

Abstract
Mounting evidence indicates that single-target drugs might be inadequate to achieve satisfactory therapeutic effects on complex diseases. Recently, increasing attention has been paid to developing drugs that can manipulate multiple targets to generate beneficial effects through potential synergy. G-protein-coupled receptors (GPCRs) become desirable targets for developing multitarget-directed ligands (MTDLs) because of their crucial roles in the pathophysiology of various human diseases and the accessibility of druggable sites at the cell surface. Herein, we review the most recent advances in the development of GPCR-targeted MTDLs in treating complex diseases, and discuss their potential therapeutic strategies to reveal current trends and shed insights into the utility of GPCR-targeted MTDLs for future drug design and development.
Introduction
Since the lock-and-key model proposed by Erlich in 1894 [1], drug discovery has mainly focused on developing selective molecules that target a single mechanism in a single disease, namely the ‘one drug, one target’ approach [2]. Following this doctrine, numerous selective drugs have been developed, demonstrating their pivotal roles in treating certain diseases [3]. However, complex diseases, also known as multifactorial diseases, are posing enormous challenges to the medicinal chemistry community because they involve multiple target proteins and/or signaling pathways associated with diseases such as cancers [4], neurodegenerative diseases [5], pain disorders [6], and infection and inflammation [7]. For these complex diseases, acting on a single target might be inadequate to achieve satisfactory therapeutic effects [8,9]. Hence, there is an emerging trend to develop MTDLs that are capable of manipulating multiple targets simultaneously, with the aim to enhance efficacy and improve safety.
GPCRs are the most intensively studied target proteins in drug design and development [10,11]. For decades, the design of bivalent and bitopic molecules has evolved as intriguing approaches to obtain highly selective ligands targeting GPCRs with desirable efficacies and/or signaling bias. As first proposed by Phil Portoghese in 1982 [12], a bivalent ligand refers to a molecule that has two pharmacophores on both ends of an appropriate linker, aiming to target the orthosteric binding site (OBS) of two adjacent or dimerized receptors to improve affinity and/or potency. Based on the identity of pharmacophores, bivalent ligands can be further branched into homobivalent and heterobivalent ones. By contrast, a bitopic ligand is proposed to improve GPCR subtype selectivity and functionality. Typically, a bitopic ligand comprises two distinct pharmacophores connected by a linker, thus conferring concomitant binding to the OBS and an allosteric binding site (ABS) of the same receptor monomer [13]. This results in favorable pharmacological properties of the molecule, through receptor selectivity, functional selectivity, and/or allostery [14]. Currently, a new avenue for GPCR drug discovery has emerged by modulating more than one receptor at once to generate beneficial effects synergistically [15], and the resulting ligands have been termed ‘GPCR-targeted MTDLs’.
Although many publications have reported promising results, there is a paucity of review literature focusing on GPCR-targeted MTDLs in treating complex diseases. Meanwhile, Class A GPCRs, the largest and most widely characterized family, has triggered the interest of medicinal chemists both in academia and pharmaceutical companies because of their roles in various diseases and their relatively facile accessibilities. Thus, we review here recent research into Class A GPCR-targeted MTDLs, and discuss their potential therapeutic strategies to reveal current trends in the field and provide insights into the utility of GPCR-targeted MTDLs for future drug design and development.
Rational combination of targets for MTDLs
The identification and validation of target combinations is pivotal to obtain an MTDL to treat complex diseases efficaciously. Generally, targets that belong to the same superfamily are easier to utilize in the design of multitarget ligands [16]. By contrast, for targets from different superfamilies, it would be realistic to perform comprehensive structure–activity relationship (SAR) studies for each of the components to exploit the relevant positions and subsequently combine pharmacophores. Three primary approaches of target combination have been applied to MTDL design and discovery [17]. First, clinical observations, especially for combination therapies, such as drug cocktails, provide increasing insight into how to improve efficacy and reduce adverse effects when more targets are involved. Target combination based on clinical observations has been typically utilized in the design of antipsychotics, several of which have been approved for marketing [18]. Second, phenotypic screening is another feasible approach for target combination;it provides visual effects of compound combinations for synergies at the cell or organism level. Moreover, the rapid development of genetic technology, such as genetic knockdown, makes it possible to validate the synergistic effect of the involved targets [19]. Third, in silico techniques are a relevantly recent approach to screen appropriate target combinations. By using machine learning, the analysis of biological target networks can be conducted to predict efficacious and safe target combinations. However, such hypothetic results require laboratorial verification to confirm their biological foundation and feasibility [20].
Strategies for designing MTDLs
There are three approaches to multitarget therapeutics utilized in clinical practice [21]: the first is multiple-medication therapy (MMT), also referred to as drug cocktails, which comprise two or three different single active drugs that combine different therapeutic mechanisms; the second approach is multi-compound medication (MCM), also known as single-pill drug combinations, which incorporates different drugs into the same formulation; the third approach is MTDLs, by which one active drug might have multiple biological properties by modulating multiple targets simultaneously [22,23]. Despite some advantages of the former two approaches, such as dose flexibility and lower treatment cost, they usually have adverse effects, such a drug–drug interactions and poor patient incompliance [24].
As defined by Morphy et al., MTDLs are compounds that are designed to treat complex diseases by acting at multiple targets associated with pathogenesis [21]. This strategy has inherent advantages over MMT or MCM [5], and also obviates redundant mechanisms caused by single target drugs. There are two well-accepted approaches to generate MTDLs: knowledge-based approaches and screening approaches [25]. Following these strategies, MTDLs have become effective therapeutics for treating complex diseases and several have advanced into clinical trials and to market [26].
Knowledge-based approach
The knowledge-based approach, also known as the pharmacophore-based approach, is the main approach used to generate MTDLs [27]. Pharmacophores from selective ligands are incorporated into a single molecule to integrate the biological activities of the ligands. Based on the degree of integration between pharmacophores, the MTDLs are categorized as linked, fused, or merged (Fig. 1a) [21].
FIGURE 1.
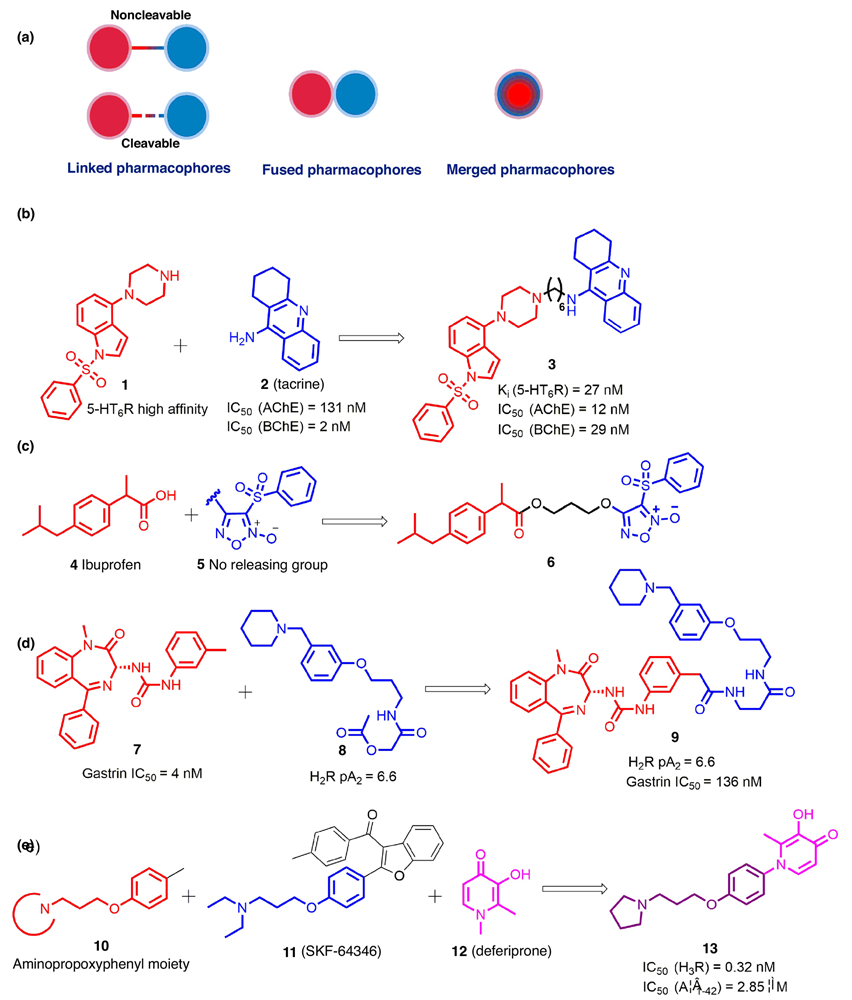
(a) Multitarget drug design strategies based on pharmacophore integration levels; (b) A representative example of noncleavable multitarget-directed ligands (MTDLs); (c) A representative example of cleavable MTDLs; (d) A representative example of fused pharmacophores; (e) A representative example of merged pharmacophores. The parts of structures colored red, blue, and pink represent different pharmacophores conferring corresponding biological activities; the parts in black and purple represent the linker and the merged scaffold between the two pharmacophores, respectively.
Linked pharmacophores
The molecular framework of a linked MTDL is typically separated by a distinct linker group that does not belong to either of the selective ligands, and can be cleavable or noncleavable [21,25]. Although this strategy appears straightforward, it is vital to select the attachment point, length, and chemical composition of linkers carefully to maintain the biological activities of the template scaffolds [28]. In our opinion, there are two approaches that can be used to determine the best positions on two pharmacophores to attach the linker. The first is the application of traditional medicinal chemistry, that is, an understanding of the SAR of each monomeric counterpart as well as consideration of the synthetic feasibility would be beneficial to identify the attachment point on each pharmacophore. In addition, the rapid development of structural biology as well as increasing molecular modeling efforts provide structure-based approaches to select appropriate attachment points on pharmacophores. Most reported linked MTDLs use noncleavable linkers, which are likely stable in vivo, whereas the linkers in cleavable MTDLs can be degraded by specific enzymes to release ligands targeting multiple proteins [25]. An example of noncleavable MTDLs is shown in Fig. 1b, where compound 3 was found to be a multifunctional ligand with well-balanced potencies against the 5-HT6 receptor (5-HT6R) and cholinesterases (ChEs). It was designed by linking a 5-HT6R antagonist pharmacophore 1 with a cholinesterase inhibitor pharmacophore 2 via a polymethylene linker. In addition to its ability to penetrate the blood–brain barrier (BBB), compound 3 also exhibited central cholinomimetic activity in a scopolamine-induced hyperlocomotion model, suggesting its potential application in treating Alzheimer’s disease (AD) [29]. By comparison, as a cleavable MTDLs, compound 6 was reported as a novel nonsteroidal anti-inflammatory drug (NSAIDs) with potent anti-inflammatory and antiaggregatory activities, which integrated ibuprofen 4 and a nitric oxide (NO)-releasing group 5 through an ester linker that can be cleaved by plasma esterases (Fig. 1b) [30].
Fused pharmacophores
Once the pharmacophores are adjacent to each other, these MTDLs can be classified as fused. This strategy is particularly advantageous when handling highly dissimilar targets, because it might be impossible to achieve multiple activities in a compact molecule in which the pharmacophores would have to be highly overlapped. One feasible option in such a situation will be to fuse pharmacophores together and explore tolerant positions in each component [21]. For example, it was suggested that introducing substitutions around the C3′ tolyl group in compound 7 would be tolerated and, thus, a histamine H2 pharmacophore 8 was fused onto the C3′-methyl group of 7 to generate novel dual-functional compound 9, which showed balanced activity at both the H2 and gastrin receptors (Fig. 1d) [31].
Merged pharmacophores
With the highest level of overlapping pharmacophores, merged MTDLs, featuring lower molecular weights and simpler structures, take advantage of commonalities in the structures of the original ligands. As exemplified by a successful case detailed in Fig. 1e, by merging the aminopropoxyphenyl moiety from both compounds 10 and 11 (SKF-64346) and additionally fusing with deferiprone 12, compound 13 was designed, synthesized, and biologically characterized [32]. Its excellent H3R affinity and portent amyloid-β (Aβ) aggregation inhibition, as well as metal ion chelation, demonstrated its potential as a multifunctional candidate to treat AD.
Screening approach
The screening approach is another important strategy in the development of MTDLs [33]. This approach can be further divided into diversity-based and focused screening [21]. Generally, in diversity-based screening, a high-throughput screening (HTS) is applied to diversified compound collections for one target protein, followed by further screening for the second target. Compared with diversity-based screening, focused screening is more commonly used. In this method, compounds are screened for another target once they have already been validated to be effective at one of the targets of interest [17]. The resulting MTDLs might show decent activity against selected targets. However, a well-balanced affinity profile towards all targets of interest is rarely obtained, while the affinity for at least one target is usually subject to further optimization. If the resulting MTDLs are nonselective over many targets, then a ‘design-out’ approach is applied, in which structural modifications are pursued to reduce the affinity for undesired targets to improve the selectivity towards the those of interest.
Recent progress in GPCR-targeted MTDLs in drug design and development
Comprehensive coverage of recently discovered GPCR-targeted MTDLs in treating all complex diseases is beyond the scope of this review. Therefore, we focus here on the application of MTDLs targeting GPCRs for several most common complex disease groups: central nervous system (CNS) diseases, cardiovascular diseases, and diabetes.
GPCR-targeted MTDLs to treat CNS diseases
MTDLs targeting serotonin and dopamine receptors to treat schizophrenia
Schizophrenia is a chronic mental disease affecting ~1% of the global population [34]. Although typical antipsychotics targeting the dopamine D2 receptor (D2R) show potency to control some positive symptoms, they can exacerbate negative symptoms and cognitive dysfunction, as well as causing extrapyramidal symptoms (EPSs) [35]. It is believed that additional interaction with serotonin 5-HT receptors (5-HTRs) could improve their therapeutic profile and reduce adverse effects in treating schizophrenia [21,36]. In this regard, Cao et al. [37] reported a series of compounds using the pharmacophore-linking approach, in which a D2R privileged structure (phenylpiperazine) and a fused tricyclic heterocycle from a reported multitarget lead compound reported by Pfizer [38] were connected by an aliphatic linker. Further SAR studies suggested that tricyclic heterocycle, as shown in compound 14 (Fig. 2), was crucial to maintain balanced affinities for both the 5-HTRs and D2R. Moreover, a four-carbon length of linker was optimal for binding at both receptors. Further in vivo studies indicated that compound 14 was efficacious in different animal models of psychoses with fewer adverse effects. Additionally, a favorable pharmacokinetics profile with oral bioavailability of 58.8% in rats was also observed.
FIGURE 2.
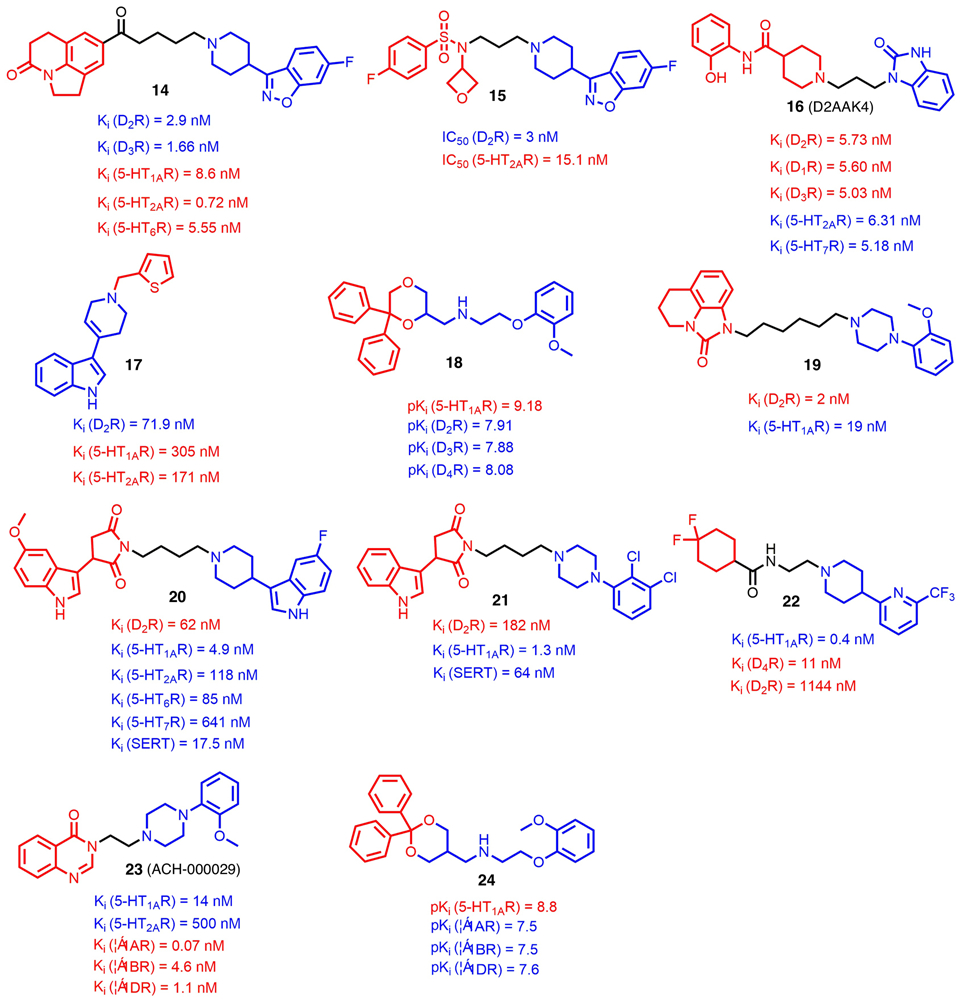
Chemical structures of recently reported G-protein-targeted receptor (GPCR)-targeted multitarget-directed ligands (MTDLs) (compounds 14–24). The parts of structures colored red and blue represent the two pharmacophores conferring corresponding biological activities, and the parts colored black represent the linkers between the two pharmacophores.
By utilizing a similar pharmacophore linking strategy, another lead compound was discovered recently (15;Fig. 2) [39], which showed potent antagonistic activities over the D2R (IC50 = 3.0 nM) and 5-HT2AR (IC50 = 15.1 nM). In addition, compound 15 showed reasonable in vivo metabolic stability with a half-life of 2 h in Institute of Cancer Research (ICR) mice and an acceptable ability to penetrate BBB with Kp value of 4.03. The incorporation of 6-fluoro-1,2-benzisoxazolyl piperidine moiety in both compounds 14 and 15 demonstrated the promising role of such a pharmacophore in the design of multitarget antipsychotics.
By adopting a D2R homology model, Kaczor et al. conducted a structure-based virtual screening from a library of 6.5 million compounds, which resulted in the discovery of another D2R/5-HT2AR antagonist (16; D2AAK4; Fig. 2) [40,41]. In vitro studies showed that compound 16 had a balanced D2R and 5-HTR binding affinity profile. Moreover, in vivo studies suggested that it decreased amphetamine-induced hyperactivity and improved memory consolidation in passive avoidance tests. As a modification of another experimentally confirmed virtual screening hit D2AAK1 [40], a series of indole derivatives was synthesized and pharmacologically studied. SAR studies revealed that a thiopheneylmethyl group attaching to the tetrahydropyridine fragment contributed favorably towards increased 5-HT2AR and D2R affinities, whereas other types of substitution on the indole ring did not significantly influence potency on either receptor. Eventually, compound 17 was found to be a dual antagonist of both 5-HT2AR and D2R with affinities in the nanomolar range (Fig. 2). Further behavioral studies revealed that it exhibited antipsychotic, procognitive, and antidepressant activities in chimney tests and forced swim tests (FST) in mice [42].
To obtain novel multitarget analogs with improved D2R affinity and high affinity for 5-HTRs, Del Bello et al. performed extensive SAR studies on a 6,6-diphenyl-1,4-dioxane scaffold, which lead to the discovery of compound 18. In vitro results indicated that, in addition to desirable binding affinity at target receptors, compound 18 showed a multitarget combination of 5-HT1A/D4 agonism and D2/D3/5-HT2A antagonism, which made it a good starting point to develop new pharmacological tools potentially useful in treating schizophrenia [43]. These results suggested that ligands that simultaneously modulate the function of several monoaminergic receptors provide beneficial therapeutic effects for patients with schizophrenia.
MTDLs targeting serotonin and dopamine receptors to treat depression
Depression remains a major burden on societies worldwide [44]. The pharmacological profile of the molecules with high affinities for 5-HT1AR and D2R is thought to improve the treatment of depression [45]. As a versatile drug template with high affinities for these two receptors, a long-chain arylpiperazine (LCAP) was linked with another tricyclic heterocycle to give compound 19, which showed a balanced affinity profile over 5-HT1AR and D2R (5-HT1AR, Ki = 19 nM; D2R, Ki = 2 nM) (Fig. 2) [46]. Further molecular docking studies showed that compound 19 orientated towards the second transmembrane helix rather than the sixth one, compared with other analogs in the D2R-binding pocket. Such a unique binding conformation conferred the largest number of hydrophobic interactions, which could explain its excellent affinity at the D2R.
Given that combining selective serotonin reuptake inhibitors (SSRIs) with agonist/antagonist activity at various 5-HTRs could significantly increase the effectiveness of depression therapy [47], Wrobel et al. [48] recently reported the discovery of compound 20 (Fig. 2), in which a 3-piperidin-3-yl-1H-indole moiety, conferring high affinity for the 5-HT1AR and serotonin transporters (SERTs), was tethered with a cyclic imide moiety through an appropriate linker. Further studies indicated that compound 20 exhibited balanced affinities for the 5-HT1AR/5-HT6R/5-HT7R/SERT/D2R and showed antidepressant-like activity in the FST model. The metabolic stability of 20 was similar to those of compounds without the fluorine substitution on the aromatic ring. In light of in silico predictions of the site of metabolism, one explanation might be that cytochrome P450 (CYP)-mediated oxidations of two-carbon atoms in the alkyl linker lead to N-dealkylation and, thus, the degradation of the molecule into two smaller parts.
After replacing the 3-piperidin-3-yl-1H-indole moiety with different LCAP moieties, a series of novel MTDLs was generated. Further SAR studies revealed that halogen substituents in the phenylpiperazine ring potently improved the binding affinity for 5-HT1AR, which could be attributed to the formation of specific halogen bonds between the ligand and the receptor. In addition, substitutions at the para-position of phenylpiperazine increased the affinity for the SERT, whereas ortho-position substitution was beneficial for 5-HT1AR. Among these ligands, compound 21 emerged with promising mixed receptor profiles for 5-HT1AR, D2R, and the serotonin transporter (Ki = 1.3 nM, 182 nM, and 64 nM, respectively) (Fig. 2) [49].
Stimulating both the 5-HT1AR and D4R simultaneously could be a promising strategy to ameliorate anxiety [50]. However, no specific drugs with selective dual agonism for both receptors have been reported. Recently, Sumitomo Dainippon Pharma Co. Ltd patented compound 22 with dual agonism for 5-HT1AR and D4R by utilizing the pharmacophore-linking strategy (Fig. 2). Besides its excellent binding affinity profile (5-HT1AR, Ki = 0.4 nM; D4R, Ki = 11 nM) and relatively long half-life in cynomolgus monkey (t1/2 = 11 h), as well as acceptable hERG inhibition activity (IC50 = 2.5 μM), compound 22 exhibited a 82.5% decrease in the cataleptic freezing reaction in a contextual fear conditioning test and a 74.7% decrease in the immobility time in a FST model [51]. This evidence demonstrates the therapeutic potential of multitarget ligands with high affinities for 5-HTRs and D2Rs in combatting depression.
MTDLs targeting serotonin and other GPCRs to treat depression
The noradrenaline (NE) and 5-HT systems have reciprocal interactions and modulate each other’s actions in the brain [52]. It has been suggested that novel molecules acting at specific 5-HT and NE receptors would achieve a higher efficacy with fewer adverse effects in treating anxiety and depression. Compound 23 (ACH-000029, Fig. 2) is a newly released compound that acts at a range of serotonergic (5-HT1A partial agonism and 5-HT2A antagonism) and α-adrenergic (α-1A, 1B and 1D antagonism) receptors (α1Rs). It also exhibited high permeability in Caco-2 cells and low CYP inhibition as well as acute anxiolytic effects in the marble-burying (MB) and light–dark box (LDB) models [53]. These properties, together with its low toxicity, supported the further development of ACH-000029 as a drug candidate for the treatment of psychiatric disorders.
Balanced modulation of 5-HTRs and α1Rs could be beneficial for the treatment of several psychiatric disorders [54]. Through ring expansion/opening and molecular elongation/simplification of a versatile 1,3-dioxolane scaffold, a series of novel compounds was obtained recently in the search for novel antidepressants. Preliminary SAR studies suggested that insertion of a methylene in the lateral chain slightly improved binding affinities for all the α1R subtypes, while leaving an unchanged affinity for 5-HT1AR. Similar results were observed when adding a methylene into the 1,3-dioxolane scaffold. The most promising compound showed potent agonism at 5-HT1AR and high binding affinities for α1Rs (24; Fig. 2). In addition, compound 24 has a high biodistribution in brain compartments when orally administered. Further open-field tests demonstrated the anxiolytic and antidepressant effects of 24, suggesting its potential as an antidepressant [55]. The above examples demonstrate that targeting 5-HTRs and another specific GPCR simultaneously could be a valuable approach to generate novel MTDLs with desirable therapeutic effects for combatting depression.
MTDLs targeting MAO-B and other GPCRs to treat Parkinson’s disease
Parkinson’s disease (PD) is one of the most common CNS disorders, with a significant impact on the aging human population. The progress of this disease is typically complicated and multifactorial, leading to degeneration of neurons and inflammatory processes in the brain [56]. As an emerging strategy, MTDLs have gained significant attention in the treatment of PD. One of the proposed strategies is the combination of monoamine oxidase B (MAO-B) inhibition and adenosine A2A receptor (A2AAR) blockade [57]. Following this strategy, Kuder et al. [58] reported a series of novel annelated xanthine derivatives bearing a N9-substituted benzyl group targeting both proteins. Follow-up SAR studies suggested that fused rings adjacent to the xanthine scaffold were tolerated towards both MAO-B and A2AAR, whereas the heterocyclic aromatic substitutions at the N9 position were detrimental to affinities. The attached benzyl group appeared to be favorable and further halogen substitutions increased the potency toward both targets. After in vitro screenings, compound 25 (A2AAR, Ki = 264 nM; MAO-B, IC50 = 243 nM) was advanced into pharmacological evaluations. Unfortunately, moderate hepatotoxicity of 25 was observed when used with the HepG2 cell line. Meanwhile, a similar phenomenon was also found in other analogs, indicating that such a tricyclic scaffold presents a risk of hepatotoxicity.
Two series of phenyl xanthine (PX) derivatives were synthesized and biologically evaluated to explore the speculation that PX derivatives have antiparkinsonian properties [59]. Among these new PX derivatives, compound 26 acted as a potent A2AAR antagonist with Ki value of 330 nM and a MAO-B inhibitor with a IC50 value of 290 nM (Fig. 3). It did not show noticeable cytotoxicity against human neuroblastoma SH-SY5Y cells at 10 μM concentration. Additionally, it also displayed significant anticatalepsy ability at a dose of 50 mg/kg in haloperidol-induced catalepsy studies. These favorable properties suggested compound 26 as a promising candidate for treating PD.
FIGURE 3.
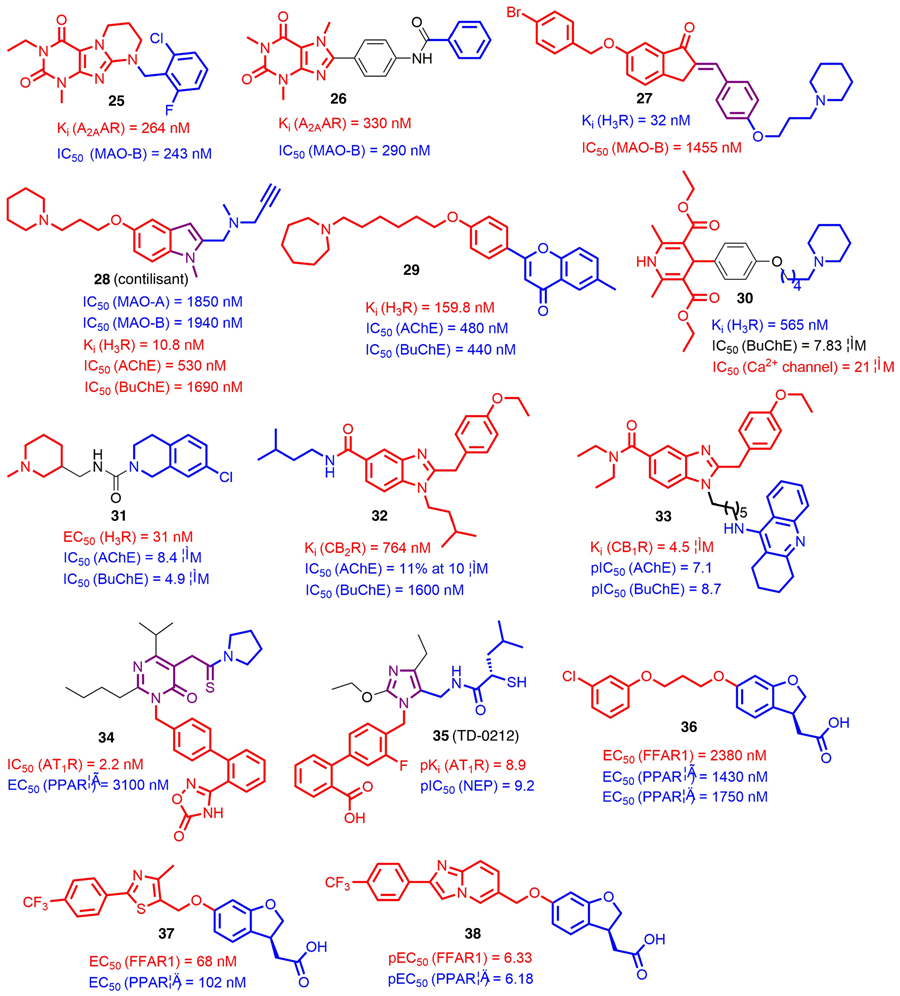
Chemical structures of recently reported G-protein-targeted receptor (GPCR)-targeted multitarget-directed ligands (MTDLs) (compounds 25–38). The parts of structures colored red and blue represent the two pharmacophores conferring corresponding biological activities, and the parts colored black and purple represent the linkers and the merged scaffolds between the two pharmacophores, respectively.
The design of MAO-B/H3R dual-targeting ligands is a potentially innovative approach to modulate the dopamine imbalance in patients with PD [60]. Three series of compounds were generated by merging a H3R pharmacophore with indanone-related MAO-B motifs [61]. The derivatives with substitutions at the C6 and C5 positions of indanone did not show the desired H3R affinity, although they did show moderate MAO-B inhibitions, and similar results were found in more lipophilic 2-benzylidene-1-indanone derivatives. After introducing a basic moiety with a polymethylene linker to the C4′ position of the benzylidene, the resulting compounds showed favorable potencies at both targets. Of these, compound 27 showed desirable binding affinity for H3R (Ki = 32 nM) and inhibition of MAO-B (IC50 = 1455 nM) (Fig. 3). Given that a reversible MAO-B inhibitor might have fewer adverse effects than an irreversible one, following dilution studies in excess of substrates revealed a reversible inhibition of compound 27 against MAO-B, suggesting it as a lead for further design of novel therapeutics against PD.
By merging structural elements of the antioxidant ASS234 and the H3R antagonist ciproxifan, a series of novel indole derivatives was designed, synthesized, and biologically evaluated [62]. SAR studies suggested that the length of the spacer between the cyclic aliphatic amines and indole core was crucial to the binding to MAO-A/B, with a two-carbon atom spacer being optimal for piperidine derivatives. From this small series, compound 28 (contilisant) was found to have inhibitory activities towards MAO-A/B and H3R as well as ChEs (Fig. 3). From dilution studies in substrate excess, contilisant was found to have improved reversible inhibition of MAO-B compared with ASS234. Additional in vitro studies showed that contilisant offered significant neuroprotection against the toxic insults assayed at 0.3 μM, which was comparable to that offered by melatonin. Further in vivo studies indicated that contilisant (at 1 mg/kg intraperitoneally) also significantly improved lipopolysaccharide (LPS)-induced cognitive deficits.
In the light of the above studies, targeting both MAO-B and relevant GPCRs appears to be a promising approach to identify novel therapeutics with high potency and desirable druggability to confront the multifactorial nature of PD neurodegeneration.
MTDLs targeting H3 receptor and other targets to treat Alzheimer’s disease
AD is a progressive neurological disorder with multifactorial etiology, and is particularly prevalent in older people [63]. Mounting evidence indicates that combining ChE inhibitory activity with H3R antagonism could provide new tools to combat the cognitive symptoms of AD [64]. By applying virtual screening from 200 structures of pitolisant and ciproxifan analogs, Bajda et al. obtained 26 hits with high ChemScore values targeting ChEs. Of these, 15 compounds were synthesized and biologically evaluated. According to preliminary SAR study results, homopiperidine with a five- or six-carbon atom linker was preferable for H3R affinity, whereas an eight-carbon atom linker was beneficial for ChE inhibition. After further in vitro screenings, compound 29 stood out and showed potent inhibitory activities toward ChEs (AChE, IC50 = 0.48 μM; BuChE, IC50 = 0.44 μM) as well as potency against H3R with Ki value of 159.8 nM [65]. Further in silico ADMET prediction by SwissADME demonstrated that 29 had properties consistent with the rule proposed by Veber [rotatable bonds ≤10 and topological polar surface area (TPSA) ≤140] [66]. These findings suggested that compound 29 could serve as a lead for further optimization and development of more potent MTDLs with more accurate pharmacokinetics profiles.
Through multicomponent synthesis, Malek et al. incorporated a typical H3R antagonist motif cycloalkylamine into 1,4-dihydropyridine (1,4-DHP) via a convenient linker to generate several new tri-target small molecules. SAR studies revealed that, when a three-carbon atom linker was attached, desirable H3R activities were always observed for synthesized compounds, whereas a longer linker appeared more favorable for BuChE inhibition. As a promising lead, compound 30 showed potent affinity for H3R (Ki = 565 nM) and moderate BuChE inhibition (IC50 = 7.83 μM) as well as a reasonable Ca2+ channel blockade activity (IC50 = 21 μM) (Fig. 3). Further in vivo studies demonstrated that compound 30 was capable of restoring cognitive impairment induced by LPS at 10 mg/kg, which was comparable to the effect of donepezil at 1 mg/kg [67].
Ghamari et al. [68] recently tested a computationally identified lead compound 31 (Fig. 3). Biological evaluations revealed that it acted as a potent H3R antagonist (EC50 = 31 nM) and a dual inhibitor against AChE and BuChE with IC50 values of 8.40 and 4.93 μM, respectively. Moreover, MTT assays suggested that compound 31 did not exhibit significant cytotoxicity in H3R-expressing CHO-K1 cells (GI50≈100 nM). Such a ligand with dual inhibitory activity on H3R and ChEs could be utilized for lead optimization to achieve higher potency and efficacy. These aforementioned observations further validate the hypothesis that hitting the multiple targets implicated in AD etiology may provide novel therapeutics with desirable pharmacological profiles.
MTDLs targeting CB2 receptor and ChE to treat Alzheimer’s disease
CBR subtype 2 (CB2R) agonists exert beneficial effect in neurodegenerative disorders [69]. By contrast, ChEs inhibition has been a well-established approach to achieve symptomatic cognition improvement in patients with AD [70]. Dolles et al. [71] merged pharmacophores from a BuChE inhibitor and a CB2R agonist into benzimidazole-based small molecules. For these novel ligands, different chains at position 5 of the benzimidazole core had varying effects on CB2R affinity, whereas it appeared that bulky and nonpolar substituents were tolerated. Among them, compound 32 was identified as a dual-acting ligand with a balanced affinity–inhibitory activity and excellent selectivity over both CB1R and AChE (Fig. 3). In vivo studies suggested that compound 32 was active with sustained prevention of Aβ25–35-induced learning deficits in passive avoidance tests at 1 mg/kg. These data showed the possibility of combining a ligand with selective and balanced GPCR activity and/or enzyme inhibition to achieve in vivo potency for AD treatment.
On the basis of these observations, the Dolles research group further adopted the pharmacophore-linking strategy and synthesized a series of novel compounds in which a ChE inhibitor tacrine and a benzimidazole-based CB2R agonist were linked by an appropriate spacer [72]. In this study, the types of linker, chain length, and connection point were extensively investigated. SAR studies indicated that if tacrine was attached to position 1 of the benzimidazole core, the affinities for both ChEs and CB2R increased with the length of the spacer. However, when switched to position 5, experimental data did not support such a correlation. Additionally, the introduction of a 2-PEG-linker or a disulfide linker had no significant effect on either targets. Among them, compound 33 emerged as a potent AChE and BuChE dual inhibitor (AChE, pIC50 = 7.10; BuChE, pIC50 = 8.7) and a partial agonist at the CB2R (Ki = 4.5 μM;EC50 = 3.05 μM; Emax = 51%) (Fig. 3). In addition to the inhibition of AChE-mediated and self-induced Aβ aggregation, compound 33 did not show any neurotoxicity at the doses tested (max. 10 μM). Further in vivo studies indicated that compound 33 significantly prevented the development of learning deficits and completely prevented Aβ25–35 impairments at a dose of 0.3 mg/kg. Given the promising in vitro and in vivo study results and its low hepatotoxicity, compound 33 could be a potential clinical candidate for treating AD. As an emerging approach, MTDLs that have a balanced affinity and desirable selectivity profile on CB2R and ChEs could be a solution for the treatment of both cognitive and pathophysiological impairments in AD.
GPCR-targeted MTDLs to treat cardiovascular diseases
Hypertension is a key risk factor for cardiovascular diseases, which directly or indirectly cause deaths of at least 9 million people globally every year [73]. Among various approaches to control hypertension, angiotensin II type 1 receptor (AT1R) antagonists with additional binding affinity and activation at peroxisome proliferator activated receptor-γ (PPARγ) might be beneficial in controlling blood pressure and lipid metabolism [74]. Choung et al. [75] applied structural modification on the basis of a Korean Food and Drug Administration (FDA)-approved PPARγ partial agonist, fimasartan. According to the docking mode of fimasartan with the AT1R crystal structure, the pyrimidinone core was maintained because it can form hydrophobic and hydrogen bonding interactions with key amino acid residues in the binding pocket. Meanwhile, rational modifications were conducted, including bioisosteric replacement of the tetrazole moiety in fimasartan and side-chain variation on the pyrimidinone core. Eventually, lead compound 34 was observed to have excellent AT1R antagonism (Ca2+ mobilization IC50 = 2.2 nM) and PPARγ agonism (EC50 = 3.1 μM) (Fig. 3). Additionally, in vivo studies indicated that compound 34 exhibited suppressive effects on blood pressure increase at a dose of 30 mpk in an angiotensin II-induced hypertension rat model and significantly suppressed blood glucose increase at a dose of 60 mpk in leptin-deficient db/db mouse diabetes model.
Dual inhibition of the renin angiotensin system (RAS) and the cardiac natriuretic peptide system (CNPS) could provide superior reductions in blood pressure [76]. To this end, McKinnell et al. designed and synthesized a series of MTDLs by merging the structures of an AT1 antagonist losartan and a neutral endopeptidase (NEP) inhibitor thiorphan (Fig. 3) [77]. SAR studies showed that introduction of an S-isobutyl group into the thiorphan moiety maintained the best balance of potency at both targets. It was notable that adding a fluorine atom ortho- to the head group maintained NEP activity and significantly boosted AT1 potency, whereas alternative fluorine substitution patterns compromised the potency at both targets. Based on these findings, compound 35 (TD-0212), a dual AT1 antagonist/NEP inhibitor, was chosen for in vivo studies. It was suggested that compound 35 resulted in blood pressure reductions similar to omapatrilat in models of renin-dependent and -independent hypertension. Additionally, it did not increase tracheal plasma extravasation (TPE) at antihypertensive doses, indicating its low risk of angioedema. These aforementioned observations highlight the importance of the approach by targeting AT1R and another target involved in hypertension pathological processes in the development of novel antihypertension agents.
GPCR-targeted MTDLs for diabetes
Type 2 diabetes mellitus is a metabolic disease with insulin resistance and/or insulin deficiency. The important role of the free fatty acid receptor 1 (FFAR1 or GPR40) in stimulating incretin secretion and mediating glucose-stimulated insulin secretion makes it a desirable target for antidiabetic drugs [78]. As mentioned earlier, PPARs have a pivotal role in metabolic regulation and are highly associated with glucose control and lipid metabolism [79]. Taken together, the multifunctional FFAR1/PPARs agonists could be a potential strategy to improve insulin secretion and sensibility. To test this hypothesis, Li et al. designed and synthesized several compounds by fusing the common scaffold of PPARs and FFAR1 agonists. Further biological assays demonstrated their multifunctional profiles targeting both the PPARs and FFAR1. First, SAR studies indicated that the linker between two pharmacophores had a strict requirement for the length and conformational flexibility, with a five-carbon atom one appeared to be preferred for the potency of both targets. Moreover, the carboxylate moiety was crucial for activities as well as for maintaining balance between FFAR1, PPARδ, and PPARγ. Among them, compound 36 had relatively balanced activities between FFAR1 and PPARs (Fig. 3) and also improved the glucose tolerance of leptin-deficient ob/ob mice in a dose-dependent manner [80].
By inserting a thiazole-based fragment into compound 36, another novel MTDL compound, 37, was generated [81]. In addition to the enhancement of tolerance of ob/ob mice for glucose loading, the introduction of a thiazole-type heterocyclic ring also conferred reasonable pharmacokinetics profiles with high plasma concentrations, a sustained half-life, and low clearance. Given the important role of the thiazole ring in optimizing pharmacological profiles of compound 37, Li et al. further conducted structural modification by incorporating bioisosteres to replace the thiazole ring [82]. In vitro study results revealed that most of the thiazole bioisosteric derivatives showed lower activities on FFAR1 and PPARδ compared with the parent compound 37, whereas thiadiazole and oxadiazole derivatives appeared to be more potent. Moreover, the introduction of fused heterocycles had varying effects on potency towards both targets. Among them, compound 38 had an improved balance between FFAR1 and PPARδ and also exerted better glucose-lowering effects and insulin sensitivity compared with compound 37. These desirable effects of multifunctional FFAR1/PPARs ligands reveal their potential applications in the development of novel antidiabetics.
Concluding remarks and future perspectives
From the above-discussed examples, it appears that a bioactive compound carrying therapeutic effects on the modulation of a single target might not be an optimal drug candidate for treating complex diseases [21]. In an effort to tackle this issue, the development of MTDLs has become a promising approach in drug design and development campaigns. Meanwhile, the pivotal roles in the pathophysiology of various human diseases and accessibilities of druggable sites at the cell surface make GPCRs desirable targets for developing MTDLs. Recent advances in MTDLs targeting GPCRs have mainly focused on treating CNS disorders, with very few for cancer treatment. However, several GPCRs have been suggested as potential cancer targets based on mRNA expression analyses of tumors [83]. Moreover, accumulating data suggest that crosstalk between GPCRs and other well-established anticancer targets [e.g., epidermal growth factor receptor (EGFR)] occur in various cancers. This reveals a promising strategy of developing novel MTDLs targeting both the GPCRs and EGFR for cancer therapy [84]. Although GPCRs are associated with nearly every aspect of human pathophysiology [15], many of them, namely untapped GPCRs, have not yet been targeted. The exploitation of these targets will further open new possibilities in GPCR-targeted MTDL discovery. Furthermore, owing to recent breakthroughs in GPCR crystallography, at least 44 distinct GPCR structures and 205 ligand–receptor complexes are now resolved, which has not only expedited the development of GPCR-targeted MTDLs [85], but also provided more opportunities to refine pharmacophores and further guide the design of multitarget ligands.
Meanwhile, drug repositioning/repurposing, referring to the exploration of new indications for marketed drugs, has gained interest over the past decade because it can reduce the time and cost of bringing a therapeutic to the market [86]. Of the approved GPCR-targeted drugs, 156 (33%) have more than one indication and the overall average is 1.5 indications per drug [15], showing the potential utility of bioactive compounds targeting GPCRs for several indications. In this sense, GPCR-targeted MTDLs could be identified as natural candidates by retargeting those off-target drugs; thus, numerous so-called ‘dirty drugs’ showing multifunctional profiles in their early stage of research could be reconsidered for new purposes [36]. Moreover, for some selective GPCR-targeted drugs already at the later stages of development, other targets outside their main therapeutic targets might be identified, which will further facilitate the development of GPCR-targeted MTDLs. With increasing understanding of the pathogenesis of complex diseases as well as new breakthroughs in the structural biology and pharmacology of GPCRs, the search for novel GPCR-targeted MTDLs with an appropriate balance of biological activities will generate major impacts in drug design, discovery, and development.
Acknowledgments
This work was partially supported by NIH/NIDA Grants R01DA024022, R01DA044855, and UG3DA050311 (Y.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
References
- 1.Fischer E (1894) Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges 27, 2985–2993 [Google Scholar]
- 2.Medina-Franco JL et al. (2013) Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 18, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins AL (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol 4, 682–690 [DOI] [PubMed] [Google Scholar]
- 4.Tao L et al. (2015) Co-targeting cancer drug escape pathways confers clinical advantage for multi-target anticancer drugs. Pharmacol. Res 102, 123–131 [DOI] [PubMed] [Google Scholar]
- 5.Cavalli A et al. (2008) Multi-target-directed ligands to combat neurodegenerative diseases.J. Med. Chem 51, 347–372 [DOI] [PubMed] [Google Scholar]
- 6.Turnaturi R et al. (2016) Multitarget opioid ligands in pain relief: new players in an old game. Eur. J. Med. Chem 108, 211–228 [DOI] [PubMed] [Google Scholar]
- 7.Grivennikov SI et al. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolognesi ML (2013) Polypharmacology in a single drug: multitarget drugs. Curr. Med. Chem 20, 1639–1645 [DOI] [PubMed] [Google Scholar]
- 9.Yang T et al. (2020) Recent advances in the rational drug design based on multi-target ligands. Curr. Med. Chem XX, YYY–ZZZ [DOI] [PubMed] [Google Scholar]
- 10.Latorraca NR et al. (2017) GPCR dynamics: structures in motion. Chem. Rev 117, 139–155 [DOI] [PubMed] [Google Scholar]
- 11.Thomsen W et al. (2005) Functional assays for screening GPCR targets. Curr. Opin. Biotechnol 16, 655–665 [DOI] [PubMed] [Google Scholar]
- 12.Erez M et al. (1982) Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. evidence for bridging between proximal recognition sites. J. Med. Chem 25, 847–849 [DOI] [PubMed] [Google Scholar]
- 13.Fronik P et al. (2017) Bitopic ligands and metastable binding sites: opportunities for G protein-coupled receptor (GPCR) medicinal chemistry. J. Med. Chem 60, 4126–4134 [DOI] [PubMed] [Google Scholar]
- 14.Newman AH et al. (2020) 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: designing bivalent or bitopic molecules for g-protein coupled receptors. the whole is greater than the sum of its parts.J. Med. Chem 63, 1779–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauser AS et al. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laubli H et al. (2018) The multi-receptor inhibitor axitinib reverses tumor-induced immunosuppression and potentiates treatment with immune-modulatory antibodies in preclinical murine models. Cancer Immunol. Immunother 67, 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J et al. (2019) Rational design of multitarget-directed ligands: strategies and emerging paradigms. J. Med. Chem 62, 8881–8914 [DOI] [PubMed] [Google Scholar]
- 18.Krause M et al. (2018) Antipsychotic drugs for patients with schizophrenia and predominant or prominent negative symptoms: a systematic review and meta-analysis. Eur. Arch. Psychiatry Clin. Neurosci 268, 625–639 [DOI] [PubMed] [Google Scholar]
- 19.Proschak E et al. (2019) Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem 62, 420–444 [DOI] [PubMed] [Google Scholar]
- 20.Chua HE et al. (2017) Synergistic target combination prediction from curated signaling networks: Machine learning meets systems biology and pharmacology. Methods 129, 60–80 [DOI] [PubMed] [Google Scholar]
- 21.Morphy R and Rankovic Z (2005) Designed multiple ligands. An emerging drug discover) paradigm. J. Med. Chem 48 (21), 6523–6543 [DOI] [PubMed] [Google Scholar]
- 22.Cavalli A et al. (2008) Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 51, 347–372 [DOI] [PubMed] [Google Scholar]
- 23.Morphy R and Rankovic Z (2009) Designing multiple ligands - medicinal chemistry strategies and challenges. Curr. Pharm. Des 15, 587–600 [DOI] [PubMed] [Google Scholar]
- 24.Eisen SA et al. (1990) The effect of prescribed daily dose frequency on patient medication compliance. Arch. Intern. Med 150, 1881–1884 [PubMed] [Google Scholar]
- 25.Morphy R et al. (2004) From magic bullets to designed multiple ligands. Drug Discov. Today. 9, 641–651 [DOI] [PubMed] [Google Scholar]
- 26.Costantino L and Barlocco D (2012) Designed multiple ligands: basic research vs clinical outcomes. Curr. Med. Chem 19, 3353–3387 [DOI] [PubMed] [Google Scholar]
- 27.Hopkins AL et al. (2006) Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol 16, 127–136 [DOI] [PubMed] [Google Scholar]
- 28.Savelieff MG et al. (2019) Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev 119, 1221–1322 [DOI] [PubMed] [Google Scholar]
- 29.Wieckowska A et al. (2016) Novel multi-target-directed ligands for Alzheimer’s disease: combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem 124, 63–81 [DOI] [PubMed] [Google Scholar]
- 30.Lolli ML et al. (2001) A new class of ibuprofen derivatives with reduced gastrotoxicity. J. Med. Chem 44, 3463–3468 [DOI] [PubMed] [Google Scholar]
- 31.Kawanishi Y et al. (1996) Synthesis and pharmacological evaluation of highly potent dual histamine H2 and gastrin receptor antagonists. Bioorg. Med. Chem. Lett 6, 1427–1430 [Google Scholar]
- 32.Sheng R et al. (2016) Novel 1-phenyl-3-hydroxy-4-pyridinone derivatives as multifunctional agents for the therapy of Alzheimer’s disease. ACS Chem. Neurosci 7, 69–81 [DOI] [PubMed] [Google Scholar]
- 33.Jenwitheesuk E et al. (2008) Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol. Sci 29, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skene NG et al. (2018) Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muench J and Hamer AM (2010) Adverse effects of antipsychotic medications. Am. Fam. Physician 81, 617–622 [PubMed] [Google Scholar]
- 36.Roth BL et al. (2004) Magic shotguns versus magic bullets: selectively nonselective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov 3, 353–359 [DOI] [PubMed] [Google Scholar]
- 37.Cao X et al. (2018) Synthesis and biological evaluation of fused tricyclic heterocycle piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics.J. Med. Chem 61, 10017–10039 [DOI] [PubMed] [Google Scholar]
- 38.Awasthi AK et al. (2008) Fused tricycle heterocycles for the treatment of schizophrenia. Journal XX, YYY–ZZZ [Google Scholar]
- 39.Zhu C et al. (2020) Discovery of aryl-piperidine derivatives as potential antipsychotic agents using molecular hybridization strategy. Eur. J. Med. Chem 193, 112214. [DOI] [PubMed] [Google Scholar]
- 40.Kaczor AA et al. (2016) Structure-based virtual screening for dopamine D2 receptor ligands as potential antipsychotics. ChemMedChem 11, 718–729 [DOI] [PubMed] [Google Scholar]
- 41.Kaczor AA et al. (2020) N-(2-hydroxyphenyl)-1-[3-(2-oxo-2,3-dihydro-1H-benzimidazol-1-yl)propyl]piperidine-4-carboxamide (D2AAK4), a multi-target ligand of aminergic GPCRs, as a potential antipsychotic. Biomolecules 10, YYY–ZZZ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondej M et al. (2019) Synthesis, pharmacological and structural studies of 5-substituted-3-(1-arylmethyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indoles as multi-target ligands of aminergic GPCRs. Eur. J. Med. Chem 180, 673–689 [DOI] [PubMed] [Google Scholar]
- 43.Del Bello F et al. (2019) Multitarget 1,4-dioxane compounds combining favorable D2-like and 5-HT1A receptor interactions with potential for the treatment of Parkinson’s disease or schizophrenia. ACS Chem. Neurosci 10, 2222–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yohn CN et al. (2017) The role of 5HT receptors in depression. Mol. Brain 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jesulola E et al. (2018) Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model - are we there yet? Behav. Brain Res 341, 79–90 [DOI] [PubMed] [Google Scholar]
- 46.Zareba P et al. (2019) New dual ligands for the D2 and 5-HT1A receptors from the group of 1,8-naphthyl derivatives of LCAP. Bioorg. Med. Chem. Lett 29, 2236–2242 [DOI] [PubMed] [Google Scholar]
- 47.Dimitriou EC and Dimitriou CE (1998) Buspirone augmentation of antidepressant therapy. J. Clin. Psychopharmacol 18, 465–469 [DOI] [PubMed] [Google Scholar]
- 48.Wrobel MZ et al. (2019) Synthesis and biological evaluation of new multi-target 3-(1H-indol-3-yl)pyrrolidine-2,5-dione derivatives with potential antidepressant effect. Eur. J. Med. Chem 183, 111736. [DOI] [PubMed] [Google Scholar]
- 49.Wrobel MZ et al. (2020) Synthesis of new 4-butyl-arylpiperazine-3-(1H-indol-3-yl) pyrrolidine-2,5-dione derivatives and evaluation for their 5–HT1A and D2 receptor affinity and serotonin transporter inhibition. Bioorg. Chem 97, 103662. [DOI] [PubMed] [Google Scholar]
- 50.Marona-Lewicka D and Nichols DE (2011) Potential serotonin 5-HT(1A) and dopamine D(4) receptor modulation of the discriminative stimulus effects of amphetamine in rats. Behav. Pharmacol 22, 508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshinaga H et al. Sumitomo Dainippon Pharma Co. 2,6-disubstitued pyridine derivative. US 20200039952 A1. [Google Scholar]
- 52.Blier P and Briley M (2011) The noradrenergic symptom cluster: clinical expression and neuropharmacology. Neuropsychiatr. Dis. Treat 7, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azevedo H et al. (2019) Preclinical characterization of ACH-000029, a novel anxiolytic compound acting on serotonergic and alpha-adrenergic receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 95, 109707. [DOI] [PubMed] [Google Scholar]
- 54.Carhart-Harris RL and Nutt DJ (2017) Serotonin and brain function: a tale of two receptors. J. Psychopharmacol 31, 1091–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franchini S et al. (2019) 1,3-Dioxane as a scaffold for potent and selective 5-HT1AR agonist with in-vivo anxiolytic, anti-depressant and anti-nociceptive activity. Eur. J. Med. Chem 176, 310–325 [DOI] [PubMed] [Google Scholar]
- 56.Kakkar AK and Dahiya N (2015) Management of Parkinsons disease: current and future pharmacotherapy. Eur. J. Pharmacol 750, 74–81 [DOI] [PubMed] [Google Scholar]
- 57.Jaiteh M et al. (2018) Docking screens for dual inhibitors of disparate drug targets for Parkinson’s Disease. J. Med. Chem 61, 5269–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuder KJ et al. (2020) Novel, dual target-directed annelated xanthine derivatives acting on adenosine receptors and monoamine oxidase B. ChemMedChem XX, YYY–ZZZ [DOI] [PubMed] [Google Scholar]
- 59.Wang X et al. (2017) Synthesis and evaluation of phenylxanthine derivatives as potential dual A2AR antagonists/MAO-B inhibitors for Parkinson’s disease. Molecules 22 (6), [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu W and Chen Z (2017) The roles of histamine and its receptor ligands in central nervous system disorders: An update. Pharmacol. Ther 175, 116–132 [DOI] [PubMed] [Google Scholar]
- 61.Affini A et al. (2018) Novel indanone derivatives as MAO B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur. J. Med. Chem 148, 487–497 [DOI] [PubMed] [Google Scholar]
- 62.Bautista-Aguilera OM et al. (2017) Multitarget-directed ligands combining cholinesterase and monoamine oxidase inhibition with histamine H3 R antagonism for neurodegenerative diseases. Angew. Chem. Int. Ed. Engl 56, 12765–12769 [DOI] [PubMed] [Google Scholar]
- 63.Querfurth HW and LaFerla FM (2010) Alzheimer’s disease. N. Engl. J. Med 362, 329–344 [DOI] [PubMed] [Google Scholar]
- 64.Khan N et al. (2016) The dual-acting H3 receptor antagonist and AChE inhibitor UW-MD-71 dose-dependently enhances memory retrieval and reverses dizocilpine-induced memory impairment in rats. Behav. Brain Res 297, 155–164 [DOI] [PubMed] [Google Scholar]
- 65.Bajda M et al. (2020) Search for new multi–target compounds against Alzheimer’s disease among histamine H3 receptor ligands. Eur. J. Med. Chem 185, 111785. [DOI] [PubMed] [Google Scholar]
- 66.Veber DF et al. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem 45, 2615–2623 [DOI] [PubMed] [Google Scholar]
- 67.Malek R et al. (2019) New dual small molecules for Alzheimer’s disease therapy combining histamine H3 receptor (H3R) antagonism and calcium channels blockade with additional cholinesterase inhibition. J. Med. Chem 62 (24), 11416–11422 [DOI] [PubMed] [Google Scholar]
- 68.Ghamari N et al. (2020) In silico and in vitro studies of two non-imidazole multiple targeting agents at histamine H3 receptors and cholinesterase enzymes. Chem. Biol. Drug Des 95, 279–290 [DOI] [PubMed] [Google Scholar]
- 69.Ehrhart J et al. (2005) Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zemek F et al. (2014) Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug. Saf 13, 759–774 [DOI] [PubMed] [Google Scholar]
- 71.Dolles D et al. (2018) Structure-activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human cannabinoid receptor 2 ligands with pro-cognitive in vivo profiles. J. Med. Chem 61, 1646–1663 [DOI] [PubMed] [Google Scholar]
- 72.Scheiner M et al. (2019) Dual-acting cholinesterase-human cannabinoid receptor 2 ligands show pronounced neuroprotection in vitro and overadditive and disease-modifying neuroprotective effects in vivo. J. Med. Chem 62, 9078–9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kitt J et al. (2019) New approaches in hypertension management: a review of current and developing technologies and their potential impact on hypertension care. Curr. Hypertens. Rep 21, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amano Y et al. (2012) Structural basis for telmisartan-mediated partial activation of PPAR gamma. Hypertens. Res 35, 715–719 [DOI] [PubMed] [Google Scholar]
- 75.Choung W et al. (2018) Discovery of the bifunctional modulator of angiotensin II type 1 receptor (AT1R) and PPARgamma derived from the AT1R antagonist, Fimasartan. Bioorg. Med. Chem. Lett 28, 3155–3160 [DOI] [PubMed] [Google Scholar]
- 76.Packer M et al. (2002) Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 106, 920–926 [DOI] [PubMed] [Google Scholar]
- 77.McKinnell RM et al. (2019) Discovery of TD-0212, an orally active dual pharmacology AT1 Antagonist and neprilysin inhibitor (ARNI). ACS Med. Chem. Lett 10, 86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Z et al. (2020) Current status of GPR40/FFAR1 modulators in medicinal chemistry (2016-2019): a patent review. Expert Opin. Ther. Pat 30, 27–38 [DOI] [PubMed] [Google Scholar]
- 79.Monsalve FA et al. (2013) Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediators Inflamm. 2013, 549627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Z et al. (2018) Design, synthesis, and biological evaluation of novel pan agonists of FFA1, PPARgamma and PPARdelta. Eur. J. Med. Chem. 159, 267–276 [DOI] [PubMed] [Google Scholar]
- 81.Li Z et al. (2019) Discovery of first-in-class thiazole-based dual FFA1/PPARdelta agonists as potential anti-diabetic agents. Eur. J. Med. Chem 164, 352–365 [DOI] [PubMed] [Google Scholar]
- 82.Li Z et al. (2019) Design, synthesis, and biological evaluation of novel dual FFA1 (GPR40)/PPARdelta agonists as potential anti-diabetic agents. Bioorg. Chem 92, 103254. [DOI] [PubMed] [Google Scholar]
- 83.Li S et al. (2005) Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int. J. Oncol 27, 1329–1339 [PubMed] [Google Scholar]
- 84.Kose M (2017) GPCRs and EGFR - cross-talk of membrane receptors in cancer. Bioorg. Med. Chem. Lett 27 (16), 3611–3620 [DOI] [PubMed] [Google Scholar]
- 85.Cooke RM et al. (2015) Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov. Today 20, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 86.Nosengo N (2016) Can you teach old drugs new tricks? Nature 534, 314–316 [DOI] [PubMed] [Google Scholar]