

Abstract
HIV-1 integrase (IN) is an enzyme which is indispensable for the stable infection of host cells because it catalyzes the insertion of viral DNA into the genome and thus is an attractive target for the development of anti-HIV agents. Earlier, we found Vpr-derived peptides with inhibitory activity against HIV-1 IN. These Vpr-derived peptides are originally located in an α-helical region of the parent Vpr protein. Addition of an octa-arginyl group to the inhibitory peptides caused significant inhibition against HIV replication associated with an increase in cell permeability but also relatively high cytotoxicity. In the current study, stapled peptides, a new class of stabilized α-helical peptidomimetics were adopted to enhance the cell permeability of the above lead peptides. A series of stapled peptides, which have a hydrocarbon link formed by a ruthenium-catalyzed ring-closing metathesis reaction between successive turns of α-helix, were designed, synthesized, and evaluated for biological activity. In cell-based assays some of the stapled peptides showed potent anti-HIV activity comparable with that of the original octa-arginine-containing peptide (2) but with lower cytotoxicity. Fluorescent imaging experiments revealed that these stapled peptides are significantly cell permeable, and CD analysis showed they form α-helical structures, whereas the unstapled congeners form β-sheet structures. The application of this stapling strategy to Vpr-derived IN inhibitory peptides led to a remarkable increase in their potency in cells and a significant reduction of their cytotoxicity.
Graphical Abstract
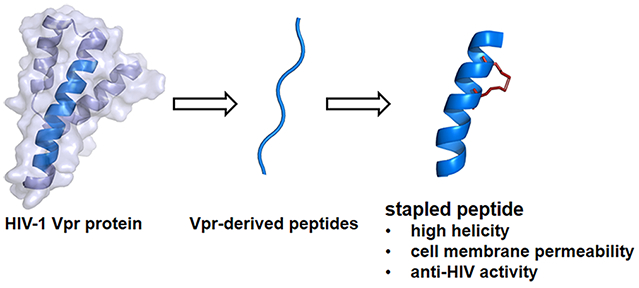
Several antiretroviral drugs have been developed to date and are now available to treat human immunodeficiency virus type 1 (HIV-1) infection and acquired immunodeficiency syndrome (AIDS).1 The main targets for antiretroviral drugs are viral enzymes such as protease, reverse transcriptase (RT) and integrase (IN) and surface proteins such as gp41 and coreceptors. New anti-HIV-1 drugs operating with different inhibitory mechanisms however, are required due to the emergence of viral strains with multidrug resistance (MDR), side effects and high costs of treatments. IN, an indispensable enzyme for the stable infection of the virus into host cells, catalyzes the insertion of viral DNA into the genome of host cells via strand transfer and 3′-end processing reactions. Thus, it is an attractive target for anti-HIV-1 drugs.2,3 It has been assumed that during the transfer of viral DNA from cytoplasm to nucleus, the activity of IN must be negatively regulated to prevent autointegration, which might abort the infection and we speculate that HIV must invoke a mechanism preventing the autointegration. The viral preintegration complex (PIC) contains viral nucleic acids; viral proteins such as RT, IN, capsids (p24CA and p7NC), matrix (p17MA), p6 and Vpr; cellular proteins HMG I (Y); and a barrier to autointegration factor (BAF).4-7 Since these proteins exist in proximity in the PIC, they might physically and functionally interact with each other, and thus PIC components may regulate one another’s function. An example of this is inhibition of IN activity by Vpr through its C-terminal domain.8,9 In our previous study, the screening of an overlapping peptide library derived from HIV-1 proteins led to the identification of several peptide motifs with inhibitory activity against HIV-1 IN,10 and the evaluation of effective inhibition of HIV-1 replication in cells using the identified peptide inhibitors possessing a cell membrane-permeable octa-arginine.11 The original Vpr-fragment (1) and its octa-arginine-conjugate with cell membrane permeability (2) have been developed as lead peptides (Figure 1), which are located in the second helix region of the whole protein Vpr. In the structure–activity relationship studies, Glu-Lys pairs were introduced into the i and i + 4 positions to increase the helicity, and Ala-scan was also performed on the lead compound to identifiy the amino acid residues responsible for the inhibitory activity.12 However, the addition of octa-arginine to these peptides led to an increase in cytotoxicity.
Figure 1.

Amino acid sequences of peptides 1 and 2.
In the current study, hydrocarbon-stapled peptides, often utilized as a new class of stabilized α-helical peptidomimetics,13-15 were adopted to enhance cell permeability in the above peptides, without octa-arginine (Figure 2). In stapled peptides, a ruthenium-catalyzed ring-closing metathesis (RCM) reaction between successive turns of α-helix causes formation of a hydrocarbon link.16,17 Several stapled peptides were designed and synthesized, and their anti-HIV assay and bioimaging experiments using living cells were performed.
Figure 2.
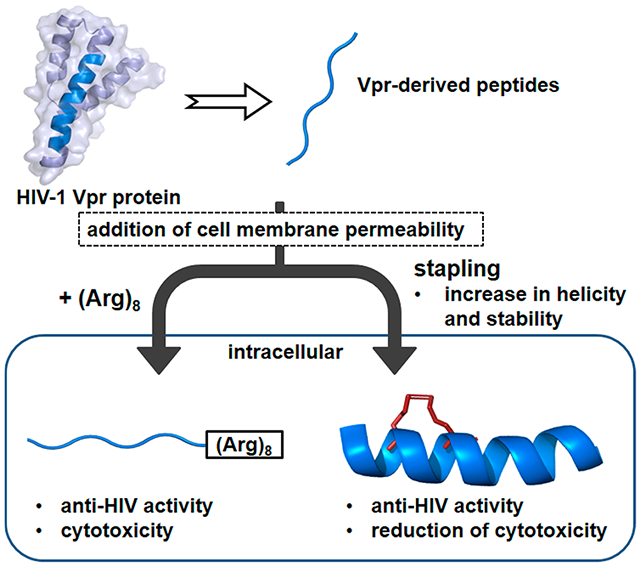
Outline of this study.
RESULTS AND DISCUSSION
Design and Synthesis of Stapled Peptides.
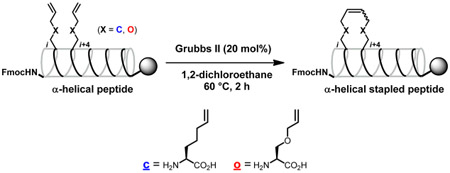
The lead peptides 1 and 2 are shown in Figure 1.10 It was found in our previous Ala-scan study that Phe12, Ile13, Phe15, and Ile17 are indispensable for IN inhibitory and anti-HIV activities.12 Avoiding substitution for these residues, several stapled peptides cross-linked between the i and i + 4 positions were designed using all-hydrocarbon staple-type18-20 and ether staple-type amino acid derivatives21 (Table 1). Okamoto et al. reported that staple positioning was indeed important in binding of BimBH3 peptides to pro-survival proteins.20 Protected linear peptides were constructed by Fmoc-solid phase peptide synthesis on the Rink amide resin using Fmoc-protected all-hydrocarbon staple-type and ether staple-type amino acid derivatives (Scheme 1). The RCM reactions of the protected linear peptides were performed on the resin by treatment with 20 mol % of the ruthenium-mediated Grubbs second catalyst in 1,2-dichloroethane at 60 °C for 2 h. The RCM reactions of the all-hydrocarbon staple-types gave high yields (83–97%), while those of the ether staple-types did not cause adequate conversion (~0–57%), probably due to their instability or hydrophilicity. The yields of the conversion in the RCM reactions from linear to cyclic peptides are shown in Table 1. With peptides that have low conversion yields, it might be difficult for the two olefin moieties to interact each other and cross-link. The configuration of the double bonds was not determined.22 After RCM, the Fmoc group at the N-terminus was removed, and the peptide was subsequently acetylated followed by the removal of the protecting groups and cleavage of the peptides from the resin using TFA. Purification of the crude peptides by preparative HPLC provided the stapled peptides as final products. In the synthesis of the corresponding linear peptides with olefinic side chains, the Fmoc group at the N-terminus was removed in the absence of RCM reactions, and the peptide was subsequently acetylated, the protecting groups were removed, and the peptide was cleaved from the resin using TFA and purified by HPLC. Since most of ether staple-type peptides had low conversion yields in the RCM reactions, only compounds 11S and 11L were synthesized. The conjugates of 6S with octa-arginine (17) and with the quartet repeat of arginine and glutamic acid (18), the conjugate of 6L with the quartet repeat of arginine and glutamic acid (19), and the conjugates of 6S with tetra-arginine (20), with penta-arginine (21), with hexa-arginine (22), and with hepta-arginine (23) were synthesized using the same method (Figure 5). For fluorescein-labeled peptides of 3S, 6S, 8S, 6L, 1, and 2, Fmoc-GABA-OH and fluorescein were condensed after the deprotection of the Fmoc group at the N-terminus (Supporting Information).
Table 1.
Sequences of Designed Stapled Peptides and Their Conversion Yields of RCM Reactions
 | ||
|---|---|---|
| peptide | sequence | conversion [%]a) |
| 3 | Ac-cAIIcILQQLLFIHFRIG-NH2 | 83 |
| 4 | Ac-EcIIRcLQQLLFIHFRIG-NH2 | 86 |
| 5 | Ac-EAcIRIcQQLLFIHFRIG-NH2 | 89 |
| 6 | Ac-EAIcRILcQLLFIHFRIG-NH2 | 85 |
| 7 | Ac-EAIIcILQcLLFIHFRIG-NH2 | 94 |
| 8 | Ac-EAIIRcLQQcLFIHFRIG-NH2 | 97 |
| 9 | Ac-EAIIRIcQQLcFIHFRIG-NH2 | 96 |
| 10 | Ac-oAIIoILQQLLFIHFRIG-NH2 | 19 |
| 11 | Ac-EoIIRoLQQLLFIHFRIG-NH2 | 57 |
| 12 | Ac-EAoIRIoQQLLFIHFRIG-NH2 | 50 |
| 13 | Ac-EAIoRILoQLLFIHFRIG-NH2 | trace |
| 14 | Ac-EAIIoILQoLLFIHFRIG-NH2 | 9 |
| 15 | Ac-EAIIRoLQQoLFIHFRIG-NH2 | 20 |
| 16 | Ac-EAIIRIoQQLoFIHFRIG-NH2 | trace |
Determined by peak areas in reverse phase HPLC after deprotection and cleavage from resins.
Scheme 1.
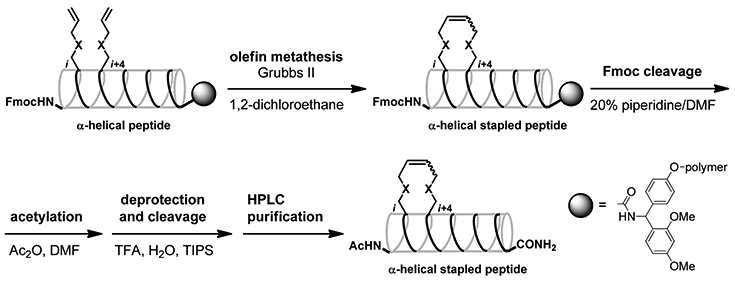
Figure 5.
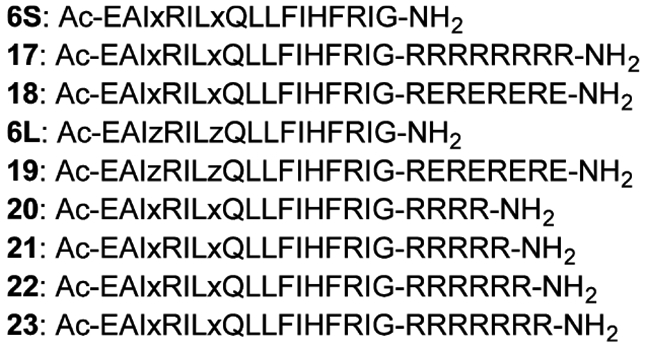
Amino acid sequences of the conjugates of 6S with octa-arginine (17) and with the quartet repeat of arginine and glutamic acid (18), the conjugate of 6L with the quartet repeat of arginine and glutamic acid (19), and the conjugates of 6S with tetra-arginine (20), penta-arginine (21), hexa-arginine (22), and hepta-arginine (23). x = cross-linked all-hydrocarbon staple-type amino acid, z = 2-aminohept-6-enoic acid.
CD Spectroscopy of Linear and Stapled Peptides.
The secondary structures of the synthetic peptides, stapled peptides, 3S–9S and 11S, and the linear peptides, 4L–6L, 8L, 9L, and 11L, were analyzed by CD spectroscopy (Figure 3). All of the stapled peptides, with the exception of 3S, showed double negative peaks at 208 and 222 nm, characteristic of an α-helical structure. In contrast, the corresponding linear peptides including compound 1 showed a broad negative peak around 215 nm, characteristic of a β-sheet structure. It is clear that stapling of the linear peptides causes a significant increase in α-helicity. Only one stapled peptide (3S) failed to show any sign of an α-helical structure, and consequently the corresponding linear peptide, 3L, was not used as a control.
Figure 3.
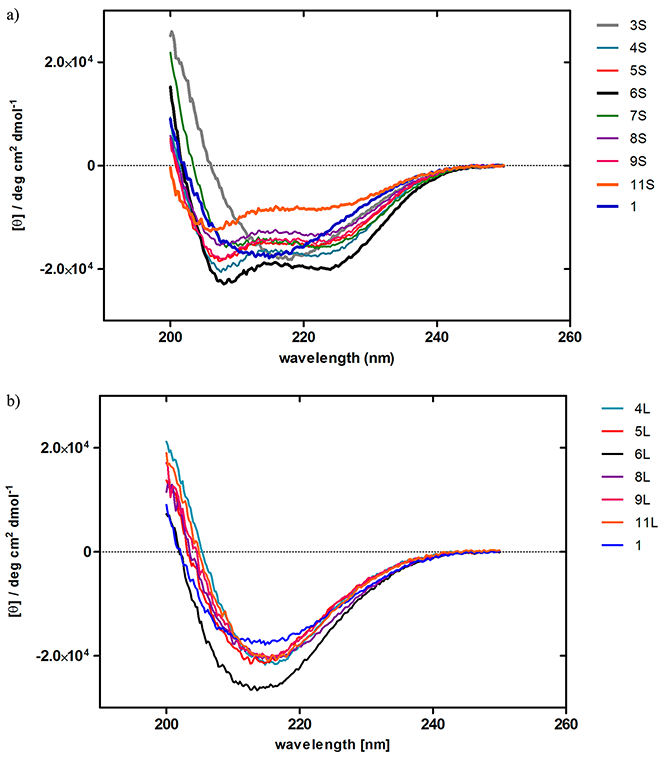
CD spectra of the stapled peptides 3S–-9S, and 11S (a) and the linear peptides 4L–6L, 8L, 9L, and 11L (b) (12.5 μM) in H2O containing 10% CH3CN and 0.625% TFE at 20 °C.
Integrase (IN) Inhibition Assays of Stapled Peptides, 3S–9S and 11S, and Linear Peptides, 4L–6L, 8L, 9L, and 11L.
The IN inhibitory activities of these compounds were evaluated in vitro by the 3′-end processing and strand transfer reactions (Table 2).23-27 Compounds 4S, 5S, 6S, 6L, 8L, and 9L showed almost the same inhibitory levels as compound 1 in both the 3′-end processing and strand transfer reactions. Compounds 3S, 8S, 9S, 4L, 5L, and 11L showed 1.3–4.0-fold lower inhibitory activities than compound 1 in both 3′-end processing and strand transfer reactions. Compound 7S showed remarkably lower inhibitory potency against both activities. Compound 11S showed lower inhibitory activity on the 3′-end processing reaction than compound 1 but almost the same inhibitory level on the strand transfer reaction as that of compound 1. Compound 3S showed a β-sheet structure and low inhibitory activity, suggesting that stapling between 1 and 5 positions is not feasible and that the original amino acid residues Glu1 and/or Arg5 are indispensable for IN inhibitory activity. Compounds 4S and 5S retained the inhibitory activity of compound 1, while compounds 4L and 5L showed low inhibitory activity. These results suggest that stapling between the 2 and 6 positions and between the 3 and 7 positions is feasible, but the introduction of pentenyl moieties into the 2 and 6 positions or into the 3 and 7 positions is not. Maintenance of inhibitory activity at the level of that of compound 1 in both compounds 6S and 6L revealed that not only stapling between the 4 and 8 positions but also the introduction of pentenyl moieties into the 4 and 8 positions is permissible. The original amino acid residue Ile4 or Gln8 is not essential for IN inhibitory activity. The considerable decrease in IN inhibitory activity of compound 7S showed that Arg5 and Gln9 are clearly important. The stapled peptide 7S showed no significant IN inhibitory activity, and consequently the corresponding linear peptide 7L was not synthesized. A decrease in activity of compounds 8S and 9S and maintenance in activity of compounds 8L and 9L suggest that Ile6, Leu7, Leu10, or Leu11 is not essential for IN inhibitory activity and that stapling by RCM between the 6 and 10 positions and between the 7 and 11 positions might have a disadvantageous effect on the other regions. A decrease in activity of compounds 11S and 11L suggests that the ether-type stapling is unsuitable, possibly because of hydrophilicity, while the reason why the inhibitory activity of 11S against the strand transfer reaction is retained is unclear. Compound 2 showed the highest inhibitory levels on both the 3′-end processing and strand transfer reactions, which were approximately 30× higher than those of compound 1. The reason for this is not clear. The IN inhibition assays against the 3′-end processing and strand transfer reactions were performed in vitro, and thus, cell membrane permeability is not relevant.
Table 2.
Inhibitory Activities (IC50 Values) of the Stapled and Linear Peptides toward the 3′-End Processing and Strand Transfer Reactions Catalyzed by HIV-1 IN
| peptide | I50
μM)> |
|
|---|---|---|
| 3′-end processing | strand transfer | |
| 3S (stapled) | 5.8 ± 0.8 | 3.8 ± 0.7 |
| 4S (stapled) | 2.5 ± 0.4 | 1.1 ± 0.08 |
| 5S (stapled) | 2.8 ± 0.3 | 1.2 ± 0.09 |
| 6S (stapled) | 2.4 ± 0.4 | 0.84 ± 0.07 |
| 7S (stapled) | >111 | 54.4 ± 9.6 |
| 8S (stapled) | 5.6 ± 0.7 | 4.1 ± 0.6 |
| 9S (stapled) | 4.9 ± 0.6 | 1.9 ± 0.2 |
| 4L (linear) | 5.2 ± 0.5 | 1.7 ± 0.3 |
| 5L (linear) | 9.6 ± 0.6 | 4.4 ± 0.4 |
| 6L (linear) | 2.6 ± 0.4 | 1.2 ± 0.09 |
| 8L (linear) | 2.5 ± 0.3 | 1.0 ± 0.07 |
| 9L (linear) | 2.1 ± 0.2 | 0.9 ± 0.06 |
| 11S (stapled) | 4.2 ± 0.7 | 0.7 ± 0.08 |
| 11L (linear) | 3.5 ± 0.3 | 1.7 ± 0.3 |
| 1 | 2.6 ± 0.2 | 1.1 ± 0.08 |
| 2 | 0.079 ± 0.007 | 0.036 ± 0.008 |
MT-4 Luc Assays of Stapled Peptides, 3S–9S and 11S, and Linear Peptides, 4L–6L, 8L, 9L, and 11L.
Anti-HIV activity of these compounds was assessed by an MT-4 Luc system, in which MT-4 cells were stably transduced with the firefly luciferase expression cassette by a murine leukemia viral vector.28 MT-4 Luc cells constitutively express high levels of luciferase, but these high levels are significantly reduced by HIV-1 infection due to the high susceptibility of MT-4 cells to HIV-1 infection. Protection of MT-4 Luc cells from HIV-1-induced cell death maintains the luciferase signals at high levels. The cytotoxicity of test compounds can be evaluated by a decrease of luciferase signals in these MT-4 Luc systems. As reported previously, the parent compound (2) showed significant anti-HIV activity at concentrations above 2.5 μM (Figure 4).10,12 Compound 1 showed no significant anti-HIV activity at concentrations below 10 μM, presumably due to lack of cell membrane permeability. Compound 6S showed significant anti-HIV activity at concentrations above 2.5 μM, its activity level being consistent with that of compound 2. Compound 8S at a concentration of 10 μM also showed significant anti-HIV activity. The corresponding linear peptides 6L and 8L did not show significant anti-HIV activity at concentrations below 10 μM, suggesting that stapling was effective for both cell membrane permeability and the expression of activity inside cells with the result that HIV-1 replication was effectively inhibited. The other stapled peptides did not show significant anti-HIV activity at concentrations below 10 μM, possibly because of the introduction of the cross-link in inappropriate positions. All of the linear peptides failed to show significant anti-HIV activity, indicating that these peptides do not enter the cytoplasm or cannot access IN. Compounds 6S and 2 have almost the same level of anti-HIV activity in cells, although 2 has much higher IN inhibitory activity in vitro than compound 6S. The additional octa-arginine sequence might play a functional role besides influencing cell membrane permeability, and thus, conjugates of compounds 6S and 6L with hydrophilic sequences were synthesized to investigate the functional role of octa-arginine.
Figure 4.
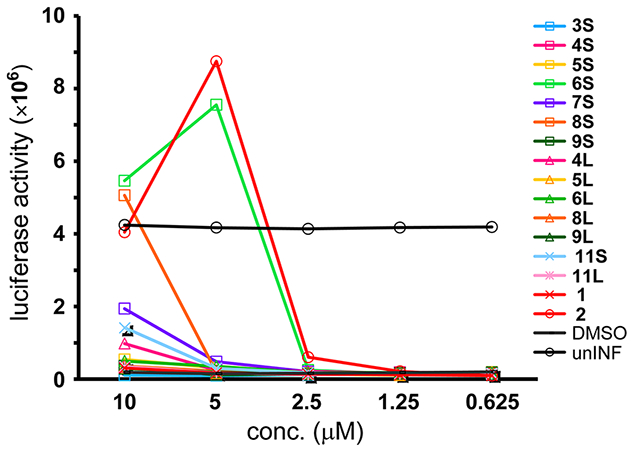
Luciferase signals in MT-4 Luc cells infected with HIV-1 in the presence of different concentrations of compounds 3S–9S, 4L–6L, 8L, 9L, 11S, and 11L. Luciferase activity is expressed as relative luciferase units (RLU). unINF: uninfected.
IN Inhibition Assays of Conjugates of Hydrophilic Sequence-containing Compounds 6S and 6L.
As a comparative study of additional hydrophilic sequences, we investigated the functional role of octa-arginine with the conjugates of compounds 6S and 6L with the quartet repeat of arginine and glutamic acid (Figure 5, Table 3). Compound 17 showed high inhibitory levels with respect to both the 3′-end processing and strand transfer reactions and its activity levels were comparable with those of compound 2. Compound 18 showed lower IN inhibitory levels, which were almost the same levels as those of compounds 19, 6S, and 1. It suggests that the addition of the quartet repeat of arginine and glutamic acid does not lead to an increase in IN inhibitory activities, and that the only addition of octa-arginine is effective.
Table 3.
Inhibitory Activities (IC50 Values) toward the 3′-End Processing and Strand Transfer Reactions Catalyzed by HIV-1 IN, DNA Binding Effects, anti-HIV Activities (EC50 Values, p24, and MTT assays) and Cytotoxicities (CC50 Values, MTT Assay) of the Peptides with Hydrophilic Sequences
| peptide | IC50 (μM) 3′-end processing |
IC50 (μM) strand transfer |
DNA binding effect (μM)a |
EC50 (μM) p24 assay |
EC50 (μM) MTT assay |
CC50(μM) MTT assay |
|---|---|---|---|---|---|---|
| 17 | 0.14 ± 0.02 | 0.056 ± 0.011 | 4 | 4.29 | 3.08 | 7.04 |
| 18 | 1.47 ± 0.18 | 0.81 ± 0.13 | 37 | 9.56 | 6.81 | >10 |
| 19 | 1.95 ± 0.25 | 1.13 ± 0.11 | 37 | >10 | >10 | >10 |
| 6S | 1.87 ± 0.14 | 0.71 ± 0.12 | 37 | 6.46 | 3.55 | >10 |
| 6L | N.T. | N.T. | N.T. | >10 | >10 | >10 |
| 8S | N.T. | N.T. | N.T. | (14.22) | 8.07 | >10 |
| 8L | N.T. | N.T. | N.T. | >10 | >10 | >10 |
| 5S | 2.6 ± 0.2 | 0.87 ± 0.14 | 37 | >10 | >10 | >10 |
| 1 | 1.62 ± 0.26 | 1.34 ± 0.2 | 37 | >10 | >10 | >10 |
| 2 | 0.119 ± 0.015 | 0.05 ± 0.004 | 4 | 3.51 | 4.54 | 5.91 |
| AZT | N.T. | N.T. | N.T. | 0.02 | 0.07 | >100 |
The lowest peptide concentrations that show retardation of DNA on wells of PAGE gels in integrase assays. That the same compounds give slightly different IC50 values, as shown in Tables 2-4 can be explained by the fact that batches of compounds were tested at different times and that overlapping compounds were used to ensure the reproducibility of the assays.
MT-4 Luc Assays of Conjugates of Compounds 6S and 6L with Hydrophilic Sequences.
Anti-HIV activity of these conjugates was assessed by the MT-4 Luc system (Figure 6). Compound 17 showed significant anti-HIV activity at concentrations above 1.25 μM but significant cytotoxicity at a concentration of 10 μM. Compound 18 showed significant anti-HIV activity at concentrations above 5 μM; its activity level was almost the same as that of compound 6S. Compound 19 showed significant anti-HIV activity at a concentration of 10 μM, whereas compound 6L did not show significant anti-HIV activity at concentrations below 10 μM. Thus the addition of the quartet repeat of arginine and glutamic acid results in a slight increase in anti-HIV activity in cells, but the increase was much lower than that resulting from the addition of octa-arginine. It is noteworthy that the octa-arginine addition has a possibility to cause cytotoxicity and also that stapled peptides have sufficient cell membrane permeability
Figure 6.
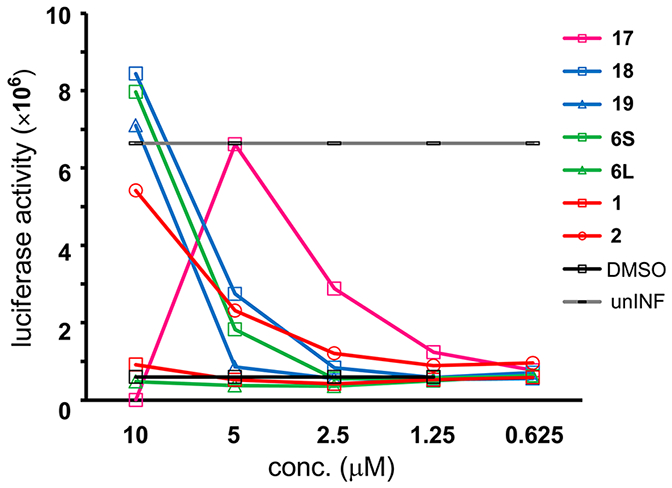
Luciferase signals in MT-4 Luc cells infected with HIV-1 in the presence of different concentrations of compounds 17, 18, and 19. Luciferase activity is expressed as relative luciferase units (RLU).
P24 ELISA and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assays of Conjugates of Hydrophilic Sequence-Containing Compounds 6S and 6L.
The MT-4 Luc assay evaluates anti-HIV activity and cytotoxicity of compounds simultaneously: HIV-1-induced MT-4 cell death and compound-derived cell death both cause reduction of the luciferase signals, and consequently, anti-HIV activity and cytotoxicity of compounds cannot be distinguished by this assay system. The p24 ELISA and MTT assays of conjugates of compounds 6S and 6L with hydrophilic sequences were performed (Table 3). EC50 values in the p24 ELISA assay were based on the reduction of the HIV-1 p24 production in MT-4 cells infected with HIV-1 (NL4-3 strain). EC50 values in the MTT assay were based on the protection of HIV-1 (NL4-3 strain)-induced cytopathogenicity in MT-4 cells, and CC50 values in the MTT assay were based on the reduction of the viability of mock infected MT-4 cells. In these tests, compound 17 showed potent anti-HIV activity (EC50 (p24 assay) = 4.29 μM, EC50 (MTT assay) = 3.08 μM), but also significant cytotoxicity (CC50 = 7.04 μM). The anti-HIV activity of compound 18 (EC50 (p24 assay) = 9.56 μM, EC50 (MTT assay) = 6.81 μM) is lower than that of compound 17. This is consistent with the results in the MT-4 Luc assay (Figure 6). Compound 19 did not show significant anti-HIV activity at concentrations below 10 μM either in the p24 assay or in the MTT assay, but compound 19 showed significant but moderate anti-HIV activity at a concentration of 10 μM in the MT-4 Luc assay. Since MT-4 cells possess luciferase, background signals can be detected in the MT-4 Luc assay whose sensitivity compared to that of the more reliable p24 or MTT assays is not therefore refined. Consequently, compound 19 might have minimal or no anti-HIV activity. Compound 6S has potent anti-HIV activity (EC50 (p24 assay) = 6.46 μM, EC50 (MTT assay) = 3.55 μM), which is consistent with that of compound 17. The corresponding linear peptide 6L did not show significant anti-HIV activity at concentrations below 10 μM in either the p24 assay or in the MTT assay, indicating the effect of stapling. Compound 6S did not show significant cytotoxicity at concentrations below 10 μM in common with all the peptides lacking octa-arginine. Only compounds 17 and 2 showed potent cytotoxicity (CC50 = 7.04 μM, CC50 = 5.91 μM, respectively). The second most potent stapled peptide (8S) and its corresponding linear peptide (8L) showed a certain level of anti-HIV activity and almost no significant anti-HIV activity, respectively. These results are consistent with those in the MT-4 Luc assay (Figures 4 and 6). Compound 5S, which did not show high anti-HIV activity in the MT-4 Luc assay, failed to exhibit significant anti-HIV activity at concentrations below 10 μM either in the p24 or the MTT assay. Compound 2 showed potent anti-HIV activity and cytotoxicity (EC50 (p24 assay) = 3.51 μM, EC50 (MTT assay) = 4.54 μM, CC50 = 5.91 μM), whereas compound 1 did not show significant anti-HIV activity or cytotoxicity at concentrations below 10 μM. These results are consistent with those from the MT-4 Luc assay. In the expression of anti-HIV activity in cells, stapling is therefore sufficient, and the addition of octa-arginine is not desirable because of cytotoxicity.
DNA Binding Effects of Conjugates of Compounds 6S and 6L with Hydrophilic Sequences.
In spite of the assay results obtained previously, the functional role of the additional octa-arginine sequence remains unclear. In the both the 3′-end processing and strand transfer reactions in vitro, the addition of the quartet repeat of arginine and glutamic acid failed to increase IN inhibitory activities, and only addition of octa-arginine was effective. Thus, the effects on DNA binding of conjugates of compounds 6S and 6L with hydrophilic sequences were investigated. Significant DNA binding effects were observed in the octa-arginine-containing peptides 17 and 2 at a concentration of 4 μM, whereas in the peptides with the quartet repeat of arginine and glutamic acid or lacking an additional sequence, DNA binding effects were observed at a concentration of 37 μM. This suggests that octa-arginine plays a critical role in DNA binding, perhaps because the arginine guanidino groups can bind to phosphonate groups of DNA and that the binding of peptides to the target DNA might lead to a significant increase in IN inhibitory activity.
IN Inhibition, P24 ELISA, MtT, and DNA Binding Assays of Conjugates of Compound 6S with Oligo-arginine Sequences.
The functional role of the additional octa-arginine sequence appears to be correlated to binding affinity for the target DNA and thus the IN inhibitory activities in vitro. The addition of the quartet repeat of arginine and glutamic acid appears not to have such a significant effect. This suggests that the hydrophilic nature of octa-arginine is not important but the sequential arginine sequence is critical. To investigate effects of the length of oligo-arginine sequences on DNA binding and IN inhibitory activities, the conjugates of compound 6S with tetra- (20), penta- (21), hexa- (22), and hepta-arginine (23) sequences were prepared (Figure 5). Compounds 20 and 21 showed 3–6× higher inhibition of both the 3′-end processing and strand transfer reactions than compound 6S (Table 4). Compound 22 showed 2× higher inhibitory levels than compounds 20 and 21 in both reactions. The activity levels of compound 23 were comparable with those of compound 17. Thus, the IN inhibitory activities appear to be positively correlated with the length of oligo-arginine sequences of the conjugates of compound 6S, and the addition of hepta-arginine is sufficient for increases in IN inhibitory activities. For DNA binding analysis, a fluorescent polarization assay was conducted. The interaction between the oligo-arginine and phosphates of DNA backbone should only be nonspecific electrostatic interactions. Although the changes in polarization did not reach equilibrium over the time-course examined, the differences in the rates of association of the peptides with DNA are positively correlated with the length of the oligo-arginine sequences of the conjugates of compound 6S (Figure 7). In the p24 ELISA and MTT assays, compounds 17 and 20–23 showed almost the same levels of anti-HIV activity (EC50 = 3.1–7.4 μM), although exact EC50 values of compounds 17 and 23 were not determined in the MTT assay because of the high cytotoxicity of the compounds. Cytotoxicity and the length of oligo-arginine sequences of the conjugates of compound 6S were inversely proportional to one another. Since compounds 20 and 21 with tetra- and penta-arginine sequences have sufficient IN inhibitory and DNA binding activities and relatively low cytotoxicity, these are useful leads in the series of stapled peptide-type IN inhibitors.
Table 4.
Inhibitory Activities (IC50 Values) toward the 3′-End Processing and Strand Transfer Reactions Catalyzed by HIV-1 IN, anti-HIV Activities (EC50 Values, p24 and MTT Assays) and Cytotoxicities (CC50 values, MTT Assay) of the Conjugates of Compound 6S with Oligo-arginine Sequences
| peptide | IC50 (μM) 3′-end processing |
IC50 (μM) strand transfer |
EC50 (μM) p24 assay |
EC50 (μM) MTT assay |
CC50 (μM) MTT assay |
|---|---|---|---|---|---|
| 20 | 0.45 ± 0.02 | 0.22 ± 0.02 | 6.5 | 3.2 | 16 |
| 21 | 0.47 ± 0.03 | 0.21 ± 0.03 | 3.2 | 3.1 | 15 |
| 22 | 0.25 ± 0.01 | 0.14 ± 0.03 | 7.4 | 4.7 | 13 |
| 23 | 0.18 ± 0.01 | 0.062 ± 0.005 | 5.6 | 27% at 5 μM | 12 |
| 17 | 0.17 ± 0.01 | 0.076 ± 0.009 | 7.1 | 28% at 5 μM | 6.9 |
| 6S | 2.6 ± 0.1 | 0.75 ± 0.08 | 8.3 | 6.4 | >20 |
| 1 | 2.0 ± 0.2 | 2.2 ± 0.2 | >20 | N.T. | N.T. |
| 2 | 0.16 ± 0.05 | 0.062 ± 0.009 | 6.4 | N.T. | N.T. |
| AZT | N.T. | N.T. | 0.29 | 0.07 | >100 |
Figure 7.
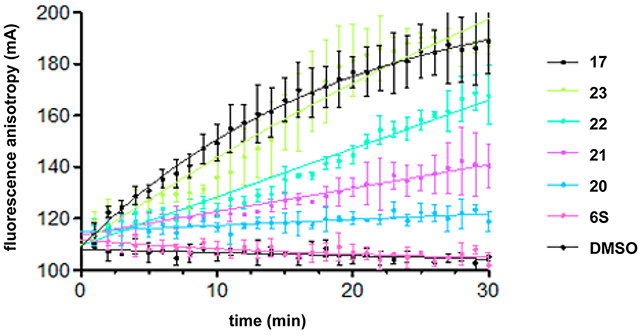
DNA binding properties of the conjugates of 6S with octa-arginine (17), with hepta-arginine (23), with hexa-arginine (22), with penta-arginine (21), and with tetra-arginine (20). Time courses at the 0.5 μM concentration of each peptide are presented. The oligonucleotide concentration is 10 nM.
Stapled and Linear Peptides Labeled with Fluorescein: Imaging Experiments.
To investigate whether stapled and linear peptides penetrate cell membranes, imaging experiments using peptides labeled with fluorescein were performed in HeLa cells (Figure 8). Compound 3S-F, the stapled peptide 3S labeled with fluorescein, was not observed inside cells, and it was concluded that compound 3S-F cannot penetrate cell membranes. According to the CD analysis, compound 3S does not form an α-helical structure (Figure 3) and thus is unable to penetrate cell membranes and did not show significant anti-HIV activity in the MT-4 Luc assay (Figure 4), although it showed significant IN inhibitory activity in vitro (Table 2). Cell membrane penetration of compound 6S-F, the stapled peptide 6S labeled with fluorescein, was observed, whereas cell membrane penetration of compound 6L-F, the linear peptide 6L labeled with fluorescein, was not. In the image of 6L-F, the high accumulation of fluorescent dyes was observed on the cell surface. The phenomenon could suggest the aggregation of peptides. It is thought that, since compound 6S forms an α-helical structure, it can penetrate cell membranes and show potent anti-HIV activity, and since compound 6L does not form an α-helical structure, it cannot penetrate cell membranes and does not show significant anti-HIV activity. Compound 8S-F, the stapled peptide 8S labeled with fluorescein, showed imaging similar to that of compound 6S-F. Compound 8S forms an α-helical structure, can penetrate cell membranes and shows significant anti-HIV activity as in the case of compound 6S. However, in the case of compound 8S-F, the accumulation of fluorescent dyes on the cell surface was also observed. Compound 1-F, the peptide 1 labeled with fluorescein, was not observed inside cells, whereas compound 2-F, peptide 2 labeled with fluorescein, was observed inside cells. The addition to the structure of octa-arginine caused the peptide 1 to penetrate the cell membrane. The concentration of each peptide was 5 μM. In the MTT assay, the CC50 value of peptide 2 was 5.91 μM (Table 3). It is possible that slightly rounded and no lamellipodia extensions of the cell image might be caused by the use of the peptide concentration similar to that of the CC50 value. More cytotoxic effects might be detected if higher concentrations of the peptides were used. As a result, stapling (which can cause an increase in helicity) enhances cell membrane penetration. All the penetrating peptides (peptides 6S-F, 8S-F, and 2-F) in the cell showed localization in cytosol and accumulation near the cell membrane.
Figure 8.
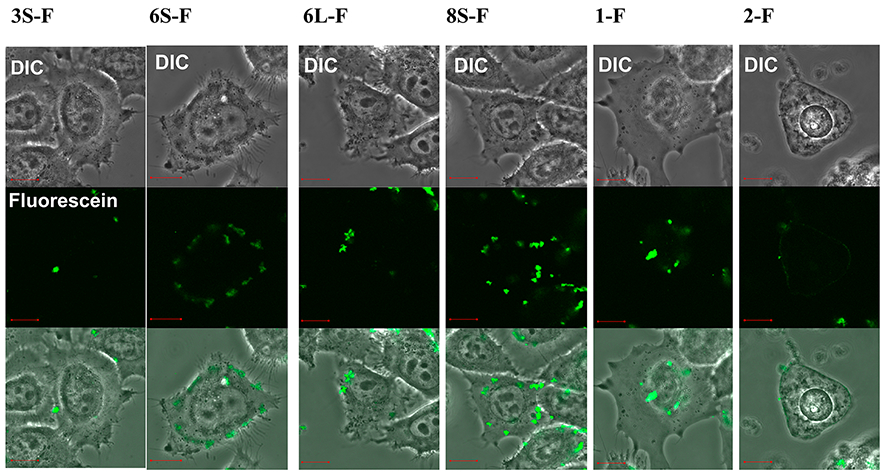
Imaging experiments for cell permeability of the stapled and linear peptides labeled with fluorescein to HeLa cells. Each panel is divided into three sections as follows: upper, differential interference contrast (DIC) image; middle, fluorescein emission; lower, merged image. The peptide concentration is 5 μM. After addition of peptides, cells were incubated at 37 °C for 30 min under 5% CO2 atmosphere. After washing three times with HBSS buffer, the fluorescent imaging was conducted by Fluoview FV10i confocal microscopy systems (Olympus). Orange bars in the panels represent 10 μm. The fluorescein-labeled peptides contain fluorescein–GABA instead of acetyl in the N-terminus.
CONCLUSIONS
Stabilized α-helical peptidomimetics, stapled peptides, were applied to Vpr-derived fragments having HIV-1 IN inhibitory activity. Stapling of these lead compounds caused a significant increase in α-helicity and cell membrane penetration, and in the expression of potent anti-HIV activity in cells. The difference in the secondary structure of peptides did not make much difference in IN inhibitory activity. Even linear peptides might form α-helix structures when they bind to the target IN. Thus, the difference in the secondary structure might affect the difference in cell membrane penetration and thereby anti-HIV activity in cells. It is noteworthy that stapling and the addition of octa-arginine caused cell membrane penetration and that stapling did not involve cytotoxicity while incorporation of octa-arginine into the structures increased the cytotoxicity of the compounds. As a result, the stapled peptides 6S and 8S were found to be potent anti-HIV agents comparable with the original lead compound (2) containing octa-arginine. Adjustment of the length of additional oligo-arginine sequences of the stapled peptide 6S led to the development of compounds 20 and 21 with relatively high IN inhibitory activity and low cytotoxicity. The ratios of anti-HIV activity/cytotoxicity still might not be enough for drugs, but in the future the use of DNA binding units besides oligo-arginine sequences would resolve this drawback. The adoption of the stapled strategy to Vpr-derived peptides was successful, and these results might be useful for the further development of potent HIV-1 IN inhibitors. To date, several anti-HIV drugs have been reported, and their cocktail therapies using two or three drugs are known as highly active antiretroviral therapy (HAART), which has brought great success and hope in the clinical treatment of HIV-infected patients.29 Recently, the first IN inhibitor, raltegravir (Merck),2,3 has appeared in a clinical setting, and several inhibitors are under development. These IN inhibitors might be useful in HAART. The present strategy, mimicking autoinhibition, would be less susceptible to resistance through mutation.
METHODS
Chemical synthesis and characterization methods for peptides and peptidomimetics are described in the Supporting Information.
CD Spectroscopy.
CD measurements were performed on a JASCO J-820 spectropolarimeter equipped with thermoregulator (JASCO Corp., Ltd.), using 12.5 μM of peptides dissolved in 20% TFE water or 0.625% TFE water containing 10% CH3CN. UV spectra were recorded at 20 °C in a quartz cell of 10.0 mm path length, a time constant of 0.25 s, and a 100 nm/min scanning speed with 0.2 nm resolution.
Integrase Assays.
Recombinant IN was expressed in E. coli and purified as previously reported.30 Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5′-end [32P]-labeled oligonucleotide substrate (full-length 21 nucleotide duplex)30 and inhibitors or DMSO (drug solvent). Reaction mixtures were incubated at 37 °C (60 min) in buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Reactions were stopped by addition of 10 μL of loading dye (10 mM EDTA, 2% SDS, 95% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were then subjected to electrophoresis in 16% polyacrylamide–7 M urea gels. Gels were dried, and reaction products were visualized with a Typhoon 8600 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analyses were performed using ImageQuant version 5.1 (Molecular Dynamics Inc.). Concentration of inhibitors allowing 50% reduction of enzyme activity (IC50) and standard deviation (SD) were determined using Prism software version 5.0c (GraphPad Software, San Diego, CA) from at least three independent experiments.
Replication Assays.
For MT-4 Luc assays, MT-4 Luc cells (2.5–5 × 103 cells) grown in 96-well plates were infected with HIV-1 NL4-3 (~50–250 pg) in the presence of varying concentrations of compounds. At 6–8 d postinfection, cells were lysed, and luciferase activity was measured using the Steady-Glo assay kits (Promega), according to the manufacture’s protocol. Chemiluminescence was detected with a Veritas luminometer.
Anti-HIV-1 activity was determined by measuring the protection against HIV-1 (NL4-3 strain)-induced cytopathogenicity in MT-4 cells by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or p24 (CA) concentrations in the culture supernatant by ELISA. Various concentrations of compounds were added to HIV-1-infected MT-4 cells at multiplicity of infection of 0.001 and placed in wells of a 96-well microplate. After 5 days’ incubation at 37 °C in a CO2 incubator, the number of viable cells was determined using the MTT method. Cytotoxicity of the compounds was determined by measurement of the reduction against the viability of mock-infected MT-4 cells. Levels of p24 antigen in the supernatants of 5-day cultures were also measured using a Retro TEK p24 antigen ELISA kit (ZeptroMetrix Corp., Buffalo, NY), according to the manufacture’s protocol. Signals were detected using an ELx808 microplate photometer.
DNA Binding Effect (Table 3).
Serial dilutions of peptides were mixed to IN and its radio-labeled DNA substrate in activity buffer (see Integrase Assays section). After 2 h at 37 °C, an equal volume of loading buffer (99% deionized formamide, 0.025% xylene cyanol, and 0.025% bromophenol blue) was added to the mixture. Samples were then loaded on 16% polyacrylamide–7 M urea gels. After 90 min migration at 2000 V, gels were dried, and migration of the DNA was visualized with a Typhoon 8600. The accumulation of signal in the well of the gel was visually investigated. The lowest concentration of peptides inducing complete retardation was reported.
DNA Binding Assays (Figure 7).
A plate-based assay using fluorescence polarization was performed to monitor DNA binding. In this experiment, the integrase DNA substrate contains a fluorescein dye on its 3′-end (cleaved strand) and fluorescence anisotropy was monitored every min for 30 min at RT using the EnVision plate reader (Perkin-Elmer). Reactions were performed in absence of IN with 10 nM of fluorescent oligonucleotide and 0.5 μM peptide in buffer containing 0.1% BSA and 0.01% tween 20, 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Each sample was assessed in triplicate, and a solution of 10% DMSO was used as control. Data analysis was done using Prism version 5.0c (GraphPad software).
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Koide and C. Yamazaki, Department of Chemistry and Biochemistry, Waseda University, for allowing access to a CD spectropolarimeter. N.O., C.H., and T.T. are supported by JSPS research fellowships for young scientists. This work was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and Health and Labour Sciences Research Grants from Japanese Ministry of Health, Labor, and Welfare.
ABBREVIATIONS:
- HIV
human immunodeficiency virus
- AIDS
acquired immunodeficiency syndrome
- RT
reverse transcriptase
- IN
integrase
- MDR
multidrug resistance
- PIC
preintegration complex
- BAF
barrier to autointegration factor
- RCM
ring-closing metathesis
- GABA
γ-aminobutyric acid
- CD
circular dichroism
- Luc
luciferase
- ELISA
enzyme-linked immunosorbent assay
- MTT
3-(4,5-dimethylthial-2-yl)-2,5-diphenyltetrazalium bromide
Footnotes
Supporting Information
Additional experimental procedures including MS data and figures: HPLC charts of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Mitsuya H, and Erickson J (1999) In Textbook of AIDS Medicine (Merigan TC, Bartlett JG, Bolognesi D, Eds.), pp 751–780, Williams & Wilkins, Baltimore. [Google Scholar]
- (2).Cahn P, and Sued O (2007) Raltegravir: A New Antiretroviral Class for Salvage Therapy. Lancet 369, 1235–1236. [DOI] [PubMed] [Google Scholar]
- (3).Grinsztejn B, Nguyen B-Y, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, and Isaacs RD (2007) Safety and Efficacy of the HIV-1 Integrase Inhibitor Raltegravir (MK-0518) in Treatment-Experienced Patients with Multidrug-Resistant Virus: A Phase II Randomised Controlled Trial. Lancet 369, 1261–1269. [DOI] [PubMed] [Google Scholar]
- (4).Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubei A, Spitz L, Lewis P, Goldfarb D, Emerman M, and Stevenson M (1993) A Nuclear-Localization Signal within HIV-1 Matrix Protein That Governs Infection of Nondividing Cells. Nature 365, 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Miller MD, Farnet CM, and Bushman FD (1997) Human Immunodeficiency Virus Type 1 Preintegration Complexes: Studies of Organization and Composition. J. Virol 71, 5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Farnet CM, and Bushman FD (1997) HIV-1 cDNA Integration: Requirement of HMG I(Y) Protein for Function of Preintegration Complexes in Vitro. Cell 88, 483–492. [DOI] [PubMed] [Google Scholar]
- (7).Chen H, and Engelman A (1998) The Barrier-to-Auto-integration Protein Is a Host Factor for HIV Type 1 Integration. Proc. Natl Acad. Sci. U.S.A 95, 15270–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gleenberg IO, Herschhorn A, and Hizi A (2007) Inhibition of the Activities of Reverse Transcriptase and Integrase of Human Immunodeficiency Virus Type-1 by Peptides Derived from the Homologous Viral Protein R (Vpr). J. Mol. Biol 369, 1230–1243. [DOI] [PubMed] [Google Scholar]
- (9).Bischerour J, Tauc P, Leh H, De Rocquigny H, Roques B, and Mouscadet JF (2003) The (52–96) C-Terminal Domain of Vpr Stimulates HIV-1 IN-Mediated Homologous Strand Transfer of Mini- viral DNA. Nucleic Acids Res. 31, 2694–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Suzuki S, Urano E, Hashimoto C, Tsutsumi H, Nakahara T, Tanaka T, Nakanishi Y, Maddali R, Han Y, Hamatake M, Miyauchi R, Pommier Y, Beutler JA, Sugiura W, Fuji H, Hoshino T, Itotani R, Nomura W, Narumi T, Yamamoto N, Romano JA, and Tamamura H (2010) Peptide HIV-1 Integrase Inhibitors from HIV-1 Gene Products. J. Med. Chem 53, 5356–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda R, and Sugiura Y (2002) Possible Existence of Common Internalization Mechanisms among Arginine-Rch Peptides. J. Biol. Chem 277, 2437–2443. [DOI] [PubMed] [Google Scholar]
- (12).Suzuki S, Maddali R, Hashimoto C, Urano E, Ohashi N, Tanaka T, Ozaki T, Arai H, Tsutsumi H, Narumi T, Nomura W, Yamamoto N, Pommier Y, Romano JA, and Tamamura H (2010) Peptidic HIV Integrase Inhibitors Derived from HIV Gene Products: Structure-Activity Relationship Studies. Bioorg. Med. Chem 18, 6771–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schafmeister CE, Po J, and Verdine GL (2000) Ai All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc 122, 5891–5892. [Google Scholar]
- (14).Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, and Debnath AK (2008) A Cell-Penetrating Helical Peptide as a Potential HIV-1 Inhibitor. J. Mol. Biol 378, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Verdine GL, and Hilinski GJ (2012) In Methods in Enzymology (Wittrup KD, Verdine GL, Eds.), Vol 503, pp 3–31, Elsevier Inc., Amsterdam.22230563 [Google Scholar]
- (16).Blackwell HE, and Grubbs RH (1998) Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem., Int. Ed 37, 3281–3284. [DOI] [PubMed] [Google Scholar]
- (17).Chatterjee AK, Choi T-L, Sanders DP, and Grubbs RH (2003) A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc 125, 11360–11370. [DOI] [PubMed] [Google Scholar]
- (18).Hung K, Harris PWR, and Brimble MA (2010) Synthesis of Methyl N-Boc-(2S,4R)-4-methylpipecolate. J. Org. Chem 75, 8728–8731. [DOI] [PubMed] [Google Scholar]
- (19).Bautista AD, Appelbaum JS, Craig CJ, Michel J, and Schepartz A (2010) Bridged β3-Peptide Inhibitors of p53–hDM2 Complexation: Correlation between Affinity and Cell Permeability. J. Am. Chem. Soc 132, 2904–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Okamoto T, Zobel R, Fedorova A, Quan C, Yang H, Fairbrother WJ, Huang DCS, Smith BJ, Deshayes R, and Czabotar PE (2013) Stabilizing the Pro-Apoptotic BimBH3 Helix (BimSAHB) Does Not Necessarily Enhance Affinity or Biological Activity. ACS Chem. Biol 8, 297–302. [DOI] [PubMed] [Google Scholar]
- (21).Boal AK, Guryanov I, Moretto A, Crisma M, Lanni EL, Toniolo C, Grubbs RH, and O’Leary DJ (2007) Facile and E-Selective Intramolecular Ring-Closing Metathesis Reactions in 310-Helical Peptides: A 3D Structural Study. J. Am. Chem. Soc 129, 6986–6987. [DOI] [PubMed] [Google Scholar]
- (22).Kim Y-W, Grossmann TN, and Verdine GL (2011) Synthesis of All-Hydrocarbon Stapled α-Helical Peptides by Ring-Closing Olefin Metathesis. Nat. Protoc 6, 761–771. [DOI] [PubMed] [Google Scholar]
- (23).Yan H, Mizutani TC, Nomura N, Tanaka T, Kitamura Y, Miura H, Nishizawa M, Tatsumi M, Yamamoto N, and Sugiura W (2005) A Novel Small Molecular Weight Compound with a Carbazole Structure That Demonstrates Potent Human Immunodeficiency Virus Type-1 Integrase Inhibitory Activity. Antivir. Chem. Chemother 16, 363–373. [DOI] [PubMed] [Google Scholar]
- (24).Marchand C, Zhang X, Pais GCG, Cowansage K, Neamati N, Burke TR Jr., and Pommier Y (2002) Structural Determinants for HIV-1 Integrase Inhibition by β-Diketo Acids. J. Biol. Chem 277, 12596–12603. [DOI] [PubMed] [Google Scholar]
- (25).Semenova EA, Johnson AA, Marchand C, Davis DA, Tarchoan R, and Pommier Y (2006) Preferential Inhibition of the Magnesium-Dependent Strand Transfer Reaction of HIV-1 Integrase by α-Hydroxytropolones. Mol. Pharmacol 69, 1454–1460. [DOI] [PubMed] [Google Scholar]
- (26).Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, and Mouscadet JF (2000) Determinants of Mg2+-Dependent Activities of Recombinant Human Immunodeficiency Virus Type 1 Integrase. Biochemistry 39, 9285–9294. [DOI] [PubMed] [Google Scholar]
- (27).Marchand C, Neamati N, and Pommier Y (2001) In Vitro Human Immunodeficiency Virus Type 1 Integrase Assays In Methods in Enzymology (Drug-Nucleic Acid Interactions) (Chaires JB, Waring MJ, Eds.), Vol 340, pp 624–633, Elsevier Inc., Amsterdam. [DOI] [PubMed] [Google Scholar]
- (28).Hazen RJ, Harvey RJ, St. Clair MH, Ferris RG, Freeman GA, Tidwell JH, Schaller LT, Cowan JR, Short SA, Romines KR, Chan JH, and Boone LR (2005) Anti-Human Immunodeficiency Virus Type 1 Activity of the Nonnucleoside Reverse Transcriptase Inhibitor GW678248 in Combination with Other Antiretrovirals against Clinical Isolate Viruses and in Vitro Selection for Resistance. Antimicrob. Agents Chemother 49, 4465–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hashimoto C, Tanaka T, Narumi T, Nomura W, and Tamamura H (2011) The Success and Failures of HIV Drug Discovery. Expert Opin. Drug Discovery 6, 1067–1090. [DOI] [PubMed] [Google Scholar]
- (30).Métifiot M, Maddali K, Naumova A, Zhang X, Marchand C, and Pommier Y (2010) Biochemical and Pharmacological Analyses of HIV-1 Integrase Flexible Loop Mutants Resistant to Raltegravir. Biochemistry 49, 3715–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.