

Abstract
Purpose:
Gene-expression-based molecular subtypes of high-grade serous tubo-ovarian cancer (HGSOC), demonstrated across multiple studies, may provide improved stratification for molecularly targeted trials. However, evaluation of clinical utility has been hindered by non-standardized methods which are not applicable in a clinical setting. We sought to generate a clinical-grade minimal gene-set assay for classification of individual tumor specimens into HGSOC subtypes and confirm previously reported subtype-associated features.
Experimental Design:
Adopting two independent approaches, we derived and internally validated algorithms for subtype prediction using published gene-expression data from 1650 tumors. We applied resulting models to NanoString data on 3829 HGSOCs from the Ovarian Tumor Tissue Analysis Consortium. We further developed, confirmed, and validated a reduced, minimal gene-set predictor, with methods suitable for a single patient setting.
Results:
Gene-expression data was used to derive the Predictor of high-grade-serous Ovarian carcinoma molecular subTYPE (PrOTYPE) assay. We established a de facto standard as a consensus of two parallel approaches. PrOTYPE subtypes are significantly associated with age, stage, residual disease, tumor infiltrating lymphocytes, and outcome. The locked-down clinical-grade PrOTYPE test includes a model with 55 genes that predicted gene-expression subtype with >95% accuracy that was maintained in all analytical and biological validations.
Conclusions:
We validated the PrOTYPE assay following the Institute of Medicine guidelines for the development of omics-based tests. This fully defined and locked-down clinical-grade assay will enable trial design with molecular subtype stratification and allow for objective assessment of the predictive value of HGSOC molecular subtypes in precision medicine applications.
Introduction:
Anatomy and histopathology have been the foundations of cancer classification for more than a century, but both are now complemented by objective assessment of underlying molecular features of disease.(1–8) The development of microarray-based gene-expression profiling of high grade serous tubo-ovarian carcinoma (HGSOC) (9,10) raised expectations for rapid advances in classification, prognostication and prediction in this most common histotype (~70%) of ovarian carcinoma, the deadliest gynecological malignancy.(11,12)
Previous studies identified four phenotypically distinct expression-based HGSOC molecular subtypes.(9,10,13–18) These subtypes have been repeatedly reproduced, with broad similarities in composite pathological characteristics. The C1/Mesenchymal (C1.MES) subtype is characterized by a desmoplastic stroma, high expression of extracellular matrix components, and poor outcomes compared to other HGSOCs; which is consistent with other solid tumors with highly desmoplastic stroma.(19–23) The C2/Immunoreactive (C2.IMM) subtype is dominated by intratumoral CD3+/CD8+ cellular infiltration, inflammatory cytokine expression, and generally more favorable outcomes. The C4/Differentiated (C4.DIF) subtype is characterized by high expression of CA125/MUC16, a subset of immuno-modulatory cytokines, modest lymphocyte infiltration, and clinical outcome indistinguishable from C2.IMM.(10,15,17,24) Finally, the C5/Proliferative (C5.PRO) are depleted for both stromal and immune elements, overexpress onco-fetal and stem cell-associated genes(24), and have unfavorable outcomes.(13–15,17,18).
Unlike modern histotype classification of ovarian carcinoma,(12,25) no agreed-on gold standard exists to define expression-based HGSOC molecular subtypes. Both analytical methods and data used for subtype assignment are fragmented, differing in algorithms and specific genes used, each defining its own brand of subtype. No methods discussed to date provide a workflow with compatibility for fixed/archival tissues that are the mainstay of modern pathology laboratories. Thus, the potential of gene-expression subtype information to guide patient management remains unrealized.(12,26)
Our motivation for the current project was driven by limitations of previous attempts, that contributed to low uptake of HGSOC subtyping in translational research and clinical trials. To optimize clinical uptake, a classification scheme needs to be cost-effective, compatible with available clinical specimens (i.e. formalin-fixed paraffin embedded; FFPE), and be technically reproducible on single patient samples. Prior methods have relied on normalization and unsupervised clustering of array based data, requiring a cohort of samples.(9,10,13–15,17,18,24,27) With few exceptions,(18) prior studies defaulted to a single method or single dataset to train models. Finally, no prior approach reviewed histotype based on the current diagnostic standards for HGSOC, which has significantly altered over the last decades, and may have contributed to significant contamination of historic datasets with non-HGSOC specimens.(28–30)
Using newly curated, previously published array data, and clinically annotated HGSOC specimens from the Ovarian Tumor Tissue Analysis (OTTA) consortium, we propose and validate(26) a Predictor of high-grade serous Ovarian carcinoma molecular subTYPE (PrOTYPE) that recapitulates previously derived gene-expression based molecular subtypes using a minimal set of genes (Figure 1). To ensure clinical applicability we adopted the NanoString platform, a highly automated processing method with tolerance to degraded RNA, typical of fixed tissue that are the mainstay of modern hospital pathology laboratories. Similar multi-gene predictors using NanoString are already in the clinic (31–34) and methods to enable single-sample analytical approaches are well established,(35) tailored to the patient-at-a-time delivery of care that is a necessity for precision medicine. The PrOTYPE assay will enable evaluation of the clinical utility of HGSOC gene-expression molecular subtypes, such as response to targeted therapies that are already emerging with a potential need for subtype information.(36)
Figure 1:
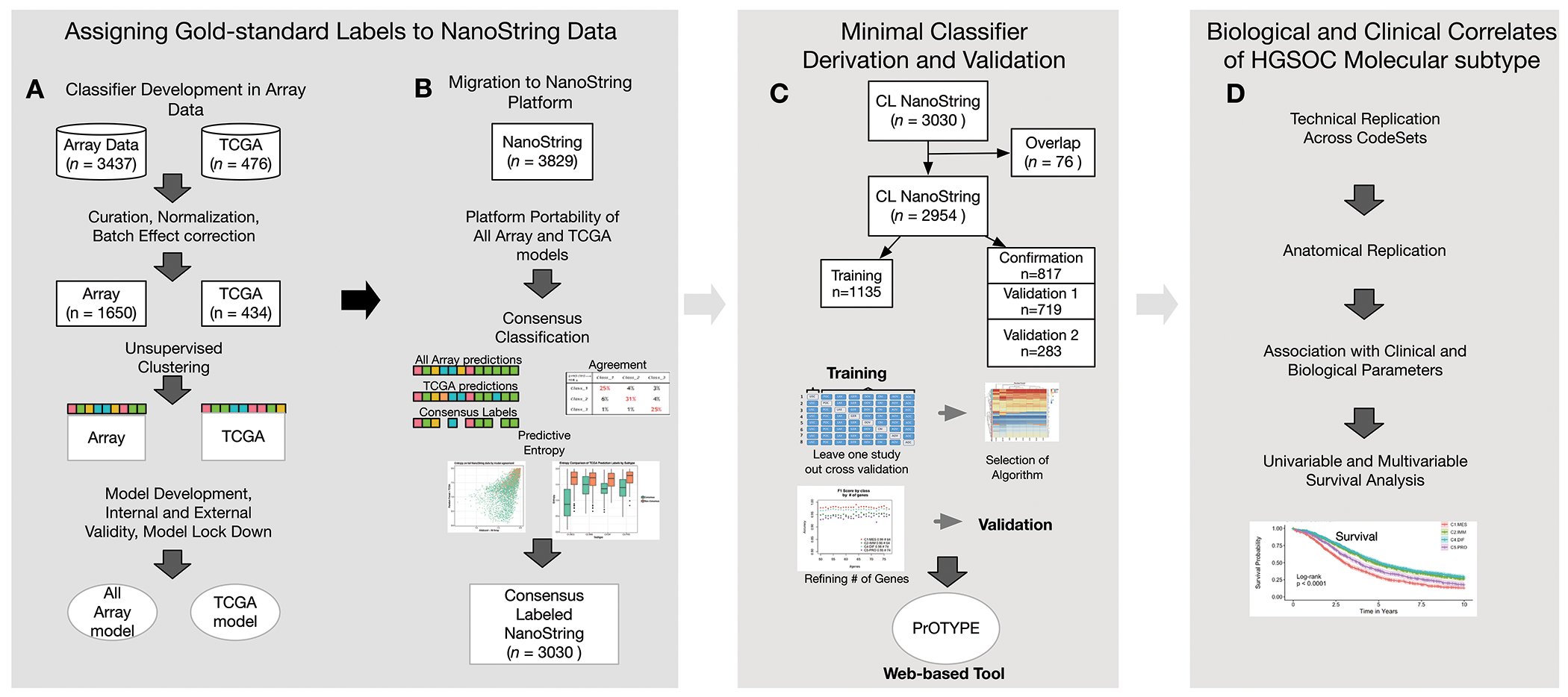
A schematic representation of the process we followed to obtain a final, clinical-grade classifier for HGSOC. Please note that the schematics above are for orientation only and are not intended to be interpreted. In the first panel we outline how de facto subtype labels were assigned to NanoString data, starting with (A) two parallel approaches to build models from array data, and (B) applying the resulting final models onto the NanoString dataset, where the consensus of the two methods became the de facto gold standard with 79% (n=3030) of our total NanoString cohort having agreement, consensus label (CL). In the second panel, (C) we provide the framework used to derive a minimal gene set classifier using the CL NanoString data after removing samples that overlapped both the NanoString and Array datasets (overlap n=76). Finally, in (D) a synopsis of the biological and clinical correlates that were investigated to confirm the biological validity of gene-expression based subtypes compared to previous work.
Methods
OTTA Consortium NanoString Study
We retrospectively analyzed FFPE tumors and clinical data from 20 OTTA consortium studies with available clinical, pathological, demographic features, and survival outcomes (Supplement A.1–A.3). Inclusion criteria (including approval through institution-specific research ethics boards), individual study settings, dates of accrual, and follow-up are described in Table SA1. Studies were asked to contribute adnexal-sourced specimens, though others were accepted when anatomical sites was defined. Expert gynecologic pathologists reviewed samples from hematoxylin and eosin (H&E) stained sections, confirmed HGSOC diagnosis(29), and marked specimens for removal non-involved organ tissues but retained infiltrating stroma.
NanoString Gene Selection and Data Processing
A NanoString CodeSet included 513 genes (plus 5 housekeeping genes), relevant for gene-expression subtyping and selected prior to beginning the analysis. We included top-ranking differentially expressed, subtype-specific genes based on prior reports;(9,10) previous supervised learning of subtype classification;(37) and manual review of literature to identify genes in commonly cited molecular pathways associated with subtype.(9,10,13,15,24) Additional genes were selected from a meta-analysis for their prognostic value and other specific hypothesis (75). To ensure representation from across the transcriptome, we tagged and included additional genes from 99%-correlated gene-expression clusters derived from previous reports, if clusters did not already have representation.(9,10,38,39)
We extracted RNA and ran NanoString assays at three sites (in Vancouver, Los Angeles, and Melbourne), as described previously.(35) We included three regularly assayed RNA reference specimens (Pool1, Pool2, Pool3) to monitor technical bias, allow for comparison of NanoString CodeSet synthesized in different lots, and integrate a single-patient data normalization strategy.(35) Additional description is in Supplement A.4–A.7; data can be found in NCBI GEO Accession GSE135820
Subtype Labels Assignment to NanoString Data
There is presently no definitive standard for gene-expression based subtypes, therefore we derived a de facto standard through application of two parallel approaches, led by independent teams (Figure 1A–B). One approach, denoted All array, aggregated gene-expression datasets to take advantage of broad sample representation and increased statistical power. The other, denoted TCGA, was conservative with respect to potential loss of signal associated with post-hoc batch correction and used the largest, optimally batch-corrected dataset(9). See also Supplement B.
All array:
One team curated data to retain only HGSOC specimens from historical datasets(30), and datasets with greater than 40 remaining unique HGSOC. This reduced 49 potential studies (n=3437) to 1650 unique HGSOC from 14 studies (Table SB1).(9,10,40–50) Individual samples where data was also available from NanoString assays were excluded (Figure SB1). The team combined and batch corrected 11/14 array studies (training 1), and used an ensemble of nine clustering algorithms(51) to re-establish previously recognized subtypes. They next restricted the data to pre-selected NanoString genes also present in all array platforms (454/513 possible NanoString genes), trained and evaluated nine supervised learning algorithms using a bootstrap approach.(52) The top five algorithms were retained and validated on the remaining three (3/14) array studies (confirmation1) with a final selection based on how well predicted subtypes correlated with previously published signatures.(13,24) The tree-based ensemble classification algorithm (AdaBoost) was selected.
TCGA:
Another team curated the TCGA data using the same criteria described above and using data and TCGA-published subtype labels,(9) retaining 434 unique HGSOC (Figure SB1). They next trained and evaluated five different supervised learning algorithms, as above using NanoString gene-restricted data (438/513 genes), using five-fold cross validation, selecting random forest. This approach was validated externally on originally published dataset and labels from Tothill et al.(10)
Minimal Gene Set Classifier
We used the above two approaches to label 3829 NanoString samples and retaining only samples with concordant labels, denoted the consensus labels (CL). We discarded previous models and started anew to rederive a minimal gene set classifier using NanoString data. Sample were randomly partitioned from the dataset into three independent groups on a per study basis: a training set (8 studies), a confirmation set (5 studies), and a validation (4 studies). A fourth partition/second validation (3 studies), comprised of clinical trial cohorts, and was set aside to validate any modifications to the predictive model after confirmation(26,53) (Figure 1C; Figure SA1). See also supplement C.
We adopted a leave-one-study-out cross-validation approach and assessed performance of three algorithms (LASSO, random forest and AdaBoost) in recovering the CL. We removed one study at a time and bootstrapped the remaining seven (500 repetitions) to train a full model that uses all the genes to predict subtype. For each bootstrap sample, we ranked the genes based on the aggregated Gini coefficients, for Random Forest and Adaboost,(54) or the proportion of non-zero coefficients for Lasso. We then ranked genes overall on the proportion of times they were included in the top 100, across bootstrap iterations. This was repeated for each study.
For n increasing from four to 100 in increments of five, we used the top n overall-ranked genes to predict the left-out study, comparing the predicted label to the CL. We selected the top algorithm based on accuracy, consistency, and stability in predictions across studies. We refined gene selection within the confirmation set by considering a smaller range of gene numbers (40-78) and repeating the previous step with one gene increments to define a minimal number of genes needed to sustain performance and we validated it in two additional datasets.
Biological Associations
We confirmed associations of predicted labels with clinical and pathological features including age, stage, residual disease, cellularity, necrosis, BRCA1/2 germline status, race/ethnicity, and CD8+ tumor infiltrating lymphocytes (TIL; Supplement A.3). We used one-way ANOVA to compare continuous variables and the chi-square test for categorical variables. We evaluated univariable survival using Kaplan-Meier survival curves and the log rank test. In multivariable models, we used the Cox proportional hazard and computed P values using an omnibus likelihood ratio test. All statistical tests were two-sided. We applied pairwise deletion (available-case analysis) on missing data, as applicable.
Results
Subtyping the NanoString Data
Parallel array-based approaches resulted in two final models: the All array (ADAboost) and TCGA (random forest) models (Supplement B). Each of these algorithms were used to generate per-subtype probabilities and predictive entropy(55) on the 3829 HGSOC samples run on the NanoString platform. The label of the subtype with the highest probability was taken as the final label from each model. The observed concordance between the two models was high (accuracy 79%; kappa 0.72) and discordance was seen mostly between C1.MES/C2.IMM and C2.IMM/C4.DIF subtypes (Figure 2A). Discordant samples were enriched for lower signal-to-noise ratio in NanoString data, consistent with lower-quality RNA (ratio < 1000 in 7.5% vs 5%, p=0.0130; Supplemental B.4). No other technical variables showed differences between concordant and non-concordant labels. In concordant samples (consensus labelled; CL), the predictive entropy was significantly lower (p < 0.0001; Figure 2B). In a set of 67 cases, repeated on both array and NanoString (and excluded from training), the CL reproduced originally published labels with 94% accuracy (kappa 0.92).(9,10) Concordant samples (n=3030) were considered the de facto standard and subsequently used for training a minimal gene set classifier.
Figure 2:
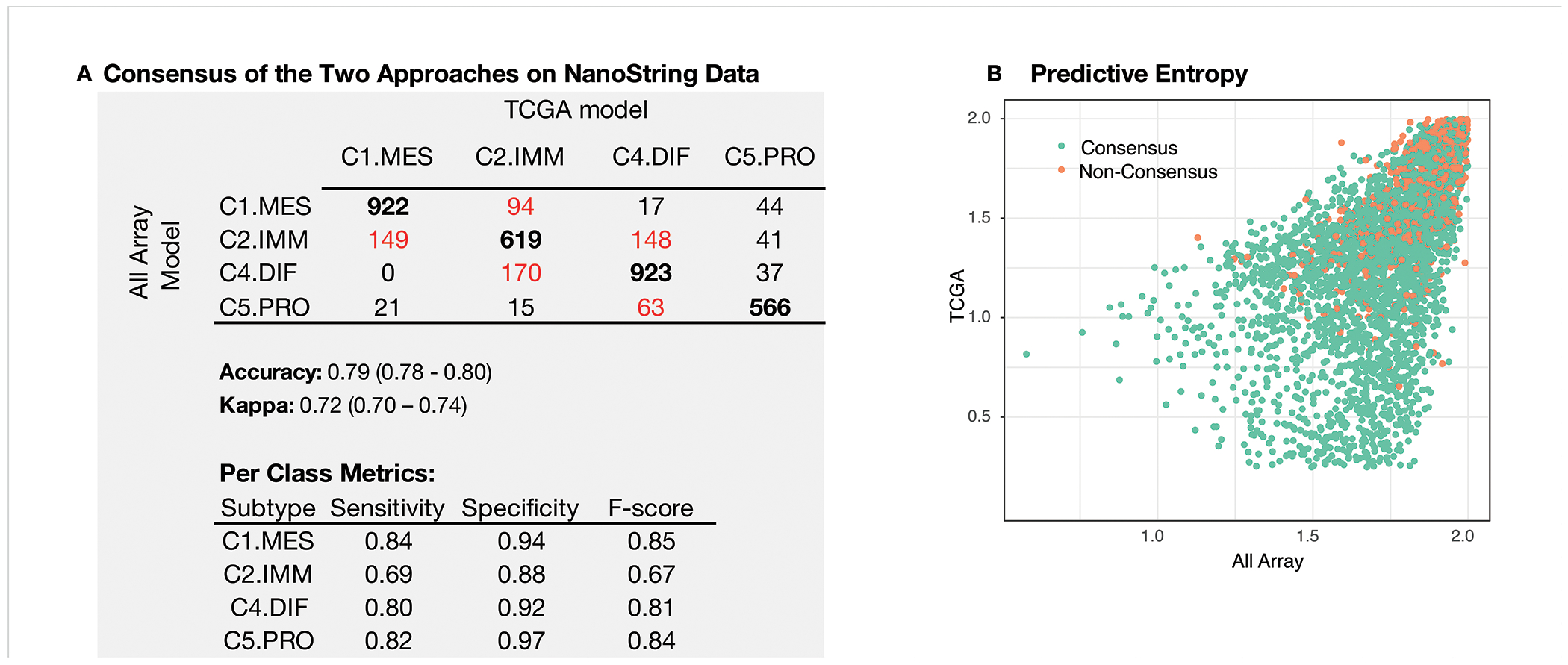
Evaluation metrics of consensus in subtype assignment between the All Array and TCGA models. (A) Confusion matrix comparing the agreement between the TCGA and the All Array approaches. In bold we present the results where there is agreement and highlighted in red are the most sizeable disagreements. We also present sensitivity, specificity, and F-score for each subtype. (B) Predictive entropy computed from per-class probabilities generated by each of the TCGA and the All Array model. When entropy approaches 0, it is indicative that the probability used to assign a sample to class is close to 1, while a high entropy (approaching 2) indicates that assignment to any class has a roughly equal probability. Overall, samples where consensus was not reached, had higher entropy in both models (p < 0.0001; Mann-Whitney U test).
Development of a NanoString Minimal-Gene Classifier
Using a leave-one-study-out cross validation design, random forest and LASSO outperformed AdaBoost (Figure 3A) in the training set (n=1135). Despite requiring more genes overall, we chose the random forest model based on stability in gene selection across studies and a less variable overall accuracy with increasing numbers of genes (Figure 3B). Accuracy of random forest in the confirmation set (n=817) ranged from 95 - 97% and achieved marginal gains after 55 genes. The locked-down assay, named PrOTYPE (Predictor of high-grade-serous Ovarian carcinoma molecular subTYPE), is represented by a final 55-gene model with specified NanoString probeset and controls, specific computational procedures, and requirements for specimen input from primary tubo-ovarian, treatment-naïve HGSOC samples as outlined in Figure 4 (see also Tables SC7; and Supplement E). Computational methods to normalize and generate predictions are available as a web application and R-script.1
Figure 3:
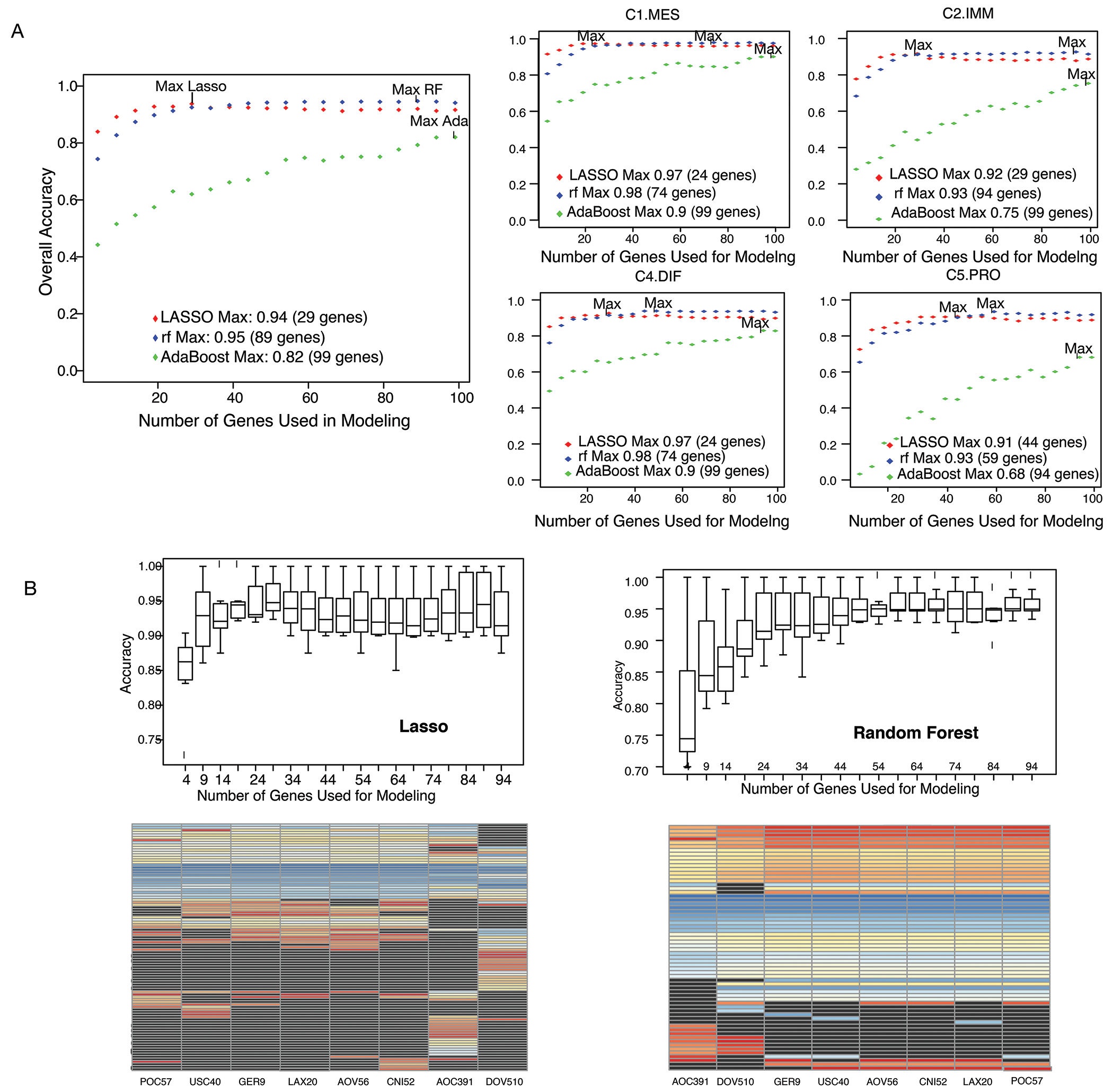
Model selection metrics for a minimal gene classifier. (A) The aggregate accuracy left and F1-score right (for all samples in all studies) obtained by increasing numbers of genes and using the top n genes from each frequency list computed above, where n varied from 4 to 100 in increments of 5. Note that the top n genes from each study were not necessarily the same. B) Top: Boxplots of the prediction accuracy by study using the LASSO and the random forest algorithm. Each point in the boxplot corresponds to the individual study prediction (when left-out). Bottom: Heatmap depicting the importance rank of the top 50 ranking genes obtained from each data partition in the leave-one-study out scheme.
Figure 4:
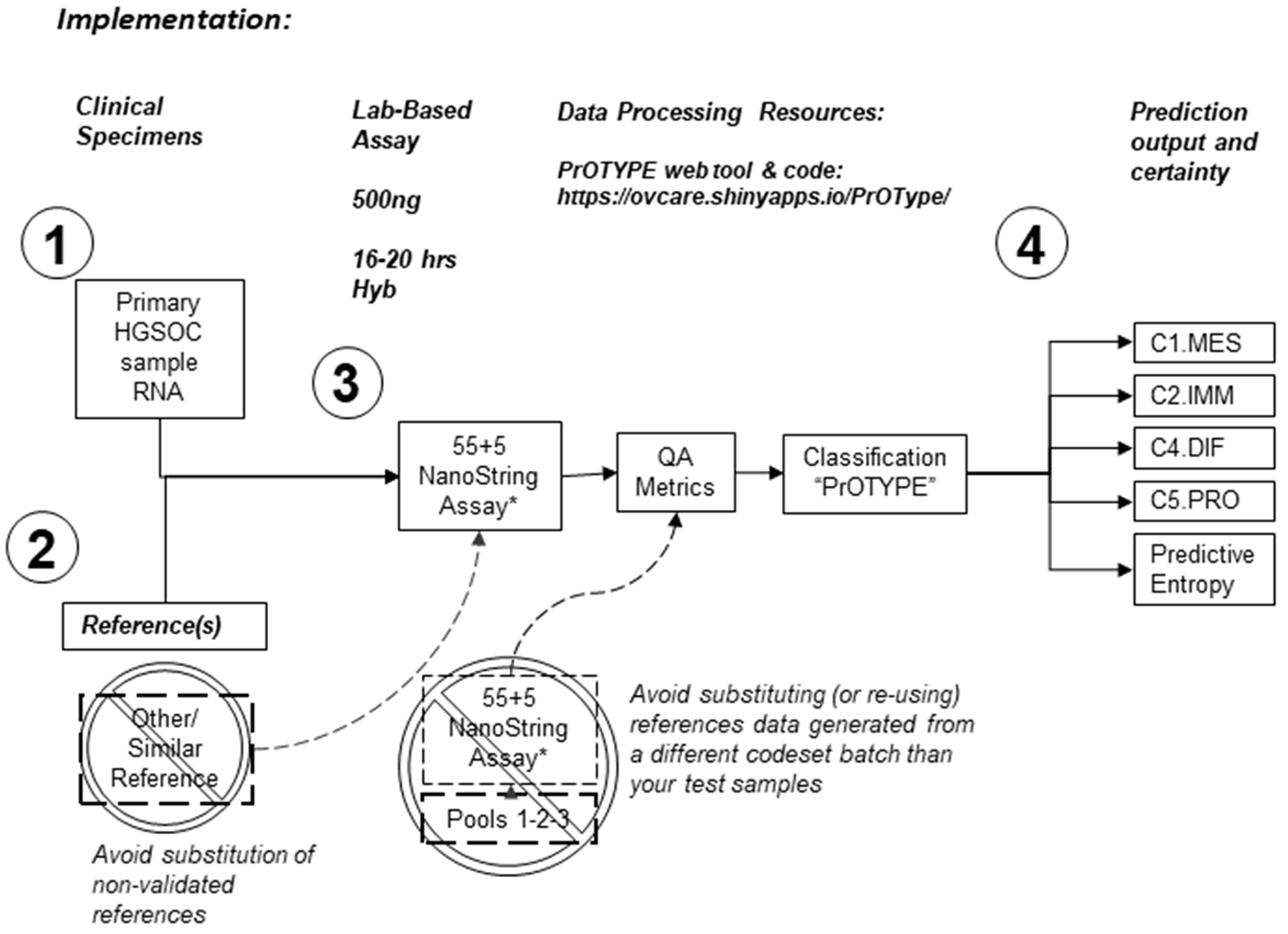
Locked-down Predictor of Ovarian carcinoma molecular subTYPE (PrOTYPE). The schematic illustrates the four critical components of the clinical-grade PrOTYPE assay (Supplement E) consisting of: (1) 500ng of total RNA from primary chemo-naïve HGSOC and (2) 100ng (each) of validated reference specimens. Each of these assayed individually by mixing specimen RNA with a (3) custom NanoString CodeSet (Supplemental Table SC7) containing 55 prediction model gene probes and 5 control gene probes (*55+5 NanoString Assay). RNA is hybridized with CodeSet, processed on a NanoString nCounter Prep-Station, and imaged at maximum fields of view on a NanoString nCounter Digital Analyzer. Resulting raw data is then normalized and HGSOC molecular subtypes predicted with our PrOTYPE computational algorithms using either a web-based tool, or R-script. This process will return (4) a prediction probability for the assayed specimen, for each subtype, and a single predictive entropy value. The latter can be used to estimate the certainty of prediction where 0 entropy corresponds to a near perfect prediction or “pure” subtype, while 2 entropy corresponds to near equal chance of assignment to any subtype.
PrOTYPE genes (55 genes plus five housekeeping genes; Table SC7) included representation from pathways previously reported as enriched in HGSOC subtypes (Figure SC9), including components of extracellular matrix (COL11A1, COL1A2, FBN1), immune cell markers (CD3D, CD3E, CD8A), surface receptors and kinases (CSF1R, CD2, AXL), cytokines and cell morphology (CXCL9, CXCL11, CCL5), and angiogenesis genes (PDGFRB, FGF1, TCF7L1). The per-subtype pattern of expression of PrOTYPE genes was near-identical between the NanoString data and the array data, used in establishing the CL standard (Figure SC10–SC12).
PrOTYPE was validated in two independent NanoString dataset partitions (n= 719 and 283 respectively) (Figure SB1). Partitions showed 95% and 94% accuracy and kappa=0.94 and 0.92 respectively, relative to the CL (Tables SB10–SB11).
In a set of 103 samples re-assayed in a newly-synthesized NanoString CodeSet, containing only the 55 PrOTYPE genes and controls, PrOTYPE predictions achieved 97% accuracy (95% CI: 92% - 99%), kappa 0.96 (0.91 - 1) in recovering the CL. We observed similar results in 100 samples that we replicated in another newly-synthesized CodeSet that included PrOTYPE genes as well as others (Tables SD2 – SD4). Of the 80 samples that overlapped all three CodeSets (original, PrOTYPE genes only, and PrOTYPE genes plus others), Fleiss’ kappa was 0.95, indicating excellent repeatability (p<0.0001). This confirmed the analytical validity of the PrOTYPE assay, our reference-based normalization, and single-sample processing strategy.
Confirmation of Subtype Signatures with Clinicopathological Associations
Patients were diagnosed between 1982-2014, with no differences in the distribution of subtypes related to year of diagnosis (Figure SD3). Omental-sourced specimens were enriched for C1.MES (72%) compared to adnexal specimens (25%), and the overall distribution of subtypes was significantly different (p<0.0001; Table 1A; Table SD7). We also noted a similar C1.MES enrichment at other anatomical sites, including the peritoneum (46%) and upper gynecological tract (50%). In tumors where anatomical site was presumed adnexal but not specifically annotated (n=1647), subtypes showed a distribution similar to those known to be adnexal (p=0.089).
Table 1:
(A) The distribution of HGSOC molecular subtype within different anatomical specimen collection sites. (B) Clinical and pathological parameters across HGSOC molecular subtype. Percentages are column wise except for totals where they are computed row wise. P values are computed using one-way analysis of variance for numerical parameters, and chi-square test for categorical ones.
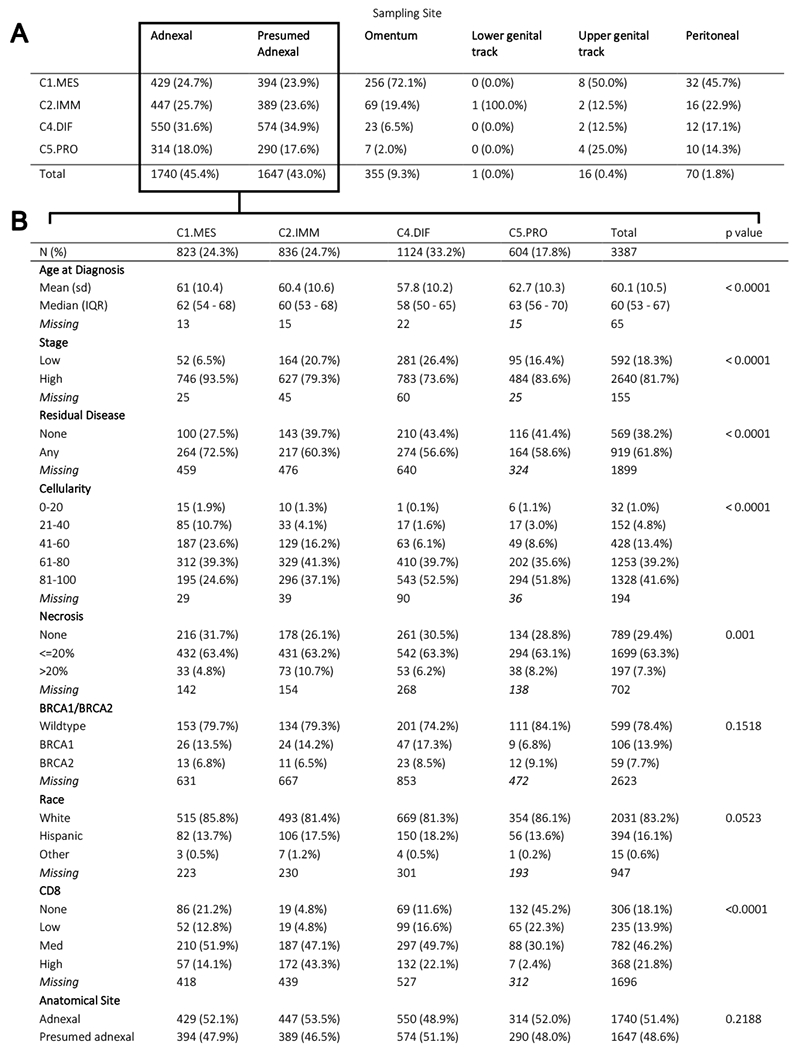 |
In 53 patients where paired adnexal and omentum samples were available, we observed poor agreement (kappa 0.06) in classification from the two sites. For 30/39 (77%) adnexal samples which were assigned non-C1.MES subtypes, their corresponding omental sample was C1.MES (Supplementary Table SD5). For all 14 adnexal specimens that were C1.MES, their omental classification was also C1.MES. As previously reported, subtype designation varies between metastatic sites within a patient, therefore we interpreted this to be a characteristic of tumors within their specific microenvironment rather than a weakness in the classification.(37,56) Heterogeneity in subtype assignment per-patient would confound clinicopathological associations; therefore, we present associations to subtype of adnexal-sourced specimen as this was the most commonly acquired specimen type (known n=1740; or presumed n=1647; Table 1B; Supplement D contains results also excluding presumed adnexal samples).
The median age at diagnosis was lowest amongst C4.DIF (58 yrs.) and highest amongst C5.PRO (63 yrs.), p<0.0001. Stage was significantly associated with subtype (p<0.0001; Table 1B): with 94% of C1.MES at high-stage and only 74% of C4.DIF. Residual disease was significantly associated with subtype, with C1.MES tumors being the most enriched. Similarly, both tumor cellularity and necrosis were associated with subtype. Lowest cellularity was in the C1.MES and highest necrosis was seen in C2.IMM. BRCA1/BRCA2 pathogenic germline mutation status was not associated with subtype. We found CD8+ TIL levels, derived from prior work,(57) highest in C2.IMM: 43% with high TIL and only 10% with absent/low CD8+ TIL. C5.PRO had the lowest CD8+ TIL, with 68% having absent/low CD8+ TIL. C4.DIF had the second highest level of CD8+ TIL at 22%.
Median follow up time was 8.1 years for overall survival (OS) and 6.5 years for progression-free survival (PFS) (reverse Kaplan-Meier), and were slightly longer for C2.IMM and C4.DIFF. Significant difference in survival was observed between subtypes for both OS and PFS (Log-rank p<0.0001; Figure 5A), as previously reported.(9,10,13–18,27) C2.IMM and C4.DIF had the best survival outcome and C1.MES had the poorest outcome. In multivariable analyses, we adjusted for risk factors known to be associated with survival: age at diagnosis, stage, residual disease, and germline deleterious BRCA1/2 status. Molecular subtypes were prognostic when adjusting for age and stage in both OS and PFS (Figure 5B). With the addition of CD8+ TIL, there was a change in the hazard ratio corresponding to subtype for both OS and PFS, but subtypes remained independently prognostic for OS only. With the addition of residual disease and/or BRCA1/2 to the model, molecular subtypes lost independent prognostic value in both OS and PFS.
Figure 5:
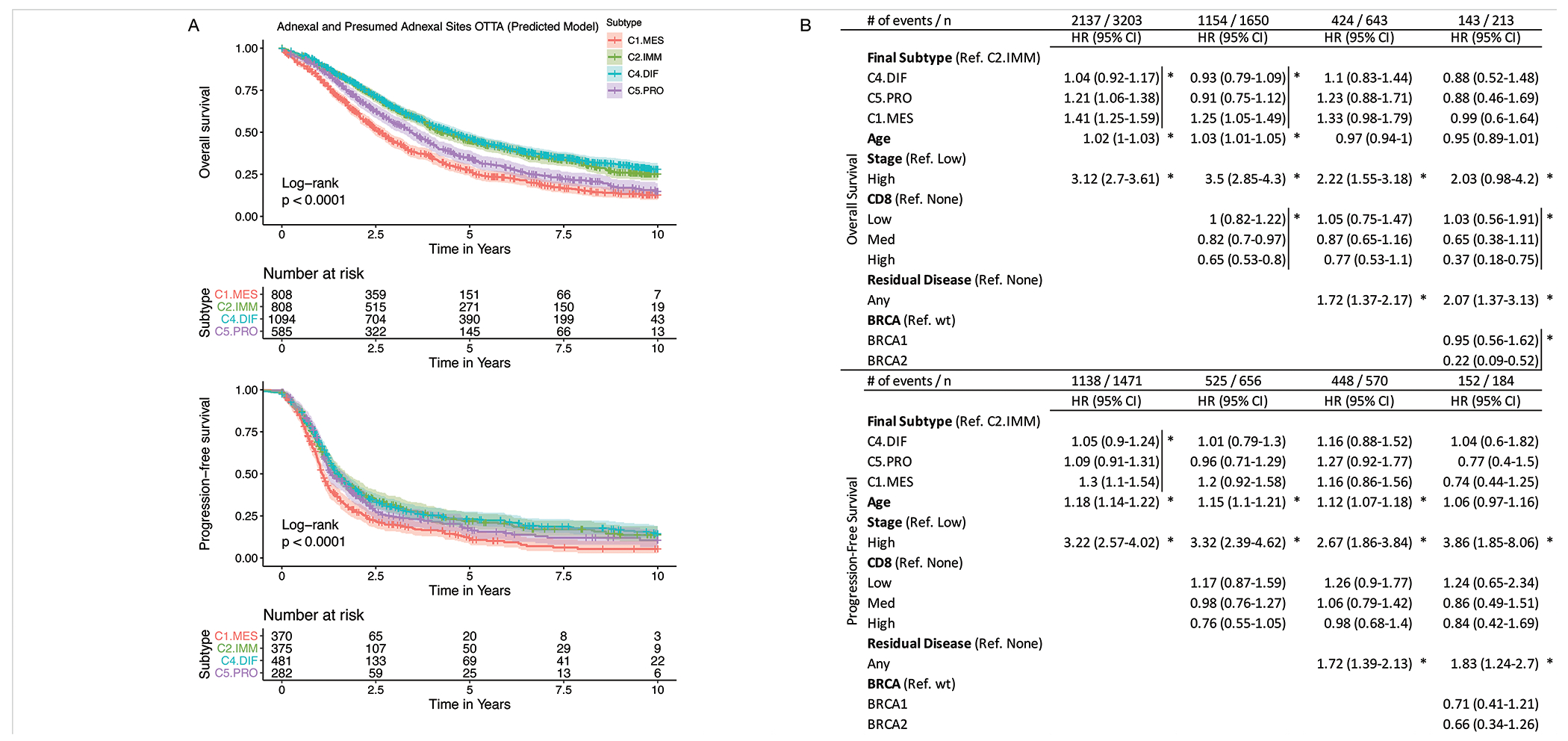
Univariable and multivariable survival analysis with PrOTYPE subtypes. (A) Kaplan-Meir survival curves for Overall and Progression-free survival by molecular subtype. C2.IMM and C4.DIF had the best survival in both OS and PFS in univariable analyses, while C5.MES had the worst survival. While C2.IMM and C4.DIF had inseparable outcomes, other clinical features were distinct between these groups (see also Table 1B). (B) Multivariable survival analysis results from Cox proportional hazard models adjusting for different known prognostic risk factors. The top table provides overall survival results while the bottom portion provides progression-free survival results. Each column in the table represents an independent model that adjusts for different risk factors. To assess the significance of a factor, we used the omnibus Likelihood Ratio Test evaluating the likelihood with and without that factor in the model. As such, the resulting P values are associated with the entire factor and not a specific level of that factor; this is indicated by a vertical bar to clarify and the asterisks (*) indicate that the omnibus Likelihood Ratio Test P value was below 0.05 for the entire marked variable. The Score Test was used to compute confidence intervals, therefore these may not always match the P value results.
Discussion:
Any potential of gene-expression subtype information to guide patient management cannot be realized without a de facto standard and validated assay that can be applied in a single-patient setting using pathology-standard fixed tissues - such as would be encountered in the clinic. Here we have defined a de facto standard for HGSOC gene-expression molecular subtypes using the consensus from two independent models derived from 1650 bona fide HGSOC samples with array data. Using these samples, we designed and validated PrOTYPE, robust and pragmatic 55-gene classifier based on the NanoString gene-expression platform. We evaluated the analytical validity of PrOTYPE by testing it in newly-synthesized CodeSets. Finally, we confirmed reported associations between subtype and clinico-pathological parameters.
We have addressed limitations of prior work including designing PrOTYPE with an established single-sample normalization and batch correction approach.(35) PrOTYPE is built on the NanoString platform, known to be tolerant to different analytes and well suited for FFPE tissues.(31,32,58,59) This particular feature is critical to implementation in modern pathology labs and may also enable retrospective re-examination of archival specimen collections and clinical trials. Our model is not derived from a single dataset but instead uses two approaches to integrate information from 14 array studies and a consortium collection of >3000 tumors. Every sample included has been curated to ensure inclusion of a pure population of HGSOC, using either central review by expert gyne-pathologists (NanoString cohort) or a proven mechanism to minimize non-HGSOCs from historical datasets (array data cohorts).(30) Using the intersection of parallel approaches as a de facto standard, we provide a first example of an HGSOC gene-expression subtype classifier derived using the step-wise best practice recommended by the Institute of Medicine.(26) The PrOTYPE assay is therefore at the so-called “bright line”, bringing gene-expression molecular subtypes to the stage at which evaluation for clinical utility and use may begin.
Similar to NanoString’s Prosigna assay for breast cancer (31,32), we use a reference based strategy for single-sample classification and batch effect correction.(35) In our development phase, one limitation is that the chosen references are finite resources and will not be sufficient for long-term, widespread distribution. Less restricted reference source material will need to be chosen and integrated into the PrOTYPE assay to ensure sustainability. PrOTYPE is designed exclusively for gene-expression HGSOC molecular subtyping, application on other histotypes is uninterpretable. Further, the relationship between subtype and effects of neoadjuvant chemotherapy, a common practice for modern management of HGSOC, are unclear. Mitigating this could be solved by using pre-treatment biopsies, however, diagnostic biopsies currently favor omentum for ease of access to the tumor mass and our data suggest the omental microenvironment strongly biases towards a C1.MES prediction. Thus, the clinical utility of PrOTYPE may relate to consistency of phenotypes predicted from multiple anatomical sites within a patient and remains to be tested.
Our dataset enables validation of biological characteristics that smaller datasets have been unable to address. Consistent with prominent desmoplastic stroma reported from metastatic disease(60–62) we noted a systematic shift of all subtypes to a C1.MES phenotype at extra-adnexal sites. In addition, few cases of C1.MES were clear of visible macroscopic residual disease, suggesting a potential application for PrOTYPE may be predicting cytoreductability. Application of PrOTYPE to biopsied specimens may provide valuable information prior to surgery and allow investigation of whether C1.MES tumors are a logical choice for neo-adjuvant or other pre-surgical targeted therapies. However, given the limitations of our retrospective cohort, with potential heterogeneity in surgical practice, a well-designed prospective study is warranted to test this hypothesis.
In multivariable models we observed waning prognostic value for molecular subtypes in the context of known age, stage, CD8+ TIL infiltration, residual disease, and germline deleterious BRCA1/2 status, albeit with reduced sample size. Previous studies have suggested there may be an overall enrichment of BRCA1 disruptions (including methylation, somatic and germline events together) within C2.IMM (63), however, data on somatic events affecting BRCA1/2, and other measures of homologous repair deficiency, are currently unavailable in our dataset. Nonetheless, subtype appears to capture some information for critical prognostic variables. However, for a disease with a generally poor prognosis, prediction may be more important.
In keeping with previous observations, only a modest proportion of cases reflect a “pure” phenotype signature.(13,18) We suggest that thresholds for subtype prediction, and implied utility, should be determined empirically - these may be specific to a given intervention. While few clinical trials have invested in HGSOC gene-expression subtyping, at least one points to differential benefits of Bevacizumab across subtypes.(36) Potential benefits to C2.IMM are presently being tested using PrOTYPE in a trial of pembrolizumab in recurrent disease (NCT03732950). Likewise, there is an ongoing investigation in targeting both the reactive stromal features of C1.MES, in the BEACON trial (NCT03363867; combined Bevacizumab, Atezolizumab and Cobimetinib), and the stem-like features of C5.PRO, in a phase II study of Vinorelbine (NCT03188159). It remains to be seen whether stringent or lax subtype thresholding is important to patient selection for these interventions. Other umbrella multicenter pragmatic studies such as INOVATe (Individualized Ovarian Cancer Treatment Through Integration of Genomic Pathology into Multidisciplinary Care) are incorporating PrOTYPE in their evaluations of guided treatment modalities.(64)
While only small improvements in HGSOC outcomes have been achieved in the past decades, an increasing number of therapeutic options are emerging with a growing need to identify response groups to targeted therapies such as angiogenesis inhibitors,(36,65) immune modulators,(66–68) and PARP inhibitors.(69–71) While In the context of these new therapeutics, PrOTYPE will enable objective testing of the clinical utility of intrinsic HGSOC gene-expression subtypes - a threshold that has previously been elusive. Similar to molecular profiling tools that are already emerging for other cancers, (31,32,72–74) the clinical-grade PrOTYPE assay is ready for integration into clinical trials as well as research applications.
Supplementary Material
Statement of translational relevance:
Outcomes for women diagnosed with high-grade serous tubo-ovarian carcinoma (HGSOC) have limited improvements over the last few decades. While novel targeted therapeutic strategies are maturing, their widespread adoption is often dependent on biomarkers that can guide management and identify women who are more likely to benefit from new compounds. For HGSOC, several previously described, near-identical gene-expression based sub-classification schemes have had little impact on practice or clinical trial design. The most prominent drawback to their implementation is that they have not been designed in a clinically applicable way. Without a de facto standard any potential clinical utility of HGSOC gene-expression subtypes cannot be determined.
Here, we develop and validate a standardized and reproducible HGSOC gene-expression subtype classifier that will enable prospective assessment of the clinical utility of HGSOC gene-expression subtypes. The Predictor of high-grade-serous Ovarian carcinoma molecular subTYPE (PrOTYPE) represents an Institute of Medicine guidelines-compliant, fully-defined, and validated assay that can be used with formalin fixed paraffin embedded (FFPE) tissues - making it practical for clinical uptake. Our report confirms the biological relevance of gene-expression subtypes in HGSOC and will facilitate the incorporation of subtype classification into ongoing and future clinical trials.
ACKNOWLEDGEMENTS
We thank all the study participants who contributed to this study and all the researchers, clinicians and technical and administrative staff who have made possible this work. This project received technical and data management support from OVCARE through the Cheryl Brown Ovarian Cancer Outcomes Unit and the Genetic Pathology Evaluation Centre. The AOV study recognizes the valuable contributions from Mie Konno, Shuhong Liu, Michelle Darago, Faye Chambers and the staff at the Tom Baker Cancer Centre Translational Laboratories. The Australian Ovarian Cancer Study gratefully acknowledges additional support from Ovarian Cancer Australia and the Peter MacCallum Foundation. The AOCS also acknowledges the cooperation of the participating institutions in Australia and acknowledges the contribution of the study nurses, research assistants and all clinical and scientific collaborators to the study. The complete AOCS Study Group can be found at www.aocstudy.org. Further the authors thank Olivier Tredan and Pierre Heudel as investigators on the TRIO14 study and Sandrine Berge-Montamat as assistant for clinical research.
Direct funding for this project was provided by the National Institutes of Health (R01-CA172404, PI: S.J. Ramus; and R01-CA168758, PIs: J.A. Doherty and M.A. Rossing), the Canadian Institutes for Health Research (Proof-of-Principle I program, PIs: D.G. Huntsman and M.S. Anglesio) and the United States Department of Defense Ovarian Cancer Research Program (OC110433, PI: D.D. Bowtell). A. Talhouk is funded through a Michael Smith Foundation for Health Research Scholar Award. M.S. Anglesio is funded through a Michael Smith Foundation for Health Research Scholar Award and the Janet D. Cottrelle Foundation Scholars program managed by the BC Cancer Foundation. J. George was partially supported by the NIH/National Cancer Institute award number P30CA034196.
In addition, other co-author, biobanks, patient-recruitment studies, and programs received funding that has indirectly supported this work. C. Wang was a Career Enhancement Awardee of the Mayo Clinic SPORE in Ovarian Cancer (P50 CA136393). D.G. Huntsman receives support from the Dr. Chew Wei Memorial Professorship in Gynecologic Oncology, and the Canada Research Chairs program (Research Chair in Molecular and Genomic Pathology). M. Widschwendter receives funding from the European Union’s Horizon 2020 European Research Council Programme, H2020 BRCA-ERC under Grant Agreement No. 742432 as well as the charity, The Eve Appeal (https://eveappeal.org.uk/), and support of the National Institute for Health Research (NIHR) and the University College London Hospitals (UCLH) Biomedical Research Centre. G.E. Konecny is supported by the Miriam and Sheldon Adelson Medical Research Foundation. B.Y. Karlan is funded by the American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124. H.R. Harris is supported by the NIH/National Cancer Institute award number K22 CA193860. OVCARE (including the VAN study) receives support through the BC Cancer Foundation and The VGH+UBC Hospital Foundation (authors AT, BG, DGH, and MSA). The AOV study is supported by the Canadian Institutes of Health Research (MOP-86727). The Gynaecological Oncology Biobank at Westmead, a member of the Australasian Biospecimen Network-Oncology group, was funded by the National Health and Medical Research Council Enabling Grants ID 310670 & ID 628903 and the Cancer Institute NSW Grants ID 12/RIG/1-17 & 15/RIG/1-16. The Australian Ovarian Cancer Study Group was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, The Cancer Council Victoria, Queensland Cancer Fund, The Cancer Council New South Wales, The Cancer Council South Australia, The Cancer Council Tasmania and The Cancer Foundation of Western Australia (Multi-State Applications 191, 211 and 182) and the National Health and Medical Research Council of Australia (NHMRC; ID199600; ID400413 and ID400281). BriTROC-1 was funded by Ovarian Cancer Action (to IAM and JDB, grant number 006) and supported by Cancer Research UK (grant numbers A15973, A15601, A18072, A17197, A19274 and A19694) and the National Institute for Health Research Cambridge and Imperial Biomedical Research Centres. Samples from the Mayo Clinic were collected and provided with support from the National Institutes of Health/National Cancer Institute P50 CA136393 (to E.L.G., G.L.K, S.H.K, M.E.S.). Samples from the German Ovarian Cancer Study were collected and provided with support from the German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401), and the German Cancer Research Center (DKFZ). M.J. Henderson received funding from Cancer Australia (1067110).
Financial Support: Core funding for this project was provided by the National Institutes of Health (R01-CA172404, PI: S.J. Ramus; and R01-CA168758, PIs: J.A. Doherty and M.A. Rossing), the Canadian Institutes for Health Research (Proof-of-Principle I program, PIs: D.G. Huntsman and M.S. Anglesio), the United States Department of Defense Ovarian Cancer Research Program (OC110433, PI: D.D. Bowtell) and others. A full list of funding support is noted at the end of the manuscript.
CONFLICTS OF INTEREST
The authors have no direct conflicts of interest to declare. B.Y. Karlan served on Invitae Corporation’s Advisory Board from 2017 to 2018. I. McNeish has acted on Advisory Boards for AstraZeneca, Clovis Oncology, Tesaro, Carrick Therapeutics and Takeda. His institution receives funding from AstraZeneca. R. Glasspool is on the Advisory Boards for AstraZeneca, Tesaro, Clovis and Immunogen and does consultancy work for SOTIO. She has received support to attend conferences from AstraZeneca, Roche and Tesaro. Her institution has received research funding from Boehringer Ingelheim and Lilly/Ignyta and she is the national co-ordinating investigator for the UK for trials sponsored by AstraZeneca and Tesaro and site principal investigator for trials sponsored by AstraZeneca, Tesaro, Immunogen, Pfizer, Lilly and Clovis. P. Fasching has received grants from Novartis, Biontech and Cepheid as well as personal fees from Novartis, Roche, Pfizer, Celgene, Daiichi-Sankyo, TEVA, Astra Zeneca, Merck Sharp & Dohme, Myelo Therapeutics, Macrogenics, Eisai and Puma during the conduct of the study. D.G. Huntsman is a co-founder and shareholder of Contextual Genomics Inc., a for-profit company that provides clinical reporting to assist in cancer patient treatment.
Footnotes
References:
- 1.The Cancer Genome Atlas RN. Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70 doi 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McAlpine J, Leon-Castillo A, Bosse T. The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses. The Journal of pathology 2018;244(5):538–49 doi 10.1002/path.5034. [DOI] [PubMed] [Google Scholar]
- 3.Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol 2012;123(4):615–26 doi 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clot G, Jares P, Gine E, Navarro A, Royo C, Pinyol M, et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 2018;132(4):413–22 doi 10.1182/blood-2018-03-838136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mottok A, Wright G, Rosenwald A, Ott G, Ramsower C, Campo E, et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018;132(22):2401–5 doi 10.1182/blood-2018-05-851154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCart Reed AE, Kalita-De Croft P, Kutasovic JR, Saunus JM, Lakhani SR. Recent advances in breast cancer research impacting clinical diagnostic practice. The Journal of pathology 2019;247(5):552–62 doi 10.1002/path.5199. [DOI] [PubMed] [Google Scholar]
- 7.Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. The New England journal of medicine 2009;360(26):2719–29 doi 10.1056/NEJMoa0902542. [DOI] [PubMed] [Google Scholar]
- 8.Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. British journal of cancer 2015;113(2):299–310 doi 10.1038/bjc.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474(7353):609–15 doi 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008;14(16):5198–208 doi 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 11.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018;68(4):284–96 doi 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Committee on the State of the Science in Ovarian Cancer Research, Board on Health Care Services, Institute of Medicine, National Academies of Sciences Engineering and Medicine, editors. Ovarian Cancers: Evolving Paradigms in Research and Care. 1 ed. Washington, D.C.: The National Academies Press; 2016. [PubMed] [Google Scholar]
- 13.Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest 2013;123(1):517–25 doi 10.1172/JCI65833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konecny GE, Wang C, Hamidi H, Winterhoff B, Kalli KR, Dering J, et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst 2014;106(10) doi 10.1093/jnci/dju249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan TZ, Miow QH, Huang RY, Wong MK, Ye J, Lau JA, et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO molecular medicine 2013;5(7):983–98 doi 10.1002/emmm.201201823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan TZ, Yang H, Ye J, Low J, Choolani M, Tan DS, et al. CSIOVDB: a microarray gene expression database of epithelial ovarian cancer subtype. Oncotarget 2015;6(41):43843–52 doi 10.18632/oncotarget.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Way GP, Rudd J, Wang C, Hamidi H, Fridley BL, Konecny GE, et al. Comprehensive Cross-Population Analysis of High-Grade Serous Ovarian Cancer Supports No More Than Three Subtypes. G3 (Bethesda) 2016;6(12):4097–103 doi 10.1534/g3.116.033514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen GM, Kannan L, Geistlinger L, Kofia V, Safikhani Z, Gendoo DMA, et al. Consensus on Molecular Subtypes of High-Grade Serous Ovarian Carcinoma. Clin Cancer Res 2018;24(20):5037–47 doi 10.1158/1078-0432.CCR-18-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest 2019. doi 10.1172/JCI96313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueno H, Kanemitsu Y, Sekine S, Ishiguro M, Ito E, Hashiguchi Y, et al. Desmoplastic Pattern at the Tumor Front Defines Poor-prognosis Subtypes of Colorectal Cancer. The American journal of surgical pathology 2017;41(11):1506–12 doi 10.1097/PAS.0000000000000946. [DOI] [PubMed] [Google Scholar]
- 21.Roman-Perez E, Casbas-Hernandez P, Pirone JR, Rein J, Carey LA, Lubet RA, et al. Gene expression in extratumoral microenvironment predicts clinical outcome in breast cancer patients. Breast Cancer Res 2012;14(2):R51 doi 10.1186/bcr3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busuttil RA, George J, Tothill RW, Ioculano K, Kowalczyk A, Mitchell C, et al. A signature predicting poor prognosis in gastric and ovarian cancer represents a coordinated macrophage and stromal response. Clin Cancer Res 2014;20(10):2761–72 doi 10.1158/1078-0432.CCR-13-3049. [DOI] [PubMed] [Google Scholar]
- 23.Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017;36(31):4457–68 doi 10.1038/onc.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helland A, Anglesio MS, George J, Cowin PA, Johnstone CN, House CM, et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS One 2011;6(4):e18064 doi 10.1371/journal.pone.0018064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobel M, Rahimi K, Rambau PF, Naugler C, Le Page C, Meunier L, et al. An Immunohistochemical Algorithm for Ovarian Carcinoma Typing. Int J Gynecol Pathol 2016;35(5):430–41 doi 10.1097/PGP.0000000000000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Institute of Medicine. EVOLUTION OF TRANSLATIONAL OMICS Lessons Learned and the Path Forward. Micheel CM, Nass SJ, Omenn GS, editors. Washington, DC: The National Academies Press; 2012. [PubMed] [Google Scholar]
- 27.Wang C, Armasu SM, Kalli KR, Maurer MJ, Heinzen EP, Keeney GL, et al. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin Cancer Res 2017;23(15):4077–85 doi 10.1158/1078-0432.ccr-17-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kommoss S, Gilks CB, du Bois A, Kommoss F. Ovarian carcinoma diagnosis: the clinical impact of 15 years of change. British journal of cancer 2016;115(8):993–9 doi 10.1038/bjc.2016.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurman RJ, Carcangiu ML, Herrington CS, Young RH, editors. WHO Classification of Tumours of Female Reproductive Organs. 4 ed. Volume 6 Lyon, France: IARC-WHO Press; 2014. 316 p. [Google Scholar]
- 30.Peres LC, Cushing-Haugen KL, Anglesio M, Wicklund K, Bentley R, Berchuck A, et al. Histotype classification of ovarian carcinoma: A comparison of approaches. Gynecologic oncology 2018. doi 10.1016/j.ygyno.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen T, Wallden B, Schaper C, Ferree S, Liu S, Gao D, et al. Analytical validation of the PAM50-based Prosigna Breast Cancer Prognostic Gene Signature Assay and nCounter Analysis System using formalin-fixed paraffin-embedded breast tumor specimens. BMC Cancer 2014;14:177 doi 10.1186/1471-2407-14-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015;8:54 doi 10.1186/s12920-015-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott DW, Chan FC, Hong F, Rogic S, Tan KL, Meissner B, et al. Gene expression-based model using formalin-fixed paraffin-embedded biopsies predicts overall survival in advanced-stage classical Hodgkin lymphoma. J Clin Oncol 2013;31(6):692–700 doi 10.1200/JCO.2012.43.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staiger AM, Ziepert M, Horn H, Scott DW, Barth TFE, Bernd HW, et al. Clinical Impact of the Cell-of-Origin Classification and the MYC/ BCL2 Dual Expresser Status in Diffuse Large B-Cell Lymphoma Treated Within Prospective Clinical Trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group. J Clin Oncol 2017;35(22):2515–26 doi 10.1200/JCO.2016.70.3660. [DOI] [PubMed] [Google Scholar]
- 35.Talhouk A, Kommoss S, Mackenzie R, Cheung M, Leung S, Chiu DS, et al. Single-Patient Molecular Testing with NanoString nCounter Data Using a Reference-Based Strategy for Batch Effect Correction. PLoS One 2016;11(4):e0153844 doi 10.1371/journal.pone.0153844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kommoss S, Winterhoff B, Oberg AL, Konecny GE, Wang C, Riska SM, et al. Bevacizumab May Differentially Improve Ovarian Cancer Outcome in Patients with Proliferative and Mesenchymal Molecular Subtypes. Clin Cancer Res 2017;23(14):3794–801 doi 10.1158/1078-0432.CCR-16-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leong HS, Galletta L, Etemadmoghadam D, George J, Australian Ovarian Cancer S, Kobel M, et al. Efficient molecular subtype classification of high-grade serous ovarian cancer. The Journal of pathology 2015;236(3):272–7 doi 10.1002/path.4536. [DOI] [PubMed] [Google Scholar]
- 38.Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Fujiwara H, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res 2012;18(5):1374–85 doi 10.1158/1078-0432.CCR-11-2725. [DOI] [PubMed] [Google Scholar]
- 39.Rudd J, Zelaya RA, Demidenko E, Goode EL, Greene CS, Doherty JA. Leveraging global gene expression patterns to predict expression of unmeasured genes. BMC Genomics 2015;16:1065 doi 10.1186/s12864-015-2250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denkert C, Budczies J, Darb-Esfahani S, Gyorffy B, Sehouli J, Konsgen D, et al. A prognostic gene expression index in ovarian cancer - validation across different independent data sets. The Journal of pathology 2009;218(2):273–80 doi 10.1002/path.2547. [DOI] [PubMed] [Google Scholar]
- 41.Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nature medicine 2011;17(12):1627–35 doi 10.1038/nm.2512. [DOI] [PubMed] [Google Scholar]
- 42.Zsiros E, Duttagupta P, Dangaj D, Li H, Frank R, Garrabrant T, et al. The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28-Costimulated T Cells Prepared for Adoptive Therapy. Clin Cancer Res 2015;21(12):2840–50 doi 10.1158/1078-0432.CCR-14-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyniel JP, Cottu PH, Decraene C, Stern MH, Couturier J, Lebigot I, et al. A genomic and transcriptomic approach for a differential diagnosis between primary and secondary ovarian carcinomas in patients with a previous history of breast cancer. BMC Cancer 2010;10:222 doi 10.1186/1471-2407-10-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong KK, et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer cell 2009;16(6):521–32 doi 10.1016/j.ccr.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferriss JS, Kim Y, Duska L, Birrer M, Levine DA, Moskaluk C, et al. Multi-gene expression predictors of single drug responses to adjuvant chemotherapy in ovarian carcinoma: predicting platinum resistance. PLoS One 2012;7(2):e30550 doi 10.1371/journal.pone.0030550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jochumsen KM, Tan Q, Hogdall EV, Hogdall C, Kjaer SK, Blaakaer J, et al. Gene expression profiles as prognostic markers in women with ovarian cancer. Int J Gynecol Cancer 2009;19(7):1205–13 doi 10.1111/IGC.0b013e3181a3cf55. [DOI] [PubMed] [Google Scholar]
- 47.Hendrix ND, Wu R, Kuick R, Schwartz DR, Fearon ER, Cho KR. Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer research 2006;66(3):1354–62 doi 66/3/1354 [pii] 10.1158/0008-5472.CAN-05-3694. [DOI] [PubMed] [Google Scholar]
- 48.Lisowska KM, Olbryt M, Dudaladava V, Pamula-Pilat J, Kujawa K, Grzybowska E, et al. Gene expression analysis in ovarian cancer - faults and hints from DNA microarray study. Front Oncol 2014;4:6 doi 10.3389/fonc.2014.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006;439(7074):353–7. [DOI] [PubMed] [Google Scholar]
- 50.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer research 2008;68(13):5478–86 doi 68/13/5478 [pii] 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiu DS, Talhouk A. diceR: an R package for class discovery using an ensemble driven approach. BMC bioinformatics 2018;19(11):1–4 doi 10.1186/s12859-017-1996-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Breiman L Out-of-bag estimation. Berkeley, CA: University of California Berkeley; 1996. 1–13 p. [Google Scholar]
- 53.Moons KG, Altman DG, Reitsma JB, Collins GS. New Guideline for the Reporting of Studies Developing, Validating, or Updating a Multivariable Clinical Prediction Model: The TRIPOD Statement. Advances in anatomic pathology 2015;22(5):303–5 doi 10.1097/PAP.0000000000000072. [DOI] [PubMed] [Google Scholar]
- 54.Nembrini S, Konig IR, Wright MN. The revival of the Gini importance? Bioinformatics (Oxford, England) 2018;34(21):3711–8 doi 10.1093/bioinformatics/bty373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKay DJC. Information Theory, Inference, and Learning Algorithms. Cambridge, UK: Cambridge University Press; 2003. [Google Scholar]
- 56.Zhang AW, McPherson A, Milne K, Kroeger DR, Hamilton PT, Miranda A, et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018;173(7):1755–69 e22 doi 10.1016/j.cell.2018.03.073. [DOI] [PubMed] [Google Scholar]
- 57.Ovarian Tumor Tissue Analysis C, Goode EL, Block MS, Kalli KR, Vierkant RA, Chen W, et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol 2017;3(12):e173290 doi 10.1001/jamaoncol.2017.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Veldman-Jones MH, Brant R, Rooney C, Geh C, Emery H, Harbron CG, et al. Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples. Cancer research 2015;75(13):2587–93 doi 10.1158/0008-5472.CAN-15-0262. [DOI] [PubMed] [Google Scholar]
- 59.Malkov VA, Serikawa KA, Balantac N, Watters J, Geiss G, Mashadi-Hossein A, et al. Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter Assay System. BMC Res Notes 2009;2:80 doi 1756-0500-2-80 [pii] 10.1186/1756-0500-2-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torres D, Kumar A, Wallace SK, Bakkum-Gamez JN, Konecny GE, Weaver AL, et al. Intraperitoneal disease dissemination patterns are associated with residual disease, extent of surgery, and molecular subtypes in advanced ovarian cancer. Gynecologic oncology 2017;147(3):503–8 doi 10.1016/j.ygyno.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torres D, Wang C, Kumar A, Bakkum-Gamez JN, Weaver AL, McGree ME, et al. Factors that influence survival in high-grade serous ovarian cancer: A complex relationship between molecular subtype, disease dissemination, and operability. Gynecologic oncology 2018;150(2):227–32 doi 10.1016/j.ygyno.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan TZ, Heong V, Ye J, Lim D, Low J, Choolani M, et al. Decoding transcriptomic intra-tumour heterogeneity to guide personalised medicine in ovarian cancer. The Journal of pathology 2019;247(3):305–19 doi 10.1002/path.5191. [DOI] [PubMed] [Google Scholar]
- 63.George J, Alsop K, Etemadmoghadam D, Hondow H, Mikeska T, Dobrovic A, et al. Nonequivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2 mutations. Clin Cancer Res 2013;19(13):3474–84 doi 10.1158/1078-0432.CCR-13-0066. [DOI] [PubMed] [Google Scholar]
- 64.deFazio A, Harnett P, Friedlander M, Balleine R, Bowtell D, Samimi G, et al. 2018 May 9, 2019. INOVATe. Westmead Institute; <https://www.westmeadinstitute.org.au/research/featured-projects/inovate-(1)/overview>. Accessed 2019 May 9, 2019. [Google Scholar]
- 65.Oza AM, Cook AD, Pfisterer J, Embleton A, Ledermann JA, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol 2015;16(8):928–36 doi 10.1016/S1470-2045(15)00086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pietzner K, Nasser S, Alavi S, Darb-Esfahani S, Passler M, Muallem MZ, et al. Checkpoint-inhibition in ovarian cancer: rising star or just a dream? J Gynecol Oncol 2018;29(6):e93 doi 10.3802/jgo.2018.29.e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oda K, Hamanishi J, Matsuo K, Hasegawa K. Genomics to immunotherapy of ovarian clear cell carcinoma: Unique opportunities for management. Gynecologic oncology 2018;151(2):381–9 doi 10.1016/j.ygyno.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Webb JR, Milne K, Kroeger DR, Nelson BH. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecologic oncology 2016;141(2):293–302 doi 10.1016/j.ygyno.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 69.Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM, et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin Cancer Res 2017;23(15):4086–94 doi 10.1158/1078-0432.CCR-16-2615. [DOI] [PubMed] [Google Scholar]
- 70.da Costa A, do Canto LM, Larsen SJ, Ribeiro ARG, Stecca CE, Petersen AH, et al. Genomic profiling in ovarian cancer retreated with platinum based chemotherapy presented homologous recombination deficiency and copy number imbalances of CCNE1 and RB1 genes. BMC Cancer 2019;19(1):422 doi 10.1186/s12885-019-5622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. The New England journal of medicine 2016;375(22):2154–64 doi 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 72.Cook DP, Vanderhyden BC. Ovarian cancer and the evolution of subtype classifications using transcriptional profiling. Biology of reproduction 2019. doi 10.1093/biolre/ioz099. [DOI] [PubMed] [Google Scholar]
- 73.Scott DW, Abrisqueta P, Wright GW, Slack GW, Mottok A, Villa D, et al. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J Clin Oncol 2017;35(15):1668–77 doi 10.1200/JCO.2016.70.7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014;123(8):1214–7 doi 10.1182/blood-2013-11-536433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Millstein J, Budden T, Goode EL, Anglesio MS, Talhouk A, Intermaggio MP, et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol 2020. doi 10.1016/j.annonc.2020.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.