

Abstract
An increasing number of studies suggest that the renal proximal tubule is a site of injury in diabetic nephropathy (DN), and progressive renal tubulointerstitial fibrosis is an important mediator of progressive kidney dysfunction in DN. In this study, we observed increased expression and activation of YAP (yes-associated protein) in renal proximal tubule epithelial cells (RPTC) in patients with diabetes and in mouse kidneys. Inducible deletion of Yap specifically in RPTC or administration of the YAP inhibitor verteporfin significantly attenuated diabetic tubulointerstitial fibrosis. EGFR-dependent activation of RhoA/Rock and PI3K-Akt signals and their reciprocal interaction were upstream of proximal tubule YAP activation in diabetic kidneys. Production and release of CTGF in culture medium were significantly augmented in human embryonic kidney (HEK)-293 cells transfected with a constitutively active YAP mutant, and the conditioned medium collected from these cells activated and transduced fibroblasts into myofibroblasts. This study demonstrates that proximal tubule YAP-dependent paracrine mechanisms play an important role in diabetic interstitial fibrogenesis; therefore, targeting Hippo signaling may be a therapeutic strategy to prevent the development and progression of diabetic interstitial fibrogenesis.
Introduction
More than 30 million people in the U.S. have diabetes mellitus, and one-third of these will develop diabetic nephropathy (DN) in their lifetime (1). Kidney dialysis and transplantation are the only available treatment options for end-stage renal disease patients (2). Glomerular injury in DN has been the focus of attention for nephrologists; however, an increasing number of studies have suggested that the renal tubule, especially the proximal tubule, is also a site of injury in DN, and progressive renal tubulointerstitial fibrosis is an important component of the pathogenesis leading to DN and ultimately end-stage renal disease (3,4). Some renal proximal tubule cell (RPTC) injury biomarkers are detectable in urine from patients with early-stage diabetes who do not yet show obvious glomerular injury, suggesting that RPTC injury is a primary lesion rather than only secondary to glomerular injury (5,6).
Overproduction and deposition of extracellular matrix (ECM) proteins in the renal tubulointerstitium, as well as thickening of tubular basement membranes, are characteristic pathological changes of DN (7,8). Collagen I is the most abundant ECM protein, although other collagens such as types III, V, VI, VII, and XV and the adhesive glycoprotein fibronectin also accumulate in the interstitial space of the fibrotic kidney (9). The activated myofibroblast is generally accepted as a primary ECM-secreting cell type in fibrotic kidney. The source of myofibroblasts in renal fibrosis is still controversial (10–13), although up to 50% of the total pool of myofibroblasts may arise from local resident fibroblasts (12). Nonetheless, induction and proliferation of myofibroblasts play key roles in kidney fibrosis, regardless of the origin(s) of the myofibroblasts (11,12), but the underlying molecular mechanisms of renal interstitial fibroblast cell proliferation and transition have not been fully elucidated.
YAP (yes-associated protein), a transcriptional coactivator for numerous target genes in the nucleus, is a crucial effector of the canonical Hippo signaling pathway that controls organ size and prevents tumorigenesis by regulating cell proliferation, apoptosis, and differentiation. Upon activation of the Hippo pathway in response to different extracellular cues, YAP is phosphorylated at specific serine/threonine residues and deactivated by cytoplasmic sequestration, followed by proteasome-mediated degradation (14). YAP is broadly expressed in adult human tissues, with moderate expression levels in adult kidney (15,16). Numerous studies suggest that YAP is involved in multiple fibrogenesis-related pathways, including the TGFβ-Smad signaling pathway, ECM protein synthetic pathways, mechano-transduction signaling pathways, and tissue remodeling and energy stress responsive pathways (14). As a critical signaling protein modulating multiple molecular pathways, YAP has been implicated in fibrogenesis in other organs (17). Increasing studies have suggested that aberrant YAP activation in kidney is also involved in kidney fibrogenesis (18–21). Interestingly, a recent study found that mice with renal tubular cell–specific Mst1/2 deletion developed renal fibrosis, which was ameliorated in Mst1/2-Yap triple knockout mice (22). Our recent study demonstrated for the first time that YAP expression and activation were upregulated in diabetic RPTC through an EGFR activation–dependent pathway (23), but the detailed underlying molecular mechanism of YAP activation and contribution to diabetic kidney injury has not previously been described.
Rho-GTPases, a member of Ras superfamily of small GTPase, exhibit both GDP/GTP binding and GTPase activity to activate ROCK (RhoA-associated coiled-coil–containing kinases), the downstream effector of the Rho-GTPases (24,25). RhoA, the most abundant Rho-GTPase expressed in the renal proximal tubules (26), and its downstream effector ROCK, have been implicated in expression of ECM proteins and tubulointerstitial fibrosis in animal models of both type 1 and type 2 diabetes (27). Inhibition of Rho/ROCK signaling has been reported to significantly ameliorate diabetic kidney injury (28,29), but the underlying mechanism of this inhibition remains unclear. Of interest, studies have suggested that Rho/ROCK signaling activation is upstream of YAP activation (30,31). Therefore, the goal of the current study was to investigate the potential role of proximal tubule activation of YAP in development of tubulointerstitial fibrosis in diabetic kidney disease and determine the underlying signaling mechanisms.
Research Design and Methods
Materials and Reagents
Erlotinib was purchased from LC Laboratories (Woburn, MA). Antibodies against EGFR (cat. no. 4060S), phosphorylated EGFR (3777S), YAP (14074S), YAP (12395S), Ki67 (9129S), phosphorylated Akt (4060S), Akt (2920S), and β-actin (4970S) were from Cell Signaling Technology (Beverly, MA). Collagen I (600-401-103-01) was purchased from Rockland Immunochemicals. CTGF (ab6992) and goat polyclonal α-SMA (ab21027) antibodies were from Abcam (Cambridge, MA). Rat monoclonal TGF-β2 antibody (MAB73461SP) was from Thermo Fisher Scientific (Waltham, MA). LY294002 were from EMD Millipore (Billerica, MA). IRDye 680RD and IRDye 800CW secondary antibodies were from LI-COR Biosciences (Lincoln, Nebraska). Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibodies for immunofluorescence (IF) staining were from Life Technologies (Grand Island, NY). Verteporfin and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Animal Studies
Inducible renal proximal tubule epithelial cell–specific YAP deletion mice (YapPTiKO) and the wild-type control mice (YapPTWT) were generated as previously described (32). Endothelial nitric oxide synthase (eNOS) knockout (eNOS−/−), FVB/NJ mice (The Jackson Laboratory, Bar Harbor, ME), or the YapPTiKO and YapPTWT mice were injected with streptozotocin (STZ) (50 mg/kg body wt i.p.) for five consecutive days to induce type 1 diabetes beginning 1 week after completion of tamoxifen injections. Prior to STZ injection, unilateral nephrectomy surgery was performed in the YapPTiKO, YapPTWT, and FVB/NJ mice that were used to analyze kidney fibrosis. Verteporfin (100 mg/kg i.p. every other day), Y27632 (10 mg/kg i.p. once/week), or their vehicles were injected in the diabetic eNOS−/− mice or the unilateral nephrectomized diabetic FVB/NJ mice starting at 1 week after STZ injection completion. Blood glucose was measured using the OneTouch Basic blood glucose monitoring system (LifeScan, Milpitas, CA) on blood samples obtained via the saphenous vein after 5 h of food deprivation. Mice were sacrificed at indicated time points, and kidney tissues were collected for analysis.
Cell Culture Studies
Human embryonic kidney (HEK)-293 cells and human renal proximal tubule epithelial cells (hRPTC) were purchased from ATCC (Manassas, VA). The HEK-293 cells were maintained in DMEM/F12 medium with 10% FBS. pcDNA4/HisMaxB-YAP1-S127A plasmid DNA (cat. no. 18988; Addgene) was transfected into HEK-293 cells using a Lipofectamine 3000 transfection kit (Thermo Fisher Scientific), and stable clones (hYAP1S127A-HEK) were selected by zeocin (600 ng/mL) and maintained in culture medium with zeocin (400 ng/mL). The hRPTC were cultured in DMEM/F12 medium supplemented with 10% FBS, 5 pmol/L triiodo-l-thyronine, 10 ng/mL recombinant human EGF, 3.5 µg/mL ascorbic acid, 5.0 µg/mL human transferrin, 5.0 µg/mL insulin, 25 ng/mL prostaglandin E1, 25 ng/mL hydrocortisone, 8.65 ng/mL sodium selenite, 0.1 mg/mL G418, and 1.2 g/L sodium bicarbonate. The hRPTC were made quiescent in DMEM medium with low glucose (1 g/L) and 0.5% FBS for 24 h before exposure to 25 mmol/L mannitol or glucose with or without Y27632, LY294002, or erlotinib treatment for another 24 h.
Assessment of Activation of Fibroblasts by the Conditioned Medium
hYAP1S127A-HEK and vector-transfected HEK-293 cells were plated into 150-mm cell culture dishes at 50% cell confluence in DMEM/F12 medium supplied with 10% FBS. Culture medium was replaced by serum-free DMEM with nonessential amino acid when cell reached 80% confluence, and the conditioned medium was harvested after 24 h incubation and concentrated with Amicon Ultra-15 centrifugal filters (3K; EMD Millipore) by centrifuge at 4,000g for 30 min at 4°C after removal of dead cells by spinning at 13,000 rpm for 2 min. Mouse or rat fibroblasts were treated with conditioned medium supplied with 0.2% FBS for 2 days, and the fibroblast-to-myofibroblast transition was assessed by α-SMA protein level or cell number was counted by using a hemocytometer.
Analysis of RT2 Profiler Human Fibrosis PCR Array
Total RNA was isolated from hYAP1S127A-HEK and their vector-transfected HEK-293 cells with use of RNeasy Mini Kit (QIAGEN) when cells reached 80% confluence. cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad) following the instruction of the products. Eighty-four key fibrosis-related genes on the RT2 Profiler human fibrosis PCR array (PAHS-120ZA) were analyzed with the QuantStudio 3 Real-Time PCR System. The threshold cycle (Ct) values of housekeeping gene ACTB (β-actin) were selected as internal control, and relative mRNA quantification is based on the 2−ΔΔCt method with normalization to either control samples. A minimum log 2-fold change ±2 was applied for statistical significance (P < 0.01).
Quantitative Real-time PCR Analysis
Relative mRNA expression levels of CTGF (CCN2), MCP-1 (CCL2), collagen type III α I (COL3A1), thrombospondin 1 (THBS1), and TGFβ2 in the hYAP1S127A-HEK and their vector-transfected HEK-293 cells were validated by quantitative real-time PCR. Relative mRNA expression level of α-SMA (Act2a) and collagen type I α I (Col1a1) in mouse fibroblasts exposed to conditioned medium treatment were also evaluated by quantitative real-time PCR. Relative gene expression was quantified with iQ SYBR Green Supermix (Bio-Rad) in QuantStudio 3 Real-Time PCR System. All raw Ct values were normalized to β-actin, and 2−ΔΔCt method was applied to compare gene fold change between experiment group and control group. All PCR primers (Supplementary Table 1) were from Integrated DNA Technologies.
Mouse Kidney Histology Analysis
Mouse kidneys were harvested and embedded in paraffin at different time points after induction of type 1 diabetes, and 5-μm tissue sections were stained with hematoxylin-eosin by standard methods. Deparaffinized sections underwent Masson trichrome staining or Sirius Red staining. The images were captured with use of an AxioCam MRc (Carl Zeiss).
IF Analysis
IF was performed on paraffin-embedded tissues or cultured hRPTC fixed by 4% paraformaldehyde with standard techniques as previously described (23,33), and images were a Nikon TE300 fluorescence microscope (Diagnostic Instruments) or an captured by Olympus FV-1000 Inverted Confocal Microscope (Vanderbilt Medical Center Cell Image Shared Resource Core).
RhoA Pull-down Activation Assay
The levels of active GTP-bound RhoA were analyzed using the RhoA Pull-down Activation Assay Biochem Kit (Cytoskeleton, Denver, CO). Specifically, the hRPTC were made quiescent for 24 h at 1 day after control or EGFR-specific siRNA transfection. The quiescent hRPTC were exposed to 25 mmol/L mannitol or glucose with or without Erlotinib (100 nmol/L) for another 24 h before being lysed in the lysis buffer. Lysates with the same protein concentration were subjected to pull-down assay following the manufacturer’s instructions, and the precipitated active RhoA was detected by Western blot after resolving on a 15% SDS-PAGE.
Transfection of siRNA in hRPTC
The control and ON-TARGETplus human siRNA SMARTpool: Nontargeting Pool (D-001810-10-05), EGFR (L-003114-00-0005), RhoA (L-003860-00-0005), AKT1 (L-003000-00-0005), CTGF (L-012633-01-0010), and TGFB2 (L-010544-00-0005) were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO). Twenty-four hours after transfection by use of Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific), the cells were made quiescent in DMEM medium with low glucose (1 g/L) and 0.5% FBS for 24 h before being exposed to 25 mmol/L mannitol or glucose. The cells were lysed in radioimmunoprecipitation assay buffer for immunoblotting analysis or fixed in 4% paraformaldehyde for IF staining.
Immunoprecipitation and Immunoblotting
Procedures were performed as we have previously described (33,34).
Statistics
Data are presented as means ± SEM for at least three separate experiments (each in triplicate). An unpaired Student t test was used for statistical analysis, and for multiple group comparisons, ANOVA with Bonferroni corrections (GraphPad Prism, version 7) were used. A P value of <0.05 compared with control was considered statistically significant.
Data and Resource Availability
The data sets generated and analyzed during the current study are available from the corresponding authors upon reasonable request.
Results
Activation of YAP in RPTC-Mediated Renal Interstitial Fibrogenesis
In our previous studies, we demonstrated that YAP was activated in diabetic mouse RPTC through an EGFR-dependent pathway (23). To determine whether YAP is also activated in RPTC of diabetic kidneys in humans, we performed IF staining of kidney samples from five patients with diabetic kidney disease and nondiabetic control subjects and observed upregulation of YAP expression and its nuclear translocation specifically in RPTC in all kidney samples, identified by the proximal tubule maker, lotus tetragonolobus lectin (LTL) (Fig. 1). Of note, there was a marked decrease in the expression levels of LTL, suggesting a certain degree of dedifferentiation of the proximal tubule cells in the diabetic kidneys.
Figure 1.

YAP expression and nuclear distribution were increased in RPTC of kidneys in patients with diabetes. Representative IF images of control biopsy kidney specimens from patients without diabetes and patients with diabetes (n = 5) (red, YAP; green, LTL, an RPTC marker; blue, DAPI) (original magnification ×600).
To define the potential role of YAP activation in diabetic RPTC, we subjected YapPTiKO (inducible conditional RPTC-specific knockout of YAP) mice to STZ injection to induce type 1 diabetes. To ensure development of obvious DN pathological alterations, we subjected some of the mice to unilateral nephrectomy (UNX) 1 week before STZ injection as we have previously described (34). Upregulation of YAP expression in the wild-type control mouse renal cortical tissue lysates was observed 1 week after development of high blood glucose and was significantly attenuated in the YapPTiKO mouse kidneys (Fig. 2A). In addition, upregulation of CTGF, a well-known YAP downstream target, and collagen I was also decreased in the diabetic YapPTiKO mouse renal cortical tissue lysates (Fig. 2A). There was also a marked decrease in collagen I deposition in diabetic YapPTiKO kidney tubulointerstitium, as indicated by Sirius Red staining (Fig. 2B), Masson trichrome staining, and collagen I IF staining (Supplementary Fig. 1A and B), although they developed comparable hyperglycemia, proteinuria, and increases in serum creatinine with a similar level of blood pressure (Supplementary Fig. 1C–F), suggesting that YAP activation in the RPTC directly contributes to diabetic tubulointerstitial fibrogenesis.
Figure 2.

YAP deletion specific in RPTC ameliorated interstitial fibrosis in diabetic mouse kidneys. A: Upregulation of expression of YAP, CTGF, and collagen I in the UNX STZ type 1 diabetic mouse cortical tissue was attenuated in YapPTiKO mice. B: Representative images of Sirius Red staining indicated that Yap deletion specific in RPTC reduced diabetic interstitial fibrosis (original magnification ×200). Values are mean ± SEM (n = 4–7 for each group). *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of YAP Activation Ameliorated Diabetic Renal Fibrogenesis
YAP is a transcriptional coactivator that activates many transcription factors, such as p73, RUNX, ERBB4, and TEADs (35–39). However, TEADs are the major YAP-interacting transcription factors (39). Our previous studies have shown that administration of verteporfin effectively inhibited YAP-TEAD association and the transcriptional activator activity of YAP in the kidney (23,32); therefore, we first treated STZ-eNOSdel mice, an accelerated DN mouse model that develops prominent DN within 16–24 weeks after STZ injection (40,41), with verteporfin for 20 weeks. As shown in Fig. 3A, verteporfin treatment effectively decreased CTGF expression and collagen I deposition in both the tubular interstitium and the glomeruli (Fig. 3B). To exclude that the possible effects of verteporfin were associated with eNOS gene deletion, we administered verteporfin to unilateral nephrectomized diabetic FVB/NJ mice and found that verteporfin similarly minimized CTGF upregulation in the diabetic renal cortical tissue lysates (Fig. 3C) and mitigated tubulointerstitial fibrosis (Fig. 3D).
Figure 3.
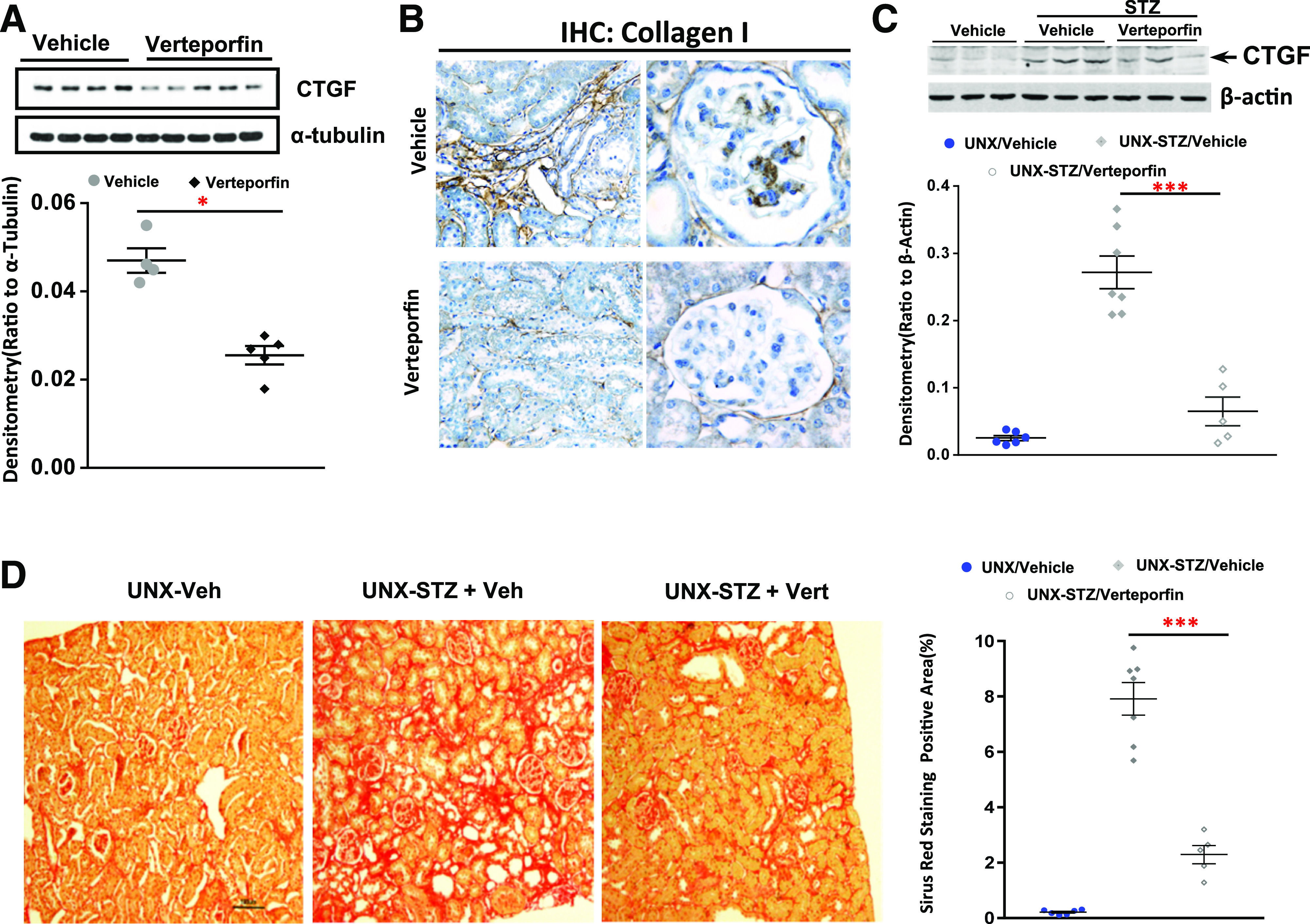
Blocking YAP-TEAD association reduced interstitial fibrosis in diabetic mouse kidneys. Nine- to 10-week old STZ diabetic eNOS−/− mice (A and B) or unilateral nephrectomized diabetic FVB/NJ mice (C and D) were administered vehicle or verteporfin (Vert) (100 mg/kg i.p. every other day) for 20 weeks: immunoblotting analysis of CTGF expression in renal cortical tissue lysates (A and C), representative immunohistochemical (IHC) staining of collagen I expression in both interstitial area and glomeruli (original magnification ×400) (B), and representative histology data of Sirius Red staining (original magnification ×200) (D). Values are mean ± SEM (n = 5–10 for each group). Veh, vehicle. *P < 0.05, ***P < 0.001.
YAP Activation in RPTC Was Critical for Induction and Proliferation of Myofibroblasts in Diabetic Kidney
Myofibroblast cells are the major source of ECM proteins, cross-linking enzymes, and inhibitors of matrix-degrading metalloproteinases in organ fibrogenesis (11,12,42). De novo synthesized α-smooth muscle actin (α-SMA) is the most widely used molecular marker for myofibroblasts in research and clinical diagnostics (43). We detected upregulation of collagen I and α-SMA in kidneys of mice with STZ-induced type 1 diabetes, with marked inhibition by administration of verteporfin (Fig. 4A and B). Deletion of YAP specifically in RPTC inhibited fibronectin, a well-known profibrotic factor, and α-SMA expression in diabetic renal cortical tissue lysates (Fig. 4C). Costaining of α-SMA with Ki-67, a marker of cell proliferation, revealed much less positive staining for both markers in tubulointerstitial areas in the diabetic YapPTiKO mice compared with their wild-type control diabetic mice (Fig. 4D). Of note, both verteporfin administration and YAP deletion in RPTC attenuated collagen I and α-SMA expression in the tubulointerstitial areas (Fig. 4B and D).
Figure 4.
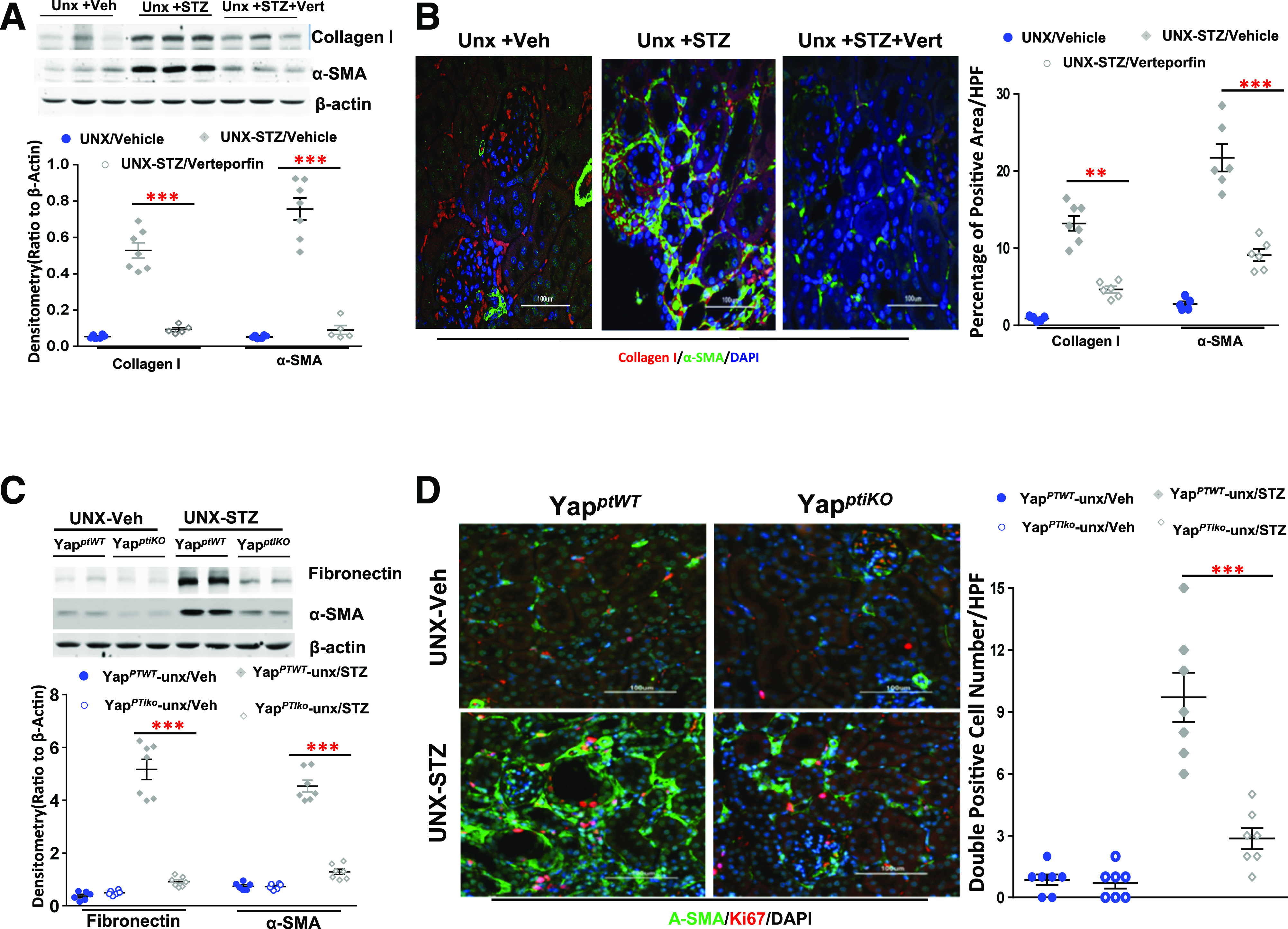
Verteporfin (Vert) or RPTC-specific YAP deletion reduced ECM and α-SMA expression in diabetic kidneys. A: Verteporfin inhibited upregulation of collagen I and α-SMA in unilateral nephrectomized diabetic FVB/NJ mouse renal cortical tissue lysates. B: Representative IF of verteporfin inhibition of collagen I and α-SMA (original magnification ×200). C: Fibronectin and α-SMA upregulation in diabetic kidneys were significantly inhibited in YapPTiKO mice. D: Representative IF indicated inhibition of α-SMA expression and Ki67-positive cells in the YapPTiKO mouse kidneys (original magnification ×200). Values are mean ± SEM (n = 4–10 for each group). **P < 0.01, ***P < 0.001. HPF, high power field.
To understand the underlying mechanism by which the aberrant YAP activation in RPTC caused fibroblast cell proliferation and transition to myofibroblasts, we transfected a constitutively active human YAP1 mutant gene (hYAP1S127A) into HEK-293 cells (hYAP1S127A-HEK). Successful transfection was confirmed by markedly increased expression of YAP and fusion protein tag Xpress (Fig. 5A). IF staining further confirmed active YAP expression and distribution in both nucleus and cytoplasm (Fig. 5B). Although it has been reported that serine 127 residue phosphorylation is a prerequisite step for YAP cytoplasmic retention (44), other possible mechanisms for the observed cytoplasmic distribution of the mutant YAP will require additional study. In addition to nuclear expression of the mutant YAP, we detected marked upregulation of CTGF in the hYAP1S127A-HEK by both immunoblotting and IF analysis (Fig. 5A and C). To test the hypothesis that resident fibroblasts are activated by profibrotic factors that are synthesized and released from injured RPTC through a YAP activation–dependent pathway, we collected conditioned medium from the hYAP1S127A-HEK cells and administered it to immortalized mouse fibroblast cells (45). We found that exposure of conditioned medium from hYAP1S127A-HEK cells but not the vector-transfected HEK cells altered the fibroblast cell morphology from an elongated spindle shape to a flattened stellate shape and markedly upregulated expression of α-SMA (Fig. 5D). Similarly, exposure of a rat kidney fibroblast cell line, NRK-49F cell, to the conditioned medium from hYAP1S127A-HEK cells also markedly increased α-SMA expression (Supplementary Fig. 3C).
Figure 5.
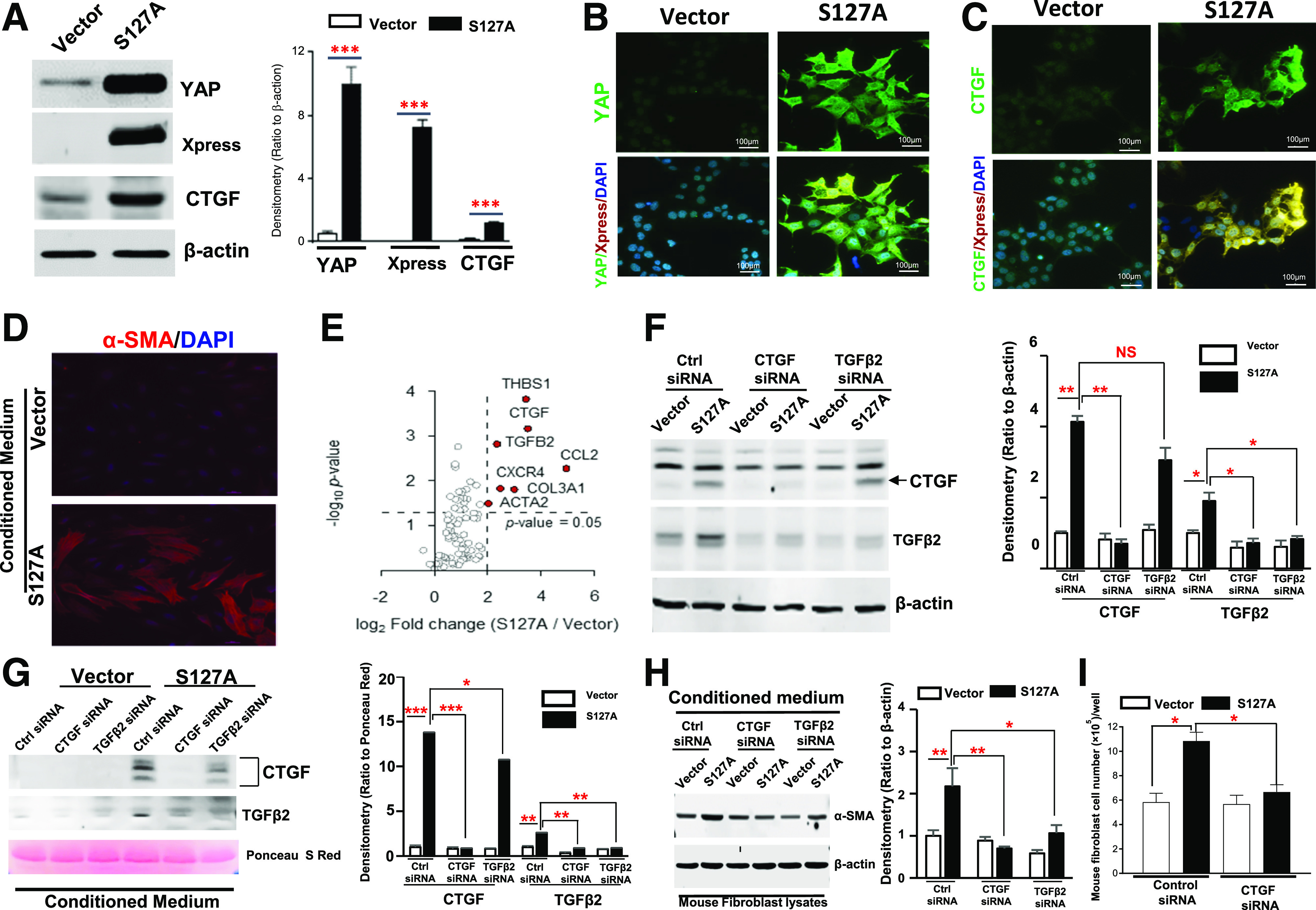
Conditioned medium from hYAP1S127A-HEK–activated fibroblasts. Generation of a stable transfectant (S127A) of a constitutively active human YAP1 mutant gene in HEK-293 cells was confirmed by immunoblotting analysis (A) and IF staining (B and C) (original magnification ×400). D: Conditioned medium from hYAP1S127A-HEK cells altered mouse fibroblast cell morphology and upregulated α-SMA expression (original magnification ×400). E: Screening results of possible profibrotic factors released from the active YAP-transfected HEK cells by using RT2 Profiler human fibrosis PCR array. F: Upregulation of CTGF and TGFβ2 expression in hYAP1S127A-HEK was effectively inhibited by transfection of specific siRNA respectively, and transfection of CTGF siRNA also blocked expression of TGFβ2. G: CTGF and TGFβ2 were detected in the cultured medium from hYAP1S127A-HEK cells; expressions were effectively abolished by transfection of their specific siRNAs. Silencing CTGF and TGFβ2 expression in hYAP1S127A-HEK cells by siRNAs inhibited conditioned medium–induced α-SMA expression in the mouse fibroblasts (H) and fibroblast cell proliferation (I). S127A, hYAP1S127A-HEK; Vector, vector-transfected HEK-293 cells. Values are mean ± SEM (the data were from at least three separate experiments). Xpress: N-terminal tag of the expression vector. *P < 0.05, **P < 0.01, ***P < 0.001. NS, nonsignificant difference. Ctrl, control; S Red, Sirius Red.
To define possible paracrine profibrotic factors released from RPTC, we screened 84 profiled human fibrosis-associated genes in the hYAP1S127A-HEK cells with RT2 Profiler human fibrosis PCR array. Forty of 84 genes were upregulated more than twofold in the hYAP1S127A-HEK cells compared with the vector-transfected HEK cells, with CTGF being one of the most predominantly upregulated (Fig. 5E and Supplementary Table 2). The most predominantly upregulated genes were confirmed by PCR analysis (Supplementary Fig. 2).
CTGF (also known as CCN2) is the second of six members of the CCN family of matricellular proteins (46) and is a secreted protein whose molecule consists of four distinct, conserved domains that can interact with growth factors, cell-surface molecules such as integrins, and ECM proteins such as fibronectin and proteoglycans (47). Immunoblotting of the conditioned medium confirmed that CTGF expression was markedly higher in the conditioned medium from hYAP1S127A-HEK compared with that from the vector-transfected HEK cells (Fig. 5F). To evaluate the effects of CTGF on fibroblast activation, we transfected CTGF-specific siRNAs in the hYAP1S127A-HEK cells for 2 days before collecting conditioned medium. We found that effectively silencing CTGF expression in the hYAP1S127A-HEK cells (Fig. 5G) markedly inhibited α-SMA, collagen I expression, and cell proliferation in mouse fibroblasts in response to conditioned medium (Fig. 5H and I and Supplementary Fig. 3A and B). In addition, our data also suggested that expression and release of TGFβ2 in the hYAP1S127A-HEK cells were partially mediated by CTGF (Fig. 5H and I).
YAP Activation in Diabetic RPTC Was Mediated by Activation of RhoA/ROCK Signal
Recent studies have suggested that YAP activation is implicated in regulation of the contractile actomyosin cytoskeleton (18,48–50). In addition, activation of Rho/ROCK signaling is also critical for mediating rearrangements of the actomyosin cytoskeleton (51–53). For evaluation of the potential role of Rho/ROCK activation in regulation of YAP activation in diabetic kidney injury, UNX diabetic FVB/NJ mice were administered a ROCK inhibitor, Y-27632. Y-27632 not only blunted upregulation of YAP and CTGF expression in the diabetic renal cortical tissue lysates (Fig. 6A) but also reduced tubulointerstitial fibrosis (Fig. 6B). RhoA is the most abundant Rho-GTPase expressed in the renal proximal tubules (26); we found increased active RhoA in the cultured hRPTC in response to high glucose treatment (Fig. 7A). Pretreatment of the cells with Y-27632 inhibited upregulation of expression of YAP and CTGF and YAP nuclear translocation in response to high glucose treatment (25 mmol/L) (Fig. 7B and C). Furthermore, upregulation of YAP and CTGF expression and YAP nuclear translocation in hRPTC in response to high glucose treatment were attenuated by silencing RhoA with siRNA transfection (Fig. 7D and E).
Figure 6.

Inhibiting RhoA/ROCK signaling prevented YAP activation and ameliorated interstitial fibrosis in diabetic mice. Administration of a RhoA/ROCK inhibitor, Y27632, to diabetic FVB/NJ mice inhibited YAP and CTGF expression in renal cortical tissue lysates (A) and significantly attenuated interstitial fibrosis (original magnification ×200) (B). Values are mean ± SEM (n = 4–7 for each group). ***P < 0.001. Veh, vehicle.
Figure 7.
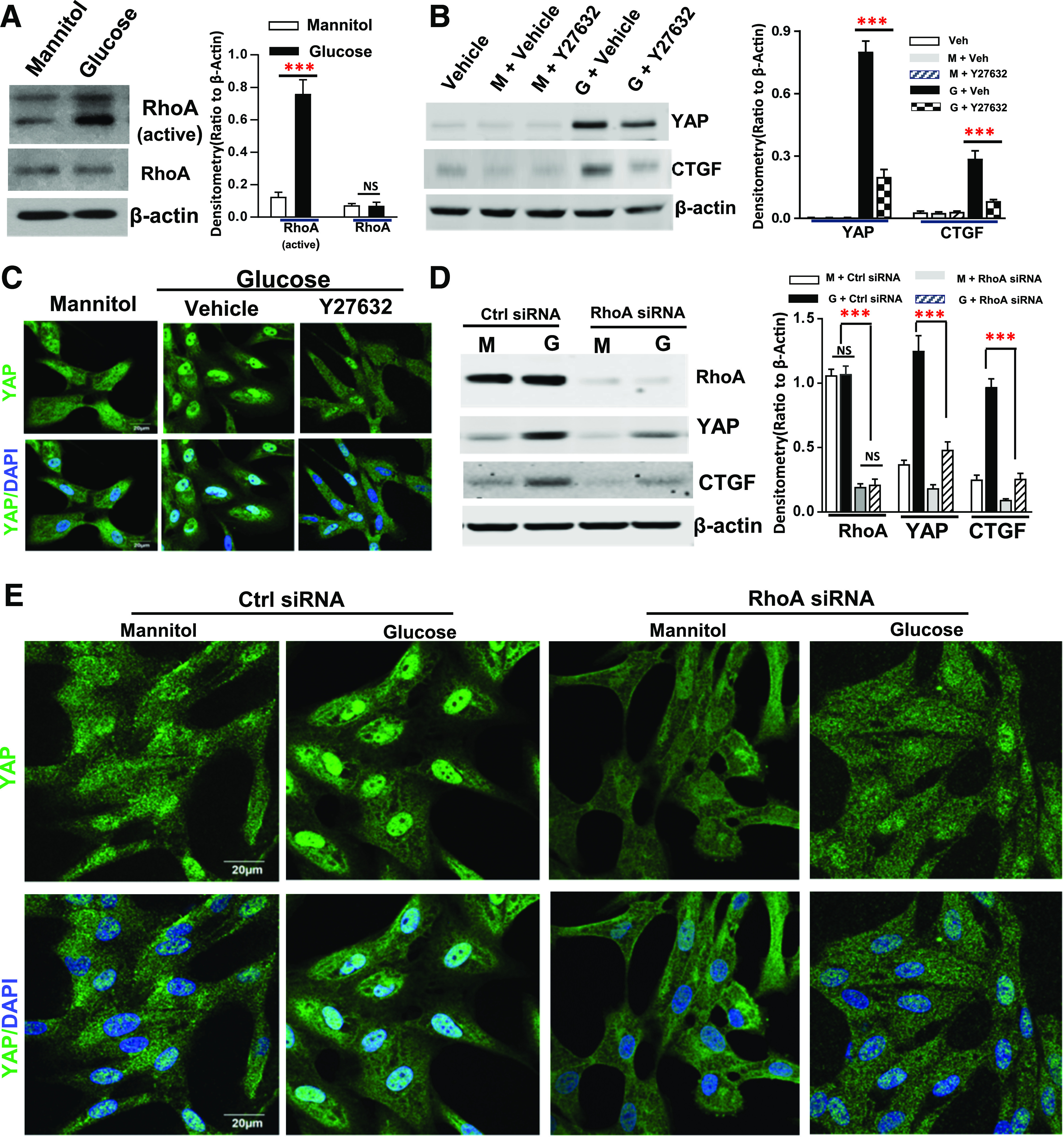
RhoA activation mediated YAP activation in response to high glucose treatment in cultured hRPTC. High glucose treatment increased RhoA activity but not its expression (A), and Y27632 inhibited YAP expression and nuclear translocation and CTGF expression in response to high glucose (original magnification ×600) (B and C). Transfection of RhoA siRNA attenuated upregulation of YAP and CTGF in response to high glucose treatment (D) and inhibited YAP nuclear translocation (E) in response to high glucose treatment (original magnification ×600). Values are mean ± SEM (the data were from at least three separate experiments). ***P < 0.001. Ctrl, control; G, glucose; M, mannitol; NS, nonsignificant difference; Veh, vehicle.
YAP Was Activated in the Diabetic RPTC by Reciprocal Interaction between EGFR-PI3K-Akt and Rho/ROCK Signaling Pathways
We have documented that EGFR-PI3K-Akt signaling pathway activation in diabetic RPTC is an upstream signal for YAP activation (23). We performed further studies to define the relationship of this pathway with RhoA/ROCK activation to mediate YAP activation in response to high glucose. Neither Y-27632 nor siRNA inhibition of RhoA expression affected EGFR tyrosine phosphorylation, but Y-27632 and siRNA both inhibited both expression and phosphorylation of Akt (Fig. 8A and B). However, either siRNA knockdown of EGFR or the EGFR tyrosine kinase inhibitor, erlotinib, markedly inhibited RhoA activity and YAP nuclear translocation in response to high glucose (Fig. 8C–E). These results suggested that RhoA/ROCK signaling was activated through an EGFR-dependent pathway and activation of RhoA/ROCK subsequently mediated Akt phosphorylation in response to high glucose treatment. Knocking down AKT1 gene expression by specific siRNA transfection inhibited both YAP and RhoA protein expression (Fig. 8F). Moreover, inhibition of Akt phosphorylation by the specific PI3K inhibitor LY290042 also decreased RhoA protein expression, which was reversed by treatment of the cells with a potent cell-permeable proteasome inhibitor, MG-132 (Fig. 8G). These results indicated that RhoA/ROCK activation is essential to mediate YAP nuclear translocation and the induction of AKT and YAP gene expression by EGFR activation in mammalian cells and the resulting AKT expression and phosphorylation are critical for RhoA protein stability, as shown in Fig. 8H.
Figure 8.
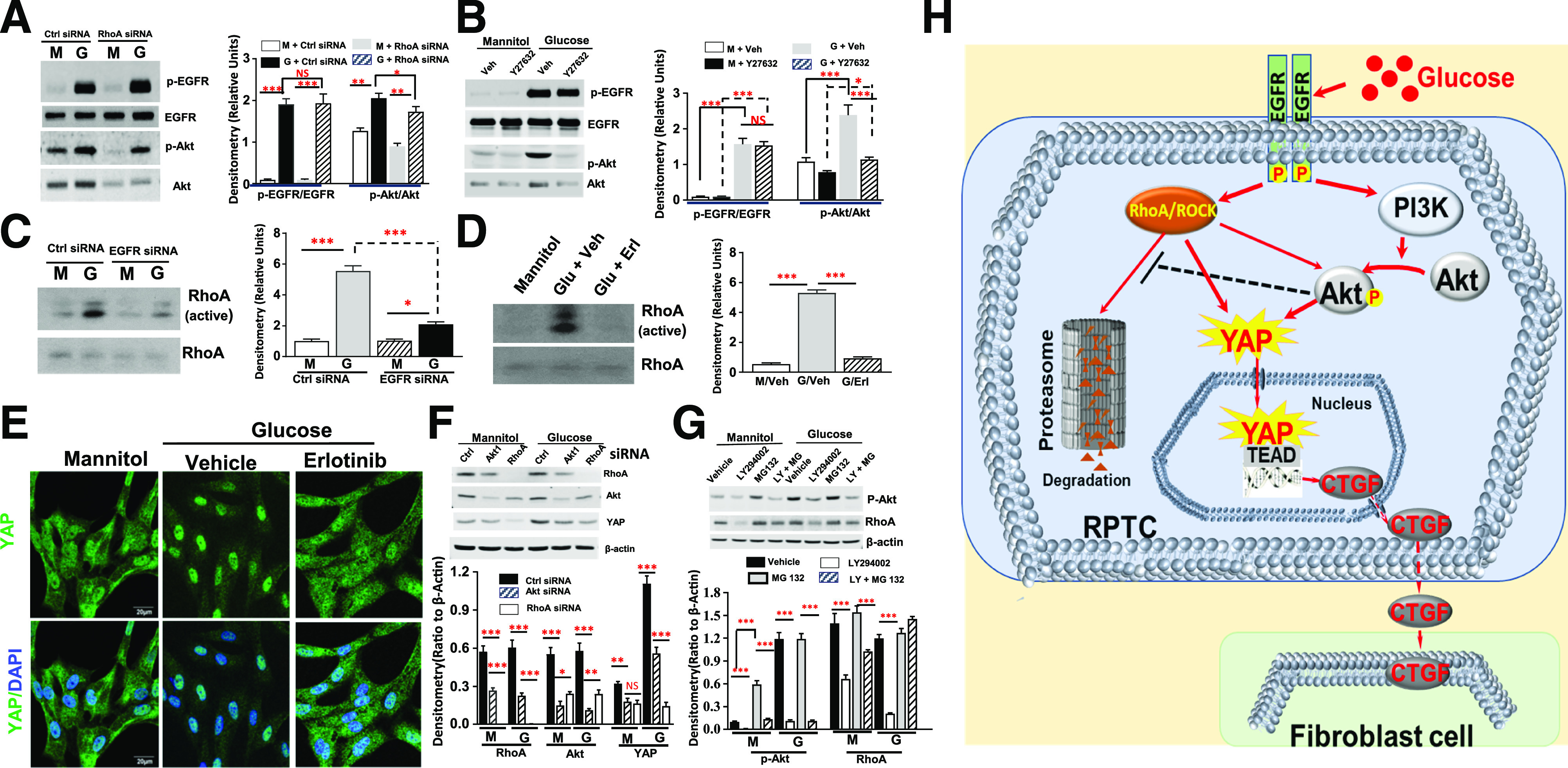
Interactions between PI3K-Akt and RhoA/ROCK mediated EGFR-dependent YAP activation in RPTC in response to high glucose. A: Inhibition of RhoA expression did not affect EGFR activation but significantly inhibited Akt expression and phosphorylation in response to high glucose treatment in hRPTC. B: Y27632 did not affect EGFR activation but also decreased Akt expression and phosphorylation in response to high glucose treatment in hRPTC. C: siRNA knockdown of EGFR did not reduce RhoA expression but inhibited its activation. D: The EGFR tyrosine kinase inhibitor erlotinib did not alter RhoA expression but inhibited its activation. E: Erlotinib inhibited YAP nuclear translocation in response to high glucose treatment in hRPTC (original magnification ×600). F: siRNA knockdown of Akt1 reduced RhoA protein expression, and siRNA knockdown expression of either Akt1 or RhoA inhibited YAP expression in response to high glucose in hRPTC. G: The PI3K inhibitor LY294002 treatment attenuated RhoA protein expression, which was reversed by treatment of the cells with a cell-permeable proteasome inhibitor, MG-132. H: In response to hyperglycemia, EGF receptor (EGFR) mediates RhoA/ROCK– and PI3K-Akt–dependent YAP activation and subsequent downstream target gene CTGF expression, which was released into peritubular space to activate fibroblast proliferation and transition to myofibroblasts. In addition, RhoA/ROCK activation is essential for induction of Akt expression and phosphorylation, and the resulting Akt activation inhibits RhoA protein degradation by proteasome system. *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl, control; G, glucose; M, mannitol; Veh, vehicle.
Discussion
In the current studies, we observed decreased renal interstitial fibrosis in diabetic mice with YAP deletion specifically in the RPTCs (YapPTiKO). Moreover, administration of verteporfin, a YAP inhibitor that acts by competitively binding with TEAD (54), also effectively ameliorated diabetic renal interstitial fibrogenesis. Further studies indicated that YAP activation–dependent expression of CTGF in RPTC and subsequent release activated fibroblast proliferation and transition to myofibroblasts, a potential mechanism for diabetic interstitial fibrogenesis. Our study also found that the beneficial effects of inhibition of RhoA/ROCK signaling on diabetic kidneys are mediated at least in part by inhibition of YAP activation. In addition, the current study indicated that both RhoA and Akt activation are downstream of EGFR activation and they reciprocally interact to regulate YAP activation in response to high glucose.
YAP/TAZ activation has been implicated in lung, liver, skin, and solid cancer fibrogenesis (55–57), but it also promotes renal recovery following ischemic injury (54) and promotes cardiomyocyte regeneration and reduces cardiac fibrosis in adult heart (58). In kidney, YAP/TAZ activation in fibroblasts has been reported to mediate the effects of TGFβ-Smad during kidney fibrogenesis, and systemic inhibition of YAP/TAZ activation attenuated interstitial fibrosis present in mice with unilateral ureteral obstruction (18,59). In addition, YAP activation by specific deletion of Sav1 in renal tubular epithelial cells has been reported to aggravate tubulointerstitial fibrosis in the unilateral ureteral obstruction mouse model (60), but definitive mechanisms by which YAP activation in kidney contributes to the development of kidney fibrosis were not elucidated in previous studies. Our current study investigated the potential role and the underlying mechanism of YAP activation in the diabetic RPTC and found that YAP activation in RPTC by high glucose is mediated by EGFR-dependent activation of RhoA/ROCK and PI3K-Akt, leading to production of profibrotic and inflammatory factors and paracrine activation of resident fibroblasts.
YAP/TAZ has been implicated as a mechanosensor and a regulator of cell-cell and cell-ECM interactions (49,50). Activation of Rho/ROCK signaling is also implicated in sensing the adjacent matrix tension or forces by regulation of the actomyosin cytoskeleton (51,53). Excessive ECM production may enhance the stiffness of peritubular matrix, which could further activate RhoA/ROCK signaling to form a vicious cycle of hyperglycemia–RhoA/ROCK–YAP–ECM–matrix stiffness–RhoA/ROCK, thereby driving the progression of fibrosis in DN.
CTGF is a well-known target of YAP activation (61). Plasma and urinary CTGF levels are elevated in patients with DN (62,63), and urinary and plasma CTGF have been suggested to be useful biomarkers of fibrogenesis in chronic kidney disease (64). Moreover, previous studies have suggested that genetic inhibition of CTGF expression protected against kidney injury in mouse models of both type 1 and type 2 diabetes and overexpression of CTGF worsened the DN (47). However, the source of plasma and urinary CTGF in patients with diabetes remains unclear. Our study indicates that CTGF and TGFβ2 were strikingly upregulated in hYAP1S127A-HEK cells and released into the conditioned medium and that silencing CTGF by siRNA in the hYAP1S127A-HEK cells not only inhibited fibroblast proliferation and myofibroblast transition induced by the conditioned media but also blocked TGFβ2 expression and release. These results suggest that YAP-dependent production and release of CTGF in diabetic RPTC may be a critical source of plasma and urinary CTGF in patients with diabetes. However, CTGF is not the only driving force in diabetic kidney fibrosis because inhibition of CTGF only attenuates, but does not halt, DN progression (47). In this regard, YAP activation also upregulates other profibrotic and inflammatory factors, such as CCL2, COL3A1, and THBS1, besides CTGF that may also drive diabetic interstitial fibrosis.
Persistent aberrant EGFR activation contributes to pathologic development of tubulointerstitial fibrosis or podocyte injury in response to hypertension or diabetes (34,40,65). Our recent study found that the beneficial effects of EGFR activation in acute kidney injury were the result of PI3K-AKT pathway–dependent YAP activation and subsequent upregulation of cyclin D expression and Rb protein phosphorylation (32). However, the current study revealed that YAP activation in diabetic RPTC through an EGFR-dependent PI3K-AKT activation contributes to renal interstitial fibrogenesis. The active YAP binds to various transcription factors and/or activates different gene sets in response to acute or chronic insult, which may explain the differential effects of YAP activation in RPTC by the same EGFR-PI3K-AKT pathway. However, elucidation of the underlying mechanism by which active YAP selectively binds to and activates different transcriptional factors following acute or chronic injury will require further study.
Article Information
Acknowledgments. IF images were captured with the aid of the Vanderbilt Cell Imaging Shared Resource.
Funding. This work was supported by funds from American Diabetes Association grant 1-18-IBS-267 (to J.C.); National Institutes of Health grants DK51265, DK62794, and DK79341 (to R.C.H. and M.-Z.Z.); Department of Veterans Affairs grant 00507969 (to R.C.H.); and the Vanderbilt O’Brien Kidney Center (DK114809).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. R.C.H. and J.C. designed the study. J.C., X.W., Q.H., N.B., and M.-Z.Z. carried out experiments. R.C.H., J.C., X.W., and A.B.F. analyzed the data. J.C. and X.W. made the figures. J.C. and X.W. drafted and R.C.H revised the manuscript, and all authors approved the final version of the manuscript. J.C. and R.C.H. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented at the 2019 American Society of Nephrology Annual Meeting, Washington, DC, 7–10 November 2019.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.12808352.
References
- 1.Martínez-Castelao A, Navarro-González JF, Górriz JL, de Alvaro F. The concept and the epidemiology of diabetic nephropathy have changed in recent years. J Clin Med 2015;4:1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkins RC, Zimmet P. Diabetic kidney disease: act now or pay later. Kidney Int 2010;77:375–377 [DOI] [PubMed] [Google Scholar]
- 3.Bonventre JV Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol 2012;32:452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chevalier RL The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 2016;311:F145–F161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miltényi M, Körner A, Tulassay T, Szabó A. Tubular dysfunction in type I diabetes mellitus. Arch Dis Child 1985;60:929–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonventre JV Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int Suppl (2011) 2014;4:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu C, Sun L, Xiao L, et al. . Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr Med Chem 2015;22:2858–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeffler I, Liebisch M, Wolf G. Collagen VIII influences epithelial phenotypic changes in experimental diabetic nephropathy. Am J Physiol Renal Physiol 2012;303:F733–F745 [DOI] [PubMed] [Google Scholar]
- 9.Genovese F, Manresa AA, Leeming DJ, Karsdal MA, Boor P. The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 2014;7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast—implications for kidney fibrosis. Nat Rev Nephrol 2015;11:233–244 [DOI] [PubMed] [Google Scholar]
- 11.Loeffler I, Wolf G. Epithelial-to-mesenchymal transition in diabetic nephropathy: fact or fiction? Cells 2015;4:631–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeBleu VS, Taduri G, O’Connell J, et al. . Origin and function of myofibroblasts in kidney fibrosis. Nat Med 2013;19:1047–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonson MS Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int 2007;71:846–854 [DOI] [PubMed] [Google Scholar]
- 14.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 2014;94:1287–1312 [DOI] [PubMed] [Google Scholar]
- 15.Sudol M, Bork P, Einbond A, et al. . Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem 1995;270:14733–14741 [DOI] [PubMed] [Google Scholar]
- 16.Macias MJ, Hyvönen M, Baraldi E, et al. . Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature 1996;382:646–649 [DOI] [PubMed] [Google Scholar]
- 17.Noguchi S, Saito A, Nagase T. YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int J Mol Sci 2018;19:3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szeto SG, Narimatsu M, Lu M, et al. . YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J Am Soc Nephrol 2016;27:3117–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J, Li PX, Wu J, et al. . Involvement of the Hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin Sci (Lond) 2016;130:349–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNeill H, Reginensi A. Lats1/2 regulate Yap/Taz to control nephron progenitor epithelialization and inhibit myofibroblast formation. J Am Soc Nephrol 2017;28:852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DH, Choi HI, Park JS, et al. . Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. FASEB J 2019;33:11109–11122 [DOI] [PubMed] [Google Scholar]
- 22.Xu C, Wang L, Zhang Y, et al. . Tubule-specific Mst1/2 deficiency induces CKD via YAP and non-YAP mechanisms. J Am Soc Nephrol 2020;31:946–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Harris RC. Interaction of the EGF receptor and the Hippo pathway in the diabetic kidney. J Am Soc Nephrol 2016;27:1689–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol 2007;50:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.David M, Petit D, Bertoglio J. Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012;11:3003–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boivin D, Béliveau R. Subcellular distribution and membrane association of Rho-related small GTP-binding proteins in kidney cortex. Am J Physiol 1995;269:F180–F189 [DOI] [PubMed] [Google Scholar]
- 27.Komers R Rho kinase inhibition in diabetic kidney disease. Br J Clin Pharmacol 2013;76:551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikuchi Y, Yamada M, Imakiire T, et al. . A Rho-kinase inhibitor, fasudil, prevents development of diabetes and nephropathy in insulin-resistant diabetic rats. J Endocrinol 2007;192:595–603 [DOI] [PubMed] [Google Scholar]
- 29.Xie X, Peng J, Chang X, et al. . Activation of RhoA/ROCK regulates NF-κB signaling pathway in experimental diabetic nephropathy. Mol Cell Endocrinol 2013;369:86–97 [DOI] [PubMed] [Google Scholar]
- 30.Regué L, Mou F, Avruch J. G protein-coupled receptors engage the mammalian Hippo pathway through F-actin: F-Actin, assembled in response to Galpha12/13 induced RhoA-GTP, promotes dephosphorylation and activation of the YAP oncogene. BioEssays 2013;35:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai J, Song X, Wang W, et al. . A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev 2018;32:781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, You H, Li Y, Xu Y, He Q, Harris RC. EGF receptor-dependent YAP activation is important for renal recovery from AKI. J Am Soc Nephrol 2018;29:2372–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J, Chen JK, Nagai K, et al. . EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 2012;23:215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Chen JK, Harris RC. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J Am Soc Nephrol 2015;26:1115–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J 1999;18:2551–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strano S, Munarriz E, Rossi M, et al. . Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem 2001;276:15164–15173 [DOI] [PubMed] [Google Scholar]
- 37.Vitolo MI, Anglin IE, Mahoney WM Jr., et al. . The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biol Ther 2007;6:856–863 [DOI] [PubMed] [Google Scholar]
- 38.Omerovic J, Puggioni EM, Napoletano S, et al. . Ligand-regulated association of ErbB-4 to the transcriptional co-activator YAP65 controls transcription at the nuclear level. Exp Cell Res 2004;294:469–479 [DOI] [PubMed] [Google Scholar]
- 39.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 2001;15:1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang MZ, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 2014;63:2063–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanetsuna Y, Takahashi K, Nagata M, et al. . Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol 2007;170:1473–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol 2013;8:241–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170:1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao B, Wei X, Li W, et al. . Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007;21:2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neelisetty S, Alford C, Reynolds K, et al. . Renal fibrosis is not reduced by blocking transforming growth factor-β signaling in matrix-producing interstitial cells. Kidney Int 2015;88:503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perbal B CCN proteins: multifunctional signalling regulators. Lancet 2004;363:62–64 [DOI] [PubMed] [Google Scholar]
- 47.Kok HM, Falke LL, Goldschmeding R, Nguyen TQ. Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nat Rev Nephrol 2014;10:700–711 [DOI] [PubMed] [Google Scholar]
- 48.Aragona M, Panciera T, Manfrin A, et al. . A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013;154:1047–1059 [DOI] [PubMed] [Google Scholar]
- 49.Calvo F, Ege N, Grande-Garcia A, et al. . Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013;15:637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dupont S, Morsut L, Aragona M, et al. . Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–183 [DOI] [PubMed] [Google Scholar]
- 51.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992;70:389–399 [DOI] [PubMed] [Google Scholar]
- 52.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005;21:247–269 [DOI] [PubMed] [Google Scholar]
- 53.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu-Chittenden Y, Huang B, Shim JS, et al. . Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noguchi S, Saito A, Mikami Y, et al. . TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci Rep 2017;7:42595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mannaerts I, Leite SB, Verhulst S, et al. . The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 2015;63:679–688 [DOI] [PubMed] [Google Scholar]
- 57.Toyama T, Looney AP, Baker BM, et al. . Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J Invest Dermatol 2018;138:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leach JP, Heallen T, Zhang M, et al. . Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017;550:260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang M, Yu M, Xia R, et al. . Yap/Taz deletion in Gli+ cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol 2017;28:3278–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leung JY, Wilson HL, Voltzke KJ, et al. . Sav1 loss induces senescence and Stat3 activation coinciding with tubulointerstitial fibrosis. Mol Cell Biol 2017;37:e00565-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao B, Ye X, Yu J, et al. . TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen TQ, Tarnow L, Jorsal A, et al. . Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care 2008;31:1177–1182 [DOI] [PubMed] [Google Scholar]
- 63.Riser BL, Cortes P, DeNichilo M, et al. . Urinary CCN2 (CTGF) as a possible predictor of diabetic nephropathy: preliminary report. Kidney Int 2003;64:451–458 [DOI] [PubMed] [Google Scholar]
- 64.Dendooven A, Gerritsen KG, Nguyen TQ, Kok RJ, Goldschmeding R. Connective tissue growth factor (CTGF/CCN2) ELISA: a novel tool for monitoring fibrosis. Biomarkers 2011;16:289–301 [DOI] [PubMed] [Google Scholar]
- 65.Chen J, Chen JK, Harris RC. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Mol Cell Biol 2012;32:981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]