

HSB-1/HSF-1 modulates longevity and mitochondrial function by mediating histone H4-dependent regulation of mtDNA gene expression.
Abstract
Heat shock factor–1 (HSF-1) is a master regulator of stress responses across taxa. Overexpression of HSF-1 or genetic ablation of its conserved negative regulator, heat shock factor binding protein 1 (HSB-1), results in robust life-span extension in Caenorhabditis elegans. Here, we found that increased HSF-1 activity elevates histone H4 levels in somatic tissues during development, while knockdown of H4 completely suppresses HSF-1–mediated longevity. Moreover, overexpression of H4 is sufficient to extend life span. Ablation of HSB-1 induces an H4-dependent increase in micrococcal nuclease protection of both nuclear chromatin and mitochondrial DNA (mtDNA), which consequently results in reduced transcription of mtDNA-encoded complex IV genes, decreased respiratory capacity, and a mitochondrial unfolded protein response–dependent life-span extension. Collectively, our findings reveal a previously unknown role of HSB-1/HSF-1 signaling in modulation of mitochondrial function via mediating histone H4-dependent regulation of mtDNA gene expression and concomitantly acting as a determinant of organismal longevity.
INTRODUCTION
The rate of aging in multicellular organisms is determined by several evolutionarily conserved cellular and metabolic processes (1, 2). Some recent studies have identified interconnectedness among these pathways in the context of longevity regulation (3–6), but the hierarchy between these hallmarks of aging at the systemic level remains less understood. Maintenance of protein homeostasis constitutes one such hallmark that undergoes impairment during normal aging and in numerous age-associated diseases (7). Misregulation in the function of chaperone complexes results in elevated misfolding and aggregation of proteins with age that is further exacerbated by metabolic and environmental stress. A major component of the protein homeostasis network in eukaryotes is the evolutionarily conserved heat shock response (HSR) pathway. HSR involves activation of members of the heat shock factor (HSF) family in response to a variety of environmental stresses such as heat, oxidative damage, proteotoxic insults, and pathogenic infection (8). While mammals have four distinct HSFs (HSF1 to HSF4), invertebrates such as Caenorhabditis elegans, Drosophila, and yeast have only one ortholog of HSF1. In the presence of stress stimuli, HSF1 binds to its target elements in the genome to induce the transcription of proteases and heat shock proteins (HSPs) (8). HSPs often function as molecular chaperones to assist in the folding of nascent polypeptides and to prevent the toxic aggregation of misfolded proteins, thus attenuating the loss of protein homeostasis induced during stress conditions.
HSF1 has also been shown to regulate longevity of animals in nonstressed physiological conditions. In the nematode worm C. elegans, overexpression of hsf-1 extends life span and delays the onset of age-related neurodegenerative diseases, while inhibition of HSF-1 activity accelerates aging (9–11). In mammals, transgenic HSF1 activation has been shown to promote longevity in a neurodegenerative mouse model (12), while knockout of HSF1 markedly shortens life span in a murine model of prion disease (13). Initial studies suggested that life-span extension associated with HSF-1 activation is at least partially due to increased expression of small HSPs (9). A recent study showed that overexpression of a truncated form of HSF-1 extended life span in worms independent of its ability to mount stress-induced activation of HSPs (14). Incidentally, prolonged life span due to transgenic HSF1 activation in a neurodegenerative mouse model did not involve increased expression of HSPs in brain tissue (12). These findings indicate that the longevity-promoting effects of HSF1 not only are attributable to chaperone induction but possibly also involve other unidentified cellular processes. HSF1 has also been ascribed regulatory functions in several physiological processes other than stress response, such as animal development, reproduction, metabolism, and cancer (8, 15).
Under normal conditions, the transcriptional activity of HSF1 is inhibited by several regulatory mechanisms that collectively dictate context-dependent modulation of HSF-1 function (8). One such negative regulator of HSF-1 is the HSF-binding protein 1 (HSB-1), which physically binds to HSF-1 to form an inhibitory multiprotein complex (3, 16). The formation of this HSF-1–inhibitory complex in C. elegans is not affected by heat stress but, instead, is facilitated by the insulin/insulin-like growth factor 1 (IGF-1)–like signaling (3), a pathway implicated in regulation of longevity in animals across evolutionary taxa (2). A loss-of-function mutation in the hsb-1 gene results in dissociation of HSF-1 from this inhibitory complex and induces a robust hsf-1–dependent extension of life span in worms (3). However, the underlying longevity-promoting mechanisms of the HSB-1/HSF-1 signaling pathway still remain elusive.
In this study, we show that the longevity achieved via HSF-1 activation by inhibiting HSB-1 is largely due to increase in the levels of histone H4 protein. Overexpression of H4 in wild-type animals phenocopies the longevity effect of hsb-1 null mutation. We also show that elevated H4 levels in hsb-1(−) worms influence the global compaction of nuclear chromatin, and markedly, also of mitochondrial DNA (mtDNA). We further elucidate how histone H4 modulates the expression of mitochondrially encoded electron transport chain (ETC) complex IV subunits and how this affects respiratory capacity in animals. Moreover, we provide evidence for the presence of histone H4 in mitochondria and how this results in a mitochondrial unfolded protein response (UPRmt)–dependent life-span extension in worms. Overall, this study connects HSB-1/HSF-1 signaling to changes in histone H4 levels and provides direct evidence for a previously unknown role of histone H4 in the regulation of mitochondrial activity.
RESULTS
Life-span extension due to HSB-1/HSF-1 signaling is suppressed by knockdown of histone H4
To identify factors that are required for HSB-1 inhibition-induced life-span extension, we performed a genome-wide RNA interference (RNAi) screen in C. elegans for genetic suppressors of the longevity phenotype of hsb-1 null worms. Our goal was to identify genes, that when knocked down, resulted in reduction of life span in the long-lived hsb-1(−) strain, without inducing a similar life-span shortening effect in wild-type animals. To avoid progeny contamination, we performed this genome-wide suppressor screen in the rrf-3(−); fem-1(−) genetic background that is sterile at 25°C. One of the suppressors identified from this screen was a histone H4-coding gene his-67 (Fig. 1A and table S1). While his-67 RNAi had minimal effect on the life span of wild-type (N2 strain) worms, it suppressed the longevity phenotype of the hsb-1 null mutant (Fig. 1A and table S1). Worms, like mammals, have only one histone H4 isotype with no known protein variants. There are 16 genes in the C. elegans genome that encode for an identical H4 protein that differs from the human H4 at only a single amino acid residue. Since these H4 genes share ~90% identity at the nucleotide sequence level (fig. S1A), we hypothesized that in addition to his-67, knockdown of other H4 genes might also affect hsb-1(−)–associated longevity. We found that knockdown of two other H4-coding genes, his-5 and his-38, resulted in complete suppression of the longevity phenotype in hsb-1(−) worms (Fig. 1, B and C, and table S1). This suppression of hsb-1(−)–associated longevity was substantially weaker when life-span analysis was performed at 20°C instead of at 25°C (fig. S1B and table S1). This suggests that temperature plays an important role in determining the H4 dependence of HSB-1–associated longevity phenotype, similar to that observed for several other longevity mechanisms (17). We found that knockdown of all three genes, his-67, his-5, and his-38, led to reduction in total H4 protein levels in both N2 and hsb-1(−) animals (Fig. 1, D and E). H4 RNAi often also resulted in slightly reduced levels of other core histone proteins (Fig. 1, D and E). This can be attributed to the cell’s ability to actively regulate protein levels of the four core histones (18), as these proteins must assemble in a stoichiometric proportion to form the histone heterooctomer in nucleosome complexes.
Fig. 1. HSB-1–associated life-span extension is suppressed by knockdown of histone H4.
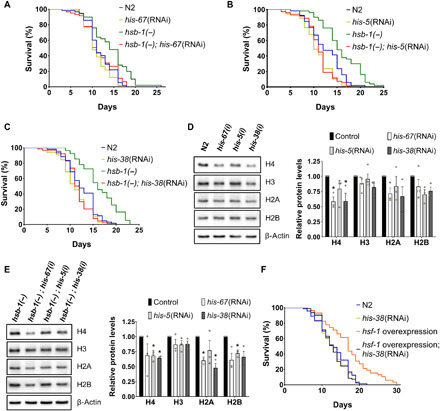
(A to C) Life-span analysis of wild-type (N2) and hsb-1(−) worms subjected to control, his-67, his-5, or his-38 RNAi at 25°C. (D and E) Immunoblots for core histones in N2 and hsb-1(−) worms subjected to control, his-67, his-5, or his-38 RNAi at 25°C. Means ± SEM for n ≥ 3. *P < 0.05 compared to corresponding control RNAi in two-tailed t test. (F) Life-span analysis of N2 and sur-5p::hsf-1 overexpression strain subjected to control or his-38 RNAi at 25°C. (A to C and F) Statistical data and additional life-span replicates are included in table S1.
Among the three H4 genes that were identified as suppressors of HSB-1–associated longevity, RNAi of his-38 resulted in the most consistent reduction of H4 levels in both N2 and hsb-1(−) strains, and hence, it was used in our subsequent H4 knockdown experiments (table S1). We tested whether knockdown of H4 also affected the life span of other well-characterized long-lived C. elegans strains. H4 RNAi completely suppressed life-span extension due to overexpression of hsf-1 (Fig. 1F and table S1) (14). This indicates that histone H4 has a major role in mediating the longevity-promoting effects of increased HSF-1 activity achieved via either of the two approaches, HSF-1 overexpression or HSB-1 inhibition (Fig. 1, C and F, and table S1). H4 RNAi had no effect on longevity associated with reduced insulin/IGF-1–like signaling in daf-2(−) worms (fig. S1C and table S1) (19) or via loss of germ line in glp-1(−) worms (fig. S1D and table S1) (20). Life-span extension due to impaired mitochondrial function in isp-1(−) worms was completely suppressed by knockdown of H4 (fig. S1E and table S1), although longevity of this strain is known to be independent of HSF-1 activity (9, 21). This suggests a potential role of histones in longevity mechanisms associated with mitochondrial inhibition.
HSB-1–associated life-span extension is due to elevated histone H4 levels in somatic tissues during development
A previous study has shown that co-overexpression of histones H3 and H4 extends replicative life span in yeast (22). We thus asked whether increased histone levels in hsb-1(−) animals is the cause of their dependence on H4 for longevity. We observed that levels of H4, but not other core histones, were significantly higher in hsb-1(−) worms compared to wild-type during early adulthood (Fig. 2A). To test whether an increase in H4 protein level can directly contribute to life-span extension in worms, we generated a strain with elevated H4 expression (Fig. 2B). In C. elegans, H4 genes exist in three different structural variants: genes that have no untranslated regions (UTRs) or introns (his-50, his-1, his-10, his-14, his-26, his-28, his-46, and his-56), genes with UTRs but no introns (his-67, his-18, his-38, his-60, his-31, his-5, and his-64), and one H4-coding gene that has UTRs and an intron (his-37). We selected one gene from each of these three categories (his-67, his-37, and his-50) and co-overexpressed them in the wild-type strain (Fig. 2B). We found that overexpression of H4 in wild-type animals is sufficient to extend their mean life span by up to 25%, and this could be suppressed by RNAi-mediated knockdown of H4 (Fig. 2C and table S1). Ectopic H4 overexpression in animals resulted in increased protein levels of the other core histones as well (fig. S2A), possibly due to cellular mechanisms that tend to maintain core histone protein levels in a stoichiometric proportion (18).
Fig. 2. HSB-1–associated life-span extension is due to higher H4 levels during development.
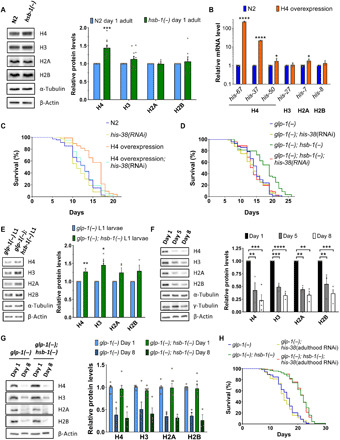
(A) Immunoblots for core histones in adult wild-type (N2) and hsb-1(−) worms. Means ± SEM for n ≥ 5. ***P < 0.001 in two-tailed t test. (B) Transcript levels of core histone genes in N2 and H4 overexpression strain. Means ± SEM for n ≥ 3. *P < 0.05 and ****P < 0.0001 in two-tailed t test. (C and D) Life-span analysis of N2, H4 overexpression strain, glp-1(−) and glp-1(−); hsb-1(−) worms subjected to control or his-38 RNAi. (E) Immunoblots for core histones in glp-1(−) and glp-1(−); hsb-1(−) L1 larvae. Means ± SEM for n ≥ 3. *P < 0.05 and **P < 0.01 in two-tailed t test. (F) Immunoblots for core histones in days 1, 5, and 8 glp-1(−) worms. Means ± SEM for n ≥ 3. **P < 0.01, ***P < 0.001, and ****P < 0.0001 in Tukey’s multiple comparisons test. (G) Immunoblots for core histones in day 1 and 8 glp-1(−) and glp-1(−); hsb-1(−) worms. Means ± SEM for n ≥ 3. Age: P < 0.0001; genotype or interaction: P > 0.05 in two-way ANOVA. (F) Life-span analysis of glp-1(−) and glp-1(−); hsb-1(−) worms subjected to control or his-38 RNAi only during adulthood. (C, D, and H) Statistical data are included in table S1.
Since transcription of histone genes in C. elegans predominantly occurs only in dividing cells (23), we tested whether H4 RNAi suppresses hsb-1(−)–associated longevity in the absence of germ line, the only proliferative tissue in adult worms. For these experiments, we used animals with a temperature-sensitive mutation in either glp-1 or glp-4, genes that are essential for proliferation and maintenance of the germ line tissue in C. elegans (24, 25). Life-span extension due to hsb-1(−) mutation in germline-deficient glp-1(−) or glp-4(−) worms was completely suppressed by knockdown of H4 (Fig. 2D, fig. S2B, and table S1), suggesting that depletion of H4 in somatic tissues alone is sufficient to abolish HSB-1 inhibition-induced longevity. We further confirmed that somatic protein levels of histones H3 and H4 were significantly elevated in hsb-1(−) animals during development (Fig. 2E), but their levels were not significantly different in somatic tissues of young adult worms (fig. S2C).
Several studies have suggested the role of histone posttranslational modifications in aging, but only few have focused on the effects of change in histone protein levels (26, 27). Histone levels decline with age in yeast and murine muscle stem cells, and this results in increased genomic instability and global transcriptional deregulation (28, 29). As previously reported for histone H3 (30), we found that protein levels of all core histones decline markedly with age in somatic tissues of worms (Fig. 2F). Moreover, this was associated with an increase in susceptibility to ultraviolet (UV)–induced DNA damage with age (fig. S2, D and E). HSB-1 inhibition provided partial resistance to DNA lesion formation at lower doses of UV (fig. S2F). We also observed that a transposon-like element called C09B7.2, which has been previously shown to accelerate aging in C. elegans (31), showed increased expression with age in somatic tissues of worms (fig. S2G), suggesting loss of silencing of repetitive elements with age. This age-associated increase in somatic C09B7.2 expression was partially rescued in hsb-1(−) animals (fig. S2G).
Unexpectedly, the decline in histone protein levels with age in somatic tissues of worms was not alleviated by HSB-1 inhibition (Fig. 2G). To confirm whether elevated histone expression only during development contributes to longevity of hsb-1(−) worms, we performed H4 knockdown in animals after they attained adulthood. Consistent with the observation that somatic H4 levels were elevated only during early development in hsb-1(−) animals (Fig. 2E), H4 RNAi started postdevelopmentally did not suppress hsb-1(−)–associated longevity in either N2 or germline-less glp-1(−) worms (Fig. 2H, fig. S2H, and table S1). Together, our findings indicate that the HSB-1/HSF-1 pathway regulates longevity by altering somatic H4 levels mainly during development.
Increased MNase protection of nuclear chromatin and mtDNA due to HSB-1 inhibition is H4-dependent
To understand how elevated levels of H4 protein might regulate the rate of aging in worms, we first tested whether global chromatin organization is altered in hsb-1(−) animals. To address this, we digested native C. elegans chromatin with micrococcal nuclease (MNase), an endonuclease that digests exposed DNA until it reaches a nucleosome or a strongly DNA-bound protein. We observed that hsb-1(−) chromatin constituted a higher amount of MNase-protected DNA, and this could be suppressed by H4 RNAi (Fig. 3A). Moreover, MNase-digested chromatin from hsb-1(−) worms showed an H4-dependent significant increase in the ratio of dinucleosomal to mononucleosomal DNA fragments (Fig. 3B). Increased proportion of dinucleosomal fragments indicates lower MNase accessibility to DNA, a property associated with higher compaction of chromatin (32). To test whether altered nucleosome positioning at specific regions of the genome contributes to the higher level of chromatin compaction in hsb-1(−) animals, we performed next-generation sequencing of MNase-digested chromatin. Analysis of global nucleosome occupancy revealed that frequency of sharp low-width peaks was slightly elevated in chromatin of hsb-1(−) worms (fig. S3A and table S2). In addition, fuzziness (or delocalization) in nucleosome positioning and frequency of low-height peaks were marginally reduced in hsb-1(−) chromatin compared to wild-type (fig. S3, C and E, and table S2). Small-scale changes in these three parameters indicate slightly better positioning of individual nucleosomes and a modest increase in sharpness of nucleosome peaks in hsb-1(−) worms, effects that were suppressed by knockdown of H4 (fig. S3, B, D, and F, and table S2) (33).
Fig. 3. HSB-1 inhibition results in increased MNase-protection of mtDNA and reduced expression of mtDNA-encoded ETC complex IV genes.
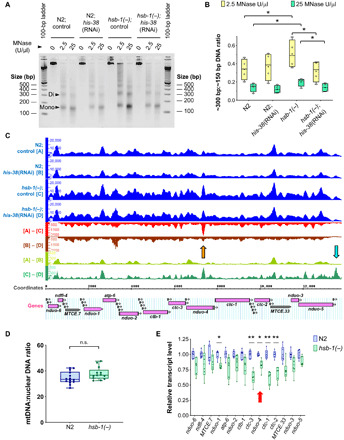
(A) Agarose gel electrophoresis for MNase-digested native chromatin of wild-type (N2) and hsb-1(−) worms subjected to control or his-38 RNAi. Arrowheads indicate mononucleosomal (~150 bp) and dinucleosomal DNA (~300 bp) bands. (B) Ratio of dinucleosomal: mononucleosomal DNA in MNase-digested chromatin samples from N2 and hsb-1(−) worms subjected to control or his-38 RNAi. Central line, box limits, “+,” and whiskers indicate median, interquartile range, mean, and data range, respectively, for n = 4. * indicates P < 0.05 in Sidak’s multiple comparisons test. (C) MNase-Seq data for entire ~13.8-kb stretch of C. elegans mtDNA from N2 and hsb-1(−) worms subjected to control or his-38 RNAi. Difference between groups, such as (A) and (B), indicates occupancy difference at each base pair. Orange arrow: most prominent peak for (A) to (C) comparison; cyan arrow: location of mitochondrial D-loop region. Mitochondrially encoded genes are shown. Labeled, protein- and ribosomal RNA (rRNA)–coding genes; unlabeled, transfer RNA (tRNA) genes. (D) mtDNA copy number relative to nuclear DNA in N2 and hsb-1(−) worms. Central line, box limits, and whiskers indicate median, interquartile range, and data range, respectively, for n ≥ 13. P = 0.09 for comparison between the two genotypes in two-tailed t test. (E) Relative transcript levels of mitochondrial rRNA (MTCE.7 and MTCE.33) and protein-coding genes in N2 and hsb-1(−) worms. Central line, box limits, and whiskers indicate median, interquartile range, and data range, respectively, for n = 6. *P < 0.05 and **P < 0.01 in two-tailed t test. Red arrow indicates the location of major H4-dependent peak in hsb-1(−) MNase-seq data.
We observed that nucleosome organization in most of the genome was unaffected in hsb-1(−) animals (fig. S3G and data file S1). Unexpectedly, the region that showed the most significant increase in MNase protection due to HSB-1 inhibition was the entire stretch of mtDNA (Fig. 3C and table S3). Moreover, the increased MNase protection of several mtDNA loci in hsb-1(−) animals was diminished after knockdown of histone H4 (Fig. 3C and table S3). Increased number of MNase sequencing (MNase-seq) reads from mtDNA of hsb-1(−) worms can be attributed to two possible contributors: (i) change in accessibility of mtDNA that protects it from endonuclease digestion or (ii) an increased copy number of mtDNA per cell in hsb-1(−) animals. We found that mtDNA copy number relative to nuclear DNA was not significantly different between hsb-1(−) and wild-type worms (Fig. 3D), indicating that the higher number of MNase-seq reads was not due to increase in mtDNA content per cell in hsb-1(−) animals. mtDNA is predicted to be nonspecifically coated by the mitochondrial transcription factor A (TFAM)–like HMG-5 nucleoid protein (34), but we observed “chromatin-like” peaks interspersed along the stretch of mtDNA (Fig. 3C). The most prominent difference in peak height between hsb-1(−) and wild-type mtDNA was observed at the reduced form of NADH-ubiquinone oxidoreductase subunit 4 (nduo-4) gene region, and this difference was partially suppressed when H4 is knocked down by RNAi (Fig. 3C, shown with orange arrow). This indicates that the increased MNase protection of the nduo-4 gene region on mtDNA of hsb-1(−) worms is dependent on the levels of histone H4.
HSB-1 inhibition leads to H4-dependent decrease in expression of mtDNA-encoded genes, reduced respiratory capacity, and UPRmt-mediated longevity
Next, we analyzed whether the altered MNase protection of mtDNA due to HSB-1 inhibition affected the expression of mitochondrially encoded genes. We observed that transcript levels of five mitochondrially encoded genes were significantly reduced in hsb-1(−) animals (Fig. 3E), while expression of others was slightly attenuated. The significant reduction in expression of these five genes due to HSB-1 inhibition was suppressed when the hsb-1(−) worms were subjected to knockdown of H4 (fig. S3, H and I). Similar to HSB-1 inhibition, H4 overexpression in worms also significantly attenuated the expression of several mtDNA-encoded genes (fig. S3J). Under normal conditions, the expression levels of three mitochondrially encoded genes, ctc-3, ctc-1, and ctc-2, were reduced by greater than 30% compared to wild-type levels in hsb-1(−) animals (Fig. 3E). These three genes are located adjacent to the MNase-seq peak in mtDNA that was most amenable to both HSB-1 inhibition and H4 knockdown (Fig. 3C, shown with orange arrow). The three ctc genes on the mitochondrial genome encode three large subunits that form the catalytic core of complex IV (i.e., cytochrome c oxidase) of mitochondrial ETC. The function of ETC complex IV is to transfer protons across the inner mitochondrial membrane concomitant with conversion of molecular oxygen to water. It has been previously demonstrated that knockdown of nuclear genome-encoded mitochondrial ETC components in worms during development is sufficient to reduce their rate of respiration and extend life span during adulthood (35). We tested whether reduced expression of mtDNA-encoded ETC complex IV components in hsb-1(−) worms affected their oxygen consumption rate (OCR). We found that maximal respiratory capacity was significantly reduced after HSB-1 inhibition, while basal mitochondrial OCR was not affected in hsb-1(−) animals compared to the wild-type level (fig. S4, A to C). In addition, spare respiratory capacity (defined as the difference between maximal and basal OCR) of hsb-1(−) worms was comparable to that of animals subjected to direct mitochondrial inhibition via knockdown of cyc-1, a nuclear genome-encoded component of ETC complex III (Fig. 4A). In contrast, the basal, maximal, and spare respiratory capacities of hsb-1(−) worms were not significantly different from the corresponding wild-type levels after RNAi-mediated knockdown of H4 in these animals (fig. S4, D to G). We further tested whether increasing histone H4 expression in worms can directly affect their mitochondrial activity similar to that observed in hsb-1(−) animals. On comparing mitochondrial respiration rates in wild-type and long-lived H4 overexpressing animals (Fig. 2, B and C), we found that overexpression of H4 significantly reduced all three mitochondrial OCR parameters (basal, maximal, and spare respiratory capacities) compared to the corresponding wild-type levels (Fig. 4B, and figs. S4C and S5, A and B). Moreover, the reduced mitochondrial respiratory capacity in H4-overexpressing worms was not due to reduced mtDNA copy number in these animals (fig. S5C). This signifies that increased H4 level in worms is sufficient to phenocopy the effects of HSB-1 inhibition on mitochondrial function.
Fig. 4. Increased level of histone H4 reduces mitochondrial respiratory capacity in worms, while UPRmt is required for H4-associated longevity.
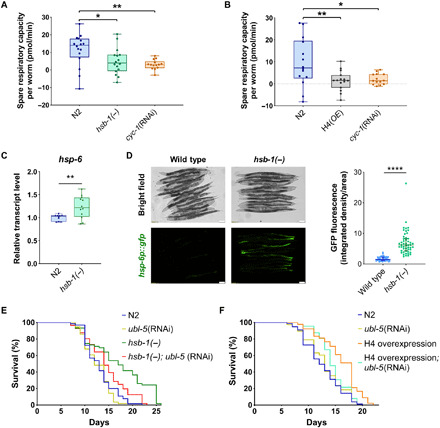
(A and B) Spare respiratory capacity per worm in wild-type (N2), hsb-1(−), H4 overexpression strain, and Cyc-1(RNAi) worms; corrected for non-mitochondrial oxygen consumption. Central line, box limits, and whiskers indicate median, interquartile range, and data range, respectively, for n ≥ 14. *P < 0.05, **P < 0.01 in Tukey’s multiple comparisons test. (C) Relative transcript levels of hsp-6 in N2 and hsb-1(−) worms. Central line, box limits, and whiskers indicate median, interquartile range, and data range, respectively, for n ≥ 10. **P < 0.01 and ****P < 0.0001 in two-tailed t test. (D) hsp-6p::gfp expression in wild-type and hsb-1(−) worms (left). Quantification of hsp-6p::gfp expression (right). Central line and error bars indicate median, and interquartile range, respectively, for n = 50 per genotype. (E and F) Life-span analysis of N2, hsb-1(−), and H4 overexpression strain subjected to control or ubl-5 RNAi. Statistical data and additional life-span replicates are included in table S1.
Disruption of mitochondrial function activates the UPRmt, an evolutionarily conserved pathway that involves transcriptional up-regulation of several genes including the mitochondrial chaperone HSP-6 (36). We found that hsb-1(−) animals have elevated hsp-6 mRNA levels compared to wild-type (Fig. 4C). We further confirmed the HSB-1 inhibition-induced transcriptional up-regulation of hsp-6 in worms using a well-characterized hsp-6p::gfp reporter (Fig. 4C) (36). Since UPRmt has been previously shown to mediate life-span extension achieved via inhibition of mitochondrial activity (37, 38), we speculated that this pathway might also contribute to the increased life span of hsb-1(−) animals. Longevity phenotypes associated with reduced function of two nuclear genome-encoded mitochondrial ETC components, isp-1 (complex III) and cco-1 (complex IV), are both dependent on the activity of UBL-5, a ubiquitin-like protein involved in UPRmt activation in worms (6, 37, 38). We found that RNAi-mediated knockdown of ubl-5 had no effect on longevity of wild-type worms, but it significantly shortened the mean life span of hsb-1(−) animals by ~15% (Fig. 5E and table S1), suggesting that UPRmt activation is required for the longevity phenotype associated with HSB-1 inhibition. Moreover, we found that ubl-5 knockdown also significantly suppressed the extension of life span in H4-overexpressing worms (Fig. 5F and table S1). This indicates that UPR signaling in the mitochondria has a crucial role in mediating the longevity-promoting effects associated with increased histone H4 levels in worms.
Fig. 5. Histone H4 is present inside mitochondria and binds to mtDNA.
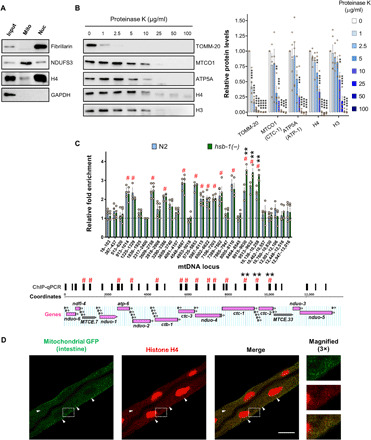
(A) Immunoblots for H4, nuclear protein FIB-1/fibrillarin, mitochondrial protein NUO-2/NDUFS3, and cytosolic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cellular fractions enriched with nuclei (Nuc) or mitochondria (Mito) obtained from wild-type worms. (B) Immunoblots for outer mitochondrial membrane protein TOMM-20, inner mitochondrial membrane proteins CTC-1/MTCO1 and ATP-1/ATP5A, and histones H3 and H4 in isolated mitochondria treated with indicated concentrations of proteinase K (left). Densitometric quantification of protein levels relative to samples not subjected to proteinase K digestion (right). Means ± SEM for n ≥ 3. **P < 0.01, ***P < 0.001, and ****P < 0.0001 in Dunnett’s multiple comparisons test. (C) Relative fold enrichment for H4 chromatin immunoprecipitation (ChIP)–qPCR performed using isolated mitochondria from N2 and hsb-1(−) worms. Means ± SEM for n ≥ 3. # indicates loci with significantly higher fold enrichment compared to the control region (2317 to 2400). ** indicates significantly higher fold enrichment in hsb-1(−) worms compared to N2. Significant differences correspond to P < 0.01 in Dunnett’s multiple comparisons test (top). Horizontal bars above mtDNA coordinates show regions amplified in ChIP-qPCR. Mitochondrially encoded genes are shown. Labeled, protein- and rRNA-coding genes; unlabeled, tRNA genes (bottom). (D) H4 immunofluorescence in worms expressing mitochondrially localized GFP in intestinal tissue. White arrowheads indicate examples of colocalization of extranuclear histone foci with intestinal mitochondria. Scale bar, 20 μM.
Histone H4 is present inside mitochondria and binds to specific regions of mtDNA
Since knockdown of H4 suppressed both the mitochondrial phenotypes of decreased MNase accessibility of several mtDNA loci (Fig. 3C) and reduced mtDNA-encoded gene expression in hsb-1(−) worms (fig. S3, H and I), we asked whether histone H4 protein physically translocates to mitochondria in these animals to regulate the function of this organelle. To test this, we used differential centrifugation to isolate intact mitochondria from worms and subjected the mitochondrial proteins to immunoblot analysis (Fig. 5A and fig. S5D). We detected all the core histones, namely H4, H3, H2A, and H2B, in isolated mitochondrial preparations (Fig. 5A and fig. S5E). In contrast, the nuclear protein FIB-1 (homolog of mammalian fibrillarin) was undetectable in the mitochondrial fraction (Fig. 5A and fig. S5E), thus validating that our mitochondrial preparations were not contaminated with nuclei. Next, we tested whether animals with elevated histone H4 levels show an increased amount of mitochondrially localized H4. For this experiment, we used the long-lived H4 overexpression strain as these animals showed a robust increase in total H4 protein levels in whole-worm lysates (Fig. 2C and fig. S2A). We found that the level of histone H4 in mitochondria of H4-overexpressing animals was noticeably higher than that in wild-type worms (fig. S5F). This indicates that an increase in total H4 protein levels can result in higher amounts of mitochondrially translocated H4 in animals.
A few studies involving mass spectrometry have previously detected core histones in mitochondria of mammalian cells (39, 40). However, since histones are among the most abundant cellular proteins, it is difficult to predict solely on the basis of biochemical fractionation whether histones are present inside the mitochondria or as impurities of the fractionation method. To address this, we treated suspensions of intact C. elegans mitochondria with increasing concentrations of proteinase K, a membrane-impermeable protease. In this assay, as the concentration of proteinase K is increased gradually, it sequentially digests proteins that are present as extramitochondrial impurities, followed by outer mitochondrial membrane proteins, while proteins present inside the mitochondria are digested at much higher protease concentrations (41). We found that both histones H3 and H4 were more protected from proteinase K digestion compared to TOMM-20, an outer mitochondrial membrane protein (Fig. 5B). While TOMM-20 was completely digested at low doses of proteinase K, the protease protection of histones H3 and H4 was comparable to that of the inner mitochondrial membrane proteins CTC-1 and ATP-1, homologs of mammalian MTCO1 (mitochondrially encoded cytochrome c oxidase I) and ATP5A, respectively (Fig. 5B). This indicates that H3 and H4 were not present as extramitochondrial impurities but, instead, were enclosed within the mitochondria.
In addition, we used chromatin immunoprecipitation to test whether mitochondrially translocated H4 directly binds to mtDNA in wild-type and hsb-1(−) animals. Numerous regions on the mitochondrial genome showed a significant ~2-fold higher binding to H4 compared to the background binding observed for other mtDNA regions tested (Fig. 5C). Among these regions, three loci spanning the ctc-1 and ctc-2 genes showed a significant ~40% higher H4-binding in hsb-1(−) worms compared to wild type (Fig. 5C). The expression of both ctc-1 and ctc-2 genes is significantly reduced in hsb-1(−) animals in an H4-dependent manner (fig. S3, H and I), suggesting that H4 binding can potentially influence gene expression from the mitochondrial genome. However, not all mtDNA-encoded genes with reduced transcript levels in hsb-1(−) worms showed altered H4 binding at the corresponding genomic loci (Figs. 3E and 5C, and fig. S3, H and I). It is plausible that the binding of H4 to a region can also influence the accessibility of adjacent genomic loci via formation of large nucleoprotein complexes on mtDNA. Together, these results provide strong evidence for the presence of histone H4 in mitochondria and suggest a potential relationship between binding of H4 to mtDNA and regulation of gene expression from the mitochondrial genome.
Histone H4 translocates to mitochondria in intestinal tissue of worms, where it is required for HSB-1–associated longevity
To obtain more conclusive evidence for the localization of histone H4 in mitochondria, we performed immunostaining for H4 in animals after formaldehyde fixation. We found H4 to be primarily localized in the nuclei of most tissues of worms, with the exception of intestinal tissue, where H4 immunostaining revealed extranuclear foci that overlapped with mitochondria (fig. S6, A to C). To further validate the mitochondrial localization of histone H4, we performed H4 immunofluorescence studies with C. elegans strains that express mitochondrially localized green fluorescent protein (GFP) in specific tissues. H4 immunostaining clearly showed that extranuclear H4 foci colocalized with mitochondrial GFP in the intestine (Fig. 5D), but not in the muscle cells of worms (fig. S6D). Moreover, we isolated intact mitochondria specifically from the intestinal tissue of worms using a recently described method called cell-specific mitochondrial affinity purification (CS-MAP) to confirm the presence of histone H4 in intestinal mitochondria (42). Briefly, TOMM-20 protein tagged with mCherry was expressed using an intestine-specific promoter in worms. Intact mitochondria were isolated from these animals and intestine-specific mitochondria were pulled down using the mCherry tag exposed outward on the outer mitochondrial membrane (42). We detected histone H4 in the intestinal mitochondria of both wild-type and hsb-1(−) animals, but the level of H4 was ~2-fold higher in the intestinal mitochondria of hsb-1(−) worms (Fig. 6A). This suggests that HSB-1 inhibition results in a particularly stronger effect on mitochondrial translocation of H4 in the intestinal tissue of worms (Fig. 6A).
Fig. 6. HSB-1 inhibition results in increased H4 levels in intestinal mitochondria.
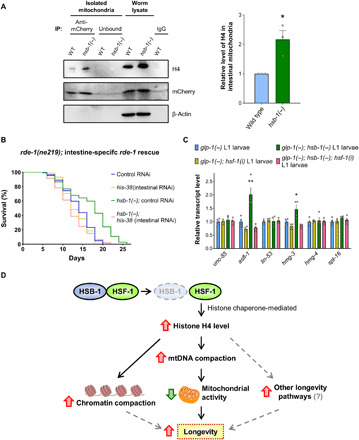
(A) Immunoblots for histone H4, mCherry-tagged outer mitochondrial membrane protein TOMM-20, and β-actin in intestinal mitochondria immunoprecipitated from wild-type (WT) and hsb-1(−) worms expressing TOMM-20::mCherry in intestinal tissue (ges-1p::tomm-20::mCherry). Protein levels in whole-worm lysates are also shown. IP, immunoprecipitation; IgG, immunoglobulin G. (B) Life-span analysis of rde-1(−) and hsb-1(−); rde-1(−) worms after intestine-specific rescue of rde-1. Worms were subjected to control or his-38 RNAi. Statistical data and additional life-span replicates are included in table S1. (C) Relative transcript levels of histone chaperone genes in glp-1(−) and glp-1(−); hsb-1(−) L1 larval worms subjected to control or hsf-1 RNAi. Means ± SEM for n = 4. *P < 0.05 and **P < 0.01 compared to all other conditions in Tukey’s multiple comparisons tests. (D) Proposed model of longevity associated with HSB-1 inhibition via increased H4 levels, mtDNA compaction, and reduced mitochondrial activity. Dashed gray arrows indicate other potential longevity pathways.
A previous study has shown that the intestine has a major role in mediating the longevity-promoting effects of increased HSF-1 activity in worms (43). In addition, the C. elegans intestine is also the target tissue for life-span extension induced via mitochondrial inhibition (37). On the basis of these evidences and our H4 immunostaining and CS-MAP data (Fig. 5D, Fig. 6A, and fig. S6A), we speculated that increased H4 levels might promote longevity via acting in the intestinal tissue of hsb-1(−) worms. To address this, we knocked down histone H4 only in the intestine and tested its effect on the longevity phenotype associated with HSB-1 inhibition. We performed this experiment in an rde-1–deficient genetic background in which RDE-1 function was restored only in the intestinal tissue of worms (Fig. 6B). The RDE-1 protein is a member of the Argonaute/PIWI family and is an essential component of the cellular RNAi machinery in C. elegans. Hence, restoring RDE-1 activity in rde-1 mutant worms using a tissue-specific promoter allows for knockdown of a gene of interest in a particular tissue of these animals. We found that RNAi-mediated knockdown of H4 in the intestine of hsb-1(−) worms is sufficient to completely suppress life-span extension in these animals (Fig. 6B and table S1). This finding, together with our H4 colocalization data, indicates that H4-dependent inhibition of mitochondrial activity in the intestinal tissue might be a major contributor to the longevity phenotype associated with HSB-1 inhibition.
DISCUSSION
HSB-1 is a conserved negative regulator of HSF-1 that limits its ability to promote organismal longevity in normal physiological conditions (3, 16). Our findings indicate that the life-span extending effects of HSB-1 inhibition are mediated via increase in histone H4 level that results in altered global compaction of both nuclear and mitochondrial genomes, and consequently inhibiting mitochondrial activity. This study unravels an unexpected role of HSB-1/HSF-1 signaling in regulating levels of the core histone protein H4 and its previously unfound functions in modulating the transcription of mitochondrial genome-encoded genes. A recent study has shown that a mild disruption of mitochondrial activity in worms induces an imbalance in the levels of core histone proteins (5). This leads to elevated protein levels of some, but not all, core histones, increased transcription of HSF-1 target genes, and extension of life span that is dependent on a chromatin remodeling protein, ISW-1 (5). In agreement with these findings, our data also support the requirement of histone H4 in mediating the longevity-promoting effects of impaired mitochondrial function (fig. S1E). This provides evidence for a bidirectional cross-talk between the nuclear and mitochondrial genomes that is mediated via HSF-1 activation and increased histone protein levels. Although previous studies have unraveled mechanisms of how stress signals originating from the mitochondria can affect the expression of nuclear-encoded genes (5, 6, 38), our study provides evidence for how signals originating from the nucleus can affect gene expression from the mitochondrial genome in the context of longevity regulation.
Although loss of histones has been identified as a conserved epigenetic hallmark of aging (26), increasing histone levels has rarely been used as a strategy to slow the rate of aging in multicellular organisms. Before this study, the only evidence of this was from budding yeast in which replenishment of histones by overexpression delays replicative senescence via counteracting the age-associated decline in histone levels (22, 28). During replicative aging, lack of sufficient histone proteins to properly package newly synthesized DNA leads to chromosomal instability and marked changes in gene expression in old yeast cells (28). Here, we show that H4 overexpression extends life span of C. elegans, a multicellular organism comprising nondividing somatic cells, via a potentially distinct mechanism. Increase in H4 levels by targeting the HSB-1/HSF-1 pathway does not significantly mitigate the age-associated decline in levels of core histones in somatic tissues of worms (Fig. 2G) but instead mediates its longevity-promoting effects primarily during development (Fig. 2, D, E, and H). A recent study has shown that prolonged heat stress in C. elegans results in transcriptional up-regulation of core histones and other genes involved in nucleosome assembly (44). Nonetheless, our results indicate that the higher H4 protein level in hsb-1(−) worms is not due to elevated transcription of H4 genes during development or adulthood (fig. S7, A and B). We further tested the possibility that inhibition of HSB-1 in worms might stabilize histone H4 posttranslationally via increasing the activity of histone chaperones, a less-studied family of proteins that functions to replace histones on chromatin in a replication-independent manner (22, 45). Although the expression of known histone chaperone genes in C. elegans was unaffected because of HSB-1 inhibition in adult worms (fig. S7C), we found elevated transcript levels of two histone chaperons, asfl-1 and hmg-3, in the somatic tissues of hsb-1(−) worms during the L1 stage of development (Fig. 6C). Moreover, the increased expression of both asfl-1 and hmg-3 was completely suppressed when HSF-1 was depleted in hsb-1(−) animals (Fig. 6C). The human homologs of C. elegans ASFL-1 and HMG-3 are ASF1A (anti-silencing function 1A histone chaperone) and the FACT (facilitates chromatin transcription) complex subunit structure-specific recognition protein 1 (SSRP1), respectively. Homologs of both these proteins have been shown to physically bind histones and stabilize them in multiple organisms across evolutionary taxa (22, 45, 46). This indicates that the elevated histone levels observed in hsb-1(−) animals are potentially a direct consequence of HSF-1–dependent up-regulation of certain histone chaperones during development.
Our findings show that knockdown of H4 suppresses the longevity phenotypes associated with HSF-1 activation and mitochondrial inhibition in C. elegans (Fig. 1, A to C and F, and fig. S1E), while overexpression of H4 extends life span of wild-type worms (Fig. 2C). In addition, depletion of H4 suppresses the higher global compaction of nuclear chromatin in hsb-1(−) animals (Fig. 3, A and B, and fig. S3, A to F). These findings collectively indicate that change in levels of a core histone can alter genome-wide chromatin organization and organismal life span. Although there are 16 H4-coding genes in C. elegans, RNAi directed against a single member of the H4 family can reduce the total protein levels of H4 in worms (Fig. 1D). This is possibly due to ~90% nucleotide sequence identity between the members of the H4 gene family in C. elegans that all code for the identical amino acid sequence (fig. S1A). Knockdown of H4 reduces the protein levels of other core histones as well (Fig. 1, D and E), while overexpression of H4 increases the protein levels of H3, H2A, and H2B (Fig. 2B and fig. S2A). This raises the possibility that change in levels of other core histones might also be partially responsible for the phenotypes observed in animals with altered H4 protein levels. We found that directly targeting other core histones results in partial or no suppression of longevity associated with HSB-1 inhibition in worms (fig. S7, D to F, and table S1). However, the biological effects solely attributable to change in protein levels of a single-core histone cannot be conclusively determined by approaches involving RNAi-mediated knockdown or ectopic overexpression. This is because, as stated previously, altering the levels of any one core histone seemingly almost always affects the levels of other core histones to some extent (Fig. 1, D and E, and fig. S2A) (5), potentially due to the presence of cellular homeostasis mechanisms (18). Hence, it is likely that many of the biological effects due to H4 RNAi or overexpression might be due to change in levels of multiple core histones and not only of H4. Nonetheless, as the triggering factor for the change in levels of the other histones was either knockdown or overexpression of H4 (Fig. 1, D and E, and fig. S2A), we can conclude that all the observed effects are either direct or indirect biological consequences of change in histone H4 levels. Even in conditions such as HSB-1 inhibition where histone levels were not manipulated exogenously, H4 might still require binding to other core histones to mediate many of its biological effects in the hsb-1(−) mutant. Hence, although the phenotypes associated with HSB-1 inhibition were found to be H4 dependent, the underlying mechanisms might involve coordinated assembly between multiple members of the histone family.
Another intriguing observation from our findings is the role of histone H4 in influencing gene expression from the mitochondrial genome. Across the animal kingdom, packaging of mitochondrial nucleoids is achieved via coating of mtDNA with TFAM, the most abundant protein constituent of the mitochondrial nucleoid complex (47). However, recent studies involving deoxyribonuclease I (DNase I) footprinting and ATAC (transposase-accessible chromatin)–seq have detected a potential chromatin-like organization of mtDNA that is labile to changes in physiological conditions but does not correlate with TFAM binding pattern (48, 49). Numerous protein-protected sites of mtDNA have been attributed to binding of primarily nuclear-localized transcription factors that have also been detected within mitochondria and several other unidentified proteins that potentially regulate transcription from the mitochondrial genome (50, 51). Before our study showing the presence of histone H4 in mitochondria of C. elegans (Fig 5, A, B, and D, and fig. S6A), a few studies have detected core histone proteins in mitochondria of mammalian tissues using mass spectrometry analysis (39, 40), although its physiological relevance was unknown. Moreover, in vitro binding studies have predicted an affinity of histone proteins for the conserved D-loop region of mtDNA (40). Our results showed that knockdown of H4 induces a significant decline in the MNase-protection of the D-loop region of mtDNA in hsb-1(−) animals (Fig. 3C, shown with cyan arrow). These evidences indicate that during the course of eukaryotic evolution, the mitochondrial genome has partially acquired chromatin-like features for context-dependent and/or locus-specific regulation of mitochondrial gene expression. Presence of H4 in C. elegans mitochondria and its binding to specific sites of the mitochondrial genome suggest an extranuclear role of mitochondrially translocated H4 in the in vivo packaging of mtDNA-nucleoid complexes (Fig 5, A to C). It is intriguing how HSB-1 inhibition results in increased H4 binding only at the ctc-1 and ctc-2 genomic loci on mtDNA (Fig 5C), but the expression of adjacent genes such as nduo-4 and ctc-3 was also altered in hsb-1(−) animals in an H4-dependent manner (Fig. 3E and fig. S3, H and I). Gene regulatory mechanisms for the mitochondrial genome remain less characterized, predominantly due to a lack of intergenic noncoding sequences in mtDNA of most organisms (51). Recent evidences have indicated that transcriptional regulatory sequences can also exist within coding sequences of mtDNA, which can potentially modulate expression of adjoining genes (51). It remains to be determined how binding of H4 to specific mtDNA loci influences the overall accessibility of different regions of the mitochondrial genome. Since the other core histones, H3, H2A, and H2B, have also been detected in mitochondria (Fig. 5B and fig. S5E) (39, 40), it remains elusive whether mitochondrial histones form a nucleosome-like structure and/or multiprotein complexes with other mitochondrially localized proteins to bind mtDNA and regulate its transcription. Overall, our findings provide new insights into the dynamic structure and expression pattern of the C. elegans mitochondrial genome and how these are amenable to longevity signals of the HSF-1/HSB-1 pathway (Fig. 6D).
Stress response, chromatin alterations, and mitochondrial function were traditionally considered to regulate longevity as three distinct hallmarks of aging (1, 2). This study establishes previously unidentified connections between these distinct cellular pathways, thus contributing to an integrated understanding of how they collectively manifest the regulation of organismal life span.
MATERIALS AND METHODS
C. elegans strains
All strains were maintained at 20°C on nematode growth medium (NGM) plates seeded with Escherichia coli OP50 strain using the standard method, unless otherwise stated. Animals were cultured for at least three generations in optimum conditions before they were used for experiments. The following strains were used in this study: wild-type (N2), EQ450: hsb-1(cg116) IV, CH116 outcrossed 4× to wild-type N2, CF512: rrf-3(b26) II; fem-1(hc17) IV, EQ196: rrf-3(b26) II; fem-1(hc17) IV; hsb-1(cg116) IV, CF1903: glp-1(e2144) III, EQ431: glp-1(e2144) III; hsb-1(cg116) IV, AGD710: uthIs235[(sur-5p::hsf-1::unc-54 3′UTR) + (myo-2p::tdTomato::unc-54 3′UTR)], CF1041: daf-2(e1370) III, MQ887: isp-1(qm150) IV, EQ435: iqEx136[(sur-5p::his-67) + (sur-5p::his-50) + (sur-5p::his-37) + (rol-6p::rol-6(su1006))], SS104: glp-4(bn2) I, EQ454: glp-4(bn2) I; hsb-1(cg116) IV, SJ4100: zcIs13[hsp-6p::gfp] V, EQ453: hsb-1(cg116) IV; zcIs13[hsp-6p::gfp] V, SJ4103: zcIs14[myo-3p::gfp(mitochondrial)], SJ4143: zcIs17[ges-1p::gfp(mitochondrial)], QK701: rde-1(ne219) V; xkIs[(ges-1p::rde-1) + (myo-2p::rfp)], EQ1404: hsb-1(cg116) IV; rde-1(ne219) V; xkIs[(ges-1p::rde-1) + (myo-2p::rfp)], EQ1426: iqIs276[(ges-1p::tomm-20::mCherry) + (rol-6p::rol-6(su1006))], and EQ1527: hsb-1(cg116) IV; iqIs276[(ges-1p::tomm-20::mCherry) + (rol-6p::rol-6(su1006))].
Wild-type (N2), CH116, MQ887, SS104, SJ4100, SJ4103, and SJ4143 strains were obtained from Caenorhabditis Genetics Center (Saint Paul, MN). AGD710 and QK701 strains were gifts from the laboratories of A. Dillin [University of California (UC), Berkeley] and J. Kim (Johns Hopkins), respectively. CF1903, EQ431, SS104, and EQ454 strains, carrying temperature-sensitive glp-1(e2144) or glp-4(bn2) mutation, were maintained at 15°C. For generation of EQ435 strain, a plasmid DNA mix consisting of pRF4[rol-6p::rol-6(su1006)] (80 ng/μl) and pAH186(sur-5p::his-67), pAH187(sur-5p::his-50), and pAH188 (sur-5p::his-37) (10 ng/μl each) was microinjected into the gonad of young adult N2 hermaphrodite animals. F1 progeny was picked on the basis of roller phenotype, and individuals from F2 progeny were isolated to generate the EQ435 strain. his-67, his-50, and his-37 genes were selected for overexpression because each of these correspond to one of the three structural variants of C. elegans histone H4 genes, all coding for the identical H4 protein. The promoter region of the ubiquitously expressed sur-5 gene was used for H4 overexpression. Microinjection of N2 worms with pRF4[rol-6p::rol-6(su1006)] alone did not affect the mean life span of wild-type animals when grown on OP50 or HT115(DE3) bacteria. For generation of EQ1426 strain, a plasmid DNA mix consisting of pRF4[rol-6p::rol-6(su1006)] (80 ng/μl) and pAH785(ges-1p::tomm-20::mCherry) (20 ng/μl) was microinjected to obtain F1 progeny with extrachromosomal arrays. Integrated lines were generated via UV irradiation followed by six generations of backcrossing to N2 worms. The mCherry signal was confirmed via fluorescence microscopy and immunoblot analysis. All experiments in this study were performed using day 1 adult worms that were obtained after transferring unhatched eggs to 25°C, unless otherwise stated.
RNAi analysis
RNAi clones used in this study were picked from J. Ahringer’s library and were confirmed by sequencing using M13 forward primer (5′-TGTAAAACGACGGCCAGT-3′). HT115(DE3) E. coli bacteria transformed with either empty vector (L4440) or plasmid expressing double-stranded RNA for desired gene were grown in lysogeny broth (LB) supplemented with carbenicillin (100 μg/ml) at 37°C overnight. Bacterial cultures were seeded on NGM or high-growth (HG) plates containing carbenicillin (100 μg/ml). Isopropyl-β-D-thiogalactopyranoside was added to the plates to a final concentration of 1 mM. Animals were subjected to RNAi by transferring unhatched eggs to RNAi plates at 25°C, unless otherwise stated. For RNAi only during adulthood, worms were transferred from L4440 plates to RNAi plates at late L4 to young adult stage.
Life-span analysis
Synchronized eggs were transferred by picking to NGM plates seeded with OP50 or HT115(DE3) bacteria (for RNAi conditions) at 25°C, unless otherwise stated. On day 1 of adulthood, worms were transferred to fresh plates with bacteria at a density of 12 to 15 worms per plate. Worms were subsequently transferred to new plates everyday until egg-laying continued. Viability of worms was scored every 1 to 2 days. Animals that were immobile and did not respond to gentle touch of a platinum pick were scored as dead. Worms that were bagged, exploded, crawled off plates, or were accidentally killed during the experiment were censored.
Genome-wide RNAi screen
For the primary screen, ~50 synchronized rrf-3(−); fem-1(−); hsb-1(−) eggs were transferred to 25°C on NGM plates seeded with double-stranded RNA-expressing HT115(DE3) bacteria (corresponding to 2700 RNAi clones from J. Ahringer’s library) at a density of ~50 eggs per plate on three plates per RNAi condition. On days 14 to 17 of adulthood, the proportion of alive worms on RNAi plates was compared to that on plates in which worms were fed empty vector–transformed bacteria. RNAi clones for which the proportion of surviving worms was at least 25% points lower than that observed in control bacteria-fed worms were selected for subsequent analyses. For the secondary screen, ~50 synchronized eggs for both rrf-3(−); fem-1(−) and rrf-3(−); fem-1(−); hsb-1(−) strains were transferred to 25°C on three plates per RNAi condition for clones that passed the criterion for primary screening. Proportion of alive worms on RNAi plates was estimated on days 12, 13, and 15 of adulthood. RNAi clones for which the proportion of surviving rrf-3(−); fem-1(−) worms was at most 10% points lower than that observed in control bacteria-fed rrf-3(−); fem-1(−) worms were selected for the tertiary screen. The purpose of the secondary screening step was to eliminate RNAi clones that induce a similar life-span shortening effect in both hsb-1(WT) and hsb-1(−) strains. For the final step of the screen, a comprehensive life-span analysis was performed with rrf-3(−); fem-1(−) and rrf-3(−); fem-1(−); hsb-1(−) worms at 25°C using RNAi clones that passed the criterion for secondary screening. RNAi clones for which the mean life-span shortening effect in rrf-3(−); fem-1(−); hsb-1(−) worms was at least 10% points greater than that observed in rrf-3(−); fem-1(−) worms were identified as suppressors of hsb-1(−)–associated life-span extension.
Immunoblot analysis using whole-worm extracts
A total of ~4000 age-synchronized young adult worms or ~ 20,000 age-synchronized L1 stage worms grown on NGM plates at 25°C were collected in M9 buffer containing 0.01% Triton X-100 and were washed three times with M9 buffer. Worms were suspended in three times the volume of 6× sample buffer [375 mM tris at pH 6.8, 12% SDS, 60% glycerol, 600 mM dithiothreitol (DTT), and 0.06% bromophenol blue], diluted to 2× final concentration, and snap-frozen in liquid nitrogen for 30 s. Samples were then boiled for 10 min, centrifuged at 16,000g for 1 min, and subjected to SDS–polyacrylamide gel electrophoresis (PAGE) using SE 250 minivertical unit (GE Healthcare). Proteins were transferred to Immobilon-P polyvinylidene difluoride membrane (Millipore Sigma) at 400 mA for 40 min using Trans-blot SD Semi-Dry Transfer Cell (Bio-Rad), and membrane was blocked in 5% blocking-grade blocker (Bio-Rad) for 1 hour at room temperature. Primary antibody incubation was performed for 12 hours at 4°C using the following antibodies: histone H4 (1:10,000; Millipore Sigma, 05-858), histone H3 (1:10,000; Abcam, ab1791), histone H2A (1:1000; Abcam, ab88770), histone H2B (1:10,000; Abcam, ab1790), β-actin (1:10,000; Abcam, ab8227), α-tubulin (1:10,000; Abcam, ab52866), γ-tubulin (1:2000; Abcam, ab50721), pan-acetyl histone H4 (acetyl K5 + K8 + K12 + K16; 1:20,000; Abcam, ab177790), histone H4 (acetyl K12; 1:10,000; Abcam, ab46983), and histone H4 (acetyl K16; 1:2000; Abcam, ab109463). Membrane was washed four times with tris-buffered saline (TBS) buffer containing 0.1% Tween 20 (TBS-T) for 5 min per wash, incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour at room temperature, and washed again four times with TBS-T buffer for 5 min per wash. Blots were developed using SuperSignal West Pico Plus chemiluminescent substrate (Thermo Fisher Scientific) and visualized using ChemiDoc MP imaging system (Bio-Rad). Relative protein levels were quantitated using ImageJ densitometry.
RNA isolation and quantitative real-time PCR
A total of ~2000 age-synchronized young adult worms or ~10,000 age-synchronized L1 stage worms grown on NGM plates at 25°C were collected in M9 buffer containing 0.01% Triton X-100 and were washed three times with M9 buffer. Total RNA was extracted with TRIzol reagent (Thermo Fisher Scientific) using the manufacturer’s protocol, and DNase treatment was performed using the TURBO DNA-free Kit (Thermo Fisher Scientific). Complementary DNA was synthesized from 5 μg of RNA with 6 μM random primer mix (New England Biolabs) using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). Real-time quantitative polymerase chain reaction (PCR) was performed with Power SYBR Green PCR master mix (Thermo Fisher Scientific), and PCR primers were listed in data file S2 using CFX96 Real-Time PCR detection system (Bio-Rad). Primers were designed using the Primer3Plus online tool. Relative transcript levels were calculated using comparative Ct method, and the housekeeping gene cdc-42 was used as internal control for normalization, unless otherwise stated. For comparison of transcript levels across age groups, the gene pmp-3 was used as internal control due to its stable expression pattern across different conditions (52). For animals of the same age group, normalization of transcript levels using either cdc-42 or pmp-3 led to similar results. For comparison across strains, expression levels of mitochondrially encoded genes were normalized to relative mtDNA copy number.
DNA damage assay
Age-synchronized young adult worms grown on NGM plates at 25°C were exposed to 254-nm wavelength UV radiation using UV Stratalinker 1800 (Stratagene). Worms were added to lysis buffer [1× standard Taq reaction buffer (New England Biolabs) containing proteinase K (1 mg/ml; Sigma-Aldrich)] in batches of γ worms/60 μl of buffer and were immediately frozen at −80°C for 15 min. Samples were incubated at 65°C for 1 hour, followed by at 95°C for 15 min, and the purified DNA was stored at −80°C. DNA lesion frequency was calculated using a previously described quantitative PCR–based DNA damage assay protocol (53). The assay is based on the principle that DNA lesions block or inhibit the progression of DNA polymerases during PCR amplification of a large genomic fragment. Briefly, a 9.3-kb fragment of the unc-2 gene was amplified from template DNA of UV-damaged worms using LongAmp Hot Start Taq 2X Master Mix (New England Biolabs), and PCR primers were listed in table S5. The PCR product was quantified using the Quant-iT PicoGreen Double-Stranded DNA Assay Kit (Thermo Fisher Scientific). To determine the starting amount of DNA for each condition, real-time quantitative PCR was performed for a 164–base pair (bp) fragment of W09C5.8 gene, as described previously, from template DNA of UV-damaged worms using PCR primers listed in data file S2. Relative amplification value for the long amplicon PCR product was quantified by normalization with starting amount of DNA determined from short amplicon PCR. Number of DNA lesions per 10 kb of DNA was estimated on the basis of inhibition of PCR amplification of the large genomic fragment from UV-treated worms compared to amplification of nondamaged template DNA from control worms, assuming a Poisson distribution of UV-induced lesions.
MNase digestion
A total of ~50,000 age-synchronized young adult worms grown on HG plates at 25°C were collected in M9 buffer containing 0.01% Triton X-100. Worms were washed three times with M9 buffer and were resuspended in an equal volume of 2× buffer A [30 mM Hepes-Na at pH 7.5, 680 mM sucrose, 120 mM KCl, 30 mM NaCl, 2 mM DTT, 1 mM spermidine (Sigma-Aldrich), 0.3 mM spermine tetrahydrochloride (Sigma-Aldrich), and 50 mM sodium bisulphite]. Samples were frozen in liquid nitrogen and stored at −80°C. For MNase digestion, frozen worm pellets were grinded in liquid nitrogen in a mortar and pestle, and CaCl2 was added to a final concentration of 1 mM. MNase (Sigma-Aldrich) was added to a final concentration of 0, 2.5, or 25 U/μl, and digestion was performed at 16°C for 12 min. The digestion reaction was stopped by addition of an equal volume of worm lysis buffer (100 mM tris at pH 8.5, 100 mM NaCl, 50 mM EDTA, and 1% SDS). Proteinase K (Sigma-Aldrich) was added to a final concentration of 1 mg/ml, and samples were incubated at 65°C for 45 min with intermittent vortexing. Samples were subjected to three sequential extraction steps using equal volumes of phenol, phenol:chloroform:isoamyl alcohol, and chloroform with centrifugation at 13,000g for 5 min. One-tenth volume of 8 M ammonium acetate solution and 2× volume of ethanol were added to precipitate DNA, and samples were centrifuged at 13,000g for 10 min. DNA pellets were washed with ethanol and resuspended in 1× TE (tris EDTA) buffer (10 mM tris at pH 8.0 and 1 mM EDTA). Ribonuclease A (Thermo Fisher Scientific) was added to a final concentration of 5 ng/μl, and samples were incubated at 37°C for 2 hours. Subsequently, glycogen (Sigma-Aldrich) was added to a final concentration of 400 μg/ml followed by addition of 3× volume of DNA extraction buffer (1 M ammonium acetate, 10 mM EDTA, and 0.2% SDS). Samples were then subjected to two sequential extraction steps using equal volumes of phenol:chloroform:isoamyl alcohol and chloroform with centrifugation at 13,000g for 6 min. DNA was precipitated by adding 2× volume of ethanol and incubation at −20°C for 1 hour, followed by centrifugation at 13,000g for 9 min. DNA pellets were resuspended in 1× TE buffer and subjected to electrophoresis on a 2% agarose gel to visualize the extent of MNase digestion. Concentration of mononucleosomal and dinucleosomal DNA fragments in the samples was quantified using the High-Sensitivity DNA Analysis Kit (Agilent Technologies) on a Bioanalyzer 2100 instrument (Agilent Technologies).
MNase sequencing and data analysis
C. elegans chromatin samples digested with MNase enzyme (25 U/μl), as described previously, were used for library preparation (four biological replicates per condition). For each sample, 20 ng of DNA was end-repaired, adapter-ligated, and PCR-amplified for 13 cycles using an Apollo 324 library preparation system (Wafergen Bio-systems). All samples were multiplexed for 150-bp paired-end sequencing on two lanes of a HiSeq 4000 platform (Illumina). For bioinformatic analysis of raw sequencing data, low-quality sequences and adapters were first trimmed using Trimmomatic tool. FastQC (available at www.bioinformatics.babraham.ac.uk/projects/fastqc) was then used for quality control of trimmed raw data. Next, Paired-End reAd mergeR (PEAR) was used to assemble read pairs. Subsequently, Bowtie 2 was used to align sequencing reads to the C. elegans genomic dataset version WS254. Nucleosome dynamics was determined from aligned sequencing reads using the Dynamic Analysis of Nucleosome Position and Occupancy by Sequencing version 2 tool (33). MNase-seq data for specific genomic regions were visualized on Integrated Genome Browser.
Relative mtDNA copy number
mtDNA content relative to nuclear DNA in worms was quantified using a previously described method (53). Briefly, age-synchronized young adult worms grown on NGM plates at 25°C were added to lysis buffer [1× standard Taq reaction buffer (New England Biolabs) containing proteinase K (1 mg/ml; Sigma-Aldrich)] in batches of eight worms/60 μl of buffer and were immediately frozen at −80°C for 15 min. Samples were incubated at 65°C for 1 hour, followed by at 95°C for 15 min. Mitochondrial and nuclear DNA content were estimated using real-time quantitative PCR, as described previously, with PCR primers listed in data file S2. Ratio of mtDNA:nuclear DNA copy number was calculated using comparative Ct method.
Mitochondrial oxygen consumption assays
Mitochondrial OCR was calculated using Seahorse Xfe96 analyzer (Agilent Technologies). Briefly, age-synchronized young adult worms grown on NGM plates at 25°C were collected in M9 buffer containing 0.01% Triton X-100. Worms were washed three times with M9 buffer and added to a 96-well plate at a density of 10 to 20 worms per well. Basal OCR measurement was performed five times, followed by measurement of maximal respiratory capacity for nine times after addition of carbonyl cyanide p-trifluoromethoxyphenylhydrazone (10 μM; Sigma-Aldrich). Subsequently, nonmitochondrial respiration was measured four times in presence of 40 mM sodium azide. OCR per worm was determined after normalization to number of worms in each well. Basal mitochondrial OCR per worm and maximal mitochondrial respiratory capacity per worm were obtained from measured OCR values after subtraction of nonmitochondrial respiration rates. Spare respiratory capacity per worm was calculated as the difference between maximal mitochondrial respiratory capacity per worm and basal mitochondrial OCR per worm for each well.
Whole-worm fluorescence intensity analysis
Age-synchronized young adult worms expressing an integrated hsp-6p::gfp transgene were anesthetized with 20 mM sodium azide and visualized under an Olympus BX63 microscope. GFP fluorescence intensity was quantified using the Fiji platform on ImageJ. For each genotype, at least 50 worms were analyzed.
Mitochondrial isolation
A total of ~60,000 age-synchronized L4 stage worms grown on HG plates at 25°C were collected in M9 buffer containing 0.01% Triton X-100 and were washed three times with M9 buffer. The worms were then washed once with ice-cold mitochondria isolation buffer (10 mM Hepes-KOH at pH 7.5, 210 mM mannitol, 70 mM sucrose, and 1 mM EDTA) with protease inhibitor cocktail (Roche), phosphatase inhibitors (Roche), and 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich). The worm pellet was resuspended in 10 times the volume of mitochondria isolation buffer and transferred in a 7-ml Dounce tissue grinder (Kimble) placed on ice. The sample was homogenized with pestle A for 20 strokes and pestle B for 40 strokes on ice and then centrifuged at 200g for 5 min to remove the debris. The supernatant was used as input and recentrifuged at 1000g for 10 min to pellet the nuclei. The supernatant obtained from the second centrifugation step was carefully filtrated using a syringe filter with a pore size of 1.2 μm (Sartorius) to remove any nuclei contamination. The filtrate was then centrifuged at 12,000g for 5 min to pellet the total mitochondria. The mitochondrial pellet was gently washed for three times with mitochondria isolation buffer and resuspended in the same buffer. The input, nuclear, and mitochondrial fractions were then subjected to SDS-PAGE followed by immunoblot analysis using the following antibodies: fibrillarin (1:1000; Novus Biologicals, NB300-269), RFP (1:10,000; Rockland 600-401-379), NDUFS3 (1:5000; Abcam, ab14711), ATP5A (1:3000; Abcam, ab14748), MTCO1 (1:3000; Abcam, ab14705), histone H3 (1:10,000; Abcam, ab1791), histone H4 (1:1000; Abcam, ab7311), histone H2A (1:1000; Abcam, ab88770), histone H2B (1:10,000; Abcam, ab1790), and glyceraldehyde-3-phosphate dehydrogenase (1:5000; Sigma-Aldrich, G8795).
Proteinase K protection assay
Freshly isolated mitochondria were digested with proteinase K as described previously (41). Briefly, the suspension of isolated mitochondria was first adjusted to a protein concentration at 1 μg/μl in mitochondria isolation buffer without protease inhibitors and was incubated with various concentrations of proteinase K (Invitrogen) for 15 min on ice. Proteinase K digestion was stopped with 20 mM PMSF (Sigma-Aldrich). Mitochondrial proteins were then subjected to SDS-PAGE followed by immunoblot analysis as described in the previous sections.
mtDNA chromatin immunoprecipitation and quantitative PCR
Freshly prepared suspensions of isolated mitochondria were cross-linked with 1% formaldehyde in mitochondria isolation buffer for 10 min at room temperature. This was followed by addition of 125 mM glycine for 10 min to quench the reaction. The mitochondria were then resuspended in 1× phosphate-buffered saline (PBS) containing 0.1% Triton X-100 and 1 mM CaCl2 and digested with MNase (4 U/μl; New England Biolabs) for 5 min at 37°C. The reaction was stopped by addition of 5 mM EDTA. The digested samples were then mixed with two times the volume of lysis buffer (25 mM Hepes-KOH at pH 7.5, 150 mM KCl, 10% glycerol, 5 mM MgCl2, 0.5 mM EDTA, and 1% Tween 20) with freshly added 1 mM PMSF and protease inhibitor cocktail (Roche). A total of 10 μg of mitochondrial protein was saved as input, while 100 μg of mitochondrial protein was immunoprecipitated with 3 μg of H4 antibody (Abcam, ab7311) by shaking at 4°C overnight. A total of 20 μl of magnetic protein A/G beads (Millipore) was added to the samples and incubated at 4°C for 2 hours to pull down the DNA-protein complexes. Beads were washed once with lysis buffer, two times with lysis buffer containing 250 mM NaCl, two times with LiCl buffer (10 mM tris at pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, and 1 mM EDTA), and two times with TE buffer (10 mM tris at pH 8.0 and 1 mM EDTA). Beads were then incubated in elution buffer (250 mM NaCl, 1% SDS, 10 mM tris at pH 8.0, and 1 mM EDTA) at 65°C for overnight. Subsequently, the samples were treated with 50 μg of proteinase K at 55°C for 2 hours to digest proteins. Both input DNA and antibody-precipitated DNA were purified using the Zymo DNA Concentrator-5 Kit (Zymo Research) and analyzed by real-time quantitative PCR using primers listed in data file S2. The PCR signal for each DNA region was normalized to the corresponding signal from input and then divided by the signal from a negative control region (mtDNA: 2317 to 2400) to determine the relative fold enrichment.
Cell-specific mitochondrial affinity purification
Intestinal mitochondria were isolated from worms expressing an integrated ges-1p::tomm-20::mCherry transgene using the previously described CS-MAP protocol (42). Briefly, mitochondria isolated from ~20,000 age-synchronized L4 stage worms were resuspended in 500 μl of ice-cold PEB buffer (1× PBS, 2 mM EDTA, and 1% bovine serum albumin) supplemented with protease inhibitor cocktail (Roche), phosphatase inhibitors (Roche), and 1 mM PMSF (Sigma-Aldrich). A preincubated mixture of 2 μg of anti-mCherry antibody (Abcam, ab183628) and 40 μl of protein G Mag Sepharose Xtra magnetic beads (GE healthcare, 28-9670-70) was added to the intact mitochondria, and the samples were incubated at 4°C for 1 hour with gentle agitation. Anti-rabbit immunoglobulin G (IgG) (Jackson ImmunoResearch) was used as a negative control antibody for immunoprecipitation. Beads were then washed three times with PEB buffer, boiled in SDS sample buffer, and subjected to SDS-PAGE followed by immunoblot analysis as described in the previous sections.
Immunofluorescence and mitochondrial labeling
A total of ~4000 age-synchronized young adult worms grown on NGM plates at 25°C were harvested in M9 buffer containing 0.01% Triton X-100. Worms were washed three times with M9 buffer and, if required, were incubated in 10 μM MitoTracker Orange CMTMRos solution (Thermo Fisher Scientific) for 1 hour at 25°C in the dark. Worms were then washed with ice-cold M9 buffer and fixed using a mixture of methanol, Bouin’s fixative [1.2% (w/v) picric acid, 37% (w/v) formaldehyde, and glacial acetic acid in 15:5:1 ratio], and β-mercaptoethanol (Sigma-Aldrich) in 50:50:1 ratio. Samples were frozen in liquid nitrogen and permeabilized by immediately thawing in a 25°C water bath for three freeze-thaw cycles. Worms were subsequently incubated on ice for 30 min, washed five times in BT buffer (50 mM H3BO3 and 0.5% Triton X-100 at pH 9.5) for 5 min per wash, and then washed five times in BT buffer containing 2% (v/v) β-mercaptoethanol for 1 hour per wash. Worms were then rinsed once with PBS buffer containing 0.1% Tween 20 and incubated in blocking solution (1% bovine serum albumin and 0.1% Tween 20 in PBS buffer) for 1 hour at room temperature. For primary antibody incubation, worms were incubated with the following antibodies for 3 days at 4°C with gentle agitation: Alexa Fluor 488–conjugated H4 antibody (1:50; Abcam, ab207387), H4 (1:50; Abcam, ab177840), and GFP (1:200; Abcam, ab1218). Subsequently, worms were washed six times in antibody wash buffer (0.1% bovine serum albumin in PBS buffer) for 5 min per wash at room temperature and with a final wash for 2 hours at 4°C. For samples treated with unconjugated primary antibodies, secondary antibody incubation was performed for 1 day at 4°C with Alexa Fluor Plus 488–conjugated goat anti-mouse IgG or Alexa Fluor Plus 555–conjugated goat anti-rabbit IgG at 1:500 dilution. Worms were again washed six times in antibody wash buffer for 5 min per wash at room temperature and then incubated in PBS containing 4′,6-diamidine-2′aphenylindole dihydrochloride (DAPI; 1 μg/ml) for 30 min at room temperature. Worms were mounted on a glass slide using Fluoroshield Mounting Medium (Abcam), and a coverslip was placed on top before imaging. Fluorescence imaging was performed on a Nikon A1 confocal microscope, equipped with diode-based laser system for 405-, 488-, 561-, and 640-nm excitation, using NIS-Elements (Nikon) imaging software. Images were processed using the Fiji platform on ImageJ.
Statistical analysis
For life-span experiments, Kaplan-Meier survival analysis and Mantel-Cox log rank test were performed using OASIS 2 (available at sbi.postech.ac.kr/oasis2). Statistical analysis of MNase-seq data was performed using “Stat” function of DANPOS2 tool (33), as described in Materials and Methods. Kolmogorov-Smirnov tests for comparison between genome-wide frequency distributions of nucleosomal parameters were performed in R. All other statistical analyses and graphing were performed using GraphPad Prism 8 for Windows. Details of statistical tests used, number of biological replicates (n), and P values for each experiment are included in figure legends.
Supplementary Material
Acknowledgments
We thank C. Mousigian for performing the genome-wide RNAi screen, Z. Pan (4S Inc.) for the help with bioinformatic analysis, W. Ding for assisting in life-span experiments, and S. Kumar for the help with Seahorse analysis. We acknowledge S. Johnson (BYU) and J. Meyer (Duke) for sharing protocols and advice, A. Dillin (UC Berkeley) and J. Kim (Johns Hopkins) for providing C. elegans strains, and S. Pletcher and D. Lombard for the helpful comments on the manuscript. Some C. elegans strains were obtained from the Caenorhabditis Genetics Center (University of Minnesota), which is supported by the NIH Office of Research Infrastructure Programs (P40 OD010440). Funding: The following funding sources supported this research: National Institute of Aging (AG028516) and Ministry of Science and Technology of Taiwan (MOST 106-2311-B-010-005 and MOST 108-2628-B-010-002). Author contributions: S.S. and A.L.H. conceptualized the project and designed the experiments. S.S., C.-Y.L., F.-Y.W., and T.-T.C. performed the experiments and analyzed data. S.S., T.-T.C., and A.-L.H. interpreted data. S.S., T.-T.C., and A.-L.H. wrote and edited the manuscript. All authors reviewed and approved the manuscript before publication. Competing interests: The authors declare that they have no competing interests. Data and materials availability: MNase-Seq data reported in this paper is deposited as NCBI Sequence Read Archive (SRA): SRP140908. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from aolinhsu@umich.edu.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaaz4452/DC1
REFERENCES AND NOTES
- 1.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riera C. E., Merkwirth C., De Magalhaes Filho C. D., Dillin A., Signaling networks determining life span. Annu. Rev. Biochem. 85, 35–64 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Chiang W.-C., Ching T.-T., Lee H. C., Mousigian C., Hsu A.-L., HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148, 322–334 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang A. B., Ryu E.-A., Artan M., Chang H.-W., Kabir M. H., Nam H.-J., Lee D., Yang J.-S., Kim S., Mair W. B., Lee C., Lee S. S., Lee S.-J., Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 111, E4458–E4467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matilainen O., Sleiman M. S. B., Quiros P. M., Garcia S. M. D. A., Auwerx J., The chromatin remodeling factor ISW-1 integrates organismal responses against nuclear and mitochondrial stress. Nat. Commun. 8, 1818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S. D., Quiros P. M., Steffen K. K., Williams E. G., Mouchiroud L., Tronnes S. U., Murillo V., Wolff S. C., Shaw R. J., Auwerx J., Dillin A., Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hipp M. S., Kasturi P., Hartl F. U., The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Gomez-Pastor R., Burchfiel E. T., Thiele D. J., Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 4–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu A.-L., Murphy C. T., Kenyon C., Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Morley J. F., Morimoto R. I., Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen E., Bieschke J., Perciavalle R. M., Kelly J. W., Dillin A., Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Fujimoto M., Takaki E., Hayashi T., Kitaura Y., Tanaka Y., Inouye S., Nakai A., Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 280, 34908–34916 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Steele A. D., Hutter G., Jackson W. S., Heppner F. L., Borkowski A. W., King O. D., Raymond G. J., Aguzzi A., Lindquist S., Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc. Natl. Acad. Sci.U.S.A. 105, 13626–13631 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baird N. A., Douglas P. M., Simic M. S., Grant A. R., Moresco J. J., Wolff S. C., Yates J. R. III, Manning G., Dillin A., HSF-1–mediated cytoskeletal integrity determines thermotolerance and life span. Science 346, 360–363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J., Labbadia J., Morimoto R. I., Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 27, 895–905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satyal S. H., Chen D., Fox S. G., Kramer J. M., Morimoto R. I., Negative regulation of the heat shock transcriptional response by HSBP1. Genes Dev. 12, 1962–1974 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller H., Fletcher M., Primitivo M., Leonard A., Sutphin G. L., Rintala N., Kaeberlein M., Leiser S. F., Genetic interaction with temperature is an important determinant of nematode longevity. Aging Cell 16, 1425–1429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh R. K., Kabbaj M.-H. M., Paik J., Gunjan A., Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat. Cell Biol. 11, 925–933 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenyon C. J., Chang J., Gensch E., Rudner A., Tabtlang R., A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Arantes-Oliveira N., Apfeld J., Dillin A., Kenyon C., Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295, 502–505 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Feng J., Bussière F., Hekimi S., Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Feser J., Truong D., Das C., Carson J. J., Kieft J., Harkness T., Tyler J. K., Elevated histone expression promotes life span extension. Mol. Cell 39, 724–735 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keall R., Whitelaw S., Pettitt J., Müller B., Histone gene expression and histone mRNA 3’ end structure in Caenorhabditis elegans. BMC Mol. Biol. 8, 51 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kodoyianni V., Maine E. M., Kimble J., Molecular basis of loss-of-function mutations in the glp-1 gene of Caenorhabditis elegans. Mol. Biol. Cell 3, 1199–1213 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beanan M. J., Strome S., Characterization of a germ-line proliferation mutation in C. elegans. Development 116, 755–766 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Sen P., Shah P. P., Nativio R., Berger S. L., Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Booth L. N., Brunet A., The aging epigenome. Mol. Cell 62, 728–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z., Chen K., Xia Z., Chavez M., Pal S., Seol J.-H., Chen C.-C., Li W., Tyler J. K., Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396–408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L., Cheung T. H., Charville G. W., Hurgo B. M. C., Leavitt T., Shih J., Brunet A., Rando T. A., Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 4, 189–204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni Z., Ebata A., Alipanahiramandi E., Lee S. S., Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11, 315–325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton B., Dong Y., Shindo M., Liu W., Odell I., Ruvkun G., Lee S. S., A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deniz Ö., Flores O., Aldea M., Soler-López M., Orozco M., Nucleosome architecture throughout the cell cycle. Sci. Rep. 6, 19729 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W., DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sumitani M., Kasashima K., Matsugi J., Endo H., Biochemical properties of Caenorhabditis elegans HMG-5, a regulator of mitochondrial DNA. J. Biochem. 149, 581–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dillin A., Hsu A.-L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A. G., Kamath R. S., Ahringer J., Kenyon C., Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Yoneda T., Benedetti C., Urano F., Clark S. G., Harding H. P., Ron D., Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Durieux J., Wolff S., Dillin A., The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houtkooper R. H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R. W., Auwerx J., Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson D. T., Harris R. A., French S., Blair P. V., You J., Bemis K. G., Wang M., Balaban R. S., Tissue heterogeneity of the mammalian mitochondrial proteome. Am. J. Physiol. Cell Physiol. 292, C689–C697 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Choi Y.-S., Jeong J. H., Min H.-K., Jung H.-J., Hwang D., Lee S.-W., Kim Pak Y., Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: Identification of mitochondrial histones. Mol. BioSyst. 7, 1523–1536 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Zhou C., Huang Y., Shao Y., May J., Prou D., Perier C., Dauer W., Schon E. A., Przedborski S., The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 105, 12022–12027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahier A., Dai C.-Y., Tweedie A., Bezawork-Geleta A., Kirmes I., Zuryn S., Affinity purification of cell-specific mitochondria from whole animals resolves patterns of genetic mosaicism. Nat. Cell Biol. 20, 352–360 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Douglas P. M., Baird N. A., Simic M. S., Uhlein S., McCormick M. A., Wolff S. C., Kennedy B. K., Dillin A., Heterotypic signals from neural HSF-1 separate thermotolerance from longevity. Cell Rep. 12, 1196–1204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jovic K., Sterken M. G., Grilli J., Bevers R. P. J., Rodriguez M., Riksen J. A. G., Allesina S., Kammenga J. E., Snoek L. B., Temporal dynamics of gene expression in heat-stressed Caenorhabditis elegans. PLOS ONE 12, e0189445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green E. M., Antczak A. J., Bailey A. O., Franco A. A., Wu K. J., Yates J. R. III, Kaufman P. D., Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol. 15, 2044–2049 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcianó G., Da Vela S., Tria G., Svergun D. I., Byron O., Huang D. T., Structure-specific recognition protein-1 (SSRP1) is an elongated homodimer that binds histones. J. Biol. Chem. 293, 10071–10083 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S. R., Han J., Mitochondrial nucleoid: Shield and switch of the mitochondrial genome. Oxidative Med. Cell. Longev. 2017, 8060949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blumberg A., Danko C. G., Kundaje A., Mishmar D., A common pattern of DNase I footprinting throughout the human mtDNA unveils clues for a chromatin-like organization. Genome Res. 28, 1158–1168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marom S., Blumberg A., Kundaje A., Mishmar D., mtDNA chromatin-like organization is gradually established during mammalian embryogenesis. iScience 12, 141–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercer T. R., Neph S., Dinger M. E., Crawford J., Smith M. A., Shearwood A.-M. J., Haugen E., Bracken C. P., Rackham O., Stamatoyannopoulos J. A., Filipovska A., Mattick J. S., The human mitochondrial transcriptome. Cell 146, 645–658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barshad G., Marom S., Cohen T., Mishmar D., Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet. 34, 682–692 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y., Chen D., Smith M. A., Zhang B., Pan X., Selection of reliable reference genes in Caenorhabditis elegans for analysis of nanotoxicity. PLOS ONE 7, e31849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.C. P. Gonzalez-Hunt, J. P. Rooney, I. T. Ryde, C. Anbalagan, R. Joglekar, J. N. Meyer, in Current Protocols in Toxicology (John Wiley & Sons Inc., 2016), vol. 67, pp. 20.11.1–20.11.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaaz4452/DC1