

Abstract
Recent advances in immuno-oncology have generated renewed optimism for therapeutic anti-tumor vaccination, and peptide vaccines in particular are being extensively employed in clinical studies. However, peptide vaccines generally suffer from poor immunogenicity. Here, we pharmacokinetically tune vaccine responses via fusion of peptide epitopes to carrier proteins to optimize vaccine potency. Antigen-carrier fusions enable three factors to be simultaneously optimized: a) efficient uptake in draining lymphatics from the site of injection, b) protection of epitope payloads from proteolytic degradation, and c) reduction of antigen presentation in uninflamed distal lymphoid organs. We find that peptides expressed as fusions with protein carriers like transthyretin can solve all three pharmacokinetic challenges, enhancing vaccine immunogenicity up to 90-fold in mice. We demonstrate the utility of this approach with several clinically relevant antigens, including viral antigens, tumor-associated antigens, oncofetal antigens, and shared neoantigens. Protein-epitope fusions represent a facile and generalizable approach to enhance T cell responses elicited by subunit vaccines.
Introduction
In recent years, immunotherapies have transformed clinical oncology, including checkpoint blockade antibodies1–3, chimeric antigen receptor T cells4,5, bispecific T cell engagers6, and oncolytic viruses7. However, vaccines, the oldest and most-studied form of immunological intervention, have had only modest clinical success, with a single FDA approval to date8, despite their theoretical potential to enhance the response rates of checkpoint inhibitors9–11. Peptide vaccines in particular have been a focus in many recent cancer therapy trials, due in part to their safety, modest cost, and capacity to be rapidly manufactured, enabling patient-specific neoantigen vaccines12,13. However, the potency of peptide vaccines remains poor, especially in humans.
A number of pharmacokinetic (PK) shortcomings of peptide vaccines have been well characterized, including inefficient antigen transport to local draining lymph nodes (dLNs)14,15 and proteolytic instability16,17. Delivery strategies have been developed to address these limitations, including the formulation of peptide antigens in synthetic particles18–20, antibody-mediated targeting of antigen to dendritic cells21,22, or direct intranodal injections23,24 to obviate the need for antigenic trafficking altogether. These approaches, while effective, are complex in terms of manufacturing and/or administration, motivating the development of additional solutions.
Here, we studied the role of vaccine antigen PK in controlling the potency of subunit vaccines, and define strategies to optimize vaccine potency via PK tuning of antigen delivery. To control antigen degradation rates and PK, we fused tumor-associated epitopes to minimally immunogenic protein carriers. By varying the identity of the carrier protein, we systematically altered the PK and biodistribution characteristics of the fusions to define key factors controlling vaccine potency. We find that immunogenicity is maximized by employing carrier proteins that exhibit prolonged residence time in local adjuvant-inflamed draining lymph nodes but short half-lives in systemic circulation, to minimize uptake in distal non-inflamed lymphoid organs. Collectively, these conclusions help identify a set of simple pharmacokinetic design criteria to aid in the engineering of molecular vaccines.
Results
Fusion of peptide antigens to albumin enhances lymphatic uptake
Upon parenteral injection, antigens can be absorbed into systemic circulation, degraded in tissue, trafficked into lymph, or captured by antigen presenting cells (APCs) locally. We hypothesized that fusion of peptide antigens to a bulky protein carrier could limit these first two potential fates to promote immune priming. Antigen levels in the blood following subcutaneous injection are a function of the systemic absorption rate (kabs) and the rate of clearance from circulation (kclear), which collectively determine antigen bioavailability in both the dLNs and distal lymphoid organs (Fig. 1a). Previous work has shown that parenterally-administered molecules < 40 kDa are systemically absorbed through capillary endothelial cell junctions, while larger molecules are size-excluded from entering the blood vasculature and instead drain to lymphatic vessels15,25, motivating the commonly implemented strategy of delivering low molecular weight vaccine components in larger particulate formulations18–20. To determine the hydrodynamic size threshold for efficient lymphatic uptake in mice, we first assessed lymph node accumulation of dextrans of varying molecular weight. Consistent with previous findings, while 4 kDa dextran (hydrodynamic radius 1.2 nm) did not access the dLN above background, dextrans over 20 kDa (radius ≥ 2.4 nm) did so effectively (Supplementary Fig. 1a–b).
Figure 1.
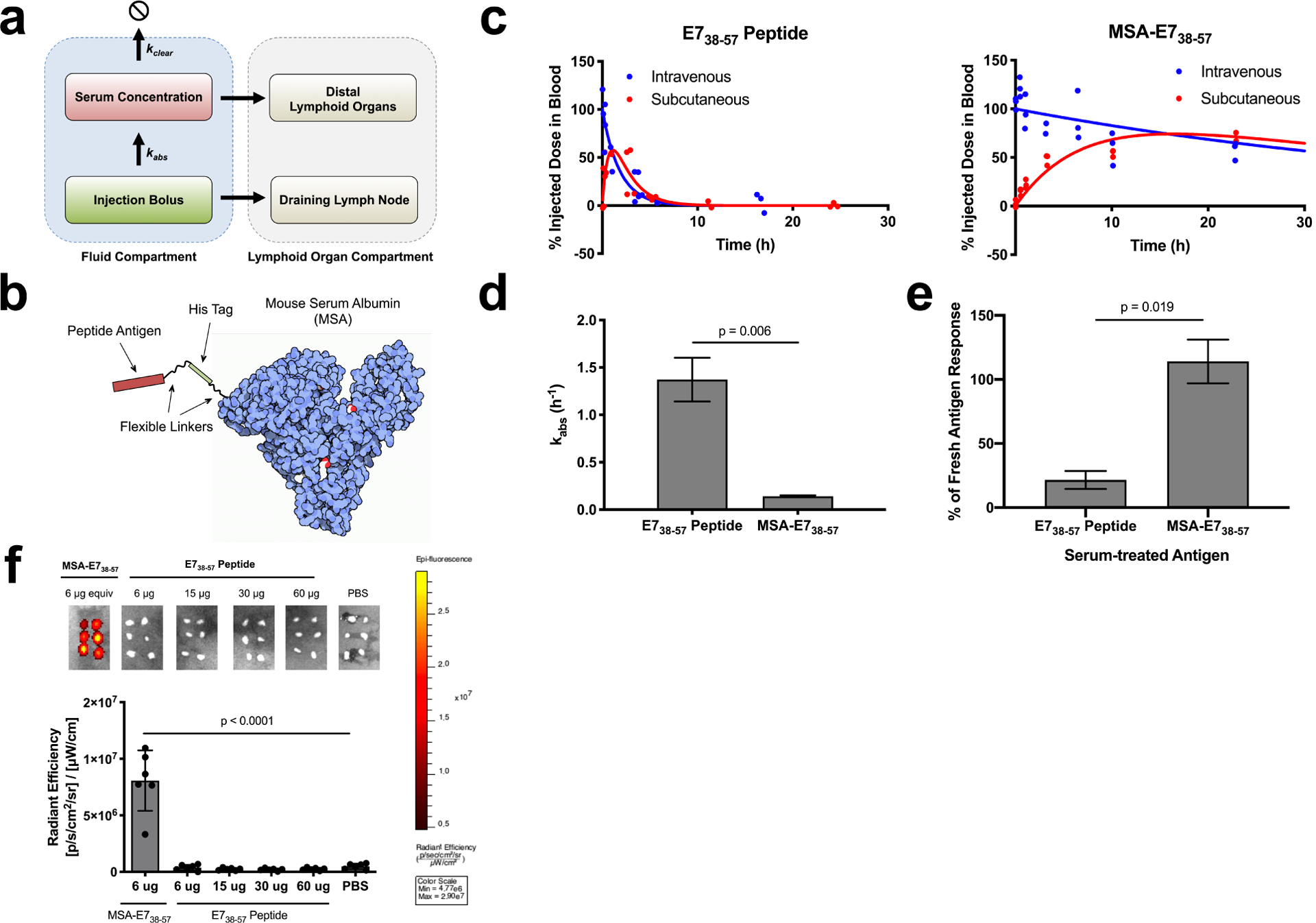
Albumin fusion enhances the bioavailability of antigen in the dLN. (a) Schematic of pharmacokinetic model describing absorbance rate (kabs) and clearance rate (kclear), which determine bioavailability in lymphoid organs. (b) Schematic of MSA-E738–57 protein design. (c) FITC labeled E738–57 or MSA-E738–57 was injected either subcutaneously or intravenously in mice (n = 3 mice per group). <10 μl blood draws were used to quantify antigen concentration in serum over the course of 24 hours following injection; data was used to determine pharmacokinetic model fits, shown with solid lines. (d) Calculated kabs rates for E738–57 peptide and MSA-E738–57 (fit data ± SE). (e) Splenocytes from E738–57-vaccinated mice were restimulated in the presence of brefeldin A with indicated antigen, either fresh or treated with 10% mouse serum for 24 hours. Shown is the percentage IFNγ response from serum-treated antigen restimulation compared to response from fresh antigen as measured by intracellular cytokine staining (n = 2 replicates). (f) FITC-labeled E738–57 or MSA-E738–57 was injected subcutaneously in mice at the indicated doses. 8 hours after injection, inguinal lymph nodes were excised and imaged on IVIS (n = 6 lymph nodes per group). Data are representative of two independent experiments. Statistical significance calculated using two-tailed t tests (d,e) or one-way ANOVA with Dunnett’s multiple comparisons test against PBS (f).
Based on these data, we hypothesized that peptide epitopes (~2 kDa) would have low lymphatic uptake due to high kabs rates, which could be reversed by fusing antigen to mouse serum albumin (MSA, 69 kDa), a highly stable and easily expressed protein, with a hydrodynamic radius of 3.3 nm (Supplementary Fig. 1a). To test this idea, we fused a long peptide from human papillomavirus (HPV) E738–57, containing the H-2Db-restricted CD8+ T cell epitope E749–57, to the C terminus of MSA26 (Fig. 1b). MSA-E738–57 was expressed in HEK cells and purified without evidence of contaminants or aggregation (Supplementary Fig. 2a). To determine kabs for this antigen fusion, we injected FITC-labeled MSA-E738–57 or free E738–57 peptide subcutaneously in mice and measured the percentage of injected dose in blood over time (Fig. 1c). These measurements revealed that kabs was greatly reduced for MSA-E738–57 compared to free peptide (0.14 h−1 vs. 1.37 h−1, respectively, Fig. 1d).
In addition to rapid systemic absorption, peptides suffer from proteolytic instability16,17. To assess whether MSA fusion could protect the peptide from degradation, we re-stimulated E738–57-vaccinated splenocytes ex vivo with either fresh or serum-treated E738–57 antigens and measured IFN-γ production by intracellular cytokine staining (ICS). Ex vivo splenocyte recall in response to free E738–57 peptide was reduced by 78% following serum treatment while stimulation by MSA-E738–57 was minimally affected by serum exposure (Fig. 1e). As a more direct measure of proteolysis, E738–57 peptides and MSA fusions were prepared with His6 and FLAG tags flanking the N- and C-terminal ends of the epitope, and a sandwich ELISA was performed to detect cleavage of the E738–57 epitope following incubation in 20% mouse plasma (Supplementary Fig. 3a); 34% of free E738–57 was cleaved in 4 h, while MSA-E738–57 remained fully intact over this time course (Supplementary Fig. 3b).
One potential disadvantage of the protein fusion strategy is that proteins poorly target the cytosolic antigen presentation pathway, hindering their immunogenicity. In spite of protection from extracellular proteases, however, we found that MSA-fused epitopes are more readily processed than epitopes buried within the polypeptide chain, as MSA-Ova251–270 could more potently stimulate OTI splenocytes than intact Ova protein (Supplementary Fig. 4). Displaying epitopes on the terminal end of MSA protects epitopes from serum degradation while preserving bioactivity in APCs.
Due to its appropriate size and improved proteolytic stability, FITC-labeled MSA-E738–57 effectively trafficked to the dLN following subcutaneous administration, while labeled E738–57 peptide failed to accumulate above background as measured by IVIS imaging, even at ten-fold higher molar doses (Fig. 1f). Notably, administering peptide in the commonly used Montanide water-in-oil emulsion adjuvant also failed to improve dLN bioavailability (Supplementary Fig. 5a) and instead led to retention at the injection site (Supplementary Fig. 5b), a phenomenon previously reported to lead to anergy/deletion of responding T cells27. Collectively, we conclude that expression as a fusion to MSA substantially improves antigen delivery to draining lymph nodes.
Albumin fusions potentiate cellular immune responses
To test the immunogenicity of MSA-E738–57 relative to free E738–57 peptide, mice were subcutaneously primed and boosted 14 days later with E738–57 or MSA-E738–57 mixed with cyclic-di-GMP, a cyclic dinucleotide (CDN) STING agonist28, as an adjuvant. H2-Db/E749–57 tetramer staining 6 days post-boost revealed that MSA fusions elicited 49-fold greater frequencies of E749–57-specific CD8+ T cells in blood compared to E738–57 peptide vaccination (Fig. 2a). Even tenfold higher doses of E738–57 peptide failed to replicate the immunogenicity of the albumin-antigen fusion protein (Fig. 2b). Importantly, antibodies were not raised against MSA (Supplementary Fig. 6a–b), and MSA-fusion vaccination elicited memory T cells that rejected a tumor challenge 2 months after vaccination (Supplementary Fig. 7). Furthermore, MSA-E738–57 outperformed E738–57 when paired with a panel of adjuvants including CpG, a TLR9 agonist; poly(I:C), a TLR3 agonist; and lipo-CpG, a potent lymph-node targeting variant of CpG14 (Fig. 2c). MSA-E738–57 fusions could also elicit therapeutic T cell responses against established subcutaneous E7+ TC-1 tumors. Following weekly vaccination, mice treated with MSA-E738–57 had a statistically significant improvement in tumor control relative to PBS-treated mice and a 40% cure rate, while the median survival of E738–57-treated mice was unchanged relative to untreated animals and only 10% of mice were cured (Fig. 2d).
Figure 2.
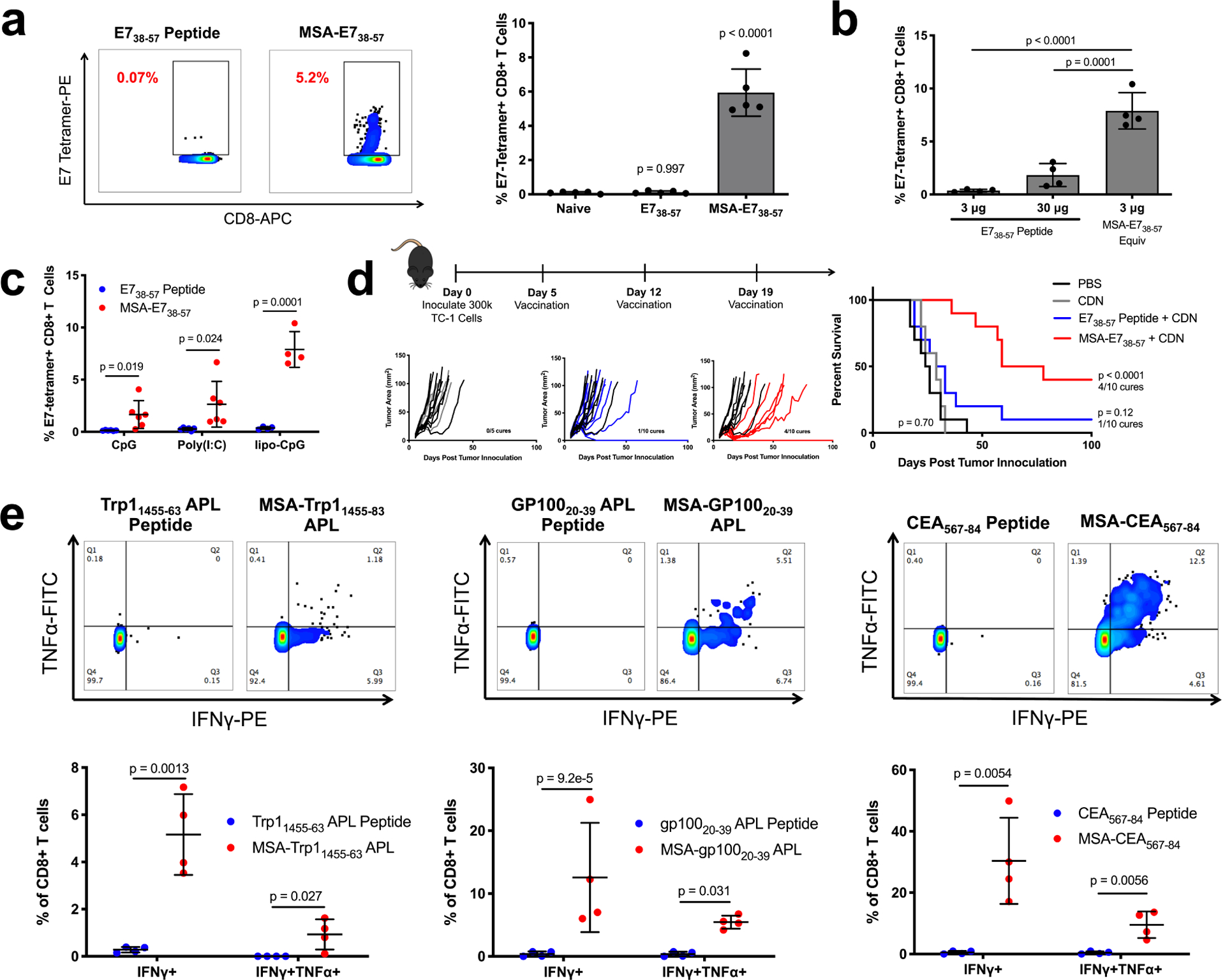
Albumin delivery of epitopes is a generalizable immunogenicity enhancement strategy. (a) Mice were primed and boosted with E738–57 or MSA-E738–57 and CDN subcutaneously. Shown are representative tetramer stain flow plots among CD8+ T cells 6 days after boost, and quantification (mean ± SD, n = 5 mice per group). Data are representative of over five independent experiments. (b) Mice were primed and boosted with E738–57 or MSA-E738–57 as in (a) at the indicated doses. Shown are tetramer stain data among CD8+ T cells 6 days after boost (mean ± SD, n = 4 mice per group). (c) Mice were primed and boosted with E738–57 or MSA-E738–57 and the indicated adjuvants subcutaneously. Shown are tetramer stain data among CD8+ T cells 6 days after boost (mean ± SD, n = 6 mice per group for CpG and poly(I:C) and n = 4 mice per group for lipo-CpG). (d) Timeline and treatment schematic of TC-1 tumor study, along with tumor growth plots and survival curves (n = 5 mice for CDN and n = 10 mice for all other groups). Data are compiled from two independent experiments. (e) Mice were primed and boosted with the indicated antigens as in (a). 6 days after boost, peripheral blood cells were stimulated for 6 hours with WT optimal antigenic peptides in the presence of brefeldin A. Shown are representative flow cytometry plots plots for intracellular cytokine staining among CD8+ T cells and quantification (mean ± SD, n = 4 mice per group). Statistical significance calculated one-way ANOVA with Dunnett’s multiple comparisons test against naïve (a), one-way ANOVA with Tukey’s multiple comparisons test between all groups (b), two-tailed t tests between groups on the x-axis with Holm-Sidak multiple comparisons test (c,e), or two-tailed log-rank (Mantel-Cox) test versus PBS (d).
The generalizability of the MSA-peptide fusion vaccine was assessed by attaching other tumor-associated epitopes, including Trp11455–63 altered peptide ligand (APL)29, gp10020–39 APL30, and oncofetal antigen CEA567–8431. Following prime and boost vaccination, ICS was used to measure IFN-γ and TNF-α in peripheral blood mononuclear cells (PBMCs) after ex vivo stimulation with appropriate peptide antigen. As measured by IFN-γ+CD8+ T cells, MSA-epitope fusion vaccines outperformed their peptide counterparts by 18-fold for Trp11455–63 APL, 39-fold for gp10020–39 APL, and 61-fold for CEA567–84 (Fig. 2e). MSA-fusion vaccines, but not peptide vaccines, also generated polyfunctional IFN-γ+TNF-α+CD8+ T cell responses.
In Batf3−/− mice that lack cross-presenting Batf3+ DCs, MSA-E738–57 immunogenicity was reduced by 70% in a prime/boost vaccination study (Supplementary Fig. 8a), and MSA-E738–57 ineffectively treated TC-1 tumors in Batf3−/− mice (Supplementary Fig. 8b–c). To test whether DC targeting would further enhance the immunogenicity of albumin fusions, we utilized yeast surface display to engineer a fibronectin clone (DEC1) that bound DEC-205, an internalizing receptor expressed on DCs, with a Kd of 0.66 nM (Supplementary Fig. 8d). DEC1 was fused to the N-terminal end of MSA-E738–57 (DEC1-MSA-E738–57). Immunization with DEC1-MSA-E738–57 increased lymph node-resident CD8+ DC uptake of antigen by 39-fold compared to MSA-E738–57, although total dLN accumulation was unaffected (Supplementary Fig. 8e–f). However, as an immunogen, DEC1-MSA-E738–57 elicited T cell responses that were not statistically different from MSA-E738–57 over a broad dose range (Supplementary Fig. 8g). Thus, at least for albumin fusion antigens, we find that specific APC targeting does not further improve T cell responses. The fact that MSA-E738–57 can elicit strong immune responses dependent on cross-presentation (Supplementary Fig. 8a) in spite of relatively poor, but non-zero, CD8+ DC uptake (Supplementary Fig. 8c) suggests that even low amounts of antigen accumulation in the right APC population can drive robust immunity.
Systemic antigen exposure promotes tolerance
Because persistent antigen presentation in the absence of inflammatory cues can induce T cell tolerization and dysfunction27,32–34, we hypothesized that the long circulating half-life of MSA-E738–57 (36h) may prolong antigen presentation in poorly inflamed distal lymphoid organs. In fact, we found that i.v.-administered MSA-E738–57 mixed with CDN failed to prime a cellular immune response (Fig. 3a). Rather, mice that were administered a single i.v. injection of MSA-E738–57, with or without adjuvant, were tolerized against subsequent s.c. prime/boost challenge. We observed a >85% drop in the magnitude of the T cell response compared to animals that had received a control i.v. injection of PBS prior to challenge (Fig. 3b). Notably, i.v. tolerization via MSA-E738–57 was significantly more effective than i.v. administration of E738–57 peptide, a commonly used strategy to induce tolerance35,36 (Fig. 3c).
Figure 3.
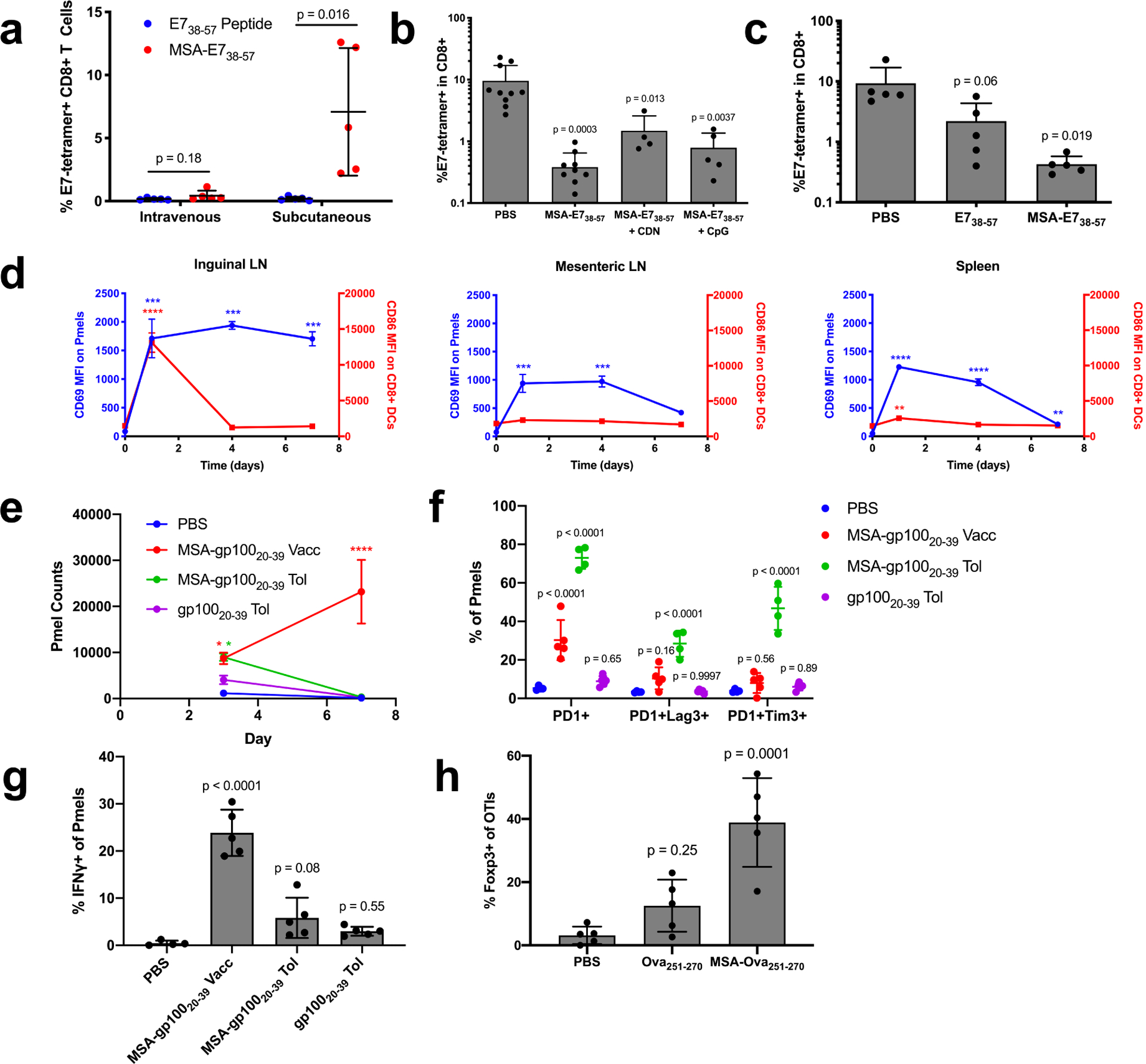
The systemic distribution of albumin fusions induces tolerance. (a) Mice were primed and boosted with E738–57 or MSA-E738–57 plus CDN either subcutaneously or intravenously. Shown are tetramer stain data among CD8+ T cells 6 days after boost (mean ± SD, n = 5 mice per group). Data are representative of two independent experiments. (b-c) Mice were intravenously administered the indicated vaccine and prime/boost challenged two weeks later subcutaneously with CDN. Shown are tetramer stain data among CD8+ T cells 6 days after challenge boost (mean ± SD, n = 10 for PBS in (b), n = 9 for MSA-E738–57 in (b), n = 4 for MSA-E738–57 + CDN in (b), n = 5 for MSA-E738–57 + CpG in (b) and n = 5 for all groups in (c)). (d) Mice were subcutaneously administered MSA-gp10020–39 APL with CDN. Following 1, 4, and 7d post-vaccination, 0.5M pmel CD8+Thy1.1+ cells were transferred into mice. 24 hours after transfer, the indicated organs were excised and CD69 MFI measured on Thy1.1+ cells by flow (blue curves). At the same time points post-vaccination, the indicated organs were excised and CD86 MFI measured on CD8+ DCs by flow cytometry (red curves) (mean ± SEM, n = 3 mice per group). (e-g) 1M pmel CD8+Thy1.1+ cells were transferred into WT mice. 24 hours later, the indicated tolerizing vaccines were administered intravenously without adjuvant (MSA-gp10020–39 and gp10020–39 tol), and an activating vaccine was administered subcutaneously with adjuvant (MSA-gp10020–39 vacc). (n = 4 mice for PBS and 5 for all other groups). (e) Shown are Thy1.1+ cell counts in the spleen 3 and 7 days after vaccination (mean ± SEM). (f) Quantification of the frequency of expression of exhaustion-associated markers on Thy1.1+ cells from inguinal LN on day 3 (mean ± SD). (g) Quantification of IFNγ production on Thy1.1+ cells from the inguinal LN on day 3 (mean ± SD). (h) 1M OT1 CD8+Thy1.1+ cells were transferred into WT mice. 24 hours later, the indicated tolerizing vaccines were administered intravenously without adjuvant (mean ± SD, n = 5). Shown is quantification of the frequency of expression of Foxp3 on Thy1.1+ cells from the inguinal LN on day 3. Statistical significance calculated using two-tailed t tests between groups on the x-axis with Holm-Sidak multiple comparisons test (a), one-way ANOVA with Dunnett’s multiple comparisons test against PBS (b,c,g,h) or compared against the day zero time point for each measurement (d), or two-way ANOVA with Dunnett’s multiple comparisons test against PBS (f). In (d), ** p < 0.01; *** p < 0.001; **** p < 0.0001.
To assess the kinetics of antigen presentation relative to inflammatory cues in different tissue sites, we subcutaneously vaccinated mice with MSA-gp10020–39 and CDN adjuvant. After 1, 4, or 7 days, 500k pmel Thy1.1+ cells expressing TCRs specific for the gp100 epitope were adoptively transferred into vaccinated animals to serve as reporters of antigen presentation. Twenty-four hours later, the local dLN (inguinal node) and distal lymphoid organs (mesenteric nodes and spleen) were excised and resident Thy1.1 cells were assessed for CD69 expression indicative of TCR triggering. At equivalent time points, CD8+ DCs were analyzed for CD86 expression indicative of DC activation. Consistent with previous reports of rapid CDN clearance37, CD8+ DCs were poorly activated in distal organs; instead, only the local dLN was inflamed. Nonetheless, antigen presentation persisted in distal non-inflamed organs: CD69 was still upregulated on pmel T cells in the mesenteric LNs when transferred 4 days after immunization or in the spleen when transferred 7 days after immunization (Fig. 3d). While the local dLN likely contributes to activating immunity due to the simultaneous presence of both antigen and inflammatory cues, persistent antigen presentation in the absence of DC activation in distal organs may blunt functional immunity.
To more specifically query the mechanisms of tolerance following systemic exposure to albumin fusions in the absence of adjuvant, we transferred 1 million Thy1.1+ pmel cells into recipient mice and subsequently administered PBS, gp10020–39 peptide, or MSA-gp10020–39 i.v. without adjuvant. MSA-gp10020–39 s.c. with adjuvant was used as a control for activating immunity. While MSA-gp10020–39 delivery both with and without adjuvant induced Thy1.1+ cell expansion by day 3, expansion continued through day 7 only in response to MSA-gp10020–39 with adjuvant; without adjuvant, Thy1.1+ cells were rapidly deleted by day 7 (Fig. 3e). Thy1.1+ cells from the inguinal LN, where the activating vaccine triggers functional immune responses, were further immunophenotyped. Only Thy1.1+ cells from mice that received MSA-gp10020–39 without adjuvant had elevated populations of PD1+Lag3+ and PD1+Tim3+ cells over background (Fig. 3f), and Thy1.1+ cells from mice that received tolerizing vaccines exhibited deficiencies in cytokine production upon ex vivo recall (Fig. 3g). CD8+ T cells that express Foxp3 are emerging as an important class of regulatory T cells CD8+ Tregs in tolerance and autoimmune disease38–40. In a similar study with transferred Thy1.1+ OTI cells and subsequently administered PBS, Ova251–270 peptide, or MSA-Ova251–270 i.v. without adjuvant, the frequency of Foxp3+ Thy1.1+ cells was elevated in the inguinal LN on day 3 most significantly in mice that received MSA fusion without adjuvant. Collectively, we conclude that the delivery of albumin fusions in the absence of adjuvant facilitates tolerization by depleting and/or exhausting antigen-specific T cells, and by stimulating the development of previously characterized Foxp3+ CD8+ Tregs.
Increasing kclear can further improve immunogenicity
The tolerizing impact of persistent systemically-distributed protein compelled us to search for an alternative protein carrier that preserved the low kabs of albumin while exhibiting a more rapid systemic kclear rate constant, to decrease systemic antigen exposure while maintaining local LN accumulation. We identified transthyretin (TTR), a ~60 kDa tetrameric endogenous hormone-trafficking serum protein as a promising candidate protein carrier, as it has a similar molecular weight as albumin but a significantly shorter systemic half life (t1/2 of ~2 days for TTR41 versus t1/2 of ~3 weeks for albumin42). Although TTR amyloidosis diseases demonstrate its propensity to form aggregates in solution, these conditions typically result from mutant rather than wild-type TTR, and fibril formation is driven by acidic rather than physiologic conditions43. After expression, we found TTR-E738–57 to be stably monomeric without evidence of fibril formation, similar to MSA-E738–57 (Supplementary Fig. 1b).
We next confirmed TTR’s similarity to MSA in all characteristics except kclear. E738–57 peptide was processed and presented by either fusion protein with similar potency during ex vivo splenocyte restimulation (Fig. 4a), and like MSA-E738–57, TTR-E738–57 was stable in serum (Supplementary Fig. 10a). Following s.c. administration, FITC-TTR-E738–57 accumulated equivalently to FITC-MSA-E738–57 in the inguinal dLN (Fig. 4b) and was taken up similarly by APCs (Fig. 4c).
Figure 4.
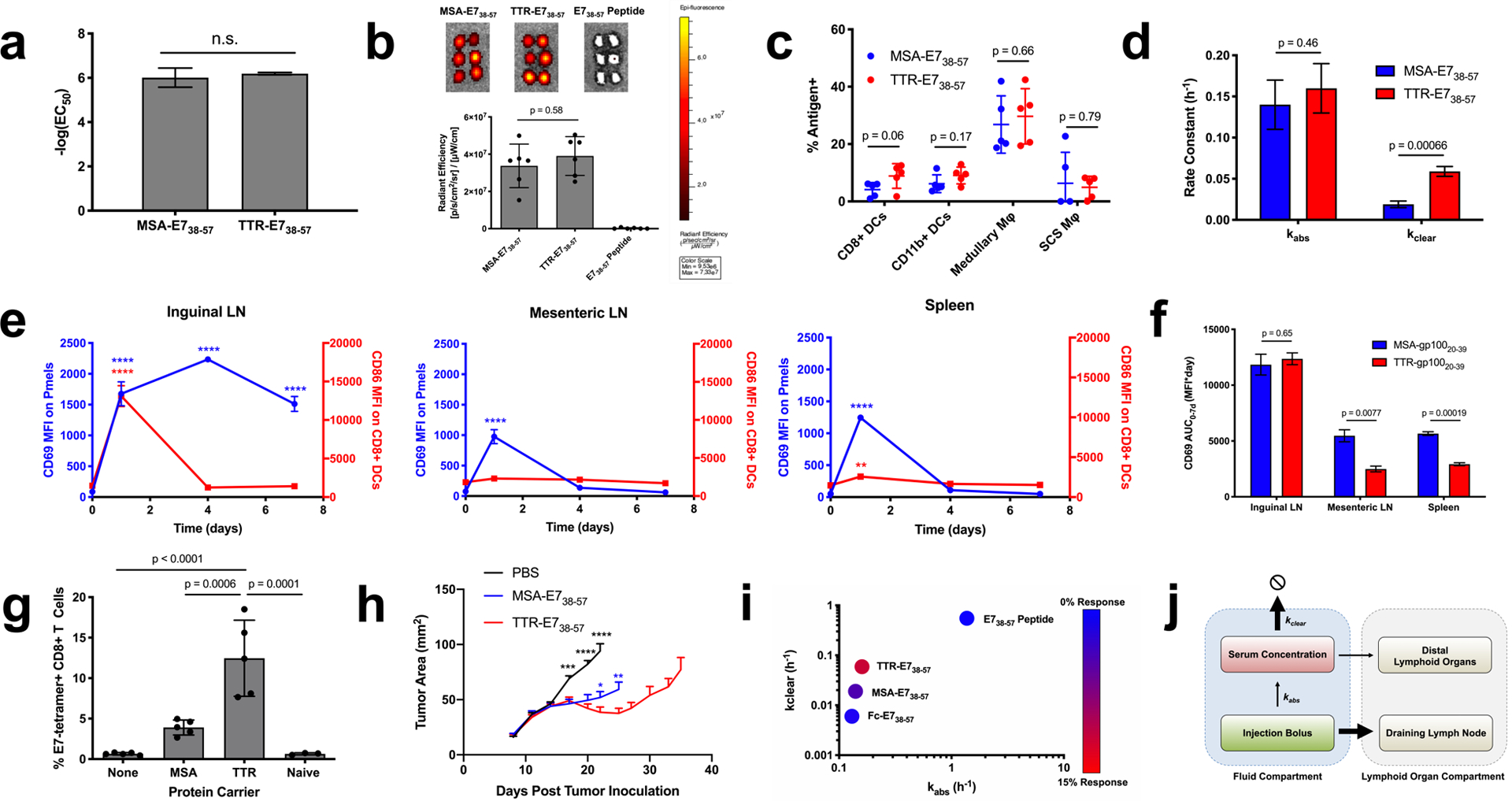
TTR fusions outperform MSA fusions due to a faster clearance rate. (a) EC50 values calculated from splenocyte restimulation studies with the indicated antigen (n = 3). (b) FITC-labeled antigen uptake in the dLN was measured as in Fig. 1f (mean ± SD, n = 6 lymph nodes per group). (c) AF488-labeled MSA-E738–57 or TTR-E738–57 was injected subcutaneously in mice. 24 hours later, the inguinal lymph node was excised and AF488 signal in APCs assessed by flow cytometry (mean ± SD, n = 5 mice per group). (d) Shown are kabs and kclear values as calculated via nonlinear regression from pharmacokinetic studies (fit data ± SE). (e) Pmel CD69 and CD8+ DC CD86 MFIs are shown (mean ± SEM, n = 3 mice per group). (f) Area under the curve calculations from CD69 MFI in Fig. 3d and Fig. 4e (calculated AUC ± SE) (g) Mice were primed and boosted with the indicated vaccine plus CDN subcutaneously. Shown are tetramer stain data among CD8+ T cells 6 days after boost (mean ± SD, n = 5 mice per group). Data are representative of over five independent experiments. (h) Mice were implanted with TC-1 tumor cells and immunized on days 8 and 15 with the indicated vaccine. Shown are tumor growth curves of the indicated groups (mean + SEM). Curves are plotted until the first mouse is euthanized in each group (n = 9 mice per group) (i) Mice were primed and boosted with the indicated vaccine plus CDN subcutaneously. Tetramer stain data are indicated by color overlaid on a kabs and kclear scatterplot. (j) Schematic of pharmacokinetic parameters that maximize bioavailability in the inflamed dLN. Statistical significance calculated using two-tailed t tests either alone (a) or with Holm-Sidak multiple comparisons test (c,d,f), one-way ANOVA with Tukey’s multiple comparisons test between all groups (b,g), or with Dunnett’s multiple comparisons test against the day zero time point for each measurement (e). In (h), two-tailed t tests were performed at each measurement comparing against TTR-E738–57. In (e, h), ** p < 0.01; *** p < 0.001; **** p < 0.0001.
By contrast, MSA-E738–57 and TTR-E738–57 had distinct pharmacokinetic properties following s.c. administration (Fig. 1c vs. Supplementary Fig. 10b). While kabs was similar between MSA-E738–57 and TTR-E738–57 (0.14 ± 0.03 h−1 and 0.16 ± 0.03 h−1 respectively), kclear was over three-fold faster for TTR-E738–57 than for MSA-E738–57 (0.019 ± 0.004 h−1 versus 0.059 ± 0.006 h−1) (Fig. 4d). When reporter pmels were used to assess T cell priming kinetics following TTR-gp10020–39 immunization, we found that TTR’s faster clearance rate led to more transient priming in distal, uninflamed lymphoid organs than observed with MSA fusions, with CD69 expression only detected in the mesenteric LN and the spleen at 1 day post-immunization with TTR-gp10020–39 (Fig. 4e). While the one week CD69 MFI area under the curve (AUC) was similar between MSA and TTR fusions in the draining inguinal LN, the AUCs in the mesenteric LN and spleen were significantly reduced for TTR fusions (Fig. 4f). Correlating with these findings, s.c. immunization with TTR-E738–57 elicited a 3.7-fold greater CD8+ T cell response than MSA-E738–57 when dosed with equimolar E7 antigen (Fig. 4g), and no immunophenotypic differences in terms of memory precursor populations, nor expression of T-bet, granzyme B, KLRG1, and CD127 were detectable (Supplementary Fig. 11). TTR-E738–57-treated TC-1 bearing mice exhibited greater tumor growth inhibition (Fig. 4h). No antibody responses against TTR were detectable (Supplementary Fig. 6c–d).
To further confirm the effect of kclear on fusion protein vaccine immunogenicity, we fused and characterized the Fc portion of an IgG2c antibody to E738–57 (Fc-E738–57) (Supplementary Fig. 1c), generating a fusion construct with a kclear value 10x slower than TTR-E738–57 and 3x lower than MSA-E738–57 (kclear = 0.006 ± 0.002 h−1) (Supplementary Fig. 10d). Following prime and boost, Fc-E738–57 and CDN vaccination primed a 1.7% E749–57-specific CD8+ T cell response, a 24-fold boost in immunogenicity compared to peptide vaccination that was nonetheless weaker than responses to MSA-E738–57 and TTR-E738–57 (Supplementary Fig. 12). Although IgG2c is an activating Fc isotype that binds to Fc receptors, mutations that reduced binding to FcγRs and FcRn did not affect Fc-E738–57’s immunogenicity (Supplementary Fig. 12). Overall, the efficacy of TTR-E738–57 > MSA-E738–57 > Fc-E738–57 >> E738–57 peptide (Fig. 4i) suggests that while lowering kabs is essential to generate vaccine responses over background, increasing kclear can further boost potency (Fig. 4j). Collectively, we conclude that size exclusion from systemic absorption, proteolytic stability, and avoidance of antigen accumulation in non-inflamed lymphoid organs all contribute to the immunogenicity of protein vaccines, which can be tuned and optimized by the judicious selection of protein fusion partner (Table 1).
Table 1 –
Overview of the pharmacokinetic determinants of vaccine immunogenicity
| 1. Size Exclusion from Systemic Absorption | 2. Proteolytic Stability | 3. Avoidance of Distal Priming | Immunogenicity | |||
|---|---|---|---|---|---|---|
| Quantity | MW(kDa) | Radius (nm) | kabs (hr−1) (fit data ± SE) | % Serum-treated antigen recall response relative to fresh | kclear(hr−1) (fit data ± SE) | Vaccine Response (% of CD8+ T Cells) (wean ± SD) |
| E738–57 Peptide | 2.2 | - | 1.37 + 0.40 | 21.5% | 0.55 + 0.11 | 0.12 + 0.07 |
| FC-E738–57 | 57.8 | 3.4 | 0.13 + 0.02 | 59.3% | 0.006 + 0.002 | 1.94 + 0.69 |
| MSA-E738–57 | 69.4 | 3.3 | 0.14 ± 0.03 | 114% | 0.019 ± 0.004 | 5.94 ± 1.38 |
| TTR-E738–57 | 68.5 | 3.5 | 0.16 ± 0.03 | 61.0% | 0.059 ± 0.006 | 12.45 ± 4.70 |
TTR as a delivery vehicle in tumor immunotherapy
We next explored the generalizability of TTR delivery in the context of other tumor-associated antigens. As measured by IFN-γ+CD8+ T cells in peripheral blood, TTR-antigen fusion vaccines outperformed MSA-antigen vaccines by 4-fold for Trp11455–63 and 2-fold for gp10020–39 APL; while TTR-CEA567–84 generated equivalent IFN-γ+ responses as MSA-CEA567–84, polyfunctional IFN-γ+TNF-α+ responses were enhanced over background only in the TTR-CEA567–84 case (Fig. 5a).
Figure 5.
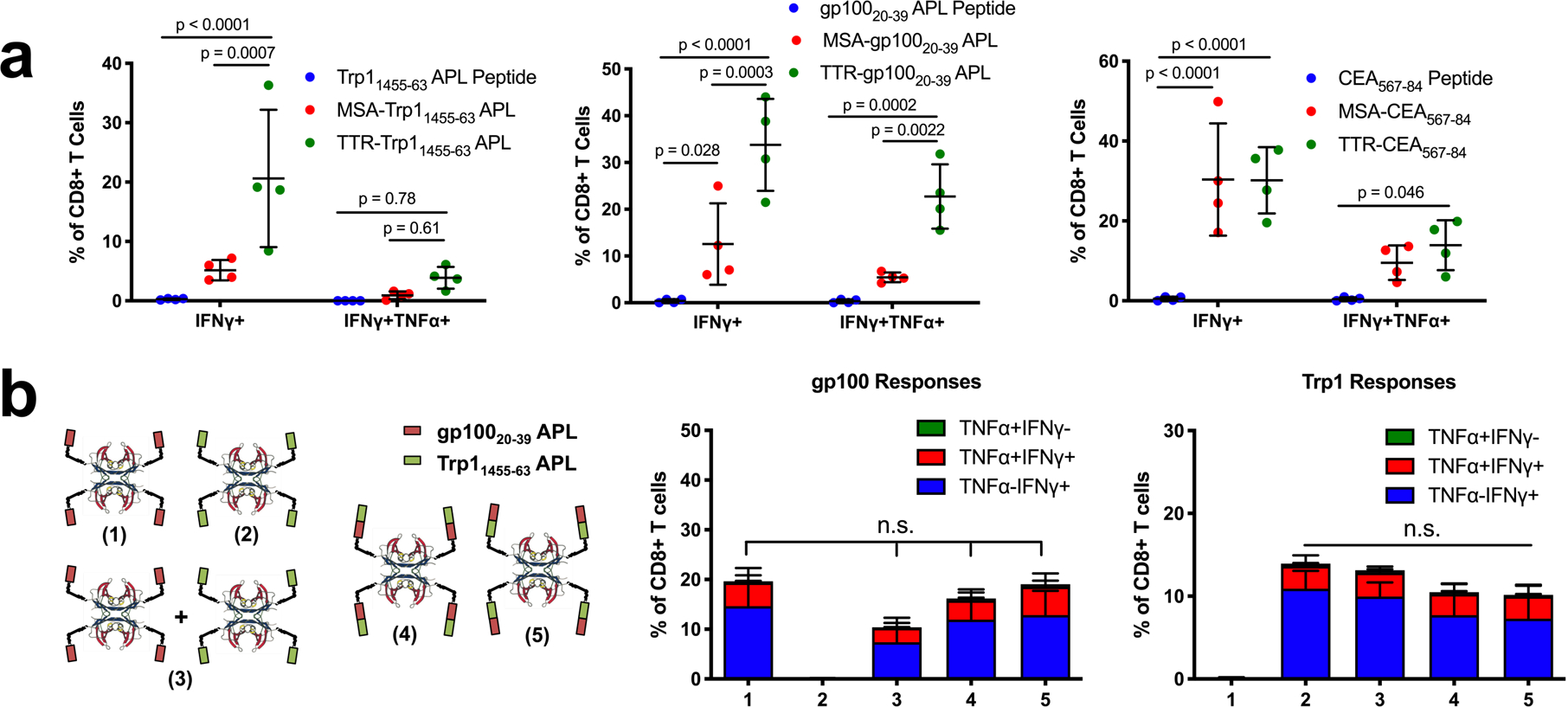
TTR-antigen fusion vaccines. (a) Mice were primed and boosted with the indicated antigens in peptide, MSA-fusion, or TTR-fusion form along with CDN subcutaneously, then assed with ICS (mean ± SD, n = 4 mice per group) (b) Schematic of the TTR constructs used to test Trp1 and gp100 orientation during co-delivery. Mice were vaccinated with constructs along with CDN. Shown are ICS data 6 days after boost (mean ± SD, n = 5 mice per group). Statistical significance calculated using two-way ANOVA with Tukey’s multiple comparisons test (a) or one-way ANOVA with Tukey’s multiple comparisons test (b).
As a tetramer, TTR also provides the opportunity to develop higher valency constructs, which offer manufacturing advantages. For example, TTR fusions carrying 2 copies of the E738–57 epitope per subunit (8 copies per tetramer) reduced by 4.7-fold the volume of production cell culture required for vaccination compared to MSA-E738–57 (Supplementary Fig. 13a), with no corresponding loss in immunogenicity (Supplementary Fig. 13b,c). We reasoned that if fusion of multiple copies of epitopes to TTR preserved immunogenicity, then the protein carrier could be designed to deliver multiple antigens simultaneously. While the co-delivery of antigens on a single molecule can simplify commercial translation, it runs the risk of biasing responses toward the most immunogenic antigen in the construct. To assess this risk, we compared TTR-gp10020–39 and TTR-Trp11455–463 vaccines administered separately or together, and compared immune responses against TTR fused to gp10020–39 and Trp11455–463 simultaneously in either orientation (Fig. 5b). TTR carriers delivering both antigens together elicited T cell responses that were not statistically different from the individual or mixed antigens, and we observed no dependence on antigen order in the fusion constructs (Fig. 5b).
To test the therapeutic efficacy of this strategy, we assessed therapeutic responses to Trp1 and gp100 vaccination in the aggressive B16F10 melanoma model. B16F10 subcutaneous tumors were vaccinated with free Trp1 and gp100 peptides or TTR-Trp1-gp100 fusions every six days with or without anti-PD1 combination therapy (Fig. 6a). Six days after the first boost, strong antigen-specific IFN-γ responses (mean > 25%) were only generated in groups vaccinated with TTR-Trp1-gp100 (Fig. 6b). As a result, TTR-Trp1-gp100-treated mice had significantly slower tumor outgrowth than mice treated with Trp1 and gp100 peptide (Fig. 6c). Anti-PD-1 therapy had no efficacy as monotherapy, but together with the TTR fusion vaccine elicited substantially enhanced anti-tumor efficacy (Fig. 6d). Statistically significant improvements in overall survival relative to PBS controls were observed only in mice receiving TTR fusion vaccines (Fig. 6e).
Figure 6.
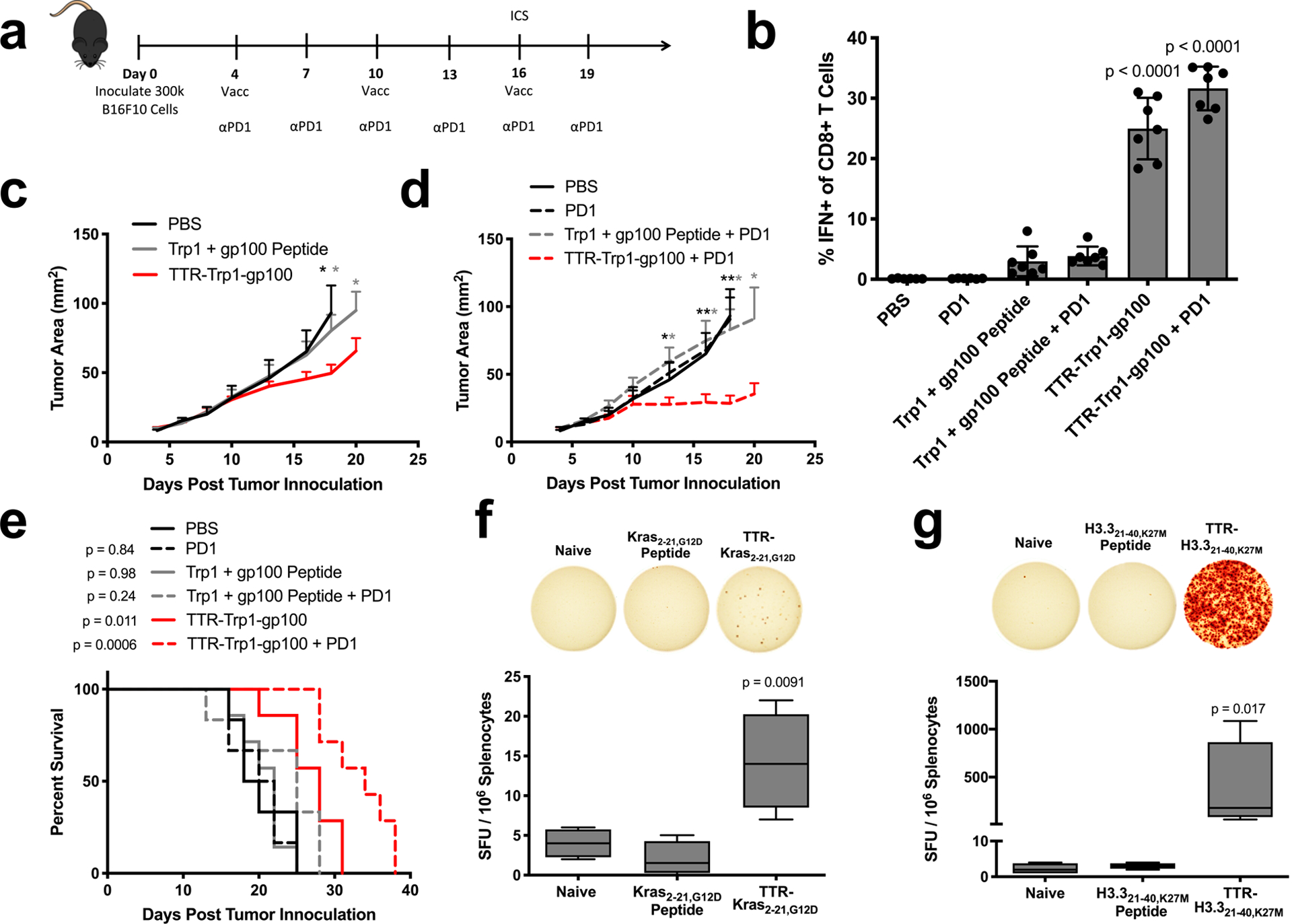
TTR-antigen fusions in cancer immunotherapy. (a) Schematic of the anti-B16F10 tumor immunotherapy study timeline used to generate panels (b-e) (n = 6 mice for PBS and Trp1 + gp100 peptide + PD1 and n = 7 mice for all other groups). (b) ICS data collected from PBMCs on day 16 of B16F10 study. Shown are pooled Trp1 and gp100 responses among CD8+ T cells in blood (mean ± SD). (c,d) Shown are tumor growth curves of the indicated groups (mean + SEM). Curves are plotted until the first mouse is euthanized in each group. (e) Survival curves. (f,g) The indicated shared neoantigen-targeting vaccines were administered with prime/boost in HLA-A11 (f) and HLA-A2 (g) transgenic mice, followed by an ELISpot read-out with splenocytes stimulated with neoantigen-containing overlapping peptides (n = 4 mice per group). Shown are boxplot representations of spot counts, with median, interquartile range, and min/max identifiers. Statistical significance calculated using two-way ANOVA with one-way ANOVA with Dunnett’s multiple comparisons test against PBS (b) or against naive (f), one-tailed t test at each measurement comparing against TTR-Trp1-gp100 (c) or against TTR-Trp1-gp100 + PD1 (d), two-tailed log-rank (Mantel-Cox) test versus PBS (e), or Kruskal-Wallis with Dunn’s multiple comparisons test against naïve (g). In (c) and (d), * p < 0.05; ** p < 0.01.
Shared neoantigens, or HLA-displayed peptides derived from mutant oncogenes, are also emerging as promising targets of vaccination. We tested two shared neoantigens with TTR fusions: Kras G12D, commonly mutated in pancreatic cancer44,45, and H3.3 K27M, a driver mutation in pediatric gliomas46–48. HLA-A11- and HLA-A2-displayed epitopes have recently been identified for Kras G12D and H3.3 K27M mutations, respectively49,50. HLA-A11 and HLA-A2 transgenic mice were prime/boosted with Kras2–21,G12D and H3.321–40,K27M peptide or TTR fusion vaccines, followed by an ELISpot read-out. Antigen-specific cellular immunity above background was observed only with TTR fusion (Fig. 6f, g). Taken together, these data demonstrate the utility of TTR-mediated delivery for viral antigens (E7), tumor-associated antigens (Trp1, gp100), oncofetal antigens (CEA), and shared neoantigens (Kras, H3.3).
Discussion
A major factor limiting the potency of peptide vaccines is their poor transport to lymph nodes following injection14. This transport limitation has spurred the development of a broad set of delivery platforms to improve antigen delivery to lymph nodes; these strategies have mostly entailed nanoparticulate formulation and/or chemical modification of synthetically produced peptide vaccines14,18,19,51–53 Here, we altered peptide biodistributions by fusing these antigens to protein-based delivery vehicles to develop a fully recombinant protein solution. We show that protein-epitope fusions are strong candidate vehicles for off-the-shelf vaccination against targets like viral antigens, tumor-associated antigens, oncofetal antigens, and shared neoantigens.
Peptides are routinely conjugated to immunogenic protein carriers like keyhole limpet hemocyanin (KLH) to increase B cell receptor crosslinking and co-deliver antigen with CD4+ T cell epitopes, boosting humoral immunity54. However, T cell responses are exclusively detected against KLH, not against the antigen of interest55; and the impacts of KLH on biodistribution have been understudied. Here, we utilize minimally immunogenic protein carriers to direct cellular responses against fused epitope payloads, and we tune immunogenicity by the modulation of pharmacokinetics. While this work focused on the delivery of CD8+ T cell epitopes, future efforts should be made to understand how the protein fusion strategy influences CD4+ T cell responses against long peptide antigens.
Analysis of the biodistribution behavior of several protein carrier-epitope fusion vaccines revealed principles for optimizing vaccine immunogenicity via pharmacokinetic tuning. We found that maximizing bioavailability in the dLN but reducing uptake in distal lymphoid organs ensures that antigen is presented at the right site at the right time. Two rate constants controlling local lymphatic vs. systemic distribution, kabs and kclear, predictably influenced antigen biodistribution, and thus had a dominant effect on vaccine immunogenicity. When these two rate constants are appropriately selected, vaccine responses can be significantly potentiated in both magnitude and functionality, delaying the growth of checkpoint-refractory tumors in mice.
In particular, appropriately bulky protein carriers reduced kabs sufficiently to reduce systemic absorption following subcutaneous injection, improving lymphatic uptake and subsequent accumulation in the dLN. But while dLN bioavailability is a prerequisite to potent vaccination, increasing kclear can improve immunogenicity even further by limiting antigen accumulation in distal lymphoid organs that have not received adjuvant stimulation. Adjuvants are typically designed to avoid systemic dissemination, preventing unacceptable toxicities14,56,57, and inflaming only the local draining lymph nodes as observed here with cyclic dinucleotides. Antigens with low kclear, however, will systemically distribute into non-inflamed distal lymphoid organs, leading to dysfunctional priming of T cells at these sites. This principle is supported by the hierarchy of immunogenicity we observed with TTR, MSA, and Fc fusions. Recognition of this phenomenon has practical implications in vaccine engineering. For example, the observation of priming in distal nodes was noted in early antibody-mediated DEC-205 targeted immunization studies, necessitating the co-administration of systemic adjuvant, which would likely be too toxic for clinical translation21. Simply matching the pharmacokinetics of antigen and adjuvant such that they similarly distribute in vivo may be a facile alternative to manufacturing antigen/adjuvant conjugates or co-encapsulations. There is opportunity for further engineering in this regard, as even TTR fusions lead to antigen presentation in the draining lymph node that outlasts inflammatory cues. The use of appropriately persistent protein-antigen fusions to treat autoimmunity should also be further explored.
Protein-epitope fusions also avoid a number of manufacturing and scale-up issues commonly associated with nanoparticulate formulations. Mass production of protein biologics is a well-developed art, and recombinant proteins can be manufactured in cGMP processes at scale to high purity. Albumin has a decades-long history of helping solubilize challenging cargo; while TTR is a less commonly utilized carrier, it can also be homogeneously produced, and as a tetramer its manufacturing efficiency is improved several-fold.
The protein-epitope fusion approach also has a potential application in nucleic acid vaccination, a cheaper, rapidly deployable modality. Intramuscular transfection with plasmid DNA and/or mRNA are both commonly studied vaccination strategies58–60. Encoding protein-epitope fusions in these systems could allow for the expression of antigen payload fused to protein delivery vehicle to shepherd antigen from the site of secretion to the dLN. Non-encodable, particulate-, or conjugate-based delivery systems are not amenable to such an approach.
Notably, the PK determinants of immunogenicity described here can be elucidated with a low number of simple, rapid, and cost-effective experiments. Data on kabs and kclear can be assessed with a single pharmacokinetic study, and proteolytic stability can be reliably measured by performing restimulation assays ex vivo on vaccinated or transgenic splenoctyes with fresh or serum-treated antigen. We propose that these two assays can be used to rapidly screen and optimize future protein carrier strategies and improve future efforts to prime functional anti-tumor immunity.
Materials and Methods
Mice
B6 mice (C57BL/6NTac) and HLA-A11 mice (B6-Tg(HLA-A*1101/H2-Kb)A11.01) were purchased from Taconic. Batf3−/− mice (B6.129S(C)-Batf3tm1Kmm/J), pmel Thy1.1+ mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J), and HLA-A2 mice (C57BL/6-Mcph1Tg(HLA-A2.1)1Enge/J) were purchased from the Jackson Laboratory. OTI mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) and Thy1.1 mice (B6.PL-Thy1a/CyJ) were purchased from Jackson Laboratory and crossed to generate an OTI Thy1.1+ colony. All animal work was conducted under the approval of the Massachusetts Institute of Technology (MIT) Division of Comparative Medicine in accordance with federal, state, and local guidelines. A summary of mouse strains used in each study is outlined in Supplementary Table 1.
Cells
HEK cells (FreeStyle 293-F) were purchased from Thermo Fisher Scientific. The TC-1 cell line, an HPV E6 and E7 expressing line derived from C57BL/6 lung epithelia, was kindly provided by T.C. Wu (Johns Hopkins University). The B16F10 cell line, a mouse model of melanoma, was purchased from ATCC. HEK cells were cultured in FreeStyle 293 Expression Medium (Thermo Fisher Scientific) and passaged every two days to a density of 300k/ml. TC-1 cells were cultured in RPMI-1640 (GE Healthcare Life Sciences) supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 4 mM L-alanyl-L-glutamine. B16F10 cells were cultured in DMEM (GE Healthcare Life Sciences) supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 4 mM L-alanyl-L-glutamine. Both tumor cells were passaged 1:10 every two days. All cells were cultured at 37°C and 5% CO2. All cells were tested regularly for mycoplasma contamination and for rodent pathogens, and none used tested positive at any point. A summary of cell lines were used for each tumor study is outlined in Supplementary Table 1.
Protein expression and purification
Codon-optimized genes encoding desired proteins were cloned into gWiz expression vectors (Genlantis) using the In-Fusion HD Cloning Kit (Clontech), prepared endotoxin free (Macherey-Nagel) and incubated with Opti-Pro (Thermo Fisher Scientific) and polyethylenimine (PEI) 25K (Polysciences) prior to drop-wise addition to HEK cell culture at 1 million cells/ml. 1 mg of DNA was mixed with 40 ml of Opti-Pro and 2 mg of PEI per 1 L of cell culture. One week after transfection, cell culture was spun down in endotoxin-free centrifuge tubes at 15000xg for 30 min, supernatant filtered, pH neutralized by addition of 10X PBS, and run through TALON metal affinity resin (Clontech) by gravity flow per manufacturer instructions. Fc-E738–57 and variants were prepared as heterodimers with one copy of E738–57 per Fc dimer by co-transfecting gWiz-Fc-His6-E738–57 and gWiz-Fc-FLAG (0.5 mg of each plasmid per 1 L of cell culture). Purification proceeded as above, but eluate from TALON resin was subsequently run through an anti-DYKDDDDK G1 affinity resin kit (Genscript) per manufacturer instructions to ensure purification of the heterodimer. In addition to wild-type Fc fusions, a pair of mutations (G236R/L328R) were introduced to knock out binding to FcγRs61. Following purification, proteins were buffer exchanged into PBS with 30 kDa molecular weight cutoff Amicon Ultra-15 centrifugal filters (Millipore), filtered using Spin-X centrifuge tube filters (Corning) or 0.2 μm syringe filters (VWR), and flash-frozen in aliquots with liquid nitrogen, then stored at −80°C. After thaw, proteins were used within one month and stored at 4°C in sterile conditions. Protein sequences are outlined in Supplementary Table 2.
Protein characterization
Protein and dextran hydrodynamic radii were assessed by DLS (DynaPro NanoStar), purity analyzed by SEC (Superdex 200 Increase), and endotoxin levels measured using a chromogenic LAL assay (Lonza). A maximum of 5 EU/kg, or 0.1 EU/mouse was accepted for all injected proteins.
PK studies
5-FAM-E738–57 was purchased from Genscript, and recombinantly expressed proteins (MSA-E738–57, TTR-E738–57, and Fc-E738–57) were labeled with NHS-5/6-FAM (Thermo Fisher Scientific) per manufacturer instructions. Following protein conjugation, the concentration of labeled protein was calculated using A493. Mice were s.c. or i.v. vaccinated with 1 nmol labeled peptide or protein. At the indicated time points, <10 μl of blood was collected from tail snip into heparin-coated microhematocrit capillary tubes (VWR). Capillaries were parafilmed and stored upright in the dark at 4°C overnight to separate serum from cellular components. On a Typhoon Trio variable mode imager, serum in capillaries was measured (526 SP filter, 532 nm laser) against a standard curve of analytes prepared in PBS and added directly into capillaries. FIJI image analysis software was used to calculate serum concentrations of analyte. I.v. curves were fit to C = C0e−kclear*t and s.c. curves were fit to C = F*C0*kabs/(kabs-kclear)*(e−kclear*t-e−kabs*t), where C0 and kclear were fixed by the i.v. fit. F is systemic absorbance, and kclear and kabs are the clearance and absorbance rates, respectively (h−1). Curves were fit using nonlinear regression on GraphPad Prism software.
Vaccinations
All activating vaccinations were performed by s.c. prime at the tail base on day 0, boost on day 14, and read-out on day 20, except for the i.v. vaccination in Fig. 3a. All studies utilized 25 μg cyclic di-GMP (InvivoGen) as the adjuvant unless otherwise specified: in Fig. 2c utilizing alternative adjuvants, 1 nmol ODN1826 (CpG) (InvivoGen), 50 μg poly(I:C) HMW (Invivogen), and 1 nmol lipo-CpG14 were instead used; in TC-1 tumor bearing animals, 50 μg cyclic di-GMP was used instead; in the B16F10 tumor study, 1 nmol lipo-CpG was used as the adjuvant. Antigen dosing (expressed as peptide equivalence): 3 μg for all E738–57 studies in non-tumor bearing mice; 10 μg E738–57 in Fig. 2e; 5 μg E738–57 in Fig. 4h; in Fig. 2e and 5a, 2.5 μg Trp11455–63 APL, and 5 μg gp10020–39 APL and CEA567–84; 3 μg of gp10020–39 APL or Ova257–265 in pmel or OTI adoptive transfer studies, respectively; for bivalent forms of TTR-Trp1-gp100, 2.5 nmol in non-tumor bearing mice and 4 nmol in tumor-bearing mice; 10 μg Kras2–21,G12D and H3.321–40,K27M. For tolerizing vaccinations (Fig. 3b–c), PBS or 3 μg peptide equivalence E738–57 or MSA-E738–57 with or without adjuvant (25 μg cyclic di-GMP or 1 nmol lipo-CpG) was i.v. administered 14 days before s.c. prime/boost with TTR-E738–57 plus cyclic di-GMP as described above. In all cases, vaccine responses were measured by tetramer stain, ICS (see flow cytometry section), or by mouse IFN-γ ELISpot (BD Biosciences) per manufacturer instructions, where 1M splenocytes were plated per well and stimulated with overlapping peptides (Kras2–21,G12D or H3.321–40,K27M) for 24 hours.
Flow cytometry
Antibodies against CD16/32 (Fc block, clone 93), CD8α (53–6.7), TNF-α (MP6-XT22), IFN-γ (XMG1.2), CD3 (17a2), B220 (RA3–6B2), CD11b (M1/70), CD11c (N418), F4/80 (BM8), CD69 (H1.2F3), CD86 (GL-1), PD1 (29F.1A12), Lag3 (C9B7W), and Tim3 (RMT3–23) were purchased from BioLegend. Cell viability was assessed with DAPI (Sigma) for tetramer stains and LIVE/DEAD fixable aqua dead cell stain (Thermo Fisher Scientific) for all other studies. Tetramer staining: E7 tetramer (iTAg Tetramer/PE - H-2 Db HPV 16 E7) was purchased from MBL; during tetramer staining, PBMCs were Fc blocked and stained with tetramer for 15 minutes prior to addition of anti-CD8α antibody, and cells were incubated with all antibodies for another 30 minutes. All labeling was performed in PBS + 0.1% BSA. Ex vivo stimulation studies: Intracellular cytokine staining (ICS) to assess the magnitude of T cell response was performed as previously reported62. Briefly, PBMCs were re-stimulated in RPMI-1640 + 10% FBS at 37°C with optimal WT peptide at 10 μg/ml (Trp11455–63, gp10025–33, CEA572–579) for 2h followed by addition of brefeldin A (Thermo Fisher Scientific) for another 4h prior to staining. In the B16F10 study, re-stimulation was performed with 10 μg/ml of both Trp11455–63 and gp10025–33. When ICS was instead used to assess stability/immunogenicity instead of a vaccine read-out, whole protein or long peptide antigen was used for stimulation (see antigenic stability analysis section), and cells were re-stimulated for 18 hours followed by addition of brefeldin A for another 6 hours prior to staining. In all cases, surface staining was performed first (Fc block, anti-CD8α) in PBS + 0.1% BSA followed by fixation and permeabilization using BD Cytofix/Cytoperm fixation/permeabilization kit per manufacturer instructions and intracellular staining (anti-IFN-γ and/or anti-TNF-α) in perm/wash buffer. To assess the immunogenicity of protein or peptide antigens at stimulating OTI cells, pooled ACK-lysed splenocytes from OTI mice were cultured in RPMI-1640 + 10% FBS and varying concentrations of SIINFEKL-containing antigen for 24 hours; after culture, cells were surface labeled with anti-CD8α and anti-CD69. Organ processing: In studies where T cells from lymph nodes were assessed by flow cytometry (Figs. 3d–h and 4e), LNs were excised, mashed through 74 μm Netwell inserts (Corning), and washed into PBS + 0.1% BSA prior to antibody labeling. In studies where dendritic cells and/or other APCs were assessed by flow cytometry (Figs. 3d, 4c, 4e, and Supplementary Fig. 8e), LNs were excised, digested in RPMI-1640 + 0.8 mg/ml collagenase/disapse (Sigma) + 0.1 mg/ml DNase I (Sigma), mashed through 74 μm Netwell inserts, and washed into PBS + 0.1% BSA prior to antibody staining. In all cases, spleens were excised and mashed through 70 μm filters (Corning), lysed in ACK buffer, and washed into PBS + 0.1% BSA prior to antibody staining. To assess CD86 expression on DCs post-immunization, mice were s.c. injected with 25 μg cyclic-d-GMP (InvivoGen) and the spleen, inguinal LN, and mesenteric LN excised and processed 1, 4, or 7d later. Cells were analyzed using BD FACSCanto and BD FACS LSR Fortessa, and data were analyzed using FlowJo.
Antigenic stability analysis
Ex vivo stimulation method:
20 days in advance, mice were primed with E738–57 fusion protein mixed with CDNs, boosted 6 days in advance, and strong peripheral antigen-specific CD8+ T cell responses confirmed on day 0. On day of experiment, spleens from vaccinated animals were excised, mashed through 70 μm filters (Corning), lysed in ACK buffer, pooled, and plated in 96 well V-bottom plates (1 spleen per 30 wells). 24 hours before ex vivo stimulation, mouse serum from naïve animals was freshly collected in collection tubes with Z-Gel to remove clotting factors (Sarstedt) and used to prepare RPMI-1640 + 10% mouse serum media. 4 μM antigen solutions were prepared in RPMI-1640 + 10% mouse serum and incubated at 37°C. After a 24 hour incubation, fresh antigen was similarly prepared at 4 μM in RPMI-1640 + 10% mouse serum and both solutions were immediately diluted to a 25 nM solution with RPMI-1640 + 10% FBS and used to re-stimulate the aforementioned splenocytes from vaccinated animals. IFN-γ+CD8+ T cell responses were measured with ICS. ELISA method: fresh mouse plasma was prepared by bleeding directly into microcentrifuge tubes and spin removal of cellular matter, maintaining functional complement and clotting factor components. 3 μM FLAG-E738–57-His6 peptide and MSA-FLAG-E738–57-His6 were incubated in PBS + 20% mouse plasma for 4 hours at 37°C and immediately quenched by addition of 100X EDTA and 100X protease inhibitor cocktail (Thermo Fisher Scientific). ELISA protocol: MaxiSorp plates (Thermo Fisher Scientific) were coated with anti-FLAG M2 antibody (Sigma) at 10 μg/ml overnight, then blocked with PBS + 2% BSA + 5% non-fat milk + .01% Tween-20. Antigen in quenched reaction solution and freshly prepared antigen at equivalent concentration were diluted 200x in blocking buffer + EDTA + protease inhibitor and incubated on coated ELISA plate for 1.5 hours at room temperature. Detection was performed using an HRP-conjugated polyclonal rabbit anti-His6 antibody (Abcam) diluted 1:1000 in blocking buffer for 1.5 hours at 4°C and developed using TMB (Thermo Fisher Scientific) and sulfuric acid. Separate standards for peptide and protein analytes were prepared. Wash buffer consisted of PBS + 0.01% Tween-20, and at least 3x washes were performed between each incubation step.
Biodistribution studies
IVIS imaging
5-FAM-conjugated peptides (GenScript), FITC-conjugated dextrans of various molecular weights (Sigma), or 5/6-FAM-conjugated proteins, in saline or Montanide ISA 51 VG ST formulation (Seppic) were s.c. injected at the tail base of B6 mice; the inguinal LN was excised 8 hours later and imaged on an IVIS Spectrum Imaging System (Caliper Life Sciences), excitation: 500 nm, emission: 540 nm. In our analysis of Montanide formulation trafficking (Supplementary Fig. 5), the injection site of the animal was imaged as well. Images were analyzed on Living Image software. Following protein conjugation, the concentration of labeled protein was calculated using A493. Dosing: In Fig. 1f, 2.7 nmol of labeled MSA-E738–57 and up to 27 nmol of E738–57; in Fig. 4b, 2.5 nmol of MSA-E738–57 and TTR-E738–57; in Supplementary Fig. 5, 1.2 nmol of labeled MSA-E738–57 and up to 6 nmol of E738–57; in Supplementary Fig. 8f, 0.5 nmol of labeled MSA-E738–57 and DEC1-MSA-E738–57. Cellular biodistribution: Alexa Fluor 488-NHS Ester (Thermo Fisher Scientific) was conjugated to recombinantly expressed proteins and injected s.c. at the tail base of B6 mice. Following protein conjugation, the concentration of labeled protein was calculated using A495. The inguinal LN was excised 24 hours later and processed for single cell analysis (see flow cytometry section). Dosing: In Fig. 4c, 2.5 nmol of MSA-E738–57 and TTR-E738–57; in Supplementary Fig. 8e, 0.5 nmol of labeled MSA-E738–57 and DEC1-MSA-E738–57.
Detection of anti-MSA and anti-TTR antibodies
Titrating serum on an analyte-coated ELISA plate is an ineffective way to detect antibodies against serum proteins because of the potential for competition between serum in solution and the analyte on the coated plate surface. Instead, we first purified IgG from serum using Pierce protein A spin plates (Thermo Fisher Scientific), per manufacturer instructions, prior to detection in ELISA format. 10 μg/ml MSA (Alpha-Diagnostic International) or TTR (Aviva Systems Biology) were coated on MaxiSorp plates (Thermo Fisher Scientific) overnight, then blocked with PBS + 0.1% bovine IgG (Sigma) + 0.1% Tween-20 for anti-MSA antibody detection or PBS + 5% non-fat milk + 0.1% Tween-20 for anti-TTR antibody detection. IgG eluate from the spin plate was diluted 3x in blocking buffer, then added to the plate for a 1.5h incubation at room temperature. Goat anti-mouse HRP detection antibody (Bio-Rad) was used at a 1:5000 dilution in blocking buffer and incubated for 1h at 4°C. Controls included IgG isolates from Ova-vaccinated animals on 10 μg/ml Ova-coated wells, and MSA/TTR-coated wells assessed with chicken polyclonal anti-MSA/TTR antibodies (Abcam) and with goat anti-chicken HRP (Abcam) detection, followed by TMB and sulfuric acid development. Wash buffer consisted of PBS + 0.01% Tween-20, and at least 3x washes were performed between each incubation step.
Yeast surface display
Fibronectin domains were engineered to bind the DEC-205 ectodomain as previously described63. Briefly, the two outermost extracellular domains of DEC-205 (the N-terminal cysteine-rich and type II fibronectin domains) were produced from HEK cells and used to select for binders from the G4 library. Four rounds of magnetic enrichment were followed by seven rounds of flow cytometry-based sorting, with additional diversity introduced via error-prone PCR in between sorting rounds. After sorting and sequencing, 4 individual clones were displayed on yeast as and binding to soluble DEC-205 was measured with flow cytometry to calculate the Kd. DEC-1 was measured to have a Kd of 0.66 nM.
Adoptive transfer
CD8+ T cells from pmel Thy1.1+ spleens were isolated using the EasySep mouse CD8+ T Cell Isolation Kit (STEMCELL Technologies). To assess the kinetics of TCR engagement, we retro-orbitally i.v. transferred 500k pmel Thy1.1+ cells into animals that had previously been s.c. vaccinated with 3 μg peptide equivalence MSA-gp10020–39 APL or TTR-gp10020–39 APL mixed with 25 μg cyclic di-GMP (InvivoGen) 1, 4, or 7 days earlier. 24 hours after transfer, the spleen, inguinal LNs, and mesenteric LNs were excised and Thy1.1+ cells assessed for CD69 expression by flow cytometry (see flow cytometry section). Area under the curve calculations were performed using the trapezoidal method. To assess T cell phenotype in response to tolerization, CD8+ T cells from pmel Thy1.1+ mice isolated as above and labeled with carboxyfluorescein succinimidyl ester (CFSE) (Thermo Fisher Scientific) at a cell density of 10M/ml and a CFSE concentration of 5 μM for 20 min at 37°C in PBS + 0.1% BSA and subsequently quenched by addition of FBS. 1M CFSE-labeled pmel Thy1.1+ cells were retro-orbitally transferred into naïve animals in PBS. 24h later, recipient mice were i.v. vaccinated with 3 μg gp10020–39 APL peptide, MSA-gp10020–39, or PBS control. 72h post-immunization, the spleen, inguinal LNs, and mesenteric LNs were excised and Thy1.1+ cells assessed by flow cytometry (see flow cytometry section). Proliferation index = log2(FInd/MFIall) where MFIall = MFI of live Thy1.1+ pmel T cells and FInd = peak fluorescence intensity of viable non-divided Thy1.1+ pmel T cells.
Tumor studies
300k TC-1 or B16F10 cells were subcutaneously administered in 50 μl sterile PBS on the right flank of shaved WT B6 mice, except in Fig. 4h, where 1M TC-1 cells were implanted. Mice bearing TC-1 tumors were treated on days 5, 12, and 19 in Fig. 2d and on days 8 and 15 in Fig. 4h; mice bearing B16F10 tumors were vaccinated on days 4, 10, and 16, and/or treated with 200 μg anti-PD1 antibody (clone 29F.1A12, BioXCell) on days 4, 7, 10, 13, 16, and 19. Mice were randomized into groups prior to treatment initiation. Tumor size was measured by area (longest dimension × perpendicular dimension), and mice were euthanized when tumor area exceeded 100 mm2. Memory from E738–57 vaccinated animals was assessed by inoculating 300k TC-1 cells in mice 66 days post-boost and assessing survival as above.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software. The specifics of the statistical test performed, p-values, and number of replicates are stated in the figure legends. For all tests, the threshold for significance was P < 0.05.31
Supplementary Material
List of abbreviations
- ACK
ammonium chloride potassium
- ANOVA
analysis of variance
- APC
antigen presenting cell
- APL
altered peptide ligand
- AUC
area under the curve
- BSA
bovine serum albumin
- CDN
cyclic di-nucleotide
- CFSE
carboxyfluorescein succinimidyl ester
- dLN
draining lymph node
- DLS
dynamic light scattering
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- ELISPOT
enzyme-linked immune absorbent spot
- FITC
fluorescein isothiocyanate
- HEK
human embryonic kidney
- HMW
high molecular weight
- ICS
intracellular cytokine staining
- IFN
interferon
- IVIS
in vivo imaging system
- KLH
keyhole limpet hemocyanin
- MFI
mean fluorescence intensity
- MSA
mouse serum albumin
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate-buffered saline
- PK
pharmacokinetic
- SEC
size exclusion chromatography
- SEM
standard error of the mean
- STING
stimulator of interferon genes
- TLR
Toll-like receptor
- TMB
3,3’,5,5’-Tetramethylbenzidine
- TNF
tumor necrosis factor
- TTR
transthyretin
Footnotes
Data availability statement
No datasets were generated or analyzed during the current study.
References
- 1.Garon EB et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Larkin J et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu SS et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantarjian H et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med 376, 836–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andtbacka RHI et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol 33, 2780–2788 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kantoff PW et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med 363, 411–422 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Snyder A et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodi FS et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci 105, 3005–3010 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ott PA et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keskin DB et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLennan DN, Porter CJH & Charman SA Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol 2, 89–96 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Brinckerhoff LH et al. Terminal modifications inhibit proteolytic degradation of an immunogenic mart-127–35 peptide: Implications for peptide vaccines. Int. J. Cancer 83, 326–334 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Moynihan KD et al. Enhancement of peptide vaccine immunogenicity by increasing lymphatic drainage and boosting serum stability. Cancer Immunol. Res canimm.0607.2017 (2018) doi: 10.1158/2326-6066.CIR-17-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon JJ et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater 10, 243–251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A & Moon JJ Designer vaccine nanodiscs for personalized cancer immunotherapy Nat. Mater advance online publication, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nembrini C et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc. Natl. Acad. Sci 108, E989–E997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonifaz LC et al. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. J. Exp. Med 199, 815–824 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kretz-Rommel A et al. In Vivo Targeting of Antigens to Human Dendritic Cells Through DC-SIGN Elicits Stimulatory Immune Responses and Inhibits Tumor Growth in Grafted Mouse Models. J. Immunother 30, 715 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Johansen P et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur. J. Immunol 35, 568–574 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Jewell CM, Bustamante Lopez SC & Irvine DJ In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci 108, 15745–15750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Supersaxo A, Hein WR & Steffen H Effect of Molecular Weight on the Lymphatic Absorption of Water-Soluble Compounds Following Subcutaneous Administration. Pharm. Res 7, 167–169 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Feltkamp MCW et al. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol 23, 2242–2249 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Hailemichael Y et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat. Med 19, 465.-+ (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burdette DL et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guevara-Patiño JA et al. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J. Clin. Invest 116, 1382–1390 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Stipdonk MJB et al. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer Res. 69, 7784–7792 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Mennuni C et al. Efficient induction of T-cell responses to carcinoembryonic antigen by a heterologous prime-boost regimen using DNA and adenovirus vectors carrying a codon usage optimized cDNA. Int. J. Cancer 117, 444–455 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Han S, Asoyan A, Rabenstein H, Nakano N & Obst R Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc. Natl. Acad. Sci 107, 20453–20458 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsdell F & Fowlkes BJ Maintenance of in Vivo Tolerance by Persistence of Antigen. Science 257, 1130–1134 (1992). [DOI] [PubMed] [Google Scholar]
- 34.Ehl S et al. Antigen persistence and time of T-cell tolerization determine the efficacy of tolerization protocols for prevention of skin graft rejection. Nat. Med 4, 1015–1019 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Warren KG, Catz I, Ferenczi LZ & Krantz MJ Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur. J. Neurol 13, 887–895 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Bielekova B et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med 6, 1167–1175 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Hanson MC et al. Nanoparticulate STING agonists are potent lymph node–targeted vaccine adjuvants. J. Clin. Invest 125, 2532–2546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Churlaud G et al. Human and Mouse CD8+CD25+FOXP3+ Regulatory T Cells at Steady State and during Interleukin-2 Therapy. Front. Immunol 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu Y et al. Recent advances in CD8+ regulatory T�cell research (Review). Oncol. Lett (2018) doi: 10.3892/ol.2018.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai S, Clemente-Casares X & Santamaria P CD8+ Tregs in autoimmunity: learning “self”-control from experience. Cell. Mol. Life Sci 68, 3781–3795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ingenbleek Y & Young V Transthyretin (Prealbumin) in Health and Disease: Nutritional Implications. Annu. Rev. Nutr 14, 495–533 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Terje Andersen J, Bekele Daba M, Berntzen G, Michaelsen TE & Sandlie I Cross-species Binding Analyses of Mouse and Human Neonatal Fc Receptor Show Dramatic Differences in Immunoglobulin G and Albumin Binding. J. Biol. Chem 285, 4826–4836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCutchen SL, Colon W & Kelly JW Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry 32, 12119–12127 (1993). [DOI] [PubMed] [Google Scholar]
- 44.Prior IA, Lewis PD & Mattos C A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 72, 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waters AM & Der CJ KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med 8, a031435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bender S et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 24, 660–672 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ochs K et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. OncoImmunology 6, e1328340 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang QJ et al. Identification of T-cell Receptors Targeting KRAS-mutated Human Tumors. Cancer Immunol. Res 4, 204–214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chheda ZS et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med 215, 141–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zom GG et al. Efficient Induction of Antitumor Immunity by Synthetic Toll-like Receptor Ligand-Peptide Conjugates. Cancer Immunol. Res 2, 756–764 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Qiu F et al. Poly(propylacrylic acid)-peptide nanoplexes as a platform for enhancing the immunogenicity of neoantigen cancer vaccines. Biomaterials 182, 82–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng L et al. Heterosubtypic influenza protection elicited by double-layered polypeptide nanoparticles in mice. Proc. Natl. Acad. Sci 115, E7758–E7767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harris JR & Markl J Keyhole limpet hemocyanin (KLH): a biomedical review. Micron 30, 597–623 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Kim SK et al. Comparison of the effect of different immunological adjuvants on the antibody and T-cell response to immunization with MUC1-KLH and GD3-KLH conjugate cancer vaccines. Vaccine 18, 597–603 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Lynn GM et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol (2015) doi: 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu TY-H et al. Rational design of small molecules as vaccine adjuvants. Sci. Transl. Med 6, 263ra160–263ra160 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Duperret EK et al. A Synthetic DNA, Multi-Neoantigen Vaccine Drives Predominately MHC Class I CD8+ T-cell Responses, Impacting Tumor Challenge. Cancer Immunol. Res (2019) doi: 10.1158/2326-6066.CIR-18-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walters JN et al. A novel DNA vaccine platform enhances neo-antigen-like T-cell responses against WT1 to break tolerance and induce anti-tumor immunity. Mol. Ther 0, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geall AJ et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci 109, 14604–14609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horton HM et al. Potent In vitro and In vivo Activity of an Fc-Engineered Anti-CD19 Monoclonal Antibody against Lymphoma and Leukemia. Cancer Res. 68, 8049–8057 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Nomura LE, Walker JM & Maecker HT Optimization of whole blood antigen-specific cytokine assays for CD4+ T cells. Cytometry 40, 60–68 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Chen TF, de Picciotto S, Hackel BJ & Wittrup KD Engineering Fibronectin-Based Binding Proteins by Yeast Surface Display. in Methods in Enzymology vol. 523 303–326 (Elsevier, 2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.