

We recently showed that perineuronal nets (PNNs) enmesh glucoregulatory neurons in the arcuate nucleus (Arc) of the mediobasal hypothalamus (MBH)1, but whether these PNNs play a role in either the pathogenesis of type 2 diabetes (T2D) or its treatment remains unclear. Here we show that compared to normoglycemic rats, PNN abundance within the Arc is markedly reduced in the Zucker Diabetic Fatty (ZDF) rat model of T2D, correlating with altered PNN-associated chondroitin sulfate (CS) glycosaminoglycan (GAG) sulfation patterns in the MBH. Each of these PNN-associated changes is reversed following a single intracerebroventricular (icv) injection of fibroblast growth factor 1 (FGF1) at a dose that induces sustained diabetes remission in male ZDF rats. Combined with previous work localizing this FGF1 effect to the Arc area2-4, our finding that enzymatic digestion of Arc PNNs markedly shortens the duration of diabetes remission following icv FGF1 injection in these animals identifies these extracellular matrix (ECM) structures as previously unrecognized participants in the mechanism underlying diabetes remission induced by the central action of FGF1.
PNNs are a distinct ECM subtype that enmesh neurons in defined circuits and interdigitate with peri-synaptic astrocytic processes to influence neurocircuit connectivity and synaptic stability5. We recently reported that in both humans and rodents, PNNs enmesh a tightly packed cluster of neurons situated at the junction of the Arc and adjacent median eminence (ME) in the MBH1. GABAergic Agouti-related peptide (Agrp) neurons are among those that are both enmeshed by PNNs and implicated in physiological control of food intake and glucose homeostasis6,7. Given the powerful influence that PNNs can exert over the neurons they enmesh, we sought to determine if obesity-associated diabetes is correlated with maladaptive changes in the formation of these Arc PNNs and if so, whether they might be targets for its treatment.
Our original report describing hypothalamic PNN matrices was based primarily on immunofluorescence (IF) labeling of PNN-associated CS-GAGs using Wisteria floribunda agglutinin (WFA) in mice1. Extending this work, we report that in normoglycemic Wistar rats, 1) Arc PNNs are enriched in the CS proteoglycan (CSPG) aggrecan8, and 2) these PNNs enmesh neurons throughout both medial Arc (ArcM) and lateral Arc/retrochiasmatic regions (ArcL) (Fig. 1a). Comprehensive stereological mapping throughout the MBH reveals that these PNNs effectively circumscribe the Arc-ME junction (Supplementary Fig. 1), as reported in mice1. Although aggrecan is a major constituent of PNNs in most brain areas, several features distinguish Arc PNNs from those located elsewhere. First, whereas PNNs in visual and motor cortices tightly enmesh both the neuronal soma and proximal axonal and dendritic processes (Supplementary Fig. 2; Supplementary Videos 1-2), PNNs in the Arc enmesh the neuron soma more loosely and show minimal extensions surrounding proximal processes (Fig. 1b; Supplementary Video 3). Second, whereas PNNs in cerebral cortex contain two extracellular PNN glycoproteins that enhance structural stability – Tenascin-R (TnR) and the hyaluronan and proteoglycan link protein 1 (HAPLN-1) – only TnR is present in Arc PNNs (Supplementary Fig. 3). This difference may explain the comparatively diffuse appearance of Arc PNNs compared to cortical PNNs, morphological differences that were also observed in human brain (Supplementary Fig. 4).
Figure 1. Diabetic ZDF rats have reduced PNN structures in the arcuate nucleus.
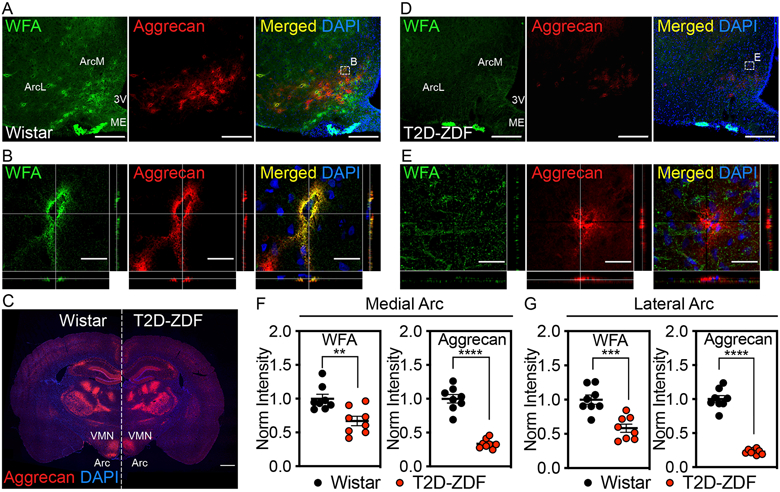
Immunofluorescent detection of WFA, a lectin that binds to the CS-GAG glycan component of PNNs1, and aggrecan, a PNN-associated CSPG core protein8, in coronal sections of rat hypothalamus (30 μm) from (A-C) normoglycemic Wistar controls and (C-E) age-matched, hyperglycemic ZDF rats. (A, D) Low-magnification view of WFA+ / aggrecan+ PNNs throughout the ArcM and ArcL areas of the mediobasal hypothalamus. Scale bar: 200 μm. (B, E) Higher-magnification orthogonal views of WFA+ / aggrecan+ PNN enmeshments. Scale bar: 25 μm. (C) Tiled panoramic image of aggrecan from a coronal section from a (left) normoglycemic Wistar control and a (right) age-matched diabetic ZDF rat shows selective loss of PNN labeling in the Arc of ZDF rats. Scale bar: 1 mm. Quantification of the mean fluorescence intensity of PNNs averaged from medial hypothalamic sections (−2.2 to −2.8 mm posterior from bregma) for (F) medial Arc and (G) lateral Arc areas from normoglycemic Wistar and diabetic ZDF rats and normalized to the Wistar mean fluorescence intensity averages (n=8 rats/group; mean ± SEM). **P < 0.01, ***P < 0.001, ****P < 0.0001 versus Wistar controls; Student’s t-test (unpaired, two-sided); (F) WFA-ArcM p=0.0.0036, aggrecan-ArcM p=<0.0001, WFA-ArcL p=0.0004, aggrecan-ArcL p=<0.0001. Two separate cohorts of diabetic ZDF and normoglycemic Wistar rats were analyzed, and the results were reproducible between studies. Arc, arcuate nucleus; ArcL, lateral Arc; ArcM, medial Arc; ME, median eminence; VMN, ventromedial nucleus; 3V, 3rd ventricle.
To determine if hypothalamic PNN abundance and/or composition differ between diabetic rats and normoglycemic controls, we performed stereological mapping of PNNs throughout the rostro-caudal extent of the MBH in ZDF rats, an established model of T2D. Compared to normoglycemic Wistar controls (Fig. 1c), the mean fluorescence intensity of both WFA and aggrecan were markedly reduced throughout the ArcM and ArcL areas of ZDF rats (Fig. 1d-g, Supplementary Fig. 5). In contrast, no such loss of PNN matrices was observed in visual or motor cortices of these same animals (Supplementary Fig. 6; Supplementary Videos 4-5), suggesting that reduced PNN formation in these animals is specific to the Arc. Further stereological mapping of Arc PNNs in ZDF rats using higher magnification and increased laser imaging power revealed the presence of aggrecan+ PNN matrices in the ArcM region, albeit at markedly reduced levels and more diffuse in appearance than in normoglycemic controls (Supplementary Fig. 7; Supplementary Video 6), whereas PNN-associated WFA immunoreactivity remained undetectable. In addition, loss of aggrecan+ PNN structures appeared to be somewhat more pronounced in the ArcL than the ArcM throughout its rostro-caudal extent. In summary, 1) ZDF rats exhibit stunted Arc PNN content and assembly compared to normoglycemic Wistar rats, and 2) the loss of PNN-associated WFA staining exceeds loss of the underlying aggrecan core protein. Because ZDF and Wistar rats are on different genetic backgrounds, we also analyzed Arc PNNs in non-diabetic Zucker Diabetic Lean (ZDL) rats, which are derived on the same background as ZDF rats. We report that in these animals, Arc PNNs are morphologically indistinguishable from those of Wistar rats (Supplementary Fig. 8), indicating that variation in background strain is unlikely to explain differences in Arc PNNs between normoglycemic Wistar and hyperglycemic ZDF rats.
The WFA-labeled CS glycan component of PNNs are long, linear polysaccharides comprised of repeating disaccharide units of glucuronic acid (GlcA) and N-acetylgalactosamine (GalNAc) (Fig. 2a)9. CS- and dermatan sulfate (DS)-GAG sulfation patterns are often referred to as a “sulfation code”, disruptions of which are associated with CNS disease states9,10. Differential sulfation of the chondroitin sulfate disaccharide unit heavily modulates the function of the underlying CSPG core protein in ways that can influence diffusional properties of the extracellular space11, the degree of matrix stiffness12, and the capacity to bind growth factors that interact with extracellular signaling molecules required for proper neurocircuit development and maintenance (Fig. 2b)13-15. Using a recently-developed liquid chromatography (LC)-tandem mass spectrometry (MS2) technique coupled with multiple reaction monitoring (MRM) analysis (Fig. 2c)16 that enables accurate quantitation of the various CS isomers from fixed brain sections, we show that CS/DS disaccharides isolated from normoglycemic Wistar hypothalamus are comprised mainly of the monosulfated ∆4S-CS isomer, but also include as minor components ∆0S-, ∆6S-, ∆2S6S-, and ∆4S6S-CS disaccharides and dermatan sulfate (Fig. 2d). While the same disaccharide species were also present in the MBH of age-matched ZDF rats, levels of the ∆4S-CS isomer were significantly increased, whereas both ∆6S- and ∆2S6S-CS disaccharides were significantly decreased, with no change in the amounts of either the ∆0S-CS or ∆4S6S-CS disaccharides or dermatan sulfate (Fig. 2d). Changes in the relative abundance of ∆4S- and ∆6S-CS isomers equated to a sharp and significant increase in the 4S/6S CS ratio (Fig. 2e), a key determinant of multiple biological processes including neurocircuit plasticity17. The MBH of ZDF rats is therefore characterized not only by reduced PNN content but also by altered CS/DS-GAG sulfation patterns.
Figure. 2. Diabetic ZDF rats exhibit abnormal hypothalamic CS/DS-GAG sulfation patterns.

(A) Schematic of chondroitin sulfate (CS) and dermatan sulfate (DS) sulfation positions. During biosynthesis, repeating CS disaccharide units can be nonsulfated (0S-S), monosulfated (4S- and 6S-CS), disulfated (2S6S- and 4S6S-CS) or epimerized and disulfated to produce dermatan sulfate (2S4S-DS). (B) The proportion of non-, mono-, and di-sulfated isomers influences the function of the underlying ECM / PNN matrices. (C-E) Isolation of CS/DS disaccharides from hyperglycemic ZDF and normoglycemic Wistar MBH tissue by ChABC can by quantitatively analyzed by LC-MS2 coupled with MRM analysis. (C) Representative MRM analysis of MBH CS/DS disaccharides from a normoglycemic Wistar rat identifies each isomer using specific channels16. The release of hyaluronan (∆HA) by ChABC is also observed in the ∆0S-CS channel. (D) Diabetic ZDF rats exhibit changes in the relative percentages of ∆4S-, ∆6S-, and ∆2S6S-CS that alter the hypothalamic CS/DS-GAG sulfation pattern and the (E) 4S/6S CS ratio compared to age-matched, normoglycemic Wistar controls (n=8 rats/group; mean ± SEM); ****P < 0.0001 versus Wistar controls; Student’s t-test (unpaired, two-sided); (D) ∆4S-CS p=<0.0001, ∆6S-CS p=<0.0001, ∆2S6S-CS p=<0.0001; (E) p=<0.0001. ChABC, Chondroitinase ABC; LC, liquid chromatography; MBH, mediobasal hypothalamus; MRM, multiple reaction monitoring; MS2, tandem mass spectrometry.
As leptin is implicated in the postnatal development of both Arc neurocircuits and Arc PNNs1,18, and since deficient leptin signaling drives the obese, diabetic phenotype of ZDF rats (owing to a leptin receptor mutation), we next sought to determine if the PNN abnormalities we detected in ZDF rats are specific to this animal model or are present in other T2D models. To this end, we first characterized hypothalamic PNN abundance and CS/DS-GAG sulfation patterns in a second rat model of T2D, the HFD/STZ Wistar rat19. Wistar rats were fed a high fat diet (HFD) for 2 wk prior to rendering them moderately diabetic by a single, low-dose injection of streptozotocin (STZ, 30 mg/kg sc). As expected, the HFD/STZ group exhibited stable hyperglycemia over the 24-day study, whereas basal glucose levels did not change in controls receiving citrate buffer rather than STZ (Extended Data Fig. 1a,b). Similar to what we observed in ZDF rats, WFA-labeled CS-GAGs were reduced in both ArcM and ArcL regions of HFD/STZ Wistar rats compared to controls (Extended Data Fig. 1c-e). In contrast, neither aggrecan expression nor CS/DS-GAG sulfation patterns were altered in the HFD/STZ group (Extended Data Fig. 1f). To investigate whether the decrease of Arc aggrecan expression and changes of CS/DS-GAG sulfation patterns observed in ZDF rats are intrinsic features of these animals (e.g., result from deficient leptin signaling), or instead are only found in rats that are hyperglycemic, we compared PNN abundance and composition in cohorts of ZDF rats both prior to (age 5 wk) and well after the onset of hyperglycemia (age 19 wk) (Supplementary Fig. 9a). Here we report that, whereas PNN-associated CS-GAG expression (detected by WFA staining) was reduced in both the ArcM and ArcL of the older, diabetic cohort of ZDF rats compared to the younger, pre-diabetic cohort (Supplementary Fig. 9b-d), no differences of either aggrecan expression or MBH CS/DS-GAG sulfation patterns were detected between the two groups (Supplementary Fig. 9e). Together, these findings indicate that whereas loss of CS-GAG abundance from Arc PNNs is linked to the diabetes phenotype in both ZDF and Wistar rat models of T2D, reduced aggrecan content and aberrant CS/DS-GAG sulfation patterns appear to be intrinsic to PNNs of ZDF rats, and are present before diabetes onset.
In ZDF rats, sustained diabetes remission can be induced by a single icv injection of FGF12−4 via an action localized to the Arc-ME area4, but the mechanisms underlying this response are unknown. To determine if this FGF1 effect is accompanied by reversal of aberrant PNN assembly, we quantified Arc PNN abundance and hypothalamic CS/DS-GAG sulfation patterns 24 d after icv injection of either FGF1 (3 μg) or vehicle in ZDF rats (Fig. 3a). As previously reported3, sustained remission of hyperglycemia was maintained throughout the 24-day study period after icv injection of FGF1, whereas baseline hyperglycemia remained unchanged in vehicle-treated controls. Meanwhile, food intake was markedly but transiently reduced following icv FGF1 injection and returned to baseline levels after 8 d, whereas intake remained unchanged in ad libitum-fed, icv vehicle-treated controls.
Figure 3. Effect of a single icv injection of FGF1 on Arc PNN assembly and composition in ZDF rats.

(A) Schematic depicting time course for central FGF1 treatment and associated PNN matrix assembly and CS/DS-GAG sulfation pattern analysis. Immunofluorescent detection of WFA (CS-GAG) and aggrecan (CSPG) in coronal sections of rat hypothalamus (30 μm) from ZDF rats treated with (B-C) icv vehicle or (D-F) icv FGF1 (3 μg). (B, D) Low-magnification view of Arc PNNs. Scale bar: 100 μm. (C, E-F) Higher-magnification orthogonal views of PNN enmeshments in the Arc imaged with higher laser intensity to maximize sensitivity. Scale bars: (E) 50 μm (C, F) 25 μm. (G-H) Quantification of the mean fluorescence intensity of PNNs averaged from medial hypothalamic sections (−2.2 to −2.8 mm posterior from bregma) for (G) medial Arc and lateral Arc areas and (H) VMN from either icv vehicle or icv FGF1 treated ZDF rats, and normalized to the icv vehicle control group. (I-J) Icv FGF1 normalizes the MBH CS/DS-GAG (I) sulfation pattern and (J) 4S/6S CS ratio compared to icv vehicle controls. Immunofluorescence analyses (A-H) and CS/DS-GAG analyses (I-J) were performed on the same cohort of rats (n=4-7 rats/group; mean ± SEM); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus icv vehicle controls; Student’s t-test (unpaired, two-sided); (G) WFA-ArcM p=0.0091, aggrecan-ArcM p=<0.0001, WFA-ArcL p=0.0436, aggrecan-ArcM p=0.0005; (I) ∆4S-CS p=0.0033, ∆6S-CS p=0.0013, ∆2S6S-CS p=0.0242; (J) p=0.0012. Two separate CS/DS-GAG isolations and mass spectrometry analyses were performed on the same cohort of icv vehicle vs icv FGF1 treated ZDF rats, and results were reproduced. Arc, arcuate nucleus; ArcL, lateral Arc; ArcM, medial Arc; MBH, mediobasal hypothalamus; ME, median eminence; VMN, ventromedial nucleus; 3V, 3rd ventricle.
The effect of icv FGF1 injection to induce sustained blood glucose lowering was associated with significant increases of both ArcM and ArcL PNN content and matrix assembly compared to vehicle controls (Fig. 3b-g). By comparison, neither WFA nor aggrecan staining were altered in the adjacent ventromedial nucleus (VMN) (Fig. 3h), suggesting that the stimulatory effect of FGF1 on PNN assembly is restricted to the Arc region. Furthermore, aberrant hypothalamic CS/DS-GAG sulfation patterns previously observed in hyperglycemic ZDF rats (Fig. 2) were also reversed by icv FGF1 injection, as ∆4S-CS isomer content decreased whereas both the ∆6S- and ∆2S6S-CS isomers increased, again with no change of either ∆0S-CS, ∆4S6S-CS or dermatan sulfate (Fig. 3i), thereby normalizing the 4S/6S CS ratio (Fig. 3j). Thus, both the reduced abundance of PNNs and associated alteration of CS/DS-GAG sulfation patterns observed in the MBH of untreated ZDF rats are effectively reversed in association with normalization of glycemia induced by icv FGF1 injection. To assess both the rapidity with which this FGF1 effect occurs and the extent to which it depends on reversal of hyperglycemia, we measured Arc PNN abundance 24 h after icv injection of FGF1 (3 μg) or vehicle in nondiabetic Wistar rats. Our finding that a robust increase of Arc PNN abundance was detected within 24 h of icv FGF1 injection in these animals (Extended Data Fig. 2) indicates both that FGF1-induced PNN formation is induced rapidly and that this induction is not dependent on normalization of glycemia (since blood glucose levels were not affected).
To test the hypothesis that the action of FGF1 on Arc PNNs contributes to the mechanism underlying sustained glucose lowering, we employed a well-established method for experimentally-induced PNN degradation via targeted microinjection of Chondroitinase ABC (ChABC), a bacterial enzyme that selectively cleaves CS/DS-GAGs into their disaccharide units while leaving other sulfated GAG species (e.g., heparan sulfate) intact20. Since ChABC also releases aggrecan core proteins from the cell surface, it induces both protein and glycan PNN disassembly21. To verify that hypothalamic PNN digestion is induced by ChABC microinjection in vivo, we performed a unilateral microinjection of ChABC targeting the MBH of Wistar rats (17.5 mU in a volume of 175 nL) and compared PNN content between the injected and uninjected sides 4 days later. As expected, WFA+ labeling of CS-GAGs was absent, and stable aggrecan+ PNN structures were reduced from the Arc on the side receiving ChABC, whereas PNNs remained intact on the uninjected side (Supplementary Fig. 10a). PNN loss was accompanied by a dramatic increase of BE-123 staining of CS “stubs” (CS neoepitope cleavage products produced on cleaved CS/DS disaccharides only after ChABC-specific digestion of CS/DS-GAGs22), which serves as a biomarker of ChABC activity. Based on the distribution of CS-stub labeling, we also showed that following a single bilateral microinjection, ChABC activity was spread throughout the full rostro-caudal extent of the Arc-ME junction in ZDF rats (Supplementary Fig. 10b).
To test whether the action of FGF1 on hypothalamic PNN assembly is required for the sustained antidiabetic effect of central FGF1 treatment, we developed and implemented a tri-injection surgical procedure in which age- and glycemia-matched ZDF rats received a single icv injection of FGF1 (3 μg) and during the same procedure underwent bilateral microinjection of either active ChABC (to disassemble PNNs) or vehicle (heat-inactivated ChABC protein control) directed to the Arc. Our finding that the FGF1-induced acute-phase (d1-d8) anorexic response and associated weight loss did not differ between groups (AUCFI1-8 and AUCBW1-8, p ≥ 0.6) indicates that the mechanism driving this anorexia is not dependent on intact Arc PNNs (Fig. 4a,b). Because our previous work shows that the initial phase of FGF1-induced glucose lowering in ZDF rats is driven largely by this anorexic response (since pair-fed controls show comparable lowering of blood glucose over the first 8-10 d)4, we anticipated that the initial phase of glucose lowering would not differ significantly between the two groups and this proved to be the case (Fig. 4c; AUCBG1-8, gray, p ≥ 0.5). However, once food intake returned to near baseline levels, the blood glucose levels began to diverge between treatment groups. Specifically, blood glucose levels increased sharply towards their baseline level of hyperglycemia during the post-acute phase (d10-d28) in ZDF rats receiving active ChABC treatment in conjunction with icv FGF1, whereas FGF1-treated animals receiving the heat-inactivated protein control remained normoglycemic throughout the 28-d study period (Fig. 4c, AUCBG10-28, p = 0.009). These findings indicate that intact Arc PNNs are required for the sustained, food intake-independent, antidiabetic action of FGF1 (but not the acute, transient reduction of intake or glycemia).
Figure 4. Impact of Chondroitinase ABC digestion of Arc PNNs on sustained blood glucose lowering induced by icv FGF1.
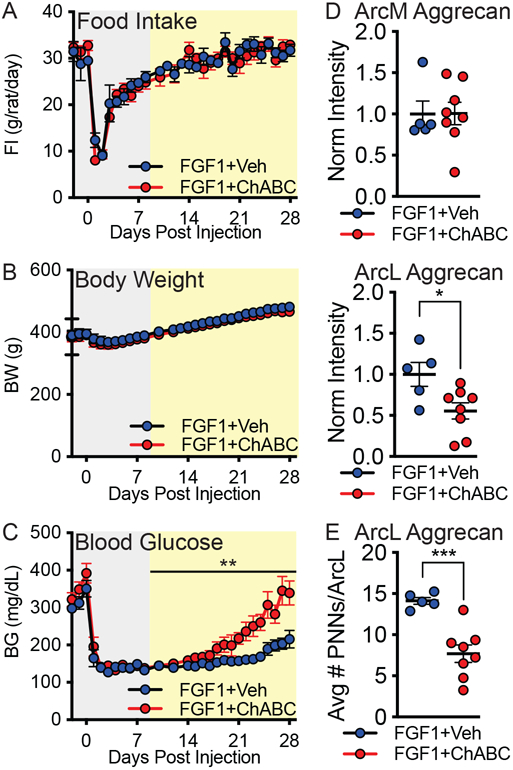
ZDF rats were treated with icv FGF1 targeting the lateral ventricle, followed immediately by a bilateral microinjection of ChABC or heat-inactivated ChABC (vehicle) targeting the MBH within the same surgical session. Levels of (A) food intake, (B) body weight, and (C) blood glucose were measured daily for 28 d following the surgical intervention. Data were analyzed separately for the acute-phase (gray) and post-acute phase (yellow) response to FGF1 treatment, since the former is known to be driven largely by reduced food intake4. (D) Quantification of mean aggrecan fluorescence intensity in medial Arc and lateral Arc regions of the MBH (−2.2 to −2.4 mm posterior from bregma) and normalized to the hypothalamic average for the vehicle control group. (E) Quantification of the total number of aggrecan+ PNN matrices in the lateral Arc region averaged from medial hypothalamic sections. In vivo manipulation (A-C) and immunofluorescence analyses (D-E) were performed on the same cohort of rats (n=5-9 rats/group; mean ± SEM). Statistics: *P < 0.05, **P < 0.01, ****P < 0.0001; (D-E) Student’s t-test (unpaired, two-sided); (D) p=0.0242, (E) p=0.0008. (A-C) Group comparisons were analyzed using AUC and the change over time, and post-acute blood glucose time series data were analyzed using a random intercept and slope linear mixed model (please see Statistical Analysis for complete details); (C) p=0.009. Arc, arcuate nucleus; ArcL, lateral Arc; ArcM, medial Arc; BG, blood glucose; BW, body weight; FI, food intake.
At the conclusion of the study, we found that among ZDF rats in which icv FGF1 injection was paired with intrahypothalamic microinjection of heat-inactivated protein vehicle, Arc PNN structures were normal in appearance. In contrast, hyperglycemia relapse in animals receiving icv FGF1 combined with active ChABC treatment was associated with loss of Arc PNN assembly resembling that of naïve ZDF rats (Supplementary Fig. 11). Extending this finding, reductions of both aggrecan fluorescence intensity (Fig. 4d) and number of aggrecan+ PNN structures (Fig. 4e) was more pronounced in the ArcL than ArcM of these animals. Similarly, we show that in a small cohort of ZDF rats that received icv FGF1 in combination with unilateral ChABC microinjection, ArcL, but not ArcM, PNNs were markedly reduced on the microinjected side, whereas FGF1-induced PNN formation remained intact on the contralateral, non-injected side (Supplementary Fig. 12). Complementing ChABC-induced loss of stable PNN complexes in the MBH was the appearance of CS “stubs”, indicative of successful ChABC targeting.
Taken together, our results show that 1) PNNs are markedly abnormal in both abundance and composition in the Arc of ZDF rats, but not in other brain areas, 2) central administration of FGF1 at a dose that induces sustained diabetes remission corrects these PNN abnormalities, and 3) ChABC-mediated disruption of these PNNs markedly shortens the duration of FGF1-induced diabetes remission. We conclude that ZDF rats are characterized by anomalous PNN assembly specifically in the Arc, and that reversal of this defect is required for the sustained antidiabetic action of FGF1 in these animals. These findings constitute the first direct evidence linking hypothalamic PNNs to the control of glucose homeostasis and identifies these PNNs as novel potential targets for diabetes treatment.
The nature of the PNN abnormality detected in the MBH of ZDF rats warrants comment. This defect is characterized by reduced labeling intensity of PNN structures, rather than their complete absence, which resembles the pattern seen in neuropsychiatric diseases such as schizophrenia23,24, and is quite distinct from the excessive PNN formation that can occur in response to head trauma or stroke10. As PNN loss in the former disease states associates with impaired synaptic connectivity, disruption of inhibitory neural networks, and aberrant synaptic refinement25, a priority for future work is to determine how anomalous PNN assembly impacts Arc neurocircuits in diabetic animals.
Loss of Arc PNNs in ZDF rats was also accompanied by decreased hypothalamic content of the Δ2S6S-CS isomer, which enhances growth factor binding and promotes neurite outgrowth26,27, along with an increase in the Δ4S-CS isomer, a decrease in the Δ6S-CS isomer, and a corresponding increase in the 4S/6S CS ratio. As these alterations were present in pre-diabetic 5-wk old rats as well as in older, diabetic animals, and since they were not detected in the genetically-normal HFD/STZ Wistar rat model of T2D, we suspect that aberrant CS/DS-GAG sulfation patterns observed in the MBH of ZDF rats are manifestations of deficient leptin signaling and are not linked to hyperglycemia per se. This interpretation is consistent with recent evidence that postnatal maturation of Arc PNNs is dependent on an intact leptin signal1. Nevertheless, because this constellation of changes is predicted to both increase matrix stiffness12 and adversely impact neurocircuit formation28, these PNN alterations could potentially impact hypothalamic glucoregulatory neurocircuit development in ways that predispose to hyperglycemia in the adult. Since these compositional changes are reversed by FGF1, studies to elucidate their role in sustained FGF1-induced blood glucose normalization are warranted.
Agrp neurons are prominent among neurons enmeshed by PNNs in the ArcM1, and excessive Agrp neuron activation is a feature of virtually all rodent diabetes models. As reduced melanocortin signaling (which results from Agrp neuron activation) is linked to T2D pathogenesis in humans as well as rodents29,30, it is possible that restored PNN assembly surrounding Agrp neurons contributes to the sustained antidiabetic action of FGF1 by increasing melanocortin signaling. Consistent with this notion is evidence that in mice with diabetes secondary to reduced central melanocortin signaling (Mc4r−/− mice), the duration of diabetes remission induced by icv injection of FGF1 is markedly shortened despite the induction of the expected anorexia31, which closely resembles the impact of Arc PNN digestion on the duration of FGF1-induced glucose lowering in ZDF rats. This evidence that Arc PNNs and melanocortin signaling must both be intact for sustained (but not acute) FGF1-induced glucose lowering supports a model in which Agrp neuron activity is constrained by the PNNs that enmesh them, and that loss of these PNNs predisposes to hyperglycemia by reducing melanocortin signaling.
Whereas Agrp neurons are confined largely to the ArcM, prominent effects of both diabetes and FGF1 on PNNs were observed in the adjacent ArcL region. Specifically, PNN loss in ZDF rat hypothalamus is, if anything, more pronounced in the ArcL than the ArcM region, and ChABC-induced disassembly of PNNs in the hypothalamus of ZDF rats receiving icv FGF1 injection was similarly more prominent in the ArcL than the ArcM. As this PNN disassembly dramatically shortened the duration of FGF1-induced blood glucose lowering, PNN-enmeshed neurons in ArcL emerge as attractive potential targets involved in this effect. Although the identity of these neurons awaits further study, non-Agrp GABAergic neurons are concentrated in the ArcL, and in mice, most of these GABAergic neurons are enmeshed by PNNs1. Moreover, a role for these ArcL non-Agrp GABAergic neurons in leptin control of energy homeostasis has been suggested32,33. Future studies are warranted to further characterize distinct subsets of PNN-enmeshed neurons in the Arc and ascertain their contributions to glucose homeostasis, diabetes pathogenesis and FGF1 action.
Methods
Study animals.
For baseline characterization of hypothalamic PNNs, naïve male Wistar (Hsd:WI, Envigo, Indianapolis, IN) and male ZDF rats (ZDF-Lepfa/Crl, 370 obese, Charles River, Wilmington, MA) were group-housed until 5 weeks or 19 weeks of age in a temperature-controlled room under 12 h: 12 h-light: dark cycle and ad-lib access to Purina 5008 diet (Animal Specialties, Inc., Hubbard, OR) and water. For PNN identification in naïve male ZDL rats (ZDF-Lep+/?, 371 lean, Charles River, Wilmington, MA), rats arrived separately and monitored in parallel housing conditions. For icv FGF1 injection studies, male ZDF rats (6-8 wk of age) were cannulated prior to the onset of hyperglycemia (see Surgery) and allowed to recover for 7 d prior to injection of either FGF1 (3 μg) or saline vehicle into the lateral ventricle immediately following hyperglycemia onset (please see Criteria for Diabetes). For studies in which icv FGF1 treatment was coupled with bilateral microinjection of ChABC targeting the MBH, male ZDF rats arrived at 8 weeks of age and again were monitored until the onset of diabetes and randomly assignment into one of the various treatment groups (detailed below). For the HFD/STZ rat model of T2D, male Wistar rats (age 8 wk) were placed on a high-fat diet prior to induction of stable, moderate hyperglycemia by administering a low dose of streptozotocin (please see STZ/HFD-Wistar rat model of T2D for additional details). All procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington (Seattle, Washington), and all experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments reported are in compliance with the ARRIVE guidelines. Please see Reporting Summary for additional details.
Criteria for diabetes and diabetes remission.
Although ZDF rats tend to develop diabetes between 8-10 wk of age, the degree of hyperglycemia at diabetes onset is variable34. In the current studies, diabetes onset was defined as morning blood glucose values >250 mg/dL lasting for at least 3 consecutive days, and icv injections were consistently given within 5 days of diabetes onset. To ensure that we captured both acute- (anorexia-induced) and post-acute phases of FGF1-induced glucose lowering2-4, morning blood glucose levels were obtained under ad-lib-feeding conditions at least 4 times/wk for a minimum of 24 d following icv FGF1 injection in ZDF rats. Blood glucose levels were measured by glucometer (NovaMax Plus meter) using the tail prick method.
Surgery.
All surgeries were performed under isoflurane anesthesia using the following stereotaxic coordinates for procedures in ZDF rats: lateral ventricle cannulation (LV; 26-ga, Plastics One, Roanoke, VA): −0.8 mm posterior to bregma, 1.5 mm lateral, and −2.6 mm below the skull surface; injection of FGF1 or vehicle into the LV under anesthesia in animals without an indwelling ventricular cannula: −0.7 mm posterior to bregma, 1.8 mm lateral, and −3.8 mm below the skull surface; MBH bilateral microinjection of ChABC (or heat inactivated ChABC protein control): −2.6 mm posterior to bregma, +/− 0.4 mm lateral, and −8.9 mm below the skull surface. For unilateral microinjection into the MBH of Wistar rats: −2.8 mm posterior to bregma, 0.4 mm lateral, and −10.3 mm below the skull surface. Animals received buprenorphine hydrochloride for post-operative pain control (Reckitt Benckiser Pharmaceuticals Inc., Richmond, VA).
Intracerebroventricular (icv) injections.
Rats were monitored for several days to ensure that mean blood glucose values were matched between study groups and fit the Criteria for Diabetes before the animals received the icv injection. For icv injections via the LV cannula in rats, saline control or recombinant mouse FGF1 (mFGF1; Prospec-Tany TechnoGene Ltd, East Brunswick, NJ), dissolved in sterile water at a concentration of 1.5 μg/μL, were injected over 60 s in a final volume of 2 μL using a 33-gauge needle extending 0.8 mm beyond the tip of the icv cannula.
Unilateral and bilateral intraparenchymal microinjections targeting the MBH.
After monitoring for several days to ensure that mean blood glucose values were matched between groups and Criteria for Diabetes was met, rats underwent targeted intraparenchymal microinjection of either active ChABC (17.5 mU) (C3667; Sigma-Aldrich, St. Louis, MO) dissolved in aCSF at a concentration of 10 U/100μL) or heat-inactivated ChABC protein vehicle control (17.5 mU) in a volume of 175 nL. Heat-inactivation was performed at 85°C for 45 min and activity loss was confirmed by in vitro digestion assays. Injections were given at a rate of 1 nL/s using the Nanoject III microinjection system (Drummond Scientific, Broomall, PA) with autoclaved glass-pulled micropipettes. Enzyme concentration and volume were optimized to ensure spread throughout the rostral-caudal extent of the MBH.
STZ/HFD-Wistar rat model of T2D.
After consuming a 60% high-fat diet (HFD; D12492, Research Diets, New Brunswick, NJ), for 2 wk, Wistar rats received either a single sc injection of streptozotocin (STZ; Sigma-Aldrich, St. Louis, MO) at a low dose (30 mg/kg body weight in a volume of 250 μL) to induce stable, moderate hyperglycemia (~300-400 mg/dL). Nondiabetic Wistar control rats were maintained on a standard chow diet and received sc vehicle (0.1 M citrate buffer; pH 4.4)19. Measurements of blood glucose levels and body weight were recorded throughout the study.
Paraformaldehyde rat brain processing.
Rats were anesthetized with ketamine and xylazine and perfused with 0.1 M PBS followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. Brains were extracted, post fixed for 48 h in 4% PFA at 4°C, cryopreserved in 30% sucrose PBS solution, and frozen in optimal cutting temperature (OCT) compound on dry ice. Brains were stored at −80°C degrees until cryosectioning.
Post-mortem human brain tissue processing.
A cohort of human brains (ages 23, 56, 59, 63, and 65 years old, with post-mortem intervals of 8, 10, 13, 4, and 18 h, respectively) were collected at autopsy. Multiple whole-mount blocks were dissected from the hypothalamus along the third ventricle and from the visual cortex at the occipital pole, and these blocks were immersion-fixed in 4% PFA at 4°C for 24-48 h. After extensive washing in PBS, the blocks were cut on a vibratome (Leica VT1200S) into coronally-oriented 50 μm floating sections and stained following the same procedures used for rat brain processing. All specimens were collected with informed consent and in ethical compliance with the St. Joseph’s Hospital and Medical Center Committee on Human Research (Institutional Review Board approval no. 10BN159). Please see Reporting Summary for additional details.
Brain cryosectioning.
Rat and human brain sections were acclimated at −20°C overnight and cut with a Leica CM1950 cryostat at 30 μm (rat) or 50 μm (human) thick serial coronal sections, divided into 6-well dishes, and stored in 0.1 M PBS + 0.02% sodium azide at 4°C as free-floating sections before processing.
Immunofluorescent labeling, confocal microscopy, and mean fluorescence intensity quantifications.
30 μm-thick sections of rat brain and 50 μm-thick sections of human brain were studied using antigen retrieval in 10 mM trisodium citrate (pH 8) and heated at 85°C for 20 min. Immunostaining was performed according to published methods1. Briefly, free-floating tissue sections were permeabilized for 25 mins at RT in 0.1 M PBS + 0.2% Triton X-100 and in blocked for 2 h at 37°C in 0.1 M PBS + 0.05% Triton X-100 (PBST) + 10% normal serum (Jackson ImmunoResearch, West Grove, PA). Sections were then incubated overnight at 4°C using 1:1,000 dilution of biotin labeled Wisteria floribunda agglutinin (WFA) (L1516; Sigma-Aldrich, St. Louis, MO), aggrecan (AB1031, Millipore, Burlington, MA), chondroitin-4-sulfate “stub” (MAB2030, Millipore, Burlington, MA), 1:250 of HAPLN-1 (AF2608, R&D, Minneapolis, MN), 1:50 tenascin R (sc-376341, Santa Cruz Biotechnology, Dallas, TX) or 1:1,000 of parvalbumin (AF5058, R&D, Minneapolis, MS) antibodies in PBST + 1% donkey serum. The following day, the sections were washed and incubated for 2 h at RT in 1:1,000 secondary antibodies in PBST + 1% serum. For studies investigating PNN changes in naïve and FGF1 treated rodent cohorts, parvalbumin was labeled with donkey anti-sheep 488 (A11015, Invitrogen, Carlsbad, CA), aggrecan was labeled with donkey anti-rabbit 555 (A31572, Invitrogen, Carlsbad, CA) and WFA was labeled with Streptavidin conjugate 647 (S32357, Invitrogen, Carlsbad, CA). Labeling was confirmed by repeating the staining using donkey anti-sheep 488 (A11015, Invitrogen, Carlsbad, CA), Streptavidin conjugate 555 (S32355, Invitrogen, Carlsbad, CA) and donkey anti-rabbit 647 (A31573, Invitrogen, Carlsbad, CA), or Streptavidin conjugate 488 (S32354, Invitrogen, Carlsbad, CA) and donkey anti-rabbit 594 (A21207, Invitrogen, Carlsbad, CA). For studies investigating ChABC targeting, aggrecan was labeled with donkey anti-rabbit 488 (A21206, Invitrogen, Carlsbad, CA), CS-stubs were labeled with donkey anti-mouse 555 (A31570, Invitrogen, Carlsbad, CA), and WFA was labeled with Streptavidin conjugate 647 (S32357, Invitrogen, Carlsbad, CA). For studies investigating changes in TnR and HAPLN-1, sectioned were stained with aggrecan using donkey anti-rabbit 488 (A21206, Invitrogen, Carlsbad, CA), TnR with donkey anti-mouse 555 (A31570, Invitrogen, Carlsbad, CA) and HAPLN-1 with donkey anti-goat 647 (A21447, Invitrogen, Carlsbad, CA). For human tissue staining, aggrecan was labeled using goat anti-rabbit 488 (A11008, Invitrogen, Carlsbad, CA) and WFA using Streptavidin conjugate 568 (S11226, Invitrogen, Carlsbad, CA). Sections were then counterstained for DAPI, mounted, and cover slipped using Floromount-G (4958-02, ThermoFisher, Waltham, MA). Confocal microscopy was performed with Nikon A1R HD confocal objectives (4X, 10X and 20X) and fluorescent microscopy was performed with Zeiss Axio Observer 7 (ZEN 2.3 pro) objectives (4x). Please see Reporting Summary for additional details.
To quantify PNN abundance, mean fluorescence intensity of PNNs was assessed using an established method for PNN quantification35 and applied using a stereological approach. We first subdivided the Arc into three regions for analysis: 1) medial Arc (Arc-M), 2) lateral Arc (Arc-L), and 3) total Arc (Arc-M+L), with ArcM and ArcL boundaries defined based on the anatomical location of Arc neuronal populations of interest (e.g. POMC, NPY)36,37. Whereas the Arc-M region is located 0.0-0.5 mm AP, the Arc-L region is located 0.5-1.2mm AP and lies lateral to the Arc-M region and extends into both the lateral Arc and retrochiasmatic areas (which we have defined as Arc-L for brevity). From each defined region of interest, we then subtract a constant background and quantified the mean fluorescence intensity (per mm2). Since PNN abundance varies throughout the rostro-caudal extent of the hypothalamus (Supplemental Figure 1), we computed the averaged normalized intensity for PNNs by first normalizing PNN abundance to the controls for each region separately and then averaging the normalized changes for each stereological region. Data were blinded, and images were processed for mean fluorescence intensity using Fiji open source imaging software38.
Hypothalamic CS/DS-GAG digestion and disaccharide isolation.
CS/DS disaccharides were isolated and quantified according to our previously published methods16. MBH sections were isolated from 30 μm-thick PFA-fixed brain sections restricted to −1.6 to −4.2 mm from Bregma. Fixed hypothalamic sections were washed 3 x in Optima LC/MS-grade water and 1 x in 50 mM ammonium bicarbonate (pH 7.6) at room temperature (RT). Chondroitinase ABC digestion of CS/DS-GAGs was performed using 500 mU/ mL of ChABC (C3667, Sigma-Aldrich, St. Louis, MO) in 50 mM ammonium bicarbonate (pH 7.6) in a Thermo Scientific MaxQ4000 orbital shaker at 80 rpm, 37°C, for 24 h. Supernatants were collected in sterile 1.7 mL microcentrifuges tubes and spun for 10 min at 14 k x g to pellet any debris. The supernatant was then dehydrated using a SpeedVac Concentrator and the lyophilized product was reconstituted in 30 μL of LC/MS-grade water.
LC-MS2 + MRM quantification of CS/DS disaccharides.
CS/DS samples were analyzed using a triple quadrupole mass spectrometer equipped with an electrospray ion source (Waters Xevo TQ-S) operated in negative mode and coupled to a Waters Acquity I-class ultra-high pressure liquid chromatography system (UPLC). Disaccharides were resolved by porous graphitic chromatography (Hypercarb column; 2.1 × 50 mm, 3 μm; ThermoFisher) as described previously16,39. Assigned MRM channels: Δ4S-CS (CS-A) m/z 458 > 300, Δ4S6S-CS (CS-E) and Δ2S4S-DS (CS-B) m/z 538 > 300, Δ0S-CS (CS-O) and ΔHA m/z 378 > 175, Δ6S-C S (CS-C) m/z 458 > 282, and Δ2S6S-CS (CS-D) m/z 268 > 282. MassLynx software version 4.1 (Waters) was used to acquire and quantify all data. Under the conditions described above, the ratios between peak areas produced from equimolar CS standard runs were normalized to the highest peak intensity and relative quantification of each CS isomer within a samples was achieved using a modified peak area normalization function16,40. Each CS/DS isomer was expressed as a relative percent of the total CS isomer composition within a brain sample.
Statistical analyses.
For each study, treatment groups were matched for sex, age, body weight and blood glucose levels. Sample size calculations (G*Power3.1) yielded estimates of 4-10/group predicated on ~80% power at alpha <0.05, 2-tailed, an end-of-treatment group glucose difference of 100 mg/dL, a pooled SD of 55, and an allocation ratio of up to 2. Pairwise comparisons involving immunofluorescent outcomes met the equality of variance assumption based on non-significant F-tests and were thus conducted using Student’s t-tests (unpaired, two-sided) in GraphPad Prism® 7.0 (Graph Pad Software, Inc., La Jolla, CA). Planned comparisons of outcomes measured at single time-points were analyzed using Student’s t-tests (unpaired, two-sided), and Extended Data 1 group by time outcomes were analyzed using linear mixed-effects analysis with Geisser-Greenhouse correction. Error bars represent the standard error of the mean (SEM). Investigators were not blinded to study conditions during the study. Investigators were blinded to study conditions during quantitative immunofluorescent analyses.
Comparison of blood glucose levels over time in icv FGF1-treated ZDF rats receiving intrahypothalamic injection of either ChABC or heat-inactivated ChABC control.
Because blood glucose declined to its lowest values between d1 and d8 in both groups, and because this initial drop in glycemia is known to result largely from the effect of icv FGF1 injection to reduce food intake in ZDF rats4, the acute (d1-d8) and post-acute (d10-d28; measurements not made on d9) phases of glucose lowering were analyzed separately. Group comparisons were made based on both the blood glucose (BG) area under the curve (AUC, computed using the trapezoidal rule41) and change of blood glucose levels over time. Group comparisons of body weight (BW) and food intake (FI) were likewise compared using AUCs, while baseline values were compared using means. Pairwise data were evaluated for equality of variance using the F-test, which was significant only for AUCBG; hence the Welch t-test was applied for that outcome because it is robust to unequal variance when the sample size differs42. To model the time series data and assess group differences in BG at discrete time points during the post-acute phase of the study, the data were analyzed using a random intercept and slope linear mixed model41; the fixed effects were group, time, time2, the group × time interaction, and the group × time2 interaction. This model was adopted because blood glucose rose in a curvilinear fashion during the post-acute phase; likelihood ratio goodness-of-fit tests comparing it to competing modelsl41; and examination of regression diagnostics. Coefficient estimates and significance tests of group differences at discrete post-acute time-points were computed by centering the interaction terms on the day in question41,42. AUC and regression analyses were performed using R (R Core Team (2016). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/). Please see Reporting Summary for additional details.
Extended Data
Extended Data Fig. 1. Diabetic Wistar rats have reduced PNN CS-GAG structures in the arcuate nucleus.
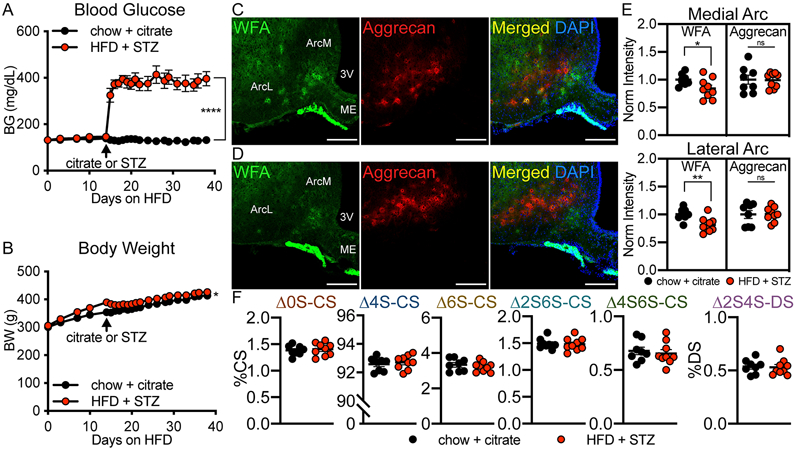
Wistar rats were rendered diabetic by 2 wk of high fat diet (HFD) followed by a single injection of low-dose streptozotocin (STZ) (30 mg/kg sc) or citrate control and (A) blood glucose and (B) body weight were monitored for 24 d after treatment. Immunofluorescent detection of WFA (PNN CS-GAGs) and aggrecan (PNN CSPG) in coronal sections of rat hypothalamus (30 μm) from (C) normoglycemic citrate Wistar controls and (D) agematched, hyperglycemic HFD/STZ Wistar rats. Scale bar: 200 μm. (E) Quantification of the mean fluorescence intensity of PNNs averaged from medial hypothalamic sections (−2.2 to −2.8 mm posterior from bregma) for ArcM and ArcL areas from citrate and HFD/STZ Wistar rats and normalized to the citrate control mean fluorescence intensity averages. (F) HFD/STZ Wistar rats exhibit no change in the relative percentages of CS disaccharides compared to age-matched, citrate controls. In vivo manipulation (A-B), immunofluorescence analyses (C-E), and CS/DS-GAG LC-MS2 analyses were performed on the same cohort of rats (n=8-9 rats/group; mean ± SEM). *P < 0.05, **P < 0.01, ****P < 0.0001, versus citrate controls; (A-B) Linear mixed-effects model analysis with Geisser-Greenhouse correction, and (E-F) Student’s t-test (unpaired, two-sided); (A) p=<0.0001, (B) p=0.0364, (E) WFA, ArcL p=0.0061; WFA, ArcM p=0.0467. Arc, arcuate nucleus; ArcM, medial Arc; ArcL, lateral Arc; ME, median eminence; sc, subcutaneous; 3V, 3rd ventricle.
Extended Data Fig. 2. Effect of a single icv injection of FGF1 on Arc PNN assembly in Wistar rats.
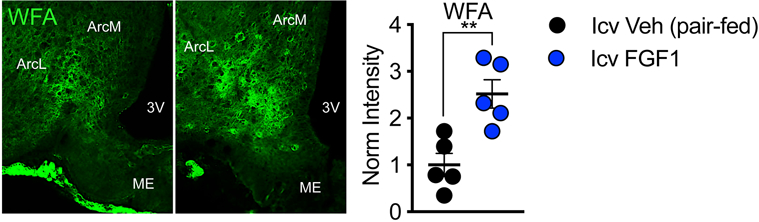
Immunofluorescence imaging of Arc PNN CS-GAGs in Wistar rats 24 h after icv treatment with FGF1 (3 μg) or vehicle, where the vehicle treated controls were pair-fed to the FGF1 group. Quantification of the mean fluorescence intensity of PNNs averaged from medial hypothalamic sections for total Arc (Arc-M+L) areas (n=5 rats/group; mean ± SEM). **P < 0.01, compared to icv vehicle controls; Student’s t-test (unpaired, two-sided); (A) p=0.0046. Arc, arcuate nucleus; ME, median eminence; 3V, 3rd ventricle.
Supplementary Material
Acknowledgements
The authors are grateful for the technical assistance provided by Trista Harvey, Miles Matsen, Nick Acharya, Hong Nguyen and Bao Anh Phan. Histochemical work was supported by Cellular and Molecular Imaging Core (CMIC) and funding to KMA and JMS by the UW Diabetes Research Center (DRC) National Institute of Diabetes and Digestive and Kidney Diseases (NIH-NIDDK) sponsored grant P30DK017047. Mass spectrometry work was supported by University of Washington School of Pharmacy Mass Spectrometry Center. This work was also supported by NIH-NIDDK grants: F32DK122662 (KMA), R01DK101997 (MWS), R01DK089056 (GJM), R01DK083042 (GJM and MWS), and K08DK114474 (JMS), the National Institute of General Medical Sciences (NIH-NIGMS) grant R01GM127579 (MG), the National Institute on Aging (NIH-NIA) grant T32AG052354 (AFL), the National Institute of Neurological Disorders and Stroke Neurosurgeon Research Career Development Program (NRCDP) K12 Award K12NS080223 (ZM), the Department of Defense Peer Reviewed Medical Research Program W81XWH-20-1-0250 (ZM), and the Barrow Neurological Foundation Grant No. 18-0025-30-05 (ZM). Work also supported by the NIH-NIDDK Diseases-funded Nutrition Obesity Research Center (NORC) P30DK035816. Additional funding to support these studies was provided to JMS by the UW Royalty Research Fund (RRF A139339). Funding was also provided to MWS by Novo Nordisk (CMS-431104) and to MAB by the Novo Nordisk Foundation (NNF17OC0024328) and the Novo Nordisk Foundation Center for Basic Metabolic Research, which is an independent research center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (NNF10CC1016515). Some figures were created with assistance from BioRender.com.
Footnotes
Conflict of Interest: Support to M.W.S. was partly funded by Novo Nordisk A/C. All other authors declare no conflict of interest.
Data Availability.
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Mirzadeh Z, et al. Perineuronal net formation during the critical period for neuronal maturation in the hypothalamic arcuate nucleus. Nat Metab 1, 212–221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scarlett JM, et al. Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat Med 22, 800–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scarlett JM, et al. Peripheral Mechanisms Mediating the Sustained Antidiabetic Action of FGF1 in the Brain. Diabetes 68, 654–664 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown JM, et al. The Hypothalamic Arcuate Nucleus-Median Eminence is a Target for Sustained Diabetes Remission Induced by Fibroblast Growth Factor 1. Diabetes, db190025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blumcke I, Eggli P & Celio MR Relationship between astrocytic processes and “perineuronal nets” in rat neocortex. Glia 15, 131–140 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Schwartz MW, Woods SC, Porte D Jr., Seeley RJ & Baskin DG Central nervous system control of food intake. Nature 404, 661–671 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Morton GJ & Schwartz MW The NPY/AgRP neuron and energy homeostasis. Int J Obes Relat Metab Disord 25 Suppl 5, S56–62 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Morawski M, Bruckner G, Arendt T & Matthews RT Aggrecan: Beyond cartilage and into the brain. Int J Biochem Cell Biol 44, 690–693 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Soares da Costa D, Reis RL & Pashkuleva I Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu Rev Biomed Eng 19, 1–26 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Testa D, Prochiantz A & Di Nardo AA Perineuronal nets in brain physiology and disease. Semin Cell Dev Biol 89, 125–135 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Hrabetova S, Masri D, Tao L, Xiao F & Nicholson C Calcium diffusion enhanced after cleavage of negatively charged components of brain extracellular matrix by chondroitinase ABC. J Physiol 587, 4029–4049 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Properzi F, et al. Chondroitin 6-sulphate synthesis is up-regulated in injured CNS, induced by injury-related cytokines and enhanced in axon-growth inhibitory glia. Eur J Neurosci 21, 378–390 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Djerbal L, Lortat-Jacob H & Kwok J Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj J 34, 363–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizumoto S, Yamada S & Sugahara K Molecular interactions between chondroitin–dermatan sulfate and growth factors/receptors/matrix proteins. Current Opinion in Structural Biology 34, 35–42 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Li Q, et al. Impaired Cognitive Function and Altered Hippocampal Synaptic Plasticity in Mice Lacking Dermatan Sulfotransferase Chst14/D4st1. Front Mol Neurosci 12(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alonge KM, et al. Quantitative analysis of chondroitin sulfate disaccharides from human and rodent fixed brain tissue by electrospray ionization-tandem mass spectrometry. Glycobiology 29, 847–860 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata S, Komatsu Y, Yoshimura Y, Taya C & Kitagawa H Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci 15, 414–422, s411–412 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Bouret SG, Draper SJ & Simerly RB Trophic action of leptin on hypothalamic neurons that regulate feeding. Science (New York, N.Y.) 304, 108–110 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Srinivasan K, Viswanad B, Asrat L, Kaul CL & Ramarao P Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res 52, 313–320 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Hamai A, et al. Two distinct chondroitin sulfate ABC lyases. An endoeliminase yielding tetrasaccharides and an exoeliminase preferentially acting on oligosaccharides. J Biol Chem 272, 9123–9130 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Miyata S & Kitagawa H Mechanisms for modulation of neural plasticity and axon regeneration by chondroitin sulphate. J Biochem 157, 13–22 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Bruckner G, et al. Acute and long-lasting changes in extracellular-matrix chondroitin-sulphate proteoglycans induced by injection of chondroitinase ABC in the adult rat brain. Exp Brain Res 121, 300–310 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Enwright JF, et al. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology 41, 2206–2214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mauney SA, et al. Developmental Pattern of Perineuronal Nets in the Human Prefrontal Cortex and Their Deficit in Schizophrenia. Biol Psychiatry 74, 427–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitanihirwe BKY & Woo T-UW Perineuronal nets and schizophrenia: The importance of neuronal coatings. Neurosci Biobehav Rev 45, 85–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadanaka S, Clement A, Masayama K, Faissner A & Sugahara K Characteristic Hexasaccharide Sequences in Octasaccharides Derived from Shark Cartilage Chondroitin Sulfate D with a Neurite Outgrowth Promoting Activity. J Biol Chem 273, 3296–3307 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Clement AM, et al. The DSD-1 carbohydrate epitope depends on sulfation, correlates with chondroitin sulfate D motifs, and is sufficient to promote neurite outgrowth. J Biol Chem 273, 28444–28453 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Koser DE, et al. Mechanosensing is critical for axon growth in the developing brain. Nat Neurosci 19, 1592–1598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi B, et al. Common polymorphism near the MC4R gene is associated with type 2 diabetes: data from a meta-analysis of 123,373 individuals. Diabetologia 55, 2660–2666 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Ellacott KL & Cone RD The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond B Biol Sci 361, 1265–1274 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bentsen MA, et al. Transcriptomic Analysis Links Diverse Hypothalamic Cell Types to Fibroblast Growth Factor 1-induced Sustained Diabetes Remission. Nat Commun 11, 4458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim ER, et al. Hypothalamic Non-AgRP, Non-POMC GABAergic Neurons Are Required for Postweaning Feeding and NPY Hyperphagia. J Neurosci 35, 10440–10450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, O’Brien WG III, Lee C-C, Myers MG Jr. & Tong Q Role of GABA Release From Leptin Receptor-Expressing Neurons in Body Weight Regulation. Endocrinology 153, 2223–2233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 34.Wang X, et al. Variability in Zucker diabetic fatty rats: differences in disease progression in hyperglycemic and normoglycemic animals. Diabetes Metab Syndr Obes 7, 531–541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slaker ML, Harkness JH & Sorg BA A standardized and automated method of perineuronal net analysis using Wisteria floribunda agglutinin staining intensity. IBRO Reports 1, 54–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elias CF, et al. Leptin Differentially Regulates NPY and POMC Neurons Projecting to the Lateral Hypothalamic Area. Neuron 23, 775–786 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Elias CF, et al. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol 423, 261–281 (2000). [PubMed] [Google Scholar]
- 38.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osago H, et al. Quantitative analysis of glycosaminoglycans, chondroitin/dermatan sulfate, hyaluronic acid, heparan sulfate, and keratan sulfate by liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal Biochem 467, 62–74 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Yu S, et al. A rapid and precise method for quantification of fatty acids in human serum cholesteryl esters by liquid chromatography and tandem mass spectrometry. J Chromatogr B 960, 222–229 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Fitzmaurice GM, Laird NM & Ware JH Applied Longitudinal Analysis. John Wiley & Sons, Inc. Hoboken, New Jersey: (2004). [Google Scholar]
- 42.Zar J Biostatistical analysis. Third Ed. Upper Saddle River, New Jersey: Prentice Hall, 1996. (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.