

Abstract
Prostate cancer (PCa) is the most common malignancy and is the second leading cause of cancer among men globally. Using a kinome‐wide lentiviral small‐hairpin RNA (shRNA) library screen, we identified phosphoinositide‐dependent kinase‐1 (PDPK1) as a potential mediator of cell survival in PCa cells. We showed that knock‐down of endogenous human PDPK1 induced significant tumour‐specific cell death in PCa cells (DU145 and PC3) but not in the normal prostate epithelial cells (RWPE‐1). Further analyses revealed that PDPK1 mediates cancer cell survival predominantly via activation of serum/glucocorticoid‐regulated kinase 3 (SGK3). Knock‐down of endogenous PDPK1 in DU145 and PC3 cells significantly reduced SGK3 phosphorylation while ectopic expression of a constitutively active SGK3 completely abrogated the apoptosis induced by PDPK1. In contrast, no such effect was observed in SGK1 and AKT phosphorylation following PDPK1 knock‐down. Importantly, PDPK1 inhibitors (GSK2334470 and BX‐795) significantly reduced tumour‐specific cell growth and synergized docetaxel sensitivity in PCa cells. In summary, our results demonstrated that PDPK1 mediates PCa cells’ survival through SGK3 signalling and suggest that inactivation of this PDPK1‐SGK3 axis may potentially serve as a novel therapeutic intervention for future treatment of PCa.
Keywords: BX‐795, GSK2334470, PDPK1, prostate cancer, RNAi screen, SGK3
1. INTRODUCTION
As the most prevalent form of non‐cutaneous cancer, prostate cancer (PCa) is the second‐highest incidence of cancer in male globally. 1 In 2018, approximately 1.3 million new patients were diagnosed with PCa, and nearly 360 000 deaths occurred globally. 1 In the early stages, PCa is mainly regulated by androgen, thus, androgen deprivation therapy (ADT) has become routine in clinical practice. However, about 10%‐20% of patients inevitably fail this therapy and progress to castration‐resistant PCa (CRPC) with a median survival ranges between 15 and 36 months. 2
Despite the fact that our understanding of the clinical, molecular and pathologic characteristics of PCa is incomplete, the androgen receptor (AR), which is regarded as the primary oncoprotein in PCa and CRPC, is regularly expressed in a heterogeneous way, even in the context of AR gene amplification. 3 In AR‐positive PCa, hormonal treatment resistance can arise via clonal selection, intracrine mechanisms or adaptation to decreased androgen (eg mutation, AR phosphorylation and bypass of the AR pathway). 4
While some of the early studies indicated that CRPC depends on AR activity, numerous new evidence suggests that other mechanisms have the capability to promote CRPC progression in a manner that is independent of AR activation. 5 For example, several studies have shown that phosphoinositide 3‐kinase (PI3K) signalling is adequate for CRPC survival when AR activity is reduced or not present. 5 , 6 , 7
In contrast to primary PCa, AR gene expression signatures were found to be inversely correlated with cell proliferation signatures in a subset of CRPC patients. 8 Furthermore, AR activities have also been shown to possess a tumour and metastasis suppressor function, suggesting PCa disease progression can be driven by AR‐independent mechanisms. 9 , 10
In order to identify genes and pathways that modify PCa growth in the context of suppressed AR signalling, we conducted in vitro high‐throughput RNA interference (RNAi) screening using a lentiviral‐shRNA library designed to target the whole kinome against the AR‐negative PCa DU145 cell line. 11
Here, we describe the results of our screen that identified the 3‐phosphoinositide‐dependent protein kinase 1 (PDPK1) as an essential kinase critical for the proliferation and viability of a subset of PCa cells.
2. MATERIALS AND METHODS
2.1. Cell lines and cell cultures
The human PCa cells (DU145, PC3 and LNCaP) and normal prostate epithelial cell (RWPE‐1) were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). DU145 and PC3 are AR‐negative while LNCaP harbours an androgen‐responsive AR mutant T877A. 11 All PCa cells were maintained in RPMI 1640 supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (Sigma‐Aldrich, St. Louis, MO, USA). The RWPE‐1 normal prostate cell line was grown in keratinocyte serum‐free medium consisting of 5 ng/mL of recombinant epidermal growth factor and 0.05 mg/mL of bovine pituitary extract (Invitrogen, Carlsbad, CA, USA). All cells were cultured at their logarithmic growth in a humidified 37°C, 5% CO2 incubator.
2.2. Lentiviral human kinase shrna library screen
The MISSION LentiExpress™ Human Kinases shRNA library (Sigma‐Aldrich, St Louis, MO, USA) was used to screen for candidate protein kinases mediating the growth of PCa cells. Briefly, the AR‐negative DU145 cells were seeded in a 384‐well plate overnight, followed by transduction of lentiviral particles at multiplicities of infection (MOI) of 1 in the presence of 7.5 μg/mL polybrene (Sigma‐Aldrich, St Louis, MO, USA). After 18h incubation, the medium containing the lentivirus particles was replaced with complete medium, and the cell viability was evaluated using the CellTiter‐Glo® assay (Promega, Madison, WI, USA) at 72 hours post‐transduction. Lentiviral particles carrying an empty vector (pLKO.1‐puro), a non‐target shRNA (NS) or a GFP expressing lentiviral construct were included as controls to examine transduction efficiency and well‐to‐well variation. All data were normalized against NS controls and hits were considered when shRNA targeting a specific gene achieved a Z‐score of less than −2. 12 , 13
2.3. Quantitative real‐time PCR (QPCR) analysis
The total RNA extraction and first‐strand cDNA synthesis were conducted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and High Capacity RNA to cDNA Master Mix (Applied Biosystems, Carlsbad, CA, USA), respectively. Gene expression levels were quantified by CFX96 PCR detector system (Bio‐Rad, Richmond, CA, USA) in the presence of FastStart Universal SYBR Green Master reagent (Roche Diagnostics, Indianapolis, IN, USA). The specific forward and reverse primer sequences are shown in Table S1. The qPCR condition that applied for all samples were: 94°C for 3 minutes, followed by 40 cycles of 94°C for 40 seconds, 60°C for 40 seconds and 72°C for 25 seconds. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was served as housekeeping gene for normalization.
2.4. Western blot analysis
All cell protein lysates were harvested using ice‐cold lysis buffer (1% NP‐40, 1 mM DTT, protease inhibitors, and phosphatase inhibitor I and II cocktails in PBS) as previously described. 14 , 15 A total of 50μg protein was loaded for immunoblotting. Monoclonal antibodies against PDPK1 and β‐actin were purchased from Santa Cruz Biotechnology, CA, USA. Primary antibodies against AR, PDPK1, p‐PDPK1 (S241), PTEN, AKT, p‐AKT (S473), p‐AKT (T308), SGK3, p‐SGK3 (T320), SGK1 and p‐SGK1 (S78) were obtained from Cell Signalling Technology, MA, USA.
2.5. Lentiviral production and transduction
Lentiviral non‐targeting shRNA (NS) and shRNA constructs targeting PDPK1, CAMKV and CKS1B were purchased from Sigma‐Aldrich, MO, United States, with target sequences shown in Table S2. To produce the lentiviral particles of interest, the target shRNA constructs were co‐transfected into HEK‐293T cells with lentiviral packaging plasmids, psPAX2 (Addgene plasmid #12260) and envelope plasmids, pMD2.G (Addgene plasmid #12259) as described previously. 16 , 17 , 18 The lentiviral particles were then collected and added with 7.5 µg/mL polybrene (Sigma‐Aldrich, St Louis, MO, USA) for transduction.
2.6. Detection of apoptosis by annexin V flow cytometry
All floating and attached cells were stained for cell apoptosis assay using a PE Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) as described previously. 19 , 20 The samples were quantitated using a FACSCalibur flow cytometer and analysed by CellQuest Pro software (version 5.1.1; BD Biosciences, San Jose, CA, USA).
2.7. Transfection
Plasmids for constitutively active myristoylated AKT and SGK3‐S486D mutant were obtained from Addgene (Addgene plasmid # 9008) and Gene Universal (Newark, DE, USA), respectively. Plasmids were transfected into target cells using X‐tremeGENE HP DNA transfection reagent (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer's protocol.
2.8. Drug combination analysis
Drug combinatory effects were analysed using the Chou‐Talalay method and Highest Single Agent (HSA) models as described previously. 21 , 22 Briefly, cells were plated at 2.5 × 103 cells/well in 96‐well format and treated with docetaxel and/or PDPK1 inhibitors (GSK2334470 and BX795) alone or in combination. The plates were terminated by MTT cell proliferation assay at 72 hours after treatment. 23 , 24 Calcusyn 2.1 software (Biosoft, Cambridge, UK) was used to generate combination index (CI) based on Chou‐Talalay method, 19 , 25 in which CI < 1, = 1 and >1 indicates synergism, additive and antagonism effect, respectively (Table S3). The dose‐response surface curves with levels of HSA synergy were plotted by Combenefit software (Cancer Research UK Cambridge Institute). 26
2.9. Statistical analysis
All results were presented as mean ± standard deviation (SD) from at least three independent experiments. SPSS (version 19.0 INC, Chicago, IL) was used to evaluate the statistical significance based on Student's independent t test. A P‐value <0.01 was considered statistically significant.
3. RESULTS
3.1. Kinome‐wide shRNA library screen identifies PDPK1 as putative target mediating PCa cell survival
To identify genes and pathways that modify PCa growth, we conducted in vitro high‐throughput RNA interference (RNAi) screening using the RNAi Consortium (TRC) kinome shRNA library consisting of 3197 lentiviruses carrying shRNA sequences targeting 506 human kinase genes. Each gene is represented by at least 3–5 individual constructs, targeting different regions of the gene sequence. A total of 45 kinases (8.9%) in the TRC kinome library were identified to induce significant growth inhibition (Z‐score < −2) in DU145 PCa cells (Figure 1A and B, Table S4). Among them include a number of proto‐oncogene (ERBB3, ERBB4, RET, SRC and YES1), pro‐survival genes involved in the PI3K signalling (AKT1, AKT3, GSK3A, GSK3B, PDPK1 and SGK3) and MAPK signalling (MAP3K4, MAPK1, MAPK4, MAPK13 and MAPK15). Importantly, knock‐down of AKT1, 27 , 28 , 29 AKT3, 27 , 28 , 29 GSK3A, 30 , 31 , 32 GSK3B, 30 , 31 , 32 , 33 , 34 , 35 MAPK1 (also known as ERK2), 36 , 37 MAPK4 (also known as ERK4) 29 and ROCK2 38 , 39 , 40 have been shown to inhibit PCa cell growth, independently validated our results.
FIGURE 1.
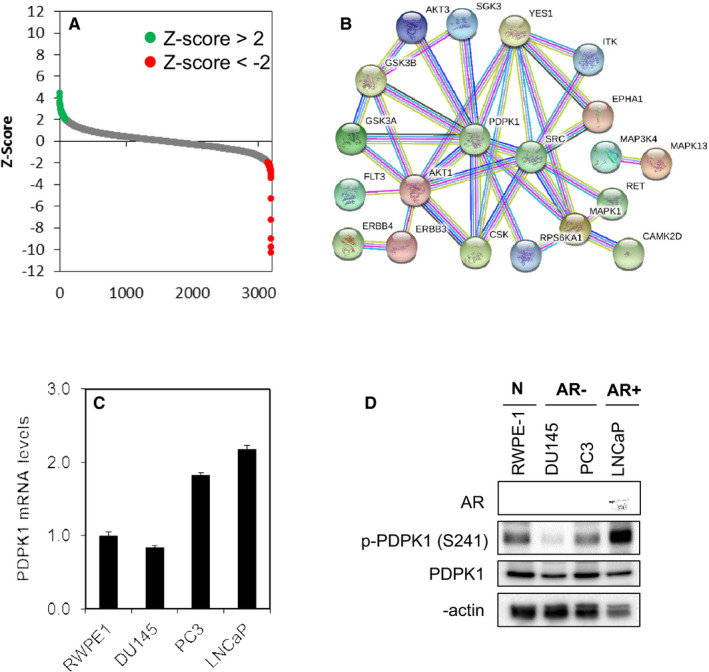
Kinome‐wide shRNA library screen identifies PDPK1 as putative target regulating the survival of PCa cells. A, Kinase shRNA screen scatter plot. Z‐scores are plotted on the y‐axis against 3109 corresponding shRNAs on the x‐axis. The red and green circled dots represent shRNA hits, which the former inhibited cell proliferation and the latter promoted cell proliferation. B, Protein‐protein interaction network of the PDPK1 target proteins. C and D, PDPK1 is expressed in a subset of PCa cells and RWPE‐1 non‐transformed prostate epithelial cells. PDPK1 mRNA expression was evaluated by qPCR with GAPDH as housekeeping gene. The level of PDPK1 protein expression was detected by immunoblotting with β‐actin as loading control
Next, we selected 3 candidate kinases (PDPK1, CAMKV and CKS1B) for further study based on the novelty and the magnitude of effect by shRNA knock‐down. Indeed, depletion of the endogenous PDPK1, CAMKV and CKS1B significantly reduced DU145 cell survival, consistent with the results obtained in the primary screen (Figure S1).
Recent studies have shown that the locus containing PDPK1 gene (16p13.3) is more frequently amplified in lymph node metastases and castration‐resistant PCa, compared to primary tumours, 41 we decided to focus on understanding the mechanism underlying PDPK1 mediated cell survival in PCa cells. We first evaluated whether PDPK1 is expressed in a panel of AR‐negative (DU145 and PC3) and AR‐positive (LNCaP) PCa cell lines, as well as non‐transformed prostate epithelial cells (RWPE‐1). Real‐time qPCR shows that PDPK1 mRNA was highly expressed in all the PCa and normal prostate epithelial cells (Figure 1C). The level of gene expression correlated well with the PDPK1 protein expression as PDPK1 proteins were highly expressed in LNCaP, PC3, RWPE‐1 and DU145 (Figure 1D). Interestingly, PDPK1 proteins were found to be phosphorylated in cells which express them, suggesting that PDPK1 proteins are constitutively active in these cells.
3.2. Depletion of PDPK1 induces tumour‐specific cell death PCa cells
To determine whether depletion of endogenous PDPK1 has any effect on the proliferation and survival of PCa cells that exhibit active PDPK1, we performed lentiviral shRNAs‐mediated knock‐down of PDPK1 in a panel of PCa and non‐transformed prostate epithelial cells. Efficient knock‐down of PDPK1 in all prostate cell lines by two independent shRNA constructs was demonstrated in Western blotting (Figure 2A). Significant reduction in cell viability of at least 80% in DU145 and PC3 was observed while no effect was observed in LNCaP and RWPE‐1 (P < .05, Student's t test) (Figure 2B and Figure S2). Consistent with the findings from cell viability assay, depletion of endogenous PDPK1 also induced significant amount of apoptosis in DU145 and PC3 cells (P < .01, Student's t test) (Figure 2C), corroborated with the induction of caspase 3 and 9, but not caspase 8 activity (Figure 2D). In contrast, no such effects were observed in RWPE‐1 nor in LNCaP cells. These results suggest that PDPK1 is required for the survival of the AR‐negative DU145 and PC3 cells, but not for the AR‐positive LNCaP cells.
FIGURE 2.
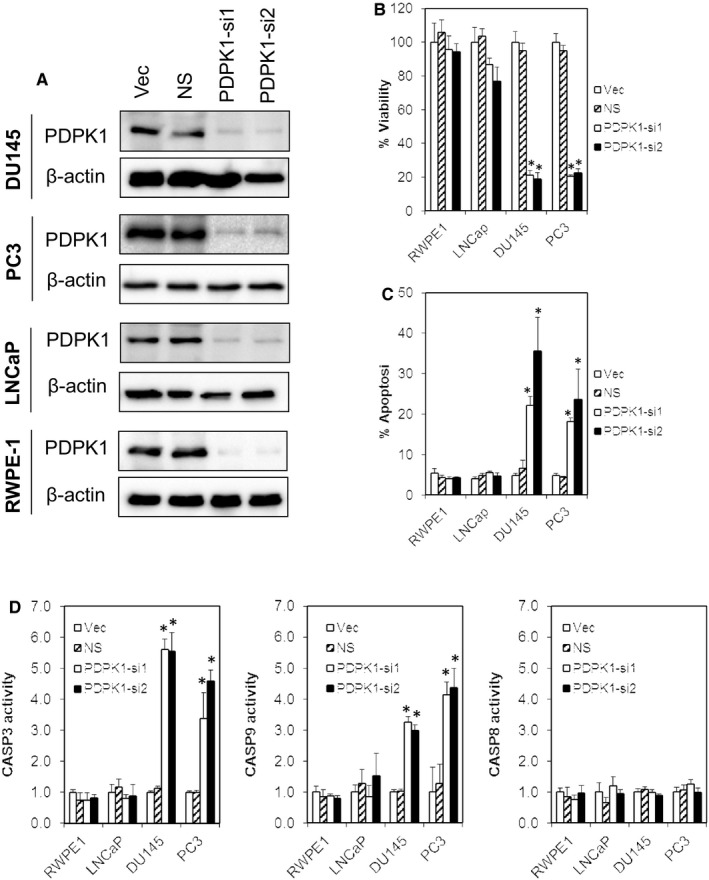
Depletion of endogenous PDPK1 induces tumour‐specific cell death in PCa cells. A, Effective PDPK1 knock‐down was achieved by two independent shRNA constructs targeting PDPK1 (PDPK1‐si1 and PDPK1‐si2). Lysates were harvested at 72 h post‐lentiviral transduction and analysed by immunoblotting. B and C, PDPK1 depletion selectively inhibited the proliferation and induced apoptosis in AR‐negative DU145 and PC3 PCa cells but not in AR‐positive LNCaP or RWPE‐1 non‐transformed prostate epithelial cells. Cell viability and apoptosis were measured using CellTiter‐Glo® assay and annexin V/7‐AAD flow cytometry at 72 h post‐transduction. D, Depletion of endogenous PDPK1 induced caspase 3 and 9 activities. Caspase 3, 8 and 9 activities were evaluated by CaspaseGlo assay at 72 h post‐transduction. Bars represent means ± SD of three independent experiments. (*) indicates statistical significance compared with NS control cells (P < .01, Student's t test)
3.3. Depletion of PDPK1 inhibits SGK3 phosphorylation
PDPK1 is known to phosphorylate AKT that regulates several signalling pathways altered in cancer. 42 However, recent studies have also shown that PDPK1 can activate many other members of AGC kinase family such as p70S6K, SGK, p90RSK and the members of PKC family, independent of AKT. 42 , 43 , 44 To evaluate whether the pro‐survival effects of PDPK1 in PCa cells are mediated through aberrant activation of downstream AKT or SGK pathways, we analysed the effects of PDPK1 knock‐down on the expression and phosphorylation of these targets. As shown in Figure 3, knock‐down of PDPK1 in DU145 and PC3 cells significantly reduced phosphorylation of SGK3 but not the phosphorylation of AKT or SGK1. The total expression of SGK3, SGK1 and AKT remained unchanged. In stark contrast, no such effects were observed in RWPE‐1 or LNCaP cells.
FIGURE 3.
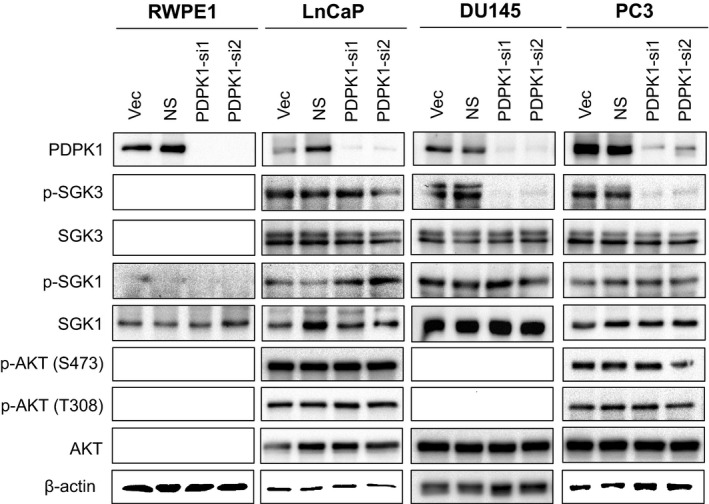
Depletion of endogenous PDPK1 reduces SGK3 phosphorylation. PDPK1 depletion down‐regulated SGK3 phosphorylation in AR‐negative DU145 and PC3 cells, but not in AR‐positive LNCaP PCa cells or RWPE‐1 non‐transformed prostate epithelial cells. The protein expression and phosphorylation of AKT, SGK1 and SGK3 Lysates were analysed by immunoblotting with β‐actin served as loading controls
3.4. PDPK1 mediates the survival of DU145 and PC3 cells via SGK3 signalling pathway
To test whether the pro‐survival effects of PDPK1 is mediated through SGK3, we transfected a constitutively active SGK3 S486D mutant in DU145 and PC3 cells followed by PDPK1 depletion. As shown in Figure 4, ectopic expression of SGK3 S486D significantly rescued the apoptosis induced by PDPK1 depletion (P < .01, Student's t test). In contrast, no such effects were observed in cells transfected with a constitutively active myristoylated AKT (Myr‐AKT), suggesting that PDPK1 regulates cell survival in PCa cells through activation of SGK3 signalling (Figure S3).
FIGURE 4.
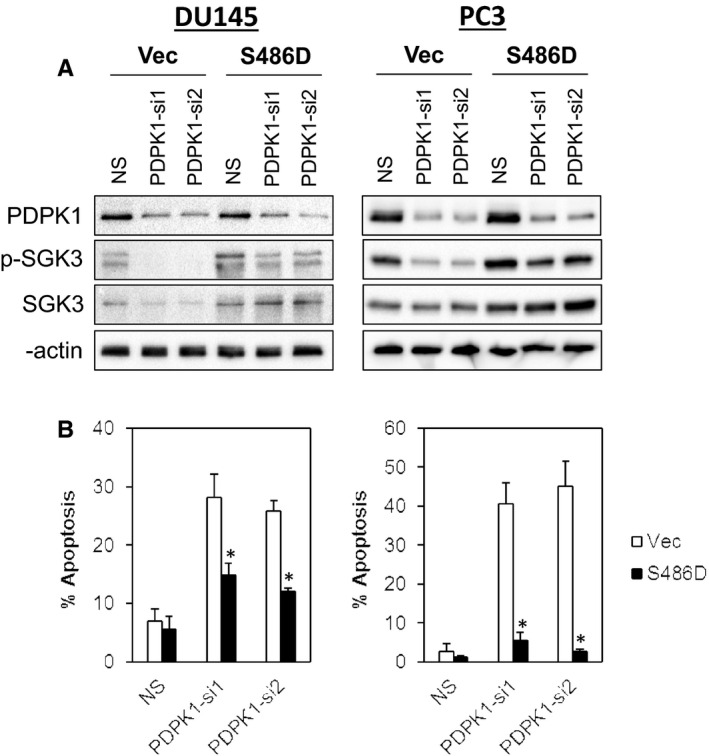
PDPK1 regulates the survival of DU145 and PC3 cells via SGK3 signalling pathway. Ectopic expression of a constitutively active SGK3 (S486D) reduced apoptotic cell death induced by PDPK1 knock‐down in DU145 cells and PC3 cells. A, The constitutively active SGK3 S486D plasmid was cotransfected with either NS or PDPK1 targeting shRNAs into DU145 and PC3 PCa cells. Whole cell protein lysates were harvested 72 h post‐transfection and analysed by immunoblotting. B, The apoptotic cell death was detected by annexin V/7‐AAD flow cytometry. Bars represent means ± SD of three independent experiments. (*) indicates statistical significance compared with Vec control cells (P < .01, Student's t test)
3.5. Inhibition of PDPK1 enhances docetaxel sensitivity in PCa cells
Since PDPK1 up‐regulation and activation have been recently shown to confer chemoresistance in breast, 45 glioblastoma, 46 neuroblastoma 47 and pancreatic cancer cells, 48 we asked whether inhibition of PDPK1 might enhance chemotherapy sensitivity in PCa cells. To test this hypothesis, we first evaluated the effects of docetaxel, a commonly used chemotherapeutic agent for PCa, 49 and PDPK1 inhibitors (GSK2334470 and BX795) on a panel of PCa cells (DU145, PC3 and LNCaP). As shown in Figure 5A and Table S5, the AR‐positive LNCaP was more sensitive to docetaxel (IC50 of 7.49 ± 2.45 nM) as compared to the AR‐negative DU145 and PC3 (IC50 of 20.00 ± 5.68 nM and > 100 nM, respectively). In contrast, both DU145 and PC3 were more sensitive to GSK2334470 (IC50 of 12.74 ± 1.22 µM and 9.16 ± 0.59 µM, respectively) and BX795 (IC50 of 11.49 ± 5.49 µM and 5.10 ± 0.45 µM, respectively) as compared to LNCaP (IC50 >100 µM), consistent with the results obtained from the PDPK1 knock‐down.
FIGURE 5.
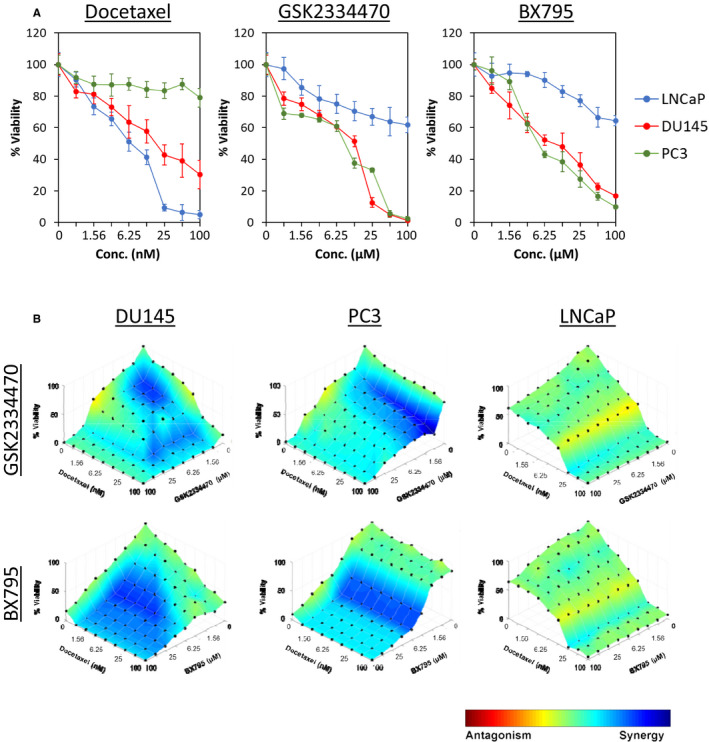
Inhibition of PDPK1 enhances docetaxel sensitivity in PCa cells. A, The PCa cells were treated with docetaxel, GSK2334470 and BX795 for 72 h, followed by evaluation of cell viability. Each data point represents mean ± SD of at least three independent experiments. B, Synergistic effects of PDPK1 inhibitors (GSK2334470 and BX795) and docetaxel in DU145, PC3 and LNCaP cells. PCa cells were treated with docetaxel and/or PDPK1 inhibitors for 72 h. The dose‐response surface curves with HSA synergy/antagonism of the combinations were generated by Combenefit software. 26 A colour scale bar is used to indicate the level of synergism (blue) or antagonism (red) for each combination. All experiments were conducted at least three times
Next, we investigated whether the inhibition of PDPK1 could synergize docetaxel sensitivity in PCa cells. As shown in Figure 5B, inhibition of PDPK1 enhances docetaxel sensitivity in DU145 and PC3 but not LNCaP cells, suggesting that PDPK1 inhibitors might potentiate sensitivity of refractory PCa cells to chemotherapy (Tables 1 and 2).
TABLE 1.
Combinatory effects of docetaxel and GSK2334470 in PCa cells
| Cell lines | Doc:GSK Ratio | Combination Index (CI) | Mean ± SD | Interactions | ||
|---|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | ||||
| DU145 | 1:250 | 0.591 | 0.479 | 0.424 | 0.498 ± 0.085 | Synergism |
| 1:500 | 0.534 | 0.513 | 0.532 | 0.526 ± 0.011 | Synergism | |
| 1:1000 | 0.606 | 0.591 | 0.607 | 0.601 ± 0.009 | Synergism | |
| 1:2000 | 0.785 | 0.759 | 0.757 | 0.767 ± 0.016 | Moderate Synergism | |
| 1:4000 | 1.060 | 0.945 | 0.857 | 0.954 ± 0.102 | Nearly additive | |
| PC3 | 1:250 | 0.232 | 0.302 | 0.441 | 0.325 ± 0.106 | Synergism |
| 1:500 | 0.369 | 0.440 | 0.524 | 0.444 ± 0.078 | Synergism | |
| 1:1000 | 0.478 | 0.467 | 0.456 | 0.467 ± 0.011 | Synergism | |
| 1:2000 | 0.643 | 0.569 | 0.505 | 0.572 ± 0.069 | Synergism | |
| 1:4000 | 0.964 | 0.746 | 0.577 | 0.763 ± 0.194 | Moderate Synergism | |
| LNCaP | 1:250 | 1.153 | 1.030 | 0.926 | 1.037 ± 0.114 | Nearly additive |
| 1:500 | 1.011 | 0.940 | 0.883 | 0.945 ± 0.064 | Nearly additive | |
| 1:1000 | 1.003 | 0.928 | 0.877 | 0.936 ± 0.063 | Nearly additive | |
| 1:2000 | 0.877 | 0.755 | 0.677 | 0.769 ± 0.101 | Nearly additive | |
| 1:4000 | 0.827 | 0.802 | 0.841 | 0.824 ± 0.020 | Nearly additive | |
TABLE 2.
Combinatory effects of docetaxel and BX795 in PCa cells
| Cell lines |
Doc:BX Ratio |
Combination index (CI) | Mean ± SD | Interactions | ||
|---|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | ||||
| DU145 | 1:250 | 0.522 | 0.277 | 0.151 | 0.317 ± 0.189 | Synergism |
| 1:500 | 0.385 | 0.195 | 0.100 | 0.227 ± 0.145 | Strong Synergism | |
| 1:1000 | 0.359 | 0.170 | 0.082 | 0.204 ± 0.142 | Strong Synergism | |
| 1:2000 | 0.280 | 0.143 | 0.073 | 0.165 ± 0.105 | Strong Synergism | |
| 1:4000 | 0.256 | 0.134 | 0.071 | 0.154 ± 0.094 | Strong Synergism | |
| PC3 | 1:250 | 0.337 | 0.272 | 0.220 | 0.276 ± 0.058 | Strong Synergism |
| 1:500 | 0.321 | 0.271 | 0.229 | 0.273 ± 0.046 | Strong Synergism | |
| 1:1000 | 0.344 | 0.279 | 0.226 | 0.283 ± 0.059 | Strong Synergism | |
| 1:2000 | 0.323 | 0.268 | 0.223 | 0.271 ± 0.050 | Strong Synergism | |
| 1:4000 | 0.240 | 0.222 | 0.205 | 0.222 ± 0.017 | Strong Synergism | |
| LNCaP | 1:250 | 1.036 | 1.048 | 1.063 | 1.049 ± 0.013 | Nearly additive |
| 1:500 | 1.056 | 1.074 | 1.097 | 1.076 ± 0.021 | Nearly additive | |
| 1:1000 | 1.033 | 0.930 | 0.846 | 0.936 ± 0.094 | Nearly additive | |
| 1:2000 | 1.000 | 0.905 | 0.833 | 0.913 ± 0.084 | Nearly additive | |
| 1:4000 | 0.991 | 0.905 | 0.880 | 0.925 ± 0.058 | Nearly additive | |
4. DISCUSSION
In this study, we identified 45 kinases that mediate PCa cell survival. These include previously identified proto‐oncogene such as ERBB3, ERBB4, RET, SRC and YES1; and pro‐survival genes involved in the PI3K and MAPK signalling such as AKT1, AKT3, GSK3A, GSK3B, PDPK1, SGK3, MAP3K4, MAPK1 (also known as ERK2), MAPK4 (also known as ERK4), MAPK13, MAPK15 and ROCK2. 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 Particularly, knock‐down of PDPK1 induced tumour‐specific cell death in both DU145 and PC3 AR‐negative cells, but not in the AR‐positive LNCaP cells nor in the RWPE‐1 non‐transformed prostate epithelial cells.
Amplification or overexpression of PDPK1 has been implicated in tumourigenesis and cancer cells survival in many human cancers including breast cancer, 50 , 51 oesophageal squamous cell carcinoma, 52 melanoma, 53 gastric carcinoma, 54 hepatocellular carcinoma 55 and acute myeloid leukaemia. 56 A recent study identified the locus containing PDPK1 gene (16p13.3) is more frequently amplified in lymph node metastases and CRPC, compared to primary tumours, suggesting PDPK1 may also support cancer metastasis. 41
PDPK1 is known to function downstream of PI3K and is required for the full activation of AKT serine/threonine kinase 1 (AKT1) and other AGC kinases such as protein kinase C (PKC), p70 ribosomal protein S6 kinase (S6K), p90 ribosomal protein S6 kinase (RSK), polo‐like kinase 1 (PLK1) and serum glucocorticoid‐dependent kinase (SGK). 57
Upon activation, PDPK1 binds to phosphatidylinositol 3,4,5 trisphosphate (PIP3), the product of PI3K, via its pleckstrin homology (PH) domain at the plasma membrane. This in turn leads to the phosphorylation AKT at T308 and activates AKT signalling. 42 , 58 In the case of PDPK1 substrates that do not possess a PH domain (eg S6K, RSK and SGK), PDPK1 can still interact and bind to the hydrophobic motif of the target kinase via the PDK1‐interacting fragment (PIF) to activate the downstream signalling pathways. 40 , 56
In an effort to define the mechanisms of PDPK1‐mediated regulation of PCa cell survival, we observed that depletion of PDPK1 reduced SGK3 phosphorylation, but have no effects on AKT phosphorylation in cell line models with low‐ and hyperactivated background levels of AKT activity (DU145 and PC3 respectively). 59 Furthermore, ectopic expression of a constitutively active SGK3 significantly abrogated the apoptotic effects induced by PDPK1 depletion, while no such effects were observed in cells expressing a myristoylated AKT. These data are consistent with accumulating evidence indicating that PDPK1 can contribute to cancer through activation of SGK3, independent of AKT. 58 , 60 For instance, it was reported that human cancers with PIK3CA mutations where AKT activity is deficient, SGK3 serves as the main PDPK1 effector to drive tumour cells survival. 43 Similarly, SGK3 was also found to be a key mediator of PDPK1‐dependent melanomagenesis and a driver for tumour formation in breast cancer cells in both PIK3CA wild‐type and mutated cells in an AKT‐independent manner. 61 , 62 , 63 Thus, our data strongly suggest that PDPK1 is mediating the survival of the AR‐negative DU145 and PC3 PCa cells through activation of SGK3, independent of AKT signalling.
Finally, our study further demonstrates that pharmacological inhibition of PDPK1, using 2 distinct chemical compounds (GSK2334470 and BX795), strongly reduced cancer cell growth in the AR‐negative DU145 and PC3 PCa cells, but not in the AR‐positive LNCaP or the RWPE‐1 non‐transformed prostate epithelial cells (data not shown). Importantly, we also report that both GSK2334470 and BX795 synergize docetaxel sensitivity in DU145 and PC3 cells, but not in LNCaP cells. Indeed, it has been observed that SGK3 can substitute for AKT in activating mTORC1, 64 which in turn has been implicated in docetaxel resistance in PCa. 65 To date, the role of PDPK1 in driving cancer and chemoresistance has been outlined in multiple cancers, including acute myeloid leukaemia, breast cancer and ovarian cancer. 44 These findings further underscore the potential of PDPK1 as a therapeutic target, as they indicate that PDPK1 or SGK3 can act as druggable targets in the treatment of hormone‐refractory PCa as single agents or in combination with chemotherapeutics as components of a multitargeted therapy regimen.
5. CONCLUSIONS
In conclusion, we identified PDPK1 as a novel potential therapeutic target in PCa and demonstrated PDPK1 is mediating PCa cells’ survival through activation of SGK3 in an AKT‐independent manner. Our data further suggest that combination of PDPK1 inhibitors with docetaxel enhances their anti‐cancer activity, possibly by targeting SGK3‐dependent resistance mechanisms. Together, our results provide a strong rationale to investigate further the use of PDPK1 inhibitors in as novel therapeutic strategies for refractory PCa patients.
AUTHORS’ CONTRIBUTION
IC, COL and AHAR designed the study. GN, FFLC, CWM, LWH, KKC and WML developed the methodology, collected the data and performed the analysis. GN, COL and IC wrote the manuscript. All authors reviewed and approved the manuscript. Geetha Nalairndran: Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal). Azad Hassan Abdul Razack: Conceptualization (equal); Funding acquisition (equal); Project administration (equal); Supervision (equal). Chun‐Wai Mai: Investigation (equal); Methodology (equal); Supervision (equal); Validation (equal). Felicia Fei‐Lei Chung: Investigation (equal); Methodology (equal); Writing‐review & editing (equal). Kok‐Keong Chan: Funding acquisition (equal); Project administration (equal); Supervision (equal). Ling‐Wei Hii: Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal). Wei‐Meng Lim: Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal). Ivy Chung: Conceptualization (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Project administration (equal); Validation (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Chee‐Onn Leong: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Project administration (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal).
CONFLICT OF INTEREST
The author(s) declare no competing interests. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
DATA STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
Supporting information
Figures S1‐S3
Tables S1‐S5
Figure Legends
ACKNOWLEDGEMENTS
This work was supported by research grant from HIGH IMPACT RESEARCH (HIR)—UNIVERSITI MALAYA (UM) (UM.C/625/1/HIR/MOHE/MED/35 to GN, IC and AHAR), the MINISTRY OF HIGHER EDUCATION, MALAYSIA (ERGS/1/2013/SKK01/IMU/02/1 to KKC and WML), and the MINISTRY OF HIGHER EDUCATION, MALAYSIA (FRGS/1/2016/SKK08/IMU/01/1 to COL, and LWH).
Nalairndran G, Hassan Abdul Razack A, Mai C‐W, et al. Phosphoinositide‐dependent Kinase‐1 (PDPK1) regulates serum/glucocorticoid‐regulated Kinase 3 (SGK3) for prostate cancer cell survival. J Cell Mol Med. 2020;24:12188–12198. 10.1111/jcmm.15876
Contributor Information
Ivy Chung, Email: ivychung@ummc.edu.my.
Chee‐Onn Leong, Email: cheeonn_leong@imu.edu.my.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Wade CA, Kyprianou N. Profiling prostate cancer therapeutic resistance. Int J Mol Sci. 2018;19(3):904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Q, Deng Q, Chao HP, et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun. 2018;9:3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang KH, Li R, Papari‐Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration‐resistant prostate cancer. Proc Natl Acad Sci USA. 2011;108:13728‐13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma NL, Massie CE, Ramos‐Montoya A, et al. The androgen receptor induces a distinct transcriptional program in castration‐resistant prostate cancer in man. Cancer Cell. 2013;23:35‐47. [DOI] [PubMed] [Google Scholar]
- 6. Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration‐resistant prostate cancer growth. Cancer Cell. 2011;19:792‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell. 2011;19:575‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niu Y, Altuwaijri S, Lai KP, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci USA. 2008;105:12182‐12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He Y, Hooker E, Yu EJ, et al. Androgen signaling is essential for development of prostate cancer initiated from prostatic basal cells. Oncogene. 2019;38:2337‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sampson N, Neuwirt H, Puhr M, Klocker H, Eder IE. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr Relat Cancer. 2013;20:R49‐64. [DOI] [PubMed] [Google Scholar]
- 12. Konig R, Chiang CY, Tu BP, et al. A probability‐based approach for the analysis of large‐scale RNAi screens. Nat Methods. 2007;4:847‐849. [DOI] [PubMed] [Google Scholar]
- 13. Tiong KH, Tan BS, Choo HL, et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget. 2016;7:57633‐57650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hii LW, Chung FF, Soo JS, Tan BS, Mai CW, Leong CO. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non‐stem breast cancer cells simultaneously. Breast Cancer Res Treat. 2020;179:615‐629. [DOI] [PubMed] [Google Scholar]
- 15. Hii LW, Chung FF, Mai CW, et al. Sphingosine kinase 1 regulates the survival of breast cancer stem cells and non‐stem breast cancer cells by suppression of STAT1. Cells. 2020;9:886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ibrahim N, He L, Leong CO, et al. BRCA1‐associated epigenetic regulation of p73 mediates an effector pathway for chemosensitivity in ovarian carcinoma. Can Res. 2010;70:7155‐7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73‐dependent apoptosis. Cancer Cell. 2006;9:45‐56. [DOI] [PubMed] [Google Scholar]
- 18. Tan BS, Tiong KH, Muruhadas A, et al. CYP2S1 and CYP2W1 mediate 2‐(3,4‐dimethoxyphenyl)‐5‐fluorobenzothiazole (GW‐610, NSC 721648) sensitivity in breast and colorectal cancer cells. Mol Cancer Ther. 2011;10:1982‐1992. [DOI] [PubMed] [Google Scholar]
- 19. Rist S, Carney Almroth B, Hartmann NB, Karlsson TM. A critical perspective on early communications concerning human health aspects of microplastics. Elsevier B.V. 2018;626:720‐726. [DOI] [PubMed] [Google Scholar]
- 20. Soo HC, Chung FFL, Lim KH, et al. Cudraflavone C induces tumor‐specific apoptosis in colorectal cancer cells through inhibition of the phosphoinositide 3‐kinase (PI3K)‐AKT pathway. PLoS One. 2017;12:e0170551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou T‐C. Drug combination studies and their synergy quantification using the Chou‐Talalay method. Can Res. 2010;70:440‐446. [DOI] [PubMed] [Google Scholar]
- 22. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27‐55. [DOI] [PubMed] [Google Scholar]
- 23. Stone EL, Citossi F, Singh R, et al. Antitumour benzothiazoles. Part 32: DNA adducts and double strand breaks correlate with activity; synthesis of 5F203 hydrogels for local delivery. Bioorg Med Chem. 2015;23:6891‐6899. [DOI] [PubMed] [Google Scholar]
- 24. Al‐Khdhairawi AAQ, Krishnan P, Mai CW, et al. A Bis‐benzopyrroloisoquinoline Alkaloid Incorporating a Cyclobutane Core and a Chlorophenanthroindolizidine Alkaloid with Cytotoxic Activity from Ficus fistulosa var. tengerensis. J Nat Prod. 2017;80:2734‐2740. [DOI] [PubMed] [Google Scholar]
- 25. Voon YL, Ahmad M, Wong PF, et al. Nutlin‐3 sensitizes nasopharyngeal carcinoma cells to cisplatin‐induced cytotoxicity. Oncol Rep. 2015;34:1692‐1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32:2866‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sasaki T, Nakashiro K, Tanaka H, et al. Knockdown of Akt isoforms by RNA silencing suppresses the growth of human prostate cancer cells in vitro and in vivo. Biochem Biophys Res Comm. 2010;399:79‐83. [DOI] [PubMed] [Google Scholar]
- 28. Cariaga‐Martinez AE, Lopez‐Ruiz P, Nombela‐Blanco MP, et al. Distinct and specific roles of AKT1 and AKT2 in androgen‐sensitive and androgen‐independent prostate cancer cells. Cell Signal. 2013;25:1586‐1597. [DOI] [PubMed] [Google Scholar]
- 29. Wang W, Shen T, Dong B, et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J Clin Investig. 2019;129:1015‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao F, Al‐Azayzih A, Somanath PR. Discrete functions of GSK3alpha and GSK3beta isoforms in prostate tumor growth and micrometastasis. Oncotarget. 2015;6:5947‐5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darrington RS, Campa VM, Walker MM, et al. Distinct expression and activity of GSK‐3alpha and GSK‐3beta in prostate cancer. Int J Cancer. 2012;131:E872‐E883. [DOI] [PubMed] [Google Scholar]
- 32. Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen‐independent prostate cancer. Oncogene. 2006;25:329‐337. [DOI] [PubMed] [Google Scholar]
- 33. Liao X, Thrasher JB, Holzbeierlein J, Stanley S, Li B. Glycogen synthase kinase‐3beta activity is required for androgen‐stimulated gene expression in prostate cancer. Endocrinology. 2004;145:2941‐2949. [DOI] [PubMed] [Google Scholar]
- 34. Schutz SV, Schrader AJ, Zengerling F, Genze F, Cronauer MV, Schrader M. Inhibition of glycogen synthase kinase‐3beta counteracts ligand‐independent activity of the androgen receptor in castration resistant prostate cancer. PLoS One. 2011;6:e25341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu Q, Yang J, Han S, et al. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate. 2011;71:835‐845. [DOI] [PubMed] [Google Scholar]
- 36. Imada K, Shiota M, Kohashi K, et al. Mutual regulation between Raf/MEK/ERK signaling and Y‐box‐binding protein‐1 promotes prostate cancer progression. Clin Cancer Res. 2013;19:4638‐4650. [DOI] [PubMed] [Google Scholar]
- 37. Hong SK, Kim JH, Lin MF, Park JI. The Raf/MEK/extracellular signal‐regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res. 2011;317:2671‐2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gong H, Zhou L, Khelfat L, et al. Rho‐associated protein kinase (ROCK) promotes proliferation and migration of PC‐3 and DU145 prostate cancer cells by targeting LIM kinase 1 (LIMK1) and matrix metalloproteinase‐2 (MMP‐2). Medical Sci Mon. 2019;25:3090‐3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu H, Hou T, Ju W, Xing Y, Zhang X, Yang J. MicroRNA122 downregulates Rhoassociated protein kinase 2 expression and inhibits the proliferation of prostate carcinoma cells. Mol Med Rep. 2019;19:3882‐3888. [DOI] [PubMed] [Google Scholar]
- 40. Kroiss A, Vincent S, Decaussin‐Petrucci M, et al. Androgen‐regulated microRNA‐135a decreases prostate cancer cell migration and invasion through downregulating ROCK1 and ROCK2. Oncogene. 2015;34:2846‐2855. [DOI] [PubMed] [Google Scholar]
- 41. Choucair KA, Guerard KP, Ejdelman J, et al. The 16p13.3 (PDPK1) genomic gain in prostate cancer: A potential role in disease progression. Transl Oncol. 2012;5:453‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gagliardi PA, Puliafito A, Primo L. PDK1: At the crossroad of cancer signaling pathways. Semin Cancer Biol. 2018;48:27‐35. [DOI] [PubMed] [Google Scholar]
- 43. Vasudevan KM, Barbie DA, Davies MA, et al. AKT‐independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emmanouilidi A, Falasca M. Targeting PDK1 for chemosensitization of cancer cells. Cancers. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castel P, Ellis H, Bago R, et al. PDK1‐SGK1 signaling sustains AKT‐independent mTORC1 activation and confers resistance to PI3Kalpha inhibition. Cancer Cell. 2016;30:229‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Velpula KK, Guda MR, Sahu K, et al. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget. 2017;8:35639‐35655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qi L, Toyoda H, Xu DQ, et al. PDK1‐mTOR signaling pathway inhibitors reduce cell proliferation in MK2206 resistant neuroblastoma cells. Cancer Cell Int. 2015;15:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li D, Mullinax JE, Aiken T, et al. Loss of PDPK1 abrogates resistance to gemcitabine in label‐retaining pancreatic cancer cells. BMC Cancer. 2018;18:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vale CL, Burdett S, Rydzewska LHM, et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone‐sensitive prostate cancer: a systematic review and meta‐analyses of aggregate data. Lancet Oncol. 2016;17:243‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mihaly Z, Kormos M, Lanczky A, et al. A meta‐analysis of gene expression‐based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. 2013;140:219‐232. [DOI] [PubMed] [Google Scholar]
- 51. Maurer M, Su T, Saal LH, et al. 3‐Phosphoinositide‐dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3‐kinase pathway in breast carcinoma. Can Res. 2009;69:6299‐6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang Z, Wu Z, Liu T, et al. Upregulation of PDK1 associates with poor prognosis in esophageal squamous cell carcinoma with facilitating tumorigenicity in vitro. Med Oncol. 2014;31:337. [DOI] [PubMed] [Google Scholar]
- 53. Scortegagna M, Ruller C, Feng Y, et al. Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf(V600E):Pten(‐/‐) melanoma. Oncogene. 2014;33:4330‐4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bai X, Li P, Xie Y, et al. Overexpression of 3‐phosphoinositide‐dependent protein kinase‐1 is associated with prognosis of gastric carcinoma. Tumour Biol. 2016;37:2333‐2339. [DOI] [PubMed] [Google Scholar]
- 55. Wang J, Liu F, Ao P, et al. Correlation of PDK1 expression with clinicopathologic features and prognosis of hepatocellular carcinoma. OncoTargets and Therapy. 2016;9:5597‐5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zabkiewicz J, Pearn L, Hills RK, et al. The PDK1 master kinase is over‐expressed in acute myeloid leukemia and promotes PKC‐mediated survival of leukemic blasts. Haematologica. 2014;99:858‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9‐22. [DOI] [PubMed] [Google Scholar]
- 58. Di Blasio L, Gagliardi PA, Puliafito A, Primo L. Serine/threonine kinase 3‐phosphoinositide‐dependent protein kinase‐1 (PDK1) as a key regulator of cell migration and cancer dissemination. Cancers. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nogueira V, Patra KC, Hay N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. eLife. 2018;7:e32213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scortegagna M, Lau E, Zhang T, et al. PDK1 and SGK3 contribute to the growth of BRAF‐mutant melanomas and are potential therapeutic targets. Can Res. 2015;75:1399‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Silva JM, Bulman C, McMahon M. BRAFV600E cooperates with PI3K signaling, independent of AKT, to regulate melanoma cell proliferation. Molecular cancer research: MCR. 2014;12:447‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gagliardi PA, di Blasio L, Orso F, et al. 3‐phosphoinositide‐dependent kinase 1 controls breast tumor growth in a kinase‐dependent but Akt‐independent manner. Neoplasia. 2012;14:719‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bago R, Sommer E, Castel P, et al. The hVps34‐SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016;35:1902‐1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Qian DZ, Rademacher BL, Pittsenbarger J, et al. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel‐induced cytotoxicity. Prostate. 2010;70:433‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S3
Tables S1‐S5
Figure Legends