

 Application of triazoles in the development of microtubule targeting agents.
Application of triazoles in the development of microtubule targeting agents.
Abstract
The triazole ring system has emerged as an exciting prospect in the optimization studies of promising lead molecules in the quest for new drugs for clinical usage. Several marketed drugs possess these versatile moieties that are used in a wide range of medical indications. This stems from the unique intrinsic properties of triazoles, which impart stability to the basic pharmacophoric unit with an added advantage of being a bioisostere of different chemical functionalities. In the last decade, the use of triazoles as bioisosteres and linkers in the development of microtubule targeting agents has been extensively investigated. The present review highlights the advances in this promising area of drug discovery and development.
1. Introduction
The transition of the drug discovery process from the isolation of active ingredients of folklore drugs to rational design addressing the clinical needs of patients has been phenomenal. In recent decades, there has been a momentous shift towards discovery and design of molecularly targeted drugs based on natural products and synthetic small molecules. A toolbox encompassing diverse areas of chemical, biological, and computational sciences is now at the disposal of researchers working in drug discovery programs.1 Unfortunately, there is a parallel evolution of new challenges, which keeps unravelling from time to time, such as the development of drug resistance in causative biological agents, mild to severe side effects of drugs, higher failure rates of new drugs in the pipeline and others. Therefore, a constant drug discovery loop is created that chases the mirage of ideal “magic bullets” for treating different diseases. Hopefully, a personalized medicine strategy tailored to the individual needs of patients will provide the answer in the future. However, in the present scenario, the well-developed drug discovery programs are essential and fit well in addressing and treating the various diseases afflicting humankind.
1.1. Importance of triazoles in drug discovery
In drug discovery and development, the lead molecules provide chemical templates that need to be fine-tuned for more desirable pharmacological and pharmacokinetic properties. This forms the crux of the lead optimization process, and different strategies like bioisosteric replacement and incorporation of suitable linkers are employed.2 Triazoles (pyrrodiazoles) are a versatile class of heterocyclic compounds with three nitrogen atoms positioned in a five-membered aromatic ring. 1,2,3-Triazole (1) and 1,2,4-triazole (2) are the two possible regio-isomers that vary in the relative position of the three nitrogen atoms in the ring with each one of them existing as tautomeric pairs (1a, b and 2a, b) depending on the nitrogen to which hydrogen is bonded. In drug design, triazoles are increasingly being employed as bioisosteres of different functionalities and as linkers to increase the efficacy of the lead molecules.3–6 Triazoles intrinsically possess a strong dipole moment, pi electron-deficient aromaticity, and hydrogen bond accepting properties. They also display stability towards both chemical and metabolic degradation and are used as bioisosteres of amide, ester, carboxylic acid, and olefinic double bonds in addition to bioisosteres of other heterocyclic rings. Most commonly, triazoles are employed as bioisosteres of the amide bond because of the similarity in size, dipole moment, and H-bond accepting capacity with an added advantage of good metabolic stability which is lacking in the amide bond. The replacement of the amide bonds with triazoles also results in enhanced activity as seen in the anticancer drug, imatinib 3, which shows an IC50 value of 0.38 μM against the K562 cancer cell line. Remarkably, the triazole isostere 4 of imatinib exhibited potent activity with an IC50 value of 0.03 μM under similar conditions (Fig. 1).7 Lead optimization studies have resulted in the development of several clinical drugs, which possess a triazole moiety in their structural frameworks. These triazole containing drugs exhibit wide therapeutic potential and are used in the treatment of cancer,8–10 fungal,11–13 viral14,15 and bacterial infections,16,17 hyperuricemia,18 chronic iron overload,19 HIV-1 strains resistant to multiple antiretroviral agents,20 diabetes,21 thrombosis,22 mydriasis,23 major depressive disorder,24 seizures,25 insomnia,26 female infertility27 and hepatitis C.15 Triazoles were first developed as antifungal agents such as fluconazole, isavuconazole, and itraconazole, followed by widening of their application in the treatment of various medicinal conditions as mentioned above. In addition to this, fused triazoles like benzotriazoles are used as sunscreen agents and fog suppressants in cosmetic products and chemical photography, respectively.28 Here, for a better understanding of their versatility, triazoles as clinical drugs are succinctly summarized (Table 1).
Fig. 1. Triazoles and bioisosteric substitution.

Table 1. Triazoles in clinical drugs.
| Entry | Drug structure | Drug name | Brand name | Chemical class | Target | Medicinal indication |
| 1. |

|
Talazoparib | Talzenna | Phenylquinoline (1H-1,2,4-triazole) | Poly [ADP-ribose] polymerase 1 | Treatment of germline BRCA mutated HER2 negative, locally advanced or metastatic breast cancer. Approved by FDA on October 16, 2018.8 |
| 2. |

|
Letrozole | Femara | Diphenylmethanes (1,2,4-triazole) | Cytochrome P450 19A1 | Adjuvant treatment of hormonally-responsive breast cancer.9 |
| Ovulation induction in women with polycystic ovarian syndrome (PCOS).27 | ||||||
| 3. |

|
Ribavirin | Copegus, Ribasphere, Rebetol | Ribonucleosides and ribonucleotides (1,2,4-triazole) | RNA binding | Treatment of hepatitis C and viral hemorrhagic fevers.15 |
| Treatment of acute myeloid leukemia is currently under investigation.10 | ||||||
| 4. |

|
Rufinamide | Banzel, Inovelon | 2-Heteroaryl carboxamide class (1,2,3-triazole) | Glutamate receptor activity | Medication to treat seizure disorders like Lennox–Gastuat syndrome.25 |
| 5. |

|
Tazobactam | NA | Alpha amino acids and derivatives (1,2,3-triazole) | Beta-lactamase SHV-1 activity | Used in combination with piperacillin to broaden the spectrum of piperacillin antibacterial action.17 |
| 6. |

|
Sitagliptin | Xelevia | Beta amino acids derivatives/triazolopyrazines (fused 1,2,4 triazole) | Dipeptidyl peptidase 4 activity | New oral hypoglycemic (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs.21 |
| 7. |

|
Lesinurad | Duzallo, Zurampic | Phenyl-1,2,4-triazoles | Urate transmembrane transporter activity | Oral uric acid transporter 1 (URAT1) inhibitor indicated for the treatment of hyperuricemia associated with gout.18 |
| 8. |

|
Isavuconazole | NA | Phenylpropanes (1,2,4 triazole) | Sterol 14-demethylase activity | Antifungal agent with a broad spectrum of activity and good safety profile. Approved by the FDA and EMA for the treatment of invasive aspergillosis and mucormycosis.11 |
| 9. |

|
Deferasirox | Exjade | Phenyl-1,2,4-triazoles | Iron | Used for the treatment of chronic iron overload due to blood transfusions (transfusional hemosiderosis).19 |
| First oral medication approved in the USA for this purpose. | ||||||
| 10. |

|
Maraviroc | Selzentry, Celsentri | Tropane alkaloids (1,2,4 triazole) | C–C chemokine receptor type 5 | Treatment-experienced adult patients infected with only CCR5-tropic HIV-1 and HIV-1 strains resistant to multiple antiretroviral agents.20 |
| 11. |

|
Lorpiprazole | Normarex | Phenylpiperazines (fused 1,2,4 triazole) | 5-Hydroxytryptamine receptor 2A | Serotonin antagonist and reuptake inhibitor used for the treatment of major depressive disorder.24 |
| 12. |

|
Dapiprazole | Rev-eyes | Phenylpiperazines (fused 1,2,4 triazole) | Alpha-1A adrenergic receptor | Alpha blocker used to reverse mydriasis after eye examination.23 |
| 13. |

|
Suvorexant | Belsomra | N,N-Dialkyl-m-toluamides (phenyl-1,2,3-triazoles) | Orexin receptor type 1 | Selective dual antagonist of orexin receptors that promote sleep by reducing wakefulness and arousal. Approved for the treatment of insomnia.26 |
| 14. |

|
Bisoctrizole | NA | Diphenylmethanes (fused 1,2,3-triazoles) | NA | Indicated for use as a sunscreen agent in cosmetic products. It is an organic UV-A filter that absorbs both UV-A and UV-B rays.28 |
| 15. |
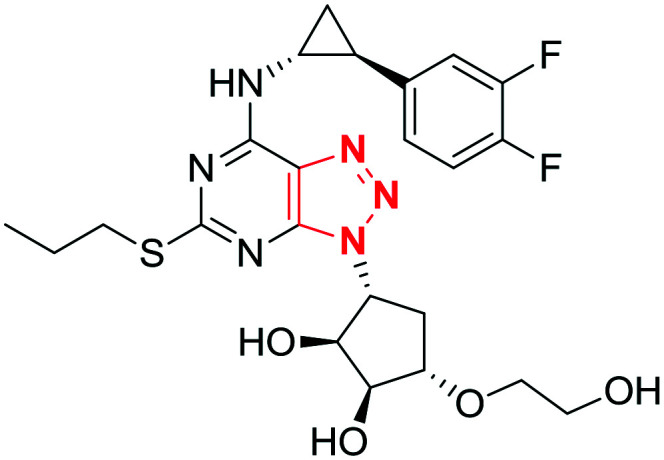
|
Ticagrelor | Brilinta, Brilique, Possia | Triazolopyrimidines (fused 1,2,3-triazoles) | P2Y purinoceptor 12 | Platelet aggregation inhibitor for the prevention of thrombotic events in patients with acute coronary syndrome or myocardial infarction with ST elevation.22 |
The promising application of triazoles in drug design is further propelled by the advent of the proposed concept of click chemistry in the synthesis of triazoles.4 Click chemistry is a powerful tool for a quick, highly selective and reliable access to a reaction product with high yields, little chromatographic separation and a wide range of applications. Earlier, 1,2,3-triazoles were accessed by Huisgen type thermal 1,3-dipolar addition, which resulted in a mixture of 1,4- and 1,5-disubstituted triazoles.29 The seminal discovery of copper(i)-catalyzed azidealkyne cycloaddition (CuAAC) in recent decades provided high yielding access to 1,4-disubstituted 1,2,3-triazole isomers under mild conditions.30 This augmented the application of triazoles in drug design and discovery, bioconjugation, polymeric material sciences and other areas.31–34 In drug discovery, triazoles are reported to exhibit a wide range of biological activities such as anticancer,5 antimicrobial,35–38 anti-human immunodeficiency virus39,40 and antitubercular properties.41 In addition to this, triazoles are also known to display other pharmacological properties like antiallergic42 anticonvulsant,43 antiprotozoal,44 selective β adrenergic receptor agonist,45 and cannabinoid CB1 receptor antagonist properties, etc.46
1.2. Microtubule targeting agents
Globally, cancer drug discovery programs are being robustly developed to fight the menace of cancer incidence, recurrence and mortality.47 Different molecular targets are identified in the treatment of cancer and one of the most promising targets is microtubules.48 The microtubular assembly comprises cytoskeletal fibres that are present abundantly in the cellular cytoplasm of eukaryotic cells. Their dynamic polarized properties make microtubules indispensable in various cellular functions such as motility, polarization, division, secretion, and maintenance. The dynamism of microtubules results from their structural units, namely α and β tubulin (heterodimers), which assemble and disassemble to elongate and shrink under appropriate cellular signals. The dynamic polymerization and depolymerization are the basis of the proper functioning of the microtubular assembly and are very susceptible to agents which can disrupt this delicate balance.49 Natural product based chemical agents are known to disturb the subtle stability of this dynamic process and act as lead molecules in the development of new tubulin inhibitors. These natural tubulin inhibitors bind at different domains of the tubulin protein at the molecular level and perturb the polymerization and depolymerization equilibrium of the microtubules.50 In cancer, uncontrollable cell divisions take place and as microtubules are essential in cell division, particularly in the segregation of the chromosomes, the inhibition of their functions results in a promising avenue in the development of potent anticancer agents. The natural tubulin inhibitors that are used as cancer-treating drugs like paclitaxel 5 and vincristine 6 bind to the taxane and vinca domain of tubulin, respectively (Fig. 2).51,52 On the other hand, natural products like colchicine 7, combretastatin (like CA-4) 8, podophyllotoxin 9, and 2-methoxyestradiol 10 are inhibitors of tubulin polymerisation that bind to the colchicine binding site and are lead molecules (Fig. 3).53 The colchicine domain binding tubulin inhibitors are relatively more studied and developed compared to other taxol and vinca domain binding tubulin inhibitors because of the simplicity of chemical scaffolds and easy synthetic access. In recent years, many synthetic small molecules have been also developed as lead molecules and are in different stages of clinical studies such as ABT-751 (E7010, 11), nocodazole 12, batabulin 13, indibulin 14, lexibulin 15, and mivobulin 16 (Fig. 4).54,55
Fig. 2. Inhibitors of tubulin function.

Fig. 3. Natural product based tubulin polymerization inhibitors.

Fig. 4. Synthetic small molecule based tubulin polymerization inhibitors.

In the development of tubulin inhibitors as anticancer agents, the application of the triazole scaffold in modulating the lead molecules has been extensively investigated. Newer natural product and small-molecule based inhibitors targeting tubulin have been developed and computational based approaches were employed in better understanding the molecular interactions at the tubulin active site.55,56 The application of click chemistry and triazoles in general as well as in drug discovery and design has been well-reviewed.3–6 In this review, we mainly focus on the application of triazoles in the development of tubulin polymerization inhibitors. For better clarity and understanding of the application of triazoles as microtubule targeting agents, we discuss 1,2,3-triazoles, 1,2,4-triazoles and fused triazoles separately. Moreover, we provide a better perspective of these diverse triazoles emphasizing on the lead molecules from which they are developed and the use of the triazole moiety as a bioisostere or linker.
2. 1,2,3-Triazole based microtubule targeting agents
1,2,3-Triazole isomers are more common than 1,2,4-triazoles because of the ease of their synthesis and relatively better pharmacological properties. Different substitution patterns on the triazole ring, which include 1,4-, 2,4-, 1,5- and 4,5-disubstitutions, were investigated. The various synthetic approaches employed for the synthesis of differently disubstituted 1,2,3-triazole based microtubule targeting agents are briefly discussed here (Table 2). The 1,5-disubstituted 1,2,3-triazoles were synthesized by reacting the appropriate azide with magnesium acetylide of alkyne or by ruthenium catalyzed azide–alkyne cycloaddition. In the synthesis of 4,5-disubstituted 1,2,3-triazoles, 1,3-diaryl-2-nitroprop-1-enes or (Z)-2,3-diarylacrylonitrile were treated with sodium azide at high temperature to access the desired triazoles. The 2,4 disubstituted 1,2,3-triazole was reported to be obtained from reacting the appropriate hydrazine derivative with hydroxylamine hydrochloride in the presence of sodium acetate. In recent years, the 1,4-disubstituted 1,2,3-triazoles were more frequently synthesized employing click chemistry involving the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction.57–62 Here, we discuss the application of 1,2,3-triazoles with different substitution patterns in the development of tubulin polymerization inhibitors.
Table 2. Synthetic approaches to access 1,2,3-triazoles.





2.1. Natural product based lead molecules
Combretastatin A-4 (CA-4) 8 is a naturally occurring compound isolated from the bark of Combretum caffrum, an African bush willow.63 It belongs to the class of phenolic stilbenes called combretastatins, which are used in the folklore treatment of scorpion sting, heart ailments and worm induced intestinal disorders.64 CA-4 exhibits high potency against various cancer cell lines and its phosphate-based prodrugs are in clinical trials for treatment of advanced solid tumour. It inhibits tubulin polymerization by binding at the colchicine domain and is a lead molecule in the design of tubulin targeting drugs. The structure–activity relationship unravelled the importance of the cis-orientation of the double bond connecting the two aromatic rings, with key trimethoxy and para-methoxy moieties. A number of studies have been carried out to conserve the rigidity of the cis-oriented double bond, thereby eliminating the undesired isomerization and in vivo degradation.65 Interestingly, triazoles are used as bioisosteres of the rigid cis-olefinic bond with or without modification of the other two aromatic rings of CA-4 and several investigations have been carried out providing insights into the versatility of the triazole moiety.
The first 1,2,3 triazole bioisostere of the cis-olefinic bond of CA-4 was reported employing the [3 + 2] dipolar cycloaddition reaction between an alkyne intermediate and azide to provide 4,5-substituted 1,2,3-triazoles. The triazoles were evaluated against the murine B16 melanoma cancer line resulting in moderate activity that provided the impetus to incorporate the triazole nucleus in CA-4. This offered enhanced solubility in physiological media and a geometry that fits well at the colchicine-binding domain. It is important to note that the CA-4 triazole analog with a trimethoxyphenyl group resulted in comparatively better activity (IC50 = 56 μM) than that with a monomethoxyphenyl group as ring A of CA-4.66 Akselsen and coworkers first studied the tubulin inhibition properties of CA-1 analogs with a triazole nucleus. CA-1 belongs to the combretastatin class of compounds and has an additional phenolic group compared to the CA-4 lead molecule. Seven 1,5-disubstituted as well as two 1,4-disubstituted 1,2,3 triazole bioisosteres of CA-1 were synthesized and the 1,5-disubstitutions were more potent than 1,4-disubstitutions. The 1,5-disubstituted triazoles were synthesized by employing either thermally induced cycloaddition or a magnesium acetylide assisted reaction between azide 17 and alkynes (18a–18b) to afford triazoles 19a–19c. Further chemical modifications provided the desired triazoles 20a, 20b. The three 1,5-disubstituted triazoles with dihydroxy, diamino and difluoro groups on ring B (20a, b, 19c) exhibited good anticancer (IC50 values 1.9–17.2 μM against the HT-29 cancer cell line) and decent tubulin inhibition properties (IC50 values 5.2–15.6 μM), though comparatively lower than those of the standard CA-4. The compounds docked successfully at the colchicine binding domain (Fig. 5).67 In another report, new cis-constrained analogs of CA-4 were synthesized by replacing the olefinic bond and one of the phenyl rings with triazole and benzyl, respectively. The CA-4 based 4,5-disubstituted 1,2,3-triazoles were produced by reacting appropriate 1,3-diaryl-2-nitroprop-1-enes 21 with sodium azide and subjected to biological evaluation. Some of these new 4,5-disubstituted 1,2,3-triazoles showed low micromolar range activity for rounding up of endothelial cells, cytotoxicity on B16 melanoma cancer cells and inhibition of tubulin polymerization. The structure–activity relationship suggested that the series with the trimethoxyphenyl group directly attached to the triazole nucleus were less potent than the 3,4,5-trimethoxybenzyl counterparts. The replacement of the meta-hydroxy group with other substituents led to good results in the order of NH2 > F > NO2. The most active triazole 22 of the series was the one that was structurally similar to CA-4 with a hydroxyl and methoxy group at the meta and para position, respectively. Triazole 22 exhibited an IC50 value of 1.6 μM against B16 melanoma cells and good tubulin inhibition properties (IC50: 1.2 μM). It was subjected to molecular modelling and in vivo testing in mice to validate the preliminary results.68
Fig. 5. 1,2,3-Triazoles in combretastatin A-4 based molecules (1).

A series of 4,5-disubstituted 2H-1,2,3-triazoles were synthesized by Madadi and coworkers as cis-restricted CA-4 analogs employing cyclization of appropriate Z-substituted diarylacrylonitriles 23. Ten analogs were identified as hits for NCI-60 cell screening resulting in the selection of six analogs for full dose–response studies. It was observed that the replacement of one of the phenyl rings with a heterocyclic ring resulted in a decrease in activity. The retention of the substitution pattern closer to CA-4 exhibited higher potency in the series and analog 24a exhibited potent activity with GI50 values of <10 nM against almost all the tested cancer cell lines. Another analog 24b displayed a better affinity for the colchicine binding domain and potent cytotoxic activity against 9LSF rat gliosarcoma cells (LD50 = 7.5 nM) by the colony formation assay.69 In a related approach, trans-cyano CA-4 was modified to enable bioisosteric substitution of the double bond with triazoles and one of the phenyl rings of CA-4 was replaced with a fused heterocyclic system namely, benzothiophene, benzofuran, and indole. The triazoles with the benzothiophene ring were more than two to six fold more potent than the benzofuran and indole counterparts and the trimethoxyphenyl ring system was crucial for enhanced activity. The most potent compound 25 with benzothiophene and trimethoxyphenyl rings showed potency in a lower nanomolar range (GI50 10 nM) against 80% of the tested cancer cell lines and inhibition of tubulin polymerization at an IC50 value of 1.7 μM.70 In a recent report, 1,2,3-triazoles and other five-membered analogs of CA-4 were evaluated for anti-tubulin properties by employing an in vivo sea urchin embryo assay as an alternative biological model and one of the triazoles caused cleavage alteration of the sea urchin embryo at 0.5 nM.71 In another report, Kamal and coworkers conserved the basic CA-4 structure and synthesized a 15-membered series of 1,2,3-triazole linked amino CA-4 conjugates by coupling amino stilbenes 26 with different substituted benzyl-1H-1,2,3-triazole-4-carboxylic acids 27 (Fig. 6). These conjugates were studied for cytotoxicity and tubulin polymerization inhibition in addition to other biological properties. The structure–activity relationship study suggested that the triazole moiety tethered to the C-3 position of ring B exhibited significantly improved activity compared with C-2 linked counterparts. Moreover, the para substitution (either electron-donating or withdrawing) on the benzyl ring attached to the triazole ring was crucial for activity. Two of the conjugates 28a, b were highly active against the lung cancer cell line (A549) displaying an IC50 value of 53 nM and 44 nM, respectively. Tubulin polymerization inhibition by these compounds was comparable to that of CA-4 and was in the range of 0.95–1.16 μM. Docking studies suggested the occurrence of binding at the colchicine domain, which was validated by a colchicine competitive binding assay.72
Fig. 6. 1,2,3-Triazoles in combretastatin A-4 based molecules (2).

The replacement of the cis-olefinic bond of CA-4 with a carbonyl group results in benzophenone derivatives with the conservation of tubulin inhibition properties. The parent molecule of this class is phenstatin, which is reported to be more stable than CA-4 with comparable activity, and in recent developments triazole based phenstatin analogs have been investigated.73 In a report, introduction of a 1,2,3-triazole moiety in between the carbonyl group and ring B of phenstatin was carried out; however the potency of the parent molecule decreased dramatically. This underlined the importance of the carbon–oxygen double bond on chalcones as it appears to be more than a mere linker, unlike the simple double bond in CA-4.74 In a more recent investigation, taking into consideration the importance of the double bond, a series of hydroxylamine derivatives of phenstatin with the triazole ring system were synthesized and studied for their stereomeric effects along with their intermediates. These triazole based hydroxylamine derivatives were synthesized employing a synthetic strategy with hydrazone derivative 29 as a key intermediate. The Z-isomer with an oxime linkage 30 was the most active of the tested compounds against selected cancer cell lines with an IC50 value in the range of 0.12–0.52 μM. Interestingly, the Z-isomer was two to five times more active than its E counterpart against the tested cell lines with comparable activity to one of the standards. Overall, the compounds with the oxime linkage were comparatively more active than the intermediates that have a simple carbonyl group and amino group on the triazole ring. The substitutions on the phenyl ring at the N-2 position of the triazole ring showed an order of potency i.e., para- > meta- > orthro- and the type of substitution on this position exhibited little change in potency. The tubulin polymerization potency of compound 30 was comparatively weaker than that of CA-4.75 In a further modification, Kamal and coworkers reported the introduction of the enone moiety in place of the cis-olefinic bond and a triazole moiety on ring B of CA-4 resulting in chalcone derivatives. Moreover, the enone moiety was incorporated into a pyrazoline ring as it was reported to possess tubulin inhibition properties. The CA-4 triazoles with the enone moiety were more potent than the pyrazoline counterparts; however, both the modifications resulted in little enhancement of potency compared to CA-4.76
Naturally occurring colchicine 7, extracted from Colchicum autumnale L., is the first chemical agent known to destabilize tubulin polymerization and the particular site where it binds to the tubulin protein is named as the “colchicine binding site”. Though colchicine inhibits mitosis efficiently, its therapeutic index is low and it is used as a lead molecule in the development of tubulin polymerization inhibition.77 To enhance its therapeutic index, a triazole ring was introduced as a bioisostere of the amide group at the C-7 position and a series of 1,4-disubstituted triazole conjugates of 7 were synthesized by diversifying the C4 position of the triazole ring with aryl, alcohol, sugar, steroid, ester (aryl and alkyl), amide, sulphonamide, ferrocene, nucleobase, and amino acid groups. All the conjugates showed significant tubulin inhibition properties and three of them (possessing alkyl, alcohol and nucleobase groups) exhibited potent activity. In an antiproliferative assay against Burkitt-like lymphoma cells, a fatty acid ester conjugate 31 was the most promising potent conjugate which inhibited BJAB cancer cells below 10 nm concentration, two fold better than the parent colchicine molecule (Fig. 7).78 Conjugate 31 was synthesized by a copper sulphate catalyzed click chemistry approach involving azide 32 and alkynyl 33 components. To confirm whether the binding is similar to that of colchicine on the tubulin protein, a colchicine binding assay was performed and seven of the conjugates caused over 80% displacement of colchicine. Further studies showed that three compounds showed induced changes in the morphology of microtubules at submicromolar concentration. In an interesting report, the low therapeutic index of colchicine was addressed by the application of the prodrug concept using lipophilic colchicine triazole analogs in liposomes. For this purpose, triazole analogs of colchicine and allocolchicine with their palmitic and oleic esters (as prodrugs) were synthesized and evaluated for their antiproliferative and tubulin polymerization inhibition potential. The triazole allocolchicine analogs exhibited a dramatic loss of activity, whereas the prodrugs of newly synthesized triazole-colchicine analogs 34a, b exhibited excellent cytotoxicity against Burkitt-like lymphoma cells (BJAB) with IC50 values of 4 and 6 nM, respectively. This was three to five fold better than the parent molecule, colchicine. Analogs 34a, b exhibited significant inhibition of tubulin polymerization at 40 μM, though the parent molecule showed complete inhibition under similar conditions. For further development, these lipophilic prodrugs were formulated for efficient delivery employing 100 nm (diameter) liposomal vehicles composed of egg phosphatidylcholine–yeast phosphatidylinositol–palmitic or oleic prodrug. Remarkably, the liposomal formulation preserved the activity of the parent molecule and the oleoyl prodrug was more efficient than that of the free prodrug. Interestingly, the formulations were stable with 1–5 μmol mL–1 of active pharmaceutical agent to achieve effective dosage per injection.79
Fig. 7. 1,2,3-Triazoles in colchicine and podophyllotoxin based molecules.

Podophyllotoxin 9, the toxic lignan isolated from Podophyllum sps., is a potent tubulin polymerization inhibitor interacting with the colchicine binding domain at the molecular level.80 The 1,2,3-triazole ring was introduced as a bioisostere of the lactone moiety of 9 using click chemistry. The bioisostere (35) was tested for antiproliferative activity against the neuroblastoma cell line (SH-SY5Y) and displayed an IC50 value of 1.5 μM, which is much lower than that of the parent molecule. However, bioisostere 35 conserved the tubulin inhibition properties of the parent molecule with a similar potency.81 In another report, 4β-[(amido-4-substituted)-1,2,3-triazol-1-yl]podophyllotoxin hybrids were investigated for cytotoxicity against selected cancer cell lines and tubulin polymerization inhibition. Structural changes were carried out on the ring attached to the C-4 position on the triazole ring. Hybrids with electron-donating groups such as tert-butyl and dimethoxy (36a, b) exhibited good activity against the HeLa cancer cell line with an IC50 value of 0.9 and 0.07 μM, respectively, which was three to fifteen fold better than that of the parent podophyllotoxin molecule. The introduction of a heteroaromatic ring such as pyridine and thiophene on the C-4 position of the triazole ring resulted in increased cytotoxic potency; however, the pyrazole and indinone rings showed no improvement. The potent hybrids exerted tubulin inhibition comparable to the parent lead molecule.82 Similarly, triazoles derivatives of other naturally occurring compounds gomisin B and endogenous estrogen metabolite 2-methoxyestradiol were synthesized with promising cytotoxicity and tubulin inhibition properties.83,84
2.2. Synthetic small molecule leads
Synthetic small molecules have emerged as an attractive alternative to complex natural product-based lead compounds because of the simplicity of their structures and ease of synthesis with the added advantage of similar therapeutic potential and receptor interaction at the molecular level.85 In the development of microtubule targeting agents, several synthetic small molecules are identified and some of them are in advanced stages of clinical evaluation. Here, the synthetic lead molecules in which the triazole moiety is incorporated are discussed.
ABT-751 (11) is a pyridine based sulphonamide which is an orally bioavailable tubulin polymerization inhibitor that has been in clinical trials alone and in combination.86 The clinical studies have been terminated due to the lack of improvement in the efficacy of the drug. However, 11 serves as a lead molecule in the development of potential anticancer agents. In a study, the diamino pyridine moiety of 11 was conserved and an amidotriazolyl unit was introduced as a bioisostere of the sulphonamide group. A large library was synthesized and the substituent effect was studied on the anilino (ring A) and benzyl group attached to the triazole moiety (ring B) for a meaningful insight into the structure–activity relationship against selected cancer cell lines. The electron-donating groups showed a pronounced increase in cytotoxicity with the exception of halogens (fluorine), which exhibited comparably lower activity. The synthesis was carried out by a click reaction between the substituted benzyl azide 37 and methyl propiolate 38 to afford triazole 39, which on ester hydrolysis and conjugation with different 2-anilino-3-aminopyridine derivatives provided the desired library. Conjugates with the 3-phenoxy group on ring B and electron-donating groups on ring A exhibited enhanced cytotoxic activity and conjugate 40 (with a 3,5-dimethoxy group on ring A) exhibited an IC50 value of 1.02 and 1.33 μM against A549 and HeLa cancer cell lines, respectively. This was better than the standard ABT-751, which showed IC50 values of 1.62 and 2.18 μM against the mentioned cancer cell lines. Interestingly, conjugate 40 exhibited tubulin inhibition at an IC50 value of 2.04 μM, which was comparable to the lead compound (Fig. 8). The structure–activity relationship studies showed that the compounds with electron-donating groups exhibited enhanced cytotoxic potency compared with ABT-751, the parent lead molecule.87 In a previous report, the amino functionality on carbon 3 of 11 was removed and an oxotriazolyl group was introduced in place of the sulphonamide group with a flanking benzyl group. A series of twenty compounds was synthesized and the structure–activity relationship was elucidated based on cytotoxic data against selected cancer cell lines. The electron-withdrawing group, fluorine, on the aryl group of the 2-aryl pyridine moiety displayed an enhanced effect compared to the electron-donating groups with the exception of the trimethoxy substituent. However, most of the conjugates with fluorine on the benzyl group attached to the triazole moiety exhibited lowered potency compared to those with electron-donating groups. Conjugate 41, with a 3-phenoxy benzyl motif and 4-fluoro 2-aryl pyridine, was the most potent compound exhibiting IC50 values in the range of 0.1 to 1.1 μM against three cancer cell lines, which was up to twenty five-fold better than the standard, ABT-751. The tubulin inhibition properties were also better than those of the parent molecule with an IC50 value of 1.84 μM. SAR and molecular docking studies suggested that the oxotriazolyl group was favorable for tubulin binding.88 The same research group also reported related ABT-751 based triazole conjugates with promising results.89 Apart from the ABT-751 based design, a series of sulphonamide–triazole hybrids with different substitutions were also synthesized and evaluated with limited success.90
Fig. 8. Small molecules with the 1,2,3-triazole ring (1).

Several other heterocyclic based arylamides with a triazole moiety were also designed based on different lead compounds and evaluated for their tubulin polymerization inhibition properties. Stefely and coworkers reported N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide as a new scaffold for tubulin inhibition which was designed based on oxazole-4-carboxamide 42, an important structural element in anti-tubercular agents. Preliminary cytotoxicity data revealed that oxazole-4-carboxamide based triazole 43 showed a promising IC50 value of 0.56 μM against the MCF cancer cell line, which was followed by short and focused structural–activity relationship study. The SAR study suggested that the m-OPh substitution on the N-1-benzyl group was crucial for cytotoxicity and different heterocyclic rings as substitutes of the aryl group on the arylamide were well tolerated. Interestingly, the replacement of the oxazole ring in the carboxamide with a pyridine ring 44 exhibited remarkable enhancement in the cytotoxic potency with an IC50 value of 0.046 and 0.58 μM against MCF-7 and U937 cancer cell lines, respectively. The potency of 44 against MCF-7 was closer to that of standard colchicine and over fifteen fold better than that of the other standard, 2-methoxyestradiol. The tested compounds also exhibited inhibition of tubulin protein with complete inhibition at 10 μM similar to the standard nocodazole. In this study, the COMPARE algorithm matrix was also utilized to measure the correlation between the different synthesized compounds.91 In a similar design to enhance the potency, pyridine and isoxazole rings were incorporated in a single chemical entity with a triazole moiety and evaluated for their cytotoxicity and anti-tubulin properties.92
Different libraries of triazole linked imidazothiazole and imidazopyridine conjugates were reported for their tubulin inhibition properties by Kamal's research group. Imidazo[2,1-b]thiazole linked triazole conjugates were synthesized and evaluated for their cytotoxicity and anti-tubulin properties. The structure–activity relationship study elucidated that the electron-donating group (4-methoxy) present on the aryl group of the imidazothiazole subunit along with the electron-rich disubstituents on the benzyl group of triazole was essential for cytotoxic properties. On the other hand, the electron-withdrawing group (fluoro) hampered the antiproliferative activity. Two of the conjugates with electron-rich substituents (45a, b) demonstrated significant cytotoxicity against the A549 cell line with IC50 values of 0.92 and 0.78 μM, respectively, which were better than those of the standard doxorubicin (IC50: 1.78 μM). Similarly, tubulin inhibition by these conjugates was promising (IC50: 1.62 and 1.43 μM) and comparable to nocodazole (Fig. 9).93 Similarly, another series of triazole linked benzimidazo[2,1-b]thiazole conjugates was also designed and evaluated for antiproliferative and tubulin inhibition properties. Conjugates with an electron-withdrawing fluorine atom on the N-benzyl unit (46a, b) exhibited potent antiproliferative activity against the MCF-7 cancer cell line with IC50 values of 0.60 and 0.78 μM, respectively (better than doxorubicin). The tubulin inhibition properties of 46a (IC50: 1.23 μM) were better than those of the standard nocodazole.94 In another study, the replacement of the imidazo[2,1-b]thiazole ring with a imidazopyridine ring in the triazole conjugates showed the conservation of the biological properties.95 In addition to this, triazole tethered indole-3-glyoxamide derivatives were also developed with limited success.96
Fig. 9. Small molecules with the 1,2,3-triazole ring (2).

In drug design and discovery, the use of the linker approach plays a promising role in conjugating different pharmacophores or duplicating a single pharmacophore for enhanced therapeutic potential of the final chemical entities. In addition, linkers are indispensable in the design of targeted drug delivery vehicles.97 The stability offered by the triazole ring provides an excellent opportunity in employing it as a linker and a number of homo and heterodimers of active pharmacophores as triazole linked conjugates have been reported.4 In the design of tubulin polymerization inhibitors, triazoles are reported as useful linkers in conjugating different pharmacophores. Sharma and coworkers employed triazole to tether monocarbonyl curcumin and isatin into a bifunctional hybrid and evaluated their cytotoxicity against selected cancer cell lines. The structure–activity relationship study provided an insight into the cytotoxicity of these hybrids and the unsubstituted or isatin unit with electron-withdrawing groups enhanced the cytotoxicity effect in the order H > F > Cl > Br > I> NO2. Moreover, a higher number of chain lengths of the carbon-bridge decreased the cytotoxicity of the hybrids. Amongst them, four of the hybrids exhibited promising cytotoxicity against the tested cancer cell lines and the most potent hybrid 47 showed an IC50 value of 0.73 and 3.04 against THP-1 (leukemia) and HCT-116 (colon) cancer cell lines, respectively. Hybrid 47 also exhibited good inhibition of tubulin polymerization (IC50 1.06 μM) which was better than that of the standard CA-4 (Fig. 10). This hybrid 47 was docked at a new curcumin binding site, which is 32 Å away from the well-known colchicine-binding site, and the results suggested that the presence of a trimethoxyphenyl group was responsible for the superior activity of 47.98 In another report, thirty-six triazole linked C5-curcuminoid-coumarin hybrids 48 were reported as novel anti tubulin agents. Two carbon bridges and a phenyl ring with methoxy substitution on ring X showed the best activity in the series. The most active hybrid possessed a trimethoxy phenyl group as ring X with a two-carbon bridge, which exhibited an IC50 value of 0.82 μM against the THP-1 cancer line and 1.55 μM against inhibition of tubulin polymerization. In addition to these reports, a library of isatin–coumarin hybrids 49 were also synthesized employing triazole as a linker and evaluated for cytotoxicity and inhibition of tubulin polymerization. The diversification was carried out on the C-5 position of the isatin moiety and the length of the carbon bridge while leaving the coumarin moiety unchanged. In this case, unsubstituted isatin with a two carbon bridge was the most active with an IC50 value of 0.73 μM against the THP-1 (leukemia) cancer line and 1.06 μM against inhibition of tubulin polymerization (better than CA-4).99,100
Fig. 10. 1,2,3-Triazole ring as a linker.

3. 1,2,4-Triazole based microtubule targeting agents
1,2,4-Triazoles are structural isomers of their 1,2,3-triazole counterparts; however, the design of tubulin polymerization inhibition based on them is comparatively fewer. The various synthetic approaches employed for the synthesis of different substituted 1,2,4-triazole based microtubule targeting agents are mentioned in Table 3. In some cases, the desired substituted triazoles were obtained by synthetic modifications on the 1,2,4 triazole ring. Combretastatin A-4 is the parent molecule based on which most of the new 1,2,4-triazole based tubulin polymerization inhibitors are designed. Ohsumi and coworkers were the first to synthesize five-membered heterocyclics such as pyrazole, triazole, tetrazole, and thiazole as cis-restricted CA-4 analogs. Amongst them, thiazole and tetrazole based analogs exhibited potent activity against one tested cancer cell line; however, the three 1,2,4-triazole analogs synthesized with the amino group on ring A and substituents on the triazole ring led to a loss of cytotoxic and polymerization inhibition properties in comparison to other analogs and the parent molecule. The most active triazole was reported to exhibit an IC50 value of 840 nM and 4 μM against the colon 26 cancer cell line and tubulin inhibition, respectively.101 After a decade, highly potent 1,2,4-triazoles as cis-restricted CA-4 mimics were reported by retention of the trimethoxyphenyl ring and simultaneous modification of ring B by introduction of groups like fluorine or dimethylamine or replacement of ring B with an indole ring. In comparison to the previous report, ring A was attached to the C-3 position of triazole rather than the N-4 position. A series 51a–f was synthesized by the cyclization of amidrazones 50 with trimethyl orthoformate under acidic conditions. Among the synthesized series, compound 51a with an N-methyl-5-indolyl moiety as the replacement of ring B was the most potent (Fig. 11). Compound 51a exhibited an IC50 value of 7.4 nM against the HCT-116 colon cancer cell line and anti-tubulin activity (IC50 3 μM) superior to the parent CA-4 molecule with docking at the colchicine binding pocket.102 To further exploit the potential of triazole 51a as a drug candidate, it was designated as T115 (51a) and subjected to advanced in vitro and in vivo studies. The studies revealed that 51a competed well with colchicine for its binding pocket in the concentration range of 0.01–100 μmol L–1, and robustly inhibited tubulin, comparable to CA-4, at the test concentration 0.1–10 μmol L–1 along with disruption of the microtubule network. In addition to this, several cancer lines including multidrug-resistant strains (breast, ovarian, cervix, prostrate, and leukemia origin) were effectively inhibited by T115 with an IC50 value in the range of 2.1 to 24 nmol L–1. To investigate the safety profile, T115 was tested against normal human cell lines (GM05659 and FBLC) at 10 μM and was non-toxic to normal cells with a selectivity index of 500–1000 times in comparison to cancer cells. Moreover, in murine models, compound 51a inhibited the tumour growth with good tolerance (400 mg kg–1).103
Table 3. Synthetic approaches to access 1,2,4-triazoles.
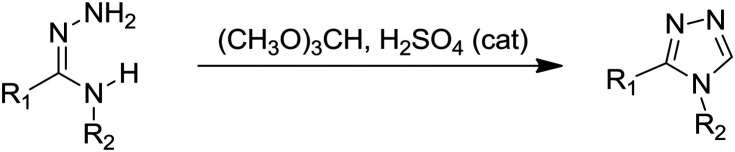




Fig. 11. 1,2,4-Triazoles as microtubule targeting agents (1).

In another report, Romagnoli and coworkers reported 1,5-disubstituted 1,2,4-triazoles as analogs of CA-4 with tubulin inhibition potential. Two different series of triazoles were synthesized by positioning the trimethoxyphenyl group on N-1 or on the C-5 atom of the triazole ring. Biological studies revealed that the trimethoxyphenyl group on N-1 was more active than the trimethoxyphenyl group on the C-5 position. It was also found that –OMe as well as –OEt groups provided good activity and the position of –OMe and –OEt groups on the phenyl ring resulted in a pronounced effect on the activity with the para position showing enhanced activity followed by meta and ortho positions. In between methoxy and ethoxy groups, the ethoxy substitution (along with the chlorine atom) showed better activity. Compounds 52a and 52b (with p-OEt substitution on the phenyl ring) were the most potent triazoles with better activity than CA-4 with 52b exhibiting IC50 values ranging from 3 to 20 nM against the tested cancer cell lines. Triazole 52a displayed excellent tubulin inhibition properties (IC50 0.76 μM) compared with the parent CA-4 lead molecule with effective colchicine binding competitiveness. Other biological assays were carried out with promising results.104 Recently, to enhance the tubulin-binding properties of 1,5-disubstituted 1,2,4-triazoles, Mustafa and coworkers introduced a carboxanilide group on the C-5 position of the triazole ring (used for cis restriction in CA-4) with phenyl and benzyl groups as rings A and B. The anisidines (ortho, meta, and para) displayed the best results compared to other substitutions on the phenyl group of carboxanilide. Compound 53 was the better compound in the series against the MCF-7 cell line (IC50 7.79 μM) and exhibited decent inhibition of tubulin polymerization by binding on the colchicine site.105 In an earlier report, this research group also reported similar 1,2,4-triazole carboxanilides with limited success.106 In addition to this, cis-restricted triazole mimics of combretastatin–benzothiazole hybrids were also reported as inhibitors of tubulin polymerization and apoptosis inducers.107
Exploiting the tubulin polymerization potency of phenstatins, Lee and coworkers envisaged variation of ring B of phenstatin by incorporating diverse heteroaromatic groups to improve its therapeutic and pharmacological properties. On ring B, substitutions were carried out on the C-2, C-4 and C-5 positions and the structural activity relationship study suggested that an appropriate combination on C-2 and C-4 substitutions was necessary for potent activity. Preliminary studies suggested that a triazole ring on the C2 position was good for activity; therefore different five-membered rings were introduced at the C-4 position and studied. This led to the identification of the most potent compound 54a with triazole and thiazole rings on C-2 and C-4 positions, respectively (Fig. 12). It showed an IC50 value of 4.8 nM comparable to CA-4 against the HL-60 cell line with good antitubulin activity (IC50 4.3 μM). Against HCT15, a P-gp overexpressing multidrug-resistant (MDR) cell line, it was better than the tested standard and caused significant cell arrest at 30 nM concentration. To improve the water solubility of 54a, amino acid prodrug CKD-516 (54b) with an l-valine unit was designed which substantially improved the aqueous solubility (930 mg mL–1). Moreover, 54b induced tumor growth inhibition in the murine model better than 54a and it displayed potent antitumor activity against the multidrug-resistant HCT15 cell line in human xenografts. The prodrug CKD-516 progressed to phase 1 clinical trial and the study demonstrated CKD-516 as a feasible, well-tolerated novel vascular disrupting agent in patients with advanced solid tumor.108,109 In a simultaneously reported investigation, the same research group studied a number of amino acid prodrugs of 54b to increase its pharmacokinetic properties and in vivo efficacy.110
Fig. 12. 1,2,4-Triazoles as microtubule targeting agents (2).

In an interesting design based on phenstatin, Romagnoli and coworkers replaced ring B of phenstatin with a 3-arylamino-5-amino-1,2,4-triazole moiety and evaluated the anticancer, anti-tubulin and cell cycle effects. Diversification was carried out on the phenyl ring of the anilino moiety present on the C-3 position of triazole to study the effect on antiproliferative activity. The structure–activity relationship study revealed that para-Me, meta, para-diMe, and para-Et groups on the phenyl ring exhibited the best results. Triazole 55 with a para-methyl substitution on the phenyl ring was the most active and exhibited an IC50 value in the range of 0.2–3.1 nM against the tested cancer cell lines, which was far superior to CA-4. Remarkably, 55 exhibited excellent potency against two tested drug-resistant cell lines (IC50 values between 0.21 and 1.2 nM) and was ineffective against the tested normal human noncancer cells (IC50 values between 8.5 and 31.2 μM) demonstrating its selectivity only towards cancer cells. Triazole 55 was almost twice as potent as CA-4 against inhibition of tubulin polymerization displaying an IC50 value of 0.75 μM and the competition assay suggested that triazole 55 inhibited colchicine binding by 92%. Moreover, additional biological studies revealed that triazole 55 was a potential drug candidate to be taken forward for further optimization.111 Recently, this research group also reported a new series of 3-aryl/heteroaryl-5-amino-1,2,4-triazoles as antiproliferative and anti-tubulin agents. The antiproliferative activity of the series was not as potent as the earlier reported series; however one of the compounds 56 strongly inhibited the tubulin assembly, with an IC50 value of 0.66 μM better than those of CA-4 and 55.112 In addition to this, some novel aldimine-type Schiff bases bearing 3,4,5-trimethoxyphenyl and 1,2,4-triazole-3-thione/thiol were also designed as tubulin polymerization inhibitors. The Schiff base 57 with thiol and 2,3-dichloro substitution of the phenyl ring provided the best results from the series with IC50 values of 12.48, 4.25, 3.33 and 9.71 μM against the HT1080, HT29, MCF-7, and A549 cell lines, respectively. It also displayed excellent tubulin inhibition properties with an IC50 value of 0.17 μM slightly better than that of colchicine (IC50 value of 0.19 μM).113
4. Fused triazole based microtubule targeting agents
Triazoles fused to a heterocyclic ring were also investigated for their tubulin polymerization inhibiting properties. The research in this direction started with a report on vinylogous analogs of CA-4, wherein the cis-olefinic bond was replaced by (Z,E)-butadiene linker 58 (Fig. 13). These analogs were reported to be potent anticancer and anti-tubulin agents; however the unstable (Z,E)-isomer was prone to convert to a more stable (E,E)-isomer, which was inactive.114 To lock the Z,E-configuration of vinylogous combretastatin A-4 which was responsible for the bioactivity, fused 1,2,4-triazole-thiadiazine and fused 1,2,4-triazole-pyridazine scaffolds were introduced to mimic the Z,E-isomer. In this direction, Xu and coworkers reported a series of 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines as inhibitors of tubulin polymerization, which exhibited moderate to potent antiproliferative activity against the tested cancer cell lines. In these analogs, the trimethoxyphenyl ring was conserved and various substitutions (both electron donating and withdrawing) were introduced at different positions of ring B, whereas the fused triazolo-pyridazine ring between the two phenyl rings mimicked the vinylogous group. The structure–activity relationship study elucidated that the electron-donating groups (CH3, OCH3, SCH3, and NH2) on the para position of ring B exhibited higher activities than the electron-withdrawing groups (CF3, NO2, CN) with the exception of halogens. The effect of disubstitution on ring B was also explored and the fused triazolo compound 59 (with a 3-amino,4-methoxyphenyl moiety) was very potent and exhibited an IC50 value in the range of 0.008–0.014 μM, against SGC-7901, A549 and HT1080 cancer cell lines, which was comparable to that of CA-4. However, the antitubulin activity of the compound was three times lower than that of CA-4 (IC50 = 1.8 μM), though it exhibited disruption of the microtubule assembly and bonded at the colchicine pocket. In addition to this, as expected the 3,4,5-trimethoxyphenyl ring provided a better activity profile than the 2,3,4-trimethoxyphenyl ring.115 Similarly, the research group also reported 3,6-diaryl-[1,2,4]triazolo[3,4-b]thiadiazines as novel antitubulin agents. The SAR study revealed an interesting aspect in that the mono and deoxygenated methoxy groups on different positions of ring A showed better activity than 3,4,5-trimethoxy substitution. Compound 60 with a 3-methoxy substitution on ring A and substitutions on the ring similar to 59 was the most active member of the series, exhibiting an IC50 value in the range of 0.011–0.015 μM against SGC-7901, A549, and HT-1080 cancer cell lines (comparable to CA-4). It also effectively inhibited tubulin polymerization at an IC50 value of 1.6 μM by binding on the colchicine site of tubulin.116 In addition to this rationalized approach of introduction of the fused triazole ring as a CA-4 olefin bond mimic, different heterocyclic fused triazoles were also designed to study their antiproliferative and tubulin polymerization inhibition properties. This includes triazolopyridinyl-acrylonitriles 61, triazolochromenes 62, triazoloquinazolinones 63 and triazoloquinoxalines 64, which exhibited moderate to good results.117–120
Fig. 13. Fused triazoles as microtubule targeting agents.

5. Conclusion and future perspective
Drug design is a complicated, multifaceted process involving huge risks with high returns. In the last seven decades, the research and development cost per new drug increased by over a hundred fold. The academic research provides a fertile ground for the industrial counterpart to plough up and develop new chemical entities for potential clinical usage in the treatment of different diseases. In this process, pharmacophores play a foundational role and fortunately, their properties can be tweaked and optimized with different chemical groups resulting in desired efficacy. Triazoles are one of the moieties used for this purpose and a number of drugs containing triazoles are being clinically used for a wide range of medical implications. The stability towards chemical and metabolic degradation and other intrinsic properties render triazole the preferred bioisostere for an amide bond, ester (lactone), olefinic double bond, carboxylic acid, and other heterocyclic rings. In addition to this, triazoles also have the potential to be used as linkers to tether two different pharmacophores, duplicate a single pharmacophore or generate bidentate inhibitors.
Microtubules are structural cytoplasmic elements with a pronounced role in cell division and are important molecular targets in the design and development of anticancer agents. These agents inhibit the basic unit tubulin by interfering with the polymerization and depolymerisation process. Both naturally occurring (combretastatin A-4, colchicine and podophyllotoxin) and synthetic small molecules (ABT-751) are employed as lead molecules in the development of tubulin polymerization inhibitors. Triazole based chemical entities are increasingly being reported as antitubulin agents harnessing the favourable pharmacokinetic properties of the triazole ring. With the advent of click chemistry, which provides robust access to 1,2,3-triazoles with several added advantages, the utilization of triazoles in the development of tubulin polymerization inhibitors is further exemplified. To the best of our knowledge, no colchicine site binding tubulin inhibitor is approved by the Food Drug and Administration though some are in clinical trials. Currently, tubulin inhibitors that are in clinical usage are natural product based complex entities binding at the taxane pocket and the development of a simple colchicine site binding drug will be a major breakthrough. The triazole moiety with its unique properties provides a good opportunity in the optimization of the lead molecules with desired pharmacokinetic properties.
Interestingly CKD-516, a triazole based tubulin inhibitor, exhibited good toleration and safety as a novel vascular disrupting agent in patients with advanced solid tumor in the initial clinical trials. The easy access to triazoles with click chemistry coupled with advances in lead optimization processes offers an optimistic scenario in the development of colchicine binding tubulin inhibitors in coming years. On the other hand, it is very surprising that there are no triazole based chemical entities used in the design of tubulin inhibitors binding at the taxane and vinca domains of tubulin. This area needs further exploration and provides a promising opportunity taking into consideration the versatility of the triazole ring system.
Conflicts of interest
The authors declare that there is no conflict of interest.
Biographies

M. Shaheer Malik
M. Shaheer Malik is currently Assistant Professor in chemistry at Umm Al-Qura University, Makkah (Saudi Arabia). He carried out his doctoral studies in organic chemistry at the Indian Institute of Chemical Technology, Hyderabad, and was awarded his Ph.D. by Osmania University, Hyderabad, India. Subsequently, he moved for a post-doctoral assignment to Yonsei University, Seoul (Korea), working in the area of biocatalysis. His research interest deals with the design and synthesis of novel chemical compounds as pharmaceutical agents and the use of enzymes in organic transformations. He regularly contributes and reviews research findings towards the advancement of science in his area of interest.

Saleh A. Ahmed
Saleh A. Ahmed received his bachelor's and master's degrees from Assiut University, Egypt, and Ph.D. in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr from Saarland University, Saarbrücken, Germany as a DAAD fellow. He has more than 18 years of experience as a postdoctoral fellow, senior researcher and visiting professor in France (CNRS Bordeaux University), Japan (JSPS, AIST-Osaka), Germany (AvH, Berlin, and Bielefeld), Italy (TEMPUS, Ferreira University), USA (Arab-Fund, Florida University) and KSA. His current research interests include synthesis and photophysical properties of novel organic compounds such as material-based gels, electronic devices, and solar energy conversion.

Ismail I. Althagafi
Ismail I. Thagafi is presently Chairman and Associate Professor at the Department of Chemistry, Faculty of Applied Sciences, Umm Al-Qura University, Makkah (Saudi Arabia). He got his bachelor's degree from Umm Al-Qura University, and master's degree and Ph.D. from Cardiff University (United Kingdom). His current research interest includes areas of physical organic chemistry, synthetic chemistry, and bioorganic chemistry.

Mohammad Azam Ansari
Dr. Mohammad Azam Ansari received his Ph.D. in Medical Microbiology from JNMCH, Aligarh Muslim University, India (2013). Presently, he is working in the Institute for Research and Medical Consultations, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, as an Assistant Professor. He has significantly contributed to the fields of green nanotechnology, nanotoxicity, and microbial biofilm. His current research interests focus on green synthesis of nanoparticles & their characterization for biomedical and cancer application. Mohammad is particularly focused on the antimicrobial properties of green nanoparticles against multidrug-resistant pathogens. He has published a significant number of papers in journals of national and international repute.

Ahmed Kamal
Ahmed Kamal graduated from Osmania University, Hyderabad (India), where he obtained his master's degree and Ph.D. in Organic Chemistry. He carried out his post-doctoral research work at the University of Portsmouth, England, and was a Visiting Scientist at the University of Alberta, Canada. For the past 30 years, he has pursued his research career at Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad, and worked in the capacity of Outstanding Scientist at this institute. Presently, he is the Pro-Vice Chancellor at Jamia Hamdard, New Delhi, India. His research interests mainly focused on the design and synthesis of gene-targeting compounds as new anti-cancer agents and biocatalytic transformations.
References
- Roughley S. D., Jordan A. M. J. Med. Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Civjan N., Chemical Biology: Approaches to Drug Discovery and Development to Targeting Disease, John Wiley & Sons, Inc., 2012. [Google Scholar]
- Bonandi E., Christodoulou M. S., Fumagalli G., Perdicchia D., Rastelli G., Passarella D. Drug Discovery Today. 2017;22:1572–1581. doi: 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Hou J., Liu X., Shen J., Zhao G., Wang P. G. Expert Opin. Drug Discovery. 2012;7:489–501. doi: 10.1517/17460441.2012.682725. [DOI] [PubMed] [Google Scholar]
- Lal K., Yadav P. Anti-Cancer Agents Med. Chem. 2018;18:21–37. doi: 10.2174/1871520616666160811113531. [DOI] [PubMed] [Google Scholar]
- Zhou C. H., Wang Y. Curr. Med. Chem. 2012;19:239–280. doi: 10.2174/092986712803414213. [DOI] [PubMed] [Google Scholar]
- Li Y. T., Wang J. H., Pan C. W., Meng F. F., Chu X. Q., Ding Y. H., Qu W. Z., Li H. Y., Yang C., Zhang Q., Bai C. G., Chen Y. Bioorg. Med. Chem. Lett. 2016;26:1419–1427. doi: 10.1016/j.bmcl.2016.01.068. [DOI] [PubMed] [Google Scholar]
- Wang B., Chu D., Feng Y., Shen Y., Aoyagi-Scharber M., Post L. E. J. Med. Chem. 2016;59:335–357. doi: 10.1021/acs.jmedchem.5b01498. [DOI] [PubMed] [Google Scholar]
- Murphy, Jr. M. J. Oncologist. 1998;3:129–130. [PubMed] [Google Scholar]
- Borden K. L. B., Culjkovic-Kraljacic B. Leuk. Lymphoma. 2010;51:1805–1815. doi: 10.3109/10428194.2010.496506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelley M. A., Zhu E. S., Thompson, 3rd G. R. Infect. Drug Resist. 2016;9:79–86. doi: 10.2147/IDR.S81416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux M. P., Denis J., Nivoix Y., Herbrecht R. J. Mycol. Med. 2018;28:15–22. doi: 10.1016/j.mycmed.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Murrell D., Bossaer J. B., Carico R., Harirforoosh S., Cluck D. Int. J. Pharm. Pract. 2017;25:18–30. doi: 10.1111/ijpp.12302. [DOI] [PubMed] [Google Scholar]
- Huggins J. W. Rev. Infect. Dis. 1989;11(Suppl 4):S750–S761. doi: 10.1093/clinids/11.supplement_4.s750. [DOI] [PubMed] [Google Scholar]
- Saab S., Rheem J., Jimenez M. A., Fong T. M., Mai M. H., Kachadoorian C. A., Esmailzadeh N. L., Bau S. N., Kang S., Ramirez S. D., Grotts J., Choi G., Durazo F. A., El-Kabany M. M., Han S. B., Busuttil R. W. J. Clin. Transl. Hepatol. 2017;5:101–108. doi: 10.14218/JCTH.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Junyent J., Benavent E., Sierra Y., El Haj C., Soldevila L., Torrejon B., Rigo-Bonnin R., Tubau F., Ariza J., Murillo O. Int. J. Antimicrob. Agents. 2019;53:612–619. doi: 10.1016/j.ijantimicag.2019.01.010. [DOI] [PubMed] [Google Scholar]
- Yang Y., Rasmussen B. A., Shlaes D. M. Pharmacol. Ther. 1999;83:141–151. doi: 10.1016/s0163-7258(99)00027-3. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Drugs. 2016;76:509–516. doi: 10.1007/s40265-016-0550-y. [DOI] [PubMed] [Google Scholar]
- Yang L. P., Keam S. J., Keating G. M. Drugs. 2007;67:2211–2230. doi: 10.2165/00003495-200767150-00007. [DOI] [PubMed] [Google Scholar]
- Abel S., Back D. J., Vourvahis M. Antiviral Ther. 2009;14:607–618. [PubMed] [Google Scholar]
- Karasik A., Aschner P., Katzeff H., Davies M. J., Stein P. P. Curr. Med. Res. Opin. 2008;24:489–496. doi: 10.1185/030079908x261069. [DOI] [PubMed] [Google Scholar]
- Tantry U. S., Bliden K. P., Gurbel P. A. Expert Opin. Invest. Drugs. 2007;16:225–229. doi: 10.1517/13543784.16.2.225. [DOI] [PubMed] [Google Scholar]
- Eltze M. J. Pharm. Pharmacol. 1997;49:1091–1095. doi: 10.1111/j.2042-7158.1997.tb06048.x. [DOI] [PubMed] [Google Scholar]
- Fagiolini A., Comandini A., Catena Dell'Osso M., Kasper S. CNS Drugs. 2012;26:1033–1049. doi: 10.1007/s40263-012-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimian S., Cheng-Hakimian A., Anderson G. D., Miller J. W. Expert Opin. Pharmacother. 2007;8:1931–1940. doi: 10.1517/14656566.8.12.1931. [DOI] [PubMed] [Google Scholar]
- Palasz A., Lapray D., Peyron C., Rojczyk-Golebiewska E., Skowronek R., Markowski G., Czajkowska B., Krzystanek M., Wiaderkiewicz R. Int. J. Neuropsychopharmacol. 2014;17:157–168. doi: 10.1017/S1461145713000552. [DOI] [PubMed] [Google Scholar]
- Casper R. F., Mitwally M. F. Clin. Obstet. Gynecol. 2011;54:685–695. doi: 10.1097/GRF.0b013e3182353d0f. [DOI] [PubMed] [Google Scholar]
- Latha M. S., Martis J., Shobha V., Sham Shinde R., Bangera S., Krishnankutty B., Bellary S., Varughese S., Rao P., Naveen Kumar B. R. J. Clin. Aesthet. Dermatol. 2013;6:16–26. [PMC free article] [PubMed] [Google Scholar]
- Huisgen R. Angew. Chem., Int. Ed. Engl. 1963;2:565–598. [Google Scholar]
- Kolb H. C., Finn M. G., Sharpless K. B. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Agalave S. G., Maujan S. R., Pore V. S. Chem. – Asian J. 2011;6:2696–2718. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- Binder W. H., Sachsenhofer R. Macromol. Rapid Commun. 2008;29:952–981. [Google Scholar]
- Hatit M. Z. C., Reichenbach L. F., Tobin J. M., Vilela F., Burley G. A., Watson A. J. B. Nat. Commun. 2018;9:4021. doi: 10.1038/s41467-018-06551-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho da Silva F., Ferreira V. F., da Silva Magalhaes Forezi L. Curr. Top. Med. Chem. 2018;18:1426–1427. doi: 10.2174/156802661817181107151553. [DOI] [PubMed] [Google Scholar]
- Glowacka I. E., Grzonkowski P., Lisiecki P., Kalinowski L., Piotrowska D. G. Arch. Pharm. 2019:e1800302. doi: 10.1002/ardp.201800302. [DOI] [PubMed] [Google Scholar]
- He S. C., Zhang H. Z., Zhang H. J., Sun Q., Zhou C. H. Med. Chem. 2020;16:104–116. doi: 10.2174/1573406414666181106124852. [DOI] [PubMed] [Google Scholar]
- Zhang B. Eur. J. Med. Chem. 2019;168:357–372. doi: 10.1016/j.ejmech.2019.02.055. [DOI] [PubMed] [Google Scholar]
- Gao F., Wang T., Xiao J., Huang G. Eur. J. Med. Chem. 2019;173:274–281. doi: 10.1016/j.ejmech.2019.04.043. [DOI] [PubMed] [Google Scholar]
- Hilimire T. A., Chamberlain J. M., Anokhina V., Bennett R. P., Swart O., Myers J. R., Ashton J. M., Stewart R. A., Featherston A. L., Gates K., Helms E. D., Smith H. C., Dewhurst S., Miller B. L. ACS Chem. Biol. 2017;12:1674–1682. doi: 10.1021/acschembio.7b00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashad A. A., Acharya K., Haftl A., Aneja R., Dick A., Holmes A. P., Chaiken I. Org. Biomol. Chem. 2017;15:7770–7782. doi: 10.1039/c7ob01448a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Xu Z., Gao C., Ren Q. C., Chang L., Lv Z. S., Feng L. S. Eur. J. Med. Chem. 2017;138:501–513. doi: 10.1016/j.ejmech.2017.06.051. [DOI] [PubMed] [Google Scholar]
- Buckle D. R., Outred D. J., Rockell C. J. M., Smith H., Spicer B. A. J. Med. Chem. 1983;26:251–254. doi: 10.1021/jm00356a025. [DOI] [PubMed] [Google Scholar]
- Song M. X., Deng X. Q. J. Enzyme Inhib. Med. Chem. 2018;33:453–478. doi: 10.1080/14756366.2017.1423068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertino M. W., Vega C., Rolon M., Coronel C., Rojas de Arias A., Schmeda-Hirschmann G. Molecules. 2017;22:369. doi: 10.3390/molecules22030369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockunier L. L., Parmee E. R., Ok H. O., Candelore M. R., Cascieri M. A., Colwell, Jr. L. F., Deng L., Feeney W. P., Forrest M. J., Hom G. J., MacIntyre D. E., Tota L., Wyvratt M. J., Fisher M. H., Weber A. E. Bioorg. Med. Chem. Lett. 2000;10:2111–2114. doi: 10.1016/s0960-894x(00)00422-4. [DOI] [PubMed] [Google Scholar]
- Szymaszkiewicz A., Zielinska M., Li K., Ramanathan M., Alam S., Hou D. R., Fichna J., Storr M. Naunyn-Schmiedeberg's Arch. Pharmacol. 2018;391:435–444. doi: 10.1007/s00210-018-1465-9. [DOI] [PubMed] [Google Scholar]
- New Endpoints, Programs Used for Drug Approvals Cancer Discovery, 2019, vol. 9, p. 160. [DOI] [PubMed] [Google Scholar]
- Ilan Y. J. Cell. Physiol. 2019;234:7923–7937. doi: 10.1002/jcp.27978. [DOI] [PubMed] [Google Scholar]
- Desai A., Mitchison T. J. Annu. Rev. Cell Dev. Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. J. Nat. Prod. 2009;72:507–515. doi: 10.1021/np800568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowinsky E. K. Annu. Rev. Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- Hamel E. Pharmacol. Ther. 1992;55:31–51. doi: 10.1016/0163-7258(92)90028-x. [DOI] [PubMed] [Google Scholar]
- Chen J., Liu T., Dong X., Hu Y. Mini-Rev. Med. Chem. 2009;9:1174–1190. doi: 10.2174/138955709789055234. [DOI] [PubMed] [Google Scholar]
- Lu Y., Chen J., Xiao M., Li W., Miller D. D. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Sun H., Xu S., Zhu Z., Xu J. Future Med. Chem. 2017;9:1765–1794. doi: 10.4155/fmc-2017-0100. [DOI] [PubMed] [Google Scholar]
- Cao Y. N., Zheng L. L., Wang D., Liang X. X., Gao F., Zhou X. L. Eur. J. Med. Chem. 2018;143:806–828. doi: 10.1016/j.ejmech.2017.11.062. [DOI] [PubMed] [Google Scholar]
- Meldal M., Tornøe C. W. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Gomes R. S., Jardim G. A. M., de Carvalho R. L., Araujo M. H., da Silva Júnior E. N. Tetrahedron. 2019;75:3697–3712. [Google Scholar]
- Zhang L., Chen X., Xue P., Sun H. H. Y., Williams I. D., Sharpless K. B., Fokin V. V., Jia G. J. Am. Chem. Soc. 2005;127:15998–15999. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]
- Rodionov V. O., Fokin V. V., Finn M. G. Angew. Chem., Int. Ed. 2005;44:2210–2215. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- Himo F., Lovell T., Hilgraf R., Rostovtsev V. V., Noodleman L., Sharpless K. B., Fokin V. V. J. Am. Chem. Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- Tornøe C. W., Christensen C., Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Pettit G. R., Singh S. B., Niven M. L., Hamel E., Schmidt J. M. J. Nat. Prod. 1987;50:119–131. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- Cirla A., Mann J. Nat. Prod. Rep. 2003;20:558–564. doi: 10.1039/b306797c. [DOI] [PubMed] [Google Scholar]
- Seddigi Z. S., Malik M. S., Saraswati A. P., Ahmed S. A., Babalghith A.A. O.O., Lamfon H. A., Kamal A. MedChemComm. 2017;8:1592–1603. doi: 10.1039/c7md00227k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pati Hari N., Wicks M., Herman L. Holt Jr., LeBlanc R., Weisbruch P., Forrest L., Lee M. Heterocycl. Commun. 2005;11:117. [Google Scholar]
- Akselsen Ø. W., Odlo K., Cheng J.-J., Maccari G., Botta M., Hansen T. V. Bioorg. Med. Chem. 2012;20:234–242. doi: 10.1016/j.bmc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Mur Blanch N., Chabot G. G., Quentin L., Scherman D., Bourg S., Dauzonne D. Eur. J. Med. Chem. 2012;54:22–32. doi: 10.1016/j.ejmech.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Madadi N. R., Penthala N. R., Howk K., Ketkar A., Eoff R. L., Borrelli M. J., Crooks P. A. Eur. J. Med. Chem. 2015;103:123–132. doi: 10.1016/j.ejmech.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penthala N. R., Madhukuri L., Thakkar S., Madadi N. R., Lamture G., Eoff R. L., Crooks P. A. MedChemComm. 2015;6:1535–1543. doi: 10.1039/C5MD00219B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova M. N., Demchuk D. V., Tsyganov D. V., Chernysheva N. B., Samet A. V., Silyanova E. A., Kislyi V. P., Maksimenko A. S., Varakutin A. E., Konyushkin L. D., Raihstat M. M., Kiselyov A. S., Semenov V. V. ACS Comb. Sci. 2018;20:700–721. doi: 10.1021/acscombsci.8b00113. [DOI] [PubMed] [Google Scholar]
- Kamal A., Shaik B., Nayak V. L., Nagaraju B., Kapure J. S., Shaheer Malik M., Shaik T. B., Prasad B. Bioorg. Med. Chem. 2014;22:5155–5167. doi: 10.1016/j.bmc.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Pettit G. R., Toki B., Herald D. L., Verdier-Pinard P., Boyd M. R., Hamel E., Pettit R. K. J. Med. Chem. 1998;41:1688–1695. doi: 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- Mesenzani O., Massarotti A., Giustiniano M., Pirali T., Bevilacqua V., Caldarelli A., Canonico P., Sorba G., Novellino E., Genazzani A. A., Tron G. C. Bioorg. Med. Chem. Lett. 2011;21:764–768. doi: 10.1016/j.bmcl.2010.11.113. [DOI] [PubMed] [Google Scholar]
- Feng D., Wu Y., Wang H., Bai Z., Wang D., Zuo D., Bao K., Wu Y., Zhang W. RSC Adv. 2017;7:29103–29111. [Google Scholar]
- Hussaini S. M., Yedla P., Babu K. S., Shaik T. B., Chityal G. K., Kamal A. Chem. Biol. Drug Des. 2016;88:97–109. doi: 10.1111/cbdd.12738. [DOI] [PubMed] [Google Scholar]
- Alkadi H., Khubeiz M. J., Jbeily R. Infect. Disord.: Drug Targets. 2018;18:105–121. doi: 10.2174/1871526517666171017114901. [DOI] [PubMed] [Google Scholar]
- Nicolaus N., Zapke J., Riesterer P., Neudorfl J. M., Prokop A., Oschkinat H., Schmalz H. G. ChemMedChem. 2010;5:661–665. doi: 10.1002/cmdc.201000063. [DOI] [PubMed] [Google Scholar]
- Kuznetsova N. R., Svirshchevskaya E. V., Sitnikov N. S., Abodo L., Sutorius H., Zapke J., Velder J., Thomopoulou P., Oschkinat H., Prokop A., Schmalz H.-G., Fedorov A. Y., Vodovozova E. L. Russ. J. Bioorg. Chem. 2013;39:543–552. [PubMed] [Google Scholar]
- Sackett D. L. Pharmacol. Ther. 1993;59:163–228. doi: 10.1016/0163-7258(93)90044-e. [DOI] [PubMed] [Google Scholar]
- Imperio D., Pirali T., Galli U., Pagliai F., Cafici L., Canonico P. L., Sorba G., Genazzani A. A., Tron G. C. Bioorg. Med. Chem. 2007;15:6748–6757. doi: 10.1016/j.bmc.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Vishnuvardhan M. V. P. S., Chandrasekhar S. R. V. K., Lakshma Nayak V., Sayeed I. B., Alarifi A., Kamal A. MedChemComm. 2017;8:1817–1823. doi: 10.1039/c7md00273d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poornima B., Siva B., Venkanna A., Shankaraiah G., Jain N., Yadav D. K., Misra S., Babu K. S. Eur. J. Med. Chem. 2017;139:441–453. doi: 10.1016/j.ejmech.2017.07.076. [DOI] [PubMed] [Google Scholar]
- Solum E. J., Vik A., Hansen T. V. Steroids. 2014;87:46–53. doi: 10.1016/j.steroids.2014.05.020. [DOI] [PubMed] [Google Scholar]
- Cañeque T., Müller S., Rodriguez R. Nat. Rev. Chem. 2018;2:202–215. [Google Scholar]
- Mauer A. M., Cohen E. E. W., Ma P. C., Kozloff M. F., Schwartzberg L., Coates A. I., Qian J., Hagey A. E., Gordon G. B. J. Thorac. Oncol. 2008;3:631–636. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]
- Prasad B., Lakshma Nayak V., Srikanth P. S., Baig M. F., Subba Reddy N. V., Babu K. S., Kamal A. Bioorg. Chem. 2019;83:535–548. doi: 10.1016/j.bioorg.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Kamal A., Subba Rao A. V., Vishnuvardhan M. V., Srinivas Reddy T., Swapna K., Bagul C., Subba Reddy N. V., Srinivasulu V. Org. Biomol. Chem. 2015;13:4879–4895. doi: 10.1039/c5ob00232j. [DOI] [PubMed] [Google Scholar]
- Kamal A., Reddy N. V., Nayak V. L., Bolla N. R., Subba Rao A. V., Prasad B. Bioorg. Med. Chem. 2014;22:3465–3477. doi: 10.1016/j.bmc.2014.04.038. [DOI] [PubMed] [Google Scholar]
- Fu D. J., Liu J. F., Zhao R. H., Li J. H., Zhang S. Y., Zhang Y. B. Molecules. 2017;22:1470. doi: 10.3390/molecules22091470. [DOI] [Google Scholar]
- Stefely J. A., Palchaudhuri R., Miller P. A., Peterson R. J., Moraski G. C., Hergenrother P. J., Miller M. J. J. Med. Chem. 2010;53:3389–3395. doi: 10.1021/jm1000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suman P., Murthy T. R., Rajkumar K., Srikanth D., Dayakar C., Kishor C., Addlagatta A., Kalivendi S. V., Raju B. C. Eur. J. Med. Chem. 2015;90:603–619. doi: 10.1016/j.ejmech.2014.11.063. [DOI] [PubMed] [Google Scholar]
- Shaik S. P., Nayak V. L., Sultana F., Rao A. V. S., Shaik A. B., Babu K. S., Kamal A. Eur. J. Med. Chem. 2017;126:36–51. doi: 10.1016/j.ejmech.2016.09.060. [DOI] [PubMed] [Google Scholar]
- Shaik S. P., Vishnuvardhan M., Sultana F., Subba Rao A. V., Bagul C., Bhattacharjee D., Kapure J. S., Jain N., Kamal A. Bioorg. Med. Chem. 2017;25:3285–3297. doi: 10.1016/j.bmc.2017.04.013. [DOI] [PubMed] [Google Scholar]
- Sayeed I. B., Vishnuvardhan M., Nagarajan A., Kantevari S., Kamal A. Bioorg. Chem. 2018;80:714–720. doi: 10.1016/j.bioorg.2018.07.026. [DOI] [PubMed] [Google Scholar]
- Naaz F., Preeti Pallavi M. C., Shafi S., Mulakayala N., Shahar Yar M., Sampath Kumar H. M. Bioorg. Chem. 2018;81:1–20. doi: 10.1016/j.bioorg.2018.07.029. [DOI] [PubMed] [Google Scholar]
- Merk D. and Schubert-Zsilavecz M., The Linker Approach,, The Linker Approach, in in Drug Selectivity, 2017, 10.1002/9783527674381.ch8. [DOI] [Google Scholar]
- Sharma S., Gupta M. K., Saxena A. K., Bedi P. M. Bioorg. Med. Chem. 2015;23:7165–7180. doi: 10.1016/j.bmc.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Singh H., Kumar M., Nepali K., Gupta M. K., Saxena A. K., Sharma S., Bedi P. M. S. Eur. J. Med. Chem. 2016;116:102–115. doi: 10.1016/j.ejmech.2016.03.050. [DOI] [PubMed] [Google Scholar]
- Singh H., Singh J. V., Gupta M. K., Saxena A. K., Sharma S., Nepali K., Bedi P. M. S. Bioorg. Med. Chem. Lett. 2017;27:3974–3979. doi: 10.1016/j.bmcl.2017.07.069. [DOI] [PubMed] [Google Scholar]
- Ohsumi K., Hatanaka T., Fujita K., Nakagawa R., Fukuda Y., Nihei Y., Suga Y., Morinaga Y., Akiyama Y., Tsuji T. Bioorg. Med. Chem. Lett. 1998;8:3153–3158. doi: 10.1016/s0960-894x(98)00579-4. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Peng Y., Wang X. I., Keenan S. M., Arora S., Welsh W. J. J. Med. Chem. 2007;50:749–754. doi: 10.1021/jm061142s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S., Wang X. I., Keenan S. M., Andaya C., Zhang Q., Peng Y., Welsh W. J. Cancer Res. 2009;69:1910–1915. doi: 10.1158/0008-5472.CAN-08-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R., Baraldi P. G., Cruz-Lopez O., Lopez Cara C., Carrion M. D., Brancale A., Hamel E., Chen L., Bortolozzi R., Basso G., Viola G. J. Med. Chem. 2010;53:4248–4258. doi: 10.1021/jm100245q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa M., Abdelhamid D., Abdelhafez E. M. N., Ibrahim M. A. A., Gamal-Eldeen A. M., Aly O. M. Eur. J. Med. Chem. 2017;141:293–305. doi: 10.1016/j.ejmech.2017.09.063. [DOI] [PubMed] [Google Scholar]
- Aly O. M., Beshr E. A., Maklad R. M., Mustafa M., Gamal-Eldeen A. M. Arch. Pharm. 2014;347:658–667. doi: 10.1002/ardp.201400096. [DOI] [PubMed] [Google Scholar]
- Subba Rao A. V., Swapna K., Shaik S. P., Lakshma Nayak V., Srinivasa Reddy T., Sunkari S., Shaik T. B., Bagul C., Kamal A. Bioorg. Med. Chem. 2017;25:977–999. doi: 10.1016/j.bmc.2016.12.010. [DOI] [PubMed] [Google Scholar]
- Lee J., Kim S. J., Choi H., Kim Y. H., Lim I. T., Yang H. M., Lee C. S., Kang H. R., Ahn S. K., Moon S. K., Kim D. H., Lee S., Choi N. S., Lee K. J. J. Med. Chem. 2010;53:6337–6354. doi: 10.1021/jm1002414. [DOI] [PubMed] [Google Scholar]
- Oh D. Y., Kim T. M., Han S. W., Shin D. Y., Lee Y. G., Lee K. W., Kim J. H., Kim T. Y., Jang I. J., Lee J. S., Bang Y. J. Cancer Res. Treat. 2016;48:28–36. doi: 10.4143/crt.2014.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Bae S., Lee S. H., Choi H., Kim Y. H., Kim S. J., Park G. T., Moon S. K., Kim D. H., Lee S., Ahn S. K., Choi N. S., Lee K. J. Bioorg. Med. Chem. Lett. 2010;20:6327–6330. doi: 10.1016/j.bmcl.2010.05.060. [DOI] [PubMed] [Google Scholar]
- Romagnoli R., Baraldi P. G., Salvador M. K., Prencipe F., Bertolasi V., Cancellieri M., Brancale A., Hamel E., Castagliuolo I., Consolaro F., Porcu E., Basso G., Viola G. J. Med. Chem. 2014;57:6795–6808. doi: 10.1021/jm5008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R., Prencipe F., Oliva P., Baraldi S., Baraldi P. G., Brancale A., Ferla S., Hamel E., Bortolozzi R., Viola G. Bioorg. Chem. 2018;80:361–374. doi: 10.1016/j.bioorg.2018.06.037. [DOI] [PubMed] [Google Scholar]
- Ameri A., Khodarahmi G., Hassanzadeh F., Forootanfar H., Hakimelahi G. H. Arch. Pharm. 2016;349:662–681. doi: 10.1002/ardp.201600021. [DOI] [PubMed] [Google Scholar]
- Kaffy J., Pontikis R., Florent J. C., Monneret C. Org. Biomol. Chem. 2005;3:2657–2660. doi: 10.1039/b505955k. [DOI] [PubMed] [Google Scholar]
- Xu Q., Wang Y., Xu J., Sun M., Tian H., Zuo D., Guan Q., Bao K., Wu Y., Zhang W. ACS Med. Chem. Lett. 2016;7:1202–1206. doi: 10.1021/acsmedchemlett.6b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Bao K., Sun M., Xu J., Wang Y., Tian H., Zuo D., Guan Q., Wu Y., Zhang W. Sci. Rep. 2017;7:11997. doi: 10.1038/s41598-017-10860-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briguglio I., Laurini E., Pirisi M. A., Piras S., Corona P., Fermeglia M., Pricl S., Carta A. Eur. J. Med. Chem. 2017;141:460–472. doi: 10.1016/j.ejmech.2017.09.065. [DOI] [PubMed] [Google Scholar]
- Kandeel M. M., Kamal A. M., Abdelall E. K., Elshemy H. A. Eur. J. Med. Chem. 2013;59:183–193. doi: 10.1016/j.ejmech.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Driowya M., Leclercq J., Verones V., Barczyk A., Lecoeur M., Renault N., Flouquet N., Ghinet A., Berthelot P., Lebegue N. Eur. J. Med. Chem. 2016;115:393–405. doi: 10.1016/j.ejmech.2016.03.056. [DOI] [PubMed] [Google Scholar]
- Alswah M., Bayoumi A. H., Elgamal K., Elmorsy A., Ihmaid S., Ahmed H. E. A. Molecules. 2018;23:48. doi: 10.3390/molecules23010048. [DOI] [PMC free article] [PubMed] [Google Scholar]