

SUMMARY
Determining protein levels in each tissue and how they compare with RNA levels is important for understanding human biology and disease as well as regulatory processes that control protein levels. We quantified the relative protein levels from over 12,000 genes across 32 normal human tissues. Tissue-specific or tissue-enriched proteins were identified and compared to transcriptome data. Many ubiquitous transcripts are found to encode tissue-specific proteins. Discordance of RNA and protein enrichment revealed potential sites of synthesis and action of secreted proteins. The tissue-specific distribution of proteins also provides an in-depth view of complex biological events that require the interplay of multiple tissues. Most importantly, our study demonstrated that protein tissue-enrichment information can explain phenotypes of genetic diseases, which cannot be obtained by transcript information alone. Overall, our results demonstrate how understanding protein levels can provide insights into regulation, secretome, metabolism, and human diseases.
Graphical Abstract
In Brief
Proteomics analysis across human tissues from the GTeX resource reveals insight into tissue-specific pathways and phenotypes arising from genetic diseases.
INTRODUCTION
Measuring RNA and proteins expressed within each tissue is fundamental for understanding human biology and disease. To date, most efforts have focused on RNA measurements. However, protein levels often correlate poorly with transcript levels (Liu et al., 2016; Payne, 2015; Vogel and Marcotte, 2012), and a detailed study of proteins, which reside downstream of transcription and participate more directly in vital cellular activities, is expected to help our molecular understanding of complex tissues. Such studies could complement transcriptomic studies and provide insights into post-transcriptional regulatory mechanisms, as well as into human biology and disease.
Previous studies of protein levels have been performed. Mass spectrometry analysis of different cell lines and human tissues has identified approximately 85% of the proteins encoded by the ~20,000 human protein-coding genes (Wilhelm et al., 2014; Kim et al., 2014; Becket al., 2011). However, most of these studies focused on in-depth protein identification and proteogenomic analysis, and the protein quantitation was primarily based on spectra counting, which is less quantitative, or on intensity-based absolute quantitation from disparate datasets (Li et al., 2012). Moreover, most samples do not have the corresponding RNA information from the same tissue, making the direct comparison of RNA and protein levels difficult. The Human Protein Atlas project (HPA) generated a tissue-based map of the human proteome on the basis of transcriptome data and antibody staining and classified tissue-specific expression based on RNA (Uhlen et al., 2015). Although antibodies can provide local and cell-type-specific information, quantitation can be challenging, isoforms are not distinguished, and antibody specificity is an important concern. A very recent study performed label-free mass spectrometry analysis of 29 different tissue samples (Wang et al., 2019a). However, only one biological sample for each tissue type was analyzed, thereby limiting the generalization of the findings, and many tissues were not analyzed. Although each of these studies has greatly advanced the tissue protein identification and provided a useful resource, a broader study with data at both protein and RNA level on the same healthy tissues, along with accurate quantitation information, is valuable to study the protein-level differences with transcripts. In addition, although valuable, none of the previous studies used proteomics data and tissue-specific protein expression to systematically examine human biological processes and diseases.
The diverse tissue resources from the GTEx project make it possible to use advanced mass spectrometry and quantitation methods to study many human tissues with multiple biological replicates and match them to RNA levels in the same tissues. By profiling 201 samples covering a wide range of tissues for which there is matched RNA data, we presented tissue-specific protein expression in many tissues not analyzed by existing studies as well as many biological insights into human biology and disease that cannot be obtained from transcript data. Notably, many RNA transcripts do not always show concordant tissue-enriched or tissue-specific patterns with their encoded proteins. Examples include many vesicular transport proteins that are involved in neurotransmitter function and cell-cell signaling highly enriched in the brain but do not exhibit RNA enrichment. Conversely, tissue-enriched/-specific RNA transcripts can be found in tissues with no corresponding protein enrichment. For the first time, we used the protein/RNA concordant enrichment information to suggest proteins that undergo constitutive or regulated secretion and the possible locations of synthesis and action of many secreted proteins.
The tissue-specific distribution of proteins also provides an in-depth view of complex biological events that require the interplay of multiple tissues. For example, the branched-chain amino acid (BCAA) pathway components that are extremely important in metabolism were found to be differentially expressed in multiple organs, indicating a coordinated systemic control of energy utilization. Examination of many tissue-specific proteins also revealed that they are associated with specific diseases and provide a molecular explanation for the underlying defects that cannot be interpreted from transcriptome level. Overall, these results provide a valuable resource and demonstrate that understanding protein levels can provide insights into metabolism, regulation, secretome, and human disease.
RESULTS
Protein Profiling across Tissues
The Genotype-Tissue Expression (GTEx) project collected samples from 54 tissues of 948 post-mortem donors and characterized their transcriptomes (Carithers et al., 2015; GTEx Consortium, 2015; Project and eGTEx Project, 2017). For this study we quantitatively profiled the proteome of 201 GTEx samples from 32 different tissue types of 14 normal individuals (Figure 1A), covering all major organs (Table S1). The proteome data were acquired with a tandem mass tag (TMT) 10plex/MS3 mass spectrometry strategy (Figure 1B), which enables 10 isotopically labeled samples to be analyzed in a single experiment (McAlister et al., 2014). To increase the proteome coverage, each TMT 10plex sample was extensively fractionated (Figure 1B). To facilitate cross-tissue comparison and to reduce the influence of technical variation between mass spectrometry runs, we randomized the tissue samples such that each TMT 10plex consists of an assortment of tissues and a reference sample.
Figure 1. Overview of Tissue Proteome Experimental Workflow and Results.

(A) Type of tissues and biological replicates analyzed in this study.
(B) TMT 10plex- and MS3-based mass spectrometry quantitative proteomics workflow.
(C) Number of proteins quantitated in each tissue (Table S2). Each dot represents data from one person.
(D) Distribution of the number of proteins quantified across different numbers of tissues. There are 6,357 proteins present in all 32 tissue types. Among them, 1,565 proteins were classified as HK proteins. See Table S2 for details.
(E) Distribution of RNA expression in log2TPM for both the identified and unidentified proteins. RNAs with log2TPM <1 were collapsed to 1.
(F) Number of proteins identified in each protein class. The predicted protein classes are based on the results from HPA study (Uhlén et al., 2015).
A database search using spectra from each TMT run separately at a peptide FDR of 1% identified proteins encoded by 13,813 genes across all tissue samples (10,196 proteins were identified when using pooled data [1% protein FDR]) (Table S1 and S4A). The relative levels of 12,627 proteins were quantified after applying strict filters (Table S2). On average, the relative abundance of more than 7,500 proteins was measured in each tissue type (Figure 1C; Table S2), and 6,357 proteins were present in all 32 tissue types (Figure 1D). These results indicate that most (85%) of the proteins detected in a given tissue are expected to be found across tissue types and that individual tissues are not characterized by the simple presence or absence of proteins but rather by their relative abundance (Wilhelm et al., 2014; Geiger et al., 2013).
To determine types of proteins that were not detected in this study, we compared the RNA abundance of both the identified and unidentified proteins. As shown in Figure 1E, low-abundance transcripts are often undetected at the protein level. However, when the RNA TPM is higher e.g., above 32, some proteins are still not detected (16% protein-coding genes); detection of these proteins is not significantly affected by RNA abundance (Table S1). Undetected proteins could be due to post-transcriptional regulation, degradation of proteins, or limitations of mass spectrometry. Membrane proteins were underdetected across the entire RNA expression abundance level; of the 5,500 predicted membrane-bound proteins and 3,000 secreted proteins (Uhlén et al., 2015), we detected 3,143 membrane and 1,848 secreted proteins, respectively (Figure 1F).
Identification of Tissue-Enriched and Tissue-Specific Proteins
To identify tissue-enriched and tissue-specific proteins, we determined their relative levels in each tissue as the normalized log ratio of each protein to the corresponding protein in the common, pan-tissue, reference. Normalization used an algorithm designed to allow for disparate compositions of proteins across different tissues (Wang et al., 2019b). Hierarchical clustering of the protein levels revealed that samples clustered by tissue types, indicating that protein variation between tissue types exceeds that between individuals (Figures 2A and S1). As expected, samples from physiologically related tissues, such as arteries from different parts of the body as well as heart and skeletal muscles, clustered together (Figures 2A and S1). The few exceptions are logical: esophagus mucosa layers, high in epithelial cells clustered with skin instead of other esophagus samples. Interestingly, lung tightly clustered with spleen despite their seemingly distinct functions (Figures 2A and S1), likely due to a common group of 78 immune proteins enriched in these tissues (Table S4). The lung has recently been found to host many immune cells (Hartl et al., 2018), likely explaining the lung-spleen clustering.
Figure 2. Quantitative Proteome Analysis across Tissues.

(A) Clustering of proteome data across tissues using t-SNE. As shown, samples are separated by tissue types not by individuals.
(B) Method for defining TS scores. As an example, for gene PHYH, the left panel shows the distribution of its TS scores across tissues fitted using AdaTiSS. The right panel shows its TS scores in each tissue.
(C) The numbers of enriched and specific protein/RNA across tissues. Enrichment categories are defined in STAR Methods.
(D) Protein enrichment across tissues and their biological functions. The enriched proteins represent tissue-specific/shared functions. The gene ontology (GO) term functional enrichment results are summarized in Table S5.
The enrichment of each protein across tissues was defined by a tissue specificity (TS) score (Figures 2B and S3; STAR Methods). We define a protein as tissue enriched if its TS score reaches 2.5 (SD from the mean of the population distribution) in at least one tissue. Similarly, if the TS score of a protein is greater than 4 in a tissue and is at least 1.5 higher than the protein’s TS scores in any other tissue, this protein is considered tissue specific. In total, we observed 3,967 (31.4%) enriched proteins and 1,595 (12.6%) tissue-specific proteins (Tables S2 and S4). Brain was found to have the largest number of enriched and specific proteins, followed by liver, heart, and muscle (Figure 2C; Tables S2 and S4). The tissue-enriched/-specific expression of 11 important proteins in diverse pathways described below was validated by western blot analysis (Figure S5).
The functional enrichment of proteins that are enriched in one tissue or tissue group is presented in Figure 2D and matches expectations (Table S5). For example, proteins involved in the nervous system and synaptic transmission are highly enriched in brain. We also found interesting common enrichment of proteins across several different tissue types. For example, a large group of proteins involved in oxidation and reduction are enriched in multiple metabolically active tissues such as heart, muscle, brain, liver, and stomach (Figure 2D; also see the metabolism section below). Although ribosomal proteins are present in almost all organs, they are enriched in organs active in protein synthesis, notably in pancreas followed by liver and stomach. The stomach has not been generally reported as metabolic active, but the enrichment of ribosomal proteins and those involved in oxidation and reduction is consistent with this interpretation.
Interestingly, even across similar tissues we found differentially enriched proteins. For example, proteins involved in mitochondrial translation are only enriched in heart, whereas proteins in proteolysis are enriched in skeletal muscle (Table S5). Sarcomere proteins, such as myosin, tropomyosin, and troponin, are highly enriched in both heart and skeletal muscle, but they utilize different gene isoforms (Table S7). The left ventricle is enriched in proteins involved in energy production, whereas the atrial appendage is abundant in peptide hormones (NPPA, NPPB) and specific myosin isoforms (Figure S5; Table S7). Surprisingly, in many of these cases the RNAs of the genes are enriched in all the relevant tissues, suggesting differential regulation at the post-transcriptional level (Table S3). Overall, the common and tissue-specific distributions of proteins across tissue types mirror the function of the proteins and tissues.
Proteins that are present in all tissues and not enriched in any of them are defined as housekeeping (HK) proteins. Of the 6,357 proteins identified in all 32 tissues, 1,565 proteins were classified as HK proteins (Figure 1D; Table S4). Functional analysis showed that the HK proteins are mainly involved in basic cell activities such as RNA processing, gene expression, and protein localization (Table S5).
Comparison with Other Studies
We sought to compare our results with those of other quantitative studies. Uhlen et al. performed tissue enrichment/specificity at the RNA level but did not provide protein tissue-specific data beyond a coarse (low, medium, high) metric based on antibody staining. We directly compared our protein-enrichment data to the most recent Wang et al. (2019a) study, which used label-free mass spectometry to quantify 13,640 proteins and classified tissue enrichment by using a fold-change metric. In our study, we quantified 12,627 proteins by using the TMT labeling method and used TS score for tissue-enrichment analysis. For proteins that are quantified in both studies, 1,080/1,438 tissue-specific proteins from our study were also enriched or enhanced in the Wang et al. (2019a) study (Table S2). However, because only 16 tissue types are in common between our study and theirs, Q11 some proteins (n = 342) that are specific in our study showed different tissue enrichment in their study due to the lack of the same tissue types in their study (skin, skeletal muscle, and arteries, etc.). For the same reason, some tissue-specific proteins (n = 497) in their study were enriched differently in our study. Although there is good agreement for the tissue-specific proteins, there is less agreement at other enrichment categories, which could be due to the different criteria we used to define tissue enrichment (detailed information is in Table S2). The fact that more biological replicates for most tissues (rather than one in Wang et al., 2019a) and TMT/MS3 quantitative measurement in our study should yield more accurate and generalizable results. Overall, our study provides extensive quantitative results concerning tissue-specific and tissue-enriched proteins in humans.
Correlation between RNA and Protein Levels
Our study offers a good opportunity to characterize the correlation between protein and RNA because the transcriptome and proteome data were generated from the same tissue specimens. We computed the correlation between the protein and RNA abundance across 32 tissues for each gene (Figure 3A; Table S4) and found the median Spearman correlation is 0.46 (interquartile range of 0.24–0.65), consistent with previous findings (Payne, 2015; Vogel and Marcotte, 2012). For 6,228/12,627 genes, their protein levels are statistically significantly and positively correlated with RNA levels (BH-adjusted p value < 0.1), and a very small number (60) showed negative correlations (Table S4).
Figure 3. Protein and RNA Correlation and Concordance Analysis across and within Tissues.

(A) Spearman Correlation of protein and RNA across 32 tissues. The significance is based on permutation test from 200 permutations (BH-adjusted p value < 0.1).
(B) The number of concordantly or discordantly enriched proteins and RNAs in each tissue.
(C) The enrichment of housekeeping RNAs at the protein level across tissues.
(D) TS-score of RAB7A across tissues for the proteome and transcriptome.
(E) Secreted proteins and their concordance to corresponding RNAs in each tissue.
(F) Concordance analysis of pituitary secreted proteins. All the peptide hormones in the anterior part of pituitary are concordantly enriched at the protein and RNA levels. Hormones in the posterior part of the pituitary are secreted from hypothalamus and stored in the pituitary.
See also Table S4.
We also compared the protein- versus RNA-enrichment pattern in each tissue. The TS score of RNA expression was calculated analogously to that of protein expression (Figures S2 and S4; Table S3). Concordance was defined as both protein and RNA enriched in the same tissue as determined by outlier analysis; for discordance, only protein or RNA was tissue enriched, and enrichment is at least 1.5 away from each other (see STAR Methods). Among 5,562 proteins that show tissue specificity or enrichment, 2,695 were concordantly enriched at the RNA level over all the tissues. Discordance was also widely observed; for example, many gene products (3,088) are highly enriched at the protein but not RNA level (Table S4; note proteins that are concordant in some tissues could be discordant in other tissues). Among all the tissues, brain hosts the largest number of genes that are only enriched at the protein level (Figure 3B). These proteins participate in many key functions in brain such as neurotransmitter transport, cell-cell communication, and signal transduction. Proteins in the oxidation reduction pathway were also only enriched at the protein level. Surprisingly, for many protein-enriched/-specific genes, their RNAs are ubiquitously distributed and defined as HK RNAs; one example is Rab proteins, which are specifically enriched in brain (TS score >4) (Figures 3C and 3D). Western blot of a few proteins such as RAB1B, VPS52, and PURA confirmed brain enrichment (Figure S5).
Another group of discordant genes (2,899/12,245) are only enriched at the RNA but not protein level (Table S4). A large number of them are in testis followed by liver. In testis, their protein level is either very low or undetectable. The failure to detect these proteins could be due to their low abundance, expression in a few cell types, and/or rapid protein degradation. In liver, most of these discordant proteins are secreted into the blood (discussed below). These different results demonstrate that tissue-specific expression is further controlled at the protein level and that tissue-specific functions cannot be distinguished on the basis of RNA information alone. The expression level of every gene in each tissue can be visualized at http://snyderome.stanford.edu/TSomics.html.
RNA and Protein Expression Differences Reveal Insights to Protein Secretion
Potential insights into protein secretion can be obtained by analyzing the discordance and concordance of RNA and protein-enrichment information. Discordance between RNA and protein in some cases could be caused by constitutive secretion of proteins to other tissues; furthermore, concordance of secretory protein levels with their RNA levels can suggest that these proteins are stored in secretory vesicles and released upon stimulation (Feizi et al., 2017) or locally secreted (Uhlén et al., 2019). We therefore systematically analyzed the concordance and discordance of RNA and protein levels of secreted proteins defined according to HPA predictions (Uhlén et al., 2015) to ascertain which proteins likely undergo regulated or constitutive secretion as well as potential sites of synthesis and action of secreted proteins (enriched only as RNA or protein, respectively). We found that many (481/1,903) predicted secretory proteins are concordantly enriched at both protein and RNA level (Figure 3E; Table S4), consistent with regulated secretion or local secretion. Examples include digestive enzymes and hormones (e.g., insulin, see below). Discordant patterns are also observed, suggestive of constitutive secretion. As Figure 3E showed, both secretion patterns exist in many tissues.
Among all tissues, liver has the highest number of secreted proteins, followed by brain, artery, pancreas, and pituitary. In liver, the largest proportion of secreted proteins are only enriched at the RNA, but not the protein, level indicative of constitutive secretion. Pathway analysis (Table S4) shows that these proteins are enriched in complement activation (e.g., C2-9, CFHRs), coagulation (e.g., CFs, SERPINs), acute phase response (e.g., CRP, HP, ITIH4, and SAA4), and lipid transport (apolipoproteins) and protein localization (e.g., TF, HRG, AGT). We found these proteins are enriched in arteries and exhibited discordant RNA expression (Table S4), indicating that these proteins are synthesized in liver and constitutively secreted into blood. However, 69 secreted proteins show concordance in their RNA and protein enrichment; these are mostly enzymes involved in drug metabolism and oxidation reduction such as CYP2 subfamily members (Table S4) and could undergo regulated secretion.
Similar to the liver, the pancreas is a major secretory organ. Uniquely, it has both exocrine and endocrine cells that secrete many digestive enzymes and multiple hormones (Dubois, 1994). Digestive enzymes in the pancreas are stored and secreted into the gut upon food ingestion. A group of major digestive enzymes showed concordant enrichment including pancreatic amylases, lipases, proteases, and others (Table S4). Multiple hormones detected in our study such as insulin, glucagon, chromogranins, secretogranins, and somatostatin are also secreted by the pancreas (Lloyd et al., 1988). Insulin and glucagon are concordantly enriched at the protein and RNA level (Table S4) and regulated by blood glucose levels. A few enzymes such as SERPINAs and SPINT1 and other proteins such as MUC6, ALB, C5, F11, and GC are enriched only at the RNA level in pancreas (Table S4). Although these proteins are well known to be secreted mainly by the liver, our data raise the possibility that the pancreas could also synthesize and secrete them into the bloodstream, although other possible post-transcriptional mechanisms exist. Nonetheless, because the pancreas and liver share a common embryological origin and some histological similarities, it is possible they also have cellular functions in common (Esrefoglu et al., 2016).
The pituitary is the master gland that secretes many hormones that regulate the secretion of other hormones (Emerald, 2016). Our data show that hormones such as TSH, ACTH, GH, PRL, CHGB, SCGs, LH, and FSH are all enriched in the pituitary at both the protein and RNA level (Table S4). These hormones are made in the anterior part of the pituitary but are stored and undergo regulated secretion by hormones produced in the hypothalamus (Emerald, 2016). Two other major hormones in the pituitary are ADH and OXT. They are both highly enriched at the protein level but not at the RNA level; these proteins are synthesized in the hypothalamus and secreted to and stored in the posterior part of pituitary (Figure 3F) (Emerald, 2016).
When examining other tissues, we also observe that the brain has many secreted proteins that are concordantly enriched (Figure 3E). Most of them are not secreted into the bloodstream but instead are brain-specific surface proteins such as receptors for signal transduction or locally secreted proteins involved in cell-cell interaction. Other tissues such as the spleen and lung have a group of secreted proteins that are discordantly enriched. These proteins are mainly involved in immune response and directly secreted into the bloodstream. In the transverse colon and small intestine, proteins are mainly secreted into the lumen or extracellular matrix (Table S4). Overall, these results indicate that proteins undergo diverse patterns of synthesis and secretion that can vary with different tissues and that protein-level analysis can provide insights into their regulated secretion and sites of action.
Insights into Metabolism through Tissue-Specific Protein Expression
In addition to secretory proteins, the analysis of a broad set of tissues provided insights into coordinated activities of metabolic biological processes across the human body. Although metabolic pathways have been mapped at the RNA level (Uhlén et al., 2015), a more direct understanding of the metabolic proteins and pathways through systematic analysis of protein levels has not been performed. Through our analysis of tissue-specific proteins, we found proteins of the same metabolic pathway (KEGG) were often present in different tissues revealing a complex interplay of multiple key organs in metabolism and energy utilization, consistent with, and extending, previously described results (Angione, 2019; Heindel et al., 2017).
Proteins from 1,434 genes annotated in the KEGG metabolism database were quantified across the 32 tissues (Table S5). Liver was found to have the largest number of enriched metabolic proteins, followed by brain, muscle, and heart. Using the proteomics data, the enrichment of metabolic pathways in each tissue is presented in Figure 4A and Table S5. Most (55/67) metabolic pathways are enriched in liver except several such as the oxidative phosphorylation pathway, which is enriched in heart, skeletal muscle, and brain, where it is likely the major energy source for tissues that function aerobically. Like the oxidative phosphorylation pathway, the glycolysis and TCA cycle pathways are also enriched in heart, skeletal muscle, and brain. Coupling of these pathways can achieve the complete oxidation of glucose and generate the maximum amount of ATP required for high energy demand (Berg et al., 2012). Previous studies have shown that, when active, skeletal muscle requires the most energy, whereas when resting, heart and kidney have the highest metabolism rate, followed by brain and liver (Wang et al., 2010). Surprisingly, oxidative phosphorylation, glycolysis, and tricarboxylic acid (TCA) cycle pathways are also enriched in the stomach, which usually is not considered a high-metabolism organ (Wang et al., 2010). However, oxidative phosphorylation in stomach tissue is necessary for acid generation by parietal cells (Suzuki et al., 2012), which is believed to break down food and serve as a biological defense that eliminates pathogens, activates endothelial NADPH oxidase, and increases endothelial RO, and the acid generation is expected to require high metabolic activity. Mitochondrial proteins and enzymes involved in energy production including NADH:ubiquinone oxidoreductase, ATP synthase, cytochrome c, and coenzymes are also highly enriched in heart, muscle, brain, and stomach tissues (Table S5). Importantly, whereas ATP synthases are enriched in muscle for energy production, the V-ATPases, which have been implicated in synaptic transmission and neurological disease (Fassio et al., 2018), are specifically enriched in brain (Bodzęta et al., 2017), which was also validated by western blot (Figure S5). Thus, these protein distributions provide insights into energy metabolism and other biology functions.
Figure 4. Analysis of Tissue-Specific Metabolism.

(A) Enrichment of a subset of metabolic pathways across different tissues. The heatmap shows the significance of the -log(p values) from the pathway enrichment test. The plot only includes the tissues having at least one significantly enriched pathway under threshold of 0.001 for the p value from Fisher’s exact test.
(B) The enrichment map of key enzymes in BCAA metabolism.
(C) Tissue enrichment of the first-step enzyme BCAT1/2 and the second-step enzyme BCKDH and its activator PPM1K.
(D) Interactive map of BCAA shuttling among tissues and the enriched enzymes.
Historically, adrenal and gonad glands (ovary, testes) have been considered as major sites for steroid hormone production, and liver has been considered as the main organ that metabolizes the hormones. In addition to these sites our data also showed that the steroid hormone biosynthesis pathway is not only enriched in the adrenal gland, testis, and liver, but also in the small intestine and transverse colon. These results support the recent view that steroid hormones can also be produced and metabolized in other tissues such as the intestine (Wittenburg et al., 2010).
BCAA Metabolic Enzymes Are Enriched in Different Organs
BCAAs are important for energy utilization and other biological processes, and their metabolism is a complex multi-tissue process coordinated through the tissue-specific distribution of key enzymes (Arany and Neinast, 2018; Holeček, 2018). By analyzing the expression pattern of these enzymes, we found tissue-specific distributions suggestive of different roles of those tissues in BCAA metabolism.
Branched-chain aminotransferase (BCAT) is the first-step enzyme in BCAA metabolism and is encoded by two genes: BCAT1 located in the cytosol and BCAT2 in the mitochondria (Conway and Hutson, 2015). Multiple studies reported that skeletal muscle is the initial site of BCAA metabolism based on the high activity of BCAT2 in skeletal muscle and lack of activity in liver (Arany and Neinast, 2018; Holeček, 2018). Surprisingly, we found that BCAT2 is not enriched in muscle at either the protein or RNA level, but it is highly expressed in heart, stomach, and adrenal glands (Figures 4B and 4C). The enrichment information was also validated by western blot as shown in Figure S5. Because the total weight of skeletal muscle is 35%–40% of body weight, even with moderate amounts of BCAT2, skeletal muscle is likely a major site for the first step of BCAA metabolism. However, the enrichment of BCAT2 in other tissues also indicates that substantial amounts of BCAAs can be metabolized in other high-energy utilization tissues (e.g., heart, stomach). Both of our mass spectrometry data and western blot results showed that BCAT1 is the most abundant in pancreas (Figures 4B, 4C, and S5), instead of brain. Although few studies have investigated BCAA metabolism in the pancreas, our data showed that pancreas is the only tissue that has high expression of both BCAT1 and BCAT2. In addition, recent studies have shown that BCAA levels spike years before pancreatic cancer, suggesting that BCAA metabolism disorder might contribute to cancer development (Katagiri et al., 2018; Mayers et al., 2014).
Liver was thought to be the major site of the second-step BCAA metabolism based on the highest activity of the second-step enzyme BCKDH (branched-chain α-ketoacid dehydrogenase). We found that BCKDH was equally enriched in heart, stomach, and liver (Figures 4B, 4C, and S5). BCKDH activity is regulated by two modifying proteins, BCKDH kinase (BCKDK, inactivator) and protein phosphatase (PPM1K, activator) (White et al., 2018). The BCKDK tissue distribution is very similar to BCKDH but the activator PPM1K showed strong enrichment in the heart and not in other tissues (Figure 4C). A high ratio of PPM1K to BCKDK greatly favors BCKA oxidative decarboxylation (White et al., 2018). It has been proposed that BCKDH and BCAT2 directly interact to achieve great efficiency of energy production during BCAA metabolism (Conway and Hutson, 2015). The concordant enrichment of these two enzymes was only observed in heart and stomach, suggesting that BCAA might be an important energy source for these two tissues; indeed, impairment of BCAA metabolism in rat and cardiomyocytes leads to the loss of cardiac contractility, premature death, and induced apoptosis (Huang et al., 2011; Du et al., 2018). Enrichment of all key enzymes in heart (Figures 4B and 4C) indicates that BCAAs are likely to be completely metabolized to provide energy for this tissue. In other tissues (e.g., liver and muscle) BCAA could be important for both energy metabolism as well as distribution of intermediates to other tissues (Figure 4D). Regardless, the tissue-specific expression of enzymes indicates that the BCAA metabolism is likely to be important in these tissues and their diverse distribution indicates a coordinated interplay of energy metabolism across multiple organs.
Protein Expression Provides Insights into Genetic Diseases and Drug Targets
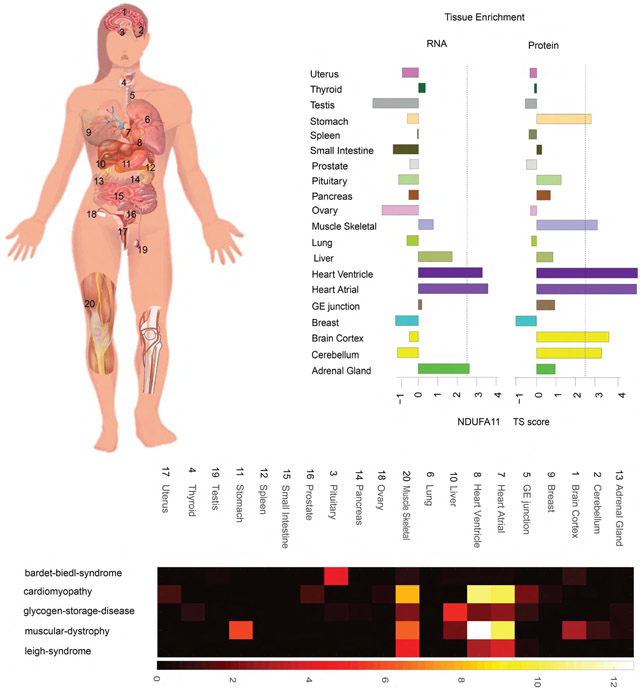
Protein expression information could provide insights into the underlying disease mechanisms that cannot be identified by using transcript localization information. We systematically investigated the protein expression pattern with genetic diseases listed in the Online Mendelian Inheritance in Man (OMIM) catalog (Amberger et al., 2015) and found many cases in which proteins known to be disrupted by disease-associated mutations are enriched in tissues that manifest disease-related pathophysiology (Figure 5A; Table S6); many of these would not be evident from RNA analysis. For example, Bardet-Biedl syndrome (BBS) is a complex genetic disorder caused by mutations in at least 14 different genes and affects many parts of the body (Khan et al., 2016; Haq et al., 2019). BBS-affiliated vision loss, polydactyly, and obesity are characteristics of BBS as well as many other abnormalities that vary among affected individuals (Foggensteiner and Beales, 2015). Some of the symptoms can be explained by specific gene mutations but many are still largely unknown; protein tissue-specific expression information might explain some of the clinic symptoms. We detected proteins from 11/14 BBS genes among which seven are enriched in the pituitary and five are enriched in brain, muscle, heart, or liver (Table S4). Abnormality of proteins highly enriched in pituitary can broadly affect developmental processes and perhaps cause obesity, diabetes, or hypogonadism observed in BBS patients (Foggensteiner and Beales, 2015). The enrichment of proteins in brain, muscle, heart, and liver might also contribute to defects such as intellectual disability, delayed motor skills, and conditions that involve the heart, liver, and digestive systems (Foggensteiner and Beales, 2015).
Figure 5. Association of Tissue-Enriched Proteins with Genetic Diseases.

(A) Heatmap of the enrichment of genetic diseases across tissues. Some genetic diseases are significantly enriched in specific tissues such as Bardet-Biedl syndrome and Leigh syndrome. The disease terms are from the OMIM database.
(B) Protein and RNA concordance heatmap for genes involved in Leigh syndrome.
Leigh syndrome is another genetic disease that is associated with mutations in as many as 75 genes (DiMauro and De Vivo, 1996; Lake et al., 2016). Most of the affected proteins are involved in oxidative phosphorylation in mitochondria. Of the 67/75 proteins we observe, 52 of them showed tissue enrichment in a few metabolically active tissues (Figures 5A and 5B). Heart has the highest, followed by muscle, brain, and stomach. Some of these proteins were enriched in all affected tissues, and some are only enriched in one or a few tissues; their different distribution might cause different tissue-related clinical symptoms. For example, the characteristic progressive loss of mental and movement abilities of Leigh syndrome are most likely related to proteins that are enriched in brain and muscle. Some affected individuals develop hypertrophic cardiomyopathy, which could be caused by mutations in proteins enriched in the heart. The first signs of Leigh syndrome in infancy are vomiting, diarrhea, and difficulty swallowing, which could be explained by the abnormality of proteins enriched in stomach. Importantly, for many genes enrichment only occurs at the protein level and not at the RNA level, demonstrating the importance of collecting protein-expression information for understanding disease phenotype. For example, the neurological and digestive symptoms are best explained by the protein-enrichment data and not the RNA data (Figure 5B).
There are 1,329 potential drug-targeted proteins identified in our data among which 421 are FDA-approved drug targets (Table S4). Among these targeted proteins, 742 are enriched in different tissues, of which about half (368) are enriched in more than one tissue Table S4). For protein drug targets enriched outside of the target organ, the drug could have unintended or side effects in the off-target tissue. For example, valproic acid is a well-known anticonvulsant exerting its effects through the inhibition of GABA transaminase (GABAT) in the brain as one of the main mechanisms of action (Chateauvieux et al., 2010). Our data showed that GABA transaminase is not only highly enriched in brain but is even more enriched in the liver and pancreas, a result validated by western blot analysis as well (Figure S5). This suggests the inhibition of GABA transaminase in these two tissues as a potential mechanism underlying the reported toxicity in liver and pancreas (Chapman et al., 2001; Gayam et al., 2018). Overall, our protein distribution studies provide insights in human disease and treatment.
Identification of Missing Proteins
Missing proteins are classified as identified on the basis of protein existence (PE) criteria by using a 1–5 tier system as defined by the Human Proteome Project (Baker et al., 2017). Our study of diverse tissues identified 374 proteins on the missing protein list provided by MissingProteinPedia and provided information on the specific tissue in which they were expressed (http://www.missingproteins.org/protein/web/; Table S1). Ninety-one proteins meet the most stringent level 5 criteria of PE: at least two unique peptides, each with a peptide length 39. Among these proteins, 23 have reliable antibody scores annotated by HPA (Uhlén et al., 2015). As examples, RASA4 and UNC13C have 11 and 15 unique peptides (39aa) identified, respectively. RASA4 was observed in a majority of the samples, and UNC13C was identified in around half of all samples. UNC13C is enriched in the brain and validated by western blot (Figure S5), whereas RASA4 is highly enriched in skeletal muscle. In total, 26 proteins showed enrichment in the brain, muscle, and a few other tissues, and most are intracellular proteins (Table S1). Among those putative proteins that do not meet PE5 criteria, 33 were supported by two unique peptides, and seven proteins have a peptide with length longer than 20.
Identification of Protein Isoforms and SNP Peptides
We also searched for protein isoforms by using the 90,203 annotated protein isoforms in the Gencode database (version 19) from 15,632 annotated genes with nonidentical proteins (Harrow et al., 2012). In total, we identified 7,368 protein isoforms from 6,963 genes that have at least one unique isoform peptide (Table S7). Six thousand six hundred and ten genes have only one protein isoform identified, and very few (353) have two or more isoforms identified (Figure 6A), consistent with other studies (Wang et al., 2019a; Tress et al., 2017; Ezkurdia et al., 2015). For all the identified protein isoforms, the majority (74%) are from the most abundant RNA isoforms (Table S7). For other protein isoforms, further analysis suggests that the inability to identify them is not caused by low RNA abundance (Figure 6B; Table S7). Analysis of the identified protein isoforms showed that most of them have the longest protein sequence, and thus more unique peptides for identification (Table S7).
Figure 6. Protein Isoform Analysis.
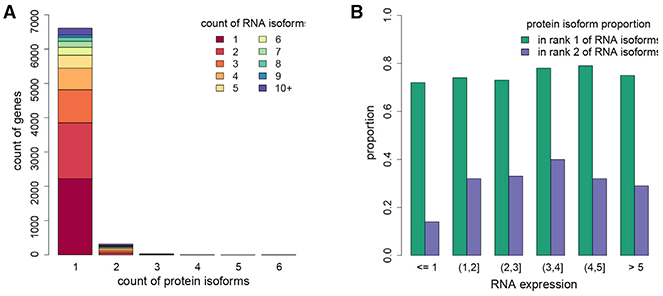
(A) Total number of genes which have different numbers of isoforms identified at the protein level. Different colors represent the number of RNA isoforms for each gene. We identified one isoform for nearly 7,000 genes, although we observe several RNA isoforms for each gene.
(B) The proportion of the rank 1 and 2 RNA isoforms identified at the protein level across RNA abundance intervals.
See also Table S7.
Tissue-enrichment analysis of the isoforms identified a total of 2,436 tissue-enriched protein isoforms (Table S7). Surprisingly, the protein enrichment in each tissue at the isoform level is very similar to the enrichment at the gene level (the sum of all protein isoforms), indicating that for these genes there is probably only one protein isoform predominantly expressed across tissues. Interestingly, however, 76 of the 1,565 HK genes that showed no enrichment in any tissues at protein level had protein isoforms that showed tissue enrichment (Table S7). In general, most of the enriched isoforms (2,070/2,436) are from the top-ranked RNA isoforms, and only 366 were from the less abundant isoforms (Table S7). Very few genes (47) have more than one protein isoform showing tissue enrichment; they can be enriched in either the same tissue or different tissues. For example, CELA2B has two isoforms that are both enriched in the pancreas at similar levels. TPM2 has two isoforms that are enriched in different tissues, with one enriched in heart and skeletal muscle and the other enriched in tissues with smooth muscles (Table S7). In summary, many proteins only have one isoform identified, and their corresponding RNAs are usually the most abundant and the longest sequence.
We also searched for single nucleotide polymorphisms (SNPs) in proteins by using a database populated with SNP peptides. We identified and quantitated 149 SNP peptides that are enriched in expected individuals based on genomic data (Table S1). The acquired spectra of the SNP peptides were compared to the corresponding synthetic peptide spectra. In total, 108 SNP peptides with multiple spectra matched to the synthetic peptide spectra with contrast angle similarity score more than 0.7 (Frewen and MacCoss, 2007) (Table S1).
DISCUSSION
In this study, we have quantitatively analyzed the proteome across 32 different normal human tissues. Proteins that are enriched in a single tissue or a group of tissues were identified and analyzed with regards to biological functions. Our platform also identified a group of proteins that have not been previously identified. It is possible that the TMT label can increase the ionization efficiency of some peptides and thereby increase their chance of detection. Multiplexing of samples from different tissues and extensive fractionation could also contribute to the identification of previously missed proteins. Although we only detected one major isoform for most proteins, our results indicate that isoform detection is mainly limited by the number of unique peptides and not their expression level.
Our protein and RNA correlation analysis showed that they have different enrichment patterns across tissues, which could be caused by multiple factors such as post-transcriptional or post-translational regulation or different turnover rate at RNA and protein levels. It is also possible that some might be caused by the intrinsic limitations of mass spectrometry technology, especially for the low-abundance proteins. It is known that post-mortem ischemia (PMI) affects RNA expression (Ferreira et al., 2018) which could de-couple protein RNA correlation. In this study, PMI effects were removed before protein RNA correlation. We also provided an interactive view of protein and RNA score in each tissue and their distribution pattern across tissues on our website (http://snyderome.stanford.edu/TSomics.html). Furthermore, our data indicated that the constitutive secretion of proteins to other tissues is one major cause of the significant negative correlation and discordance of protein and RNA. Our analyses on secretory proteins provided a special approach for deciphering sites of synthesis and potential action of secreted proteins.
The tissue-specific distribution of proteins can provide an in-depth view of complex biological processes that require the interplay of multiple tissues. Enrichment analysis of enzymes in BCAA metabolism revealed different roles of each tissue as well as new tissues (heart, stomach, pancreas) that are important for metabolic control. We envision this kind of analysis can shed light onto the understanding of many biological processes. Lastly, for genetic diseases caused by mutations in protein-coding regions, the protein-enrichment information across tissues can suggest the affected tissues and explain specific disease symptoms. As such, the proteomic information generated in this study is expected to provide valuable insights into human biology and disease.
There are several limitations to our study. First, very lowly expressed but highly tissue-specific proteins might not be detected. However, by analyzing many different tissues we were able to study widely expressed proteins and the variation in their abundance as well as tissue-specific proteins expressed at moderate and high levels. Second, we note that the GTEx tissue samples represent mixtures of cell types. Hence, the protein enrichment/specificity we observed reflects a composite of the different cell types. Cell type similarities among tissues account for similar tissue function to what was observed in the esophageal mucosa to skin samples, both of which contain abundant epithelial cells. Similarly, some of the smooth muscle cell-rich tissues such as sigmoid colon, esophagus muscle, and artery share similar functions (Figure 2D). As single-cell proteomic technology develops, future studies could characterize cell-type-specific proteomes. A third limitation is that we did not account for sex and age. We are underpowered to do so but note that the tissues cluster by tissue type (Figures 2A and S4), indicating that sex and age are minor contributors to the variance observed. Lastly, there are unbalanced biological samples for each tissue. Enrichment analysis will be mostly affected in tissues that have a limited number of samples. However, empirically we have found that many highly tissue-specific proteins can be robustly identified from a limited number of biological samples. For example, brain samples were from two to three individuals, but many brain-specific proteins were clearly identified. The proteins that could be affected by unbalanced tissue sample sizes are the ones that are slightly enriched in one/more tissue(s). Despite these limitations we believe the data provide a valuable scientific resource.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Michael P. Snyder (mpsnyder@stanford.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Raw proteome data are deposited to ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016999. RNA and Protein TS-score across tissues can be checked at: http://snyderome.stanford.edu/TSomics.html.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tissue proteome profiling was performed in house as part of the GTEx project. Paxgene fixed human tissue samples were provided by NIH GTEx consortium. Detailed information about donor enrollment, tissue collection, sample fixation and histopathological review methods are described in (Carithers et al., 2015) (GTEx Consortium 2015). There are in total 201 samples from 32 major organs from 14 different individuals (details in Table S1) obtained from the GTEx consortium.
METHOD DETAILS
Sample Preparation
There are in total 201 Paxgene fixed samples from 32 major organs from 14 different individuals. The sample preparation method was as described before with modifications (Kulak et al., 2014). About 30mg tissue samples were cut into small pieces on ice and further disrupted using beat beating and sonication in lysis buffer (6 M guanidine, 10mM TCEP, 40mM CAA, 100mM Tris pH 8.5). The supernatant was collected and heated at 95°C for 5min. After protein reduction and alkylation, protein concentration was measured using the BCA kit (ThermoFisher). Since Paxgene fixed samples have a high amount of PEG contamination, protein extract was cleaned up by acetone precipitation at −20°C overnight (1:4 sample to acetone volume ratio). The protein pellet was washed with acetone 3 times and air-dried. The pellet was resuspended in 6 M guanidine and 100mg sample was used for digestion using LysC (1:100 protease to protein ratio) for 2 h at room temperature followed by trypsin (1:50) digestion overnight at 37°C. Peptides were cleaned up using Waters HLB column and subsequently labeled using TMT 10Plex (ThermoFisher) in 100mM TEAB buffer according to manufacturer’s recommendations. An equal amount of proteins from each tissue were pooled together as a reference sample.
TMT Experimental Design
In this study, we used TMT10plex which can label up to 10 samples in one experiment. We randomized tissue samples so that each TMT10plex consists of an assortment of tissues. To facilitate cross-tissue comparison and reduce the technical variation among mass-spectrometry runs, two pooled reference samples were added into each TMT10plex experiment. Equal amounts of eight tissue samples and the two common reference samples were multiplexed into one TMT10plex run. TMT126 and 131 were used for the same reference sample in each run. Note that the use of two reference samples was used as a quality check for ensuring reproducibility of the sample measurements in each run. For 201 samples, we designed 28 TMT10plex runs (Table S1). Due to low sample availability, there are only 9 TMT samples in 14 TMT10plex runs. Also in 9 cases samples were repeated in different runs mainly for the purpose of filling the channels, resulting in three technical replicates. In total, from 28 runs, we acquired data from 210 samples. We also performed at least two technical replicates for all samples. In the replicate, samples have the same TMT labels as in the initial run but they were re-randomized to mix with different samples in each TMT10plex run. In total, there are 56 runs with data from 420 samples. Detailed information for each run, sample composition and their corresponding TMT tags can be found in Table S1 sheet A. The number of biological and technical replicates for each tissue type and individual was listed in Table S1 sheet B. To ensure equal amount of sample material was used for each run, we mixed a small amount of each sample first and adjusted the sample amount for the final run based on the mass spectrometry results of the small mix.
Two Dimensional Liquid Chromatography Separation
We used the Waters online nano 2D LC system for fractionation using approximately 15ug of multiplexed sample. Peptides were separated by reverse-phase chromatography at high pH in the first dimension, followed by an orthogonal separation at low pH in the second dimension. In the first dimension, the mobile phases were buffer A: 20mM ammonium formate at pH10 and buffer B: Acetonitrile. Peptides were separated on an Xbridge 300μm × 5 cm C18 5.0μm column (Waters) using 12 discontinuous steps of buffer B at 10.8%, 13.1%, 14.9%, 16.7%, 17.7%, 18.9%, 19.9%, 20.4%, 22.2%, 25.8%, 28.9% and 45% at 2 μl/min flow rate. For each step, a 5 min isogradient of %B was used. In the second dimension, peptides were loaded to an in-house packed 75μm ID/15μm tip ID × 25cm Sepax GP-C18 1.8μm resin column with buffer A (0.1% formic acid in water). Peptides were separated with a linear gradient from 5% to 30% buffer B (0.1% formic acid in acetonitrile) at a flow rate of 300 nL/min in 180 min. The LC system was directly coupled in-line with an Orbitrap Fusion (Thermo Fisher Scientific).
Mass Spectrometry Data Acquisition and Analysis
The Orbitrap Fusion was operated in a data-dependent mode for both MS2 and MS3. MS1 scan was acquired in the Orbitrap mass analyzer with resolution 120,000 at m/z 400. Top speed instrument method was used for MS2 and MS3. For MS2, the isolation width was set at 0.7 Da and isolated precursors were fragmented by CID at a normalized collision energy (NCE) of 35% and analyzed in the ion trap using “turbo” scan. Following the acquisition of each MS2 spectrum, a synchronous precursor selection (SPS) MS3 scan was collected on the top 5 most intense ions in the MS2 spectrum. SPS-MS3 precursors were fragmented by higher energy collision-induced dissociation (HCD) at an NCE of 65% and analyzed using the Orbitrap at a resolution of 60,000. Each sample was run again on another Orbitrap Fusion in the same lab with the exact same settings for technique replicates.
We used SEQUEST in ProteomeDiscoverer 2.1 (ThermoFisher Scientific) for protein identification. Raw files from 12 fractions of each sample were combined together for a single search against GENCODE V19 (GRCh37.p13 https://www.gencodegenes.org/human/release_19.html) human proteome database (Harrow et al., 2012). Mass tolerance of 10ppm was used for precursor ion and 0.6 Dalton for fragment ions. The search included cysteine carbamidomethylation as a fixed modification. Peptide N-terminal and lysine TMT 10plex modification, protein N-terminal acetylation and methionine oxidation were set as variable modifications. Up to two missed cleavages were allowed for trypsin digestion. The peptide false discovery rate (FDR) was set as < 1% using Percolator. For protein identification, at least one unique peptide with a minimum 6 amino acid length was required. In this study, we did not include any peptides with post translational modifications for quantitation. For protein quantitation, only unique peptides with reporter ion mass tolerance of less than 10ppm were used. Peptide precursor ion isolation purity should be > 50%, signal-to-noise (S/N) >15 and the summed S/N of all channels > 200. Peptides passing these criteria were summed to represent protein abundance, thereby giving more weight to the most-intense peptides. We also pooled together all the spectra in this study for a single search at protein FDR of 1%. For SNP peptide search, we reconstructed the protein database by adding all the structure variant peptides to the database. The SNP peptides were extracted based on the SNP information provided by the GTEx consortium.
Validation of SNP Peptides by Synthetic Reference Peptides
To validate the identification of the SNP peptides, we synthesized 121 synthetic peptides without cysteine in the sequence through Pierce. Synthetic peptides were TMT labeled and run on Orbitrap Lumos using the same instrument methods. Spectral Libraries were generated using the Trans Proteomic Pipeline (TPP) in conjunction with the BiblioSpec software suite. Briefly, results from DDA acquisitions containing synthetic peptides of interest were converted to mzXML and searched through the Trans Proteomic Pipeline using Comet against a database specific for these synthetic peptides of interest and randomized decoy peptides. Target-decoy modeling of peptide spectral matches was performed with PeptideProphet and peptides with a probability score of > 95% from the entire experimental dataset were included in subsequent peptide library building. A spectral library was then generated using the BlibBuild, a tool in the BibloSpec software suite which parses the results to include only the best peptide spectral match for each identified peptide in the library. Spectral libraries were then searched using the BlibSearch. Briefly, query spectra were compared to peptide spectral matches in the library generated via BlibBuild and scored on similarity. Peptides with contrast angle similarity score > 0.7 are considered matches. The matched results were further validated by inspecting if they came from the expected raw files (Table S1).
Protein Enrichment Validation Using Western Blot
We validated some of our protein tissue enrichment information using western blot analysis. We selected 11 proteins that are involved in important biological pathways or not well characterized by Uhlén et al. (2015). Twenty eight representative tissue samples were selected for interrogation with antibodies as listed in Key Resources Table. Both GAPDH and ACTIN were used as control because neither of them were consistently expressed across tissues. For each western blot, 4x LDS Sample Buffer and 1M DTT (to final added concentration of 125mM) were mixed with 15ug protein lysate from each tissue ample, and subsequently heated at 70°C for 10 min. Protein lysates were then loaded and separated on 4%–12% acrylamide SDS-PAGE 15 well gels and transferred to nitrocellulose membranes. After the transfer, the nitrocellulose membranes were blocked with 3% BSA in TBST for 1 h and washed 3x with TBST. Next, the nitrocellulose membranes were stained with the primary antibody diluted according to the antibody manufacturer’s instructions in 3% BSA in TBST overnight at 4°C. The next day, the nitrocellulose membranes were washed 3x with TBST, and followed by secondary antibody staining for 2 h at room temperature. The nitrocellulose membranes were visualized on an Odyssey CLx.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-ATP6V1E1 | Abcam | Cat# ab111733; RRID: AB_10861729 |
| Rabbit Anti-BCAT1 | Abcam | Cat# ab197941; RRID: AB_2858260 |
| Rabbit Anti-BCAT2 | Abcam | Cat# ab111733; RRID: AB_10677595 |
| Rabbit Recombinant Anti-ABAT/GABA-T | Abcam | Cat# ab216465; RRID: AB_2801302 |
| Rabbit Anti-BCKDHB | Abcam | Cat# ab201225; RRID: AB_2858261 |
| Rabbit Anti-PURA | Abcam | Cat# ab79936; RRID: AB_2253242 |
| Rabbit Anti-VPS52 | Aviva Systems Biology | Cat# ARP57644_P050; RRID: AB_10714467 |
| Mouse Anti-β-Actin monoclonal | Cell Signaling Technology | Cat# 3700; RRID: AB_2242334 |
| Rabbit Anti-β-Actin monoclonal | Cell Signaling Technology | Cat# 4970; RRID: AB_222317 |
| IRDye 680RD Donkey anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68072; RRID: AB_10953628 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-32211; RRID: AB_621843 |
| Mouse Anti-GAPDH | Millipore | Cat# CB1001; RRID: AB_2107426 |
| Rabbit Anti-FHOD1 | Novus Biologicals | Cat# NBP1-83900; RRID: AB_11043001 |
| Mouse Anti-MYL7 | Novus Biologicals | Cat# NBP2-03891; RRID: AB_2858262 |
| Rabbit Anti-UNC13C | Sigma-Aldrich | Cat# HPA041516; RRID: AB_10795124 |
| Rabbit Anti-GAPDH | Sigma-Aldrich | Cat# G9545; RRID: AB_796208 |
| Biological Samples | ||
| Table S1A | GTEx (Genotype-Tissue Expression) Consortium | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile, Optima LC/MS Grade, Fisher Chemical | Fisher Scientific | Cat# A955 |
| Water, Optima LC/MS Grade, Fisher Chemical | Fisher Scientific | Cat# W6500 |
| Lysyl Endopeptidase Mass Spectrometry Grade (Lys-C) | Fujifilm Wako Pure Chemical | Cat# 12505061 |
| 2-Chloroacetamide (CAA) | Sigma Aldrich | Cat# 22790 |
| Acetone | Sigma Aldrich | Cat# 179124 |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) | Sigma Aldrich | Cat# C4706 |
| Trizma® base | Sigma Aldrich | Cat# 93350 |
| Guanidine-HCl | Thermo Fisher Scientific | Cat# 24110 |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | Cat# 90059 |
| TMT10plex Isobaric Label Reagent Set, 1 × 0.8 mg | Thermo Fisher Scientific | Cat# 90110 |
| Critical Commercial Assays | ||
| TBS W TWEEN (TBST) 20X SOL 1L | Alfa Aesar | Cat# J77500K2 |
| Nitrocellulose Membrane, Precut, 0.2 μm, 7 × 8.4 cm | Bio-Rad | Cat# 1620146 |
| Extra thick blot filter paper 30x15x15 cm sheets | Bio-Rad | Cat# 1703959 |
| Amersham ECL Plex Fluorescent Rainbow Markers | Cytiva | Cat# RPN851E |
| Lysing matrix D tubes | MP Biomedicals | Cat# 6913100 |
| Bovine Serum Albumin | Sigma Aldrich | Cat# A5611 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| MOPS SDS Running Buffer (20X) | Thermo Fisher Scientific | Cat# NP0001 |
| NuPAGE 4-12% Bis-Tris Protein Gels, 1.0 mm, 15 well |
Thermo Fisher Scientific | Cat# NP0323BOX |
| NuPAGE 4x LDS Buffer | Thermo Fisher Scientific | Cat# NP0008 |
| NuPAGE Transfer Buffer (20X) | Thermo Fisher Scientific | Cat# NP00061 |
| Oasis HLB 1 cc Vac Cartridge 10 mg Sorbent per Cartridge 30 μm 100/pk | Waters | Cat# 186000383 |
| Deposited Data | ||
| GENCODE v26 | GENCODE | https://www.gencodegenes.org/human/release_26.html |
| Proteome Data | This study | ProteomeXchange Consortium: PXD016999 |
| OMIM | Amberger et al., 2015 | https://omim.org/ |
| RNA-seq TPM data v8 | GTEx portal | https://www.gtexportal.org/home/ |
| TSomics | This study | http://snyderome.stanford.edu/TSomics.html |
| RNA isoform data v8 | GTEx portal | https://www.gtexportal.org/home/ |
| Software and Algorithms | ||
| Proteome Discoverer (Version 2.1) | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30945?SID=srch-hj-OPTON-30945#/OPTON-30945?SID=srch-hj-OPTON-30945 |
| Xcalibur (Version 4.0.27.42) | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30965#/OPTON-30965 |
| Orbitrap Fusion Tune (Version 2.1.1565.24) | Thermo Fisher Scientific | https://www.thermofisher.com/ |
| MassLynx (Version 4.1) | Waters | https://www.waters.com/waters/en_US/MassLynx-Mass-Spectrometry-Software-/nav.htm?cid=513164&locale=en_US |
| BlibSearch | BiblioSpec 2.0 | https://skyline.ms/wiki/home/software/BiblioSpec/page.view?name=default |
| Gephi (Version 0.9.2) | Gephi | https://gephi.org/ |
| AdaReg | Wang et al., 2019c | https://github.com/mwgrassgreen/AdaReg |
| AdaTiSS | Wang et al., 2019d | N/A |
| ggplot2 R package v | Wickham H, 2016 | https://ggplot2.tidyverse.org |
| R(v3.6.1) | R core team | https://www.r-project.org/ |
| RobNorm | Wang et al., 2019b | https://github.com/mwgrassgreen/RobNorm |
| Rtsne R package (v0.15) | van der Hinton Geoffrey, 2008 | https://cran.r-project.org/web/packages/Rtsne/Rtsne.pdf |
| STRINGdb R package (v10) | Franceschini et al., 2013 | https://www.bioconductor.org/packages/release/bioc/html/STRINGdb.html |
QUANTIFICATION AND STATISTICAL ANALYSIS
Proteomics Data Processing
Proteomics Dataset
As mentioned above, in total, we have data from 56 mass spectrometry runs containing data from 420 samples and 112 references. Run 29-56 is the technical replicate of run 1-28. We combined the 56 result files into one excel sheet (Table S2B). The specific sample information and its corresponding TMT tag can be found in Table S1A. The number of biological and technical replicates for each tissue type and individual was listed in Table S1B. In brief, in each run, there are 10 samples with 126 and 131 as the same pooled reference sample and the other 8 channels representing different tissue samples. Due to the availability of samples, 14 runs in each replicate have 9 instead of 10 samples. The blank TMT channels were removed in the final data table. In the replicate run, each sample has the same TMT tag as in the initial run but was remixed with different samples in each 10plex.
Quantification of Each Gene at Protein Level
Protein abundances in each sample were first rescaled so that the total sum of the peptide abundances in each channel was the same as the average of the total sum abundance of the two reference channels in the same run. Since low abundance peptides have more variations, we filtered them out before quantitative analysis. In each 10plex sample, for each peptide, if the reporter ion abundance in one channel was less than 15, the value in this channel was set as NA. If the total sum of 10 channels was less than 200, values in all channels were set as NA. The abundances of peptides that are unique to a gene were summed to represent the protein expression level of the gene. In total, we identified 13,813 proteins and quantified 12,627 after peptide level filtering. The protein expression data combined from 56 runs is in Table S2.
Quantification from a Single Peptide versus Multiple Peptides
We investigated the correlation between the intensity of a single peptide and the sum of all the peptides for each protein. The single peptide was selected in two ways: the most abundant peptide or the peptide with the median abundance. The Pearson correlation was obtained based on the relative abundance. In our data, 10,442 proteins were quantified from at least two peptides. The median of correlation from the most abundant peptide is 0.94 and from the median abundance peptide is 0.89. The 25%-percentile is 0.86 for the most abundant peptide and 0.71 for median abundance peptide and the 75%-percentile is 0.99 for both types of peptides. Low correlation may be due to the large variation of the observed peptide abundance or outlier peptides. Thus, overall by mass spectrometry, the results are reproducible. More discussion on the enrichment of the proteins quantified from a single peptide will be discussed in the following sections.
Robust Normalization
Since there are two reference replicate samples in each run, batch effects were removed by using the relative abundance of each sample to the average of the reference samples. NAs in the reference channels (126, 131 channels) were imputed using a minimum value of 15. The relative abundance of each sample was logarithm transformed at base 2. Different from traditional case-control study or a study with a few conditions, our samples were from 32 different types of tissues, which are highly heterogeneous. The majority of previous normalization methods cannot guarantee a robust and tissue-sample adaptive correction. Here, we applied our data-driven robust normalization method (RobNorm) which took into account sample heterogeneities (Wang et al., 2019b). To robustly estimate the sample effects, we implemented the density-power-weight to down weigh the outliers for the structured data. Our algorithm automatically detected the sample inliers (stable abundances) which were used for the robust normalization and at the same time kept the genuine heterogeneities from outliers. To avoid the bias from missing values, the estimation for sample effects was based on the genes with less than 50% missing values, in total, 6,320 genes. We set density power parameter γ = 1, and took zero vectors as the standard sample to implement RobNorm (Wang et al., 2019b). The sample effects were then corrected for all the genes on the relative abundances in logarithm scale. After normalization, the log ratio values were transformed back to the absolute abundances for missing value imputation in the next step. The boxplots of relative abundances in log2 scale of tissue samples before and after robust normalization are shown in Figure S1 (A-B). The normalized absolute abundances in the protein profile are summarized in Table S2.
Missing Value Imputation
Proteins are missing due to one or two reasons: they are not detected by the mass spectrometry analysis or they are highly tissue-specific. For a single TMT 10plex experiment, if only a few of the channels but not all had missing values, we imputed a signal-to-noise value of 15 as a minimum value for these channels. For proteins which were quantified in less than 28 out of 56 runs, they were considered as highly missing proteins. Fisher’s exact test was used to determine if the chance of the detection of these proteins was associated with specific tissue types. Since only 32 tests were performed for each protein, the level for controlling the family-wise error rate (FWER) was set as 0.2. Tissues that are significantly associated with the chance of protein detection are the ones having p value < 0.2/32 after Bonferroni’s correction. For these highly missing proteins (871), we imputed a signal-to-noise value of 15 as a minimum value for missing values across runs. For the rest of the highly missing proteins without tissue association, imputation was not employed. After missing value imputation, protein tissue enrichment was further determined by the tissue specificity analysis.
Tissue Level Filtering and Technical Replicates Combination
After robust normalization and tissue sample imputation, we then took the non-missing absolute abundance < 15 to be 15 and obtained the relative abundance in log scale at base 2. To combine technical replicates, we first filtered the dataset on the tissue level. For each gene and each tissue, if a technical replicate is 2.5 median absolute deviation (MAD) away from the corresponding tissue median, its value was set as NA. To maintain the individual variation, if all replicates from the same individual were outliers, we still kept those replicates. Among 237 pairs of technical replicates, the median Pearson correlation was 0.84, the 1st percentile was 0.81 and the 3rd percentile was 0.88. After filtering for each gene, we took an average of technical replicates of each tissue and finally constructed a protein expression matrix from 12,627 genes and 201 samples. The following tissue specificity analysis was based on this expression matrix, which is summarized in Table S1.
Removing PMI Effect in proteome
(Ferreira et al., 2018) showed the impact of the post-mortem interval (PMI) on gene expression in each individual tissue using the data from the GTEx project. In our analysis, we investigated the PMI effect on protein expression (PMI information are obtained from GTEx portal https://storage.googleapis.com/gtex_analysis_v8/annotations/GTEx_Analysis_v8_Annotations_SampleAttributesDS.txt). After combining the technical replicates for each protein, we modeled the protein expression with linear relationship to the covariates of tissue type and the PMI effect, i.e., linear.model (protein abundance ~tissue type + PMI effect). Due to the small sample size for each tissue type in each time interval, we did not incorporate the interaction terms between the tissue types across the PMI time intervals. However, we tested the effect of PMI on a single tissue using data from an unpublished ongoing GTEx study. Based on RNA study, PMI has the biggest impact on colon samples (Ferreira et al., 2018). We sampled 20 colon protein samples and tested the significance of PMI and there were only 6 proteins significantly affected.
We tested the significance of the PMI effect for each protein, by comparing the model with and without PMI effect from F-test. From Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995) under FDR < 0.05, there were 400 proteins significantly affected by PMI, from a total of 11,608 proteins (which were observed in at least two samples in two PMI time intervals). We corrected the protein expression by subtracting the fitted PMI effect from the least-squares estimate. The hierarchical plot of the cleaned protein expressions based on pairwise Euclidean distance from Ward’s method is shown in Figure S1.
Protein Abundance in Each Tissue
Based on the cleaned technical combined protein profile, we took the sample median (in log scale) for each tissue as tissue level abundance for each gene, which is summarized in Table S2.
Transcriptome Data Processing
Dataset
The RNA-seq data in Transcripts Per Kilobase Million (TPM) were obtained from the GTEx portal in version 8 based on the GENCODE v26. The GTEx v8 data, the large cohort, contained 19,291 protein-coding genes from 17,382 samples. There were 12,245 genes quantified at the protein level and 182 RNA-seq samples matched to the 14 individuals in our study, the sub-cohort.
There were 1,330 genes with all the tissue median TPMs less than 1 based on the large cohort data. They were excluded from our tissue enrichment analysis. There were 9,412 genes with all the tissue median TPMs > 1, where 7,806 were quantified at both RNA and protein levels.
Robust Normalization
The RNA expression level (TPM) was logarithm transformed at base 2 from the corresponding 32 tissues in the large cohort, in total 12,461 samples. To avoid taking the log of zero, small values were added to genes with TPM close to zero. When a TPM lay within the range of [0, 0.01], it was replaced by a value randomly picked from [0.001, 0.01]. Our normalization method RobNorm in (Wang et al., 2019b) was applied. The sample effects were estimated based on 9,711 genes having 0.05-quantile > 0 in log scale. The density-power parameter g was set as 1 and the sample medians were used as the standard sample. The boxplots of RNA abundances of tissue samples before and after robust normalization are shown in Figure S2.
Each RNA’s expression was modeled with respect to a linear relationship to the covariates of tissue effect, the PMI effect and the interaction effects between tissue types and PMI based on the normalized data, i.e., linear.model (RNA abundance ~tissue type + PMI effect + tissue type × PMI effect). Similarly as in protein expression correction, we corrected the RNA expression by subtracting the fitted global PMI effect and the tissue-specific PMI effect from the least-squares estimates. The following analysis was based on the adjusted expression from the matched samples in our sub-cohort. The hierarchical plot of RNA tissue samples based on pairwise Euclidean distance from Ward’s method is shown in Figure S2.
RNA Abundance in Each Tissue
We took the sample median of the cleaned RNA abundances (in log scale) for each tissue as tissue level abundance for each gene, which is summarized in Table S3.
Protein and RNA Isoform Data Processing
Each protein isoform must have at least one isoform unique peptide. Based on GENCODE v26, in total, we identified 7,371 protein-coding isoforms at the protein level corresponding to 6,966 genes. To quantify protein isoforms, the same filter criteria was used on the peptides as we did for protein abundance at the gene level. After data filtering, we quantified 6,311 protein-coding isoforms in the protein level corresponding to 6,044 genes. We applied the same procedures as the data processing for proteins to obtain normalized and cleaned protein isoform expression data.
At the RNA level, due to the fact that some isoforms share the same CDs, we collapsed their transcript IDs as one based on GENCODE v26 and combined their abundances. The identified protein-coding isoform names in both protein and RNA levels are summarized in Table S4. We retrieved isoform expression from the GTEx portal in version 8 and applied the same procedures as the data processing for RNAs to obtain normalized and cleaned RNA isoform expression data.
Tissue Specificity Score for Proteins
Different proteins exhibit different distributions across tissues: concentrated or spreadout and with or without tissue specific expression. The protein distribution across tissues is a mixture model of population distribution and outlier distribution. We considered the population in Gaussian shape but did not assume a particular distribution for outliers. Our algorithm AdaTiSS (Wang et al., 2019d) robustly estimated the population information. Once we obtained the population parameters, the TS score is a robust version of z-transformation, which measures the distance of expression for a particular tissue relative to the population mean in units of standard deviation from the population. Such robust z-transformation is scale-free, thereby normalizing the relative protein expression using the same metric (z-score). Below we detailed the discussion on defining protein population and calculating the TS scores for our data.
Protein Population for defining TS scores
In previous studies, several methods have been developed to define tissue specificity (TS) scores (Wang et al., 2019d). As discussed in (Wang et al., 2019d), the key for defining tissue specificity is to distinguish inlier and outlier tissues. Due to the complexity of the tissue-specific outliers, we focused on the inliers and defined the concept of a population level of expression in the majority of samples. When comparing samples from multiple tissues in our data, the main effect in the population was the tissue effect, which was confirmed by the t-SNE plot (van der Hinton Geoffrey, 2008) in Figure 2A in the main text and the hierarchical cluster plot in Figure S1, where the tissue samples are clustered by tissue types. From our website resource TSomics (http://snyderome.stanford.edu/TSomics.html), for most of the proteins, their abundances across tissues form unimodal density as the population, while some samples outside the majority population may indicate tissue specificity. In most studies, it is conventional to take the logarithm transformation assuming Gaussian noise in the analysis of expression data. Based on our experience and other previous work (Ipsen 2017) (Hill et al., 2008), we modeled the population distribution as Gaussian but we did not assume outlier distribution. We took into account heterogeneities of various proteins and developed a data-adaptive and robust estimation method for the population fitting, where the data itself can select a tuning parameter to adapt its heterogeneous expression and thus the method better fits the population. The statistical analysis for this procedure can be found in AdaReg (Wang et al., 2019c). Its application and comparison to other methods are discussed in AdaTiSS (Wang et al., 2019d).
We applied our population fitting algorithm (AdaTiSS) to the preprocessed data to obtain population mean, population standard deviation and population proportion (the proportion of samples contributing to the population distribution). Due to the presence of missing values, we can only fit the population from AdaTiSS for proteins that were observed (after imputation) in at least 50 samples (out of 201) which were greater than zero in the log scale, in total 8,412 proteins (out of 12,627). For the rest of the proteins that had more than 50 samples below zero, we obtained the population mean from the sample median, the population standard deviation from the sample median of absolute deviation (MAD), and the population proportion from the proportion of the observed samples within ± 2MAD from the median. The fitted population standard deviation less than 0.01 was set to be 0.01. All the fitted population information for proteins can be found in Table S2.
We compared our fitted population results from AdaTiSS to the standard sample median and sample MAD on the 8,412 proteins; sample median and MAD are commonly used practices for identifying outlier and estimating enrichment. The protein-wise comparison is shown in Figures S3A and S3B. There are 142 proteins in which the ratio of the fitted SD from AdaTiSS over MAD is greater than 1.2. Such cases mainly occur when the fitted variance is too small resulting in a local concentration, which may come from our imputation step. There are 795 proteins in which that ratio is less than 0.8. As shown in S5 (C), there are samples in the tails of the population. This is one of the advantages of our method because it maintains robustness under heavy outliers. For some proteins, there are two comparable density peaks close to each other in the majority population. In these cases, our algorithm tends to fit one large single density. More discussion and details are in AdaReg (Wang et al., 2019c) and AdaTiSS (Wang et al., 2019d). As a resource, we provided all the population information and population fitting on our website and in Table S1.
Protein TS Scores
Once we robustly captured the population information, the TS score was obtained based on the robust z-score. Let X be the expression matrix with proteins in rows and samples in columns. Define the sample robust z-score Z for protein i sample j by
then the TS score for tissue t is
where jt is the sample index belonging to tissue t. The z-based TS score measures the distance of protein expression in a particular tissue relative to the population mean in units of standard deviation from the population. It standardizes protein expression across proteins and tissues, making protein expression comparable. Note that since our score is compared to the population, it can be positive or negative.
TS Scores for Highly Missing Proteins
From Fisher’s exact test in the step of imputation in the data processing for proteins, we obtained the protein-detection-associated tissues. If none of those tissues has the TS score ≥ 2.5, the corresponding protein will not get across-run imputation (169 proteins) then we reran the procedures for these proteins without across-run imputation to obtain their final TS scores.
Among those highly missing proteins, we detected 271 tissue enriched proteins quantified from a single peptide. Their reporter ion S/N on average was greater than 200 which usually is confident for ion statistics. About 65% of these enriched peptides have higher S/N than the third quantile of the most abundant single peptides selected.
Protein TS Score Filtering
If a protein was only detected once in a tissue type, NA was assigned and the tissue score was marked as “NA_one_rep_in_raw.” The protein TS scores and standard z-scores based on sample median and MAD are summarized in Table S2 with filter information.
Protein Enrichment Category
We defined four enrichment categories based on protein TS scores: tissue-specific, tissue-enriched-but-not-specific, house-keeping and others. We generally assigned proteins as tissue-enriched if they were tissue-specific or tissue-enriched-but-not-specific. If the TS score ≥ 4 and there were no other tissues with scores in the interval (2.5, 4), the protein was classified as tissue-specific (in total 1,595). If there was at least one TS score ≥ 2.5 but the protein was not tissue-specific, the protein was classified as tissue-enriched-but-not-specific (in total 3,967). If a protein was observed in at least one sample before imputation in each tissue type and all its TS scores were non-NAs and less than 2, the protein was classified as house-keeping (in total 1,565). The rest proteins belonged to the “others” category (in total 5,500). As a requirement, at least three non-NA tissue scores were needed for a protein to be categorized as tissue-enriched The protein enrichment information is summarized in Table S2.
Tissue Specificity Score for RNA
RNA Population for Defining TS Scores
Similar to our protein expression analysis, when comparing samples from multiple tissues in RNA expression, the main effect is the tissue effect, which is confirmed from the hierarchical cluster plot shown in Figure S2. Like several previous studies (Melé et al., 2015) (Li et al., 2017), we assumed Gaussian noise on the population samples. We applied our algorithm (AdaTiSS) on each gene in RNA expression to obtain population information. The fitting results in RNA can be found in Table S3.
In addition, we compared our population fitting to the standard sample median and sample MAD. Figures S4A and 4B shows the gene-wise comparisons for the population mean and population standard deviation (SD) on 12,245 protein-and-RNA commonly quantified genes. We found there are 47 genes that have the ratios of the fitted SD over MAD greater than 1.2 and 1,178 genes have the ratios less than 0.8. Examples of population fittings can be found in Figure S4C. We provided all the population information and population fitting for RNA on our website and Table S2.
RNA TS Scores
The same definition as for protein TS scores.
RNA TS Score Filtering
For genes with low expression (all tissue median TPMs ≤ 1), we assigned the TS scores as NAs and marked them as “NA_all_tissues_tpm_less_1.” We filtered the tissue score as NA if the tissue has score greater than 2.5 but its raw sample median TPM is less than 1, and marked it as “NA_raw_tpm_less_1.” The RNA TS scores and the standard z-scores based on sample median and MAD are summarized in Table S3 with filtering information.
RNA Enrichment Category
The same definition as for the protein enrichment category. Among 12,245 protein and RNA commonly quantified genes, there are 980 tissue-specific genes, 5,302 tissue-enriched-but-not-specific genes, and 2,533 house-keeping genes.
Protein and RNA expression comparison
Protein and RNA Correlation Across Tissues
Across tissues, we obtained the Spearman correlation between the protein TS scores and the RNA TS scores for each gene. For the analysis, at least five non-NA tissues in both protein and RNA were required. The p values were obtained from the permutation test by randomly permuting tissue labels based on 200 permutations and were adjusted from the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). The results are summarized in Table S3. 10,349 genes were compared. Under FDR less than 0.05, we found there are 6,228 genes positively correlated in protein and RNA scores across tissues, 60 negatively correlated genes, and 4,061 remaining genes non-significantly correlated.
Concordance of Protein and RNA Enrichment in Each Tissue
In each tissue, if the protein and its corresponding RNA both have TS scores greater than 2.5, they are defined as concordantly enriched in that tissue. If the RNA TS score is less than 2.5 and the protein TS score is greater than 2.5 and at least 1.5 higher than its RNA TS score, we defined the gene as discordantly enriched at protein level in that tissue. Vice versa, if protein TS score is less than 2.5 and the RNA TS score is greater than 2.5 and at least 1.5 higher than its protein TS score, the gene is discortantly enriched at RNA level in the tissue. The protein/RNA concordance and discordance results in each tissue are summarized in Table S4.
Enrichment Threshold Justification
To obtain the TS scores, we introduced the concept of population and assumed a Gaussian population when we applied our algorithm (we do recognize this assumption is not true for all proteins, but this does appear to be the case for most proteins). For a random variable in the standard Gaussian distribution, the chance of its value greater than 2.5 is less than 1%. The 2.5 threshold is a common cutoff for outliers in Gaussian theory (Rousseeuw and Leroy, 1987).
In addition, based on our obtained data, we justified the threshold. We varied several enrichment thresholds from 1.5 to 4.5 with an increment of 0.5. A tissue is categorized as enriched if its score is above the threshold. Under a threshold, we calculated the Jaccard index on the co-enriched tissue sets for each RNA-and-protein co-quantified gene, where the Jaccard index measures similarity of the enriched tissue sets from RNA and protein levels. In formula, the Jaccard index under a threshold l for gene i is
where
and ∣A ∣ is the cardinality of a set. We compared the number of genes whose Jaccard index di,j is greater than or equal to 0.9 under different thresholds. From the comparison, we found that RNAs and proteins exhibited the largest number of genes in co-tissue enrichment under threshold 2.5.
Tissue Specificity Score for Isoforms
We calculated TS scores for protein isoforms following the same procedures as in the gene level analysis. The protein and RNA isoform TS scores and their enrichment comparison results are summarized in Table S7.
Analysis of PMI Effect on tissue enrichment
At the protein level, we compared protein enrichment before and after removing the PMI effect. The two sets of scores were highly correlated (the median Spearman correlation is 0.9999). In the 12,627 proteins quantified, there were 243 proteins that changed from the category of “enriched” to “non-enriched” or vice versa, and these changes happened when the scores were near the tissue enrichment threshold.
We compared the protein and RNA enrichment categories before and after removing the PMI effect. There were 595 genes (before) versus 574 genes (after) in both protein and RNA house-keeping categories, and 2387 genes (before) versus 2283 genes (after) in both protein and RNA enrichment categories. Thus, the PMI effect is small when all the tissue samples were jointly compared.
Gene Function Analysis
We performed an enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) (Gene Ontology Consortium, 2015) terms based on R package “STRINGdb” (Franceschini et al., 2013). Table S5 summarizes the results for the GO term enrichment and pathway enrichment for tissue-enriched proteins for each tissue type. We obtained the enriched genes in the metabolism pathway by using the sub-pathway IDs from KEGG under the “Metabolism” category. These findings are also summarized in Table S4.
Analysis of Unidentified Proteins
The Human Protein Atlas project (HPA) has annotated protein classes for 19,628 genes (Uhlén et al., 2015). Our data had 12,909 genes mapped to their list. We applied Fisher’s exact test to see whether the unidentified proteins are enriched in a certain predicted protein class. The results (summarized in Table S1) showed that the unidentified proteins are significantly enriched in the membrane protein class, but not significantly enriched in the intercellular nor the secreted protein classes.
Association Analysis of Protein Identification and RNA Expression Level
We investigated whether a gene that is identified at the protein level is associated with its RNA expression abundance. In our analysis, we only considered the genes whose RNA expression level had a raw TPM > 1 in at least one tissue. In total, there are 17,976 such genes. The mean RNA expression level (in log scale) across 32 tissues was used as the gene’s RNA level expression. The RNA expression abundances were grouped into 10 bins: ( − ∞,1],(1,2], …, (8,9], (9, + ∞). We applied the chi-square test for independence of gene expression in RNA and protein identification. We found that when we performed this analysis on all the bins, the p value is less than 2.2 × 10−16, but if we only tested on the bins starting from (5, 6], the p value is 0.21. This indicates when the average RNA expression is higher than 32 in raw TPM, the identification of the corresponding protein is not significantly associated with its RNA expression. The contingency table is summarized in Table S1.
Association Analysis of Protein Isoform Identification and RNA Isoform Expression Level
We further investigated whether the identification of protein isoforms is associated with RNA abundances using a similar approach as in our gene analysis. Genes that have at least two isoforms in RNA are included in the study. The mean RNA expression (in log scale) from 32 tissues is used as the RNA isoform level expression. The RNA isoform expressions are grouped into six bins: ( − ∞,1],(1,2], (2,3], (3,4], (4,5], (5, + ∞). We counted the number of rank 1 RNA isoforms that had corresponding protein evidence in each expression bin and summarized the information in Table S7. In addition, the same analysis was performed for the rank 2 RNA isoforms and the information is summarized in Table S7. We applied the chi-square test for testing independence of RNA isoform expression and protein identification. For the rank 1 isoforms, protein identification is not significantly associated with the RNA expression at significance level of 0.05. For the rank 2 isoforms, RNA isoform expression may affect the identification of the protein isoform (p value < 2.2 × 10−16).
Tissue Enriched Proteins and Genetic Diseases Analysis
For this analysis, we investigated the link between tissue enriched proteins and genetic diseases. The enriched proteins in each tissue were compared to the OMIM disease gene list (Amberger et al., 2015) with disease-relevant tissue abnormality. Fisher’s exact test was applied to assess the significance of the overlap. The results are summarized in Table S6.
Single-Nucleotide Polymorphism (SNP) Peptides Analysis
The spectra were all searched against the GENCODE V19 database populated with SNP peptides at peptide FDR of 1%. The SNP peptides were generated based on the genomic information available from the 14 individuals in our study. We tested whether the SNP peptides were identified in the expected individual based on the known genetic information from GTEx consortium. Since SNP candidate peptides have high missing values, we took (sample median + 2.5 MAD) as the threshold based on the observed samples and the outliers were the ones whose abundances exceeded the threshold. Then we applied Fisher’s exact test for testing the enrichment of SNP peptides in the samples of the expected individual. With controlled FDR under level 0.1, there are 149 significantly enriched SNP peptides. The results are summarized in Table S1.
Protein Enrichment Comparison to Previous Studies
We directly compared our protein enrichment data to Wang et al. (2019a). Wang et al. studied the tissue specificity based on fold change of the expression and categorized proteins into five classes in the same classification scheme as in (Uhlén et al., 2015). Wang et al. quantified 13,640 proteins and our study quantified 12,627. The number of identified proteins that overlapped is 11,221, which is 88.9% of our results. For proteins that are quantified in both studies, 1080/1438 tissue specific proteins from our study, were also enriched or enhanced in Wang et al. (2019a) study (Table S2). However, since only 16 tissue types are in common between our study and theirs, some proteins (n = 342) that are specific in our study showed different enrichment in the (Wang et al., 2019a) study due to the lack of these samples in their study (skin, skeletal muscle and arteries etc.). For the same reason, some tissue specific proteins (n = 497) in their study were enriched differently in our study. The differences may be due to the different enrichment criteria we used to define tissue enrichment or different tissue types included in each study. We also used a different quantitation method and had more biological samples. The summarized comparison result was listed in Table S2.
Supplementary Material
Figure S1. Quality Control for Proteomics Data, Related to Figure 2 and Table S2
A. Protein relative abundances (in log2 scale) in each tissue type before robust normalization.
B. Protein relative abundance after robust normalization.
C. Dendrogram of the hierarchical relationship between tissue samples in protein expression based on pairwise Euclidean distance from Ward’s method. The sample names are labeled by the people index p01 – p14 followed by the tissue names.
Figure S2. Quality Control for Transcriptomic Data, Related to Figure 2 and Table S3
A. RNA TPM (in log2 scale) in each tissue type before robust normalization.
B. RNA TPM after robust normalization.
C. The dendrogram of the hierarchical relationship between tissue samples in RNA expression for the RNA-and-protein co-quantified genes based on pairwise Euclidean distance from Ward’s method. The sample names are labeled by the people index p01 – p14 followed by the tissue name.
Figure S3. Population Fitting for Proteins, Related to Figure 2 and Table S2
A. Protein-wise comparison between standard sample median versus fitted mean using AdaTiSS for estimating population mean.
B. Sample median of absolute deviation (MAD) versus fitted mean using AdaTiSS for estimating population standard deviation. Each protein is represented by a single dot. The identity line y = x is shown in red. The blue and green lines are y = 1.2x and y = 0.8x, respectively.
C. Histograms of standardized protein expression. Protein expression is standardized by subtracting the fitted population mean and divided by the fitted population standard deviation. The first row shows three examples of proteins in which the ratios of the fitted standard deviation from AdaTiSS to MAD are between 0.8 and 1.2. The middle row shows proteins in which the ratios are greater than 1.2. The bottom row shows the ones in which the ratios are less than 0.8. The red curve is the standard Gaussian density (scaled by a factor of fitted population proportion).
Figure S4. Population Fitting for RNAs, Related to Figure 2 and Table S3
A. RNA-wise comparison between standard sample median versus fitted mean using AdaTiSS for estimating population mean.
B. Standard sample MAD versus fitted mean using AdaTiSS for estimating population standard deviation. Each protein is represented by a single dot. The identity line y = x is shown in red. The blue and green lines are y = 1.2x and y = 0.8x, respectively.
C. Histograms of RNA standardized expression. The RNA expression is standardized by subtracting the fitted population mean and divided by the fitted population standard deviation. The first row shows the examples of the RNAs in which the ratios of the fitted standard deviation from AdaTiSS to MAD are between 0.8 and 1.2. The middle row shows the ones in which the ratios are greater than 1.2. The bottom row shows the ones in which the ratios are less than 0.8. The red curve is the standard Gaussian density (scaled by a factor of fitted population proportion)
Figure S5. Western Blot Results of 11 Key Proteins across Tissues, Related to Figure 4
In this study we used both GAPDH and ACTIN as control because neither of them are consistently expressed across tissues. Tissue names are numbered as listed: 1.Adrenal Gland 2.Artery – Aorta 3.Artery – Coronary 4. Brain – Cerebellum 5.Brain – Cortex 6.Colon – Sigmoid 7.Colon – Transverse 8.Esophagus – Mucosa 9.Esophagus – Muscularis 10.Heart - Atrial Appendage 11.Heart - Left Ventricle 12.Liver 13.Lung 14.Muscle – Skeletal 15.Nerve – Tibial 16.Ovary 17.Pancreas 18.Pituitary 19.Prostate 20.Skin - Not Sun Exposed (Suprapubic) 21.Skin - Sun Exposed (Lower leg) 22.Small Intestine - Terminal Ileum 23.Spleen 24.Stomach 25.Testis 26.Thyroid 27.Uterus 28.Vagina. The corresponding mass spectrometry results can be checked here: http://snyderome.stanford.edu/TSomics.html.
Highlights.
Quantified more than 12,000 proteins across 32 normal human tissues
Discordance of RNA and protein enrichment provides evidence of protein secretion
Tissue-specific distribution of enzymes indicates a coordinated control of metabolism
Tissue-enriched proteins provide insights into phenotypes of genetic diseases
ACKNOWLEDGMENTS
We acknowledge the GTEx donors and families for donating organs to the GTEx Consortium. We thank K. Ardlie for coordinating the distribution of tissue samples. We also thank A. Breschi for discussion on RNA-seq data analysis and R. Tibshirani for suggestions on statistical analysis. We thank H. Tang for her help with experimental design and data analysis. Funding was provided by the NIH eGTEx grant (1U01HG00761101-01) and CEGS (Center for Personal Dynamic Regulomes) grant 3P50HG00773505S1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.036.
DECLARATION OF INTERESTS
M.P.S. is a cofounder and is on the scientific advisory board of Personalis, Filtircine, SensOmics, Qbio, January, Mirvie, Oralome, and Proteus. He is also on the scientific advisory board (SAB) of Genapsys and Jupiter. The other authors declare no competing interests.
REFERENCES
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, and Hamosh A (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angione C (2019). Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. BioMed Res. Int 2079, 8304260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, and Neinast M (2018). Branched Chain Amino Acids in Metabolic Disease. Curr. Diab. Rep 18, 76. [DOI] [PubMed] [Google Scholar]
- Baker MS, Ahn SB, Mohamedali A, Islam MT, Cantor D, Verhaert PD, Fanayan S, Sharma S, Nice EC, Connor M, and Ranganathan S (2017). Accelerating the search for the missing proteins in the human proteome. Nat. Commun 8, 14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, and Aebersold R (2011). The quantitative proteome of a human cell line. Mol. Syst. Biol 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L, and Gatto GJ Jr. (2012). Biochemistry. [Google Scholar]
- Bodzęta A, Kahms M, and Klingauf J (2017). The Presynaptic v-ATPase Reversibly Disassembles and Thereby Modulates Exocytosis but Is Not Part of the Fusion Machinery. Cell Rep. 20, 1348–1359. [DOI] [PubMed] [Google Scholar]
- Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, et al. ; GTEx Consortium (2015). A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv. Biobank. 13, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman SA, Wacksman GP, and Patterson BD (2001). Pancreatitis associated with valproic acid: a review of the literature. Pharmacotherapy 21, 1549–1560. [DOI] [PubMed] [Google Scholar]
- Chateauvieux S, Morceau F, Dicato M, and Diederich M (2010). Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol 2010, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway ME, and Hutson SM (2015). The Cytosolic and Mitochondrial Branched Chain Aminotransferase. In Branched Chain Amino Acids in Clinical Nutrition, pp. 25–40. [Google Scholar]
- DiMauro S, and De Vivo DC (1996). Genetic heterogeneity in Leigh syndrome. Ann. Neurol 40, 5–7. [DOI] [PubMed] [Google Scholar]
- Du X, Li Y, Wang Y, You H, Hui P, Zheng Y, and Du J (2018). Increased branched-chain amino acid levels are associated with long-term adverse cardiovascular events in patients with STEMI and acute heart failure. Life Sci. 209, 167–172. [DOI] [PubMed] [Google Scholar]
- Dubois PM (1994). The Exocrine and Endocrine Pancreas: Embryology and Histology. In Radiology of the Pancreas, pp. 1–8. [Google Scholar]
- Emerald M (2016). Pituitary Gland: Pituitary Hormones. In Encyclopedia of Food and Health, pp. 392–400. [Google Scholar]
- Esrefoglu M, Taslidere E, and Cetin A (2016). Development of Liver and Pancreas. Bezmialem Science 5, 30–35. [Google Scholar]
- Ezkurdia I, Rodriguez JM, Carrillo-de Santa Pau E, Vázquez J, Valencia A, and Tress ML (2015). Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res 14, 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassio A, Esposito A, Kato M, Saitsu H, Mei D, Marini C, Conti V, Nakashima M, Okamoto N, Olmez Turker A, et al. ; C4RCD Research Group (2018). De novo mutations of the ATP6V1A gene cause developmental encephalopathy with epilepsy. Brain 141, 1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feizi A, Gatto F, Uhlen M, and Nielsen J (2017). Human protein secretory pathway genes are expressed in a tissue-specific pattern to match processing demands of the secretome. NPJ Syst. Biol. Appl 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PG, Muñoz-Aguirre M, Reverter F, Sá Godinho CP, Sousa A, Amadoz A, Sodaei R, Hidalgo MR, Pervouchine D, Carbonell-Caballero J, et al. (2018). The effects of death and post-mortem cold ischemiaon human tissue transcriptomes. Nat. Commun 9, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foggensteiner L, and Beales P(2015). Bardet-Biedl syndrome and other ciliopathies (Oxford Medicine Online). [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, and Jensen LJ (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frewen B, and MacCoss MJ (2007). Using BiblioSpec for creating and searching tandem MS peptide libraries. Curr. Protoc. Bioinformatics Chapter 13, 7. [DOI] [PubMed] [Google Scholar]
- Gayam V, Mandal AK, Khalid M, Shrestha B, Garlapati P, and Khalid M (2018). Valproicacid induced acute liver injury resulting in hepatic encephalopathy-a case report and literature review. J. Community Hosp. Intern. Med. Perspect 8, 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T, Velic A, Macek B, Lundberg E, Kampf C, Nagaraj N, Uhlen M, Cox J, and Mann M (2013). Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 12, 1709–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq N, Schmidt-Hieber C, Sialana FJ, Ciani L, Heller JP, Stewart M, Bentley L, Wells S, Rodenburg RJ, Nolan PM, et al. (2019). Loss of Bardet-Biedl syndrome proteins causes synaptic aberrations in principal neurons. PLoS Biol. 17, e3000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D, Tirouvanziam R, Laval J, Greene CM, Habiel D, Sharma L, Yildirim AÖ, Dela Cruz CS, and Hogaboam CM (2018). Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J. Innate Immun 10, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, Nadal A, Palanza P, Panzica G, Sargis R, et al. (2017). Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, and Schey KL (2008). A statistical model for iTRAQ data analysis. J. Proteome Res 7, 3091–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeček M (2018). Branched-chain aminoacids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. (Lond.) 15, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhou M, Sun H, and Wang Y (2011). Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc. Res 90, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsen A (2017). Derivation of the Statistical Distribution of the Mass Peak Centroids of Mass Spectrometers Employing Analog-to-Digital Converters and Electron Multipliers. Anal. Chem 89, 2232–2241. [DOI] [PubMed] [Google Scholar]
- Katagiri R, Goto A, Nakagawa T, Nishiumi S, Kobayashi T, Hidaka A, Budhathoki S, Yamaji T, Sawada N, Shimazu T, et al. (2018). Increased Levels of Branched-Chain Amino Acid Associated With Increased Risk of Pancreatic Cancer in a Prospective Case-Control Study of a Large Cohort. Gastroenterology 155, 1474–1482.e1. [DOI] [PubMed] [Google Scholar]
- Khan SA, Muhammad N, Khan MA, Kamal A, Rehman ZU, and Khan S (2016). Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet 90, 3–15. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. (2014).A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, and Mann M (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Lake NJ, Compton AG, Rahman S, and Thorburn DR (2016). Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol 79, 190–203. [DOI] [PubMed] [Google Scholar]
- Li Z, Adams RM, Chourey K, Hurst GB, Hettich RL, and Pan C (2012). Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res 11, 1582–1590. [DOI] [PubMed] [Google Scholar]
- Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, Hess GT, Zappala Z, Strober BJ, Scott AJ, et al. ; GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)–Analysis Working Group; Statistical Methods groups–Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site–NDRI; Biospecimen Collection Source Site–RPCI; Biospecimen Core Resource–VARI; Brain Bank Repository–University of Miami Brain Endowment Bank; Leidos Biomedical-Project Management; ELSI Study; Genome Browser Data Integration &Visualization–EBI; Genome Browser Data Integration &Visualization–UCSC Genomics Institute, University of California Santa Cruz (2017). The impact of rare variation on gene expression across tissues. Nature 550, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. [DOI] [PubMed] [Google Scholar]
- Lloyd RV, Cano M, Rosa P, Hille A, and Huttner WB (1988). Distribution of chromogranin A and secretogranin I (chromogranin B) in neuroendocrine cells and tumors. Am. J. Pathol 130, 296–304. [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, et al. (2014). Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med 20, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. ; GTEx Consortium (2015). Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SH (2015). The utility of protein and mRNA correlation. Trends Biochem. Sci 40, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Project E; eGTEx Project (2017). Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat. Genet 49, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw PJ, and Leroy AM (1987). Robust Regression and Outlier Detection (Wiley Series in Probability and Statistics). [Google Scholar]
- Suzuki H, Nishizawa T, Tsugawa H, Mogami S, and Hibi T (2012). Roles of oxidative stress in stomach disorders. J. Clin. Biochem. Nutr 50, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Abascal F, and Valencia A (2017). Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci 42, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Karlsson MJ, Hober A, Svensson A-S, Scheffel J, Kotol D, Zhong W, Tebani A, Strandberg L, Edfors F, et al. (2019). The human secretome. Sci. Signal 12, 12. [DOI] [PubMed] [Google Scholar]
- van der Hinton Geoffrey ML (2008). Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, Heymsfield SB, and Müller MJ (2010). Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr 92, 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Eraslan B, Wieland T, Hallström B, Hopf T, Zolg DP, Zecha J, Asplund A, Li L-H, Meng C, et al. (2019a). A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol 15, e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Jiang L, Jian R, Chen J, Snyder MP, and Tang H (2019b). RobNorm: Model-Based Robust Normalization for High-Throughput Proteomics from Mass Spectrometry Platform. bioRxiv. 10.1101/770115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Jiang L, and Snyder MP (2019c). AdaTiSS: A Novel Data-Adaptive Robust Method for Quantifying Tissue Specificity Scores. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Jiang L, and Snyder MP (2019d). AdaReg: Data Adaptive Robust Estimation in Linear Regression with Application in GTEx Gene Expressions. bioRxiv. [DOI] [PubMed] [Google Scholar]
- White PJ, McGarrah RW, Grimsrud PA, Tso S-C, Yang W-H, Haldeman JM, Grenier-Larouche T, An J, Lapworth AL, Astapova I, et al. (2018). The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab. 27, 1281–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. (2014). Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587. [DOI] [PubMed] [Google Scholar]
- Wittenburg H, Tennert U, and Mössner J (2010). [Hormonal and metabolic functions of the small intestine]. Internist (Berl.) 51, 695–701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Quality Control for Proteomics Data, Related to Figure 2 and Table S2
A. Protein relative abundances (in log2 scale) in each tissue type before robust normalization.
B. Protein relative abundance after robust normalization.
C. Dendrogram of the hierarchical relationship between tissue samples in protein expression based on pairwise Euclidean distance from Ward’s method. The sample names are labeled by the people index p01 – p14 followed by the tissue names.
Figure S2. Quality Control for Transcriptomic Data, Related to Figure 2 and Table S3
A. RNA TPM (in log2 scale) in each tissue type before robust normalization.
B. RNA TPM after robust normalization.
C. The dendrogram of the hierarchical relationship between tissue samples in RNA expression for the RNA-and-protein co-quantified genes based on pairwise Euclidean distance from Ward’s method. The sample names are labeled by the people index p01 – p14 followed by the tissue name.
Figure S3. Population Fitting for Proteins, Related to Figure 2 and Table S2
A. Protein-wise comparison between standard sample median versus fitted mean using AdaTiSS for estimating population mean.
B. Sample median of absolute deviation (MAD) versus fitted mean using AdaTiSS for estimating population standard deviation. Each protein is represented by a single dot. The identity line y = x is shown in red. The blue and green lines are y = 1.2x and y = 0.8x, respectively.
C. Histograms of standardized protein expression. Protein expression is standardized by subtracting the fitted population mean and divided by the fitted population standard deviation. The first row shows three examples of proteins in which the ratios of the fitted standard deviation from AdaTiSS to MAD are between 0.8 and 1.2. The middle row shows proteins in which the ratios are greater than 1.2. The bottom row shows the ones in which the ratios are less than 0.8. The red curve is the standard Gaussian density (scaled by a factor of fitted population proportion).
Figure S4. Population Fitting for RNAs, Related to Figure 2 and Table S3
A. RNA-wise comparison between standard sample median versus fitted mean using AdaTiSS for estimating population mean.
B. Standard sample MAD versus fitted mean using AdaTiSS for estimating population standard deviation. Each protein is represented by a single dot. The identity line y = x is shown in red. The blue and green lines are y = 1.2x and y = 0.8x, respectively.
C. Histograms of RNA standardized expression. The RNA expression is standardized by subtracting the fitted population mean and divided by the fitted population standard deviation. The first row shows the examples of the RNAs in which the ratios of the fitted standard deviation from AdaTiSS to MAD are between 0.8 and 1.2. The middle row shows the ones in which the ratios are greater than 1.2. The bottom row shows the ones in which the ratios are less than 0.8. The red curve is the standard Gaussian density (scaled by a factor of fitted population proportion)
Figure S5. Western Blot Results of 11 Key Proteins across Tissues, Related to Figure 4
In this study we used both GAPDH and ACTIN as control because neither of them are consistently expressed across tissues. Tissue names are numbered as listed: 1.Adrenal Gland 2.Artery – Aorta 3.Artery – Coronary 4. Brain – Cerebellum 5.Brain – Cortex 6.Colon – Sigmoid 7.Colon – Transverse 8.Esophagus – Mucosa 9.Esophagus – Muscularis 10.Heart - Atrial Appendage 11.Heart - Left Ventricle 12.Liver 13.Lung 14.Muscle – Skeletal 15.Nerve – Tibial 16.Ovary 17.Pancreas 18.Pituitary 19.Prostate 20.Skin - Not Sun Exposed (Suprapubic) 21.Skin - Sun Exposed (Lower leg) 22.Small Intestine - Terminal Ileum 23.Spleen 24.Stomach 25.Testis 26.Thyroid 27.Uterus 28.Vagina. The corresponding mass spectrometry results can be checked here: http://snyderome.stanford.edu/TSomics.html.
Data Availability Statement
Raw proteome data are deposited to ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016999. RNA and Protein TS-score across tissues can be checked at: http://snyderome.stanford.edu/TSomics.html.