

Abstract
Breast cancer remains one of the leading causes of cancer mortality in the US. Elevated cholesterol is a major risk factor for breast cancer onset and recurrence, while cholesterol-lowering drugs, such as statins, are associated with a good prognosis. Previous work in murine models showed that cholesterol increases breast cancer metastasis, and the pro-metastatic effects of cholesterol were due to its primary metabolite, 27-hydroxycholesterol (27HC). In our prior work, myeloid cells were found to be required for the pro-metastatic effects of 27HC, but their precise contribution remains unclear. Here we report that 27HC impairs T cell expansion and cytotoxic function through its actions on myeloid cells, including macrophages, in a Liver × receptor (LXR) dependent manner. Many oxysterols and LXR ligands had similar effects on T cell expansion. Moreover, their ability to induce the LXR target gene ABCA1 was associated with their effectiveness in impairing T cell expansion. Induction of T cell apoptosis was likely one mediator of this impairment. Interestingly, the enzyme responsible for the synthesis of 27HC, CYP27A1, is highly expressed in myeloid cells, suggesting that 27HC may have important autocrine or paracrine functions in these cells, a hypothesis supported by our finding that breast cancer metastasis was reduced in mice with a myeloid specific knockout of CYP27A1. Importantly, pharmacologic inhibition of CYP27A1 reduced metastatic growth and improved the efficacy of checkpoint inhibitor, anti-PD-L1. Taken together, our work suggests that targeting the CYP27A1 axis in myeloid cells may present therapeutic benefits and improve the response rate to immune therapies in breast cancer.
Keywords: 27-hydroxycholesterol, cholesterol, breast cancer, myeloid cell, macrophage, T cell, immune suppression
Introduction
Among women in the United States, breast cancer is the most frequently diagnosed cancer and the second leading cause of cancer-related death [1]. Worldwide, over 500,000 female lives were lost to breast cancer and 1.7 million incidences were recorded in 2016 [2]. While the mortality rate of breast cancer has significantly declined since 1989, mainly due to screen-detection and improved therapeutics, the overall 5-year survival rate of stage IV metastatic breast cancer remains low [3, 4]. Therefore, there is a continued need to identify targetable drivers of breast cancer, especially for late stage and therapy-refractory disease. Although immune checkpoint blockade (anti-programmed death-ligand 1; αPD-L1) in combination with nab-paclitaxel has recently been approved for the treatment of patients with advanced triple-negative breast cancer (TNBC) staining positive for PD-L1, a majority of these patients do not respond in a clinically meaningful way [5, 6]. Furthermore, immune interventions for the treatment of the other breast cancer subtypes have shown very limited success [7]. Although undoubtedly multifactorial, one major obstacle to immune-therapy is the highly immunosuppressive microenvironment of breast tumors – a phenomenon that is strongly maintained by myeloid immune cells, including macrophages [8, 9]. However, myeloid cells are also critical for a robust antitumor response [10]. Thus, strategies to ‘re-educate’ myeloid cells away from being pro-tumorigenic are warranted and starting to be explored, at least preclinically [11–14].
Cholesterol and control of its metabolism have emerged as potential therapeutic targets. An elevated level of circulating cholesterol has been associated with breast cancer recurrence both pre-clinically and clinically [15–17]. Conversely, several studies have found that patients taking cholesterol-lowering drugs, such as statins, had improved prognosis, as demonstrated by decreased breast cancer recurrence and increased progression-free survival [18–21]. Thus, clinical observations strongly indicate that cholesterol is important for the progression of breast cancer.
Previously, work has shown that a primary metabolite of cholesterol, 27-hydroxycholesterol (27HC), has the capacity to activate the estrogen receptors (ERs) and can directly promote the growth of ERα-positive tumors [22]. Notably, we also found that 27HC can promote the metastatic colonization and progression of breast cancer, regardless of the expression of ER or breast cancer subtype [23]. Furthermore, ER-negative models of ovarian cancer, 27HC was essential for growth [24]. In the lung metastatic site of breast cancer, it was found that neutrophils and γδ T cells were required for the full pro-metastatic effects of 27HC, while in ovarian cancer monocytic myeloid-derived suppressor cells (M-MDSCs, with the same surface markers as inflammatory monocytes) were required [23, 24]. These observations suggest that 27HC is a critical factor in shaping myeloid cell function, directly or indirectly, and that the type of myeloid cell required for its pro-tumorigenic actions is dependent on the metastatic site and type of cancer.
Within the tumor microenvironment, macrophages are one of the most abundant subsets of the myeloid cell lineage [25, 26]. Accumulation of this cell type at the tumor site is often associated with poor prognosis [27–29]. Tumor-associated macrophages (TAMs) are known to have a diverse set of polarities, with an M2-like phenotype dominating [30, 31]. TAMs are known to promote tumor progression via a multitude of mechanisms, including providing angiogenesis-inducing factors (VEGF, proMMP-9), impairing effector T cells via reactive oxygen species, and recruiting T regulatory cells by producing interleukin (IL)-10, IL-2 and transforming growth factor (TGF)-β [32]. For both metastatic breast cancer and ovarian cancer models, 27HC resulted in decreased T cell abundance, indicating that immune suppression is a putative role for 27HC-myeloid cell-mediated pro-tumor actions [23, 24]. A recent report showed that the use of statins to lower cholesterol led to reduced number of TAMs and better prognosis in human lung adenocarcinoma [33]. This is particularly intriguing as macrophages highly express the enzyme responsible for the synthesis of 27HC, CYP27A1, suggesting that their pro-tumor function may require the conversion of cholesterol to 27HC.
Given the evidence suggesting that 27HC is critical for promoting the pro-tumorigenic properties of myeloid cells, and the abundance and plastic nature of macrophages within the tumor microenvironment, the goals of this work were to (1) establish the effects of 27HC on macrophages and (2) elucidate the mechanisms by which 27HC impacts macrophage regulation of T cell activity.
Materials and Methods
Reagents
27HC (purity >95%) and dendrogenin A (purity >98%) were synthesized by Sai Life (Hyderabad, India). 25HC, 4βHC and Desmosterol were synthesized by Steraloids (Newport, RI) (purity >98%). 22(R)HC was synthesized by Avanti (Alabaster, AL) (purity >99%). 24(S)HC and 24,25-epoxycholesterol (24,25EC) were synthesized by Enzo (Farmingdale, NY) (purity ≥98%). GW3965, T0901317, GSK2033 and 7-ketocholesterol (7KC) were synthesized by Cayman (Ann Arbor, MI) (purity ≥98%). 20(S)HC was synthesized by Tocris (Bristol, UK) (purity >95%). 17β-Estradiol (E2) was synthesized by MP Biomedicals (Irvine, California) (purity ≥98%). 27HC, dendrogenin A, 24(S)HC, 20(S)HC, GW3965, T0901317 and GSK2033 were dissolved in dimethyl sulfoxide (DMSO) and stored in −20°C. 7KC, 4βHC, 22(R)HC, 24,25EC and 25HC were dissolved in ethanol and stored in −20°C. Desmosterol was dissolved in M-pyrol and stored in −20°C. Antibodies for flow cytometry were purchased from BD Biosciences and used at a working dilution of 1:100 in FACS buffer (2% fetal bovine serum (FBS) in phosphate buffered saline (PBS)). Catalogue numbers were as follows: CD3: 555275, CD4: 553729, CD8: 557682. Annexin V-FITC/propidium iodide apoptosis assay was purchased from Thermo Fisher (catalogue number: V13242), and FAM-DEVD-FMK/propidium iodide apoptosis assay was purchased from Immunochemistry (catalogue number: 94).
Cell lines
E0771 and 4T1-luc cells were a gift from Mark Dewhirst (Duke University) and cultured in RPMI supplemented with 10% FBS (Corning), 1% non-essential amino acids (Corning), 1% sodium pyruvate (Corning) and 1% penicillin/streptomycin (Corning). E0771-OVA cells were a gift from Jun Yan (University of Louisville) and grown in the above-described condition. EL4 and E.G7-OVA cells were from American Type Culture Collection (ATCC) and cultured with RPMI medium supplemented with 10% FBS, 1% non-essential amino acids, 1% sodium pyruvate, 1% penicillin/streptomycin and 0.05 mM 2-mercaptoethanol. E.G7-OVA cells were additionally cultured with 0.4 mg/ml G418 (Fisher). We did not culture any of these cell lines longer than 3 months after thawing the stocks. Cell lines were tested for mycoplasma.
Preparation of bone marrow-derived macrophages (BMDMs)
Bone marrow cells were collected from mouse tibia and femur. Cells were passed through 70-micron cell strainer and subsequently cultured in the presence of 25 ng/mL of M-CSF (Novoprotein). Complete RPMI media supplemented with M-CSF was replaced every 3–4 days. On day 10 or 14, BMDMs were harvested for further experiments. For subsequent experiments, BMDMs were treated with DMSO or 27HC (solvent at a 1:1000 dilution) for 24hrs. Treatments were washed off prior to co-culturing with T cells.
T cell proliferation and apoptosis assay
CD3+ T cells were enriched from the spleen of wildtype, OT-I or OT-II mice, using Dynabeads Untouched mouse T cells kit (Thermo Fisher) according to the manufacturer’s instructions. Naïve CD4+ T cells were isolated from wildtype mice, using mouse Naive CD4+ T Cell Isolation Kit (Miltenyi Biotec) according to manufacturer’s instruction. T cells were labeled with the vital dye CFSE according to the manufacturer’s instructions (BioLegend). Wildtype T cells activated chemically were treated with 10 nM PMA and 1 μM ionomycin and cultured with washed BMDMs [as described in 34]. OT-I or OT-II T cells activated by antigen presentation were also labeled and cultured with LPS- and OVA-primed BMDMs [35–37]. BMDMs activating T cells from OT-I mice were primed with 50 μg/ml OVA257–264 (Bachem) and BMDMs activating T cells from OT-II mice were primed with 50 μg/ml OVA323–339 (Sigma). The proliferation of T cells was assessed by flow cytometry 72hrs after co-culture. Some OT-I mouse-derived T cells were harvested 48 or 72hrs after co-culture for RNA extraction (Thermo Fisher) and mRNA expression measurement by qPCR (BioRad). Some OT-I mouse-derived T cells were harvested 48 or 72hrs after co-culture for apoptosis assay using Annexin V-FITC and propidium iodide (Thermo Fisher), and some OT-I mouse-derived T cells were harvested 24, 48 or 72hrs after co-culture for apoptosis assay using FAM-DEVD-FMK (FAM-FLICA) against active caspase-3/7 and propidium iodide (ImmunoChemistry Technologies). Naïve CD4+ T cells were isolated from wildtype mice, chemically activated and cultured in conditioned media from treated BMDMs, harvested after 24 or 48hrs and assayed for apoptosis (FAM-FLICA + PI) as described above.
T cell cytolytic assay
T cells were isolated from the spleen of OT-I mice and activated for 72hrs by LPS- and OVA-primed BMDMs. E0771 and E0771-OVA or EL4 and E.G7-OVA cells were labeled with CFSE or Far Red cell tracker (Far Red) (Thermo Fisher), respectively, and then plated in a 96-well plate. Subsequently, activated T cells were harvested and counted to be co-cultured with cancer cell lines of interest at various ratios. 24hrs after co-culture, the cytolytic activity of activated T cells was assessed by the ratio between the OVA-expressing lines and the parental cell lines by flow cytometry. The level of granzyme B secreted in the co-culture was measured by ELISA according to the manufacture’s instruction (Thermo Fisher).
Gene silencing
For gene knockdown using siRNA, negative control and LXR/ER-complementary siRNA (Sigma, siRNA ID: SASI_Mm01_00189872, SASI_Mm01_00115295) were delivered to BMDMs for 48hrs using DharmaFECT transfection reagent 4 (GE) as previously described [38].
For gene knockdown using shRNA, negative control and a set of 3 LXR-complementary shRNA (Horizon) were delivered to BMDMs using lentiviral particles following the manufacture’s instruction at MOI of 20 for 96hrs. The shRNA sequences targeting LXRα were: GTGAGCCTTCGCCATGTGG, GCGCCTGTTACACTGTTGC and GCAGGCGAAGGGCAAACAC. The shRNA sequences targeting LXRβ were: ATGATGGCTAGCTCGGTGA, CTGCACATTAGGCCGATCG and AACCATTCCCAGGCACGGG. Expression of the target genes after the knockdown was assessed using qPCR.
Animal and animal studies
All protocols involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois Urbana-Champaign. Wildtype C57BL/6 and BALB/C mice were purchased from Charles River Laboratory. Founder OT-I, OT-II and LysMCre mice were purchased from Jackson Laboratory and bred in-house. Founder CYP27A1fl/fl mice were a kind gift from Donald P. McDonnell (Duke University). Fig. 7A: CYP27A1fl/fl;LysMCre+ and age-matched LysMCre+ mice were orthotopically grafted with 500,000 E0771-OVA cells in Matrigel (Corning) into the mammary fat pad. The resulting tumor growth was measured by caliper 5 times a week. After the tumor size reached ~200 mm3 in either group, 7 million T cells from OT-I mice were grafted retro-orbitally into each tumor-bearing mouse in that particular group. One day before T cell adoptive transfer, each tumor-bearing mouse was immunized intraperitoneally with 25 μg of OVA256–264 in Complete Freund’s Adjuvant (CFA) (Sigma). For Fig. 7B–C: 200,000 E0771 cells were grafted intravenously as described previously [23]. Mice were euthanized 3 weeks post-graft, and exterior, visible metastatic nodules were assessed and scored based number and size. The metastatic lesions were quantified with the following scoring criteria: 1) The metastatic nodules were counted and then scored according to their diameters using photos taken. Specifically, a nodule with diameter greater than or equal to 0.5 in, 0.3 in, 0.2 in and 0.1 in received a score of 4, 3, 2 and 1, respectively. 2). If a metastatic lesion was discovered outside the lung (such as liver), the lesion would also receive a score of 4. For Fig. 7D: BALB/C mice were grafted intravenously with 4T1-luc cells, which are 4T1 cells stably expressing a luciferase plasmid construct. Bioluminescent signal was used to quantify resulting metastatic burden [39]. GW273297X treatment commenced 3d post-graft at 100mg/kg/day [23]. αPD-L1 treatment started at the same time and followed the same regimen as described previously [24].
Figure 7: Absence or inhibition of CYP27A1 promotes T cell function, decreased tumor growth and enhanced efficacy of αPD-L1.
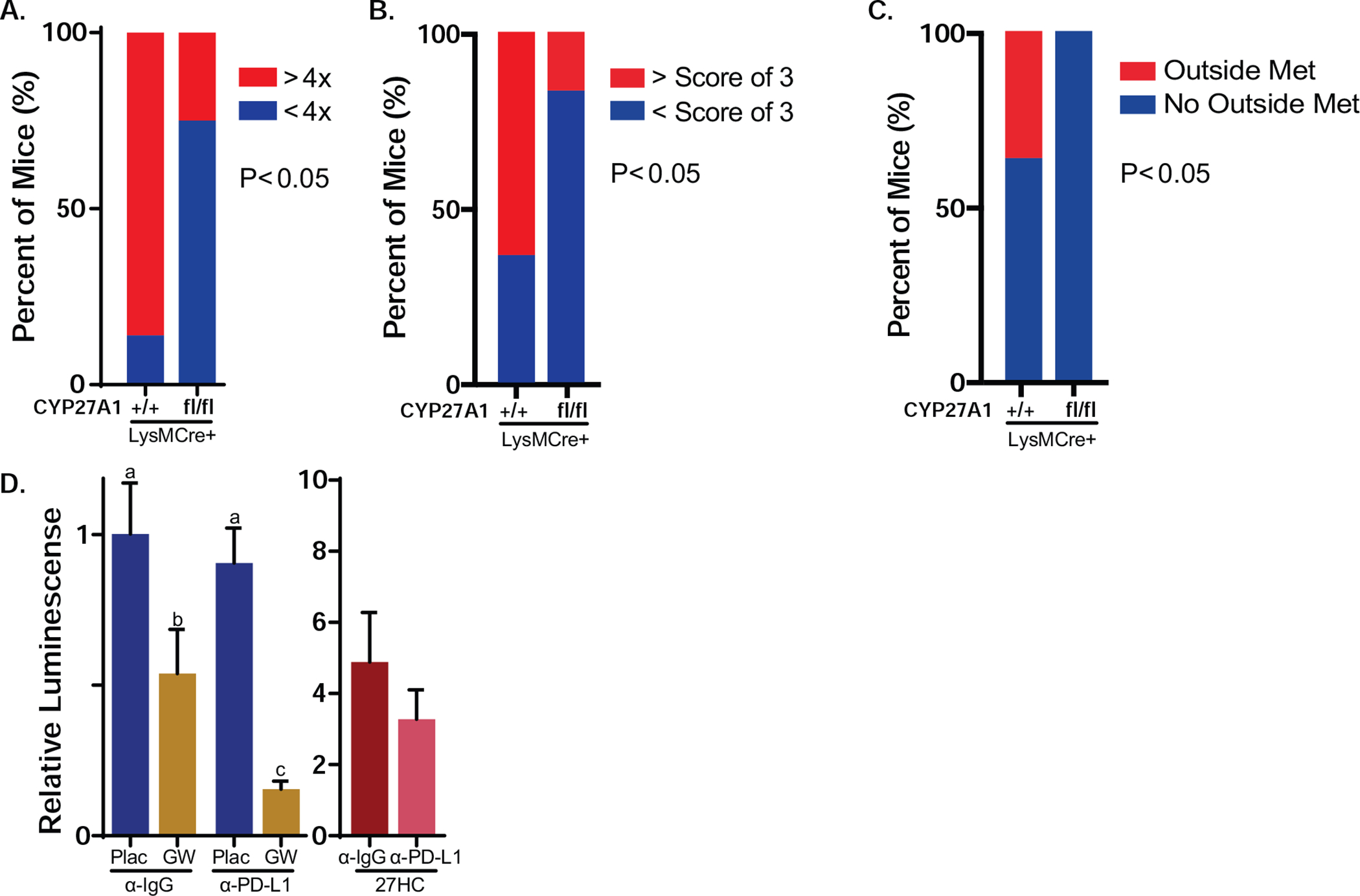
(A) Mice with myeloid cell knockout of CYP27A1 (CYP27A1fl/fl;LysMCre+) exhibited lower level of antigen-specific primary tumor growth. CYP27A1fl/fl;LysMCre+ mice (N=8) and their replete controls (N=7) were grafted orthotopically with E0771-OVA cells and tumors allowed to grow to ~200 mm3. Mice then received an adoptive transfer of CD3+ T cells isolated from OT-I mice. The difference between final tumor volume and the established tumor volume at adoptive transfer was calculated as a ratio between the two. The ratio used for comparison of response between two groups was 4. Macrophage-selective knockout illustrated in Supplemental Fig. 11). Growth curves are depicted in Supplemental Fig. 12A–B. (B) Myeloid cell CYP27A1 knockout mice exhibited lower level of metastatic burden, after intravenous graft of E0771 cells (CYP27A1fl/fl;LysMCre+ mice, N=12; CYP27A1+/+;LysMCre+ mice N=11. The mice were euthanized 3 weeks after injection and metastatic nodules counted and then scored according to their diameters using photos taken. Specifically, a nodule with diameter greater than or equal to 0.5 in, 0.3 in, 0.2 in and 0.1 in received a score of 4, 3, 2 and 1, respectively. If the metastatic lesion was discovered outside lung (e.g. liver), the lesion would also receive a score of 4. Representative pictures of metastatic lungs are in Supplemental Fig. 12C. (C) CYP27A1fl/fl;LysMCre+ mice exhibited decreased extra-plural metastases. Many controls, but no myeloid cell CYP27A1 knockout mice, had metastases outside the lung (same mice as in B). (D) Small molecule inhibition of CYP27A1 reduced metastatic growth and improved checkpoint inhibitor efficacy. BALB/C mice were grafted intravenously with 4T1-luc cells, and lesions allowed to establish for 3d. At this time, mice were treated with indicated ligands, including the CYP27A1 inhibitor, GW273297X (GW). αPD-L1 refers to a monoclonal antibody against murine PD-L1. (N=10/group except for 27HC treatments which had an N of 7–8/group). Data in (A-C) are presented as contingency table bar graph and data in (D) are presented as mean ± SEM (Fisher’s exact test in (A-C), one-way ANOVA followed by Newman-Keuls multiple comparison test for the first 4 groups in (D), p < 0.05).
Data Analysis
RNA sequencing data (RSEM, batched-normalized) for breast invasive carcinoma that were generated from the Cancer Genome Atlas (TCGA, https://www.cell.com/pb-assets/consortium/pancanceratlas/pancani3/index.html) were downloaded from the cBioPortal (http://www.cbioportal.org) using R package cgdsr [40, 41]. Data included 1084 samples in Fig. 3I–N and Supplemental Fig. 3. Correlations between gene expression were assessed by log2 transformation and Pearson’s correlation in Fig 3. I–N and Supplemental Fig. 3, and the best fit line was determined by linear regression between the explanatory (x axis) and the response variable (y axis). Two-tailed t-test followed by Benjamini-Hochberg false discovery rate correction was performed for comparing cytokine secretion by BMDMs in Supplemental Table 1A–B [42]. Two-way ANOVA followed by Bonferroni’s multiple comparisons test was used in Fig. 6B. Fisher’s exact test was performed for proportion testing in Fig 7. A–C. For Fig. 7D, luminescence was normalized to the mean placebo + α-IgG, and one-way ANOVA was performed followed by the Student Newman-Keuls post-hoc test for significance. For the rest of the results, two-tailed t-test or nonparametric Mann-Whitney U-test was performed to compare the effect of vehicle and 27HC on BMDMs and, subsequently, T cells.
Figure 3: T cells co-cultured 27HC-treated macrophages have decreased cytotoxic activity.
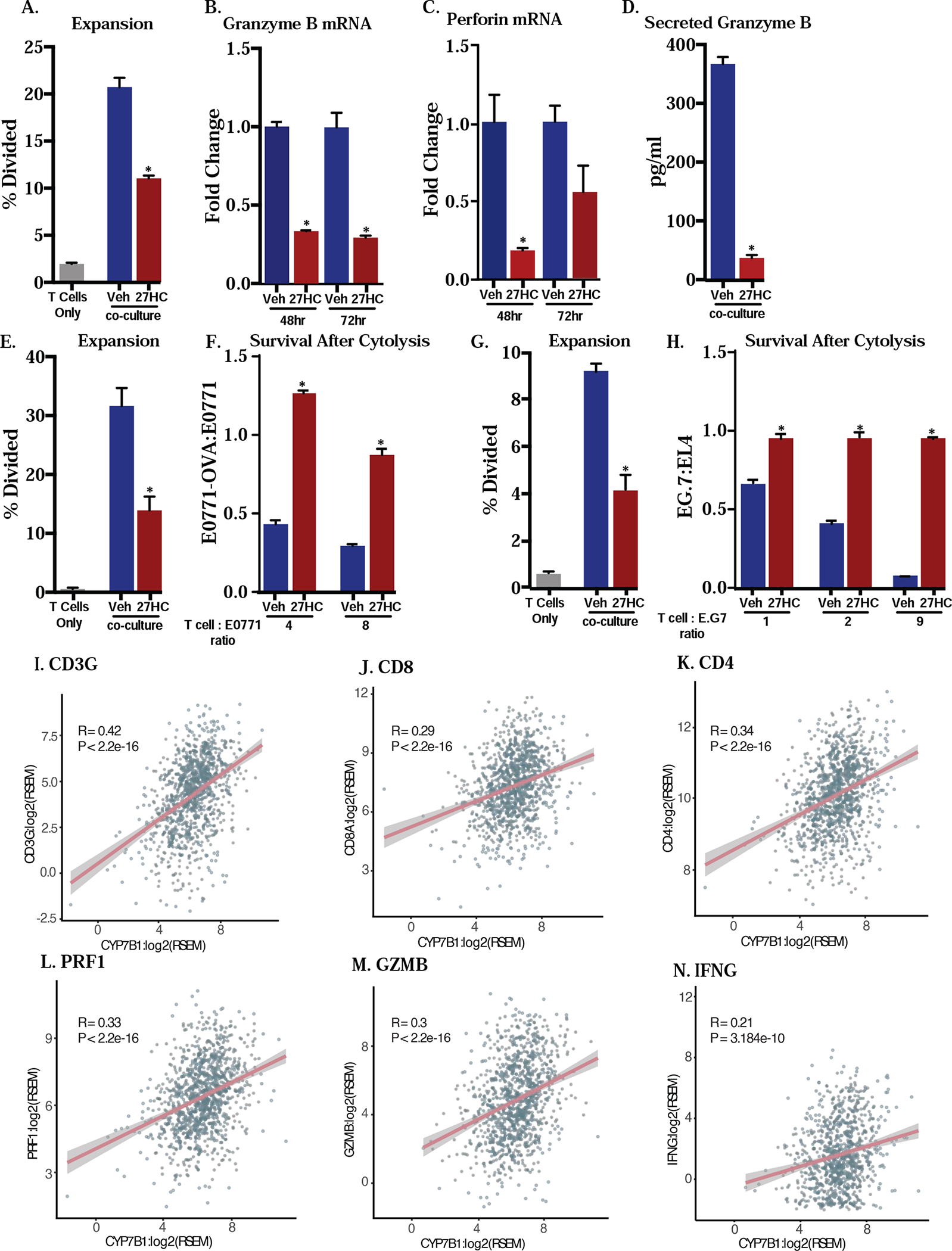
T cells co-cultured with BMDMs treated with 27HC for 72hrs have decreased expansion (A) and the resulting expanded T cells also have decreased granzyme B and perforin mRNA expression through time after co-culture as measured by qPCR (B-C) and secretion of granzyme B as measured by ELISA (D). Data in (A-C) are presented as normalized to respective vehicle control. (E and G) When OT-I T cells are co-cultured with BMDMs treated with indicated ligands as well as OVA, 27HC treatment results in decreased expansion, and these T cells have decreased cytolytic activity against (F) E0771 mammary cancer cells or (H) EG.7-OVA lymphoma cells. Both of these lines were engineered to express OVA. Specifically, the parental cell lines (E0771 or EL4) were labeled with CFSE and derivative cell lines (E0771-OVA and E.G7-OVA) were labeled with Far Red. Equivalent cell number of the respective parental and derivative cell lines were plated and co-cultured with OT-I T cells activated by vehicle- or 27HC-treatd BMDMs at indicated ratios. The cytolytic activity of T cells was assessed via flow cytometry as represented by the ratio of derivative (Far Red)/parental (CFSE) cell lines. (I-N) Human RNA sequencing data from TCGA Pan Cancer for invasive breast carcinoma indicated a moderately positive and significant correlation between CYP7B1, an enzyme responsible for catabolizing 27HC, expression and the expression of 6 T cell genes (CD3G, CD8A, CD4, perforin 1, granzyme B, INF-γ) that are related to immune competence. mRNA expressions were log2 transformed and Pearson’s correlation was assessed (N=1059, 1081, 1082, 1082, 1077 and 905, respectively). Data in (A-H) are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (two-tailed t-test in (A-H), p < 0.05). A: N=3/group for control and N=6/group for co-culture conditions, B: N=3–4/group, C: N=3–4/group, D: N=5–6/group, E: N=4/group, F: N=4/group, G: N=4/group, H: N=5/group.
Figure 6: 27HC-treated macrophages induce T cell apoptosis.
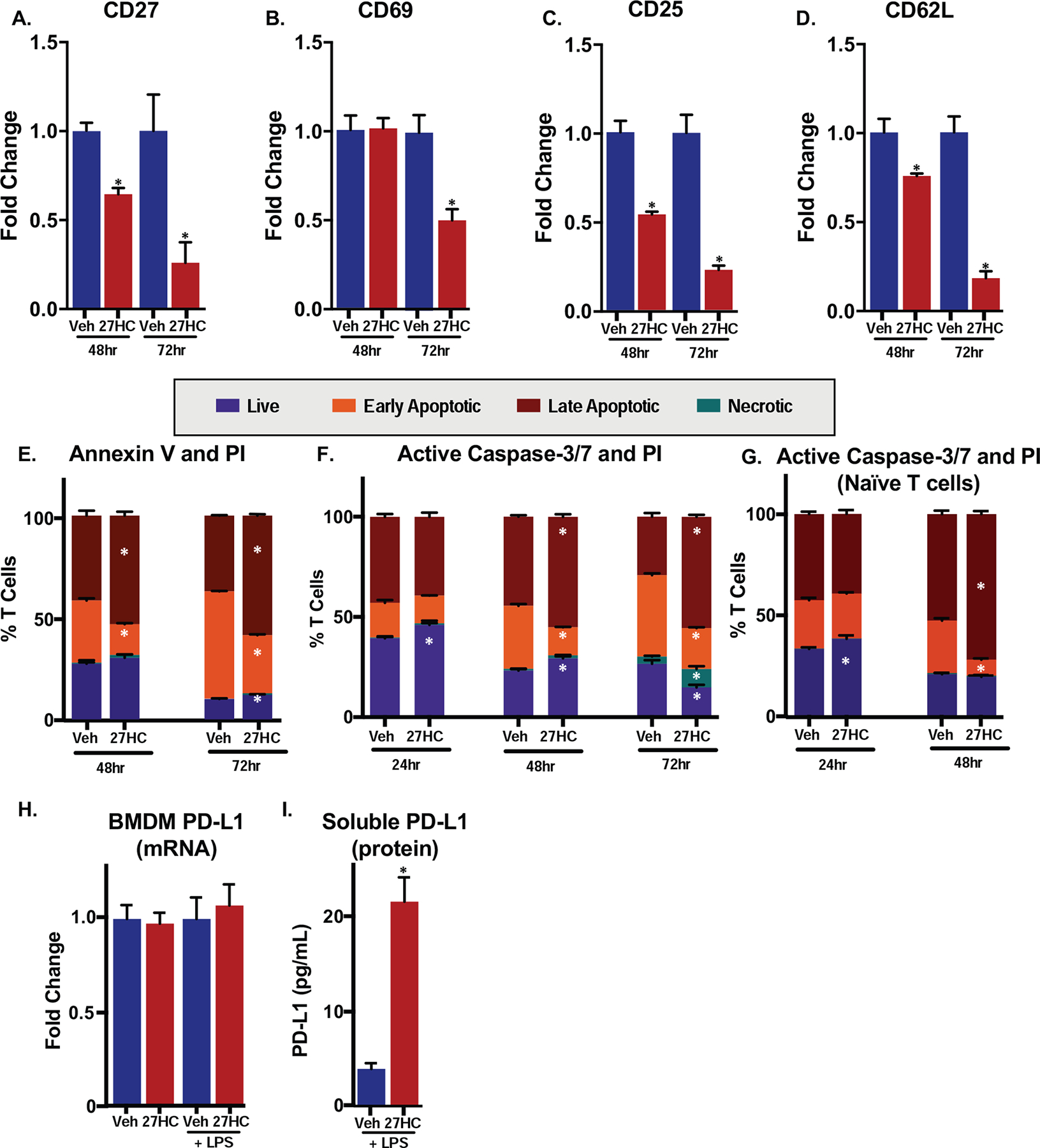
(A-D) Gene expression of markers of T cell activation was downregulated when T cells are co-cultured with BMDMs treated with 27HC. qPCR on T cells, 48 or 72hrs after co-culture (N=3–4/group). Data are presented as normalized to respective vehicle control. (E) OT-I T cells 48 or 72 hours after co-culture with vehicle- or 27HC-treated BMDMs exhibited enhanced apoptosis. T cells were harvested from the co-culture at two different time points and then were labeled with Annexin V-FITC and PI. Flow cytometry analysis was performed to measure the cell populations (N=4/group). (F) OT-I T cells in co-culture with 27HC-treated BMDMs exhibit increased apoptosis. T cells were harvested from the co-culture (as in E) at three different time points and then labeled with FLICA-FAM, a caspase-3/7 inhibitor that covalently binds to active forms of caspase-3/7, and PI. Flow cytometry analysis was performed to measure the cell populations (N=4/group). (G) Naïve CD4+ T cells were isolated from the spleen, chemically activated and cultured with conditioned media from vehicle or 27HC treated BMDMs. Subsequent analysis was similar to (F) (N=4/group). (H) PD-L1 mRNA expression in BMDMs; see Fig. 1 for details. (I) Soluble PD-L1 is increased in media from LPS-primed BMDMs treated with 27HC for 24hrs (similar experimental setup to Fig. 4D (N=4/group). Data are presented as mean ± SEM, and asterisks (*) indicate statistical significance from the respective vehicle control. (two-tailed t-test in (A-D), two-way ANOVA followed by Bonferroni’s multiple comparison test in (E) and (F), p < 0.05)
Results
27HC modulates gene expression in macrophages.
Oxysterols are a large group of compounds, with a diverse spectrum of biological activities [43]. Many oxysterols including 27HC have been described as ligands of the liver × receptors (LXRs), the activation of which promotes cholesterol homeostatic machinery, such as cholesterol efflux and cholesterol metabolism [44]. LXR activation, by various ligands including synthetic agonists, has also been implicated in regulating various aspects of the immune system. In general, many reports indicate that LXRs promote an anti-inflammatory phenotype, although these conclusions are largely based on synthetic ligands. This has led the field to believe that LXR agonists are generally anti-inflammatory. However, endogenous oxysterols and other LXR agonists are likely to have diverse, or even opposing actions [44–46]. It is well established that LXR induction in T cells impairs proliferative expansion and activity [47]. However, the actions of LXRs on the immune function of myeloid cell are less clear. Therefore, to further explore these putative immunomodulatory effects on macrophages, we investigated the effect of 27HC on the expression of genes associated with cholesterol homeostasis and inflammation. In order to simulate an inflammatory state, we evaluated the effects of 27HC in the absence as well as presence of lipopolysaccharide (LPS). LPS would be expected to polarize the macrophages towards an M1-like phenotype.
Bone marrow-derived macrophages (BMDMs) were prepared from female C57BL/6 mice. As expected, a 24-hour treatment of BMDMs with 27HC resulted in an induction of the LXR target genes ABCA1 and ABCG1, regardless of the presence of the inflammatory stimulus LPS (Fig. 1A–B). IL-10, a cytokine previously implicated in immune suppression in the tumor microenvironment [48] was found to be induced by 27HC in the absence of LPS, but modestly suppressed in the presence of LPS, suggesting that 27HC may selectively regulate genes depending on the inflammatory state of BMDMs (Fig. 1C). Similarly, 27HC induced the expression of MYC, a known factor involved in the alternative M2 polarization of macrophages [49], and this induction was most pronounced in the absence of LPS (Fig. 1D). Interestingly, 27HC decreased the expression of antigen-presentation/co-stimulation makers, namely MHCII (H2-Aa, H2-Ab1 and H2-Eb1) and B7 (CD80 and CD86) (Fig. 1E–I). MHCI proteins (H2-K1, H2-D1 and H2-L1) were also decreased by 27HC, regardless of whether the BMDMs were exposed to LPS (Fig. 1J–L). Because macrophages serve as antigen-presenting cells bridging innate and adaptive immunity, such suppression of antigen-presentation and co-stimulation markers indicates that 27HC may decrease the capacity of macrophages to present and activate T cells.
Figure 1: 27HC results in altered expression of genes associated with macrophage function and interaction with T cells.
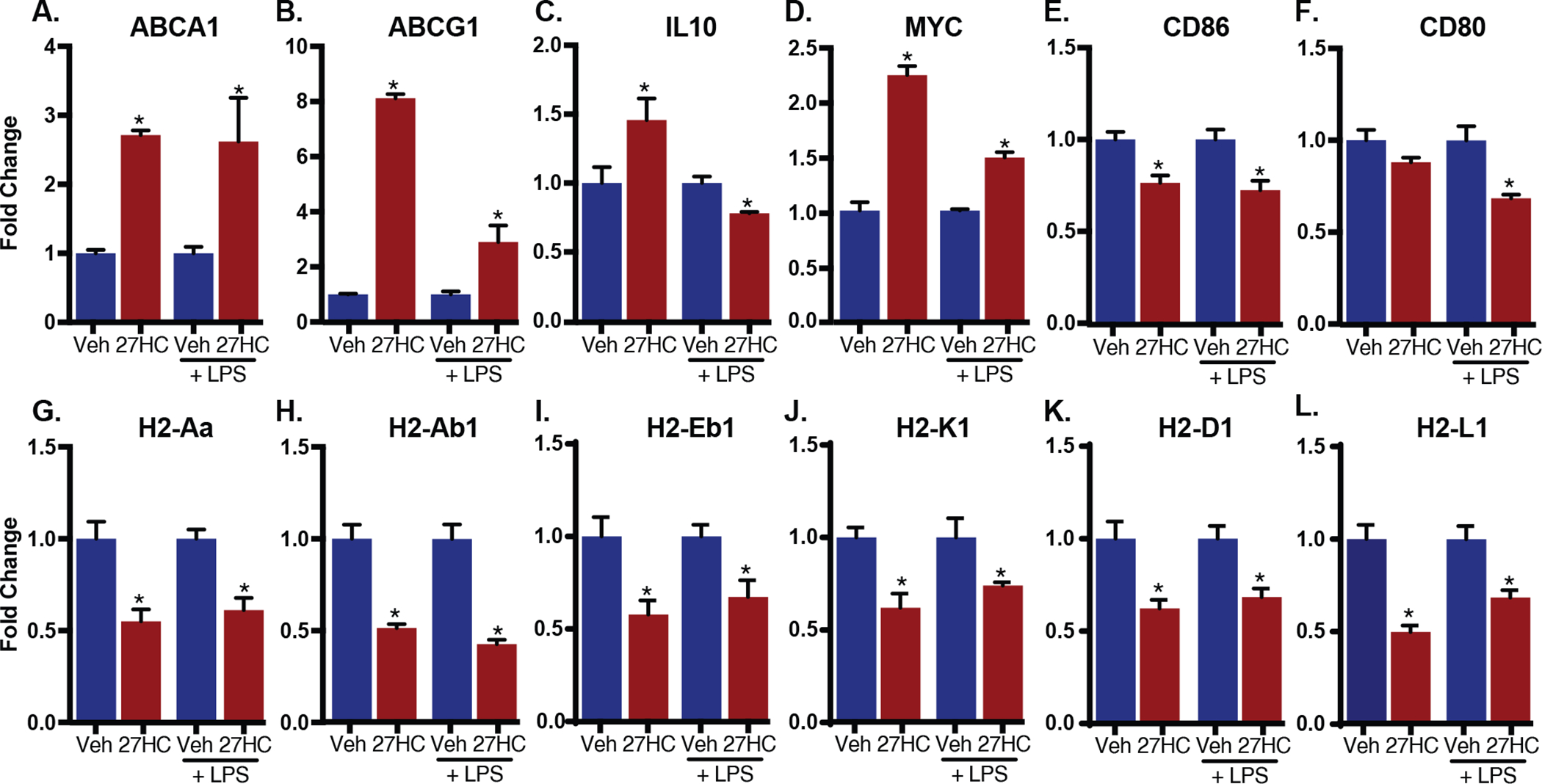
Murine BMDMs were treated with vehicle (DMSO) or 10 μM 27HC in the absence or presence of LPS. Transcripts were detected and quantified by qPCR and displayed as mean ± SEM. Data are presented as normalized to respective vehicle control. Asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for all samples treated with LPS, N=6/group for all samples treated with vehicle only, N=5 for all samples treated with 27HC only). (two-tailed t-test, p < 0.05)
27HC-treated macrophages inhibit T cell expansion.
The regulation by 27HC of genes in BMDMs that are associated with antigen presentation and cross-presentation to T cells (Fig. 1) suggests that 27HC likely influences myeloid cell-T cell crosstalk. Therefore, we investigated whether treating BMDMs with 27HC impacts T cell activation and function. When T cells are activated, they undergo rapid clonal expansion, with proliferative activity therefore, being a biological readout of activation.
For these experiments, we utilized T cells isolated from OT-I or OT-II mice. These mice have been genetically modified so that their T cell receptors (TCRs) recognize specific ovalbumin peptides in the context of the major histocompatibility class (MHC) I and II molecules (H2-Kb or I-Ab) respectively [50]. T cells were enriched from the spleen of these mice, with a purity of 60–80% CD3+ T cells as assessed by flow cytometry (data not shown). When the cells were purified from OT-I mice, 50–60% of all cells were CD8+ (data not shown). Wildtype murine BMDMs were cultured in the presence of the activating agent LPS and ovalbumin peptides (OVA257–264 or OVA323–339), which serve as defined antigens. Simultaneously, BMDMs were treated with either vehicle or 27HC for 24hrs. At this time, BMDMs were washed to preclude the direct effects of 27HC on T cells, and subsequently co-cultured with CFSE-labeled CD3+ T cells isolated from OT-I or OT-II mice.
Intriguingly, flow cytometry showed that 27HC-treated BMDMs failed to promote antigen-specific T cell expansion, a hallmark of T cell activation. Impaired expansion was observed regardless of the T cell subtype considered: CD4+ helper T cells (OT-II) or CD8+ cytotoxic T cells (OT-I) (Fig. 2A and B, respectively; representative histograms in Supplemental Fig. 1A). Therefore, 27HC-treated BMDMs result in decreased T cell expansion.
Figure 2: 27HC-treated macrophages decrease T cell activation and expansion.
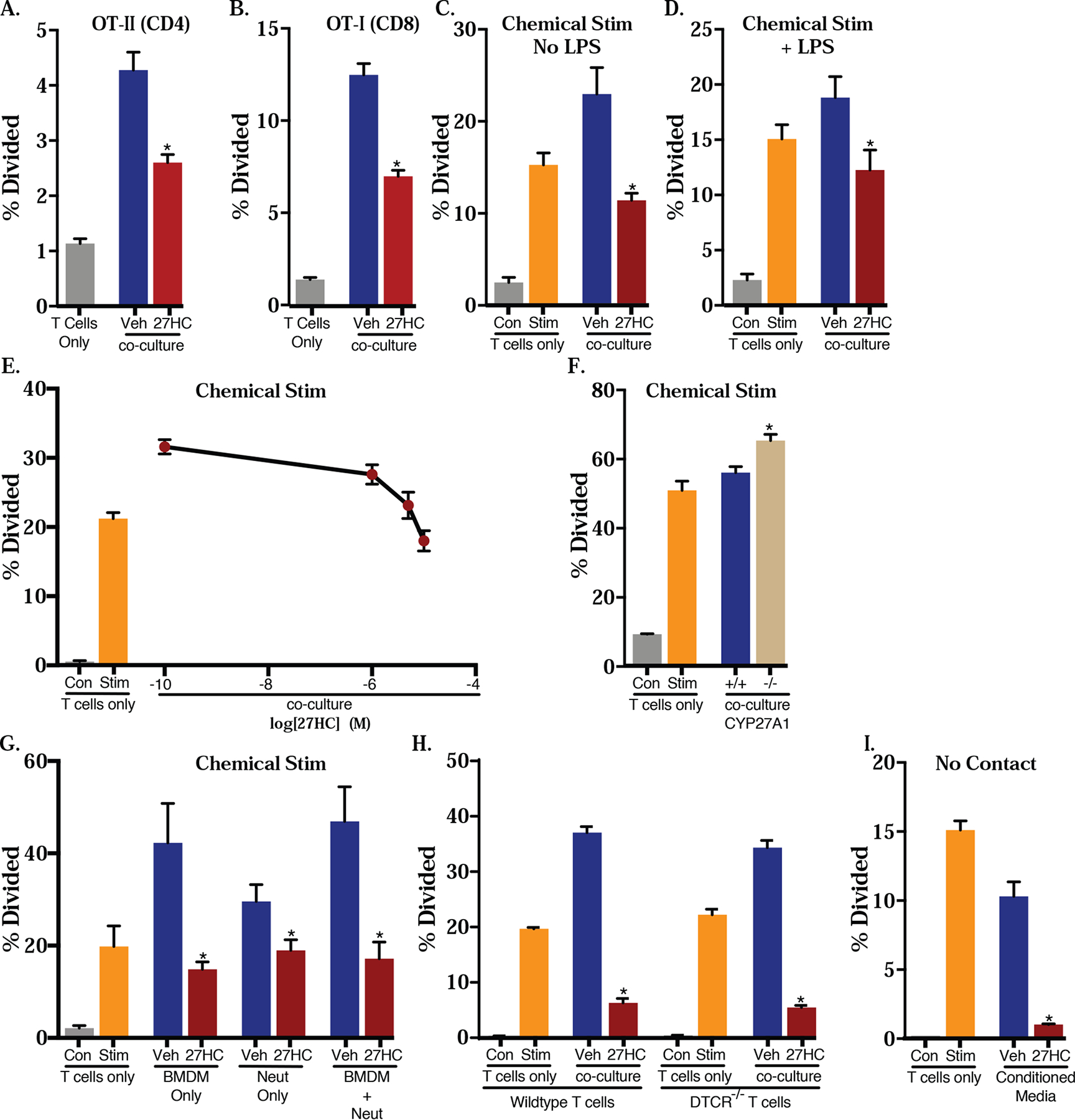
Murine BMDMs were cultured with OVA peptide in the presence or absence of indicated ligands in A and B. The media was then replaced and BMDMs were co-cultured with CFSE-labeled T cells from (A) OT-II mice (where TCRs of CD4+ T cells are engineered to recognize OVA), or (B) OT-I mice (where TCRs of CD8+ T cells are engineered to recognize OVA) for 72hrs. Subsequent T cell proliferation was quantified by flow cytometry for CFSE dilution. For subsequent panels (C-I), T cells from wildtype mice were activated by chemical stimulation with 10 nM PMA and 250 nM ionomycin and co-cultured with BMDMs. (F) BMDMs from wildtype or CYP27A1−/− were used for co-culture with T cells. (G) The same inhibitory effects of 27HC-treated BMDMs were observed with 27HC-treated neutrophils (PMNs). (H) γδ T cells are dispensable for the effects of 27HC-treated BMDMs on T cell expansion; T cells isolated from DTCR−/− mice do not have γδ T cells. (I) Direct contact between 27HC-treated BMDMs and T cells is not required for its inhibitory effects. Conditioned media from treated BMDMs was used to culture chemically activated T cells. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3 for all T cell only controls. For co-culture conditions, N=4/group in (A-D), N=5/group in (E), and N=6/group in (F-I)). (two-tailed t-test, p < 0.05)
Although the ability of 27HC to regulate MHCII and B7 expression (Fig. 1) is consistent with the decreased expansion of T cells resulting from a deficit in antigen presentation or co-stimulation, it is well known that macrophages can also impair T cell activity via several distinct mechanisms, including those independent of antigen presentation [51]. To assess this possibility, we evaluated the impact of 27HC-treated BMDMs on the expansion of ‘chemically’ activated T cells. Specifically, BMDMs were treated with vehicle or 27HC, along with or without stimulation signals, LPS and BSA. After the treatments were washed off, those BMDMs were then co-cultured with CFSE-labeled T cells that were activated chemically by phorbol 12-myristate 13-acetate (PMA) and ionomycin [as described in 34]. These activated T cells should be highly proliferative due to the activation of the PKC pathway without the need for specific antigen-presentation. The induction of T cell proliferation by PMA and ionomycin was confirmed in assays with T cells only (verified in each experiment, Fig. 2C–H).
Under these co-culture conditions, we found that 27HC treatment of BMDMs resulted in suppression of the expansion of chemically activated T cells (Fig. 2C). This impaired expansion was observed whether or not the BMDMs were previously stimulated by LPS and BSA (Fig. 2C–D). The suppression was found to be dose-dependent: when higher concentrations of 27HC were used to treat BMDMs, there was a concomitant decrease in subsequent T cell expansion (Fig. 2E). The decreased expansion was also apparent when conditioned media was cultured with naïve CD4+ T cells (Supplemental Fig. 1B). Therefore, the effects of 27HC on the macrophage-mediated impairment of T cell expansion did not require MHC-cognate antigen-TCR ligation, indicating that any macrophage exposed to 27HC within proximity to a T cell could suppress its expansion. Furthermore, this would suggest that 27HC-induced macrophages could impair T cell expansion regardless of how they are activated (as in, even if they were activated by dendritic cells), since the chemical activation strategy targets downstream cell surface interactions.
Given that the enzyme responsible for the synthesis of 27HC, CYP27A1 is highly expressed in macrophages, it was of interest whether an autocrine source of 27HC was sufficient to impair T cell expansion. Therefore, we evaluated the capacity of BMDMs isolated from CYP27A1−/− mice to influence T cell expansion. In support of our hypothesis, we found that compared to BMDMs from wildtype mice, co-culture of T cells with CYP27A1−/− BMDMs resulted in increased expansion (Fig. 2F). This finding is important, given that CYP27A1 is amenable to targeting with a small molecule inhibitor, and one such molecule was previously shown to have anti-metastatic effects [23].
Since we previously demonstrated the requirement of neutrophils and γδ T cells for the pro-metastatic effects of 27HC [23], we were interested in whether these cell types could influence the 27HC-mediated BMDM impairment of T cell expansion. Thus, we co-cultured 27HC-treated BMDMs, bone marrow-derived neutrophils or both cell types with chemically activated T cells. We found that 27HC-treated neutrophils alone could impair T cell expansion (Fig. 2G). There was no obvious additive or synergistic effect of co-culturing both neutrophils and BMDMs on T cell expansion (Fig. 2G). Culture of T cells from mice incapable of producing γδ T cells (DTCR−/− mice) continued to be suppressed by 27HC-treated BMDMs, suggesting that, at least in vitro, this cell type is dispensable for the immunosuppressive activities of 27HC (Fig. 2H). Collectively, these results indicate that (A) γδ T cells are not required, and (B) provide support for our hypothesis that 27HC results in impaired acquired immunity, regardless of what myeloid cell subtype it is working through, and the specific myeloid cell of importance is likely tumor type, tumor site and tumor-stage dependent [23, 24].
Our observations that BMDMs treated with 27HC impaired the ability of chemically activated T cells to proliferate, suggest that antigen-presentation is not required for 27HC-treated BMDMs to inhibit T cell expansion. Therefore, it was of interest as to whether direct contact between these two cell types was required and whether a soluble factor was mediating these effects. To address this question, we treated BMDMs with vehicle or 27HC for 24hrs and then washed the treatments off. BMDMs were then cultured in fresh media for another 72hrs, before we harvested supernatant from them. The conditioned media was subsequently used in culture with chemically activated T cells. Intriguingly, even without the presence of BMDMs, conditioned media alone exerted T cell suppression (representative experiments in Fig. 2I and Supplemental Fig. 1C). This is consistent with our findings that 27HC-pretreated mice continue to have increased metastatic burden post intravenous graft of mammary cancer cells, even when MHCII has been neutralized by an antibody (Supplemental Fig. 2). Thus, the 27HC-macrophage-mediated T cell impairment does not require cell-cell contact, suggesting that the effects of 27HC are mediated by a soluble factor(s).
T cells co-cultured with 27HC-treated macrophages have decreased cytotoxic activity.
Our data indicate that 27HC-treated BMDMs severely impair T cell expansion. However, it was also of interest whether those T cells that did expand had altered functional activity. In this regard, when we controlled for input post exposure to 27HC-treated BMDMs (Fig. 3A), we observed downregulated mRNA expression of cytotoxic T cell effector proteins, granzyme B and perforin (Fig. 3B–C). Subsequent ELISA analysis of T cell-conditioned media confirmed that cytotoxic T cells exposed to 27HC-treated BMDMs had decreased secretion of granzyme B (Fig. 3D). These findings indicate that the effector function of T cells was impaired after exposure to 27HC-treated BMDMs.
To specifically test whether anti-cancer T cell function was impaired by 27HC-treated BMDMs, we performed tumor cytolysis assays. T cells derived from OT-I mice were co-cultured with BMDMs exposed to LPS and OVA peptide, in the presence or absence of 27HC. As expected, 27HC resulted in decreased T cell expansion (Fig. 3E). The resulting T cells were harvested, counted, and then co-cultured with E0771 murine mammary cancer cells (controlling for T cell input) for 24hrs. Two different sublines of E0771 were used: the parental line that was stained with CFSE, and E0771-OVA cells that ectopically express OVA257–264 and were stained with Far Red [development of E0771-OVA cells described in 52]. In this way, antigen-specific cytolytic activity could be determined as a ratio between E077-OVA (Far Red) : parental E0771 (CFSE). A low ratio indicates strong cytolytic activity of T cells, while a high ratio suggests otherwise. Importantly, T cells harvested from co-culture with 27HC-treated BMDMs were less efficient at killing E0771-OVA cells (Fig. 3E–F). In order to ensure that these effects were not specific to E0771 cells, we repeated this experiment using the murine lymphoma cell line EL4 and its OVA257–264 expressing derivative, E.G7-OVA [53]. As expected, T cells harvested from co-culture with 27HC-treated BMDMs had significantly reduced capacity to eliminate this cell type, as the ratio between E.G7-OVA and EL4 remained high and unchanged, even with increasing numbers of antigen-specific T cells added to the co-culture (Fig. 3G–H). Collectively, our data indicate that 27HC-treated BMDMs not only impair T cell expansion, but also their functional activity, potentially explaining the robust increase in metastasis observed in 27HC-treated mice [23].
Elevated level of CYP7B1, the enzyme responsible for catabolizing 27HC, expression has been associated with improved progression-free survival among breast cancer patients, and is reflective of intra-tumoral 27HC concentrations [54, 55]. In an effort to demonstrate the relevance of our findings to human health, we surveyed The Human Cancer Genome Atlas (TCGA) as accessed through the Invasive Breast Carcinoma from Pan-Cancer Atlas. We assessed tumoral CYP7B1 mRNA, the enzyme responsible for the catabolism of 27HC, expecting that higher CYP7B1 levels would result in decreased 27HC levels, as recently demonstrated previously for human thyroid tumors [55]. We found a modest, positive correlation between CYP7B1 and CD3G, a component of the T cell receptor CD3 family (Fig. 3I). Likewise, there is a modest, positive correlation between CYP7B1 and both CD8 and CD4 expression (Fig. 3J–K). Finally, CYP7B1 had a modest, positive correlation with both T cell effector proteins, perforin 1, granzyme B, and IFN-γ (Fig. 3L–N). Even after normalizing for CD8A expression, we still observed a weak but significant positive correlation between granzyme B or perforin and CYP7B1 (Supplemental Fig. 3). All breast cancer subtypes were used for these analyses, which would be expected to exhibit heterogeneity. Therefore, data from human breast tumor specimens support our pre-clinical findings that 27HC impairs T cell expansion and function.
27HC alters cytokine secretion in macrophages (BMDMs), potentially contributing to its suppressive effect.
Since direct contact between BMDMs and T cells was dispensable for the suppressive effects of 27HC, we next examined the involvement of potential soluble factors, focusing on those previously reported to mediate the pro-tumor or immunosuppressive effect of macrophages [51, 56]. In this regard, TAMs have been shown to upregulate arginase 1 (ARG1) and indoleamine 2,3-dioxygenase 1 (IDO1) to deprive T cells of arginine and/or tryptophan, and thereby inhibit their activity (T cells being auxotrophic for these amino acids) [57]. However, supplementation with either arginine or tryptophan in our BMDM-T cell co-culture failed to attenuate the immunosuppressive effect of 27HC-treated BMDMs (Fig. 4A, B). CD8+ activity has been shown to be impaired by increased cholesterol content, and given that oxysterols would result in reverse-cholesterol transport in BMDMs, it was possible that nearby T cells were subject to elevated cholesterol [43, 58]. However, expansion of T cells isolated from LDLR−/− mice (and thus lacking the receptor to take up LDL-cholesterol) continued to be impaired by 27HC-treated BMDMs, although their baseline expansion was also decreased compared to wildtype T cells (Supplemental Fig. 4). It is important to note that LDLR−/− T cells may still be able to internalize cholesterol via scavenger receptors, so it is still possible that cholesterol mediates these effects.
Figure 4: T cell suppression resulting from 27HC treated macrophages is not mediated by common mechanisms.
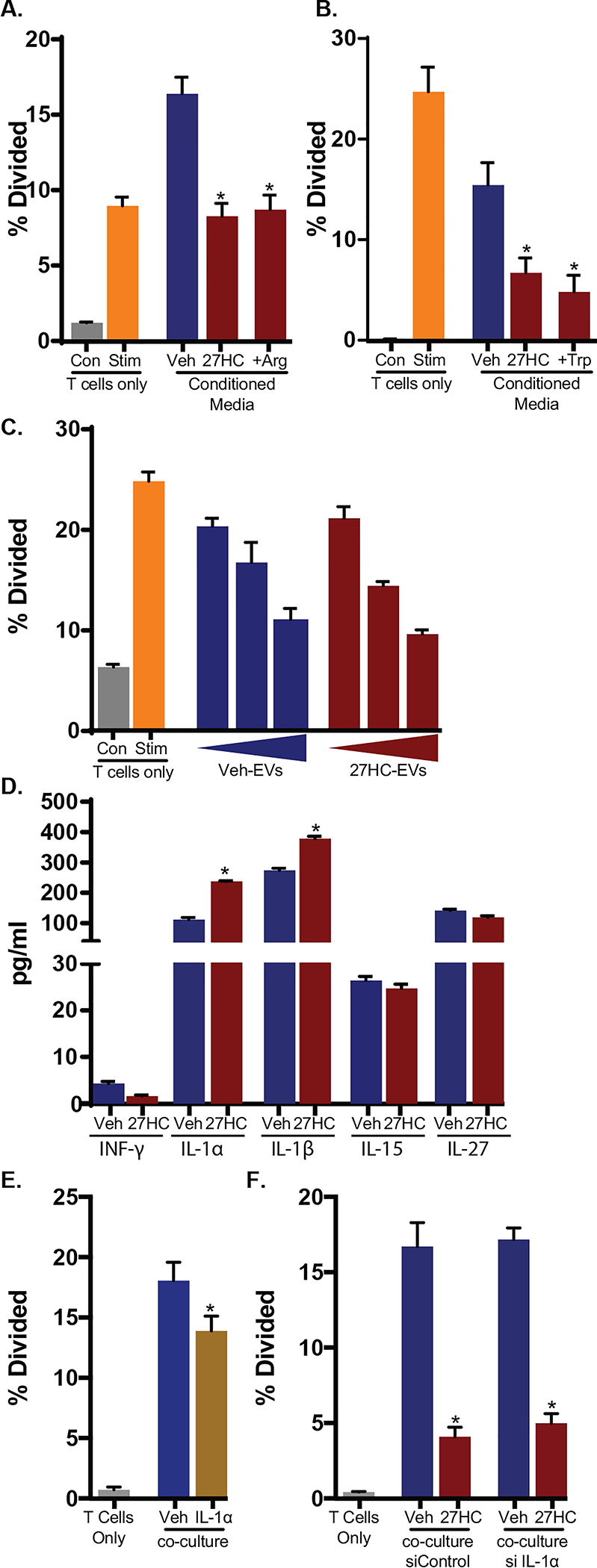
(A) Arginine (Arg) depletion by BMDMs is not likely the cause of impaired T cell expansion as supplementation with exogenous Arg failed to rescue the phenotype (N=3 for all controls, N=6/group for co-culture conditions). BMDMs were treated with vehicle or 27HC. Those cells were then co-cultured with chemically activated wildtype T cells. In the co-culture, 0.2 g/L of Arg or equivalent volume of PBS was supplemented. T cell expansion was measured by dilution of CFSE as assessed by flow cytometry. (B) Tryptophan (Trp) depletion by BMDMs is not likely the cause of impaired T cell expansion as supplementation with 0.005 g/L of exogenous Trp failed to rescue the phenotype (N=3 for all controls, N=4/group for co-culture conditions). (C) Extracellular vesicles derived from BMDMs are not likely the cause of impaired T cell expansion. Direct treatment of T cells with extracellular vesicles decrease expansion, but there was no difference between extracellular vesicles isolated from vehicle or 27HC-treated BMDMs (N=3). Extracellular vesicles were isolated from the conditioned media of vehicle- or 27HC-treated BMDMs at three increasing densities, using Total Exosome Isolation Reagent (Thermo Fisher, Cat no. 4478359) and then cultured with chemically activated T cells at three increasing doses. The increasing densities of starting BMDMs were 1x, 2x, or 3x the number of macrophages used in other experiments. T cell expansion was measured. (D) A cytokine array found modest changes in certain cytokines secreted from LPS-primed vehicle- or 27HC-treated BMDMs (full panels indicated in Supplemental Table 1) (N=3/group). (E) In line with previous reports, IL-1α, at the same concentrations induced by 27HC as determined in (D), resulted in decreased T cell expansion (N=5/group, Mann-Whitney U test). (F) IL-1α is not likely a sufficient cause of impaired T cell expansion as siRNA knockdown within BMDMs failed to reverse the effects of 27HC (N=3 for all controls, N=4–5/group for co-culture conditions). Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control. (two-tailed t-test unless otherwise specified, p < 0.05)
There are recent reports of extracellular vesicles and their miRNA cargo being important mediators of cholesterol homeostasis [59, 60]. Extracellular vesicles, including exosomes are known to be secreted by myeloid cells, and have been implicated in immune suppression [61, 62]. Therefore, it is possible that 27HC-treated BMDMs released extracellular vesicles that mediated the decreased T cell expansion and function. To test this possibility, we isolated extracellular vesicles from vehicle or 27HC-treated BMDMs and cultured them with chemically activated T cells. Although there was an inhibition of T cell expansion by extracellular vesicles isolated from increasing numbers of BMDMs, there was no difference between extracellular vesicles derived from vehicle or 27HC-treated BMDMs (Fig. 4C). Therefore, it is unlikely that the inhibitory effects of 27HC-treated BMDMs are mediated through extracellular vesicles.
Several cytokines secreted by BMDMs have been described to inhibit T cell activation or proliferation. Therefore, we assessed a multiplex panel of murine cytokines at two time points: 24hrs after treatment with vehicle or 27HC and 72hrs after co-culture with T cells. The majority of changes observed with the treatment of 27HC were apparent after 24hrs (Supplemental Table 1A, B). Under basal conditions, many cytokines were below the detection thresholds of the assay, although 13 were detected and of those, only two were significantly regulated by 27HC after 24hrs (MIP-1a and MIP-1b), but these were no longer significantly different after 72hrs in culture (Supplemental Table 1A). When stimulated with LPS, several cytokines reached detectable concentrations after 24hrs, and of those, only IL-1α and IL-1β were significantly regulated by 27HC, although IL-6, IFN-γ, IL-18 and MIP-1b had false discovery rates <0.15 (Fig. 4D and Supplemental Table 1B). TGFβ was not included in the multiplex panel, but of potential relevance, and thus was assessed independently by ELISA (Supplemental Fig. 5). Of those cytokines regulated by 27HC, IL-1α was of interest as it has previously been shown to influence the growth and activity of T cells [63–66], and was consistently upregulated by 27HC under both time points. We confirmed this observation, as addition of IL-1α at a concentration reflective of measured levels in our co-culture, resulted in reduced proliferation of T cells activated by antigen presentation (Fig. 4E). However, siRNA mediated knockdown of IL-1α in BMDMs, did not significantly impair the ability of 27HC-treated BMDMs to inhibit T cell expansion (Fig. 4F). These observations indicate that while IL-1α may play a role in suppressing T cells, it is unlikely to be only one factor among several that mediate the effects of 27HC. It is very possible that 27HC modulates the phenotype of BMDMs through several diverse mechanisms, which converge to exhibit a strong immunosuppressive effect on T cells.
Reduced T cell expansion by 27HC-treated macrophages is mediated by the LXR.
Since 27HC is an abundant oxysterol, and its presence and metabolism have been clinically associated with breast, ovarian and thyroid cancers [24, 54, 55, 67], it was of importance to determine the mechanism by which 27HC alters macrophages to impair T cell expansion and function. Previously reported pathways involved in macrophage function or cholesterol signaling (PI3K, AKT, NOTCH and CaMKK2) were ruled out through the use of pharmacologic inhibitors of these enzymes (Supplemental Fig. 6). Thus, to provide initial insight into potential mechanisms, we assessed the ability of 12 different oxysterols, cholesterol metabolites and synthetic LXR ligands to inhibit T cell expansion via treatment of BMDMs (Supplemental Table 2). Most oxysterols and cholesterol metabolites tested impaired T cell expansion to varying degrees (Fig. 5A). Dendrogenin A at 1 μM was a notable exception, with no noticeable effect on T cell expansion. This finding is in line with previous reports indicating that this metabolite induces tumor cell differentiation and immune cell infiltration and thus is associated with a good prognosis [68, 69]. Moreover, when BMDMs were co-treated, dendrogenin A exhibited dose-dependent attenuation of the actions of 27HC (Fig. 5A). This indicates that the effects of 27HC are likely receptor-mediated, and that dendrogenin A has the capacity to out-compete 27HC for binding.
Figure 5: LXR mediates the myeloid cell immunosuppressive effect of 27HC.
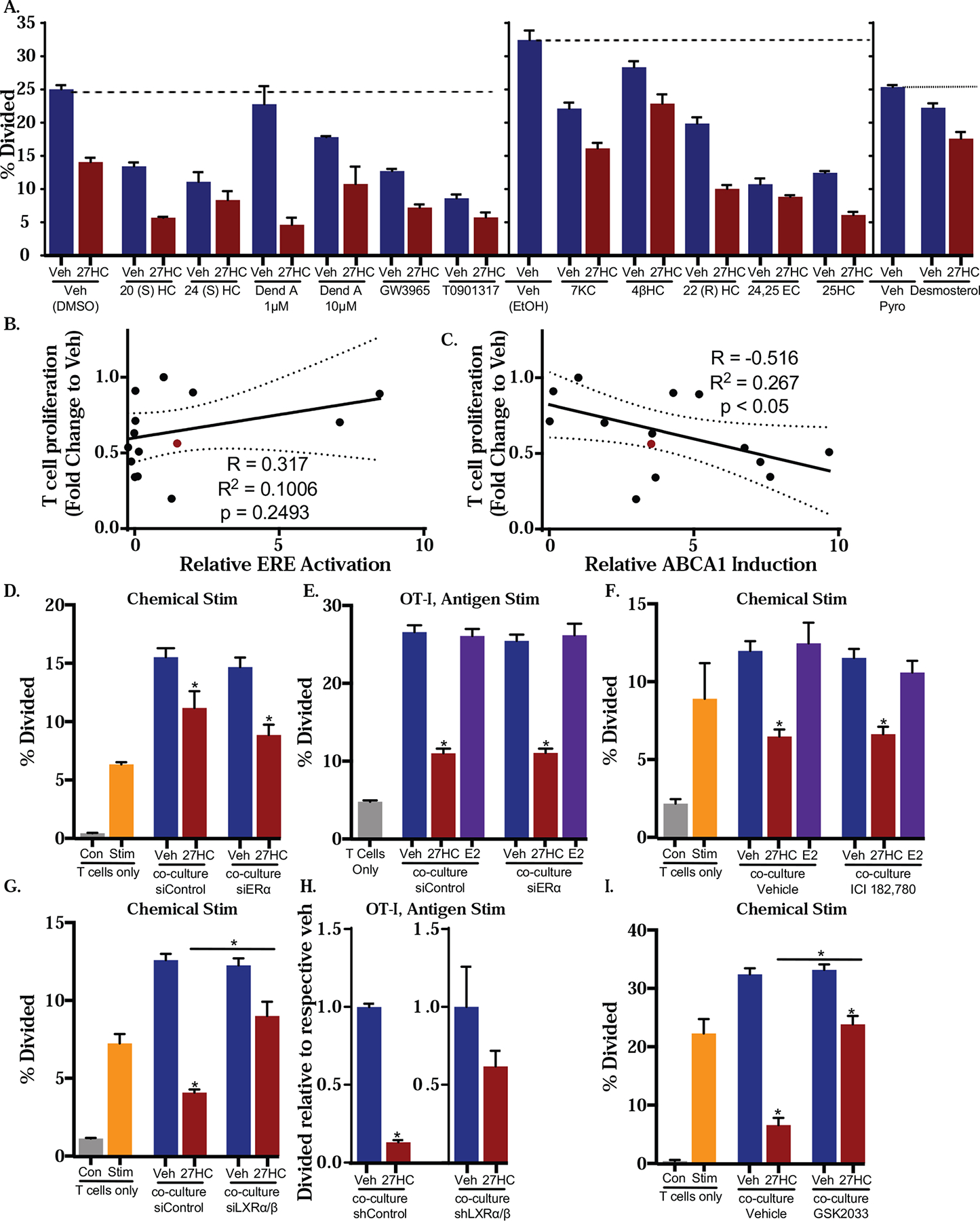
(A) BMDMs were treated with 12 cholesterol metabolites, LXR ligands or indicated vehicle controls for 24hrs prior to co-culture with chemically activated T cells for 72hrs, and proliferation was assessed. (B-C) Two cohorts of BMDMs were treated with ligands for 24hrs. Expression of ABCA1 mRNA was assessed in the first cohort. BMDMs from the second cohort were co-cultured with chemically activated T cells for 72hrs post treatment, at which point T cell proliferation was measured. (B) T47D cells stably transfected with a ERE luciferase reporter were treated as in (A) and the activation of ER was measured by luciferase activity. Pearson’s correlation between T cell proliferation and ER activity is presented. (C) Pearson’s correlation between T cell proliferation and induction of ABCA1 mRNA is presented. (D) BMDMs were transfected with siControl or siERα for 48hrs and then treated with vehicle or 27HC. After 24hrs, the treatments were washed off, and media was replaced. Those BMDMs were co-cultured with chemically activated T cells for 72hrs. (E) BMDMs transfected with siControl or siERα for 48hrs were primed with OVA and treated with vehicle or 27HC for 24hrs. BMDMs were then co-cultured with CD3+ T cells from OT-I mice for 72hrs and proliferation assessed. (F) BMDMs were treated with vehicle, 27HC, ICI (an ER antagonist), E2, ICI+27HC or E2+27HC. BMDMs were then co-cultured with chemically activated wildtype T cells as in (D). (G) BMDMs were transfected with siControl or siLXRα/β for 48hrs. Those BMDMs were then harvested, treated and co-cultured with chemically activated T cells as in (D). (H) BMDMs were transduced with lentiviral shControl or shLXRα/ββplasmids for 96hrs and then treated and co-cultured as in (E). Data presented as normalized to respective control. (I) LXR antagonist (GSK2033) attenuates effects of 27HC. BMDMs were treated and co-cultured with T cells as in (D). All T cells were labeled with CFSE and the proliferation was assessed by flow cytometry. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3 for all T cell only controls. For co-culture conditions: N=4 for A, E, H and I. N=3 for B. N=5–6 for D and F. N=6 for G. (two-tailed t-test, p < 0.05). For luciferase activity in (C), N=4)
27HC is known to bind to two nuclear receptors, ER and LXR, both receptors being implicated in immunomodulatory effects [43, 70]. Since several oxysterols have been shown to modulate the activities of the ERs and LXRs to different extents, we first evaluated capacity of the same panel of oxysterols and cholesterol metabolites (Supplemental Table 2) to activate the ERs and LXRs. We then assessed whether there was a correlation between this receptor activation and impaired T cell expansion. T47D cells, engineered to stably express an ER-reporter (estrogen response element upstream of a luciferase construct) [71], were treated with different doses of the ligands. Several oxysterols were found to have estrogenic activity, including those not previously been reported to do so (Fig. 5B, Supplemental Fig. 7 and Supplemental Table 2). Importantly, there was no notable correlation between the ability to activate ER-mediated transcription and inhibition of T cell expansion (Fig. 5B). Furthermore, no impact on T cell expansion was observed after treatment of BMDMs with either E2, a naturally occurring ER agonist, or ICI 182,780 (ICI, Fulvestrant) an ER antagonist and degrader (Fig. 5B, F). These results indicate that impairment of T cell expansion upon 27HC treatment of BMDMs is unlikely to be ER-mediated.
In order to assess the ability of different ligands to activate the LXRs, we evaluated the extent to which they could induce ABCA1, a well-known LXR target gene, in BMDMs. As expected, 27HC and many of the oxysterols could induce ABCA1 as well as another LXR target gene, ABCG1, while E2 did alter basal expression of ABCA1 (Supplemental Fig. 8). Interestingly, the ability of a ligand to induce ABCA1 expression was moderately and significantly correlated with its ability to inhibit T cell proliferation after treatment of BMDMs (Fig. 5C). This suggests that modulation of the LXR is mediating the effects of 27HC and other oxysterols. To definitively test this, we used complementary strategies of RNA-interference and pharmacologic antagonists.
ERα has been reported to be the predominant subtype in macrophages [72–74]. Therefore, we first evaluated the impact of knocking down ERα on the 27HC-mediated decrease in T cell expansion and function. Although siRNA-mediated knockdown resulted in an over 80% decrease in ERα mRNA, 27HC treatment of these BMDMs continued to inhibit the expansion of chemically activated T cells (Fig. 5D). Similarly, siRNA against ERα in BMDMs did not impact the ability of 27HC to decrease OT-I T cell proliferation after antigen-mediated activation (Fig. 5E). Furthermore, the cytotoxic capacity of T cells was reduced by 27HC-treated BMDMs, regardless of ERα expression (not shown). We confirmed these findings using the pharmacological antagonist and ER degrader, ICI 182,780 (ICI; Fulvestrant; Fig. 5F) [75]. ICI antagonizes both ERα and β subtypes. As expected, ICI treatment failed to ‘rescue’ the inhibitory effects of 27HC-treated BMDMs on T cell proliferation or cytotoxic capacity (Fig. 5F and not shown). Collectively, these data strongly indicate that the mechanism by which 27HC modulates macrophages to impair T cell expansion and function is independent of the ER.
Treatment of BMDMs with a synthetic LXR agonist, GW3965, resulted in impaired T cell expansion (Fig. 5A, B), suggesting that this receptor may mediate the effects of 27HC. Both subtypes of LXRs (α and β) are expressed in BMDMs and whether they play specific roles is unclear. Therefore, we elected to knockdown both subtypes simultaneously. mRNA expression shows that siRNA-mediated knockdown of LXRα was marginal in several attempts, while LXRβ knockdown was moderate (around 10% and 50%, respectively, Supplemental Fig. 9A). However, even in the context of this partial knockdown of LXR, the immunosuppressive effect of 27HC-treated BMDMs on chemically activated T cells was significantly attenuated (Fig. 5G). shRNA against the LXRs administered by lentivirus infection of BMDMs (Supplemental Fig. 9B) also lead to the attenuation of 27HC-mediated impairment of OT-I T cell expansion after antigen-activation (Fig. 5H). The involvement of the LXRs was corroborated as co-treatment with the small molecule LXR antagonist, GSK2033 [76–80], restored T cell proliferation when cultured with 27HC-treated BMDMs (Fig. 5I). In this assay, GSK2033 was also able to ‘rescue’ the inhibitory effect of BMDMs treated with a synthetic ligand of the LXRs, GW3965 (Supplementary Fig. 9C). GSK2033 has been implicated in modulating the activity of other receptors (such as RORγ and ERRα) [81], and this cannot be ruled out here, although the effects of siRNA against the LXRs would support the conclusion that LXRs are mediating the effects of 27HC.
27HC-treated macrophages impair T cell function by promoting T cell apoptosis
Mechanistically, it was important to investigate whether the impaired T cell activity was a result of impairment of initial activation or post-activation proliferation, as myeloid-derived suppressor cells and macrophages have been previously shown to inhibit T cell activation [82, 83]. Consistent with this notion, we observed down-regulation of several markers of T cell activation (CD25, CD27 and CD69) 48 and 72hrs after co-culture with 27HC-treated BMDMs (Fig. 6A–C). However, down-regulation of CD62L expression was also observed (Fig. 6D). Shedding of CD62L is commonly observed in activated T cells [84]. A lower level of CD62L in T cells cultured with 27HC-treated BMDMs would suggest that those cells were indeed being activated, despite decreased expression of early activation markers and effector proteins (Fig. 6A–C, 3B–D). While mRNA expression is not necessarily correlated to surface protein concentration, one explanation for this apparent contradiction and the consistent, unidirectional change in gene expression is that 27HC-treated BMDMs induce T cell death, thereby leading to global downregulation of gene expression [85–88]. To investigate this possibility, we labeled OT-I T cells 48 or 72hrs after co-culture with Annexin V-FITC and propidium iodide. Annexin V binds to phosphatidylserine, a membrane lipid that becomes externalized and accessible during apoptosis. Propidium iodide (PI) enters cells with disrupted membranes and binds DNA, indicative of cell necrosis. Through this assay, we found that at 48hr time point, there was no significant difference in live cell (Annexin V−/PI−) and necrotic cell (Annexin V−/PI+) populations (Fig. 6E). However, at both 48 and 72hr time points, 27HC-treated BMDMs induced a significantly greater number of Annexin V+/PI+ (indicative of late apoptotic/necrotic) T cells (Fig. 6E and Supplemental Fig. 10A). It is important to note that the 27HC group also had lower levels of early apoptotic cell population (Annexin V+/PI−; Fig. 6E), which could be a result of increasing number of cells moving into late apoptosis. Given that Annexin V is capable of entering ruptured membrane and binding to phosphatidylserine in necrotic cells and PI also interacts with DNA of cells during late apoptosis, we could not definitively tell whether the Annexin V+/PI+ population was a result of late apoptosis, necrosis or both [89, 90]. In order to conclusively identify the type of BMDM-driven T cell death, we alternatively labeled T cells, harvested from 3 time points (24, 48, 72hrs) post co-culture with vehicle- or 27HC-treated BMDMs, with fluorescent caspase-3/7 inhibitor probe FAM-DEVD-FMK (FLICA-FAM) and propidium iodide. FAM-DEVD-FMK covalently binds to activated caspase-3/7 through the fluoromethylketone (fmk) moiety [91, 92]. Because caspase-3 is an executioner in both the intrinsic and extrinsic apoptosis pathways but has no role in necrosis, presence of active caspase-3 in T cells after co-culture with 27HC-treated BMDMs more clearly distinguishes between apoptosis and necrosis. Specifically, FLICA+/PI− and FLICA+/PI+ population indicate early and late apoptosis respectively, while FLICA−/PI+ suggests necrosis. Consistent with the Annexin V/PI assay, results from this approach support the conclusion that 27HC-treated BMDMs induce T cell apoptosis between 24 and 48 hours after co-culture (Fig. 6F, Supplemental Fig. 10B). Notably, T cells in contact with 27HC-treated BMDMs entered late apoptosis at a much higher rate and, 72 hours after co-culture, there were less than half the live cells when cultured with 27HC-treated BMDMs compared to control (Fig. 6F). Since splenic T cells include different forms such as memory T cells and thus may not be representative of a normal T cell immune response, we isolated CD4+ naïve T cells, chemically activated them, and cultured them in the presence of conditioned media from either vehicle or 27HC treated BMDMs. Under these conditions, naïve T cells had increased late apoptotic cells after 48hrs of exposure to conditioned media from 27HC-treated BMDMs (Fig. 6G), mirroring results from pan-splenic T cells. Therefore, our results suggest that 27HC modulated BMDMs promote accelerated T cell apoptosis, where the T cells rapidly (within 48hrs) progressed from an early into a late apoptotic stage. Admittedly, at 72hrs, a small yet significant increase in a population of necrotic T cells was observed after T cells were co-cultured with 27HC-treated macrophages, and therefore we cannot rule out the possibility that caspase-3/7-independent pathways could also play a role in mediating 27HC-driven T cell dysfunction.
The immune checkpoint molecule, PD-L1 is known to induce apoptosis in effector T cells [93]. Therefore, we assessed PD-L1 mRNA in BMDMs treated with 27HC (same experiment as in Fig. 1). No changes in PD-L1 transcript were observed (Fig. 6H). Intriguingly however, there was a five-fold increase in soluble PD-L1 in conditioned media from 27HC-treated BMDMs, as assessed by ELISA (Fig. 6I). The biological relevance of soluble PD-L1 is still unclear, but emerging reports suggest that it too can induce apoptosis [94], suggesting that this may be the putative factor mediating the pro-apoptotic effects of 27HC.
Absence or inhibition of CYP27A1 results in increased antigen-specific T cell cytolysis, decreased tumor growth and enhanced efficacy of immune checkpoint inhibition.
To determine whether the impaired T cell expansion and function upon BMDM treatment with 27HC occurred within the complex in vivo microenvironment, we orthotopically grafted E0771-OVA cells into mice that had CYP27A1 genetically knocked out specifically in the myeloid immune lineage (CYP27A1flfl;LysMCre+) and respective control mice (CYP27A1+/+;LysMCre+) (genotyping and selective knockout demonstrated in Supplemental Fig. 11). A myeloid-specific knockout approach was adopted, because introduction of exogenous 27HC would likely have direct effects on cancer cells, potentially confounding interpretation our results [22, 24]. After tumors were established, we then grafted all mice with OT-I T cells and measured resulting tumor growth; OT-I T cells were expected to specifically target OVA-expressing cells after presentation and activation by myeloid cells. Since we hypothesized that loss of myeloid cell CYP27A1 would result in increased T cell function, the OT-I T cells were grafted at a suboptimal concentration and would not be expected to fully eradicate the tumor. As expected, loss of myeloid CYP27A1 resulted in an increased number of mice whose tumors grew at a reduced rate post OT-I T cell graft compared to those grown in CYP27A1 replete mice (using a quadrupling of tumor volume as our threshold) (Fig. 7A and Supplemental Fig. 12A–B).
We have previously shown that 27HC robustly increases the metastatic colonization and growth of breast cancer cells in a myeloid cell dependent manner [23]. Given the results of the current study indicating that 27HC-treated BMDMs significantly impair T cell expansion and anti-cancer function, we therefore wanted to determine whether loss of myeloid cell expression of CYP27A1 would itself result in altered metastatic colonization and growth. Therefore, mice were grafted intravenously with E0771 cells, and subsequent metastatic lesions were quantified 3 weeks later. Intriguingly, significantly reduced lung metastatic burden was observed in CYP27A1flfl;LysMCre+ mice compared to control mice (Fig. 7B and Supplemental Fig. 12C). Furthermore, the only mice exhibiting macro-metastases outside the lung were from CYP27A1 intact controls (Fig. 7C). Since LysM promoter is active in all myeloid cells, one cannot ascribe the effects solely to the loss of CYP27A1 to macrophages, but rather to all types of myeloid cells. This is likely the case, as neutrophils also displayed inhibitory effects when treated with 27HC (Fig. 2G).
Immune checkpoint inhibition is, at best, modestly effective for the treatment of metastatic breast cancer, even within the patient cohort that is approved for its use. Since myeloid cells highly express CYP27A1 and are abundant in breast tumors and metastatic lesions, we speculated that inhibition of 27HC synthesis may increase the efficacy of this therapeutic strategy. Thus, we grafted mice intravenously with the highly aggressive 4T1 model of TNBC and allowed lesions to establish. At this point, mice were randomized and treated with αPD-L1 (checkpoint inhibitor), GW273297X (a small molecule inhibitor of CYP27A1), or both. As expected, treatment with αPD-L1 alone had no significant effects on metastatic burden (Fig. 7D). In this model, exogenous 27HC treatment robustly increased metastatic burden, regardless of whether mice were treated with αPD-L1, indicating that the pro-metastatic effects of 27HC are likely a combination of enhanced colonization [23] as well as growth (Fig. 7D). Interestingly however, treatment with GW273297X as a single agent modestly reduced metastatic burden (Fig. 7D). Importantly, co-treatment with GW273297X and αPD-L1 further reduced metastatic burden, suggesting that myeloid-cell synthesized 27HC does impede the actions of checkpoint inhibition.
Discussion
Despite recent improvements in the treatment of breast cancer, advanced-stage and metastatic disease remain major contributors to breast cancer mortality. Although success using checkpoint inhibition in advanced TNBC have been reported and is now approved in combination with nab-Paclitaxel for a subset of patients, its overall response rate in breast cancer remains low [6]. Therefore, developing a clear understanding of the checkpoint-independent immune suppression biology within breast cancer is critical to the future development of curative therapies. The current work indicates that the actions of cholesterol metabolites such as 27HC on myeloid cells results in severe T cell impairment, both in terms of expansion as well as function.
Clinically, elevated cholesterol has been shown to be associated with breast cancer recurrence, while patients taking cholesterol-lowering medication (statins) have increased time to recurrence [95]. In preclinical models, a high cholesterol diet increases tumor growth and metastasis of mammary cancer [95]. However, our previous work has found that the pathophysiology of cholesterol on breast cancer progression is largely attributed to its primary metabolite, 27HC, as the growth and metastatic effects of a high cholesterol diet are absent in CYP27A1−/− mice [22, 23]. 27HC promotes primary tumor growth through a cancer-cell intrinsic mechanism by being an ER agonist and activating the ERs [22]. However, exogenous 27HC does not require the expression of ERs in cancer cells to robustly increase metastatic burden in mice [22, 23]. Engagement of the LXR by 27HC did result in increased EMT and modest increases in metastatic capacity of mammary cancer cell lines [22, 96]. However, we have shown that 27HC also acts in a cell extrinsic manner to robustly promote metastatic colonization [23]. Specifically, it was found that myeloid cells were required for these effects. Immune depletion of neutrophils ablated the pro-metastatic effects of 27HC, suggesting involvement of this cell type. This is perhaps because neutrophils dominate the myeloid cell landscape at the lung metastatic site of mice [97, 98]. On the other hand, the ovarian tumor microenvironment required 27HC, which appeared to mediate its effects through MDSCs [24]. Thus, it is likely that while 27HC exerts its effects on all myeloid cells, the extent to which it does so is dependent on the tumoral and microenvironmental context. Therefore, we explored the biology of 27HC in the prototypical myeloid cell, the macrophage.
Here, we found that antigen-primed, 27HC-exposed macrophages not only failed to promote the expansion of T helper cells and cytotoxic T cells, likely through induction of apoptosis, but also severely impaired the cytolytic capacity of cytotoxic T cells against cancer cells. We further showed that the suppression of T cells by 27HC-treated macrophages is not limited to the antigen-presentation pathway, as unprimed macrophages could also suppress chemically activated T cell expansion. The general suppression of T cells by 27HC-modulated macrophages suggests even though T cells are already active, their proliferation and effector functions could still be severely impaired by 27HC-exposed macrophages in the tumor microenvironment. This is in line with a previous report that an oxysterol mixture could promote ‘M2’-like macrophages [99].
The ability of media-conditioned with macrophages treated with 27HC to suppress T cells indicates that certain soluble factors mediate the effects. Through our experiments, we ruled out obvious metabolic regulators (arginine and tryptophan) and extracellular vesicles. We have also identified several cytokines that are regulated by 27HC in time-dependent manners. Of those that are consistently altered by 27HC and known to result in T cell impairment, the involvement of IL-1α [63–66] was specifically tested, with our results indicating that this cytokine alone was not mediating the effects of 27HC.
27HC-treated BMDMs were found to increase T cell apoptosis. Conditioned media from 27HC-BMDMs also increased apoptosis in naïve T cells, in line with our other evidence suggesting that a soluble factor was responsible for the T cell suppressive effects of 27HC. Since the immune checkpoint molecule, PD-L1 is known to induce apoptosis in effector T cells [93], we explored whether this factor was regulated by 27HC. Its transcript levels were not altered in BMDMs post-treatment with 27HC (Fig. 6). To our surprise however, soluble PD-L1 was significantly increased in conditioned media from 27HC-treated BMDMs. The soluble form of PD-L1 is an active area of study, with several isoforms (splice variants) being described [100–102], and its precise biological functions and relevance being unclear. However, it has been found to induce apoptosis, similar to its membrane-bound form [94]. This suggests that soluble PD-L1 may be the putative factor mediating the pro-apoptotic effects of 27HC. Our results provide strong rationale for future studies exploring whether this is indeed mediating the effects of 27HC, how 27HC alters its secretion but not its mRNA expression, and whether 27HC alters different splice variants. The specific soluble factor(s) from myeloid cells responsible for mediating the suppressive effects of 27HC remain to be determined.
Our results are supported by preclinical models as metastatic lesions from mice treated with 27HC had decreased CD8+ T cells [23]. Furthermore, human patient tissue from breast cancer tumors with decreased CYP7B1 expression and therefore expected increased 27HC concentrations [55] had decreased expression of CD3G, CD4, CD8, perforin and granzyme B ([23] and Fig. 3I–M). This highlights the relevance of our results to human health and provides context to a recent report indicating that the recruitment of monocytes and subsequent epigenetic silencing of CYP7B1 results in the accumulation of intratumoral 27HC in breast cancer, and corroborated by another report that breast tumors had increased 27HC compared to normal-adjacent material [103, 104].
There have been several reports indicating that the synthesis/secretion of oxysterols is altered upon inflammatory stimuli [105, 106], and in turn, oxysterols exert modulatory activities on the immune system and/or inflammation [43, 107]. Mechanistically, we have shown that, macrophage LXRs, but not ERs, are required for the immunosuppressive effect of 27HC, as knocking down LXRs or blocking them with a pharmacological antagonist attenuates the suppressive effects of 27HC on T cells. Further supporting this finding are revelations of a positive correlation between the ability of different oxysterols, cholesterol metabolites and synthetic LXR agonists to induce the LXR target gene ABCA1, and their ability to suppress T cell expansion. The involvement of LXR in the immunosuppressive activities of 27HC provides a contrast to certain inflammatory actions of 27HC being mediated through the ER, at least with respect to cardiovascular disease [108], although LXR activation by 27HC has been implicated in the protective effects of vitamin D on atherosclerosis in a preclinical model [109]. Estrogen has been shown to increase hematopoietic stem cell proliferation [110] and myelopoiesis [70]. 27HC was also found to increase hematopoietic stem cell mobilization but not proliferation [110]. Estrogen was found to enhance the immunosuppressive activities of MDSCs [70]. However, here, we show that despite 27HC being an ER ligand, it is its activities through the LXRs in myeloid cells that result in T cell suppression.
Several previous studies have demonstrated that oxysterols and the LXR are involved in management of the innate immune system [99, 111–114]. Interestingly, in apparent contrast with previous reports indicating that macrophages lacking LXRs had increased apoptosis [115], or were more susceptible to LPS-induced liver injury and resistant to inflammatory responses caused by treatment with α-galactosylceramide or concanavalin-A [113], treatment of mice and humans with synthetic LXR agonists decreased MDSC abundance within tumors by decreasing their [MDSC] survival [116]. Specifically, MDSCs isolated from tumors of LXR-treated mice were less able to suppress T cell expansion compared to those isolated form vehicle-treated mice [116]. This apparent discrepancy with our work indicating that myeloid cells treated directly with LXR agonists impair T cell function may be due to either: (1) Differential kinetics and compensatory mechanisms, it is possible that MDSCs isolated from in vivo treated mice lose their LXR signal and overcompensate before returning to baseline. (2) Different myeloid cell types may behave differently. Our data indicate that 27HC induces an immune suppressive phenotype on both macrophages and neutrophils, and as such ‘MDSCs’ may not behave similarly. (3) LXR agonists may exert their effects in a ‘selective’ fashion, similar to selective estrogen receptor modulators. In other words, different LXR agonists may behave differentially, in a context-dependent \ specific manner. Even though the exact immunomodulatory pathway downstream of LXR is currently unknown, previous reports have shown that the LXR agonists suppress NLRP3 inflammasome activation, this downregulation being associated with poorer clinical outcome in hepatocellular carcinoma [117–119]. Regardless of mechanism, our work presented here, along with emerging evidence of cancer-cell intrinsic roles for the LXRs [22, 96], would suggest that caution should be taken prior to the development of LXR as an immune-modulatory target for the treatment of solid tumors. Furthermore, these results provide a strong rationale for studies to elucidate the precise signaling events downstream of LXR.
One potential limitation to our study is the reliance on BMDMs, which may not be reflective of macrophages or other myeloid cells within the tumor microenvironment. Indeed, microenvironmental factors can significantly alter macrophage response. Since 27HC is likely one of these factors, it is still relevant that naïve BMDMs are ‘entrained’ by this oxysterol. Furthermore, bone marrow-derived neutrophils treated with 27HC also exhibited an anti-T cell response (Fig. 2). Finally, our in vivo data (Fig. 7) and correlational data from human tumors (Fig. 3) support an immune-suppressive role for 27HC within the tumor microenvironment.
Collectively, we have shown that 27HC-treated macrophages strongly impair the expansion of T cells, suggesting that inhibition of 27HC synthesis should improve the efficacy of immune therapy. Indeed, in a preclinical model of metastatic breast cancer, blocking 27HC synthesis with a small molecule inhibitor of CYP27A1 resulted in a positive response to αPD-L1. The expression profile of CYP27A1, which is abundant in myeloid cells the liver and within cancer cells of high-grade tumors [22, 103, 120] suggests that targeting this enzyme will have a low risk of unwarranted side effects. Our work also reinforces the broader strategy of reeducating myeloid cells to be pro-immune and anti-cancer, rather than ablating them altogether [11, 23, 121–123].
Supplementary Material
Supplemental Figure 1. 27HC-treated macrophages suppress T cell expansion. (A) Representative histograms from flow cytometry. CD3+ T cells were isolated from the spleen of OT-1 mice, stained with CFSE, were co-cultured with BMDMs treated with OVA257–264 and either vehicle- or 27HC. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. This data is from Fig. 3E, and is also representative of Figs. 2D&E. (B) Naïve T cells were isolated from the spleen of wildtype C57BL/6 micese, chemically activated and cultured in the presence of conditioned media harvested from vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the vehicle control (N=3/group for T cell only control, N=6/group for conditioned media two-tailed t-test, p < 0.05). (C). CD3+ T cells were isolated from the spleen of wildtype C57BL/6 mice, labeled with CFSE, chemically activated and cultured with conditioned media harvested from vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for T cell only controls, for conditioned-media conditions, N=5–6/group). (two-tailed t-test, p < 0.05). This sub-figure is an independent repeat of Fig. 1I.
Supplemental Figure 2. Blockade of MHCII does not attenuate pro-colonizing effects of 27HC. Mice were pretreated with placebo or 27HC (20mg/kg/d) for 5d, as in [23]. 36h and 12h prior to cell graft, mice were injected (i.p) with an isotype control antibody (IgG) or an antibody against murine I-A/I-E. Mice were then grafted with Met1 cells expressing iRFP. Resulting cancer colonies within the lung were quantified by ex vivo imaging for iRFP fluorescence. Different letters denote statistical significance, N=8/group. (one-way ANOVA followed by Neuman-Kuels multiple comparison test, p < 0.05).
Supplemental Figure 3. Expression of granzyme B (GZMB) and perforin (PRF1) normalized to CD8A expression was modestly but significantly correlated with CYP7B1 expression. mRNA expressions from Human RNA sequencing data from TCGA Pan Cancer for invasive breast carcinoma were log2 transformed and Pearson’s correlation was assessed (N=1077 in (A) and 1081 in (B)). These data partner with Fig. 3L and M.
Supplemental Figure 4. Cholesterol efflux from macrophages unlikely to mediate the suppressive effect of 27HC on T cells. CD3+ T cells were isolated from wildtype C57BL/6 or LDLR−/− mice, labeled with CFSE and chemically activated. They were then co-cultured with vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for all T cell only controls, for co-culture conditions, N=5/group). (two-tailed t-test, p < 0.05).
Supplemental Figure 5. ELISA analysis of TGFβ after macrophages were treated with LPS and either vehicle or 27HC. BMDMs were treated with LPS and either vehicle or 27HC for 24hrs. Conditioned media was harvested and TGFβ was quantified by ELISA (R&D, DY1679–05).
Supplemental Figure 6. Previously reported pathways involved in myeloid cell suppression of T cells (PI3K, AKT, NOTCH and CaMKK2) were ruled out from the suppressive mechanism of 27HC-treated macrophages. BMDMs were treated with vehicle ± indicated inhibitors or 27HC ± indicated inhibitors for 24hrs. CD3+ T cells were then isolated from wildtype C57BL/6 mouse, labeled with CFSE and chemically activated at the time of co-culture with pre-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for all T cell only controls, for co-culture conditions, N=3–4/group). (two-tailed t-test, p < 0.05)
Supplemental Figure 7. Estrogenic activity of 13 ligands. T47D cells stably expressing a ERE luciferase reporter were treated with indicated compounds and the activation of ER was measured by luciferase activity. Data are presented as mean ± SEM (N=4/treatment).
Supplemental Figure 8. LXR-activating activity of 12 cholesterol metabolites, synthetic LXR ligands and E2. BMDMs were treated with indicated compounds and mRNA expression of (top) ABCA1 (N=3/group for all cholesterol metabolites and synthetic LXR ligands, N=5 or 6/group for E2) and (bottom) ABCG1 (N=3/group), were quantified by qPCR. Data are presented as mean ± SEM relative to the respective vehicle control.
Supplemental Figure 9. Knockdown or inhibition of LXR isoforms attenuates immunosuppressive effects of 27HC and the synthetic LXR agonist, GW3965 (A) Representative LXRα and β expression after siRNA knockdown for 48hrs as measured by qPCR (N=3/group; This corresponds with Fig. 5G). (B) LXRα and β expression after lentivirus-delivered shRNA knockdown for 96hrs as measured by qPCR (N=2/group; this corresponds with Fig. 5H). (C) T cell proliferation was restored with the LXR antagonist, GSK2033. After 24hrs, the treatments were washed off and treated BMDMs were co-cultured with chemically activated wildtype T cells for 72hrs. T cells were labeled with CFSE and proliferation measured by flow cytometry (N=3/group for T cell only controls, for co-culture conditions, N=4/group). Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control. (two-tailed t-test, p < 0.05). This corresponds with Fig. 5I.
Supplemental Figure 10. 27HC-treated macrophages induce T cell apoptosis. (A) Representative flow cytometry dot plots of Fig. 6B (48hrs), where T cells were harvested and labeled with Annexin V and PI, after co-culture with vehicle- (top) or 27HC-treated BMDMs (bottom) for 48hrs. (B) Representative flow cytometry dot plots of Fig. 6C (48hrs), where T cells were harvested and labeled with FLICA-FAM and PI, after co-culture with vehicle- (top) or 27HC-treated BMDMs (bottom) for 48hrs.
Supplemental Figure 11. CYP27A1fl/fl;LysMCre+ mice do not express CYP27A1 in bone marrow-derived macrophages. Tissues were harvested from 3 separate CYP27A1fl/fl;LysMCre+ and 3 CYP27A1+/+;LysMCre+ (control) male mice. Tissue was harvested from a traditional (global) CYP27A1−/− mouse as a control. Bone marrow was harvested and used to derive macrophages. (A) Genotyping analysis indicating presence of floxed alleles in the CYP27A1 gene (smaller PCR amplicon). (B) Protein was extracted in RIPA buffer and run on SDS-page for Western analysis using anti-CYP27A1 (Abcam 126785) or cyclophilin B (Cyp B; Santa Cruz Biotechnology sc-517566) as an internal control. As can be noted, CYP27A1 was not expressed in macrophages, but continued to be expressed in the liver, spleen and lungs of CYP27A1fl/fl;LysMcre+ mice.
Supplemental Figure 12. Absence of myeloid CYP27A1 decreased tumor growth post T-cell graft. Mice with myeloid cell knockout of CYP27A1 (CYP27A1fl/fl;LysMCre+) exhibited lower level of antigen-specific primary tumor growth. CYP27A1fl/fl;LysMCre+ mice (N=8) and their replete controls (N=7) were grafted orthotopically with E0771-OVA cells and tumors allowed to grow to ~200 mm3. Control (CYP27A1+/+;LysMCre+) mice received an adoptive transfer of CD3+ T cells isolated from OT-I mice 12 days post-cancer graft (first dashed line), and CYP27A1fl/fl;LysMCre+ mice received an adoptive transfer of CD3+ T cells isolated from OT-I mice 13 days post-cancer graft. Control and CYP27A1fl/fl;LysMCre+ mice were euthanized 25 days after initial cancer cell graft respectively. (A) Tumor volumes were measured through time by digital calipers and plotted as mean ± SEM. Two way ANOVA followed by Sidak’s multiple comparison test on data up to and including day 25 found a statistically significant difference between the groups on day 25 (p < 0.05, denoted by asterisk). (B) Growth of tumors post T cell transfer. This is the same data from (A), but adjusted for time post T cell transfer. Data were then fit to a curve with non-linear regression (four parameter variable slope). This corresponds with Fig. 7A.
HIGHLIGHTS:
The cholesterol metabolite, 27-Hydroxycholesterol (27HC), works on myeloid cells to suppress T cell activity
27HC-LXR activity mediates myeloid cell-driven immune suppression
Myeloid cell-driven T cell inhibition likely results from accelerated T cell apoptosis
Pharmacologic inhibition of 27HC synthesis enhances immune check-point inhibition
The immune suppression is likely a dominant mechanism by which 27HC promotes breast cancer progression.
Acknowledgements
This work was supported by grants to ERN from the National Cancer Institute of the National Institutes of Health (R01CA234025), METAvivor, American Institute of Cancer Research (579732), and Department of Defense Breast Cancer Research Program (BC171214). This work is also supported by grants to DJS from National Institutes of Health (R01DK071909) and the E. Howe Scholar Award. We would like to thank David M. Kranz (University of Illinois) for his thoughtful insight to the design and interpretation of experiments, Jaume Amengual (University of Illinois) and Ivan Pinos Cabezas (University of Illinois) for providing LDLR−/− mice, and Natalia Krawczynska for her constructive feedback on the manuscript. We would like to thank Sarah Y. Adams, Kelly Bradham, and Lea Ann Carson for serving as advocates on this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: The authors have no conflicts of interest to disclose.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020, CA Cancer J Clin, 70 (2020) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Sharma R, Breast cancer incidence, mortality and mortality-to-incidence ratio (MIR) are associated with human development, 1990–2016: evidence from Global Burden of Disease Study 2016, Breast Cancer, 26 (2019) 428–445. [DOI] [PubMed] [Google Scholar]
- [3].DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, Siegel RL, Breast cancer statistics, 2019, CA Cancer J Clin, 69 (2019) 438–451. [DOI] [PubMed] [Google Scholar]
- [4].Munoz D, Near AM, van Ravesteyn NT, Lee SJ, Schechter CB, Alagoz O, Berry DA, Burnside ES, Chang Y, Chisholm G, de Koning HJ, Ali Ergun M, Heijnsdijk EA, Huang H, Stout NK, Sprague BL, Trentham-Dietz A, Mandelblatt JS, Plevritis SK, Effects of screening and systemic adjuvant therapy on ER-specific US breast cancer mortality, J Natl Cancer Inst, 106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Polk A, Svane IM, Andersson M, Nielsen D, Checkpoint inhibitors in breast cancer - Current status, Cancer Treat Rev, 63 (2018) 122–134. [DOI] [PubMed] [Google Scholar]
- [6].Swoboda A, Nanda R, Immune Checkpoint Blockade for Breast Cancer, Cancer Treat Res, 173 (2018) 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S, Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients?, Am Soc Clin Oncol Educ Book, 39 (2019) 147–164. [DOI] [PubMed] [Google Scholar]
- [8].Vonderheide RH, Domchek SM, Clark AS, Immunotherapy for Breast Cancer: What Are We Missing?, Clin Cancer Res, 23 (2017) 2640–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Basu A, Ramamoorthi G, Jia Y, Faughn J, Wiener D, Awshah S, Kodumudi K, Czerniecki BJ, Immunotherapy in breast cancer: Current status and future directions, Adv Cancer Res, 143 (2019) 295–349. [DOI] [PubMed] [Google Scholar]
- [10].Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, Krummel MF, Tuning the Tumor Myeloid Microenvironment to Fight Cancer, Front Immunol, 10 (2019) 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Racioppi L, Nelson ER, Huang W, Mukherjee D, Lawrence SA, Lento W, Masci AM, Jiao Y, Park S, York B, Liu Y, Baek AE, Drewry DH, Zuercher WJ, Bertani FR, Businaro L, Geradts J, Hall A, Means AR, Chao N, Chang CY, McDonnell DP, CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer, Nat Commun, 10 (2019) 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kowal J, Kornete M, Joyce JA, Re-education of macrophages as a therapeutic strategy in cancer, Immunotherapy, 11 (2019) 677–689. [DOI] [PubMed] [Google Scholar]
- [13].Di Mitri D, Mirenda M, Vasilevska J, Calcinotto A, Delaleu N, Revandkar A, Gil V, Boysen G, Losa M, Mosole S, Pasquini E, D’Antuono R, Masetti M, Zagato E, Chiorino G, Ostano P, Rinaldi A, Gnetti L, Graupera M, Martins Figueiredo Fonseca AR, Pereira Mestre R, Waugh D, Barry S, De Bono J, Alimonti A, Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer, Cell Rep, 28 (2019) 2156–2168. e2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, Joyce JA, Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas, Oncogene, 36 (2017) 6049–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Baek AE, Nelson ER, The Contribution of Cholesterol and Its Metabolites to the Pathophysiology of Breast Cancer, Horm Cancer, 7 (2016) 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Llaverias G, Danilo C, Mercier I, Daumer K, Capozza F, Williams TM, Sotgia F, Lisanti MP, Frank PG, Role of cholesterol in the development and progression of breast cancer, Am J Pathol, 178 (2011) 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bahl M, Ennis M, Tannock IF, Hux JE, Pritchard KI, Koo J, Goodwin PJ, Serum lipids and outcome of early-stage breast cancer: results of a prospective cohort study, Breast Cancer Res Treat, 94 (2005) 135–144. [DOI] [PubMed] [Google Scholar]
- [18].Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, Sorensen HT, Lash TL, Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study, J Natl Cancer Inst, 103 (2011) 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brewer TM, Masuda H, Liu DD, Shen Y, Liu P, Iwamoto T, Kai K, Barnett CM, Woodward WA, Reuben JM, Yang P, Hortobagyi GN, Ueno NT, Statin use in primary inflammatory breast cancer: a cohort study, Br J Cancer, 109 (2013) 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Borgquist S, Broberg P, Tojjar J, Olsson H, Statin use and breast cancer survival - a Swedish nationwide study, BMC Cancer, 19 (2019) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beckwitt CH, Brufsky A, Oltvai ZN, Wells A, Statin drugs to reduce breast cancer recurrence and mortality, Breast Cancer Res, 20 (2018) 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP, 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology, Science, 342 (2013) 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, Sullivan PM, Kemper JK, Gunn MD, McDonnell DP, Nelson ER, The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells, Nat Commun, 8 (2017) 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He S, Ma L, Baek AE, Vardanyan A, Vembar V, Chen JJ, Nelson AT, Burdette JE, Nelson ER, Host CYP27A1 expression is essential for ovarian cancer progression, Endocr Relat Cancer, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Morita Y, Zhang R, Leslie M, Adhikari S, Hasan N, Chervoneva I, Rui H, Tanaka T, Pathologic evaluation of tumor-associated macrophage density and vessel inflammation in invasive breast carcinomas, Oncol Lett, 14 (2017) 2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Soncin I, Sheng J, Chen Q, Foo S, Duan K, Lum J, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C, The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma, Nat Commun, 9 (2018) 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ai L, Mu S, Wang Y, Wang H, Cai L, Li W, Hu Y, Prognostic role of myeloid-derived suppressor cells in cancers: a systematic review and meta-analysis, BMC Cancer, 18 (2018) 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Toor SM, Syed Khaja AS, El Salhat H, Faour I, Kanbar J, Quadri AA, Albashir M, Elkord E, Myeloid cells in circulation and tumor microenvironment of breast cancer patients, Cancer Immunol Immunother, 66 (2017) 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K, Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression, Front Immunol, 9 (2018) 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quatromoni JG, Eruslanov E, Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer, Am J Transl Res, 4 (2012) 376–389. [PMC free article] [PubMed] [Google Scholar]
- [31].Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim H, Oh J, Bae H, Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide, J Immunother Cancer, 7 (2019) 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D, Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice, J Clin Invest, 121 (2011) 4015–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dujaily EA, Baena J, Das M, Sereno M, Smith C, Kamata T, Officer L, Pritchard C, Le Quesne J, Reduced pro-tumourigenic tumour-associated macrophages with statin use in premalignant human lung adenocarcinoma, JNCI Cancer Spectrum, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shahoei SH, Kim YC, Cler SJ, Ma L, Anakk S, Kemper JK, Nelson ER, Small Heterodimer Partner Regulates Dichotomous T Cell Expansion by Macrophages, Endocrinology, 160 (2019) 1573–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, Tsichlis PN, TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway, Cell, 103 (2000) 1071–1083. [DOI] [PubMed] [Google Scholar]
- [36].Xiao L, Ornatowska M, Zhao G, Cao H, Yu R, Deng J, Li Y, Zhao Q, Sadikot RT, Christman JW, Lipopolysaccharide-induced expression of microsomal prostaglandin E synthase-1 mediates late-phase PGE2 production in bone marrow derived macrophages, PLoS One, 7 (2012) e50244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC, CD14 controls the LPS-induced endocytosis of Toll-like receptor 4, Cell, 147 (2011) 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nelson ER, DuSell CD, Wang X, Howe MK, Evans G, Michalek RD, Umetani M, Rathmell JC, Khosla S, Gesty-Palmer D, McDonnell DP, The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver × receptors, Endocrinology, 152 (2011) 4691–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nelson ER, Li S, Kennedy M, Payne S, Kilibarda K, Groth J, Bowie M, Parilla-Castellar E, de Ridder G, Marcom PK, Lyes M, Peterson BL, Cook M, Pizzo SV, McDonnell DP, Bachelder RE, Chemotherapy enriches for an invasive triple-negative breast tumor cell subpopulation expressing a precursor form of N-cadherin on the cell surface, Oncotarget, 7 (2016) 84030–84042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci Signal, 6 (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov, 2 (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing, Journal of the Royal Statistical Society. Series B (Methodological), 57 (1995) 289–300. [Google Scholar]
- [43].Ma L, Nelson ER, Oxysterols and nuclear receptors, Molecular and cellular endocrinology, 484 (2019) 42–51. [DOI] [PubMed] [Google Scholar]
- [44].Ma L, Nelson ER, Oxysterols and nuclear receptors, Molecular and cellular endocrinology, (2019). [DOI] [PubMed] [Google Scholar]
- [45].Shi Y, Xu X, Tan Y, Mao S, Fang S, Gu W, A liver-X-receptor ligand, T0901317, attenuates IgE production and airway remodeling in chronic asthma model of mice, PloS One, 9 (2014) e92668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Thomas DG, Doran AC, Fotakis P, Westerterp M, Antonson P, Jiang H, Jiang X-C, Gustafsson J-Å, Tabas I, Tall AR, LXR Suppresses Inflammatory Gene Expression and Neutrophil Migration through cis-Repression and Cholesterol Efflux, Cell reports, 25 (2018) 3774–3785. e3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, Tontonoz P, LXR signaling couples sterol metabolism to proliferation in the acquired immune response, Cell, 134 (2008) 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR, Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy, Immunol Res, 51 (2011) 170–182. [DOI] [PubMed] [Google Scholar]
- [49].Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, Mantovani A, Locati M, Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology, Blood, 119 (2012) 411–421. [DOI] [PubMed] [Google Scholar]
- [50].Barnden MJ, Allison J, Heath WR, Carbone FR, Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements, 76 (1998) 34–40. [DOI] [PubMed] [Google Scholar]
- [51].Noy R, Pollard JW, Tumor-associated macrophages: from mechanisms to therapy, Immunity, 41 (2014) 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Qi C, Cai Y, Gunn L, Ding C, Li B, Kloecker G, Qian K, Vasilakos J, Saijo S, Iwakura Y, Yannelli JR, Yan J, Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived beta-glucans, Blood, 117 (2011) 6825–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhou F, Rouse BT, Huang L, Prolonged survival of thymoma-bearing mice after vaccination with a soluble protein antigen entrapped in liposomes: a model study, Cancer Res, 52 (1992) 6287–6291. [PubMed] [Google Scholar]
- [54].Nelson ER, The significance of cholesterol and its metabolite, 27-hydroxycholesterol in breast cancer, Molecular and cellular endocrinology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Revilla G, Pons MP, Baila-Rueda L, Garcia-Leon A, Santos D, Cenarro A, Magalhaes M, Blanco RM, Moral A, Ignacio Perez J, Sabe G, Gonzalez C, Fuste V, Lerma E, Faria MDS, de Leiva A, Corcoy R, Carles Escola-Gil J, Mato E, Cholesterol and 27-hydroxycholesterol promote thyroid carcinoma aggressiveness, Sci Rep, 9 (2019) 10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zou W, Immunosuppressive networks in the tumour environment and their therapeutic relevance, Nat Rev Cancer, 5 (2005) 263–274. [DOI] [PubMed] [Google Scholar]
- [57].Mondanelli G, Iacono A, Allegrucci M, Puccetti P, Grohmann U, Immunoregulatory Interplay Between Arginine and Tryptophan Metabolism in Health and Disease, Front Immunol, 10 (2019) 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CC, Chang TY, Zhang T, Zhou P, Song BL, Liu W, Sun SC, Liu X, Li BL, Xu C, Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism, Nature, 531 (2016) 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Reiss AB, Vernice NA, Siegart NM, De Leon J, Kasselman LJ, Exosomes in Cholesterol Metabolism and Atherosclerosis, Cardiovasc Hematol Disord Drug Targets, 17 (2017) 185–194. [DOI] [PubMed] [Google Scholar]
- [60].Castano C, Kalko S, Novials A, Parrizas M, Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice, Proc Natl Acad Sci U S A, 115 (2018) 12158–12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang T, Nasser MI, Shen J, Qu S, He Q, Zhao M, Functions of Exosomes in the Triangular Relationship between the Tumor, Inflammation, and Immunity in the Tumor Microenvironment, J Immunol Res, 2019 (2019) 4197829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, Carbone CJ, Yu Q, Katlinski KV, Katlinskaya YV, Handa S, Haas V, Volk SW, Brice AK, Wals K, Matheson NJ, Antrobus R, Ludwig S, Whiteside TL, Sander C, Tarhini AA, Kirkwood JM, Lehner PJ, Guo W, Rui H, Minn AJ, Koumenis C, Diehl JA, Fuchs SY, An Interferon-Driven Oxysterol-Based Defense against Tumor-Derived Extracellular Vesicles, Cancer Cell, 35 (2019) 33–45 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner-Hurevich M, Berzofsky JA, Nir-Paz R, Paul WE, IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells, J Exp Med, 210 (2013) 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Di Paolo NC, Shayakhmetov DM, Interleukin 1alpha and the inflammatory process, Nat Immunol, 17 (2016) 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Santarlasci V, Cosmi L, Maggi L, Liotta F, Annunziato F, IL-1 and T Helper Immune Responses, Front Immunol, 4 (2013) 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lichtman AH, Chin J, Schmidt JA, Abbas AK, Role of interleukin 1 in the activation of T lymphocytes, Proc Natl Acad Sci U S A, 85 (1988) 9699–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP, 27-Hydroxycholesterol Links Hypercholesterolemia and Breast Cancer Pathophysiology, Science (New York, N.Y.), 342 (2013) 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dalenc F, Poirot M, Silvente-Poirot S, Dendrogenin A: A Mammalian Metabolite of Cholesterol with Tumor Suppressor and Neurostimulating Properties, Curr Med Chem, 22 (2015) 3533–3549. [DOI] [PubMed] [Google Scholar]
- [69].de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F, Lacroix-Triki M, Filleron T, Pont F, Saati TA, Morisseau C, Hammock BD, Silvente-Poirot S, Poirot M, Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties, Nat Commun, 4 (2013) 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Svoronos N, Perales-Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, Nguyen JM, Curiel TJ, Cadungog MG, Singhal S, Eruslanov EB, Zhang P, Tchou J, Zhang R, Conejo-Garcia JR, Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells, Cancer Discov, 7 (2017) 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Andruska N, Mao C, Cherian M, Zhang C, Shapiro DJ, Evaluation of a luciferase-based reporter assay as a screen for inhibitors of estrogen-ERalpha-induced proliferation of breast cancer cells, J Biomol Screen, 17 (2012) 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Murphy AJ, Guyre PM, Wira CR, Pioli PA, Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages, PLoS One, 4 (2009) e5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bolego C, Cignarella A, Staels B, Chinetti-Gbaguidi G, Macrophage function and polarization in cardiovascular disease: a role of estrogen signaling?, Arterioscler Thromb Vasc Biol, 33 (2013) 1127–1134. [DOI] [PubMed] [Google Scholar]
- [74].Calippe B, Douin-Echinard V, Delpy L, Laffargue M, Lelu K, Krust A, Pipy B, Bayard F, Arnal JF, Guery JC, Gourdy P, 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo, J Immunol, 185 (2010) 1169–1176. [DOI] [PubMed] [Google Scholar]
- [75].Wardell SE, Marks JR, McDonnell DP, The turnover of estrogen receptor alpha by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy, Biochem Pharmacol, 82 (2011) 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Patel R, Magomedova L, Tsai R, Angers S, Orellana A, Cummins CL, Separating the Anti-Inflammatory and Diabetogenic Effects of Glucocorticoids Through LXRβ Antagonism, Endocrinology, 158 (2017) 1034–1047. [DOI] [PubMed] [Google Scholar]
- [77].Bruschi FV, Claudel T, Tardelli M, Starlinger P, Marra F, Trauner M, PNPLA3 I148M Variant Impairs Liver × Receptor Signaling and Cholesterol Homeostasis in Human Hepatic Stellate Cells, Hepatol Commun, 3 (2019) 1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fan J, Zhao RQ, Parro C, Zhao W, Chou HY, Robert J, Deeb TZ, Raynoschek C, Barichievy S, Engkvist O, Maresca M, Hicks R, Meuller J, Moss SJ, Brandon NJ, Wood MW, Kulic I, Wellington CL, Small molecule inducers of ABCA1 and apoE that act through indirect activation of the LXR pathway, J Lipid Res, 59 (2018) 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bang BR, Li M, Tsai KN, Aoyagi H, Lee SA, Machida K, Aizaki H, Jung JU, Ou JJ, Saito T, Regulation of Hepatitis C Virus Infection by Cellular Retinoic Acid Binding Proteins through the Modulation of Lipid Droplet Abundance, J Virol, 93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zuercher WJ, Buckholz RG, Campobasso N, Collins JL, Galardi CM, Gampe RT, Hyatt SM, Merrihew SL, Moore JT, Oplinger JA, Reid PR, Spearing PK, Stanley TB, Stewart EL, Willson TM, Discovery of tertiary sulfonamides as potent liver × receptor antagonists, Journal of medicinal chemistry, 53 (2010) 3412–3416. [DOI] [PubMed] [Google Scholar]
- [81].Griffett K, Burris TP, Promiscuous activity of the LXR antagonist GSK2033 in a mouse model of fatty liver disease, Biochem Biophys Res Commun, 479 (2016) 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI, Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards, Nat Commun, 7 (2016) 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA, PI3Kgamma is a molecular switch that controls immune suppression, Nature, 539 (2016) 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gerberick GF, Cruse LW, Miller CM, Sikorski EE, Ridder GM, Selective modulation of T cell memory markers CD62L and CD44 on murine draining lymph node cells following allergen and irritant treatment, Toxicol Appl Pharmacol, 146 (1997) 1–10. [DOI] [PubMed] [Google Scholar]
- [85].Munn DH, Pressey J, Beall AC, Hudes R, Alderson MR, Selective activation-induced apoptosis of peripheral T cells imposed by macrophages. A potential mechanism of antigen-specific peripheral lymphocyte deletion, J Immunol, 156 (1996) 523–532. [PubMed] [Google Scholar]
- [86].Saio M, Radoja S, Marino M, Frey AB, Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide, J Immunol, 167 (2001) 5583–5593. [DOI] [PubMed] [Google Scholar]
- [87].Daigneault M, De Silva TI, Bewley MA, Preston JA, Marriott HM, Mitchell AM, Mitchell TJ, Read RC, Whyte MK, Dockrell DH, Monocytes regulate the mechanism of T-cell death by inducing Fas-mediated apoptosis during bacterial infection, PLoS Pathog, 8 (2012) e1002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Vassena L, Giuliani E, Koppensteiner H, Bolduan S, Schindler M, Doria M, HIV-1 Nef and Vpu Interfere with L-Selectin (CD62L) Cell Surface Expression To Inhibit Adhesion and Signaling in Infected CD4+ T Lymphocytes, J Virol, 89 (2015) 5687–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sachet M, Liang YY, Oehler R, The immune response to secondary necrotic cells, Apoptosis, 22 (2017) 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ, Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry, Cold Spring Harb Protoc, 2016 (2016). [DOI] [PubMed] [Google Scholar]
- [91].Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z, Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation, Exp Cell Res, 259 (2000) 308–313. [DOI] [PubMed] [Google Scholar]
- [92].Darzynkiewicz Z, Pozarowski P, Lee BW, Johnson GL, Fluorochrome-labeled inhibitors of caspases: convenient in vitro and in vivo markers of apoptotic cells for cytometric analysis, Methods Mol Biol, 682 (2011) 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, Sahebkar A, PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy, Journal of cellular physiology, 234 (2019) 16824–16837. [DOI] [PubMed] [Google Scholar]
- [94].Zhu X, Lang J, Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer, Oncotarget, 8 (2017) 97671–97682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nelson ER, Chang CY, McDonnell DP, Cholesterol and breast cancer pathophysiology, Trends Endocrinol Metab, 25 (2014) 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Torres CG, Ramirez ME, Cruz P, Epunan MJ, Valladares LE, Sierralta WD, 27-hydroxycholesterol induces the transition of MCF7 cells into a mesenchymal phenotype, Oncol Rep, 26 (2011) 389–397. [DOI] [PubMed] [Google Scholar]
- [97].Yu YR, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, Gunn MD, A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues, PLoS One, 11 (2016) e0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, Bado I, Lo HC, Toneff MJ, Nguyen T, Bu W, Jiang W, Arnold J, Gu F, He J, Jebakumar D, Walker K, Li Y, Mo Q, Westbrook TF, Zong C, Rao A, Sreekumar A, Rosen JM, Zhang XH, Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms, Nature cell biology, 21 (2019) 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Marengo B, Bellora F, Ricciarelli R, De Ciucis C, Furfaro A, Leardi R, Colla R, Pacini D, Traverso N, Moretta A, Pronzato MA, Bottino C, Domenicotti C, Oxysterol mixture and, in particular, 27-hydroxycholesterol drive M2 polarization of human macrophages, BioFactors (Oxford, England), 42 (2016) 80–92. [DOI] [PubMed] [Google Scholar]
- [100].Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, Besse B, Yanagitani N, Friboulet L, Nishio M, Takeuchi K, Kawamoto H, Fujita N, Katayama R, Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer, J Exp Med, 216 (2019) 982–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, Eisenhaure TM, Pendergraft WF 3rd, Hua P, Pham HC, Bu X, Zhu B, Hacohen N, Fritsch EF, Boussiotis VA, Wu CJ, Freeman GJ, A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression, Cancer immunology, immunotherapy: CII, 68 (2019) 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].He XH, Xu LH, Liu Y, Identification of a novel splice variant of human PD-L1 mRNA encoding an isoform-lacking Igv-like domain, Acta Pharmacol Sin, 26 (2005) 462–468. [DOI] [PubMed] [Google Scholar]
- [103].Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA, Umetani M, Euhus DM, Xie Y, Shaul PW, 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth, Cell reports, 5 (2013) 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shi SZ, Lee EJ, Lin YJ, Chen L, Zheng HY, He XQ, Peng JY, Noonepalle SK, Shull AY, Pei FC, Deng LB, Tian XL, Deng KY, Shi H, Xin HB, Recruitment of monocytes and epigenetic silencing of intratumoral CYP7B1 primarily contribute to the accumulation of 27-hydroxycholesterol in breast cancer, American journal of cancer research, 9 (2019) 2194–2208. [PMC free article] [PubMed] [Google Scholar]
- [105].Mutemberezi V, Buisseret B, Masquelier J, Guillemot-Legris O, Alhouayek M, Muccioli GG, Oxysterol levels and metabolism in the course of neuroinflammation: insights from in vitro and in vivo models, J Neuroinflammation, 15 (2018) 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ahsan F, Maertzdorf J, Guhlich-Bornhof U, Kaufmann SHE, Moura-Alves P, IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis, Sci Rep, 8 (2018) 1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shahoei SH, Nelson ER, Nuclear receptors, cholesterol homeostasis and the immune system, The Journal of steroid biochemistry and molecular biology, 191 (2019) 105364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Umetani M, Ghosh P, Ishikawa T, Umetani J, Ahmed M, Mineo C, Shaul PW, The cholesterol metabolite 27-hydroxycholesterol promotes atherosclerosis via proinflammatory processes mediated by estrogen receptor alpha, Cell Metab, 20 (2014) 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yin K, You Y, Swier V, Tang L, Radwan MM, Pandya AN, Agrawal DK, Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine, Arteriosclerosis, thrombosis, and vascular biology, 35 (2015) 2432–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Oguro H, McDonald JG, Zhao Z, Umetani M, Shaul PW, Morrison SJ, 27-Hydroxycholesterol induces hematopoietic stem cell mobilization and extramedullary hematopoiesis during pregnancy, J Clin Invest, 127 (2017) 3392–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ma L, Nelson ER, Oxysterols and nuclear receptors, Mol Cell Endocrinol, 484 (2019) 42–51. [DOI] [PubMed] [Google Scholar]
- [112].Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P, LXR-dependent gene expression is important for macrophage survival and the innate immune response, Cell, 119 (2004) 299–309. [DOI] [PubMed] [Google Scholar]
- [113].Endo-Umeda K, Nakashima H, Umeda N, Seki S, Makishima M, Dysregulation of Kupffer Cells/Macrophages and Natural Killer T Cells in Steatohepatitis in LXRalpha Knockout Male Mice, Endocrinology, 159 (2018) 1419–1432. [DOI] [PubMed] [Google Scholar]
- [114].Gargiulo S, Gamba P, Testa G, Rossin D, Biasi F, Poli G, Leonarduzzi G, Relation between TLR4/NF-kappaB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability, Aging Cell, 14 (2015) 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P, LXR-Dependent Gene Expression Is Important for Macrophage Survival and the Innate Immune Response, Cell, 119 (2004) 299–309. [DOI] [PubMed] [Google Scholar]
- [116].Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, Takeda S, Tafreshian KN, Rowinsky E, Szarek M, Waltzman RJ, McMillan EA, Zhao C, Mita M, Mita A, Chmielowski B, Postow MA, Ribas A, Mucida D, Tavazoie SF, LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer, Cell, 172 (2018) 825–840 e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yu SX, Chen W, Hu XZ, Feng SY, Li KY, Qi S, Lei QQ, Hu GQ, Li N, Zhou FH, Ma CY, Du CT, Yang YJ, Liver × receptors agonists suppress NLRP3 inflammasome activation, Cytokine, 91 (2017) 30–37. [DOI] [PubMed] [Google Scholar]
- [118].Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, Zhao W, Huai W, Guo P, Han L, Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression, Lab Invest, 94 (2014) 52–62. [DOI] [PubMed] [Google Scholar]
- [119].Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A, Role of the NLRP3 inflammasome in cancer, Mol Cancer, 17 (2018) 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kimbung S, Chang CY, Bendahl PO, Dubois L, Thompson JW, McDonnell DP, Borgquist S, Impact of 27-hydroxylase (CYP27A1) and 27-hydroxycholesterol in breast cancer, Endocr Relat Cancer, 24 (2017) 339–349. [DOI] [PubMed] [Google Scholar]
- [121].Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, Das R, Silvin C, Van Waes C, Horn LA, Schlom J, Palena C, Maeda D, Zebala J, Allen CT, Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy, JCI Insight, 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T, Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells, Nature, 539 (2016) 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, Singh R, Schall TJ, Datta M, Jain RK, Mitchell DA, Harrison JK, CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas, Proc Natl Acad Sci U S A, 117 (2020) 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ, An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha, Nature, 383 (1996) 728–731. [DOI] [PubMed] [Google Scholar]
- [125].Kim WK, Meliton V, Tetradis S, Weinmaster G, Hahn TJ, Carlson M, Nelson SF, Parhami F, Osteogenic oxysterol, 20(S)-hydroxycholesterol, induces notch target gene expression in bone marrow stromal cells, J Bone Miner Res, 25 (2010) 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ, Structural requirements of ligands for the oxysterol liver × receptors LXRalpha and LXRbeta, Proc Natl Acad Sci U S A, 96 (1999) 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wang CW, Huang CC, Chou PH, Chang YP, Wei S, Guengerich FP, Chou YC, Wang SF, Lai PS, Soucek P, Ueng YF, 7-ketocholesterol and 27-hydroxycholesterol decreased doxorubicin sensitivity in breast cancer cells: estrogenic activity and mTOR pathway, Oncotarget, 8 (2017) 66033–66050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lappano R, Recchia AG, De Francesco EM, Angelone T, Cerra MC, Picard D, Maggiolini M, The cholesterol metabolite 25-hydroxycholesterol activates estrogen receptor alpha-mediated signaling in cancer cells and in cardiomyocytes, PLoS One, 6 (2011) e16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ, 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen, Nat Med, 13 (2007) 1185–1192. [DOI] [PubMed] [Google Scholar]
- [130].Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG, 27-hydroxycholesterol is an endogenous ligand for liver × receptor in cholesterol-loaded cells, J Biol Chem, 276 (2001) 38378–38387. [DOI] [PubMed] [Google Scholar]
- [131].Crestani M, De Fabiani E, Caruso D, Mitro N, Gilardi F, Vigil Chacon AB, Patelli R, Godio C, Galli G, LXR (liver × receptor) and HNF-4 (hepatocyte nuclear factor-4): key regulators in reverse cholesterol transport, Biochem Soc Trans, 32 (2004) 92–96. [DOI] [PubMed] [Google Scholar]
- [132].Poirot M, Silvente-Poirot S, The tumor-suppressor cholesterol metabolite, dendrogenin A, is a new class of LXR modulator activating lethal autophagy in cancers, Biochem Pharmacol, 153 (2018) 75–81. [DOI] [PubMed] [Google Scholar]
- [133].Silvente-Poirot S, Segala G, Poirot MC, Poirot M, Ligand-dependent transcriptional induction of lethal autophagy: A new perspective for cancer treatment, Autophagy, 14 (2018) 555–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH Jr., Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW, Glass CK, Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses, Cell, 151 (2012) 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, Seidman JS, Henke A, Roland JT, Ozeki KA, Thompson BM, McDonald JG, Bahadorani J, Tsimikas S, Grossman TR, Tremblay MS, Glass CK, Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages, Proc Natl Acad Sci U S A, 115 (2018) E4680–e4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. 27HC-treated macrophages suppress T cell expansion. (A) Representative histograms from flow cytometry. CD3+ T cells were isolated from the spleen of OT-1 mice, stained with CFSE, were co-cultured with BMDMs treated with OVA257–264 and either vehicle- or 27HC. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. This data is from Fig. 3E, and is also representative of Figs. 2D&E. (B) Naïve T cells were isolated from the spleen of wildtype C57BL/6 micese, chemically activated and cultured in the presence of conditioned media harvested from vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the vehicle control (N=3/group for T cell only control, N=6/group for conditioned media two-tailed t-test, p < 0.05). (C). CD3+ T cells were isolated from the spleen of wildtype C57BL/6 mice, labeled with CFSE, chemically activated and cultured with conditioned media harvested from vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for T cell only controls, for conditioned-media conditions, N=5–6/group). (two-tailed t-test, p < 0.05). This sub-figure is an independent repeat of Fig. 1I.
Supplemental Figure 2. Blockade of MHCII does not attenuate pro-colonizing effects of 27HC. Mice were pretreated with placebo or 27HC (20mg/kg/d) for 5d, as in [23]. 36h and 12h prior to cell graft, mice were injected (i.p) with an isotype control antibody (IgG) or an antibody against murine I-A/I-E. Mice were then grafted with Met1 cells expressing iRFP. Resulting cancer colonies within the lung were quantified by ex vivo imaging for iRFP fluorescence. Different letters denote statistical significance, N=8/group. (one-way ANOVA followed by Neuman-Kuels multiple comparison test, p < 0.05).
Supplemental Figure 3. Expression of granzyme B (GZMB) and perforin (PRF1) normalized to CD8A expression was modestly but significantly correlated with CYP7B1 expression. mRNA expressions from Human RNA sequencing data from TCGA Pan Cancer for invasive breast carcinoma were log2 transformed and Pearson’s correlation was assessed (N=1077 in (A) and 1081 in (B)). These data partner with Fig. 3L and M.
Supplemental Figure 4. Cholesterol efflux from macrophages unlikely to mediate the suppressive effect of 27HC on T cells. CD3+ T cells were isolated from wildtype C57BL/6 or LDLR−/− mice, labeled with CFSE and chemically activated. They were then co-cultured with vehicle- or 27HC-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for all T cell only controls, for co-culture conditions, N=5/group). (two-tailed t-test, p < 0.05).
Supplemental Figure 5. ELISA analysis of TGFβ after macrophages were treated with LPS and either vehicle or 27HC. BMDMs were treated with LPS and either vehicle or 27HC for 24hrs. Conditioned media was harvested and TGFβ was quantified by ELISA (R&D, DY1679–05).
Supplemental Figure 6. Previously reported pathways involved in myeloid cell suppression of T cells (PI3K, AKT, NOTCH and CaMKK2) were ruled out from the suppressive mechanism of 27HC-treated macrophages. BMDMs were treated with vehicle ± indicated inhibitors or 27HC ± indicated inhibitors for 24hrs. CD3+ T cells were then isolated from wildtype C57BL/6 mouse, labeled with CFSE and chemically activated at the time of co-culture with pre-treated BMDMs. 72hrs after co-culture, T cells were harvested for flow cytometry analysis. Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control (N=3/group for all T cell only controls, for co-culture conditions, N=3–4/group). (two-tailed t-test, p < 0.05)
Supplemental Figure 7. Estrogenic activity of 13 ligands. T47D cells stably expressing a ERE luciferase reporter were treated with indicated compounds and the activation of ER was measured by luciferase activity. Data are presented as mean ± SEM (N=4/treatment).
Supplemental Figure 8. LXR-activating activity of 12 cholesterol metabolites, synthetic LXR ligands and E2. BMDMs were treated with indicated compounds and mRNA expression of (top) ABCA1 (N=3/group for all cholesterol metabolites and synthetic LXR ligands, N=5 or 6/group for E2) and (bottom) ABCG1 (N=3/group), were quantified by qPCR. Data are presented as mean ± SEM relative to the respective vehicle control.
Supplemental Figure 9. Knockdown or inhibition of LXR isoforms attenuates immunosuppressive effects of 27HC and the synthetic LXR agonist, GW3965 (A) Representative LXRα and β expression after siRNA knockdown for 48hrs as measured by qPCR (N=3/group; This corresponds with Fig. 5G). (B) LXRα and β expression after lentivirus-delivered shRNA knockdown for 96hrs as measured by qPCR (N=2/group; this corresponds with Fig. 5H). (C) T cell proliferation was restored with the LXR antagonist, GSK2033. After 24hrs, the treatments were washed off and treated BMDMs were co-cultured with chemically activated wildtype T cells for 72hrs. T cells were labeled with CFSE and proliferation measured by flow cytometry (N=3/group for T cell only controls, for co-culture conditions, N=4/group). Data are presented as mean ± SEM and asterisks (*) indicate statistical significance from the respective vehicle control. (two-tailed t-test, p < 0.05). This corresponds with Fig. 5I.
Supplemental Figure 10. 27HC-treated macrophages induce T cell apoptosis. (A) Representative flow cytometry dot plots of Fig. 6B (48hrs), where T cells were harvested and labeled with Annexin V and PI, after co-culture with vehicle- (top) or 27HC-treated BMDMs (bottom) for 48hrs. (B) Representative flow cytometry dot plots of Fig. 6C (48hrs), where T cells were harvested and labeled with FLICA-FAM and PI, after co-culture with vehicle- (top) or 27HC-treated BMDMs (bottom) for 48hrs.
Supplemental Figure 11. CYP27A1fl/fl;LysMCre+ mice do not express CYP27A1 in bone marrow-derived macrophages. Tissues were harvested from 3 separate CYP27A1fl/fl;LysMCre+ and 3 CYP27A1+/+;LysMCre+ (control) male mice. Tissue was harvested from a traditional (global) CYP27A1−/− mouse as a control. Bone marrow was harvested and used to derive macrophages. (A) Genotyping analysis indicating presence of floxed alleles in the CYP27A1 gene (smaller PCR amplicon). (B) Protein was extracted in RIPA buffer and run on SDS-page for Western analysis using anti-CYP27A1 (Abcam 126785) or cyclophilin B (Cyp B; Santa Cruz Biotechnology sc-517566) as an internal control. As can be noted, CYP27A1 was not expressed in macrophages, but continued to be expressed in the liver, spleen and lungs of CYP27A1fl/fl;LysMcre+ mice.
Supplemental Figure 12. Absence of myeloid CYP27A1 decreased tumor growth post T-cell graft. Mice with myeloid cell knockout of CYP27A1 (CYP27A1fl/fl;LysMCre+) exhibited lower level of antigen-specific primary tumor growth. CYP27A1fl/fl;LysMCre+ mice (N=8) and their replete controls (N=7) were grafted orthotopically with E0771-OVA cells and tumors allowed to grow to ~200 mm3. Control (CYP27A1+/+;LysMCre+) mice received an adoptive transfer of CD3+ T cells isolated from OT-I mice 12 days post-cancer graft (first dashed line), and CYP27A1fl/fl;LysMCre+ mice received an adoptive transfer of CD3+ T cells isolated from OT-I mice 13 days post-cancer graft. Control and CYP27A1fl/fl;LysMCre+ mice were euthanized 25 days after initial cancer cell graft respectively. (A) Tumor volumes were measured through time by digital calipers and plotted as mean ± SEM. Two way ANOVA followed by Sidak’s multiple comparison test on data up to and including day 25 found a statistically significant difference between the groups on day 25 (p < 0.05, denoted by asterisk). (B) Growth of tumors post T cell transfer. This is the same data from (A), but adjusted for time post T cell transfer. Data were then fit to a curve with non-linear regression (four parameter variable slope). This corresponds with Fig. 7A.