

Abstract
Immunotherapy has become the mainstay for lung cancer treatment, providing sustained therapeutic responses and improved prognosis compared with those obtained with surgery, chemotherapy, radiotherapy, and targeted therapy. It has the potential for anti-tumor treatment and killing tumor cells by activating human immunity and has moved the targets of anti-cancer therapy from malignant tumor cells to immune cell subsets. Two kinds of immune checkpoints, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed death-1 (PD-1)/programmed death ligand 1 (PD-L1), are the main targets of current immunotherapy in lung cancer. Despite the successful outcomes achieved by immune checkpoint inhibitors, a small portion of lung cancer patients remain unresponsive to checkpoint immunotherapy or may ultimately become resistant to these agents as a result of the complex immune modulatory network in the tumor microenvironment. Therefore, it is imperative to exploit novel immunotherapy targets to further expand the proportion of patients benefiting from immunotherapy. This review summarizes the molecular features, biological function, and clinical significance of several novel checkpoints that have important roles in lung cancer immune responses beyond the CTLA-4 and PD-1/PD-L1 axes, including the markers of co-inhibitory and co-stimulatory T lymphocyte pathways and inhibitory markers of macrophages and natural killer cells.
Keywords: Lung cancer, Immunotherapy, Targets
Introduction
Lung cancer still has the highest morbidity and mortality in all types of cancer worldwide.[1] Monoclonal antibodies (mAbs), such as nivolumab, pembrolizumab, atezolizumab, and durvalumab, which are targeted to key immune checkpoint proteins of programmed death-1 (PD-1) and programmed death ligand 1 (PD-L1), have been approved by the US Food and Drug Administration as second-line or first-line therapy for patients with non-small cell lung cancer (NSCLC).[2] However, despite the recent success of immunotherapy against a variety of cancer types, it was estimated that only 12.46% of patients with advanced or metastatic tumors respond to these immune checkpoint inhibitors.[3] Clinical trial data have revealed that the responsive proportion of NSCLC patients to this treatment is between 15% and 25%,[2] and thus a proportion of lung cancer patients still cannot benefit from current immunotherapy. Various approaches have been implemented to maximize the therapeutic effect of immune checkpoint inhibitor therapy; these include the identification of predictive biomarkers for patient stratification based on responsiveness. PD-1/PD-L1 antibodies have shown encouraging results in combination with chemotherapy or radiotherapy in several phase III trials.[4] Despite this, many patients inevitably become resistant or acquire resistance to these treatments, and this may be attributed to the complex and dynamic nature of the tumor immune microenvironment.[2] Therefore, there is an increasing need to identify alternative immune checkpoints for anti-cancer treatment. The emerging targets include not only markers of co-inhibitory and co-stimulatory T lymphocyte pathways [Figure 1], but also inhibitory markers of macrophages and natural killer (NK) cells [Figure 2].
Figure 1.
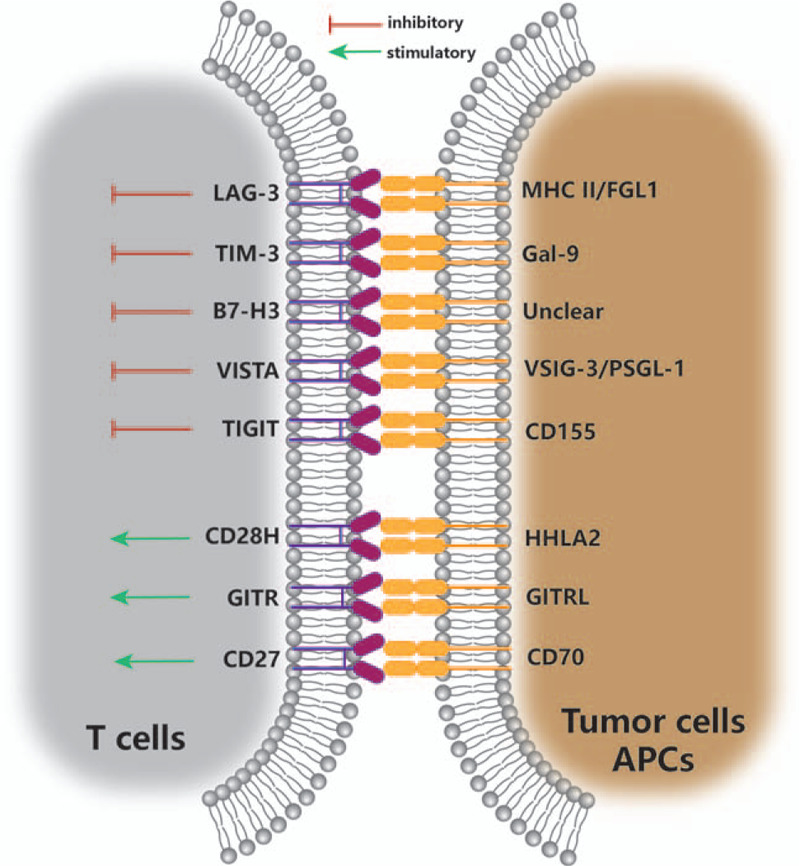
Emerging checkpoints of T lymphocytes in lung cancer immunotherapy. APC: Antigen presenting cell; B7-H3: B7 homolog 3; CD: Cluster of differentiation; CD28H: CD28 homolog; FGL1: Fibrinogen-like protein 1; Gal-9: Galectin-9; GITR: Glucocorticoid-induced tumor necrosis factor receptor (TNFR)-related receptor; GITRL: GITR ligand; HHLA2: Human endogenous retrovirus-H long terminal repeat-associating protein 2; LAG-3: Lymphocyte activation gene-3; MHC II: Major histocompatibility class II; PSGL-1: P-selectin glycoprotein ligand-1; TIGIT: T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain; TIM-3: T-cell immunoglobulin- and-mucin-domain-containing molecule 3; VISTA: V-domain immunoglobulin suppressor of T cell activation; VSIG-3: V-Set and immunoglobulin domain containing-3.
Figure 2.
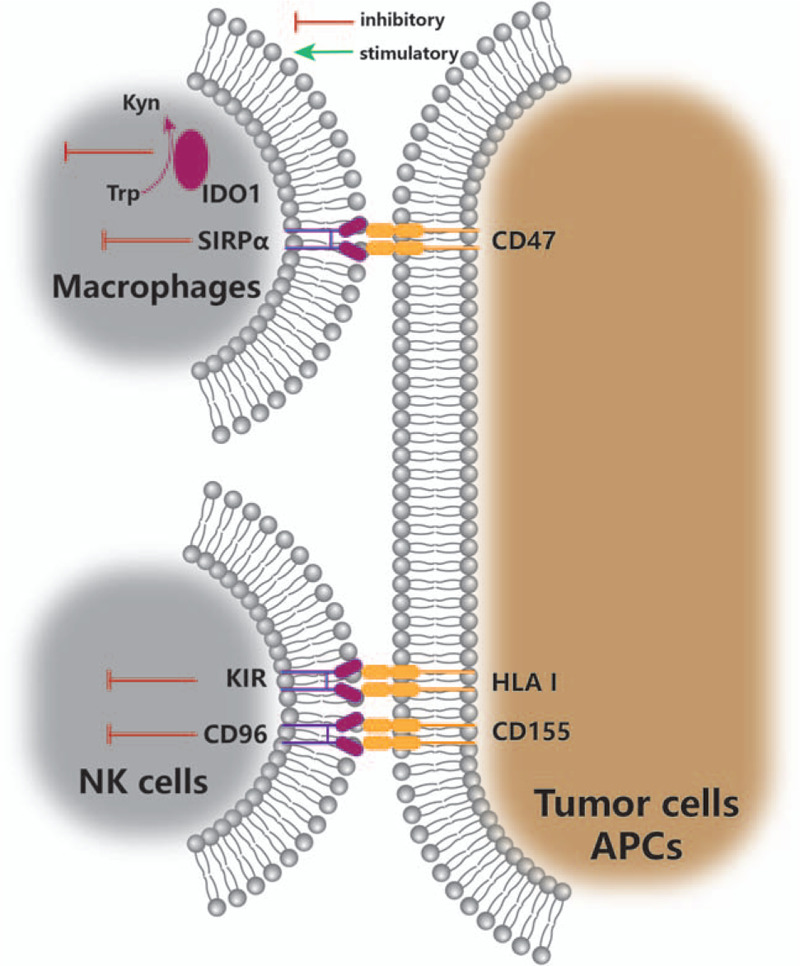
Emerging checkpoints of macrophages and NK cells in lung cancer immunotherapy. APC: Antigen presenting cell; CD: Cluster of differentiation; HLA I: Human leukocyte antigen class I; IDO1: Indoleamine 2,3-dioxygenase 1; KIR: Killer immunoglobulin-like receptor; Kyn: Kynurenine; NK: Natural killer; SIRPα: Signal regulatory protein α; Trp: Tryptophan.
Co-inhibitory T Lymphocyte Checkpoints
Lymphocyte activation gene-3 (LAG-3)
LAG-3 (CD223) was first discovered as a membrane protein in activated NK cells and T lymphocytes and the gene is located close to the CD4 gene on chromosome 12.[5] LAG-3 is also present on the surface of dendritic cells (DCs), B cells, tumor-infiltrating lymphocytes (TILs), and regulatory T cells (Tregs).[6] LAG-3 functions as a negative regulator in the stimulation of CD4+ T lymphocytes and is essential for the suppressive function of Tregs according to early studies.[7,8] LAG-3 protein can bind to major histocompatibility (MHC) class II with high affinity due to its highly structural homology with CD4, in spite of sharing only approximately 20% amino-acid sequence homology.[5,9] LAG-3 selectively recognizes stable complexes of peptide and MHC class II (pMHC II) and suppresses the reaction of T cells to pMHC II through its intracellular region rather than a direct interference with the interaction between CD4/T cell receptor (TCR) and MHC class II.[10] In addition, a novel ligand of LAG-3, fibrinogen-like protein 1 (FGL1), can inhibit T-cell activation independent from MHC class II. FGL1 is secreted from healthy hepatocytes and its production is elevated in tumor cells.[11] LAG-3 also inhibits CD4+ T cell expansion by suppressing metabolism and mitochondrial biogenesis.[12]
In a recent cohort of 139 surgically resected tumor specimens from NSCLC patients, the LAG-3 protein was found to be associated with poor prognosis and its expression is correlated with PD-1/PD-L1 expression.[13] However, LAG-3 was found to be expressed on TILs in tumor tissues of some patients in a study including 553 primary NSCLC tumors as well as 143 metastatic lymph nodes, and LAG-3+ TILs were related to improved survival.[14] The differences between the two studies reflect the complexity of LAG-3 function, indicating further research is required. A pre-clinical study using a novel LAG-3 antibody, LBL-007, blocked the binding of LAG-3 to MHC class II molecules and downstream signaling of LAG-3. This significantly controlled tumor growth and was considerably more effective in combination with a PD-1 antibody.[15] Another LAG-3 antibody, TSR-033, has also demonstrated its potent therapeutic efficacy in boosting anti-PD-1 therapy.[16]
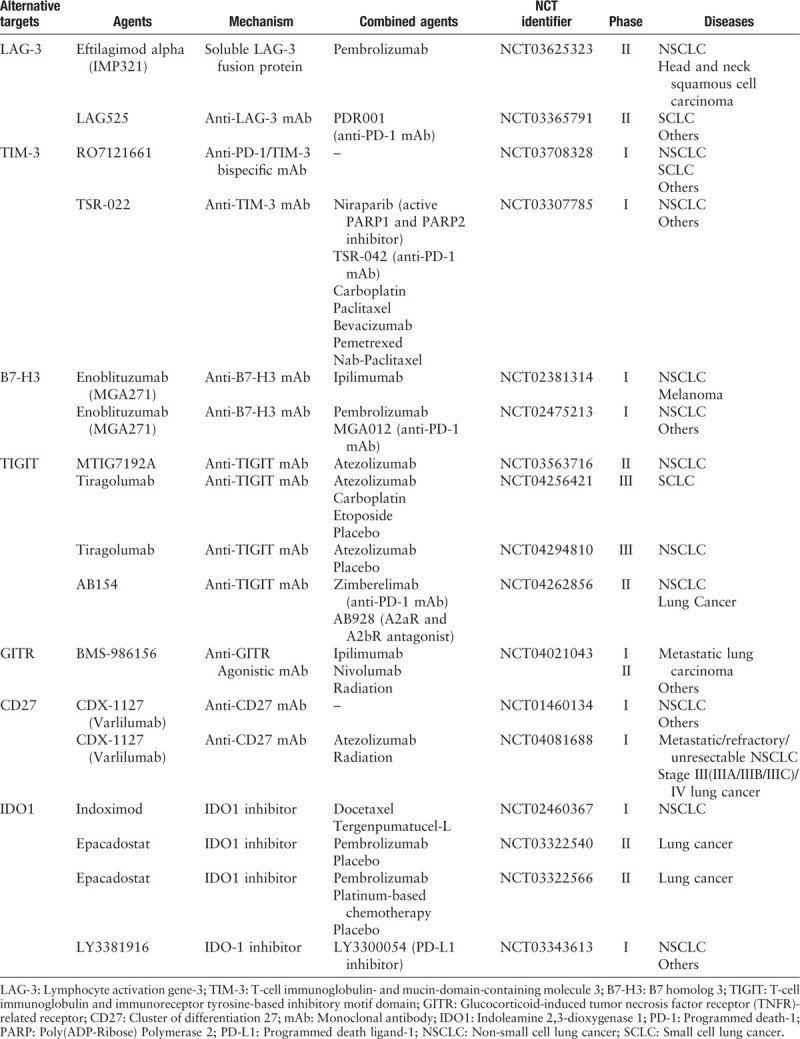
Currently, a soluble LAG-3 fusion protein IMP321, and several LAG-3 mAbs, such as BMS-986016, LAG525, MK-4280, REGN3767, and TSR-033 are being tested in clinical trials. IMP321 demonstrated favorable tolerability in completed early phase I clinical trials on different types of tumors with promising outcomes in tumor immune microenvironment, and clinical benefits were observed in breast carcinoma.[17] Currently, there are two phase II clinical trials that are recruiting eligible NSCLC and small cell lung cancer (SCLC) patients, separately, to test the combination of IMP321/LAG525 and anti-PD-1 antibody [Table 1].
Table 1.
Summary of clinical trials of emerging immunotherapy targets in lung cancer.
T-cell immunoglobulin- and mucin-domain-containing molecule 3 (TIM-3)
TIM-3 works as an inhibitory receptor suppressing the function of effector T helper 1 (Th1) cells and cytotoxic T-lymphocytes (CTLs) and has a pivotal role in maintaining the tumor immunosuppressive microenvironment.[18] The TIM gene family (TIM-1, TIM-3, and TIM-4 in humans) is located on chromosome 5q33.2 and all members are associated with immune-related diseases.[19] The interaction between TIM-3 and its alternative ligand C-type lectin galectin-9 (Gal-9) causes TIM-3-dependent Th1 cell death by inducing an intracellular calcium influx and suppression of Th1 cell responses.[20] In addition to being highly expressed on CD4+ and CD8+ TILs in tumor tissues, TIM-3 has also been identified on the surface of NK cells, DCs, macrophages, Tregs, and even tumor cells.[18,21,22]
TIM-3 has been suggested to be highly expressed on CD4+ T cells in NSCLC patients with lymph node metastasis and advanced tumor stages.[21] Similarly, increased expression of TIM-3 on NK cells in circulating blood was also correlated with several high risk clinicopathological parameters and independently demonstrated a shorter overall survival of lung adenocarcinoma patients.[22] Tumor cells release more exosomes than physiological conditions, and they mirror the molecular features of their parental cells. Gao et al[23] discovered that upregulated TIM-3 and Gal-9 in exosomes isolated from peripheral blood may be potential biomarkers correlated to poor clinicopathological parameters in NSCLC, similar to expression of TIM-3 on T cells and NK cells. In another study, the TIM-3 positive group revealed poorer survival performance, and approximately half of the resected lung adenocarcinoma samples demonstrated a strong association of TIM-3 expression with PD-1 expression.[24] Co-expression of both TIM-3 and PD-1 on TILs resulted in more exhausted immune cells than expressing of either TIM-3 or PD-1 alone in mouse models, indicating that a complex cross-regulation may exist between TIM-3 and PD-1 pathways in the immunosuppressive environment.[25] In addition, a TIM-3 blockade may play a much more important role in converting the immunosuppressive environment into an immunoactive state, as TIM-3 is significantly expressed on the surface of Tregs and is crucial to the tumor immune microenvironment.[21] Therefore, combined targeting of both the two pathways could be a promising strategy for anti-tumor therapy. Additionally, up-regulation of TIM-3 observed in the progress of anti-PD-1 therapy indicated that blockading TIM-3 following acquired resistance to present immunotherapy would be an effective therapeutic solution.[26] Most importantly, TIM-3 is selectively expressed on T cells at the tumor site, while the expression of TIM-3 on T cells in circulating blood is negligible.[21] This means that TIM-3 targeting therapy may avoid the immune-related adverse events (irAEs) commonly observed in patients treated with PD-1 antibodies and CTL-associated antigen 4 (CTLA-4) antibodies.
A TIM-3 antibody, TSR-022, and a bispecific antibody, RO7121661, are currently undergoing safety and tolerability evaluation in a phase I study [Table 1].
B7 homolog 3 (B7-H3)
B7-H3 protein is a member of the B7 family, also known as CD276. B7-H3 was initially discovered in 2001 as a co-stimulator of both CD4+ and CD8+ T cells, enhancing the proliferation of cytotoxic T cells and stimulating interferon gamma (IFN-γ) production in activated T cells.[27] However, a later study demonstrated that the B7-H3-mediated inhibition was overwhelmed by the stimulatory effect of CD28. B7-H3 has been shown to play an inhibitory role on the activities of CD4+ and CD8+ T cells and reduces the production of interleukin (IL)-2 and IFN-γ.[28] B7-H3 is constitutively expressed in many normal tissues and overexpressed in various types of tumors.[29] Tumor-associated macrophages could promote the expression of membrane B7-H3 on tumor cells, and up-regulated B7-H3 could then induce DCs to an immunosuppressive phenotype in lung cancer.[30,31]
B7-H3 is expressed in 70% to 80% of NSCLC cases.[32,33] High expression of this protein is correlated with smoking history and a shortened overall survival in patients.[32] Soluble B7-H3 in malignant pleural effusions is a potential biomarker for its correlation with the stage of NSCLC.[34] In addition, B7-H3 is associated with refractoriness to PD-1 antibodies and effective anti-tumor activity by increasing CD8+ T cell infiltration, which was shown in the dual blockade of B7-H3 and PD-1.[33] Another study also reported that inhibition of B7-H3 could enhance the sensitivity to cisplatin chemotherapy.[35] Higher B7-H3 expression on tumors in human immunodeficiency virus (HIV)-infected lung cancer patients as compared with uninfected patients may provide clues for the treatment of HIV-infected lung cancer patients.[36] It is reported that B7-H3 could regulate lipid metabolism by hijacking sterol regulatory element-binding protein 1/fatty acid synthase signaling in lung cancer, indicating a possible mechanism of B7-H3 in tumor development.[37] Interestingly, the combination of a B7-H3-targeting agent and a PD-1 antibody remarkably suppressed the growth and metastases of tumors as B7-H3 is not only overexpressed on tumor cells and on vascular endothelial cells in tumor tissues but finitely expressed in normal tissue vessels.[38] This may enable it to distinguish tumor vessels from physiological vessels.
Two phase I clinical trials are ongoing for the safety of enoblituzumab (MGA271), a B7-H3 mAb, in combination with antibodies to either PD-1 or CTLA-4, in NSCLC patients [Table 1].
V-domain immunoglobulin (Ig) suppressor of T cell activation (VISTA)
VISTA is a novel B7 family member, mainly expressed in hematopoietic cells and overexpressed in myeloid antigen-presenting cells (APCs) and T cells. VISTA is also known as PD-1 homology and its Ig-V domain is highly homologous to PD-L1.[39] VISTA plays an important role in regulating immune responses acting both as a receptor and a ligand.[40] The engagement of VISTA and its recently identified ligand, V-Set and immunoglobulin domain containing-3 suppresses proliferation of activated T cells and further reduces the production of several cytokines and chemokines.[41] Additionally, as a ligand, VISTA inhibits T cells by interacting with receptor P-selectin glycoprotein ligand-1 preferentially in acidic environments usually found in tumor beds.[42] Furthermore, the expression of VISTA on Tregs is crucial to maintain the Treg pool size by promoting Treg differentiation and stabilization.[43]
VISTA was detected in the majority of tumor samples from 758 NSCLC cases in different stages and displayed differential distribution in stromal and tumor cells. Predominately, these cases exhibited expression of VISTA on stromal cells, and only one fifth of the cases demonstrated expression in both stromal and tumor cells.[44] Although VISTA preferentially inhibits CD4+ T cells-mediated immune responses,[45] it is more highly expressed on CD8+ T cells than CD4+ helper T lymphocytes, and the expression of VISTA is generally correlated with CD8+ T cell infiltration rather than CD4+ T cell infiltration.[44] This may be related to its main inhibitory effects on CD4+ T cell in early stage and down-regulation on activated CD4+ T cells in vivo, which is a unique regulatory pattern absent from other inhibitory B7 family members.[39] Interestingly, VISTA is found to be reduced by IL-10 and IFN-γ, in a similar fashion to PD-L1.[46] The co-expression of VISTA, PD-1, and PD-L1 potentially contributes to a cooperative tumor immune evasion procedure and indicates a promising immunotherapy strategy by simultaneously blocking both the VISTA and PD-1/PD-L1 pathways.[44] Additionally, the limited association between VISTA levels and tumor mutational burden is also consistent with PD-L1 expression.[44,47] Most importantly, the expression of VISTA predicted a positive outcome with a longer 5-year survival rate.[44]
The increased expression of VISTA after ipilimumab (CTLA-4 antibody) therapy in prostate cancer suggests that VISTA may represent a significant potential mechanism of acquired resistance to immune checkpoint inhibitor therapy. This response, and the heightened infiltration of VISTA positive lymphocytes noted after anti-PD-1 therapy in melanoma, indicates that VISTA may be an important immunotherapy target following these therapies.[48,49] However, this possible role of VISTA in lung cancer requires further exploration.
T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT)
TIGIT and CD96 are both members of immunoglobulin superfamily. These two inhibitory receptors bind to CD155 and share the ligand with the CD226 co-stimulatory receptor. Both TIGIT and CD96 have a higher affinity for CD155 than CD226, which is analogous to the relationship between CTLA-4 and CD28 interacting with B7 ligands.[50] Moreover, research shows that TIGIT could influence CD226 homodimerization and disturb its stimulation signaling.[51] TIGIT and CD96 are expressed on the surface of effector T cells and NK cells, and TIGIT is also expressed on Tregs.[50]
TIGIT was identified on the surface of 58% of CD8+ TILs in NSCLC, and the majority of these cells also have high levels of PD-1 expression, although the proportion of CD4+ TILs that express both TIGIT and PD-1 is minor in comparison. Elevated TIGIT expression on CD8+ and CD4+ T cells is also present in peripheral blood.[51] Blockade of TIGIT has been shown to enhance therapy with PD-L1 antibody and the expression of TIGIT is associated with the exhaustion of NK cells. Blockading TIGIT could avoid this situation and restore anti-tumor immunity through evoking T cell immunocompetence in an NK cell-dependent pattern.[52] Additionally, TIGIT directs Tregs to a highly suppressive phenotype and marks the most dysfunctional CD8+ T cell subset. However, the anti-tumor immune milieu suppressed by TIGIT relies primarily on the expression on Tregs rather than on CD8+ T cells.[53] This study also demonstrated a synergistic effect of TIGIT and TIM-3 indicated by the co-expression of the two receptors in tumor tissues.
MTIG7192A, AB154, and tiragolumab are three experimental anti-TIGIT antibodies. A number of studies are currently evaluating them, including a phase II study of MTIG7192A combined with atezolizumab in chemotherapy-naïve patients with locally advanced or metastatic NSCLC, a phase II study of AB154 in lung cancer patients, and two phase III studies of tiragolumab combined with atezolizumab in participants with SCLC or NSCLC [Table 1].
Co-stimulatory T Lymphocyte Checkpoints
Human endogenous retrovirus-H long terminal repeat-associating protein 2 (HHLA2)
HHLA2 is also a member of B7 family, and it is only found in higher primates.[54] The most studied receptor of HHLA2 is transmembrane and immunoglobulin domain containing 2 (TMIGD2), also known as CD28 homolog (CD28H) or immunoglobulin-containing and proline-rich receptor-1.[55] HHLA2 is constitutively expressed on APCs, whereas CD28H is primarily expressed on endothelial cells and all naive T cells; the HHLA2 gene of human is located in chromosome 3 q13.13 close to the B7-1 and B7-2 genes.[54,56] The interaction of HHLA2 and CD28H was reported to co-stimulate T cell growth and cytokine production in a protein kinase B (AKT)-dependent pattern.[56] However, other studies have demonstrated that HHLA2 and CD28H binding is co-inhibitory in the company of TCR signaling.[54,57]
HHLA2 was detected in approximately 65% of NSCLC tumor tissues and in a greater proportion in PD-L1 negative cases, but was not expressed in the majority of healthy lung tissue.[58,59] The expression of HHLA2 is significantly correlated with high TILs and epidermal growth factor receptor mutation in lung adenocarcinoma.[58] Contradictorily, HHLA2 has greater potency in inhibition of T cell proliferation than PD-L1 and may mediate immune evasion in PD-L1 negative environment.[59] A comprehensive analysis to evaluate the prognostic significance of HHLA2 in human pan-cancer suggested that HHLA2 is an independent predictive factor for prognosis of many human tumors.[60] Unfortunately, this comprehensive study did not indicate the prognostic value in lung cancer. In an independent study on NSCLC, the patients with high HHLA2 expression tended to have a poorer survival, but this was not statistically significant.[58]
HHLA2 is emerging as a potential therapeutic target for lung cancer as it also has an angiogenesis effect via an interaction with TMIGD2 on endothelium cells.[55] However, due to a lack of understanding of the precise mechanism of HHLA2-induced tumor immune suppression, especially the contradictory phenomenon on the co-inhibitory and co-stimulatory effect between HHLA2 and its ligand, more progress is required before clinical application of HHLA2 immunotherapy.
Tumor necrosis factor receptor superfamily (TNFRSF)
Members of TNFRSF, such as CD27, glucocorticoid-induced tumor necrosis factor receptor-related receptor (GITR), OX-40 (CD134), and 4-1BB (CD137), can promote immune responses by stimulating conventional T cells and APCs, suppressing Tregs, and mediating communication between CD4+ and CD8+ T cells,[61,62] making them attractive targets for cancer immunotherapy.
GITR is a type I transmembrane receptor expressed on both immune cells and human tissues. Among the two ligands of GITR, the GITR ligand (GITRL) and secreted and transmembrane protein 1A (SECTM1A), the GITR/GITRL signaling plays a critical role in immune modulation.[62] An in vitro study has shown that an SCLC cell line expressing GITR was induced to cell death through increased apoptosis inducing factor production in the presence of mesenchymal stromal cells expressing GITRL.[63] GITR-agonistic antibody promoted tumor cell apoptosis and increased the activity of NK cells, T lymphocytes, and APCs, accompanied with decreased levels of pro-angiogenesis chemokines and increased levels of anti-angiogenesis chemokines in lung cancer mice.[64] Interestingly, a completed phase I trial of GITR-agonistic antibody TRX518 (NCT01239134) in human advanced tumors found that peripheral Tregs are a reliable biomarker in GITR agonist therapy and were reduced to the same extent with Tregs at the tumor site. Patients were reported to have decreased Treg numbers and elevated effector T cell to Treg (Teff:Treg) ratios, but failed to show obvious clinical benefit from the GITR agonist monotherapy, indicating that stimulated GITR alone is unable to restore the anti-tumor immunity sufficiently due to excessive exhaustion of effector T cells.[65] Similarly, cell co-culture in vitro also revealed that even under co-stimulation of GITR or CD27, the activation of T cells was inhibited by PD-1 effectively.[66] These results demonstrated that the combination of blockading PD-1 and triggering TNFRSF molecules is a rational therapeutic strategy of tumor treatment. Indeed, preclinical study of NSCLC mouse models revealed that a GITR agonist combined with anti-PD-1 and radiation therapy improved the survival significantly and produced tumor-free status in half the mice tested.[67] Diminished Tregs in the tumor tissue also indicated that GITR agonist treatment may help overcome Treg immunosuppression induced by radiation therapy. A phase I/II trial has been launched to evaluate the safety and efficiency of BMS-986156 (agonistic anti-GITR mAb) combined with PD-1/CTLA-4 blockade in metastatic lung carcinoma and other tumors [Table 1].
CD27 is constitutively expressed on naïve and activated CD4+ and CD8+ T cells, and the expression of its ligand CD70 is induced on macrophages, DCs, and B cells. An agonistic mAb of CD27, varlilumab, showed both clinical and biologic activity with favorable safety and tolerance in melanoma and renal cell carcinoma patients.[68] The anti-tumor efficacy of CD27 targeted therapy is attributed to both stimulation of effector T cells and depletion of Tregs.[69] Chemokines and IFN-γ released by activated T cells further stimulated macrophages and increased myeloid infiltration.[70] However, persistent CD27-CD70 co-stimulatory signaling can directly contribute to T cell exhaustion and lethal immunodeficiency.[71] In addition, recent results reported that high levels of soluble CD27 predicted a poor prognosis in NSCLC, and may be supporting an immune exhaustion state in these patients.[72] Therefore, it is necessary to vary the dosage of CD27 agonists to achieve the optimal therapeutic effect in treatment. Two phase I trials are underway in lung cancer patients to evaluate anti-CD27 antibody alone or in combination with atezolizumab and radiation therapy [Table 1].
Co-inhibitory Macrophage Checkpoints
CD47 and signal regulatory protein α (SIRPα)
CD47 is widely known as the “don’t eat me” signal and is expressed on the surface of all cells in the human body while its receptor, SIRPα, is expressed restrictively on all myeloid cells and also on neuronal cells in the central nervous system. Binding of CD47 to SIRPα restricts phagocytic and cytotoxic function of SIRPα-expressing cells to CD47 positive healthy and malignant cells.[73] Interestingly, CD47 is also a receptor for thrombospondin-1, which is a negative regulator of angiogenesis expressed on tumor stromal endothelial cells, and treatment with antibody against CD47 could lead to tumor progression by enhancing angiogenesis.[74]
The overexpression of CD47 is detected in NSCLC cell lines and is associated with tumor stages, and distant lymph node metastases. The downstream molecule cell division cycle 42 plays a critical role in prompting tumor invasion and metastasis.[75] CD47 is highly expressed on the membrane of human SCLC cells and blockade with CD47 antibody or dysfunction of the CD47 gene effectively suppressed SCLC tumor growth by inducing phagocytosis of macrophages.[76] Moreover, analysis of CD47 expression in NSCLC patients indicated that high levels of CD47 were correlated with a shorter overall survival, delayed apoptosis or necrosis, and impaired macrophage-mediated clearance of CD47 expressing neutrophils. The latter may contribute to neutrophilia in NSCLC, which is a negative predictor for patients.[77] A CD47-targeting fusion protein SIRPα-Fc was reported to activate macrophages in NSCLC xenograft mice, probably mediated by inactivation of mammalian target of rapamycin and reactive oxygen species, and was enhanced by simultaneously targeting autophagy.[78] Caspase-3/7 were poorly activated in NSCLC,[77] while caspase-3 was activated in CD47-targeting therapy, indicating an acceleration of apoptosis.[78] An anti-CD47 small interfering RNA (siRNA) was also shown to be effective in inhibiting tumor invasion and metastasis in NSCLC cell lines.[75] In addition, Wu et al[79] explored a novel strategy to deliver anti-CD47 siRNA to kill lung cancer cells precisely without adverse effects on CD47-expressing platelets and red blood cells, exploiting the excessive glutamine demand of tumor cells. In summary, the CD47/SIRPα axis is a potential immunotherapeutic target for both NSCLC and SCLC.
There are presently two types of CD47-targeting agents being evaluated in clinical trials, soluble recombinant fusion protein TTI-621 (SIRPα- immunoglobulin G1 [IgG1] Fc) and CD47 antibodies, such as Hu5F9-G4 (magrolimab), SRF231, AO-176, CC-90002, ZL-1201, IBI188, and SGN-CD47M.
Indoleamine 2,3-dioxygenase 1 (IDO1)
IDO1 is an enzyme expressed in DCs, endothelial cells, and tumor cells. The IDO1 pathway has an important role in immunosuppression through the metabolism of tryptophan (trp) to kynurenine (kyn). The decreased amino acid level results in the activation of the general control nonderepressible 2 kinase that can inhibit T cell proliferation and promote the differentiation from naïve CD4+ T cells to Tregs. In addition, the binding of IDO1 to aryl hydrocarbon receptor also results in the generation of immunosuppressive DCs and macrophages, as well as Tregs, which jointly foster a tumor immune microenvironment tolerant to tumor cells.[80]
In a study of lung adenocarcinoma, analysis of IDO1 and PD-L1 expression on resected tumor specimens found that co-expression of IDO1 and PD-L1 was associated with a poor prognosis of patients.[81] Similar results were obtained from lung squamous cell carcinoma.[82] The frequent expression of IDO1 and its significant co-expression with PD-L1 in NSCLC indicated that IDO1 and PD-1/PD-L1 may be promising targets of combined immunotherapy. Furthermore, IDO1 activity, expressed by the trp/kyn ratio, is correlated with responsiveness of anti-PD-1 treatment. Resistance to anti-PD-1 agents could be predicted by a high trp/kyn ratio.[83] These results may optimize the treatment of NSCLC patients by assessing their trp/kyn ratio. In another study, IDO1 inhibition also shows a bright future in lung cancer patients non-responsive to anti-PD-1 therapy, as an IDO1 inhibitor restricted tumor growth and metastases by reducing myeloid derived suppressor cells in anti-PD-1-resistant lung cancer model.[84] Additionally, IDO1 activity, as measured by the trp/kyn ratio in the serum, was found to be influenced by radiotherapy. IDO1 was also associated with reduced overall survival of these NSCLC patients and may be a meaningful biomarker guiding personalized radiotherapy.[85]
Among the three IDO1 inhibitors (epacadostat, LY3381916, and indoximod) under investigation in current clinical trials for lung cancer patients [Table 1, epacadostat is the most studied and a completed phase I study has reported that this agent was well tolerated in previously treated stage IIIB/IV NSCLC patients, with limited clinical effect.[86] In addition, an IDO1 peptide vaccine was observed with durable clinical responses in stage III-IV NSCLC patients.[87]
Co-inhibitory NK Cell Checkpoints
Al Omar SY et al[88] reported that NK cells could be stimulated to increase anti-tumor immunity and to reduce tumor growth, and impressively, activated NK cells further recruited T cells to exhibit high immunological functioning inside tumors, indicating NK cells as an promising target in cancer immunotherapy.
The killer immunoglobulin-like receptor (KIR) family are encoded by a cluster of genes on chromosome 19 and have both co-inhibitory and co-stimulatory effects on NK cells and a portion of T cells.[89] Interaction between inhibitory members of the KIR family and human leukocyte antigen (HLA) class I molecules plays an indispensable role in educating NK cells to program NK responsiveness, similar to Ly49 (KIR in mice)-binding MHC class I in mice.[90] A signature feature of educated NK cells is that they respond effectively to HLA class I negative targets, which is central to the maintenance of self-tolerance. NK cells highly expressed KIR CD158b in NSCLC patients and this suppressed NK cell-mediated cytotoxicity against tumor cells expressing HLA class I.[88] The expression of KIR 2D (L1, L3, L4, and S4) and KIR 3DL1 was associated with poor prognosis of NSCLC patients.[91] The significant co-expression of inhibitory KIR and PD-1 in tumor cells and TILs indicated that simultaneous blockade of KIR and PD-1/PD-L1 pathways could make a breakthrough in NSCLC immunotherapy.[92]
CD96 is another inhibitory marker of NK cell. Although TIGIT and CD96 are both expressed on the surface of effector T cells and NK cells, they play a major role in T cells and NK cells, respectively.[50] Blake et al[93] reports that CD96 blockade could inhibit lung metastases, which is dependent on NK cells, CD226, and IFN-γ, instead of T cells. This could be enhanced by combining anti-PD-1, anti-CTLA-4, or chemotherapy. Tumor metastasis has always been the main cause of terminal pain and death in cancer patients. CD96 mAb may provide a promising immunotherapy target to reduce tumor metastases.
Conclusions
Recent years have seen the successful application of the immune checkpoint inhibitors in anti-tumor treatment. However, T cell-targeted therapies are effective against lung cancer only in a fraction of patients, highlighting a need for supplementary approaches. Combined therapy with chemotherapy or radiotherapy and stratification of patients into responder classification have also further expanded the proportion of clinical benefited patients. However, the resistance to checkpoint immunotherapy prevents more patients from benefiting from the current immunotherapy. In addition, acquisition of resistance in the treatment progress hinders a more durable clinical response in patients. This resistance to immunotherapy, therefore, seriously challenges both clinicians and patients. It has been gradually realized that the inhibitory and stimulatory pathways together form a complex immunoregulatory network that shapes and maintains the suppressive immune homeostasis of the tumor microenvironment. Moreover, there is a continuous remodeling progress in this microenvironment during tumor progression and acquisition of resistance. Therefore, well-designed combination strategies could be a feasible approach to yield a synergistic effect in refractory cancer patients. There is no doubt that a comprehensive understanding of pre-treatment and post-treatment tumor microenvironment could help uncover the mechanism of the immunoregulatory network at the tumor site. In conclusion, a new generation of immune modulators could be exploited to increase response rates of current immunotherapeutic agents and achieve more effective responses to the refractory cancers, including lung cancer.
However, the anti-tumor immune response is just like proper flow of a pipeline. Restoring flow of the pipeline (effective anti-tumor immune response) by unblocking specific blockage (immune impairment) rather than increasing flow or pressure at the risk of pipe rupture (adverse effects) may be a more attractive strategy in cancer immunotherapy.[94] Apparently, in the last decade, “immune normalization” strategies, such as anti-PD-1/PD-L1/CTLA-4 therapy, have achieved higher objective response rates in patients with much fewer irAEs, compared with rare objective responses and frequent irAEs encountered in “immune enhancement” strategies. And there are more trials evaluating the inhibitory checkpoints for clinical applications.
Funding
This work was supported by a grant from the WU JIEPING Medical Foundation (No. 320.6750.19071).
Conflicts of interest
None.
Footnotes
How to cite this article: Zhu HH, Feng Y, Hu XS. Emerging immunotherapy targets in lung cancer. Chin Med J 2020;20:2456–2465. doi: 10.1097/CM9.0000000000001082
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Pu X, Wu L, Su D, Mao W, Fang B. Immunotherapy for non-small cell lung cancers: biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer 2018; 18:1082.doi: 10.1186/s12885-018-4990-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open 2019; 2:e192535.doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, et al. Advances in cancer immunotherapy 2019 - latest trends. J Exp Clin Cancer Res 2019; 38:268.doi: 10.1186/s13046-019-1266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med 1990; 171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamura T, Fujio K, Sumitomo S, Yamamoto K. Roles of LAG3 and EGR2 in regulatory T cells. Ann Rheum Dis 2012; 71: Suppl 2: i96–100. doi: 10.1136/annrheumdis-2011-200588. [DOI] [PubMed] [Google Scholar]
- 7.Huard B, Tournier M, Hercend T, Triebel F, Faure F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur J Immunol 1994; 24:3216–3221. doi: 10.1002/eji.1830241246. [DOI] [PubMed] [Google Scholar]
- 8.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity 2004; 21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci U S A 1997; 94:5744–5749. doi: 10.1073/pnas.94.11.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol 2018; 19:1415–1426. doi: 10.1038/s41590-018-0217-9. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 2019; 176:334–347.e12. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Previte DM, Martins CP, O’Connor EC, Marre ML, Coudriet GM, Beck NW, et al. Lymphocyte activation gene-3 maintains mitochondrial and metabolic quiescence in naive CD4+ T cells. Cell Rep 2019; 27:129–141.e4. doi: 10.1016/j.celrep.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol 2017; 12:814–823. doi: 10.1016/j.jtho.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Hald SM, Rakaee M, Martinez I, Richardsen E, Al-Saad S, Paulsen EE, et al. LAG-3 in non-small-cell lung cancer: expression in primary tumors and metastatic lymph nodes is associated with improved survival. Clin Lung Cancer 2018; 19:249–259.e2. doi: 10.1016/j.cllc.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs 2019; 11:1139–1148. doi: 10.1080/19420862.2019.1629239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S, Sharma G, Travers J, Kumar S, Choi J, Jun HT, et al. TSR-033, a novel therapeutic antibody targeting LAG-3, enhances T-cell function and the activity of PD-1 blockade in vitro and in vivo. Mol Cancer Ther 2019; 18:632–641. doi: 10.1158/1535-7163.MCT-18-0836. [DOI] [PubMed] [Google Scholar]
- 17.Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med 2010; 8:71.doi: 10.1186/1479-5876-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev 2017; 276:97–111. doi: 10.1111/imr.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med 2005; 11:362–369. doi: 10.1016/j.molmed.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 2005; 6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 21.Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One 2012; 7:e30676.doi: 10.1371/journal.pone.0030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol 2015; 29:635–641. doi: 10.1016/j.intimp.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Qiu X, Li X, Fan H, Zhang F, Lv T, et al. Expression profiles and clinical value of plasma exosomal Tim-3 and Galectin-9 in non-small cell lung cancer. Biochem Biophys Res Commun 2018; 498:409–415. doi: 10.1016/j.bbrc.2018.02.114. [DOI] [PubMed] [Google Scholar]
- 24.Su H, Xie H, Dai C, Ren Y, She Y, Xu L, et al. Characterization of TIM-3 expression and its prognostic value in patients with surgically resected lung adenocarcinoma. Lung Cancer 2018; 121:18–24. doi: 10.1016/j.lungcan.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016; 7:10501.doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol 2001; 2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 28.Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol 2003; 4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- 29.Janakiram M, Shah UA, Liu W, Zhao A, Schoenberg MP, Zang X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol Rev 2017; 276:26–39. doi: 10.1111/imr.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider T, Hoffmann H, Dienemann H, Schnabel PA, Enk AH, Ring S, et al. Non-small cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating B7-H3. J Thorac Oncol 2011; 6:1162–1168. doi: 10.1097/JTO.0b013e31821c421d. [DOI] [PubMed] [Google Scholar]
- 31.Chen C, Shen Y, Qu QX, Chen XQ, Zhang XG, Huang JA. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp Cell Res 2013; 319:96–102. doi: 10.1016/j.yexcr.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Altan M, Pelekanou V, Schalper KA, Toki M, Gaule P, Syrigos K, et al. B7-H3 expression in NSCLC and its association with B7-H4, PD-L1 and tumor-infiltrating lymphocytes. Clin Cancer Res 2017; 23:5202–5209. doi: 10.1158/1078-0432.CCR-16-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, et al. B7-H3 negatively modulates CTL-mediated cancer immunity. Clin Cancer Res 2018; 24:2653–2664. doi: 10.1158/1078-0432.CCR-17-2852. [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Zhang G, Sheng S, Zhou Q, Pan Y, Guan S. Upregulation of soluble B7-H3 in NSCLC-derived malignant pleural effusion: a potential diagnostic biomarker correlated with NSCLC staging. Clin Chim Acta 2016; 457:81–85. doi: 10.1016/j.cca.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 35.He CS, Liu YC, Xu ZP, Dai PC, Chen XW, Jin DH. Astragaloside IV enhances cisplatin chemosensitivity in non-small cell lung cancer cells through inhibition of B7-H3. Cell Physiol Biochem 2016; 40:1221–1229. doi: 10.1159/000453175. [DOI] [PubMed] [Google Scholar]
- 36.Scilla KA, Zandberg DP, Bentzen SM, Mainor C, Heath J, Ioffe OB, et al. Case-control study of PD-1, PD-L1 and B7-H3 expression in lung cancer patients with and without human immunodeficiency virus (HIV) infection. Lung Cancer 2018; 123:87–90. doi: 10.1016/j.lungcan.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 37.Luo D, Xiao H, Dong J, Li Y, Feng G, Cui M, et al. B7-H3 regulates lipid metabolism of lung cancer through SREBP1-mediated expression of FASN. Biochem Biophys Res Commun 2017; 482:1246–1251. doi: 10.1016/j.bbrc.2016.12.021. [DOI] [PubMed] [Google Scholar]
- 38.Bao R, Wang Y, Lai J, Zhu H, Zhao Y, Li S, et al. Enhancing anti-PD-1/PD-L1 immune checkpoint inhibitory cancer therapy by CD276-targeted photodynamic ablation of tumor cells and tumor vasculature. Mol Pharm 2019; 16:339–348. doi: 10.1021/acs.molpharmaceut.8b00997. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med 2011; 208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev 2017; 276:66–79. doi: 10.1111/imr.12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019; 156:74–85. doi: 10.1111/imm.13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019; 574:565–570. doi: 10.1038/s41586-019-1674-5. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, He J, Flies DB, Luo L, Chen L. Programmed death one homolog maintains the pool size of regulatory T cells by promoting their differentiation and stability. Sci Rep 2017; 7:6086.doi: 10.1038/s41598-017-06410-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villarroel-Espindola F, Yu X, Datar I, Mani N, Sanmamed M, Velcheti V, et al. Spatially resolved and quantitative analysis of VISTA/PD-1H as a novel immunotherapy target in human non-small cell lung cancer. Clin Cancer Res 2018; 24:1562–1573. doi: 10.1158/1078-0432.CCR-17-2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J Clin Invest 2014; 124:1966–1975. doi: 10.1172/JCI74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bharaj P, Chahar HS, Alozie OK, Rodarte L, Bansal A, Goepfert PA, et al. Characterization of programmed death-1 homologue-1 (PD-1H) expression and function in normal and HIV infected individuals. PLoS One 2014; 9:e109103.doi: 10.1371/journal.pone.0109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018; 36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med 2017; 23:551–555. doi: 10.1038/nm.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw R, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol 2017; 30:1666–1676. doi: 10.1038/modpathol.2017.89. [DOI] [PubMed] [Google Scholar]
- 50.Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017; 276:112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 51.Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014; 26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 2018; 19:723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 53.Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 2015; 125:4053–4062. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Zhao R, Chinai JM, Buhl S, Scandiuzzi L, Ray A, Jeon H, et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc Natl Acad Sci U S A 2013; 110:9879–9884. doi: 10.1073/pnas.1303524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janakiram M, Chinai JM, Zhao A, Sparano JA, Zang X. HHLA2 and TMIGD2: new immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology 2015; 4:e1026534.doi: 10.1080/2162402X.2015.1026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Y, Yao S, Iliopoulou BP, Han X, Augustine MM, Xu H, et al. B7-H5 costimulates human T cells via CD28H. Nat Commun 2013; 4:2043.doi: 10.1038/ncomms3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rieder SA, Wang J, White N, Qadri A, Menard C, Stephens G, et al. B7-H7 (HHLA2) inhibits T-cell activation and proliferation in the presence of TCR and CD28 signaling. Cell Mol Immunol 2020; doi: 10.1038/s41423-020-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng H, Janakiram M, Borczuk A, Lin J, Qiu W, Liu H, et al. HHLA2, a new immune checkpoint member of the B7 family, is widely expressed in human lung cancer and associated with EGFR mutational status. Clin Cancer Res 2017; 23:825–832. doi: 10.1158/1078-0432.CCR-15-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng H, Borczuk A, Janakiram M, Ren X, Lin J, Assal A, et al. Wide expression and significance of alternative immune checkpoint molecules, B7x and HHLA2, in PD-L1-negative human lung cancers. Clin Cancer Res 2018; 24:1954–1964. doi: 10.1158/1078-0432.CCR-17-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang B, Ran Z, Liu M, Ou Y. Prognostic significance of potential immune checkpoint member HHLA2 in human tumors: a comprehensive analysis. Front Immunol 2019; 10:1573.doi: 10.3389/fimmu.2019.01573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 2009; 9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Riccardi C, Ronchetti S, Nocentini G. Glucocorticoid-induced TNFR-related gene (GITR) as a therapeutic target for immunotherapy. Expert Opin Ther Targets 2018; 22:783–797. doi: 10.1080/14728222.2018.1512588. [DOI] [PubMed] [Google Scholar]
- 63.Kopru CZ, Cagnan I, Akar I, Esendagli G, Korkusuz P, Gunel-Ozcan A. Dual effect of glucocorticoid-induced tumor necrosis factor-related receptor ligand carrying mesenchymal stromal cells on small cell lung cancer: a preliminary in vitro study. Cytotherapy 2018; 20:930–940. doi: 10.1016/j.jcyt.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 64.Zhu LX, Davoodi M, Srivastava MK, Kachroo P, Lee JM, St John M, et al. GITR agonist enhances vaccination responses in lung cancer. Oncoimmunology 2015; 4:e992237.doi: 10.4161/2162402X.2014.992237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med 2019; 25:759–766. doi: 10.1038/s41591-019-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mizuno R, Maruhashi T, Sugiura D, Shimizu K, Watada M, Okazaki IM, et al. PD-1 efficiently inhibits T cell activation even in the presence of co-stimulation through CD27 and GITR. Biochem Biophys Res Commun 2019; 511:491–497. doi: 10.1016/j.bbrc.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Schoenhals JE, Cushman TR, Barsoumian HB, Li A, Cadena AP, Niknam S, et al. Anti-glucocorticoid-induced tumor necrosis factor-related protein (GITR) therapy overcomes radiation-induced Treg immunosuppression and drives abscopal effects. Front Immunol 2018; 9:2170.doi: 10.3389/fimmu.2018.02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol 2017; 35:2028–2036. doi: 10.1200/JCO.2016.70.1508. [DOI] [PubMed] [Google Scholar]
- 69.Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol 2017; 199:4110–4123. doi: 10.4049/jimmunol.1700606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Turaj AH, Hussain K, Cox KL, Rose-Zerilli M, Testa J, Dahal LN, et al. Antibody tumor targeting is enhanced by CD27 agonists through myeloid recruitment. Cancer Cell 2017; 32:777–791.e6. doi: 10.1016/j.ccell.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tesselaar K, Arens R, van Schijndel GM, Baars PA, van der Valk MA, Borst J, et al. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol 2003; 4:49–54. doi: 10.1038/ni869. [DOI] [PubMed] [Google Scholar]
- 72.Kashima J, Okuma Y, Hosomi Y, Hishima T. High serum soluble CD27 level correlates with poor performance status and reduced survival in patients with advanced lung cancer. Oncology 2019; 97:365–372. doi: 10.1159/000502441. [DOI] [PubMed] [Google Scholar]
- 73.Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol Rev 2017; 276:145–164. doi: 10.1111/imr.12527. [DOI] [PubMed] [Google Scholar]
- 74.Gao L, Chen K, Gao Q, Wang X, Sun J, Yang YG. CD47 deficiency in tumor stroma promotes tumor progression by enhancing angiogenesis. Oncotarget 2017; 8:22406–22413. doi: 10.18632/oncotarget.9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao H, Wang J, Kong X, Li E, Liu Y, Du X, et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep 2016; 6:29719.doi: 10.1038/srep29719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest 2016; 126:2610–2620. doi: 10.1172/JCI81603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barrera L, Montes-Servín E, Hernandez-Martinez JM, MLÁ G, Montes-Servín E, Herrera-Martínez M, et al. CD47 overexpression is associated with decreased neutrophil apoptosis/phagocytosis and poor prognosis in non-small-cell lung cancer patients. Br J Cancer 2017; 117:385–397. doi: 10.1038/bjc.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Fan J, Wang S, Li Y, Wang Y, Li S, et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol Res 2017; 5:363–375. doi: 10.1158/2326-6066.CIR-16-0398. [DOI] [PubMed] [Google Scholar]
- 79.Wu J, Li Z, Yang Z, Guo L, Zhang Y, Deng H, et al. A glutamine-rich carrier efficiently delivers anti-CD47 siRNA driven by a “glutamine trap” to inhibit lung cancer cell growth. Mol Pharm 2018; 15:3032–3045. doi: 10.1021/acs.molpharmaceut.8b00076. [DOI] [PubMed] [Google Scholar]
- 80.Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol 2016; 37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kozuma Y, Takada K, Toyokawa G, Kohashi K, Shimokawa M, Hirai F, et al. Indoleamine 2,3-dioxygenase 1 and programmed cell death-ligand 1 co-expression correlates with aggressive features in lung adenocarcinoma. Eur J Cancer 2018; 101:20–29. doi: 10.1016/j.ejca.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 82.Takada K, Kohashi K, Shimokawa M, Haro A, Osoegawa A, Tagawa T, et al. Co-expression of IDO1 and PD-L1 in lung squamous cell carcinoma: potential targets of novel combination therapy. Lung Cancer 2019; 128:26–32. doi: 10.1016/j.lungcan.2018.12.008. [DOI] [PubMed] [Google Scholar]
- 83.Botticelli A, Cerbelli B, Lionetto L, Zizzari I, Salati M, Pisano A, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC. J Transl Med 2018; 16:219.doi: 10.1186/s12967-018-1595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li A, Barsoumian HB, Schoenhals JE, Cushman TR, Caetano MS, Wang X, et al. Indoleamine 2,3-dioxygenase 1 inhibition targets anti-PD1-resistant lung tumors by blocking myeloid-derived suppressor cells. Cancer Lett 2018; 431:54–63. doi: 10.1016/j.canlet.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang W, Huang L, Jin JY, Jolly S, Zang Y, Wu H, et al. IDO immune status after chemoradiation may predict survival in lung cancer patients. Cancer Res 2018; 78:809–816. doi: 10.1158/0008-5472.CAN-17-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hellmann MD, Gettinger S, Chow L, Gordon M, Awad MM, Cha E, et al. Phase 1 study of epacadostat in combination with atezolizumab for patients with previously treated advanced nonsmall cell lung cancer. Int J Cancer 2020; doi: 10.1002/ijc.32951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kjeldsen JW, Iversen TZ, Engell-Noerregaard L, Mellemgaard A, Andersen MH, Svane IM. Durable clinical responses and long-term follow-up of stage III-IV non-small-cell lung cancer (NSCLC) patients treated with IDO peptide vaccine in a phase i study-a brief research report. Front Immunol 2018; 9:2145.doi: 10.3389/fimmu.2018.02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al Omar SY, Marshall E, Middleton D, Christmas SE. Increased killer immunoglobulin-like receptor expression and functional defects in natural killer cells in lung cancer. Immunology 2011; 133:94–104. doi: 10.1111/j.1365-2567.2011.03415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manser AR, Weinhold S, Uhrberg M. Human KIR repertoires: shaped by genetic diversity and evolution. Immunol Rev 2015; 267:178–196. doi: 10.1111/imr.12316. [DOI] [PubMed] [Google Scholar]
- 90.Boudreau JE, Hsu KC. Natural killer cell education and the response to infection and cancer therapy: stay tuned. Trends Immunol 2018; 39:222–239. doi: 10.1016/j.it.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He Y, Bunn PA, Zhou C, Chan D. KIR 2D (L1, L3, L4, S4) and KIR 3DL1 protein expression in non-small cell lung cancer. Oncotarget 2016; 7:82104–82111. doi: 10.18632/oncotarget.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He Y, Liu S, Mattei J, Bunn PA, Jr, Zhou C, Chan D. The combination of anti-KIR monoclonal antibodies with anti-PD-1/PD-L1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in NSCLC. Drug Des Devel Ther 2018; 12:981–986. doi: 10.2147/DDDT.S163304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov 2016; 6:446–459. doi: 10.1158/2159-8290.CD-15-0944. [DOI] [PubMed] [Google Scholar]
- 94.Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 2018; 175:313–326. doi: 10.1016/j.cell.2018.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]