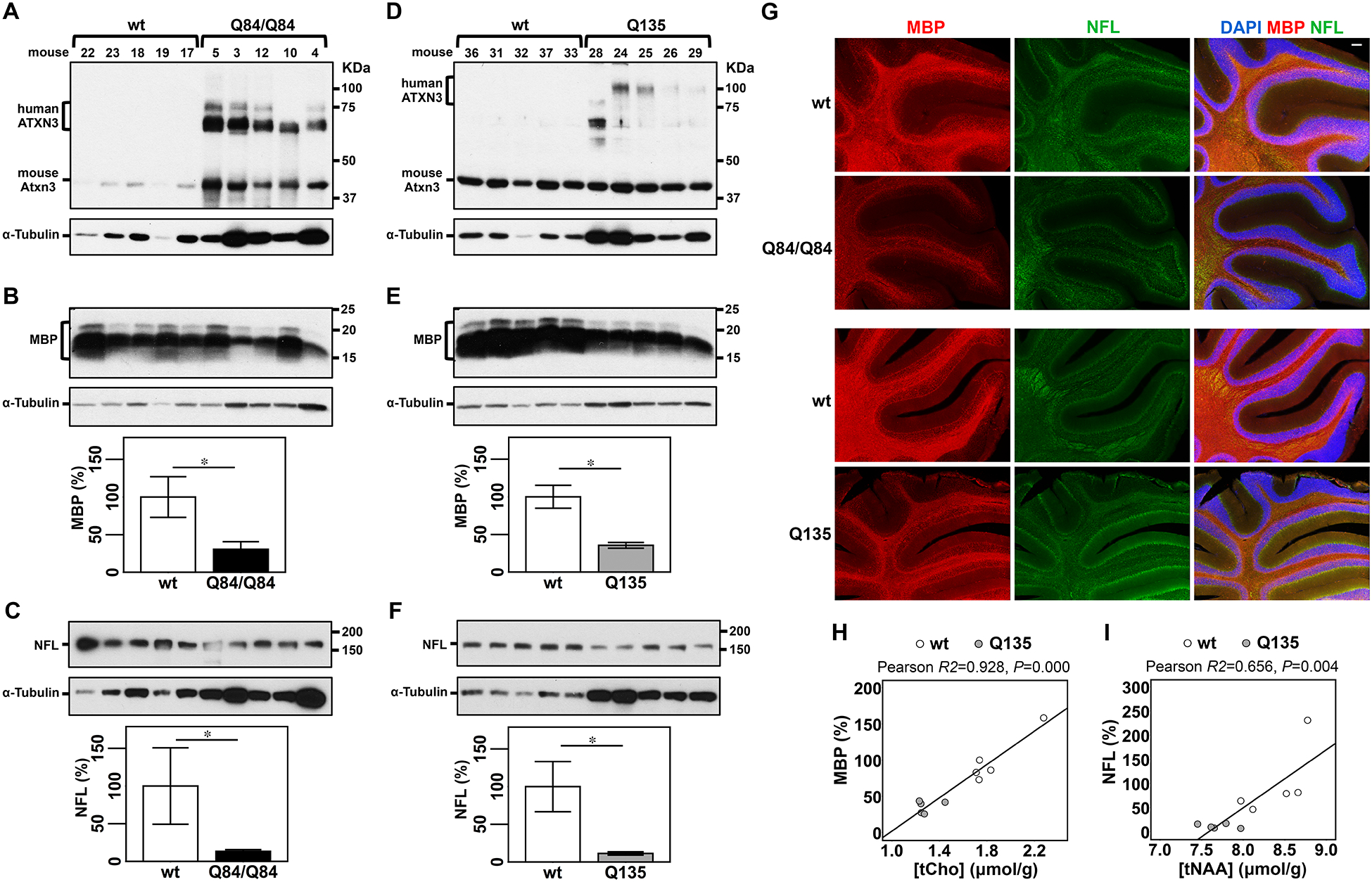
A,D) Anti-ATXN3 immunoblot (anti-MJD antibody) detecting mutant human ATXN3 and endogenous mouse Atxn3 in cerebellar soluble protein extracts of the subset of Q84/Q84 (A), Q135 (D) and corresponding littermate wild type mice used to assess levels of MBP (B,E) and NFL medium (NFL) (C,F). Western blots show decreased levels of MBP and NFL in cerebella of Q84/Q84 (B,C) and Q135 (E,F) compared with controls. Graphs show quantification of protein bands by densitometry. Bars represent the average percentage of protein relative to respective wild type controls, normalized for a-Tubulin (± SEM). Statistical significance determined by Student’s t-test is indicated as *P<0.05. G) Representative images of immunofluorescent labelling of MBP (red) and NFL (green) show decreased signal of these two proteins in cerebella of Q84/Q84 and Q135 mice compared with corresponding litter mates (N=4 mice per group). Nuclei were labeled with DAPI (blue). Scale bar, 100 μm. H, I) Plots showing Pearson correlations of levels of MBP with tCho (H), and NFL with tNAA (I) in Q135 (grey circles), and their respective wt littermate mice (white circles).
Immunoblot analysis of cerebellar protein extracts from a subset (N=5 per group, 3F/2M) of the mice used to obtain neurochemical profiles (Figure 3A,D) revealed that the levels of MBP were indeed reduced to 31% of wt levels in Q84/Q84 mice (Figure 3B) and to 35% of wt levels in Q135 mice (Figure 3E). NFL abundance was also reduced to 13% of wt levels in cerebella of Q84/Q84 mice (Figure 3C) and to 11% of wt levels in Q135 mice (Figure 3F). Decreased cerebellar levels of MBP and NFL in Q84/Q84 and Q135 mice compared to controls were further evidenced by immunolabeling of these two proteins in cerebellar slices of these mice (N=4 animals per group) (Figure 3G). Triple immunofluorescence staining of MBP, NFL, and ATXN3 in cerebella of this subset of mice further revealed significantly thinner molecular layer in Q84/Q84 and Q135 mice than wt littermates (Supplementary Figure 4A), and a similar number of Purkinje cells in the primary fissure folia of mice from all genotypes (Supplementary Figure 4B).
In vivo myelin and neuronal markers measured by MRS correlate with post-mortem myelin and neuronal markers in SCA3 transgenic mice
We next sought to determine if levels of tCho and/or myo-Ins are indicators of the status of myelination and if tNAA levels reflect axon integrity in the cerebellum. Hence, we investigated correlations between the levels of these neurochemicals with abundance of MBP and NFL proteins in the two SCA3 mouse models (Figure 3H,I, Supplementary Figure 5). Because of the small sample size per genotype group, correlation analyses were performed combining data from transgenic mice and corresponding wt littermates for each SCA3 transgenic mouse strain. Concentration of tCho correlated significantly with MBP levels (R2=0.928, P<0.001) (Figure 3H) and levels of tNAA correlated significantly with NFL expression in Q135 mice (R2=0.656, P=0.004) (Figure 3I). To test if the significance of tCho-MBP and tNAA-NFL correlations in Q135 mice was due to a genotype clustering effect we repeated the correlation analyses adjusting for the genotype. While both correlations remained significant adjusting for genotype (tCho-MBP, R2=0.929, P=0.000; tNAA-NFL, R2=0.658, P=0.023), we verified that MBP and tCho concentrations correlated independently of the genotype, and that NFL correlated with tNAA levels due to a combined effect of tNAA and genotype. No correlation was found between tCho and MBP levels, and tNAA and NFL concentrations in Q84/Q84 mice (Supplementary Figure 5B,D), or between myo-Ins and MBP levels in either mouse model (Supplementary Figure 5A,C).
MBP and NFL are lower in the cerebellar cortex of patients with SCA3 than healthy controls
To investigate whether dysregulation of cerebellar MBP and NFL in the two SCA3 transgenic mouse models reflects the human SCA3 condition, we assessed levels of these proteins in samples from cerebellar cortex of patients with SCA3 and healthy controls (Supplementary Table 2). Levels of MBP were decreased to 9% of control levels and NFL to 19% of control levels in cerebellar cortex of patients with SCA3 (Figure 4A–C) indicating demyelination and axonopathy in this brain region. Luxol fast blue staining of brains from patients with SCA3 and aged-matched unaffected individuals confirmed extensive demyelination in cerebella of patients with SCA3 that is more evident in aged patients (Figure 4D).
Figure 4: Cerebellar lobules of patients with SCA3 show decreased levels of MBP and NFL medium and demyelination.
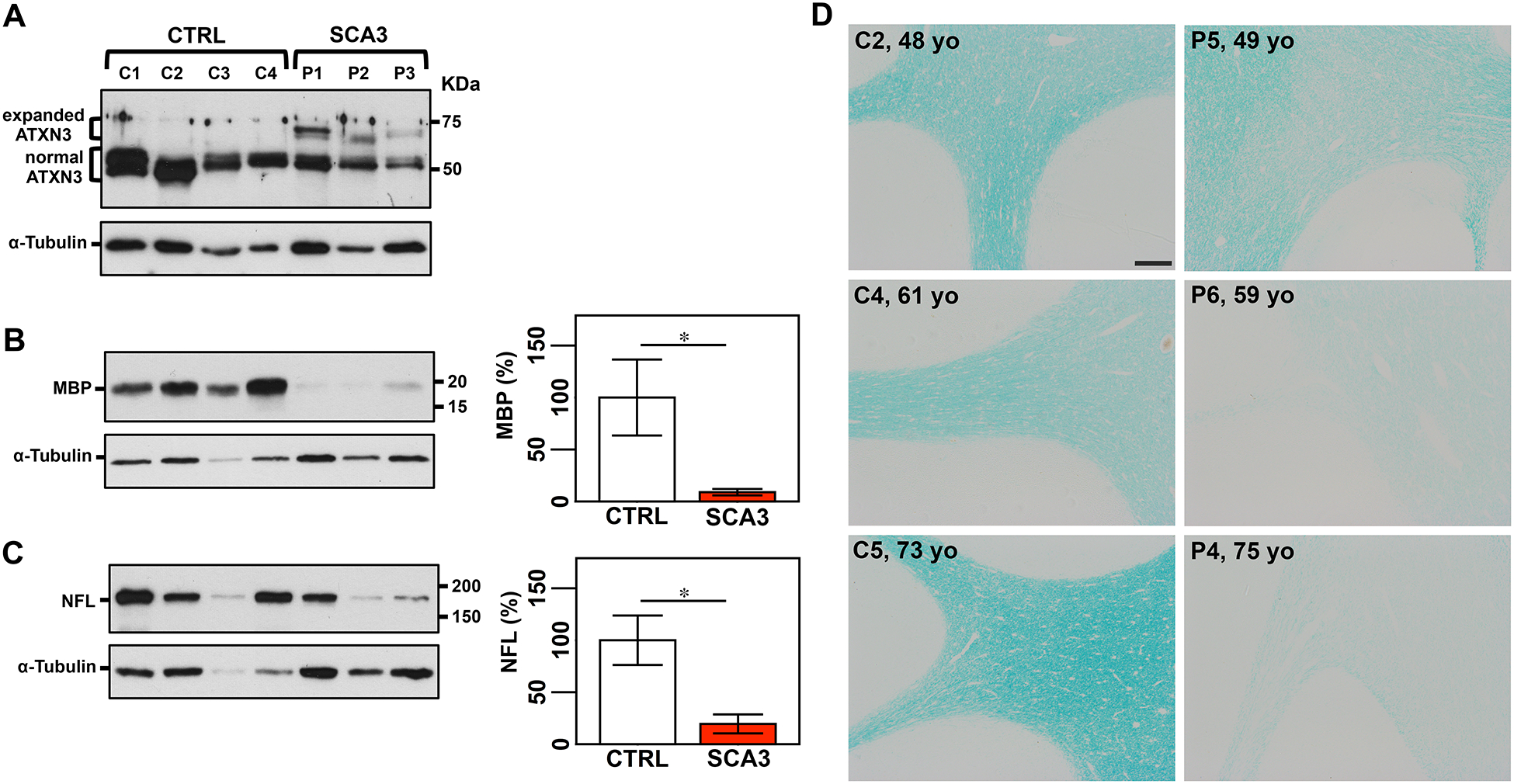
A) Anti-ATXN3 immunoblot (anti-MJD antibody) detecting expanded and normal ATXN3 in soluble protein extracts from cerebellar cortex of patients with SCA3 and control individuals. Detection of MBP (B) and NFL medium (C) by Western blotting shows lower levels of both proteins in patients compared with controls. Graphs show quantification of protein bands by densitometry. Bars represent the average percentage of protein relative to respective wild type controls, normalized for a-Tubulin (± SEM). Statistical significance determined by Student’s t-test is indicated as *P<0.05. D) Luxol fast blue staining of cerebellar sections shows signs of demyelination in patients with SCA3 compared with aged-matched unaffected individuals, which is more evident in older patients. Scale bar, 200 μm.
Long-term sustained reduction of mutant ATXN3 rescues cerebellar myelination and neurofilament biomarkers in homozygous Q84/Q84 mice
Because approaches to reduce levels of ATXN3 gene products are currently a promising candidate therapy for SCA3, we sought to evaluate whether long-term sustained RNAi-mediated decrease of mutant human ATXN3 (hATXN3) abundance in Q84/Q84 cerebella would prevent reduction of MBP and NFL. Therefore, we used cerebellar protein extracts from end-stage Q84/Q84 mice that were injected at 6–8 weeks-of-age in the DCN with an AAV encoding an artificial microRNA targeting mutant ATXN3 (miRATXN3)31. We previously showed that miRATXN3 reduced hATXN3 levels to ~40% of vehicle-injected Q84/Q84 mice31. Cerebella of miRATXN3-treated Q84/Q84 mice in fact showed MBP and NFL levels of, respectively, 137% and 177% than their levels in vehicle-treated Q84/Q84 mice (Figure 5A,B). Whereas no association was detected between MBP and hATXN3 levels (P=0.218) (Figure 5C), NFL levels were significantly correlated with levels of mutant hATXN3, demonstrating an association between the level of gene silencing and the extent of rescue of neuroaxonal pathology in miRATXN3-treated Q84/Q84 mice (P=0.006) (Figure 5D).
Figure 5: Cerebella from miRATXN3-treated Q84/Q84 show higher levels of MBP and NFL medium compared to vehicle-treated Q84 animals.
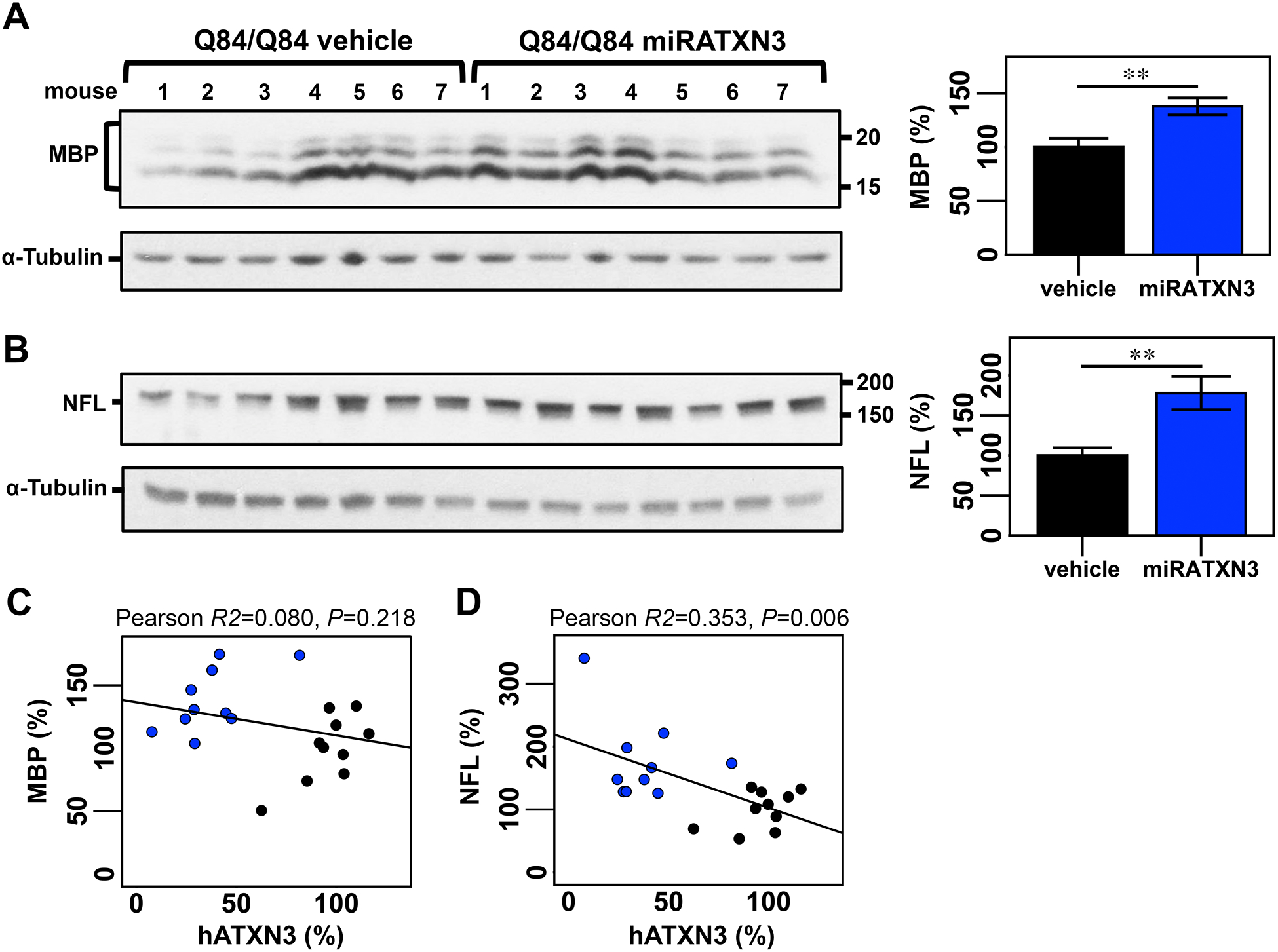
Immunoblotting to detect MBP (A) and NFL (B) in cerebellar soluble protein extracts from end-stage Q84/Q84 mice injected with an adeno-associated virus delivering an artificial microRNA targeting ATXN3 transcripts (miRATXN3) or injected with vehicle. Graphs show quantification of protein bands by densitometry. Bars represent the average percentage of protein relative to levels in vehicle-injected Q84/Q84 mice, normalized for a-Tubulin (± SEM). Black and blue bars indicate, respectively, vehicle-injected and miRATXN3-injected Q84/Q84 mice. Statistical significance determined by Student’s t-test is indicated as **P<0.01. Plots showing Pearson correlations of levels of MBP with hATXN3 (C) and of NFL with hATXN3 (D) in vehicle (black circles) or miRATXN3 (blue circles) treated Q84/Q84 mice.
Discussion
Future human clinical trials that seek to successfully translate preclinical animal studies would be facilitated by validated imaging biomarkers of cerebellar pathology. Here we identified shared neurochemical abnormalities in the homozygous Q84/Q8431, 42 and hemizygous Q13543 transgenic mice. Both of these models are well characterized models, reproduce several aspects of SCA3, and are extensively used in preclinical trials. We have further demonstrated associations of these noninvasive MR markers with protein markers of neuroaxonal pathology and demyelination, as well as the reversibility of these pathologies with mutant ATXN3 gene silencing.
Previously cerebral and cerebellar metabolic alterations were only reported in one study that used in vitro 1H-MRS in brain tissue obtained from hemizygous Q84 mice62. The cerebellar neurometabolite spectra of homozygous Q84/Q84 mice showed similarities to the ones previously described in the in vitro study, namely higher Gln and lower NAA, tCho and myo-Ins in Q84/Q84 mice than wt controls62. Furthermore, the lower levels of tNAA observed in both models, lower levels of Glu in Q135 and higher levels of Gln in Q84/Q84 mice compared with controls, recapitulate prior observations in patients with SCA339, 41. Decrease of tNAA in the cerebellum is a common biomarker of neuronal dysfunction or loss displayed by patients affected with several SCAs (SCA1, SCA2, SCA3/MJD, SCA6, and SCA7)39, 41, 63, 64, SCA1 mouse models44, 45, 65, and hemizygous Q84 mice62. The decreased Glu levels detected suggests dysfunction of glutamatergic granule cells, unipolar brush cells (UBCs), and/or parallel and climbing fibers in the cerebellar cortex66. While the number or function of these glutamatergic cells has not been investigated in cerebellar lobules of Q135 or Q84/Q84 mice, Purkinje cells (PCs) show decreased calbindin staining in Q135 mice35 and electrophysiological alterations in Q84 mice53 indicating that these cells are somehow dysfunctional. Because PCs receive excitatory input from parallel and/or climbing fibers, it is possible that Q135 PCs are impaired due to decreased input from these fibers into the molecular layer. End-stage Q84/Q84 cerebella did not show decreased levels of Glu, but displayed increased concentration of Gln, which could suggest abnormal Glu-Gln cycle between glutamate cells and Bergmann glia in the molecular layer, and/or a higher number of glial cells that produce the bulk of Gln in the brain. While cerebellar cell loss in aged SCA3 mice needs to be further investigated, thinning of the molecular layer accompanied by preserved Purkinje cell number in these mice suggests that dysregulated levels of tNAA, Glu and Gln reflect neuronal dysfunction and dendritic atrophy rather than cell loss in these models, similar to prior observations in SCA1 models44.
In addition to the above described neurochemical changes in SCA3 mice, each of the transgenic mouse groups showed a clear separation from their respective wild type controls in concentrations of tNAA, myo-Ins and tCho in the cerebellum (Figures 1C, 2C, Supplementary Figure 3). NAA is a validated marker of neuronal viability61 and a source of acetate for myelin lipid synthesis in oligodendrocytes67, myo-Ins is a structural sugar of membrane and myelin phospholipids39, and changes in tCho levels reflect disturbances in phospholipid metabolism67. Because all three metabolites were decreased in spectra of both SCA3 mouse models, we hypothesized that these mice would display cerebellar demyelination and axon atrophy. Demyelination, white matter and axon dysfunction and axonopathy have indeed been reported histopathologically in brain tissue obtained from patients with SCA317, 40, 41, and primary oligodendrocyte/myelin dysfunction indicated by diffusion MRI in SCA314. Furthermore, altered function of oligodendrocytes was recently reported in SCA3 models including the Q8468. Here, by showing that aged Q84/Q84 and Q135 cerebella and post-mortem cerebellar cortex from patients with SCA3 display reduced levels of MBP and NFL compared to their respective controls, we confirmed demyelination and axon loss in SCA3 cerebella. The magnitude of reduction that we detected in NFL in SCA3 cerebella relative to controls was markedly higher than the reduction in the in vivo neuronal marker, tNAA. Similarly, the magnitude of reduction in MBP in SCA3 was substantially higher than the % reduction in the MRS markers myo-Ins and tCho. This is likely because these neurochemicals have multiple roles, such as serving as metabolic intermediates and osmolytes, and therefore are less specific for neuroaxonal and myelin pathology than the post-mortem protein markers. However, importantly, the associations between tCho with MBP levels and tNAA with NFL abundance in the Q135 mice suggest that tCho and tNAA levels reflect the status of myelination and axon integrity in the cerebellum of these mice. This evidence supports the inclusion of MRS evaluation of these neurochemicals in preclinical trials using Q84/Q84 and Q135 mice to evaluate the efficacy of therapeutic agents in rescuing SCA3 pathology.
While MBP levels were higher in miRATXN3-treated Q84/Q84 cerebella than wt controls, concentrations of MBP and hATXN3 did not correlate, possibly indicating that AAV2/1 miRATXN3 poorly transduced oligodendrocytes, as expected for this virus serotype69. Cerebellar axon loss seems to be, however, a consequence of expression of toxic expanded ATXN3 since reduction of NFL concentration in Q84/Q84 cerebella was suppressed by long-term decrease of mutant ATXN3 levels in miRATXN3-treated Q84/Q84 mice. NFL levels significantly correlated with hATXN3 abundance in the cerebella of Q84/Q84 mice suggesting that AAV2/1 miRATXN3 effectively transduced and reduced hATXN3 in the majority of cerebellar neurons. Importantly, the reversal of the neuroaxonal pathology with RNAi-mediated reduction of ATXN3 levels supports the use of noninvasive imaging markers of this pathology in future pre-clinical and clinical trials that evaluate similar gene silencing strategies. No MRS measurements are available for this set of RNAi-treated SCA3 mice31. Whether gene silencing will rescue the abnormal levels of the myelin and neuronal markers measured by MRS, similar to prior observations in SCA1 mice70, remains to be determined in future studies.
The ultra-high field strength MRI scanner we used in this study is not widely available for human trials that will like utilize the widely available 3T platform. We previously demonstrated the feasibility of detecting neurochemical alterations in patients with SCAs at field strengths ranging from 3–7T39, 41, 63, 64, 71. Specifically, neurochemical abnormalities were reliably detected in SCA3 at 3T39. Thus, the current observations in SCA3 models can be obtained with 3T technology when similar optimized methods are used for MRS data acquisition and analysis. We have further shown good test-retest reproducibility of cerebellar neurochemical concentrations at 3T72 and the two-site reproducibility of cerebellar and brainstem neurochemical concentrations at 3T73. Finally, these MRS methods are now being validated for clinical trial readiness in SCA3 in an international, multi-site setting (https://readisca.org/).
In summary, we have identified novel neurochemical and molecular similarities between end-stage homozygous Q84/Q84, aged hemizygous Q135 mice, and patients with SCA3, further supporting the use of these mouse models in preclinical trials of SCA3. Like patients with SCA3, both transgenic models show neurochemical signatures indicative of neuronal dysfunction, dysregulation of glutamatergic systems, and myelin and neurite loss. We also provide molecular confirmation of cerebellar myelin and axon damage in SCA3 mice and patients and establish significant associations of select neurochemical markers with invasive measures of myelin and axon damage. It will be important to perform future longitudinal MRS, molecular and pathological studies in parallel to evaluate the progression of neurometabolite dysregulation and associated molecular and pathological outcomes in the cerebellum and other SCA3-affected brain areas of transgenic mice. These novel neurochemical alterations in SCA3 transgenic mice reflect important aspects of pathology and should enable noninvasive monitoring of pathology reversal in future preclinical trials of therapeutic agents for SCA3.
Supplementary Material
Acknowledgements
The authors thank Dr. Sujith V. Weerasinghe and Dr. M. Bishr Omary for help with the liver studies, the Michigan Brain Bank (5P30 AG053760 University of Michigan Alzheimer’s Disease Core Center) and its coordinator Mathew Perkins for providing brain samples of MJD patients and control individuals, and Dr. Lynn Eberly for statistical consulting. This work was funded by: Portuguese National funds, through the Foundation for Science and Technology (FCT) - projects UIDB/50026/2020 and PTDC/NEU-NMC/3648/2014, and by FEDER funds, through the Competitiveness Factors Operational Programme (COMPETE) and the Northern Portugal Regional Operational Program (NORTE 2020), under the Portugal 2020 Partnership Agreement - projects NORTE-01-0145-FEDER-000013 and POCI-01-0145-FEDER-016818 to P.M.; NIH/NINDS R01NS03871 to H.L.P; NIH/NINDS R21 NS111154 to H.S.M. and G.Ö; and a Becky Babcox Research Fund pilot research award, University of Michigan, to M.C.C.. The Center for Magnetic Resonance Research is supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant P41 EB027061, the Institutional Center Cores for Advanced Neuroimaging award P30 NS076408 and the W.M. Keck Foundation.
Footnotes
Financial disclosures
The authors declare no competing interests.
References
- 1.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 2010;11(4):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- 3.Ashizawa T, Oz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol 2018;14(10):590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karam A, Trottier Y. Molecular Mechanisms and Therapeutic Strategies in Spinocerebellar Ataxia Type 7. Adv Exp Med Biol 2018;1049:197–218. [DOI] [PubMed] [Google Scholar]
- 6.Lane RM, Smith A, Baumann T, et al. Translating Antisense Technology into a Treatment for Huntington’s Disease. Methods Mol Biol 2018;1780:497–523. [DOI] [PubMed] [Google Scholar]
- 7.Singh K, Roy I. Nucleic acid therapeutics in Huntington’s disease. Recent Pat Biotechnol 2019. [DOI] [PubMed] [Google Scholar]
- 8.Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med 2019;380(24):2307–2316. [DOI] [PubMed] [Google Scholar]
- 9.Da Silva JD, Teixeira-Castro A, Maciel P. From Pathogenesis to Novel Therapeutics for Spinocerebellar Ataxia Type 3: Evading Potholes on the Way to Translation. Neurotherapeutics 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duarte-Silva S, Maciel P. Pharmacological Therapies for Machado-Joseph Disease. Adv Exp Med Biol 2018;1049:369–394. [DOI] [PubMed] [Google Scholar]
- 11.Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci 2017;18(10):613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawaguchi Y, Okamoto T, Taniwaki M, et al. CAG expansions in a novel gene for Machado- Joseph disease at chromosome 14q32.1. Nat Genet 1994;8(3):221–228. [DOI] [PubMed] [Google Scholar]
- 13.Costa Mdo C, Paulson HL. Toward understanding Machado-Joseph disease. Prog Neurobiol 2012;97(2):239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guimaraes RP, D’Abreu A, Yasuda CL, et al. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov Disord 2013;28(8):1125–1132. [DOI] [PubMed] [Google Scholar]
- 15.Linnemann C, Tezenas du Montcel S, Rakowicz M, et al. Peripheral Neuropathy in Spinocerebellar Ataxia Type 1, 2, 3, and 6. Cerebellum 2016;15(2):165–173. [DOI] [PubMed] [Google Scholar]
- 16.Rezende TJR, de Paiva JLR, Martinez ARM, et al. Structural signature of SCA3: From presymptomatic to late disease stages. Ann Neurol 2018;84(3):401–408. [DOI] [PubMed] [Google Scholar]
- 17.Rub U, Schols L, Paulson H, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 2013;104:38–66. [DOI] [PubMed] [Google Scholar]
- 18.Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012;124(1):1–21. [DOI] [PubMed] [Google Scholar]
- 19.Suga N, Katsuno M, Koike H, et al. Schwann cell involvement in the peripheral neuropathy of spinocerebellar ataxia type 3. Neuropathol Appl Neurobiol 2014;40(5):628–639. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Liao X, Zhan Y, et al. Microstructural Alterations in Asymptomatic and Symptomatic Patients with Spinocerebellar Ataxia Type 3: A Tract-Based Spatial Statistics Study. Front Neurol 2017;8:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLoughlin HS, Moore LR, Paulson HL. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol Dis 2019;134:104635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coutinho P, Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology 1978;28(7):703–709. [DOI] [PubMed] [Google Scholar]
- 23.Lima L, Coutinho P. Clinical criteria for diagnosis of Machado-Joseph disease: report of a non-Azorena Portuguese family. Neurology 1980;30(3):319–322. [DOI] [PubMed] [Google Scholar]
- 24.Paulson H Spinocerebellar Ataxia Type 3. 1993.
- 25.Riess O, Rub U, Pastore A, Bauer P, Schols L. SCA3: neurological features, pathogenesis and animal models. Cerebellum 2008;7(2):125–137. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg RN. Machado-Joseph disease: an autosomal dominant motor system degeneration. Mov Disord 1992;7(3):193–203. [DOI] [PubMed] [Google Scholar]
- 27.McLoughlin HS, Moore LR, Chopra R, et al. Oligonucleotide therapy mitigates disease in Spinocerebellar Ataxia Type 3 mice. Ann Neurol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toonen LJA, Rigo F, van Attikum H, van Roon-Mom WMC. Antisense Oligonucleotide-Mediated Removal of the Polyglutamine Repeat in Spinocerebellar Ataxia Type 3 Mice. Mol Ther Nucleic Acids 2017;8:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kourkouta E, Weij R, Gonzalez-Barriga A, et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol Ther Nucleic Acids 2019;17:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore LR, Rajpal G, Dillingham IT, et al. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol Ther Nucleic Acids 2017;7:200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.do Carmo Costa M, Luna-Cancalon K, Fischer S, et al. Toward RNAi Therapy for the Polyglutamine Disease Machado-Joseph Disease. Mol Ther 2013;21(10):1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobrega C, Nascimento-Ferreira I, Onofre I, et al. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in machado-joseph disease transgenic mice. PLoS One 2013;8(1):e52396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashraf NS, Duarte-Silva S, Shaw ED, et al. Citalopram Reduces Aggregation of ATXN3 in a YAC Transgenic Mouse Model of Machado-Joseph Disease. Mol Neurobiol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esteves S, Oliveira S, Duarte-Silva S, Cunha-Garcia D, Teixeira-Castro A, Maciel P. Preclinical Evidence Supporting Early Initiation of Citalopram Treatment in Machado-Joseph Disease. Mol Neurobiol 2018. [DOI] [PubMed] [Google Scholar]
- 35.Teixeira-Castro A, Jalles A, Esteves S, et al. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado-Joseph disease. Brain 2015;138(Pt 11):3221–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adanyeguh IM, Perlbarg V, Henry PG, et al. Autosomal dominant cerebellar ataxias: Imaging biomarkers with high effect sizes. Neuroimage Clin 2018;19:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Abreu A, Franca MC Jr, Yasuda CL, Campos BA, Lopes-Cendes I, Cendes F. Neocortical Atrophy in Machado-Joseph Disease: A Longitudinal Neuroimaging Study. J Neuroimaging 2011:doi: 10.1111/j.1552-6569.2011.00614.x. [DOI] [PubMed] [Google Scholar]
- 38.Reetz K, Costa AS, Mirzazade S, et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain 2013;136(Pt 3):905–917. [DOI] [PubMed] [Google Scholar]
- 39.Adanyeguh IM, Henry PG, Nguyen TM, et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov Disord 2015;30(5):662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D’Abreu A, Franca M Jr., Appenzeller S, Lopes-Cendes I, Cendes F. Axonal dysfunction in the deep white matter in Machado-Joseph disease. J Neuroimaging 2009;19(1):9–12. [DOI] [PubMed] [Google Scholar]
- 41.Joers JM, Deelchand DK, Lyu T, et al. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann Neurol 2018;83(4):816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cemal CK, Carroll CJ, Lawrence L, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet 2002;11(9):1075–1094. [DOI] [PubMed] [Google Scholar]
- 43.Silva-Fernandes A, Duarte-Silva S, Neves-Carvalho A, et al. Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease. Neurotherapeutics 2014;11(2):433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oz G, Nelson CD, Koski DM, et al. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 2010;30(10):3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oz G, Vollmers ML, Nelson CD, et al. In vivo monitoring of recovery from neurodegeneration in conditional transgenic SCA1 mice. Exp Neurol 2011;232(2):290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hennig J, Nauerth A, Friedburg H. RARE imaging: a fast imaging method for clinical MR. Magn Reson Med 1986;3(6):823–833. [DOI] [PubMed] [Google Scholar]
- 47.Gruetter R, Tkac I. Field mapping without reference scan using asymmetric echo-planar techniques. Magn Reson Med 2000;43(2):319–323. [DOI] [PubMed] [Google Scholar]
- 48.Garwood M, DelaBarre L. The return of the frequency sweep: designing adiabatic pulses for contemporary NMR. J Magn Reson 2001;153(2):155–177. [DOI] [PubMed] [Google Scholar]
- 49.Near J, Harris AD, Juchem C, et al. Preprocessing, analysis and quantification in single-voxel magnetic resonance spectroscopy: experts’ consensus recommendations. NMR Biomed 2020:e4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klose U In vivo proton spectroscopy in presence of eddy currents. Magn Reson Med 1990;14(1):26–30. [DOI] [PubMed] [Google Scholar]
- 51.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 1993;30(6):672–679. [DOI] [PubMed] [Google Scholar]
- 52.Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed 2001;14(4):260–264. [DOI] [PubMed] [Google Scholar]
- 53.Shakkottai VG, do Carmo Costa M, Dell’orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 2011;31(36):13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duarte-Silva S, Neves-Carvalho A, Soares-Cunha C, et al. Lithium Chloride Therapy Fails to Improve Motor Function in a Transgenic Mouse Model of Machado-Joseph Disease. Cerebellum 2014. [DOI] [PubMed] [Google Scholar]
- 55.Esteves S, Oliveira S, Duarte-Silva S, Cunha-Garcia D, Teixeira-Castro A, Maciel P. Preclinical Evidence Supporting Early Initiation of Citalopram Treatment in Machado-Joseph Disease. Mol Neurobiol 2019;56(5):3626–3637. [DOI] [PubMed] [Google Scholar]
- 56.McLoughlin HS, Moore LR, Chopra R, et al. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol 2018;84(1):64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cudalbu C, McLin VA, Lei H, et al. The C57BL/6J mouse exhibits sporadic congenital portosystemic shunts. PLoS One 2013;8(7):e69782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emir UE, Brent Clark H, Vollmers ML, Eberly LE, Oz G. Non-invasive detection of neurochemical changes prior to overt pathology in a mouse model of spinocerebellar ataxia type 1. J Neurochem 2013;127(5):660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisher SK, Novak JE, Agranoff BW. Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J Neurochem 2002;82(4):736–754. [DOI] [PubMed] [Google Scholar]
- 60.Mountford CE, Stanwell P, Lin A, Ramadan S, Ross B. Neurospectroscopy: the past, present and future. Chem Rev 2010;110(5):3060–3086. [DOI] [PubMed] [Google Scholar]
- 61.Demougeot C, Garnier P, Mossiat C, et al. N-Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: its relevance to studies of acute brain injury. J Neurochem 2001;77(2):408–415. [DOI] [PubMed] [Google Scholar]
- 62.Griffin JL, Cemal CK, Pook MA. Defining a metabolic phenotype in the brain of a transgenic mouse model of spinocerebellar ataxia 3. Physiological Genomics 2004;16:334–340. [DOI] [PubMed] [Google Scholar]
- 63.Oz G, Hutter D, Tkac I, et al. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord 2010;25(9):1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oz G, Iltis I, Hutter D, Thomas W, Bushara KO, Gomez CM. Distinct neurochemical profiles of spinocerebellar ataxias 1, 2, 6, and cerebellar multiple system atrophy. Cerebellum 2011;10(2):208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oz G, Kittelson E, Demirgoz D, et al. Assessing recovery from neurodegeneration in spinocerebellar ataxia 1: Comparison of in vivo magnetic resonance spectroscopy with motor testing, gene expression and histology. Neurobiol Dis 2015;74:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoshino M Neuronal subtype specification in the cerebellum and dorsal hindbrain. Dev Growth Differ 2012;54(3):317–326. [DOI] [PubMed] [Google Scholar]
- 67.Xu H, Zhang H, Zhang J, Huang Q, Shen Z, Wu R. Evaluation of neuron-glia integrity by in vivo proton magnetic resonance spectroscopy: Implications for psychiatric disorders. Neurosci Biobehav Rev 2016;71:563–577. [DOI] [PubMed] [Google Scholar]
- 68.Ramani B, Panwar B, Moore LR, et al. Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes. Hum Mol Genet 2017;26(17):3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lawlor PA, Bland RJ, Mouravlev A, Young D, During MJ. Efficient gene delivery and selective transduction of glial cells in the mammalian brain by AAV serotypes isolated from nonhuman primates. Mol Ther 2009;17(10):1692–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friedrich J, Kordasiewicz HB, O’Callaghan B, et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018;3(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deelchand DK, Joers JM, Ravishankar A, et al. Sensitivity of Volumetric Magnetic Resonance Imaging and Magnetic Resonance Spectroscopy to Progression of Spinocerebellar Ataxia Type 1. Mov Disord Clin Pract 2019;6(7):549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terpstra M, Cheong I, Lyu T, et al. Test-retest reproducibility of neurochemical profiles with short-echo, single-voxel MR spectroscopy at 3T and 7T. Magn Reson Med 2016;76(4):1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deelchand DK, Adanyeguh IM, Emir UE, et al. Two-site reproducibility of cerebellar and brainstem neurochemical profiles with short-echo, single-voxel MRS at 3T. Magn Reson Med 2015;73(5):1718–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.