

Abstract
Botulinum neurotoxins have remarkable persistence (~weeks to months in cells), outlasting the small molecule inhibitors designed to target them. To address this disconnect, inhibitors bearing two pharmacophores — a zinc binding group and a Cys-reactive warhead — were designed to leverage both affinity and reactivity. A series of 1st generation bifunctional inhibitors was achieved through structure-based inhibitor design. Through X-ray crystallography, engagement of both the catalytic Zn2+ and Cys165 was confirmed. A 2nd generation series improved on affinity by incorporating known reversible inhibitor pharmacophores; the mechanism was confirmed by exhaustive dialysis, mass spectrometry, and in vitro evaluation against the C165S variant. Finally, a 3rd generation inhibitor was shown to have good cellular activity and low toxicity. In addition to our findings, an alternative method of modeling time-dependent inhibition that simplifies assay set-up and allows comparison of inhibition models is discussed.
Graphical Abstract
1. INTRODUCTION.
Botulinum neurotoxins (BoNTs), the etiological agents of botulism, are the most potent known human poisons, with the BoNT/A serotype exhibiting a lethal dose (LD50) of ~1–2 ng/kg of body weight.1–2 Although pharmaceutical preparations of BoNT/A are routinely used at very low doses in medical procedures, including the treatment of facial wrinkles, migraines and strabismus among others, the implications of BoNT/A misuse, either through medical malpractice or as a bioterrorism agent, are dire.1–4 Adverse events, while rare, are associated with extended use and misuse of pharmaceutical BoNTs and include muscle weakness, depressed respiration, dysphagia, and necrotizing fasciitis.5 Because of its remarkable toxicity, ease of procurement and absence of effective post-exposure interventions, the Centers for Disease Control (CDC) classifies BoNT/A as a Tier 1 Category A agent, emphasizing its high risk to public health safety.2, 6–8 In humans, botulism causes long lasting muscle paralysis, which in severe cases may progress to paralysis of the diaphragm, inhibiting normal respiration, causing death. Even with extensive and prolonged supportive therapy including nutritional and digestive support and in severe cases respiratory support, mortality is about 5%. Widespread dissemination of aerosolized or foodborne BoNT/A would overwhelm the country’s healthcare infrastructure, particularly the need for mechanical ventilators as life support. Botulism also occurs naturally, though infrequently. Many U.S. botulism cases in adults result from unintentional exposure to BoNT produced by Clostridium botulinum, the anaerobic, Gram positive, BoNT/A-producing bacteria, in mishandled food.9–10 The most common type of botulism in the US occurs through Clostridium botulinum colonization of infants’ immature intestines (infant botulism).10 Additionally, botulism can be contracted through wound infections, and has found an unexpected connection to the opioid epidemic through association with the use of black-tar heroin.11–13
The pathology of BoNT/A toxicity results from intoxication of peripheral neurons, predominantly motor-neurons. The BoNT/A heavy chain (HC) binds to neuronal cell receptors and facilitates translocation of the BoNT/A light chain (LC) into the neuron terminal.14–15 The LC, a Zn2+-dependent endopeptidase, cleaves soluble SNARE proteins; BoNT/A specifically cleaves SNAP-25.16 Cleavage of SNAP-25 impairs the process of vesicular fusion at neuronal termini, thus inhibiting the release of acetylcholine into neuromuscular junctions.17
Currently, the only available treatment for botulism is an equine-derived heptavalent antitoxin that is severely limited by adverse effects and a restricted window of utility.18 The antibody-based antitoxin targets and neutralizes BoNT/A holotoxin (intact HC + LC) but cannot reach the intracellular LC; thus, effectiveness is lost once BoNT/A LC enters neuronal cells (12–24 h post-exposure), where it persists for several weeks to months.6, 19–22 Unlike immunotherapies, small molecules have the ability to gain entry to the neuronal compartment and inhibit BoNT/A after nerve intoxication. Thus, the discovery of clinically relevant pharmacotherapeutic BoNT/A inhibitors remains of urgent need to deliver the first post-exposure treatment for botulism.
Extensive efforts over the last two decades have produced several in vitro and in vivo inhibitors of BoNT/A LC translocation and enzyme activity.23 The most potent class of BoNT/A inhibitors remains zinc-chelating molecules, particularly those inclusive of a hydroxamic acid pharmacophore.24–33 Zinc-chelating inhibitors bind competitively at the BoNT/A active site, directly preventing the association of the substrate with the enzyme catalytic site. While best-in-class reversible inhibitors have achieved sub-micromolar potency and modest protection against BoNT/A challenge in murine models, no drug candidate has reached clinical trials.
An important consideration for the development of small molecule inhibitors of BoNT/A is the toxin’s inherently long half-life, both in circulation and in nerve cells. In particular, BoNT/A persists intraneuronally on a timescale of several months to >1 year, remaining fully active.19 Comparatively, the majority of pharmacotherapeutic countermeasures being studied for BoNT/A inhibition exhibit half-lives that are a fraction of this timescale (hours to days). The design of reversible BoNT/A inhibitors is limited by this transient mechanism-of-action, even with perfectly optimized binding affinity and drug metabolism and pharmacokinetic (DMPK) properties. Irreversible inhibition using an electrophilic small molecule offers notable benefits for BoNT/A drug design, particularly as the toxin is non-regenerative.19 However, irreversible inhibitors for metalloproteases are rare.24, 34
To date, five types of irreversible BoNT/A inhibitors have been reported (Figure 1 A). These covalent inhibitors putatively target Cys165, a catalytically important residue approximately 7–8 Å from the catalytic zinc. Mutagenesis of Cys165 to serine reduces BoNT/A catalytic activity 50-fold, highlighting the importance of this amino acid.35 The first class of covalent inhibitors included rationally-designed benzylidene cyclopentenediones (1), reported by our group in 2010.36 Although impressively potent in vitro (520–580 M−1s−1), we found that the cyclopentenedione pharmacophore was completely consumed within five minutes when incubated with bovine serum and glutathione. Despite no possibility for in vivo translation, this report demonstrated that irreversible inhibition of BoNT/A is possible. The second disclosure of an irreversible BoNT/A inhibitor leveraged a benzimidazole acrylonitrile pharmacophore, which was identified through high-throughput screening.37 While the lead inhibitor (2) also exhibited preferable in vitro inhibition kinetics (326–979 M−1·s−1) and in silico modelling showed a mechanism of covalent adduct formation at Cys165, no further development of this class has been reported.38 Stura et al. accomplished the first X-ray co-crystal structure of covalent BoNT/A inhibition at Cys165 using well-characterized, small sulfonyl-containing probes, namely (2-aminoethyl) methanethiosulfonate (MTSEA, 3) and (3-aminopropyl) methanethiosulfonate (MTSPA, 4), that specifically react with cysteine residues.35 As a follow up to the previous report, a library of electrophilic fragments was screened and benzoquinone was identified as an active warhead for irreversible inhibition of BoNT/A LC.39 Efforts to elaborate upon the benzoquinone pharmacophore (5), including an attempt to deliver the first bifunctional (zinc-chelating and covalent) inhibitor, significantly compromised covalent inhibition. The selenium-containing compound Ebselen (6), initially investigated in 2010 by Saunders et al. and more recently by the Bogyo group, was shown to irreversibly inhibit BoNT/A by reaction with Cys165, with a reported kinact/Ki of 23,000 M−1s−1.40–41 Ebselen, however, is a well-known PAINS scaffold with extensive activity against a number of biological targets, including the recently identified main protease Mpro of COVID-19.42–43
Figure 1.
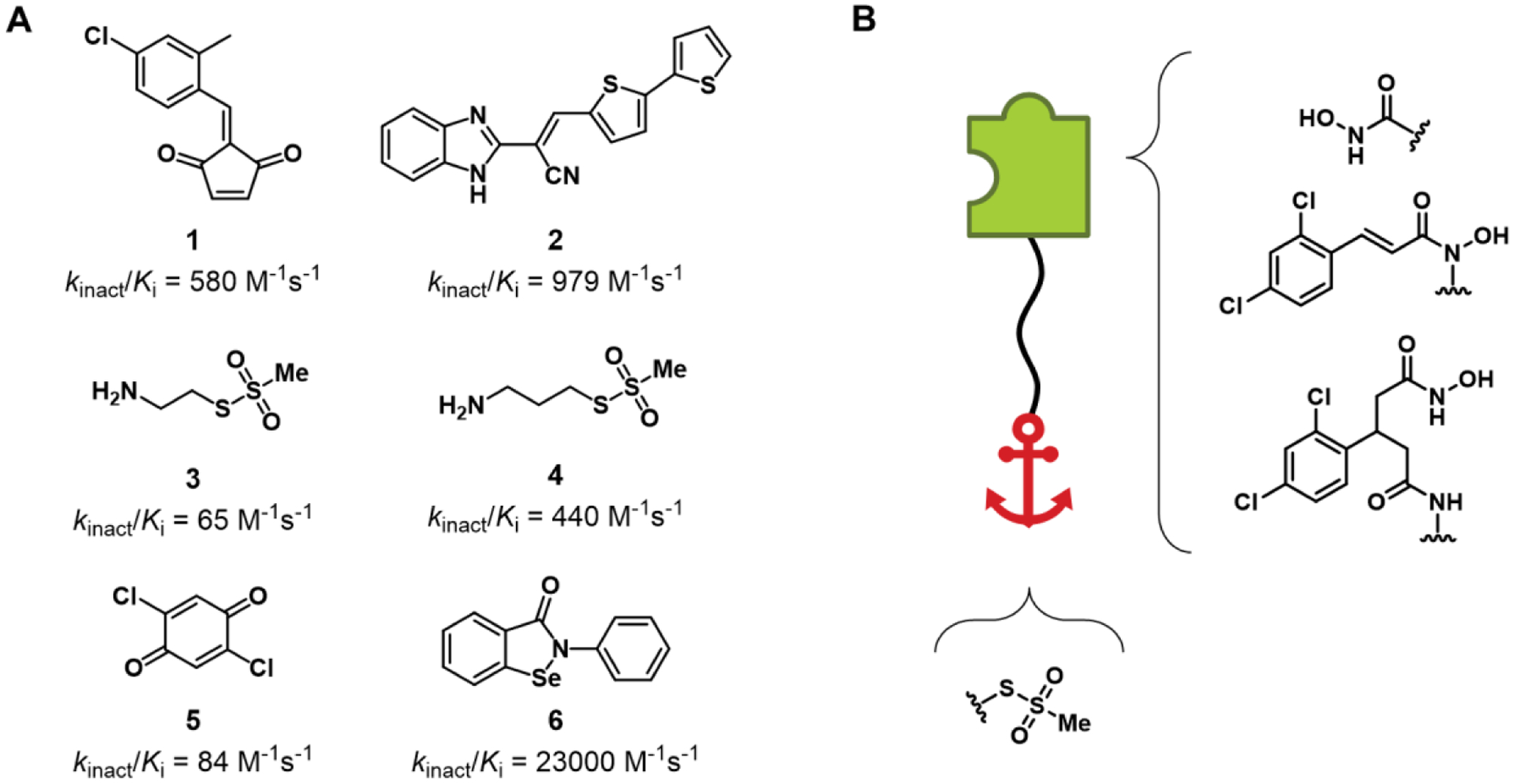
Summary of past efforts to develop irreversible BoNT/A LC inhibitors, and this work. (A) Previously reported covalent inhibitors of BoNT/A LC. kinact/Ki values are presented as reported. (B) Graphical representation of the bifunctional inhibitor strategy, shown with the fragments used in this manuscript. The green puzzle piece represents a reversible zinc-chelating active-site inhibitor, which “catches” the enzyme, and is connected to a red anchor representing the Cys-reactive warhead.
In this effort, we highlight a new strategy to address toxin persistence; namely, the rational design of irreversible inhibitors of BoNT/A LC with dual mechanism-of-action (bifunctional) scaffolds (Figure 1 B). To accomplish this strategy, previously optimized BoNT/A inhibitory scaffolds with non-covalent, zinc-chelating mechanism-of-action were synthetically linked to electrophilic moieties with irreversible inhibitory potential. An important aspect of this strategy is the use of a competitive inhibitor that will inhibit fully to prevent substrate depletion; as will be demonstrated in this work, partial covalent inhibition may not be sufficient. This work provides proof-of-concept that Cys165 and the catalytic zinc of BoNT/A can be simultaneously engaged by a small molecule, offering a strategy for the future development of irreversible, bifunctional BoNT/A inhibitors that irrevocably inhibit toxin, decrease intraneuronal half-life and accelerate nerve cell recovery. Notably, this strategy is somewhat serotype-selective; examination of available X-ray crystal structures of other human-relevant BoNTs B, C, E and F show that only BoNT/F LC possesses Cys residues (C165, C166) proximal to the catalytic Zn.44–47 In addition, BoNTs D, G and X, which have not been conclusively linked to botulism pathology in humans, also lack an active-site proximal Cys.48–50
2. RESULTS & DISCUSSION.
2.1. 1ST GENERATION BIFUNCTIONAL INHIBITORS.
The first bifunctional inhibitors of BoNT/A LC were designed by leveraging privileged pharmacophores; specifically, we hypothesized that the combination of previously optimized zinc-binding groups (ZBG) with cysteine reactive electrophiles would afford an irreversible inhibitor with improved potency and selectivity. As discussed by Bremer et al., this inhibitor design strategy could increase the effective molarity of an electrophilic moiety for Cys165, in turn enhancing kinact.39 Although initial attempts to design a bifunctional inhibitor with a benzoquinone electrophile and an amino acetamide ZBG were mostly unsuccessful, the importance of overcoming BoNT/A LC persistence dictates a deeper investigation into this design strategy. BoNT/A LC endopeptidase activity is dependent on zinc-mediated hydrolysis of SNAP-25 between residues Gln197 and Arg198. The catalytic, tetrahedral zinc is coordinated by a water molecule and a consensus zinc-binding motif of His-Glu-X-X-His, where X represents any amino acid and the His and Glu residues coordinate the zinc metal (Figure 2).51 The most successful inhibitor design for BoNT/A LC, both in vitro and in vivo, has targeted this zinc-binding region and is headlined by hydroxamates, quinolinols, and recently, steroidal 4-aminoquinolines.24–33, 52–56 Despite their well-known liabilities, hydroxamates remain the most potent BoNT/A LC inhibitors. As such, the hydroxamate functional group was selected for our bifunctional inhibitor design. For the electrophile, methanethiosulfonate (MTS) was selected due to its ligand efficiency, notable BoNT/A inhibition kinetics (Figure 1), and available crystallographic structural information (Figure 2 B, C).35
Figure 2.
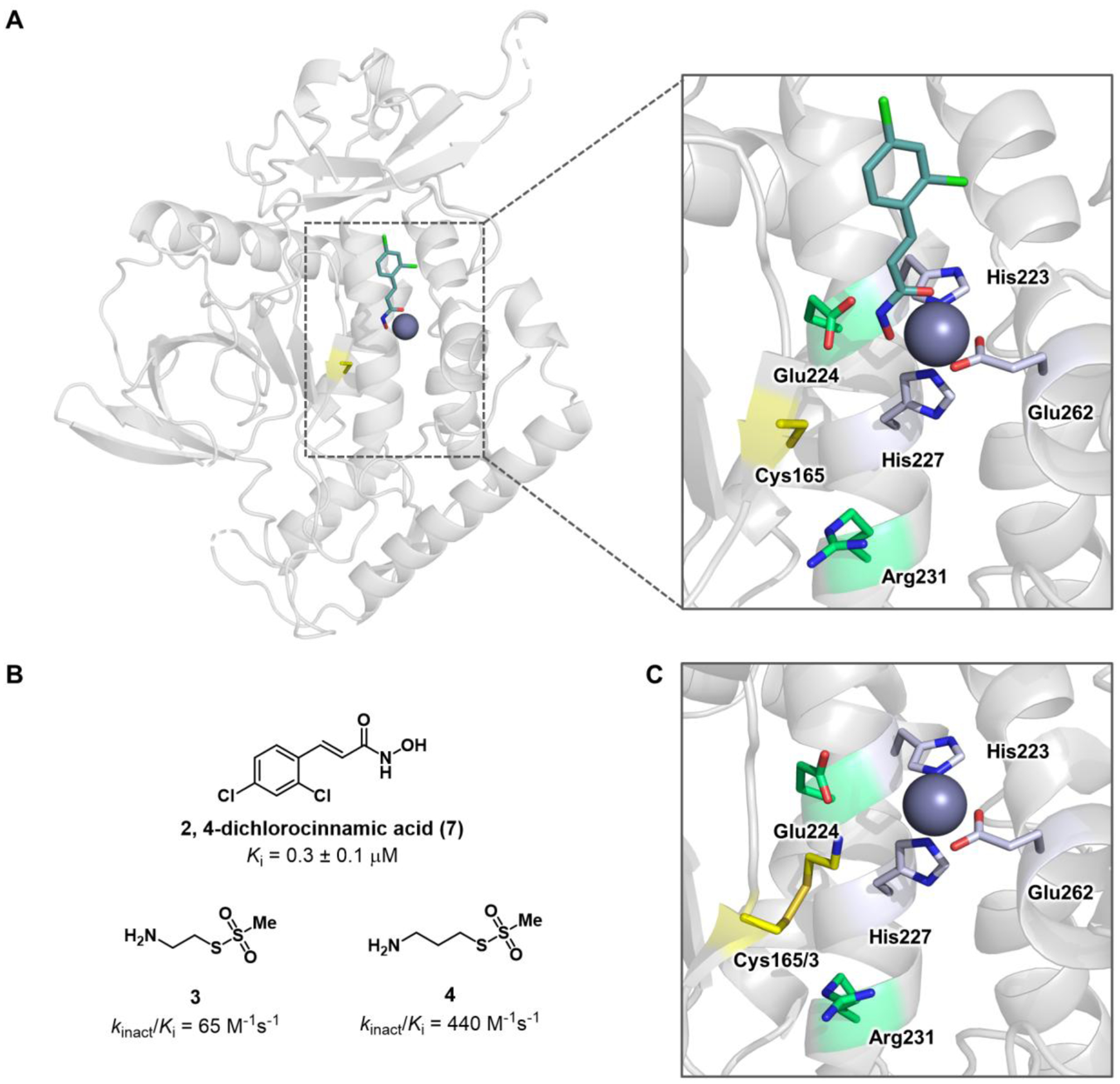
Visualizing the bifunctional design strategy with X-ray co-crystal structures. (A) Rendering of the active site of BoNT/A LC in complex with 2,4-dichlorocinnamic hydroxamic acid (7) (PDB 2IMA). The inhibition of BoNT/A LC by 7 is driven by hydroxamate-mediated Zn2+-chelation and displacement of the active site water. Cys165 is ~6–8 Å from the catalytic Zn2+ metal and is separated by a shallow, unrestricted cleft. (B) Structure and kinetic properties of reversible inhibitor 7, and MTS covalent inhibitors MTSEA (3) and MTSPA (4). (C) X-ray crystal structure of the active site of BoNT/A LC with 3 covalently bound to Cys165 (PDB 4ELC). The zinc ion is depicted as a purple sphere. Residues coordinating zinc are colored light blue. Catalytically important residues are shown in green, and the Cys165 covalent adduct is colored yellow.
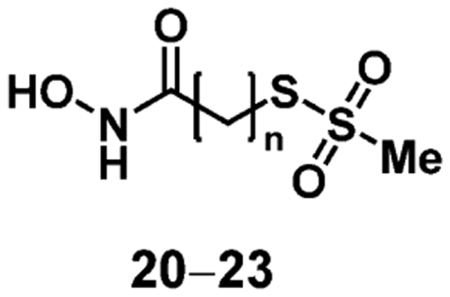
Initial proof-of-concept studies investigated the simplest bifunctional scaffold, comprised of a hydroxamic acid, aliphatic carbon linker and MTS electrophile. Because Cys165 is separated from the catalytic Zn by ~6–8 Å through a shallow, hydrophobic cleft, carbon linkers of lengths 4–7 were synthesized. A linker length of six carbons between the ZBG and electrophile was hypothesized to exhibit superior inhibitor activity because the linker length was closest to 7 Å. Synthesis of the 1st generation bifunctional inhibitor series (Scheme 1) began with bromoaliphatic carboxylic acids (8–11). The bromo-containing acids were subjected to nucleophilic substitution with sodium methanethiosulfonate (NaMTS) at elevated temperatures to give 12–15 in good yields. Subsequent amide coupling with O-tetrahydropyran (THP)-protected hydroxylamine (OTX) afforded 16–19 in good yields. Finally, these were subjected to mild deprotecting conditions to give the final hydroxamate-MTS compounds 20–23.
Scheme 1.
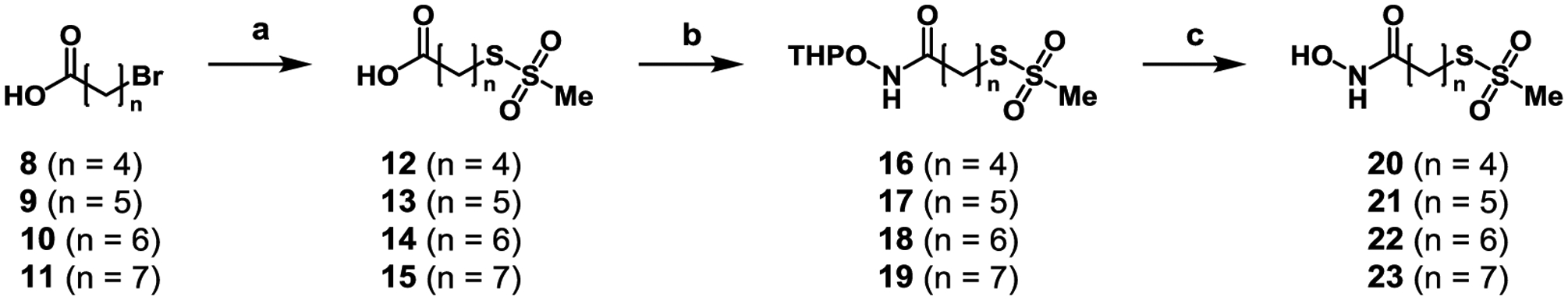
Synthesis of 1st generation bifunctional BoNT/A LC inhibitors. Reagents and Conditions: (a) NaMTS, DMF, 80 °C, 86–92%; (b) OTX, HATU, DIPEA, DMF, 68–86%; (c) PPTS, EtOH, 65 °C, 3–49%.
This series of putative, bifunctional inhibitors was evaluated using a time-dependent assay using the FRET substrate SNAPtide.57 Enzyme and inhibitor were pre-incubated for 0.5–30 min, followed by 100-fold dilution into substrate solutions to initiate the enzymatic reaction, while effectively diluting the inhibitor and halting further covalent inhibition. Quantitative analysis of BoNT/A LC activity was determined as previously described.58 The kinetic parameter kinact/Ki was determined by plotting the first order rate constant (kobs) versus inhibitor concentration, and fitting to a hyperbolic equation.59 Ultimately, all four of the putative bifunctional inhibitors demonstrated irreversible inhibition of BoNT/A LC with kinact/Ki = 1–10 M−1·s−1 (Table 1). As predicted, the bifunctional inhibitor with the six-carbon linker (22) was approximately five to ten-fold more potent than analogues with four (20), five (21) and seven-carbon (23) linkers. Although irreversible inhibition among this series was modest, this 1st generation design demonstrated that irreversible inhibition with a putative bifunctional scaffold was possible.
Table 1.
Kinetic Parameters of 1st Generation Bifunctional BoNT/A Inhibitors
 |
||||
|---|---|---|---|---|
| Compound | n | Ki (μM) | kinact (s−1) | kinact/Ki (M−1s−1) |
| 20 | 4 | 66 | 0.00007 | 1.0 |
| 21 | 5 | 88 | 0.00013 | 1.5 |
| 22 | 6 | 41 | 0.00041 | 10.1 |
| 23 | 7 | 998 | 0.00191 | 1.9 |
Furthermore, we obtained X-ray crystal structures of 20, 21, and 22 in complex with BoNT/A LC at 1.78, 1.90, and 1.92 Å resolution, respectively. Co-crystallization was achieved by simultaneously mixing solutions of the protein, inhibitor, seed solution and crystallization reagents as described in the Experimental Section. Phases were calculated by molecular replacement using the apoprotein (PDB 3BON) as the search model. Overall, there were no large conformational changes of the protein compared to the unliganded protein (rmsd 0.19–0.32 Å). The structures show formation of the respective Cys165-adduct in the BoNT/A LC active site with binding of the hydroxamate moiety to the Zn2+ cofactor, confirming the bifunctional mechanism of action. The X-ray crystal structure for 22 is shown in Figure 3, and structures of complexes with 20 and 21 can be found in Figure S3 of the supplemental text. Notably, the structure of BoNT/A LC/21 revealed that suboptimal linker length leads to a noticeable difference in the positioning of Cys165 in that there is electron density corresponding to partial occupancy of the rotamer typically observed in the unmodified protein (Figure S3B). In this conformation, the Cys165 side chain is rotated ~180° and no covalent attachment can be formed. This observation complements kinetic data showing decreased potency of 21 compared with 22. With these results in-hand, a 2nd generation series with expanded functionality for molecular recognition was explored.
Figure 3.
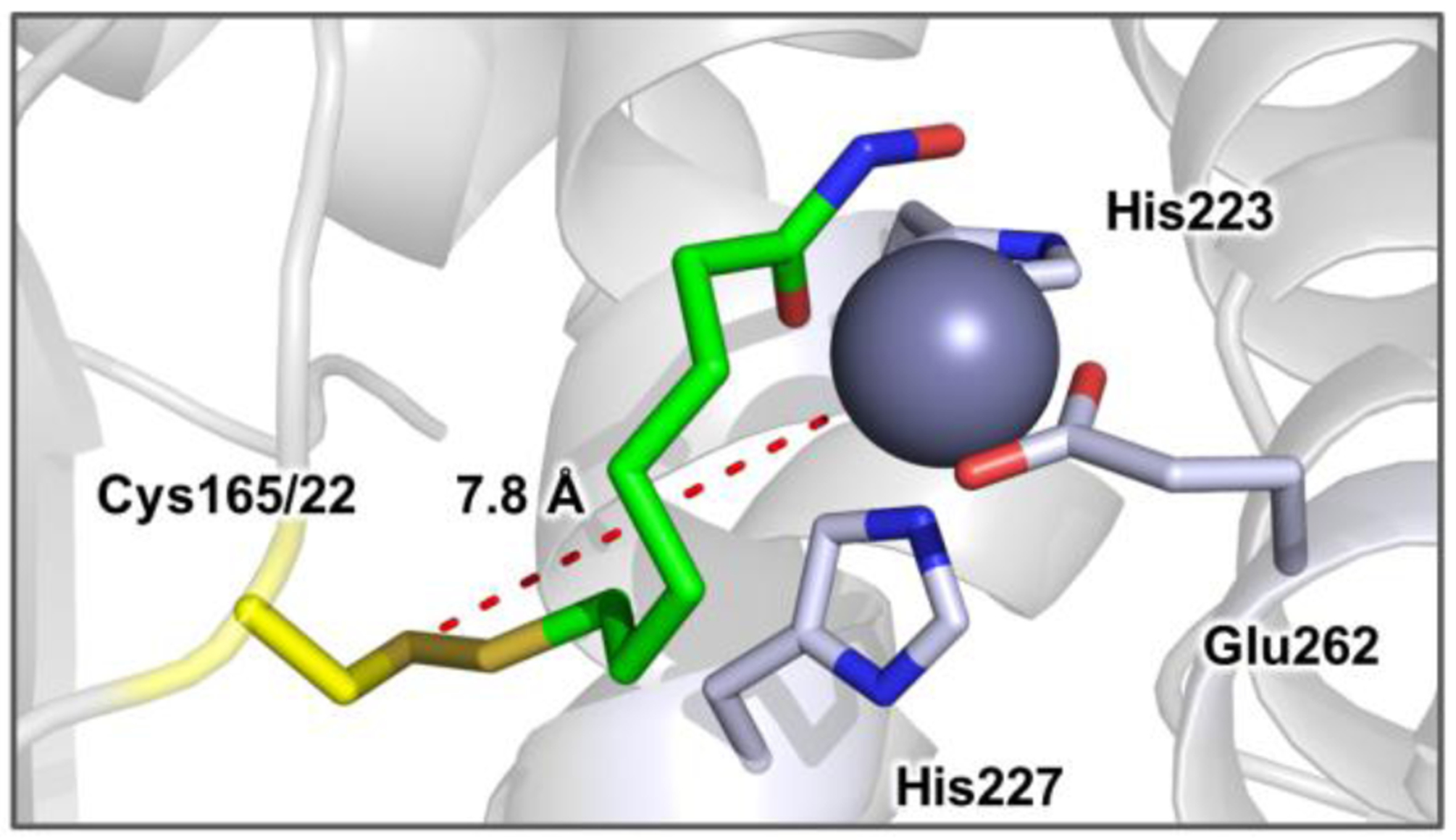
X-ray crystal structure of BoNT/A LC in complex with 22 show as green sticks. The Zn2+ ion is depicted as a purple sphere and residues coordinating Zn2+ are colored light blue. The flexible nature of the active site accommodates simultaneous interaction with the catalytic zinc and Cys165. The distance between the cysteine thiol and Zn2+ is shown as red dashes with an interatomic distance of 7.8 Å.
2.2. A CONTINUOUS ASSAY FOR COVALENT INHIBITION.
Concurrent with our exploration of 1st generation bifunctional inhibitors, we sought to improve the method by which covalent inhibitors are evaluated. The previously described assay can be considered the conventional method of determining covalent kinetic parameters, dependent on back-calculating kinact and Ki from kobs. Although theoretically straightforward, reproducibility issues can arise from the time-sensitive nature of the assay being affected by physical limitations of equipment, reagent sensitivity, and order of operations. Importantly, the data analysis methods are dependent on understanding the mode of inhibition; incorrect assumptions about the inhibition mechanism can lead to the use of incorrect models, and outdated analysis methods can have significant impact on the accuracy of data.60 Our own difficulties with replicating previously reported covalent kinetic parameters using both identical and modified assay protocols motivated us to develop an alternative method.
Rather than measuring activity remaining as a function of reaction time after the “removal” of pre-incubated covalent inhibitors, described vide supra, the reduction in enzyme activity as a function of time in the presence of inhibitor can be directly observed. Equation 1, which describes product formation as a function of time, inhibitor concentration, and inhibitor affinity and rate-constant for inactivation, was obtained by integrating the rate equations for a system containing a covalent inhibitor and a substrate (Figure S5A). Integration of the rate equations of an alternative scenario—a system in which the enzyme is covalently modified by an inhibitor that does not fully prevent substrate binding (partial inhibition, Figure S5B)—gives rise to Equation 2. The ability to distinguish between these two scenarios is crucial to our bifunctional strategy, which links a covalent warhead to an active-site directed inhibitor; lack of warhead reactivity may simply result in a reversible inhibitor, but disruption of the active site inhibitor portion may be harder to detect.
| (Equation 1) |
| (Equation 2) |
Using our current FRET substrate and assay conditions, we determined that the linear range of activity extends up to 50 minutes. This time frame allows us to confidently observe reaction half-lives (t1/2) from 10–30 minutes, which is the expected range for Cys-reactive warheads.61–63 Fitting fluorescent data obtained in kinetics mode to either Equations 1 or 2, constraining total enzyme ([E]T), inhibitor concentration ([I]), substrate concentration ([S]), and substrate KM to defined values, yield values for kinact and Ki. To validate these models, comparison of kinact and Ki values obtained from this method and previously described assay methods is reported, vide infra. In addition to the facile determination of inhibition kinetics, this assay setup allows qualitative differentiation of reversible and covalent inhibitors during the assay by observing the linearity of fluorescence, as demonstrated in Figure 4 .
Figure 4.
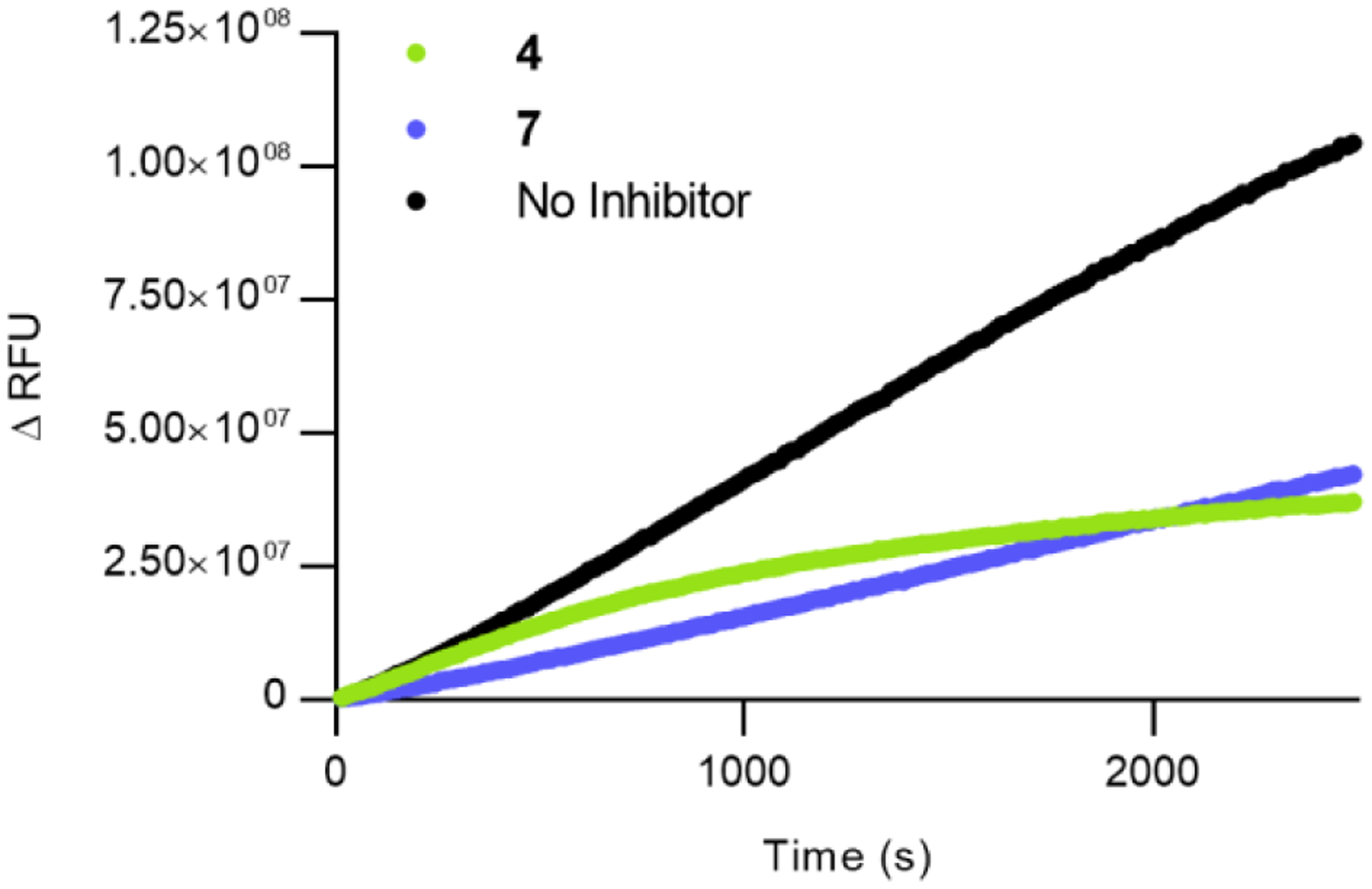
Fluorescence output of the SNAPtide FRET assay. Black represents no inhibitor added, green represents 50 μM of 4, blue represents 0.5 μM of 7. Reversible inhibitor 7 shows a linear increase in fluorescence, whereas covalent inhibitor 4 shows a non-linear response.
Notably, when MTSPA (4) was tested, the reported Ki of 7.7 μM could not be replicated via either the continuous method or the conventional method. A comparison of fits was made between Equations 1 and 2 using the extra sum-of-squares F-test, and it was found that the irreversible partial inhibitor model was preferred (P < 0.0001) (Figure S6). Furthermore, with this model, the inhibition kinetic constants of MTSPA (4) were determined to be kinact = 0.00114 s−1 and Ki = 52 μM. Examination of the crystal structure of the related compound MTSEA (3, Figure 2 C) is consistent with this model; even with an additional carbon, the small probe does not occupy the same area as other reported active site inhibitors. Considering the documented flexibility of the active site, it is possible that substrate-binding in a reduced capacity may be tolerated even in the presence of this covalent inhibitor.29, 58 Therefore, covalent modification alone is not sufficient in depleting enzyme activity—full occupancy of the enzyme active site and covalent modification is required to ensure complete inhibition. This outlines the need to develop more effective bifunctional inhibitors.
2.3. 2ND Generation Bifunctional Inhibitors.
Despite being among the first reported small molecule BoNT/A LC inhibitors, 2,4-dichlorocinnamic hydroxamic acid (7) remains one of the most potent.25, 27, 31 Inhibitor 7 exhibits an IC50 against BoNT/A LC of 0.41 μM, Ki = 300 ± 12 nM, and at ~10 mg/kg rescued 16% (5/31) of mice challenged with ~2.5–5x the intraperitoneal LD50 of BoNT/A in a post-exposure toxicity model.25, 32 In the surviving animals, no signs of botulism were observed and importantly, there was no small molecule-associated toxicity upon treatment.64–65 The mechanism of BoNT/A inhibition by 7 was determined using X-ray crystallography of enzyme-inhibitor complexes (Figure 2).51 Inhibition of protease activity was driven by hydroxamate chelation of the active site zinc. The dichlorophenyl ring of the inhibitor binds within a tight, hydrophobic pocket comprised of Ile161, Phe163, Phe194, and Phe369 in the 370 loop. Notably, the dichlorophenyl ring completely fills this hydrophobic pocket, explaining previous structure-activity relationship (SAR) studies which found that bulky substituents at the 3- and 4-positions translated to significantly reduced potency.25 This hydrophobic pocket is oriented away from Cys165; thus, no modification to the 2,4-dichlorocinnamic functional group is required. Alternatively, the hydroxamate amide nitrogen, which faces the shallow hydrophobic cleft towards Cys165, may be modified with a linked electrophile. The goal of this 2nd generation bifunctional series was to leverage the excellent in vitro and promising in vivo performance of 7 to accomplish a potent and irreversible BoNT/A inhibitor.
Biochemical evaluation of the 1st generation series identified the six-carbon linker (22) as the optimal spacer between the ZBG and the Cys-reactive warhead. Despite the added cinnamoyl moiety, the optimal spacing between the pharmacophores was expected to remain the same. As such, three putative inhibitors were synthesized in the 2nd generation series, with linkers of five to seven carbons. As with the 1st generation series (20–23), the six-carbon linked compound was expected to have significantly enhanced inhibitory potency over its five- and seven-carbon counterpart. Synthesis of the 2nd generation series (Scheme 2) first proceeded with SN2 displacement of bromoalkanes (24–26) using NaMTS to afford 27–29 in moderate yields. Subsequent substitution of the remaining bromine with OTX was carried out; we found that THP-protected intermediates were highly reactive and therefore required immediate coupling to acyl chloride (30). Upon purification, an in situ deprotection was observed affording putative, 2nd generation bifunctional inhibitors 31–33.
Scheme 2.
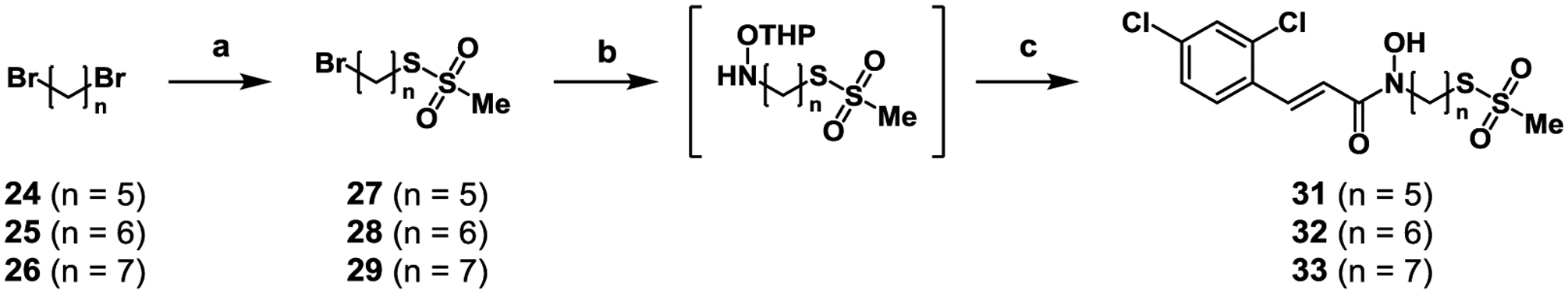
Synthesis of 2nd generation bifunctional BoNT/A inhibitors. Reagents and conditions: (a) NaMTS, DMF, 70 °C, 1 h, 62–66%; (b) OTX, DMF, 48 h; (c) 2,4-dichlorocinnamoyl chloride (30), DCM, 0.1–10% over three steps.
Using the continuous, time dependent SNAPtide assay described vide supra, 31–33 were evaluated for irreversible inhibition (Table 2). Compound 32 was also evaluated in the conventional assay, with observed kinetic parameters of Ki = 2 μM and kinact = 0.00035 s−1, in agreement with the results of the continuous assay. As expected, compound 32 was not only more potent than its five-carbon and seven-carbon analogues (31 and 33, respectively), but also the 1st generation bifunctional inhibitors (20–23), with a kinact/Ki = 418 M−1s−1. The improvement in inhibition kinetics between the 1st and 2nd generation series can be explained by the increased molecular recognition between the 2,4-dichlorocinnamoyl substituent and the small hydrophobic pocket adjacent to the active site, defined by the 370 loop. Although the Ki value of 32 (2.0 μM) is significantly less potent when compared to that of 7 (Ki = 300 ± 12 nM), the irreversible mechanism of inhibition compensates for any potency losses by catalyzing an irreversible event.
Table 2.
Kinetic parameters of 2nd generation bifunctional BoNT/A inhibitors.
 |
||||
|---|---|---|---|---|
| Compound | n | Ki (μM) | kinact (s−1) | kinact/Ki (M−1s−1) |
| 31 | 5 | 8.9 | 0.000297 | 33.5 |
| 32 | 6 | 2.0 | 0.000836 | 418 |
| 33 | 7 | 88 | 0.00013 | 1.5 |
To demonstrate the irreversible mechanism-of-action, and further validate the need for the bifunctional inhibition, we incubated BoNT/A LC with 4, 7, and 32. N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN) was included as a control compound which irreversibly inhibits BoNT/A LC via zinc stripping. Bifunctional inhibitor 32 exhibited a large reduction in BoNT/A LC activity that remained constant after exhaustive dialysis/re-addition of zinc to the enzyme, indicating that the loss of activity was due to permanent occupation of the active site through irreversible covalent modification (Figure 5). By contrast, enzyme incubated with reversible parent compound 7 showed ~50% inhibition before dialysis, which is a result of the 10-fold assay dilution not completely diluting out inhibitor, but recovered ~20% activity upon exhaustive dialysis, indicating a reversible mechanism-of-action. Covalent inhibitor 4 showed a much smaller reduction in activity when compared to 32 before dialysis, remaining constant after dialysis — indicating covalent modification. The difference in BoNT/A LC activity between 32 and 4 is consistent with the hypothesis that full occupation of the active site is needed in order to fully perturb enzyme activity. These results highlight the strength of the bifunctional strategy and underscore the need for active-site engagement for potency, and covalent modification for longevity.
Figure 5.
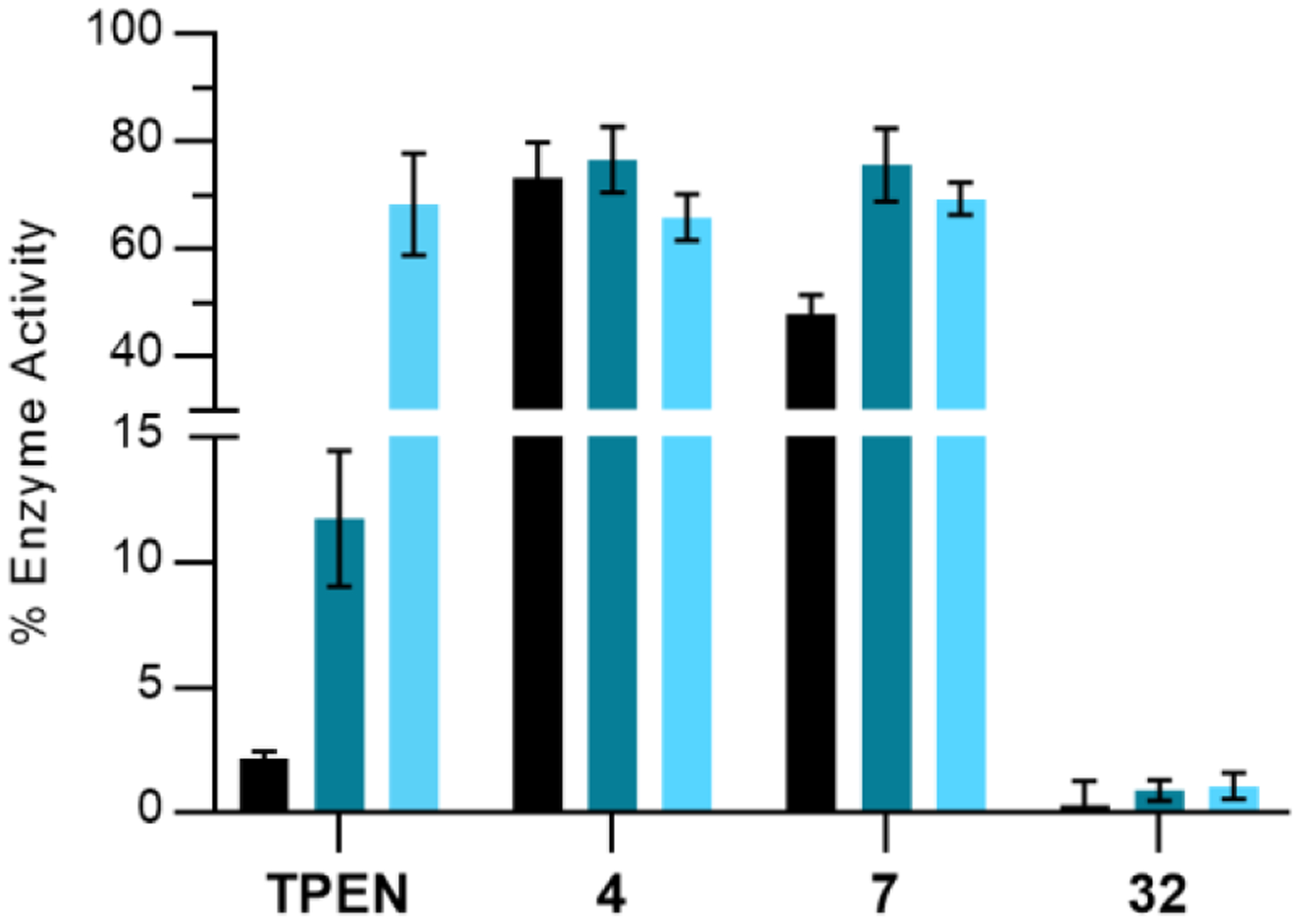
Dialysis of various BoNT inhibitors. Error bars represent standard deviation (n = 3). BoNT/A LC activity pre-dialysis, post-dialysis, and post-dialysis after addition of ZnCl2 is represented by black, dark-teal, and light-blue bars, respectively. Incubation concentrations: BoNT/A LC (10 nM), TPEN (100 μM), 4 (20 μM), 7 (10 μM) and 32 (20 μM).
To further validate the necessity for bifunctional inhibition, we synthesized additional compounds masking either the ZBG or the Cys165 reactive warhead (Scheme 3). N-Boc-6-bromohexylamine (34) was substituted with NaMTS at elevated temperature, followed by deprotection with TFA to furnish 36. This amine was coupled to 2,4-dichlorocinnamic acid to give 37, which no longer possesses a ZBG. Free thiol analogue 38 was obtained through reduction of 32 using NaBH4. In addition to the control compounds, a BoNT/A LC variant, which possesses a serine in place of Cys165, was expressed to further validate the mechanism-of-action of compounds 31–33.
Scheme 3.
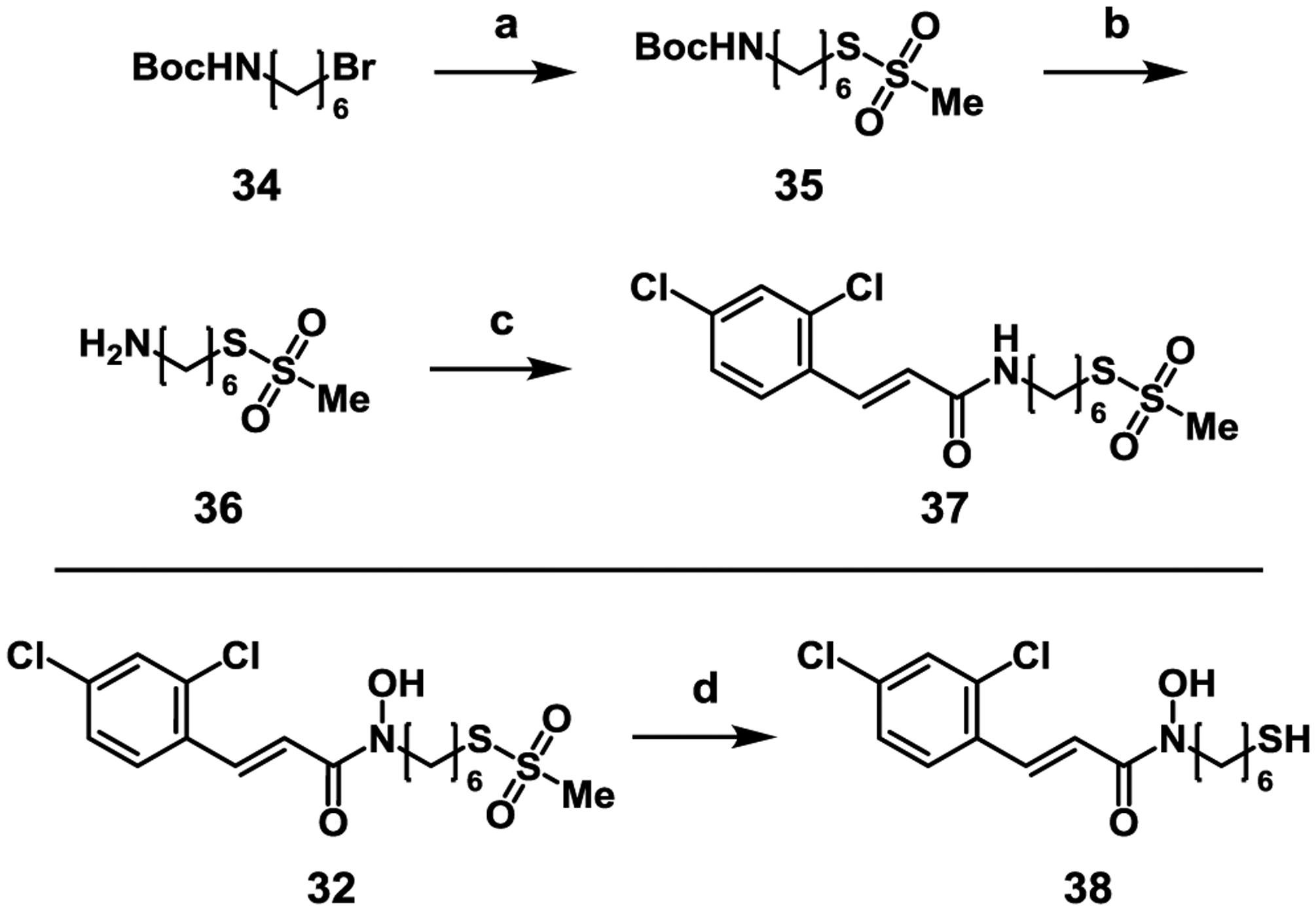
Synthesis of control compounds. Reagents and conditions: a) NaMTS, EtOH, reflux, 16 h, 40%; b) TFA, DCM, r.t., 1 h, quant.; c) 2,4-dichlorocinnamic acid, HATU, DIPEA, DMF, 46%; d) NaBH4, EtOH, 29%.
Wild type (WT) BoNT/A LC and the C165S variant were each incubated with 32, 37, and 38, then analyzed by high-resolution mass spectrometry (HR-MS) to observe changes in mass upon covalent adduct formation (Figure S7). In addition to Cys165, BoNT/A LC possesses a second cysteine, Cys134, that is located away (~34 Å) from the active site. As might be expected, a mass difference corresponding to the addition of two adducts was observed when either 32 or 37 was incubated with BoNT/A LC WT. The C165S variant, however, only showed the addition of one adduct. Notably, when 38 was incubated with either BoNT/A LC WT or C165S, no mass change was observed. The free thiol lacks the reactivity necessary to form a disulfide bond with BoNT/A, even when oriented by the hydroxamic acid group, which increases the thiol’s effective molarity with respect to Cys165.
To further examine the activity of compounds 32 and 38, a cursory screen for inhibition of BoNT/A LC C165S was performed. At 100 μM, 32 inhibited the enzyme by 49%, and 38 inhibited the enzyme by 65% (Figure S8). As expected, no time-dependent irreversible inhibition was observed. The difference in inhibitory potency can likely be attributed to the smaller thiol being more easily accommodated in the active site compared to the much larger MTS group.
Despite significant efforts to obtain an X-ray structure of the complex between 32 and BoNT/A LC, no structural data was obtained. Instead, we performed covalent docking66 to visualize the binding mode of 32. As expected from the shape complementarity and potent binding, the 2,4-dichlorocinnamoyl moiety of 32 in the docked structure overlaid well with that in the previously determined structure of the complex with 7 (Figure 6A). Thus, occupation of the hydrophobic pocket by the 2,4-dichlorocinnamoyl moiety not only results in more favorable binding, but also allows for better structural guidance than the 1st generation bifunctional inhibitors. This additional reversible molecular recognition directs the MTS-moiety towards the shallow, hydrophobic cleft, increasing proximity to Cys165. Notably, upon docking both 31 and 33, we observed that the flexibility of the linker allowed similar accommodation in the binding pocket upon formation of the covalent adduct (Figure S4). Thus, the binding preference of BoNT/A LC for the six-carbon linker over the five-carbon or seven-carbon analogues likely stems from enhanced reactivity resulting from the orientation and fit of the MTS group near Cys165.
Figure 6.
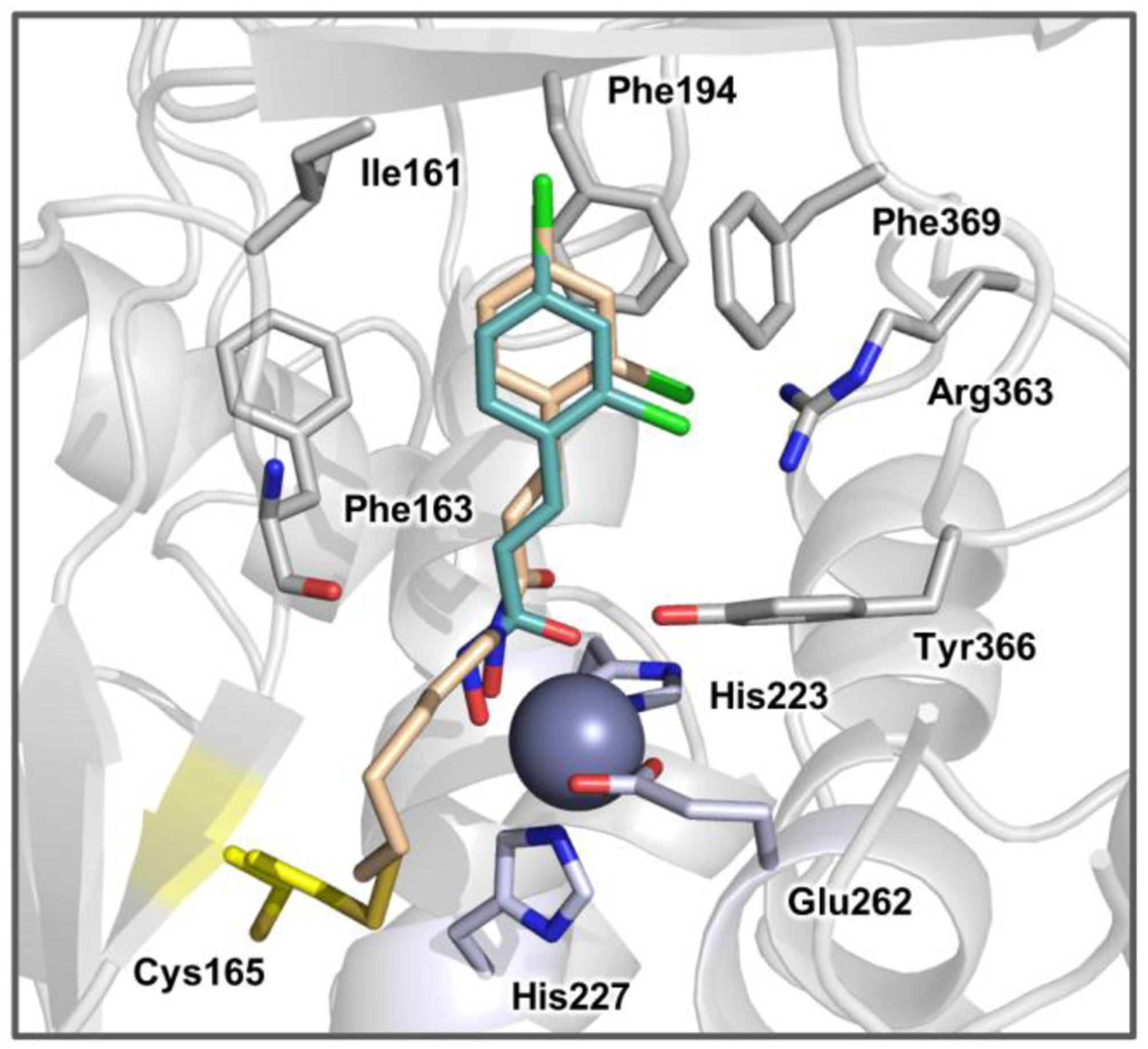
The predicted binding mode resulting from covalent docking of 32 into the BoNT/A LC active site (PDB 2IMA). Covalently bound 32 is depicted as beige sticks and noncovalently bound 7 is depicted in cyan sticks. Residues making interactions with inhibitor are shown as grey sticks. The catalytic zinc metal is rendered as a purple sphere and Zn2+-coordinating residues are colored light blue. The covalent target, Cys165, is colored yellow. The hydrophobic binding pocket is comprised of Ile161, Phe194, and Phe369 (loop 370).
2.4. 3RD GENERATION BIFUNCTIONAL INHIBITORS.
A 3rd generation series of bifunctional inhibitors was pursued with the goal of identifying an agent with improved activity and more drug-like properties.27 The most potent BoNT/A inhibitory hydroxamate is an (R)-hydroxy ethyl analogue (39) of 7 that was designed to extend the inhibitor within the polar space of the enzyme’s active site and incorporate chirality. This best-in-class hydroxamate exhibits a Ki = 0.16 ± 0.02 μM for BoNT/A LC inhibition. Presuming that this hydroxamate would also offer the most potent bifunctional inhibitor, a series of putative leads was synthesized (Scheme 4). For ease, the bifunctional inhibitor was first synthesized as the racemic mixture starting from 2,4-dichlorobenzaldehye (48). The aldehyde was condensed with ethyl acetoacetate (49) in a Knoevenagel condensation, followed by saponification to give dicarboxylic acid 50. Ring closure of 50 to its corresponding anhydride (51) by reflux in acetic anhydride proceeded with excellent yield before ring opening with OTX, giving protected hydroxamate 52. Linkers of three to six carbons in length (4, 36, 46–47) were then installed via standard coupling conditions. Finally, deprotection of the THP protecting group afforded the 3rd generation bifunctional compounds (53–56).
Scheme 4.
Synthesis of 3rd generation bifunctional inhibitors with structural similarity to 39. Reagents and conditions: a) NaMTS, DMF, 70 °C, 57–79%; b) TFA, DCM, r.t., 1 h, quant.; c) piperidine; KOH (20 M), H2O, 78%; d) Ac2O, 120 °C, 2 h, 89%; e) OTX, CHCl3, r.t., 77%; (f) 4 or 36 or 46–47, HATU, DIPEA, DMF, 5–32%.
The series of 3rd generation bifunctional inhibitors was evaluated for time dependent inhibition using the continuous SNAPtide assay (Table 3). All analogues exhibited inferior inhibition kinetics compared to 32 (Table 3). In fact, inhibitors of three, five, and six carbon lengths (53, 55–56) exhibited inhibition kinetics reflecting the 1st generation of bifunctional inhibitors (20–23. Inhibitor 54, with a four-carbon linker, showed a slightly improved kinact/Ki (19.38 M−1ˑs−1) but remained lesser than the 2nd generation inhibitors.
Table 3.
Kinetic parameters of 3rd generation bifunctional inhibitors.
 |
||||
|---|---|---|---|---|
| Compound | n | Ki (μM) | kinact (s−1) | kinact/Ki (M−1s−1) |
| 53 | 3 | 17 | 0.000130 | 7.6 |
| 54 | 4 | 32 | 0.00062 | 19.38 |
| 55 | 5 | 48 | 0.00008 | 1.67 |
| 56 | 6 | 38 | 0.00006 | 1.55 |
We confirmed the mechanism of time-dependent inhibition by incubating 53 with the BoNT/A LC C165S variant. We found that 53 only inhibited 32% of the enzyme’s activity at 100 μM and that the inhibition did not appear to be time dependent in nature (Figure S8). This result indicates that covalent inhibition is dependent on Cys165. HR-MS analysis of the WT and C165S variant incubated with 53 further supports this conclusion; an increase in mass consistent with two adducts was observed in the WT, whereas an increase in mass of only one adduct was observed with the C165S variant (Figure S7).
In order to further explore the requirements for successful covalent inhibition, we synthesized control compounds 57 and 59, which lack the hydroxamic acid ZBG and the MTS group, respectively (Scheme 5). Compound 57 was obtained by the reaction of MTSPA (4) with anhydride 51, while compound 59 was obtained by coupling 52 with 1-aminopentane. Both compounds were tested in the continuous SNAPtide enzyme assay, and no inhibition by 57 was observed up to a concentration of 500 μM; 59 showed only reversible inhibition (IC50 = 5.7 ± 0.6 μM, Figure S9). We concluded from this result that the Cys-reactive MTS group itself is not capable of mediating covalent inhibition; in other words, the hydroxamic acid ZBG is likely required to position the electrophile in proximity to Cys165 for successful adduct formation.
Scheme 5.
Synthesis of 3rd generation control compounds 57 and 59. Reagents and conditions: a) 4, CHCl3, 12%; b) 1-aminopentane, HATU, DMF, r.t., 1 h, 76%; c) PPTS, EtOH, 60 °C, 16 h, 42%.
X-ray crystal structures of lead 53 and control compound 59 were resolved to 2.5 and 1.67 Å resolution, respectively (Figures S1 and S2). Inhibitor complexes were co-crystallized as described for complexes 20–22. Unexpectedly, electron density corresponding to the zinc atom was not present in the active site of the BoNT/A LC/53 X-ray crystal structure, although zinc was present in the crystallization milieu. As a result, the inhibitor ZBG is oriented away from the Zn2+ binding motif (HEXXH). Notably, this structure shows the covalent adduct of 53 reacted with Cys165. This unique orientation in the crystal structure is consistent with the inhibition kinetics, showing decreased potency of the 3rd generation inhibitors compared to the 2nd generation. In addition, the X-ray crystal structure of BoNT/A LC/59 shows that the alkyl chain of 59 fits into a hydrophobic pocket resulting in a rotation of Phe194; this hydrophobic pocket may be employed to improve inhibitor affinity if future iterations of this scaffold are pursued. A more detailed discussion of the X-Ray crystal structures of BoNT/A LC complexed with 53 or 59 is included in the Supporting Information. Together, these structures demonstrate the plasticity of the active site and is consistent with the induced-fit model of specificity,29 providing a structural framework for the design of future, highly specific inhibitors of BoNT/A LC.
Optimization of reversible binding interactions while maintaining an optimal vector/placement of the Cys-warhead is challenging and current efforts are focused on improving both these aspects in unison. The barriers to creating optimal binding interactions and Cys-warhead vector clearly impact affinity and reactivity, the latter of which is sensitive to warhead placement, as illustrated with the 1st and 2nd generation inhibitors.
2.5. CELLULAR EVALUATION OF BIFUNCTIONAL INHIBITORS
The promising in vitro data of the 2nd and 3rd series prompted evaluation for BoNT/A LC inhibition in cells. In this assay, human induced pluripotent stem cell (hiPSC) derived matured neurons were intoxicated with BoNT/A1 before inhibitor was added. After incubation, hiPSC derived neurons were lysed and analyzed by Western blot using an anti-SNAP-25 antibody. Absence of the cleaved substrate band demonstrates successful BoNT/A inhibition.
Bifunctional 2nd generation compounds 31 and 32 were found to have IC50 values of 59 ± 15 μM and 34 ± 4 μM, respectively, and 3rd generation compound 53 was found to have an IC50 of 35 ± 6 μM (Figure 7). Interestingly, none of the other analogues in the 3rd generation series demonstrated inhibitory activity in cells (Figure S12). Despite the difference in inhibition constants, 32 and 53 are equipotent in cells, and nearly two-fold more potent than 31. Although 53 may have lower affinity and reactivity against the enzyme (Table 3), its cellular activity compensates for its shortcomings. Overall, cellular activity of these proof-of-concept inhibitors is modest; in the past, we have found that hydroxamates may perform better when dosed as pro-drugs.28
Figure 7.
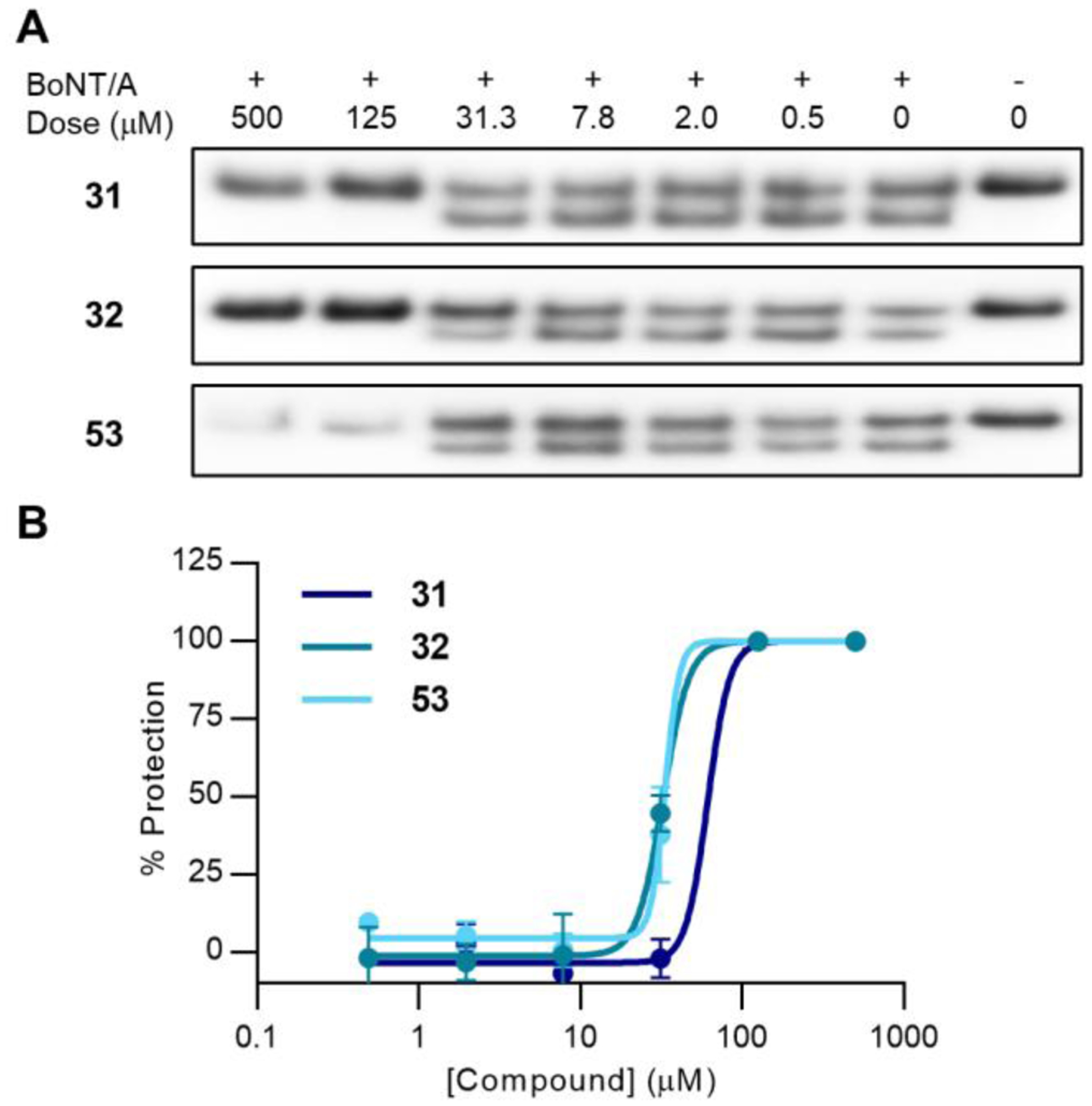
Cellular evaluation of 2nd and 3rd generation inhibitors. A) Western blot evaluation of 31, 32, and 53 against post-exposure BoNT/A1 in hiPSCs. BoNT/A cleavage of the SNAP-25 substrate is measured by quantifying the lower molecular weight band corresponding to the SNAP-25 breakdown product. Cell rounding as a result of toxicity was observed at higher concentrations, resulting in lower SNAP-25 signal. Full immunoblots are presented in Figure S12. B) Dose-response curves of 31, 32, 53 (n = 3).
Following the observation of signs of toxicity at high concentrations in the hiPSC assay, the effect of the compounds on cellular proliferation was tested (Figure 8A). Compound 59 was included to probe for toxicity stemming from the MTS group. Notably, the 2nd generation compounds inhibited HEK293 proliferation more than 3rd generation compounds 53 and 59, which did not exhibit cellular toxicity up to a concentration of 100 μM. Compound 32 inhibited cells with an IC50 of 60 ± 5 μM; compound 31 did not inhibit cells to the extent that an IC50 value could be calculated. Moreover, hiPSC derived neurons treated with high concentrations of 2nd generation inhibitors did not exhibit cell rounding and lower intact SNAP-25 signal, but 3rd generation inhibitors did.
Figure 8.
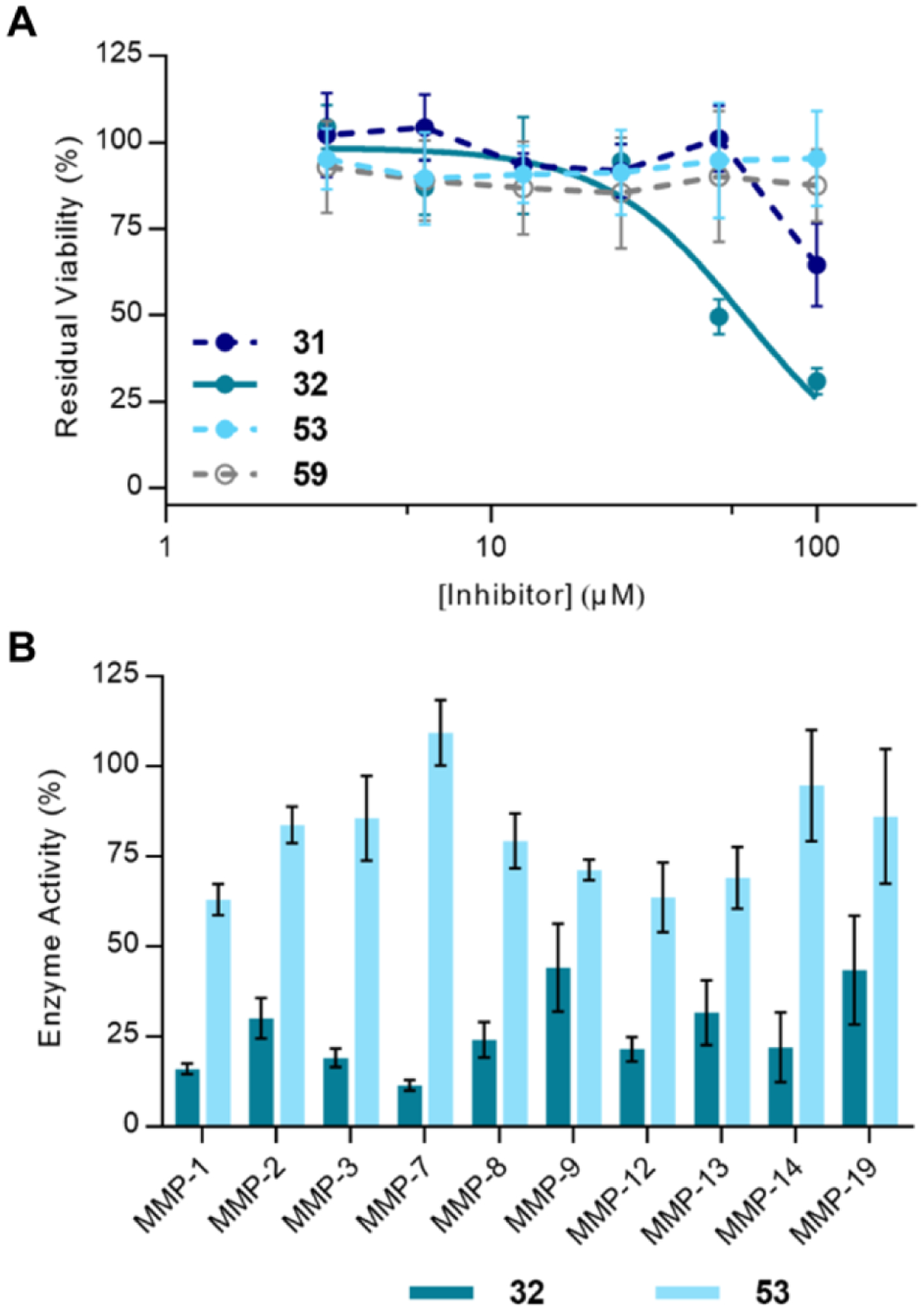
Cellular toxicity and MMP specificity of 2nd and 3rd generation compounds. A) Cellular viability of HEK293 cells after incubation with 31, 32 (IC50 = 60 ± 5 μM, n = 3), 53, and 59. Results for compounds 54 and 55 can be found in the supplemental text, Figure S13. B) MMP panel screen of 32 and 53, (n = 3). Compounds were screened at 25 μM.
Because most potent BoNT/A LC inhibitors employ a zinc-chelating motif, it is also crucial to ensure that the inhibitor has sufficient specificity to avoid off-target effects on matrix metalloproteinases (MMPs). Hydroxamates in particular tend to display broad-spectrum MMP inhibitory activity.67 Thus, 32 and 53 were screened against a panel of MMPs at a concentration of 25 μM (Figure 8B). Importantly, no covalent inhibition was observed for either inhibitor on any MMP, likely due to the absence of a Cys residue proximal to the active site Zn. Due to the covalent nature of BoNT/A LC inhibition by the bifunctional inhibitors, direct comparisons about potency are difficult to make. However, it is clear that overall, 53 inhibits MMPs significantly less than 32 ((Figure 8B, S10). Although 53 has poorer enzyme inhibition kinetics than 32, the two inhibitors have equipotent cellular activity (Figure 7); use of the 3rd generation inhibitor scaffold may limit off-target effects. MMP selectivity can also be dependent on the ZBG, thus a possible avenue for improving selectivity in future bifunctional inhibitors would be to incorporate different ZBGs.68–69 Importantly, the bifunctional inhibitors allow for covalent inhibition of only BoNT/A LC, which is non-regenerative in motor neurons. Thus, long-term off-target side effects are not expected as the need for long-term dosing would not be required.
CONCLUSION AND FUTURE OUTLOOK.
The work described herein reports the first bifunctional, irreversible inhibitors of BoNT/A LC, which incorporate a two-pharmacophore design of a ZBG and Cys165-targeting electrophile. These inhibitor series provide proof-of-concept that the BoNT/A active site can be bridged between the catalytic Zn, Zn2+-binding motif, 370 loop and the catalytically-important Cys165 residue. In addition to defining the kinetic parameters of irreversible, bifunctional BoNT/A inhibitors, studies were performed to validate the dual mechanism of inhibition, and demonstrate the necessity of this strategy. A continuous method of obtaining irreversible kinetic inhibition constants was developed and validated in order to minimize errors arising from data analysis or experimental procedure. The best-in-class compounds were also found to exhibit cell-based BoNT/A inhibitory activity. Additionally, five novel BoNT/A LC X-ray co-crystal structures were reported which provide valuable insight into the mechanism of this strategy. This work provides a clear foundation for further development of this inhibitor design strategy to accomplish more potent and “drug-like” scaffolds. Our current endeavors are focused on leveraging structural information to improve affinity and reactivity, focusing on improving reversible binding interactions and warhead variation, and vector/orientation. Following this proof-of-concept work, we will examine the effect of chirality on 3rd generation inhibitors, incorporation of other reversible inhibitor scaffolds, and implementation of more drug-like electrophiles en route to a post-exposure BoNT/A pharmacotherapy that can match toxin persistence.
EXPERIMENTAL DATA
Biology
Enzyme Expression
Standard Protocols
Media and appropriate equipment were sterilized in an SG-120 autoclave (AMSCO Scientific) at 121 °C for 45 min. Sterile conditions were maintained using a bench-top Bunsen burner. Bacterial cultures were incubated in an Innova® 44 incubator (New Brunswick Scientific). Cell mixtures and protein samples were stored on ice throughout all steps of purification. Sonication of cell mixtures was carried out on ice using a Branson Digital 450 cell disruptor. Spectrophotometric readings were measured using a NanoDrop 1000 spectrophotometer (Thermo Fischer Scientific). Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using a Novex Bolt Mini Gel Tank using Bolt™ 4–12% Bis-Tris Plus (Invitrogen) polyacrylamide gels that were imaged using a Fluorchem™ 8900 apparatus. Mass spectrometry (MS) analysis of proteins was carried out using an Agilent 6230 TOF LC/MS mass spectrometer with a Dual AJS ESI ion source. Analytical grade reagents were supplied from commercial sources and used without further purification.
Media and buffers were made in-house unless otherwise stated. Buffers used in purification of protein were filtered through a vacuum-driven, 0.22 μm membrane (Corning).
BoNT/A LC wild type and C165S variant expression
The BoNT A/LC WT and C165S variant constructs were prepared as described previously.70 A glycerol stock of BL-21 RIL (DE3) cells (15 μL) containing the desired construct was used to inoculate lysogeny broth (LB) (5 mL) containing carbenicillin (CB) and chloramphenicol (CAM) (50 μg/ mL). The mini culture was incubated overnight at 37 °C. LB (2 × 100 mL), containing CB and CAM at the same concentrations as before, was inoculated with the overnight culture (1 mL) and the cells grown at 30 °C until an OD600 of ~0.8 was reached (~8 h). Protein expression was induced using isopropyl ß-D-1-thiogalactopyranoside (IPTG) ([ ]f = 0.75 mM) and the culture incubated overnight at 16 °C. Cells were harvested by centrifugation at 7700 rpm for 10 min and the supernatant discarded. Cell pellet was stored at -80 °C until needed.
BoNT/A LC wild type and C165S Variant purification
The cell pellet was resuspended in 4 mL of lysis buffer (20 mM tris-(hydroxymethyl)-aminomethane HCl (Tris), 500 mM NaCl, 10 mM imidazole, and 0.05% Tween-20, pH 7.4) with freshly prepared lysozyme (500 μL, [ ]f = 1 mM) and three EDTA-free protease inhibitor tablets (Sigma). The cell mixture was sonicated and then DNAseI ([ ]f = 2.5 μg/ mL) was added and incubated for 10 min. The cell mixture was centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatant was carefully decanted and loaded directly onto pre-equilibrated Ni-NTA spin columns (Qiagen, 2 × 500 μL loads per column). The columns were washed successively with 600 μL of 20, 40, and 60 mM imidazole solutions prepared in 20 mM Tris pH 7.9, 500 mM NaCl, 0.05% Tween-20, before elution with 3 × 300 μL of 250 mM imidazole prepared in the same buffer. Purity of eluted fractions were analyzed by SDS-PAGE (InstantBlue-Coomassie stain) and pure fractions pooled and transferred to 10 kDa molecular weight cut-off (MWCO) 0.5–3.0 mL dialysis cells (3 mL per cell) (Thermo Fisher Scientific) and dialyzed in phosphate-buffered saline (PBS) (pH 7.4), 10% glycerol overnight, changing buffer after the initial 4 h. The sample was further dialyzed for 8 hours in 20 mM potassium 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), 0.01% Triton X-100 (added fresh), changing buffer after 4 h. Protein samples were centrifuged to remove precipitate and the supernatant/precipitate analyzed for purity by SDS-PAGE (Figure S11) and the concentration determined using a NanoDrop and a calibration curve of BoNT/A LC.
Protein expression for crystallography
A gene containing BoNT/A LC (1–425; 13 amino acids removed from the C-terminus)71 was cloned into the pET-15(+) vector, containing a C-terminal 6xHis tag, as previously described.29–30 This plasmid was transformed into E. coli strain BL21(DE3) competent cells (New England Biolabs), plated and incubated overnight at 37 °C. Single colonies were picked and used to inoculate LB containing CB (100 μg/mL) and were grown at 37 °C until OD600 = 0.6–0.8 was reached. Protein expression was induced using IPTG ([ ]f = 1 mM) and the culture incubated overnight at 16 °C. Cells were harvested by centrifugation at 5,000 g for 15 min and the supernatant discarded. Cell pellet was stored at -80 °C until needed.
Protein purification for crystallography
The cell pellet was resuspended in 50–60 mL of lysis buffer (25 mM Tris pH 8.0, 250 mM NaCl and 25 mM imidazole) along with 600 μL of DNase I (10 mg/mL) and one half of EDTA-free protease inhibitor tablet (Sigma) and stirred gently at r.t. for 1 h. Cells were lysed using microfluidization at 18,000 psi and debris was removed using ultracentrifugation at 100,000 g for 35 min. The supernatant was loaded onto a Ni Sepharose High Performance column (GE Healthcare Life Sciences) using lysis buffer to bind and a linear gradient of elution buffer (25 mM Tris pH 8.0, 250 mM NaCl and 250 mM imidazole) to elute the enzyme. Alternatively, for some purifications, a HiTrap affinity column charged with Zn2+ was used in place of the Ni sepharose column, although the overall yield and protein purity did not significantly differ between the two methods. BoNT/A LC was further purified by a second round of affinity purification or using size-exclusion chromatography (HiPrep 26/60 Sephacryl S-100 HR, GE Healthcare Life Sciences). The BoNT/A LC-containing fractions were confirmed using SDS-PAGE (Coomassie), pooled and dialyzed overnight in buffer containing 50 mM HEPES pH 7.5, 200 mM NaCl and 2 mM EDTA. The enzyme was concentrated using 10 kD Amicon Ultra centrifugal filter device; 5% glycerol was added to 200 μL aliquots, which were flash-frozen in liquid nitrogen and stored at -80 °C. Protein yield was approximately 4–5 mg/g wet cell pellet.
Crystallization Studies
Co-crystallization of BoNT/A LC with Inhibitors
Initial crystallization conditions were determined using high-throughput screens (Hampton Research PEG/Ion) as previously described.72 Crystals of BoNT/A LC were grown using the vapor-diffusion method with hanging-drop geometry at a 1:1 ratio of protein (6–8 mg/ mL) and well solution. The protein solution was reconstituted in buffer containing 50 mM HEPES, pH 7.5, 200 mM NaCl, 2 mM EDTA, and 5% glycerol. The condition comprising 0.2 M ammonium tartrate dibasic (pH 7.0) and 20% PEG 3,350 produced square, plate-like crystals. Crystal morphology was improved by microseeding using the Seed Bead (Hampton Research) and addition of seed solution, diluted 1:104 from the seed stock, to the crystallization drop. This resulted in three-dimensional rectangular prism-shaped crystals of approximately 0.05 × 0.01 × 0.01 mm final dimensions, although crystal size varied significantly with the inhibitor used in co-crystallization. All inhibitor-bound structures were determined using both co-crystallization and soaking with individual components detailed in Table S1. All inhibitor solutions were provided as 10–50 mM DMSO stocks and were diluted to the working concentrations using the solvents listed in Table S1 and protein buffer forming the inhibitor solution. All concentrations listed in Table S1 are working concentrations prior to addition to the crystallization drop. The crystallization drop consisted of 1 μL of protein, 1 μL reservoir, and 0.5 μL each of additives (seed solution, inhibitor solution, and metal), so that the final drop volume was 3.5 μL. Reservoir solution volume was 300 μL and consisted of 0.3–0.45 M ammonium tartrate dibasic (pH 7.0) and 15–25% PEG 3,350. Crystals were transferred to a solution of 12–20% glycerol in mother liquor and subsequently soaked with inhibitor (concentration and solvent as listed in Table S1) for 1–2 h prior to flash-freezing in liquid nitrogen at 100 K.
Data collection and structure refinement
For the BoNT/A LC/20, BoNT/A LC/21, and BoNT/A LC/22 complex structures, diffraction data were collected on beamline 17-ID-1 (AMX) at the Brookhaven National Laboratory at the National Synchrotron Light Source II (Upton, NY) using an Eiger 9M detector. For the BoNT/A LC/53 structure, data were collected on the Bruker X8 Dual Incoatec Microsource at the X-ray Diffraction Facility at the Massachusetts Institute of Technology (Cambridge, MA) using an APEX2 area detector. For the BoNT/A LC/59 structure, data were collected on beamline 9–2 at the Stanford Synchrotron Light Source at the SLAC National Accelerator Laboratory (Menlo Park, CA) using a Dectris Pilatus 6M PAD detector. All diffraction data were collected at 100 K under nitrogen gas flow and were processed, integrated and scaled using HKL2000,73 unless otherwise indicated. All datasets collected belonged to the space group P21 and contained one molecule in the asymmetric unit. The unit-cell dimensions, along with data collection and refinement statistics for all BoNT/A LC inhibitor complexes are detailed in Table S2. Initial phases were calculated using a previously-determined structure of BoNT/A LC (PDB 3BON) with all ligands and waters removed as a search model for molecular replacement in PHENIX Phaser-MR.74 Multiple rounds of refinement in PHENIX Refine75 using individual B factors, TLS parameters and addition of waters, along with manual model building were used to general the final structures. All inhibitors were built in manually using Coot76 and validated using omit F0–Fc and Polder electron density maps in PHENIX.77 All images were generated in PyMOL.78
Covalent docking studies
Molecular modeling was performed with modules from the Schrödinger Small Molecule Drug Discovery Suite (Maestro), release 2018–3, using the OPLS3 force field for parameterization.79 X-ray co-crystal structures were imported from the PDB80 and prepared using the Protein preparation wizard using default settings and ligands were prepared using LigPrep.81 Covalent docking was carried out in confined receptor grids built 30 Å around the receptor-bound ligand: (7 for PDB 2IMA) and simulated using CovDock66 (Reaction type: Disulfide formation to Cys165) using default settings. As the CovDock module does not recognize the MTS-warhead for disulfide formation, the results were computed from the corresponding thiol. Figures were generated in PyMOL.78
BoNT/A LC Activity Assays
Reversible inhibition assay
BoNT/A LC (20 μL of 25 nM, final assay concentration = 10 nM) was added in 40 mM HEPES, 0.01% Triton X-100, pH 7.4 in a black half-volume 96-well plate (Corning, cat. #3964). 5 μL of 20× inhibitor stocks of varying concentrations were added, dissolved in assay buffer and DMSO, resulting in a final concentration of 1% DMSO. A negative control sample containing enzyme with no inhibitor in 1% DMSO was included. The assay mixtures were allowed to incubate at r.t. for 30 min, before 25 μL of SNAPtide substrate flP6 (List Labs) dissolved in assay buffer was added to give a final substrate concentration of 4 μM, initiating the reaction. The assay plate was immediately inserted into a Spectramax i3x plate reader (Molecular Devices), and the fluorescence monitored in kinetics mode for 20 min, λex = 490 nm, λem = 523 nm, cutoff = 495 nm. Initial rate of enzyme activity (RFU/s) was obtained by calculating the slope of the fluorescence versus time. Initial rates of inhibitor-samples were then normalized to the initial rate of the negative control sample to give relative enzyme activity expressed as a percentage of full activity. Data was analyzed in Excel, then GraphPad Prism 7. Dose response curves were fitted with four-parameters least squares fit.
Conventional covalent activity assay
A low-binding 96-well plate (Corning, cat. #3964) was plated with 22.5 μL of inhibitor stocks dissolved in assay buffer (1.11×final concentration). Negative control wells containing only assay buffer were included. BoNT/A LC (2.5 μL of 5000 nm, assay concentration = 500 nM) was added at the following timepoints: -30 min, -15 min, -10 min, -5 min, -1 min, -0.5 min. In between time points, assay plate was incubated at 37 °C. At time = 0, the inhibitor was diluted out by pipetting 10 μL of each assay well quickly into a prepared plate containing 90 μL of assay buffer, then withdrawing 10 μL per well from that plate and diluting it into 90 μL of SNAPtide flP6 substrate, giving final concentrations of 1% inhibitor, 5 nM BoNT/A LC, 4 μM substrate. The fluorescence was then monitored as described previously. Data analysis was performed in Excel and GraphPad Prism 7. First, the relative activity was determined as described previously, and plotted versus incubation time for that given inhibitor concentration. These data were fit with an exponential decay to determine kobs for each inhibitor concentration. Plotting kobs as a function of inhibitor concentration allows the following equation to be fit, thus determining kinact and Ki:
Continuous covalent activity assay
Assay was performed using the same concentrations and setup as the reversible inhibition assay, with the following alterations: 1) substrate and inhibitor were plated first, then enzyme was added to initiate the reaction with no incubation period; 2) the fluorescence was monitored for at least 40 min. Data analysis was performed in GraphPad Prism 7. Background fluorescence of the substrate was subtracted from each time point and fluorescence (RFU) was plotted versus time (s). The data was then fit to custom equations corresponding to Equation 1 or 2, constraining substrate concentration [S], Km, and inhibitor concentration [I] to defined values. If necessary, comparisons between the two models were made using the extra-sum-of-squares comparison feature in GraphPad Prism 7.
Exhaustive dialysis of BoNT/A LC
Inhibitors were incubated with 20 nM BoNT/A LC for 30 min at a concentration of 10 μM in 40 mM HEPES, pH 7.4, 1% DMSO for 30 min at r.t. Control samples containing no inhibitor (negative control) and 100 mM TPEN (positive control) were also prepared. After incubation, an aliquot of enzyme was tested in the BoNT/A LC enzyme activity assay as described above. 100 μL remainder of the samples were injected into 0.1 mL Slide-a-Lyzer MINI dialysis units (10K MWCO, Thermo Fisher Scientific), and dialyzed in 500 mL assay buffer for 4 h, changing dialysis buffer at the 1 and 2 h time points. An aliquot of enzyme was withdrawn from each sample and its activity tested. To the remainder of the enzyme samples was added ZnCl2 at a final concentration of 800 nM. The enzyme samples were incubated at r.t. for 30 min, and then tested for activity. Data analysis proceeded as previously described.
Hi-resolution mass spectrometry of covalent adducts
Inhibitors were incubated with BoNT/A LC WT or C165S for at least 4 h at r.t. at a concentration of 100 mM inhibitor, 4 μM enzyme in 40 mM HEPES, 0.01% Triton X-100, pH 7.4, 1% DMSO. Control samples containing only enzyme were also analyzed. Putative masses of the covalent adducts were determined in ChemDraw 18. The difference between the masses of inhibitor-incubated protein and control protein samples were found, and then divided by the covalent adduct mass to give the number of compounds bound.
BoNT/A cell activity
The hiPSC-derived GABA Neurons and culture medium (Cellular Dynamics International (Madison, WI)) were cultured in PLO-matrigel coated 96-well TPP plates (Midsci) for 12 days prior to the assay. 200 LD50 Units of BoNT/A1 (150 kDa purified as previously described,82 specific activity 1.7×108 U/mg) was added to the cells in 50 μL stimulation medium (modified neurobasal containing 2.2 mM CaCl2 and 56 mM KCl (Invitrogen) and supplemented with B27 and Glutamax, and the cells were incubated at 37°C in a humidified 5% CO2 atmosphere for 7.5 min. Toxin was removed, cells were washed twice in 300 μL of culture medium, and further incubated in fresh culture medium at 37 °C in a humidified 5% CO2 atmosphere. At 30 min post first toxin addition, the inhibitors were added at the indicated concentrations and with a final DMSO concentration of 1%, in duplicate. Positive control (+C) was toxin without inhibitor in culture media, and negative control (-C) was culture media, both with 1% DMSO added. Cells were incubated for 7 h post toxin addition at 37 °C, 5% CO2 to allow for SNAP-25 cleavage. Inhibitor mixtures were then aspirated and cells lysed in 50 μL of 1× LDS lysis buffer (Invitrogen). The samples were analyzed by Western blot using a monoclonal anti-SNAP-25 antibody (Synaptic Systems, Germany) as described previously,83 and bands were visualized using Phosphaglo chemiluminescent reagent (KPL) on a Azure C600 imaging system equipped with a CCD camera (Azure Biosystems). Gel images were quantified by densitometry using the Azure spot software, and IC50 values were determined using GraphPad Prism 7 by dose response curves fitted with four-parameters least squares fit.
Cell viability assay (MTT Assay84)
HEK293T cells (ATCC, #CRL-11268) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, cat. #10566) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) Pen Strep (Gibco, cat. #15140), and routinely passaged every 2 days. Cells were plated in a 96-well plate (Corning; cat. #3596) at a density of 5,000 cells/well, and incubated at 37 °C, 5% CO2 for 24 h. Compounds, dissolved in DMSO, were diluted in cell media and added to wells (2-fold dilution from 100–3.13 μM); final volume of 100 μL and 1% DMSO per well) and incubated for 24 h at 37 °C, 5% CO2. 10 μL Thiazolyl Blue Tetrazolium Bromide (2.41 mM in PBS, pH 7.4; Sigma, cat. #M2128) was added to each well, and the assay plate incubated for a further 4 h at 37 °C, 5% CO2. 100 μL ice-cold isopropanol containing 0.04 M HCl was added to each well and solid formazan dye was dissolved by vigorous pipetting. The absorbance was then read on a plate reader (Spectramax i3x, Molecular Devices) at 570 nm with background subtracted at 630 nm. The absorbance of each well was then normalized to the DMSO control to calculate % cell viability and IC50 values were determined using GraphPad Prism 7 by dose-response curves fitted with four-parameters least squares fit.
MMP screening panel
MMP inhibitor profiling kit was purchased from Enzo Life Sciences (cat. #BML-AK308–0001), and used per manufacturer instructions. Briefly, MMP stocks were diluted in the assay buffer (50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, pH 7.5), and 20 μL per well was plated in a black half-volume 96-well plate (Corning, cat. #3964) at supplier-recommended concentrations. Assay buffer was added to each well to bring the total volume to 70 μL. The plate was equilibrated by incubating it for 10 min at 37 °C. Then, 20 μL of inhibitor was pipetted per well, to a final assay concentration of 100 μM, 1% DMSO. A negative control containing only DMSO was added, as well as the supplied positive control inhibitor (NNGH peptide, data not shown). The assay plate was then incubated for 30 min at 37 °C. Fluorogenic substrate (10 μL, final concentration = 0.75 μM) was added to initiate the reaction, and immediately placed in a plate reader (Spectramax i3x, Molecular Devices). Fluorescence was monitored in kinetics mode for 20 min, λex = 545 nm, λem = 576 nm, cutoff = 570 nm. Data analysis was performed in Excel by determining the rate of fluorescence increase versus time to give the initial reaction rate in RFU/s, then normalizing it to the initial reaction rate of the negative control to give relative activity remaining of each MMP.
Interference compound filters
All final compounds were examined using publicly accessible PAINS filters: ZINC patterns search (http://zinc15.docking.org/patterns/home) and Shoichet’s aggregation advisor (https://advisor.bkslab.org/).85 No compounds were indicated by these filters to have interference potential.
Synthesis of compounds
General protocols and instrumentation
Reactions were carried out under atmospheric conditions unless otherwise stated and all reagents obtained from commercial sources and used without further purification. Anhydrous DCM was distilled from calcium hydride under positive pressure of nitrogen and stored over 4 Å molecular sieves. Reactions were monitored using thin layer chromatography (TLC) or high performance liquid chromatography-mass spectrometry (HPLC-MS). TLC was performed using Merck precoated analytical plates (0.25 mM thick, silica gel 60 F254), and visualized under UV light and/or by dipping and heating in either: potassium permanganate stain, ninhydrin stain, bromocresol green stain, or ferric chloride stain. HPLC-MS analysis was performed on an Agilent 1260 Infinity II instrument coupled to a single quadrupole InfinityLab LC/MSD instrument running a gradient of eluent I (0.1% formic acid in H2O) and eluent II (0.1% formic acid in MeCN) rising linearly from 0% to 95% of II during t = 0.00−6.00 min and then with eluent II from t = 6.00–10.0 min, at a flow rate of 0.5 mL/min on a Zorbax 300SB-C8 column at 35 °. Flash automated column chromatography (ACC) was performed using a CombiFlash Rf+ Lumen (Teledyne Isco) purification system with flash silica RediSep Rf columns for normal phase (NP) or RediSep Rf Gold C18 HP columns for reverse phase (RP). Purity of all compounds is >95% as determined by HPLC-MS.
Nuclear magnetic resonance (NMR) spectra were recorded on either a Bruker AVIII HD 600 NMR equipped with a 5 mM CPQCl CryoProbe/ 5 mM CPDCH CryoProbe or a Bruker AV NEO at 400, 500, or 600 MHz for 1H NMR, and 101, 126, or 151 MHz for 13C NMR. Chemical shifts are reported in ppm and are reported with reference to the residual solvent peak (δH CDCl3 7.26 ppm; δC CDCl3 77.16 ppm; δH DMSO-d6 2.50 ppm; δC DMSO-d6 39.52 ppm; δH MeOD-d4 3.31 ppm; δC MeOD-d4 49.00 ppm). Multiplicities are reported with coupling constants and are given to the nearest 0.1 Hz. High resolution mass spectrometry (HRMS) was carried out using an Agilent 1260 Infinity II instrument coupled to an Agilent 6230 TOF-MS spectrometer using electro spray ionization (ES+/-), giving masses correct to four decimal places.
General methods
Method A: Sodium methanethiosulfonate (NaMTS) SN2 displacement
NaMTS (1.0–2.0 equiv.) was dissolved in the minimum amount of DMF (~1–2 mL) and heated at 70 °C, or EtOH (4–10 mL) and heated to 80 °C. The appropriate bromo reagent (1.0–6.0 equiv.) was added in one portion and the reaction stirred at the same temperature for 1–16 h. See individual compounds for purification.
Method B: Amide coupling
The chosen carboxylic acid (1.0 equiv.) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (1.1–1.2 equiv.) were dissolved in anh. DMF (3–4 mL). N,N-diisopropylethylamine (DIPEA) (1.5–2.5 equiv.) was added and stirred for 2 min. The chosen amine (1.1–1.5 equiv.) was added and the reaction stirred at r.t. for 2 h. The DMF was blown dry to give the crude product as a yellow oil. See individual compounds for purification.
Method C: THP deprotection
The appropriate THP-protected compound (1.0 equiv.) was dissolved in abs. EtOH (3 mL) and pyridinium p-toluenesulfonate (PPTS) (0.5 equiv.) was added and the reaction stirred at 60–65 °C for 2–16 h. The reaction mixture was cooled to r.t and then reduced in vacuo to reveal the crude product as a colorless semi-solid. See individual compounds for purification.
Method D: One-pot OTX SN2 displacement/amide coupling
The chosen bromo-alkyl-MTS compound (1.0 equiv.) was dissolved in DMF (2–3 mL) and OTX (1.3 equiv.) was added and stirred at r.t. for 48 h. The crude reaction mixture was added directly to a solution of 30 (1.3 equiv.) in anh. DCM (2–5 mL) and stirred for 1 h. HPLC-MS confirmed formation of the protected THP product – however, upon purification the deprotected product was obtained. See individual compounds for purification.
Method E: Boc deprotection
The chosen Boc-protected compound (1.0 equiv.) was dissolved in DCM (0.4–5 mL) and TFA (0.2–2 mL) was added dropwise and the reaction stirred for 1 h. The reaction mixture was reduced in vacuo to afford the crude product as an oil. See individual compounds for deviations.
Method F: One-pot amide coupling/THP deprotection
The chosen amine (1.3–2.4 equiv.) and 52 (1.0 equiv.) were dissolved in DMF (2 mL) and HATU (1.7 equiv.) and DIPEA (3.0 equiv.) were added the mixture stirred at r.t. for 16 h. The reaction mixture was poured into sat. NaHCO3 and extracted with EtOAc. The organic layer was separated, dried (MgSO4) and concentrated in vacuo to reveal the crude product as oil. The crude product was purified by NP ACC (gradient: 0–30% MeOH–DCM). Appropriate fractions were collected and reduced in vacuo to give an oil. The obtained oil was dissolved in ethanol (2 mL) and PPTS (5 mg) added and the reaction mixture stirred at 60 °C for 16 h. The reaction mixture was reduced in vacuo and the crude oil purified by NP ACC (gradient: 0–30% MeOH–DCM). The title compounds were collected as colorless oils.
1st generation bifunctional inhibitors
5-((methylsulfonyl)thio)pentanoic acid (12)
Synthesized by Method A using NaMTS (167 mg, 1.24 mmol, 1.5 equiv.), 8 (150 mg, 0.83 mmol, 1.0 equiv.), and DMF (2 mL) and the reaction stirred for 1 h. The DMF was blown dry and the resulting oil dissolved in DCM (10 mL) and partitioned with water (10 mL) and the organic layer separated. The aqueous layer was extracted with DCM (2 × 10 mL) and the combined organic layers dried (Na2SO4) and reduced in vacuo to give a colorless oil which solidified over time under high vacuum. The title compound (152 mg, 0.72 mmol, 86%) was collected as an off-white solid.
1H NMR (500 MHz, DMSO-d6) δ 12.06 (s, 1H), 3.51 (s, 3H), 3.21 (t, J = 7.3 Hz, 2H), 2.25 (t, J = 7.3 Hz, 2H), 1.75–1.67 (m, 2H), 1.63–1.55 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 174.2, 50.2, 35.2, 32.9, 28.3, 23.3. HRMS (ES−) m/z calcd for [C6H11O4S2]−: 211.0104; found 211.0111.
6-((methylsulfonyl)thio)hexanoic acid (13)
Synthesized by Method A using NaMTS (155 mg, 1.15 mmol, 1.5 equiv.), 9 (150 mg, 0.77 mmol, 1.0 equiv.), and DMF (2 mL) and the reaction stirred for 1 h. The work up procedure was identical to that performed in the synthesis of 12. The title compound (158 mg, 0.70 mmol, 91%) was collected as an off-white solid.
1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H), 3.51 (s, 3H), 3.19 (t, J = 7.3 Hz, 2H), 2.21 (t, J = 7.3 Hz, 2H), 1.71–1.70 (m, 2H), 1.58–1.47 (m, 2H), 1.41–1.32 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 174.3, 50.2, 35.4, 33.4, 28.6, 27.4, 23.9. HRMS (ES−) m/z calcd for [C7H13O4S2]−: 225.0261; found 225.0257.
7-((methylsulfonyl)thio)heptanoic acid (14)
Synthesized by Method A using NaMTS (144 mg, 1.08 mmol, 1.5 equiv.), 10 (150 mg, 0.72 mmol, 1.0 equiv.), and DMF (2 mL) and the reaction stirred for 1 h. The work up procedure was identical to that performed in the synthesis of 12. The title compound (159 mg, 0.66 mmol, 92%) was collected as an off-white solid.
1H NMR (500 MHz, DMSO-d6) δ 11.98 (s, 1H), 3.51 (s, 3H), 3.19 (t, J = 7.4 Hz, 2H), 2.20 (t, J = 7.4 Hz, 2H), 1.72–1.65 (m, 2H), 1.52–1.45 (m, 2H), 1.40–1.25 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 174.4, 50.1, 35.4, 33.5, 28.7, 27.9, 27.6, 24.3. HRMS (ES−) m/z calcd for [C8H15O4S2]−: 239.0417; found 239.0428.
8-((methylsulfonyl)thio)octanoic acid (15)
Synthesized by Method A using NaMTS (135 mg, 1.01 mmol, 1.5 equiv.), 11 (150 mg, 0.67 mmol, 1.0 equiv.), and DMF (2 mL) and the reaction stirred for 1 h. The work up procedure was identical to that performed in the synthesis of 12. The title compound (155 mg, 0.61 mmol, 91%) was collected as a white solid.
1H NMR (500 MHz, DMSO-d6) δ 11.96 (s, 1H), 3.51 (s, 3H), 3.19 (t, J = 7.4, 2H), 2.19 (t, J = 7.4 Hz, 2H), 1.73–1.64 (m, 2H), 1.53–1.45 (m, 2H), 1.40–1.22 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 174.5, 50.1, 35.5, 33.6, 28.8, 28.3, 28.1, 27.7, 24.4. HRMS (ES−) m/z calcd for [C9H17O4S2]−: 253.0574; found 253.0574.
S-(5-oxo-5-(((tetrahydro-2H-pyran-2-yl)oxy)amino)pentyl) methanesulfonothioate (16)
Synthesized by Method B using 12 (300 mg, 1.41 mmol, 1.0 equiv.), HATU (645 mg, 1.70 mmol, 1.2 equiv.), OTX (248 mg, 2.12 mmol, 1.5 equiv.), DIPEA (369 μL, 2.12 mmol, 1.5 equiv.) and DMF (4 mL). The crude residue was purified by RP ACC (gradient: 10–20%–hold MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and extracted with DCM, dried (Na2SO4), and reduced in vacuo to give the title compound (304 mg, 0.98 mmol, 69%) as a pale-yellow viscous oil.
H NMR (400 MHz, MeOD-d4) δ 4.89 (br.t, J = 3.0 Hz, 1H), 4.00 (td, J = 11.0, 2.8 Hz, 1H), 3.64–3.52 (m, 1H), 3.39 (s, 3H), 3.22 (t, J = 6.9 Hz, 2H), 2.21–2.10 (m, 2H), 1.86–1.50 (m, 10H), NH not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.1, 103.2, 63.0, 50.7, 36.7, 33.0, 30.0, 28.9, 26.2, 25.4, 19.4. HRMS (ES+) m/z calcd for [C11H21NNaO5S2]+: 334.0759; found: 334.0765.
S-(6-oxo-6-(((tetrahydro-2H-pyran-2-yl)oxy)amino)hexyl) methanesulfonothioate (17)
Synthesized by Method B using 13 (300 mg, 1.33 mmol, 1.0 equiv.), HATU (606 mg, 1.59 mmol, 1.2 equiv.), OTX (233 mg, 1.99 mmol, 1.5 equiv.), DIPEA (347 μL, 1.99 mmol, 1.5 equiv.) and DMF (4 mL). The crude residue was purified by RP ACC (gradient: 10–20%–hold–40% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and extracted with DCM, dried (Na2SO4), and reduced in vacuo to give the title compound (292 mg, 0.90 mmol, 68%) as a pale-yellow viscous oil.
1H NMR (400 MHz, MeOD-d4) δ 4.89 (br.t, J = 2.9 Hz, 1H), 4.00 (td, J = 10.9, 2.9 Hz, 1H), 3.64–3.57 (m, 1H), 3.39 (s, 3H), 3.21 (t, J = 7.3 Hz, 2H), 2.14 (t, J = 7.3 Hz, 2H), 1.87–1.53 (m, 10H), 1.51–1.42 (m, 2H), NH not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.4, 103.2, 63.0, 50.6, 37.0, 33.4, 30.2, 28.9, 28.8, 26.2, 25.9, 19.4. HRMS (ES+) m/z calcd for [C12H23NNaO5S2]+: 348.0915; found: 348.0921.
S-(7-oxo-7-(((tetrahydro-2H-pyran-2-yl)oxy)amino)heptyl) methanesulfonothioate (18)
Synthesized by Method B using 14 (300 mg, 1.25 mmol, 1.0 equiv.), HATU (570 mg, 1.50 mmol, 1.2 equiv.), OTX (219 mg, 1.87 mmol, 1.5 equiv.), DIPEA (326 μL, 1.87 mmol, 1.5 equiv.) and DMF (4 mL). The crude residue was purified using the same protocol as outlined in the synthesis of 17. The title compound (366 mg, 1.08 mmol, 86%) was collected as a pale-yellow viscous oil.
1H NMR (500 MHz, MeOD-d4) δ 4.89 (br.t, J = 3.0 Hz, 1H), 4.00 (td, J = 11.0, 2.9 Hz, 1H), 3.63–3.57 (m, 1H), 3.38 (s, 3H), 3.20 (t, J = 7.4 Hz, 2H), 2.13 (t, J 7.3 Hz, 2H), 1.85–1.72 (m, 5H), 1.68–1.54 (m, 5H), 1.50–1.35 (m, 4H), NH not observed. 13C NMR (126 MHz, MeOD-d4) δ 172.6, 103.1, 63.0, 50.6, 37.0, 33.6, 30.4, 29.3, 29.1, 28.9, 26.3, 26.2, 19.4, DCM present–trapped in viscous oil. HRMS (ES+) m/z calcd for [C13H25NNaO5S2]+: 362.1072; found: 362.1078.
S-(8-oxo-8-(((tetrahydro-2H-pyran-2-yl)oxy)amino)octo) methanesulfonothioate (19)
Synthesized by Method B using 15 (300 mg, 1.18 mmol, 1.0 equiv.), HATU (538 mg, 1.42 mmol, 1.2 equiv.), OTX (207 mg, 1.77 mmol, 1.5 equiv.), DIPEA (308 μL, 1.77 mmol, 1.5 equiv.) and DMF (4 mL). The crude residue was purified using the same protocol as outlined in the synthesis of 17. The title compound (337 mg, 0.95 mmol, 81%) was collected as a pale-yellow viscous oil.
1H NMR (500 MHz, MeOD-d4) δ 4.89 (br.t, J = 3.0 Hz, 1H), 4.00 (td, J = 11.0, 2.9 Hz, 1H), 3.62–3.57 (m, 1H), 3.38 (s, 3H), 3.20 (t, J = 7.4 Hz, 2H), 2.12 (t, J 7.4 Hz, 2H), 1.85–1.71 (m, 5H), 1.67–1.54 (m, 5H), 1.48–1.32 (m, 6H), NH not observed. 13C NMR (126 MHz, MeOD-d4) δ 172.7, 103.1, 63.0, 50.6, 37.1, 33.7, 30.5, 29.8, 29.6, 29.3, 28.9, 26.5, 26.2, 19.4, DCM present–trapped in viscous oil. HRMS (ES+) m/z calcd for [C14H27NNaO5S2]+: 376.1228; found: 376.1235.
S-(5-(hydroxyamino)-5-oxopentyl) methanesulfonothioate (20)
Synthesized by Method C using 16 (122 mg, 0.39 mmol, 1.0 equiv.), PPTS (49 mg, 0.20 mmol, 0.5 equiv.) and EtOH (3 mL) and the reaction stirred at 65 °C for 2 h. The crude product was purified by NP ACC (gradient: 0–8% MeOH–DCM). Appropriate fractions were collected and reduced in vacuo to reveal a colorless oil. The oil was further purified using NP ACC (stepwise: 0→2→4% MeOH–DCM in 0.1% AcOH). The title compound (6 mg, 0.03 mmol, 7%) was collected as a colorless semi-solid. Poor yield attributed to troublesome purification.
1H NMR (600 MHz, MeOD-d4) δ 3.39 (s, 3H), 3.22 (t, J = 7.1 Hz, 2H), 2.14 (t, J = 7.1 Hz, 2H), 1.84–1.71 (m, 4H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.3, 50.7, 36.7, 33.0, 30.0, 25.5. HPLC tR = 2.45 min (>98%, UV214). HRMS (ES+) m/z calcd for [C6H14NO4S2]+: 228.0359; found 228.0360.
S-(6-(hydroxyamino)-6-oxohexyl) methanesulfonothioate (21)
Synthesized by Method C using 17 (171 mg, 0.53 mmol, 1.0 equiv.), PPTS (66 mg, 0.26 mmol, 0.5 equiv.) and EtOH (3 mL) and the reaction stirred at 65 °C for 2 h. The crude residue was purified using the same protocol as outlined in the synthesis of 20. The title compound (4 mg, 0.02 mmol, 3%) was collected as a colorless semi-solid. Poor yield attributed to troublesome purification.
1H NMR (600 MHz, MeOD-d4) δ 3.39 (s, 3H), 3.21 (t, J = 7.3 Hz, 2H), 2.11 (t, J = 7.4 Hz, 2H), 1.83–1.75 (m, 2H), 1.69–1.62 (m, 2H), 1.49–1.42 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.7, 50.6, 36.9, 33.5, 30.2, 28.9, 26.0. HPLC tR = 3.73 min (>98%, UV214). HRMS (ES+) m/z calcd for [C7H16NO4S2]+: 242.0515; found 242.0521.
S-(7-(hydroxyamino)-7-oxoheptyl) methanesulfonothioate (22)
Synthesized by Method C using 18 (115 mg, 0.34 mmol, 1.0 equiv.), PPTS (42 mg, 0.17 mmol, 0.5 equiv.) and EtOH (3 mL) and the reaction stirred at 65 °C for 2 h. The crude product was purified by NP ACC (gradient: 0–8% MeOH–DCM). Appropriate fractions were collected and reduced in vacuo to reveal a colorless oil. The oil was further purified using RP ACC (gradient: 0% hold–20% hold MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and reduced in vacuo to give the title compound (42 mg, 0.16 mmol, 49%) as a colorless semi-solid.
1H NMR (600 MHz, MeOD-d4) δ 3.38 (s, 3H), 3.20 (t, J = 7.4 Hz, 2H), 2.10 (t, J = 7.4 Hz, 2H), 1.81–1.75 (m, 2H), 1.66–1.59 (m, 2H), 1.49–1.42 (m, 2H), 1.40–1.34 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.8, 50.6, 37.0, 33.6, 30.4, 29.4, 29.1, 26.5. HPLC tR = 4.32 min (>98%, UV214). HRMS (ES+) m/z calcd for [C8H18NO4S2]+: 256.0672; found 256.0679.
S-(8-(hydroxyamino)-8-oxooctyl) methanesulfonothioate (23)
Synthesized by Method C using 19 (122 mg, 0.35 mmol, 1.0 equiv.), PPTS (43 mg, 0.17 mmol, 0.5 equiv.) and EtOH (3 mL) and the reaction stirred at 65 °C for 2 h. The crude residue was purified using the same protocol as outlined in the synthesis of 22. The title compound (46 mg, 0.17 mmol, 49%) was collected as a colorless semi-solid.
1H NMR (600 MHz, MeOD-d4) δ 3.38 (s, 3H), 3.20 (t, J = 7.4 Hz, 2H), 2.09 (t, J = 7.4 Hz, 2H), 1.80–1.74 (m, 2H), 1.65–1.59 (m, 2H), 1.48–1.41 (m, 2H), 1.41–1.32 (m, 4H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 172.9, 50.6, 37.1, 33.7, 30.5, 29.9, 29.6, 29.3, 26.6. HPLC tR = 4.75 min (>98%, UV214). HRMS (ES+) m/z calcd for [C9H20NO4S2]+: 270.0828; found 270.0834.
2nd generation bifunctional inhibitors and controls
S-(5-bromopentyl) methanesulfonothioate (27)
Synthesized by Method A using NaMTS (500 mg, 3.73 mmol, 1.0 equiv.), 24 (3.06 mL, 22.36 mmol, 6.0 equiv.) and DMF (2 mL) and the reaction heated for 1 h. The reaction mixture was filtered and washed with DCM and the filtrate reduced in vacuo and the DMF blown dry. The crude oil was purified by NP ACC (gradient: 0–40% EtOAc–hexane). The title compound (641 mg, 2.47 mmol, 66%) was collected as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 3.42 (t, J = 6.6 Hz, 2H), 3.33 (s, 3H), 3.18 (t, J = 7.2 Hz, 2H), 1.94–1.86 (m, 2H), 1.85–1.77 (m, 2H), 1.63–1.53 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 50.8, 36.3, 33.3, 32.1, 28.9, 27.2. Compound degrades upon ionization.
S-(6-bromohexyl) methanesulfonothioate (28)
Synthesized by Method A using NaMTS (500 mg, 3.73 mmol, 1.0 equiv.), 25 (3.44 mL, 22.36 mmol, 6.0 equiv.) and DMF (2 mL) and the reaction heated for 1 h. The product was purified using the same protocol as outlined in the synthesis of 27. The title compound (679 mg, 2.47 mmol, 66%) was collected as a colorless oil.
1H NMR (500 MHz, CDCl3) δ 3.41 (t, J = 6.7 Hz, 2H), 3.32 (s, 3H), 3.18 (t, J = 7.6 Hz, 2H), 1.91–1.84 (m, 2H), 1.83–1.76 (m, 2H), 1.52–1.43 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 50.8, 36.4, 33.7, 32.5, 29.5, 27.8, 27.6. Compound degrades upon ionization.
S-(7-bromoheptyl) methanesulfonothioate (29)
Synthesized by Method A using NaMTS (500 mg, 3.73 mmol, 1.0 equiv.), 26 (3.82 mL, 22.36 mmol, 6.0 equiv.) and DMF (2 mL) and the reaction heated for 1 h. The product was purified using the same protocol as outlined in the synthesis of 27. The title compound (667 mg, 2.31 mmol, 62%) was collected as a colorless oil.
1H NMR (500 MHz, CDCl3) δ 3.41 (t, J = 6.8 Hz, 2H), 3.32 (s, 3H), 3.17 (t, J = 7.4 Hz, 2H), 1.89–1.82 (m, 2H), 1.81–1.74 (m, 2H), 1.49–1.40 (m, 4H), 1.39–1.32 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 50.8, 36.5, 33.9, 32.7, 29.5, 28.5, 28.2, 28.0. Compound degrades upon ionization.
(E)-3-(2,4-dichlorophenyl)acryloyl chloride (30)
Trans-2,4-dichlorocinnamic acid (1.20 g, 5.53 mmol, 1.0 equiv.) was suspended in SOCl2 (5 mL) and refluxed for 45 min. The reaction was cooled to r.t. and excess SOCl2 blown dry. The title compound 30 (1.30 g, 5.53 mmol, quant.) was collected as a colorless solid.
1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 15.6 Hz, 1H), 7.59 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 2.1 Hz, 1H), 7.32 (ddd, J = 8.5, 2.1, 0.6 Hz, 1H), 6.64 (d, J = 15.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 166.0, 144.9, 138.4, 136.8, 130.6, 130.0, 129.0, 128.1, 125.2. HRMS (ES+) m/z calcd for methyl ester [C10H9Cl2O2]+: 230.9974; found 230.9973.
(E)-S-(5-(3-(2,4-dichlorophenyl)-N-hydroxyacrylamido)pentyl) methanesulfonothioate (31)
Synthesized by Method D using 27 (641 mg, 2.45 mmol, 1.0 equiv.), OTX (376 mg, 3.21 mmol, 1.3 equiv.), DMF (3 mL), 30 (756 mg, 3.21 mmol, 1.3 equiv.) and anh. DCM (5 mL). The reaction mixture was filtered under vacuum and the filtrate reduced in vacuo with excess DMF blown dry. The crude product was purified by RP ACC (gradient: 20–70% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and extracted with DCM, dried (Na2SO4) and reduced in vacuo to give a colorless semi-solid. The product was further purified by NP ACC (gradient: 30–70% EtOAc–hexane). The title compound (98 mg, 0.24 mmol, 10%) was collected as a colorless semi-solid which solidified upon standing.
1H NMR (600 MHz, DMSO-d6) δ 10.04 (s, 1H), 7.88 (br.d, J = 8.4 Hz, 1H), 7.76–7.70 (m, 2H), 7.48 (dd, J = 8.5, 2.2 Hz, 1H), 7.31 (br.d, J = 15.8 Hz, 1H), 3.63 (br.t, J = 7.0 Hz, 2H), 3.51 (s, 3H), 3.20 (t, J = 7.4 Hz, 2H), 1.76–1.70 (m, 2H), 1.65–1.59 (m, 2H), 1.41–1.34 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 164.4, 134.9, 134.6, 134.2, 131.8, 129.4, 129.2, 128.0, 121.2, 50.1, 47.3, 35.4, 28.6, 25.7, 25.1. HPLC tR = 6.50 min (>96%, UV254). HRMS (ES+) m/z calcd for [C15H20Cl2NO4S2]+: 412.0205; found 412.0213.
(E)-S-(6-(3-(2,4-dichlorophenyl)-N-hydroxyacrylamido)hexyl) methanesulfonothioate (32)
Synthesized by Method D using 28 (555 mg, 2.44 mmol, 1.0 equiv.), OTX (371 mg, 3.17 mmol, 1.3 equiv.), DMF (3 mL), 30 (746 mg, 3.17 mmol, 1.3 equiv.) and anh. DCM (5 mL). The product was purified using the same protocol as outlined in the synthesis of 31. The title compound (84 mg, 0.20 mmol, 9%) was collected as a colorless semi-solid.
1H NMR (600 MHz, DMSO-d6) δ 10.03 (s, 1H), 7.88 (br.d, J = 8.5 Hz, 1H), 7.75–7.70 (m, 2H), 7.47 (dd, J = 8.5, 2.2 Hz, 1H), 7.31 (br.d, J = 15.8 Hz, 1H), 3.61 (br.t, J = 7.1 Hz, 2H), 3.51 (s, 3H), 3.19 (t, J = 7.3 Hz, 2H), 1.75–1.65 (m, 2H), 1.62–1.55 (m, 2H), 1.43–1.36 (m, 2H), 1.34–1.26 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 164.4, 134.8, 134.6, 134.2, 131.8, 129.4, 129.2, 128.0, 121.3, 50.1, 47.4, 35.5, 28.8, 27.6, 26.1, 25.5. HPLC tR = 6.67 min (>95%, UV254). HRMS (ES+) m/z calcd for [C16H22Cl2NO4S2]+: 426.0362; found 426.0368.
(E)-S-(7-(3-(2,4-dichlorophenyl)-N-hydroxyacrylamido)heptyl) methanesulfonothioate (33)
Synthesized by Method D using 29 (121 mg, 0.42 mmol, 1.0 equiv.), OTX (64 mg, 0.54 mmol, 1.3 equiv.), DMF (2 mL), 30 (127 mg, 0.54 mmol, 1.3 equiv.) and anh. DCM (2 mL). The reaction mixture was blown dry and crude product purified by RP ACC (gradient: 10–100% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and extracted with DCM, dried (Na2SO4), and reduced in vacuo to give a red semi-solid. The semi-solid was further purified by NP ACC (stepwise: 0→1→2% MeOH–DCM). The title compound (2 mg, 4.54 μmol, 0.1%) was collected a colorless semi-solid.
1H NMR (600 MHz, DMSO-d6) δ 10.02 (s, 1H), 7.88 (br.d, J = 8.6 Hz, 1H), 7.75–7.70 (m, 2H), 7.48 (dd, J = 8.5, 2.1 Hz, 1H), 7.31 (br.d, J = 15.9 Hz, 1H), 3.63–3.58 (m, 2H), 3.50 (s, 3H), 3.19 (t, J = 7.4 Hz, 2H), 1.75–1.65 (m, 2H), 1.61–1.55 (m, 2H), 1.39–1.24 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 164.4, 134.8, 134.6, 134.2, 131.8, 129.4, 129.2, 128.0, 121.3, 50.1, 47.5, 35.5, 28.8, 28.0, 27.8, 26.2, 25.9. HPLC tR = 6.86 min (>97%, UV254). HRMS (ES+) m/z calcd for [C17H24Cl2NO4S2]+: 440.0518; found. 440.0522.
S-(6-((tert-butoxycarbonyl)amino)hexyl) methanesulfonothioate (35)
Synthesized using Method A using NaMTS (479 mg, 3.56 mmol, 2.0 equiv.), 34 (500 mg, 1.78 mmol, 1.0 equiv.) and EtOH (10 mL) and the reaction heated for 16 h. Water (10 mL) was added and the reaction mixture extracted with EtOAc (3 × 30 mL), washed with brine (30 mL), dried (Na2SO4) and reduced in vacuo to reveal the crude product as a colorless oil. The crude product was purified by NP ACC (gradient: 0–100% EtOAc–hexane). The title compound (222 mg, 0.71 mmol, 40%) was collected as a colorless oil.
1H NMR (600 MHz, CDCl3) δ 4.52 (br.s, 1H), 3.31 (s, 3H), 3.16 (t, J 7.3 Hz, 2H), 3.13–3.08 (m, 2H), 1.79–1.73 (m, 2H), 1.51–1.40 (m, 13H), 1.38–1.32 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 156.1, 79.3, 50.8, 40.5, 36.4, 30.0, 29.6, 28.6, 28.3, 26.2. HRMS (ES+) m/z calcd for [C12H25NNaO4S2]+: 334.1117; found 334.1124.
6-((Methylsulfonyl)thio)hexan-1-aminium 2,2,2-trifluoroacetate (36)
Synthesized using Method E using 35 (210 mg, 0.67 mmol, 1.0 equiv.), TFA (0.2 mL) and DCM (0.4 mL). The crude product was collected as a colorless oil (256 mg) and used without further purification.
1H NMR (600 MHz, MeOD-d4) δ 3.39 (s, 3H), 3.22 (t, J = 7.5 Hz, 2H), 2.92 (t, J = 7.7 Hz, 2H), 1.84–1.77 (m, 2H), 1.70–1.63 (m, 2H), 1.53–1.41 (m, 4H), NHs not observed. 13C NMR (151 MHz, MeOD-d4) δ 50.5, 40.6, 36.9, 30.4, 28.9, 28.4, 26.8, TFA peaks not observed. HRMS (ES+) m/z calcd for [C7H18NO2S2]+: 212.0773, obsd 212.0777.
(E)-S-(6-(3-(2,4-dichlorophenyl)acrylamido)hexyl) methanesulfonothioate (37)
Synthesized by Method B using 2,4-dichlorocinnamic acid (217 mg, 1.00 mmol, 1.0 equiv.), HATU (475 mg, 1.25 mmol, 1.3 equiv.), 36 (232 mg, 1.10 mmol, 1.1 equiv.), DIPEA (435 μL, 2.50 mmol, 2.5 equiv.) and DMF (3 mL). The crude yellow oil was dissolved in EtOAc (20 mL) and the organic layer washed water (3 × 20 mL), 1 M HCl (20 mL), dried (Na2SO4) and concentrated in vacuo to give a crude yellow oil. The oil was purified on RP ACC (gradient: 10–90% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled, MeCN blown off, and the title compound (189 mg, 0.46 mmol, 46%) collected as a fluffy white solid after lyophilization.
1H NMR (600 MHz, CDCl3) δ 7.90 (d, J = 15.6 Hz, 1H), 7.50 (d, J = 8.5 Hz, 1H), 7.42 (d, J = 2.1 Hz, 1H), 7.24 (ddd, J = 8.5, 2.1, 0.6 Hz, 1H), 6.38 (d, J = 15.6 Hz, 1H), 5.81–5.74 (m, 1H), 3.39 (q, J = 7.0 Hz, 2H), 3.32 (s, 3H), 3.17 (t, J = 7.3 Hz, 2H), 1.81–1.76 (m, 2H), 1.62–1.56 (m, 2H), 1.50–1.44 (m, 2H), 1.44–1.37 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 165.3, 135.9, 135.8, 135.4, 132.0, 130.1, 128.4, 127.5, 124.2, 50.8, 39.7, 36.4, 29.5, 29.5, 28.1, 26.3. HPLC tR = 6.75 min (>98%, UV254). HRMS (ES+) m/z calcd for [C16H22Cl2NO3S2]+ 410.0413; found 410.0425.
(E)-3-(2,4-dichlorophenyl)-N-hydroxy-N-(6-mercaptohexyl)acrylamide (38)
32 (21 mg, 0.05 mmol, 1.0 equiv.) was dissolved in abs. EtOH (3 mL) and NaBH4 (6 mg, 0.15 mmol, 3.0 equiv.) added and stirred for 5 min. HCl (1 M, 3 mL) was added dropwise until pH ~1 and the reaction mixture was reduced in vacuo to give the crude product as a colorless semi-solid. The crude product was purified by RP ACC (gradient: 20–90% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled and extracted with DCM, dried (Na2SO4) and reduced in vacuo to reveal the title compound (5 mg, 0.01 mmol, 29%) as a colorless semi-solid.
1H NMR (400 MHz, MeOD-d4) δ 7.93 (d, J = 15.9 Hz, 1H), 7.78 (br.d, J = 8.5 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.40–7.30 (m, 2H), 3.73 (br.t, J = 7.1 Hz, 2H), 2.50 (t, J = 6.8 Hz, 2H), 1.75–1.66 (m, 2H), 1.66–1.57 (m, 2H), 1.51–1.42 (m, 2H), 1.42–1.32 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 167.4, 137.7, 137.0, 136.3, 133.5, 130.8, 129.9, 128.9, 121.3, 35.1, 29.0, 27.6, 27.2, 24.9, one aliphatic C not observed. HPLC tR = 6.95 min (>98%, UV254). HRMS (ES+) m/z calcd for [C15H20Cl2NO2S]+: 348.0586; found 348.0594.
3rd Generation Bifunctional Inhibitors and Controls
S-(3-((tert-butoxycarbonyl)amino)propyl) methanesulfonothioate (43)
Synthesized by Method A using NaMTS (248 mg, 1.85 mmol, 1.1 equiv.), 40 (400 mg, 1.68 mmol, 1.0 equiv.) and DMF (5 mL) and the reaction was heated for 16 h. Water (10 mL) was added and the reaction mixture extracted with EtOAc (3 × 30 mL), washed with brine (30 mL), dried (Na2SO4) and reduced in vacuo to reveal the crude product as an orange oil. The crude product was purified by NP ACC (gradient: 0–30% EtOAc–hexane). The title compound (356 mg, 1.32 mmol, 79%) was collected as a viscous colorless oil.
1H NMR (500 MHz, DMSO-d6) δ 6.90 (br.t, J = 5.8 Hz, 1H), 3.51 (s, 3H), 3.18 (t, J = 7.2 Hz, 2H), 3.05–2.98 (m, 2H), 1.85–1.76 (m, 2H), 1.38 (s, 9H). 13C NMR (151 MHz, DMSO) δ 155.7, 77.6, 50.1, 38.4, 33.1, 29.4, 28.2. HRMS (ES+) m/z calcd for [C9H19NNaO4S2]+: 292.0648; found 292.0647.
S-(4-((tert-butoxycarbonyl)amino)butyl) methanesulfonothioate (44)
Synthesized using Method A using NaMTS (265 mg, 1.98 mmol, 2.0 equiv.), 41 (250 mg, 0.99 mmol, 1.0 equiv.) and EtOH (6 mL) and the reaction was heated for 16 h. The reaction was worked up using the same protocol outlined in the synthesis of 43. The crude product was purified by NP ACC (gradient: 0–100% EtOAc–hexane). The title compound (223 mg, 0.79 mmol, 79%) was collected as a colorless oil.
1H NMR (600 MHz, CDCl3) δ 4.58 (br.s, 1H), 3.32 (s, 3H), 3.21–3.12 (m, 4H), 1.84–1.77 (m, 2H), 1.64–1.58 (m, 2H), 1.43 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 156.1, 79.5, 50.8, 39.8, 36.1, 29.2, 28.5, 26.9. HRMS (ES+) m/z calcd for [C10H21NNaO4S2]+: 306.0804; found 306.0812.
S-(5-((tert-butoxycarbonyl)amino)pentyl) methanesulfonothioate (45)
Synthesized using Method A using NaMTS (161 mg, 1.20 mmol, 1.9 equiv.), 42 (168 mg, 0.63 mmol, 1.0 equiv.) and EtOH (4 mL) and the reaction was heated for 16 h. The reaction was worked up and purified using the same protocol outlined in the synthesis of 44. The title compound (115 mg, 0.39 mmol, 61%) was collected as a colorless solid.
1H NMR (600 MHz, CDCl3) δ 4.54 (br.s, 1H), 3.32 (s, 3H), 3.16 (t, J = 7.3 Hz, 2H), 3.14–3.09 (m, 2H), 1.82–1.76 (m, 2H), 1.55–1.48 (m, 2H), 1.48–1.40 (m, 11H). 13C NMR (151 MHz, CDCl3) δ 156.1, 79.4, 50.8, 40.3, 36.4, 29.6, 29.3, 28.6, 25.8. HRMS (ES+) m/z calcd for [C11H23NNaO4S2]+: 320.0961; found 320.0964.
3-((Methylsulfonyl)thio)propan-1-aminium 2,2,2-trifluoroacetate (4)
Synthesized by Method E using 35 (317 mg, 1.18 mmol, 1.0 equiv.), TFA (2 mL) and DCM (5 mL). The crude oil was purified by RP ACC (H2O in 0.1% formic acid). The title compound (190 mg, 0.67 mmol, 57%) was collected as a colorless solid.
1H NMR (500 MHz, DMSO-d6) δ 7.89 (br.s, 3H), 3.54 (s, 3H), 3.28 (t, J = 7.2 Hz, 2H), 2.90 (t, J = 7.4 Hz, 2H), 2.05–1.97 (m, 2H). 13C NMR (151 MHz, DMSO) δ 158.2 (q, J 30.6 Hz), 50.0, 37.4, 32.2, 27.2, one TFA q missing. HRMS (ES+) m/z calcd for [C4H12NO2S2]+: 170.0304; found 170.0307.
4-((Methylsulfonyl)thio)butan-1-aminium 2,2,2-trifluoroacetate (46)
Synthesized by Method E using 43 (209 mg, 0.74 mmol, 1.0 equiv.), TFA (0.2 mL) and DCM (0.4 mL). The crude product was collected as a colorless oil (213 mg) and used without further purification.
1H NMR (600 MHz, MeOD-d4) δ 3.41 (s, 3H), 3.26 (t, J = 7.1 Hz, 2H), 2.97 (t, J = 7.6 Hz, 2H), 1.91–1.85 (m, 2H), 1.85–1.76 (m, 2H), NHs not observed. 13C NMR (151 MHz, MeOD-d4) δ 50.6, 40.1, 36.3, 27.7, 27.3, TFA peaks not observed. HRMS (ES+) m/z calcd for [C5H14NO2S2]+: 184.0460; found 184.0464.
5-((Methylsulfonyl)thio)pentan-1-aminium 2,2,2-trifluoroacetate (47)
Synthesized using Method E using 44 (105 mg, 0.35 mmol, 1.0 equiv.), TFA (0.2 mL) and DCM (0.4 mL). The crude product was collected as a colorless oil (137 mg) and used without further purification.
1H NMR (600 MHz, MeOD-d4) δ 3.39 (s, 3H), 3.23 (t, J = 7.3 Hz, 2H), 2.94 (t, J = 7.7 Hz, 2H), 1.87–1.81 (m, 2H), 1.73–1.66 (m, 2H), 1.56–1.50 (m, 2H), NHs not observed. 13C NMR (151 MHz, MeOD-d4) δ 50.5, 40.5, 36.7, 30.1, 27.9, 26.2 TFA peaks not observed. HRMS (ES+) m/z calcd for [C6H16NO2S2]+: 198.0617; found 198.0620.
3-(2,4-dichlorophenyl)pentanedioic acid (50)
48 (8.75 g, 50 mmol, 1.0 equiv.) was suspended in ice cold 49 (15.9 mL, 125 mmol, 2.5 equiv.) and piperidine (0.5 mL, 50 mmol, 1.0 equiv.) was added dropwise at 0 °C. The reaction mixture was warmed to r.t. and stirred for 42 h. The reaction mixture was reduced in vacuo and the crude brown oil suspended in 20 M KOH (30 mL) and heated to 110 °C for 2 h. The reaction mixture was poured carefully into ice and washed with EtOAc (2 × 50 mL). The aqueous phase was separated and then carefully adjusted to pH 1 using concentrated HCl. The resulting white precipitate was filtered under vacuum and the title compound (10.8 g, 39 mmol, 78%) collected as a white solid.
1H NMR (600 MHz, DMSO-d6) δ 12.19 (br.s, 2H), 7.55 (d, J = 2.2 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.39 (dd, J = 8.4, 2.2 Hz, 1H), 3.88 (quint, J = 7.4 Hz, 1H), 2.66–2.57 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 172.4, 139.8, 133.9, 131.5, 129.6, 128.7, 127.4, 38.4, 33.6. HRMS (ES−) m/z calcd for [C11H9Cl2O4]−: 274.9883; found 274.9896.
3-(2,4-dichlorophenyl)glutaric anhydride (51)
50 (2.77 g, 10.0 mmol, 1.0 equiv.) was suspended in acetic anhydride (20 mL) and the reaction mixture heated to 120 °C for 2 h. The reaction mixture was cooled slowly to allow the product to crystallize. The resulting crystals were filtered under vacuum and washed with ice-cold Et2O. The title compound (2.32 g, 8.95 mmol, 90%) was collected as colorless crystals.
1H NMR (600 MHz, DMSO-d6) δ 7.65 (d, J = 2.3 Hz, 1H), 7.50 (dd, J = 8.4, 2.2 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 3.97 (tt, J = 12.1, 4.4 Hz, 1H), 3.06 (dd, J = 17.0, 12.1 Hz, 2H), 2.94 (dd, J = 16.9, 4.5 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.9, 137.0, 133.8, 132.7, 129.2, 128.8, 128.0, 34.7, 29.6. HRMS (ES+) m/z calcd for [C11H9Cl2O3]+: 258.9923; found 258.9914.
3-(2,4-dichlorophenyl)-5-oxo-5-(((tetrahydro-2H-pyran-2-yl)oxy)amino)pentanoic acid (52)
51 (920 mg, 3.55 mmol, 1.0 equiv.) was suspended in CH3Cl (20 mL) and OTX (832 mg, 7.10 mmol, 2.0 equiv.) was added and the reaction stirred at r.t. for 30 min. The reaction mixture was reduced in vacuo to give the crude product as a yellow semi-solid. The crude product was purified by NP ACC (5% MeOH–DCM in 0.1% AcOH). Appropriate fractions were collected and reduced in vacuo and the subsequent oil resuspended in DCM (30 mL). The solution was washed with 0.1 M NaHCO3 (15 mL) and the organic layer separated¸ dried (Na2SO4) and reduced in vacuo to reveal the title compound (820 mg, 2.18 mmol, 61%) as a fluffy white solid.
1H NMR (400 MHz, DMSO-d6) δ 12.22 (br.s, 2H), 10.97 (br.s, 2H), 7.54 (s, 2H), 7.38 (d, J = 3.0 Hz, 4H), 4.74* (br.s, 1H), 4.60* (br.s, 1H) 3.97–3.79 (m, 4H), 3.52–3.37 (m, 2H), 2.61 (d, J = 7.4 Hz, 4H), 2.35 (d, J = 7.4 Hz, 4H), 1.69–1.41 (m, 12H). 13C NMR (151 MHz, DMSO-d6) δ 172.4, 166.6, 139.6, 133.9**, 131.5**, 129.6**, 128.7, 127.3**, 101.0**, 61.4**, 38.3, 36.7**, 33.9, 27.7**, 24.6, 18.4**. HRMS (ES+) m/z calcd for [C16H18Cl2NO5]−: 374.0568; found 374.0580.
* Anomeric protons distinct in diastereomeric mixture – all other peaks overlap.
** Diastereomeric carbons visible as doublets.
S-(3-(3-(2,4-dichlorophenyl)-5-(hydroxyamino)-5-oxopentanamido)propyl) methanesulfonothioate (53)
Synthesized by Method F using 52 (75 mg, 0.30 mmol, 1.3 equiv.), 4 (87 mg, 0.23 mmol, 1.0 equiv.), HATU (141 mg, 0.37 mmol, 1.6 equiv.), DIPEA (120 μL, 0.69 mmol, 3.0 equiv.) and DMF (2 mL). The title compound (33 mg, 0.07 mmol, 32%) was collected as a colorless oil. 1H NMR (600 MHz, MeOD-d4) δ 7.43 (d, J = 2.2 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.30 (dd, J = 8.4, 2.2 Hz, 1H), 4.17–4.10 (m, 1H), 3.37 (s, 3H), 3.25–3.20 (m, 1H), 3.18–3.12 (m, 1H), 3.00 (t, J = 7.3 Hz, 2H), 2.65 (dd, J = 14.2, 6.5 Hz, 1H), 2.58 (dd, J = 14.2, 8.8 Hz, 1H), 2.50–2.47 (m, 2H), 1.84–1.78 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 173.4, 170.1, 139.9, 135.9, 134.1, 130.8, 130.5, 128.5, 128.4, 50.7, 41.5, 38.6, 38.4, 36.6, 34.4, 30.6. HPLC tR = 5.04 min (>98%, UV230). HRMS (ES+) m/z calcd for [C15H21Cl2N2O5S2]+: 443.0263; found 443.0266.
S-(4-(3-(2,4-dichlorophenyl)-5-(hydroxyamino)-5-oxopentanamido)butyl) methanesulfonothioate (54)
Synthesized by Method F using 52 (107 mg, 0.58 mmol, 2.4 equiv.), 46 (90 mg, 0.24 mmol, 1.0 equiv.), HATU (146 mg, 0.38 mmol, 1.6 equiv.), DIPEA (125 μL, 0.72 mmol, 3.0 equiv.) and DMF (2 mL). The title compound (17 mg, 0.04 mmol, 15%) was collected as a colorless oil. 1H NMR (600 MHz, MeOD-d4) δ 7.43 (d, J = 2.1 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.29 (dd, J = 8.4, 2.2 Hz, 1H), 4.18–4.08 (m, 1H), 3.38 (s, 3H), 3.17–3.11 (m, 3H), 3.09–3.03 (m, 1H), 2.63 (dd, J = 14.2, 6.4 Hz, 1H), 2.57 (dd, J = 14.2, 8.9 Hz, 1H), 2.48 (d, J = 7.4 Hz, 2H), 1.62–1.56 (m, 2H), 1.50–1.44 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 173.2, 170.1, 139.9, 136.0, 134.0, 130.9, 130.5, 128.4, 50.6, 41.5, 39.3, 38.5, 36.7, 29.2, 27.7, one aliphatic C not observed. HPLC tR = 5.17 min (>98%, UV230). HRMS (ES+) m/z calcd for [C16H23Cl2N2O5S2]+: 457.0420; found 457.0424.
S-(5-(3-(2,4-dichlorophenyl)-5-(hydroxyamino)-5-oxopentanamido)pentyl) methanesulfonothioate (55)
Synthesized by Method F using 52 (111 mg, 0.56 mmol, 2.3 equiv.), 47 (90 mg, 0.24 mmol, 1.0 equiv.), HATU (146 mg, 0.38 mmol, 1.6 equiv.), DIPEA (125 μL, 0.72 mmol, 3.0 equiv.) and DMF (2 mL). The title compound (19 mg, 0.04 mmol, 17%) was collected as a colorless oil. 1H NMR (600 MHz, MeOD-d4) δ 7.43 (d, J = 2.2 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.29 (dd, J = 8.4, 2.2 Hz, 1H), 4.17–4.10 (m, 1H), 3.38 (s, 3H), 3.15 (t, J 7.3 Hz, 2H), 3.13–3.08 (m, 1H), 3.04–3.00 (m, 1H), 2.63 (dd, J = 14.1, 6.4 Hz, 1H), 2.57 (dd, J = 14.1, 8.8 Hz, 1H), 2.48 (d, J = 7.4 Hz, 2H), 1.73–1.67 (m, 2H), 1.40–1.35 (m, 2H), 1.29–1.23 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 173.1, 170.1, 140.0, 136.0, 134.0, 130.9, 130.4, 128.4, 50.7, 41.5, 39.9, 38.5, 37.0, 30.2, 29.7, 26.6, one aliphatic C not observed. HPLC tR = 5.36 min (>97%, UV230). HRMS (ES+) m/z calcd for [C17H25Cl2N2O5S2]+: 471.0576; found 471.0581.
S-(6-(3-(2,4-dichlorophenyl)-5-(hydroxyamino)-5-oxopentanamido)hexyl) methanesulfonothioate (56)
Synthesized by Method F using 52 (117 mg, 0.36 mmol, 1.5 equiv.), 36 (90 mg, 0.24 mmol, 1.0 equiv.), HATU (146 mg, 0.38 mmol, 1.6 equiv.), DIPEA (125 μL, 0.72 mmol, 3.0 equiv.) and DMF (2 mL). The title compound (6 mg, 0.01 mmol, 5%) was collected as a colorless oil. 1H NMR (600 MHz, MeOD-d4) δ 7.42 (d, J = 2.2 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.29 (dd, J = 8.5, 2.2 Hz, 1H), 4.17–4.10 (m, 1H), 3.38 (s, 3H), 3.18 (t, J = 7.4 Hz, 2H), 3.12–3.07 (m, 1H), 3.03–2.98 (m, 1H), 2.62 (dd, J = 14.1, 6.4 Hz, 1H), 2.56 (dd, J = 14.1, 8.9 Hz, 1H), 2.47 (d, J = 7.4 Hz, 2H), 1.75–1.69 (m, 2H), 1.40–1.31 (m, 4H), 1.21–1.14 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, MeOD-d4) δ 173.1, 170.1, 139.9, 136.0, 134.0, 130.9, 130.4, 128.4, 50.6, 41.5, 40.0, 38.9, 38.5, 37.1, 30.5, 30.1, 29.2, 27.1. HPLC tR = 5.56 min (>97%, UV230). HRMS (ES+) m/z calcd for [C18H26Cl2N2NaO5S2]+: 507.0552; found 507.0555.
3-(2,4-dichlorophenyl)-5-((3-((methylsulfonyl)thio)propyl)amino)-5-oxopentanoic acid (57)
51 (259 mg, 1.00 mmol, 1.0 equiv.) and 4 (253 mg, 1.50 mmol, 1.5 equiv.) were suspended in anh. CHCl3 (15 mL) and heated to reflux for 2 h. The reaction mixture was cooled to r.t and the precipitate filtered and washed with DCM. The filtrate was reduced in vacuo the crude product purified by RP ACC (gradient: 10–100% MeCN‒H2O in 0.1% formic acid). Appropriate fractions were pooled, MeCN blown off, and the title compound (51 mg, 0.12 mmol, 12% yield) collected as a fluffy white solid after lyophilization.
1H NMR (600 MHz, DMSO-d6) δ 7.91 (t, J = 5.7 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.36 (dd, J = 8.4, 2.1 Hz, 1H), 3.94–3.87 (m, 1H), 3.49 (s, 3H), 3.11–3.01 (m, 4H), 2.60 (d, J = 7.4 Hz, 2H), 2.42 (d, J = 7.5 Hz, 2H), 1.75–1.69 (m, 2H), heteroatom Hs not observed. 13C NMR (151 MHz, DMSO-d6) δ 172.6, 169.9, 140.0, 133.9, 131.4, 129.6, 128.7, 127.2, 50.1, 38.6, 36.9, 34.1, 33.0, 29.1. HPLC tR = 5.52 min (>98%, UV230). HRMS (ES−) m/z calcd for [C15H18Cl2NO5S2]−: 426.0009; found 426.0011.
3-(2,4-dichlorophenyl)-N1-pentyl-N5-((tetrahydro-2H-pyran-2-yl)oxy)pentanediamide (58)
52 (78 mg, 0.21 mmol, 1.0 equiv.) and 1-aminopentane (73 μL, 0.62 mmol, 3.0 equiv.) were dissolved in anh. DMF (3 mL) and the reaction cooled to 0 °C. HATU (87 mg, 0.23 mmol, 1.1 equiv.) was added and stirred for 10 min and the reaction allowed to warm to r.t., stirring for 1 h. DMF was blown dry and the crude yellow oil resuspended in DCM (15 mL) and water (15 mL) and the organic layer separated. The aqueous layer was extracted with DCM (2 × 15 mL) and the combined organic layers dried (Na2SO4) and reduced in vacuo to give the crude product as a colorless oil. The crude product was purified by RP ACC (gradient: 10–55% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled, MeCN blown off, and the title compound (70 mg, 0.16 mmol, 76%) collected as a fluffy white solid after lyophilization.
1H NMR (600 MHz, DMSO-d6) δ 10.95 (d, J = 17.1 Hz, 2H), 7.75 (t, J = 5.7 Hz, 2H), 7.51 (d, J = 1.8 Hz, 2H), 7.38–7.30 (m, 4H), 4.72* (br.s, 1H), 4.57* (br.s, 1H), 3.96–3.80 (m, 4H), 3.48–3.38 (m, 2H), 2.98–2.84 (m, 4H), 2.47–2.28 (m, 8H), 1.66–1.44 (m, 12H), 1.27–1.14 (m, 8H), 1.10–1.02 (m, 4H), 0.81 (t, J = 7.3 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 169.7, 166.9**, 139.7**, 134.1**, 131.5**, 129.9, 128.7**, 127.2**, 101.2**, 61.6**, 38.3, 37.0**, 34.6, 28.8, 28.5, 27.8**, 24.7, 21.9, 18.5**, 14.0, one aliphatic C not observed. HRMS (ES+) m/z calcd for [C21H30Cl2N2NaO4]+: 467.1475; found 467.1486.
* Anomeric protons distinct in diastereomeric mixture – all other peaks overlap.
** Diastereomeric carbons visible as doublets.
3-(2,4-dichlorophenyl)-N1-hydroxy-N5-pentylpentanediamide (59)
Synthesized by Method C using 58 (35 mg, 0.08 mmol, 1.0 equiv.), PPTS (10 mg, 0.04 mmol, 0.5 equiv.) and EtOH (3 mL) and the reaction stirred at 60 °C for 16 h. The crude product was purified by RP ACC (gradient: 10–40% MeCN–H2O in 0.1% formic acid). Appropriate fractions were pooled, MeCN blown off, and the title compound (12 mg, 0.03 mmol, 42%) collected as a fluffy white solid after lyophilization.
1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.69 (br.s, 1H), 7.74 (t, J = 5.7 Hz, 1H), 7.50 (d, J = 2.1 Hz, 1H), 7.35 (dd, J = 8.4, 2.1 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 3.98–3.91 (m, 1H), 2.97–2.85 (m, 2H), 2.44–2.31 (m, 3H), 2.26 (dd, J = 14.6, 7.0 Hz, 1H), 1.27–1.14 (m, 4H), 1.09–1.02 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.5, 166.8, 139.8, 134.0, 131.3, 129.6, 128.7, 127.1, 39.9, 38.2, 36.8, 34.4, 28.7, 28.4, 21.8, 13.9. HPLC tR = 5.67 min (>98%, UV230). HRMS (ES+) m/z calcd for [C16H23Cl2N2O3]+: 361.1080; found 361.1087.
Supplementary Material
ACKNOWLEDGMENT
Research was supported by the National Institutes of Health grants R01 AI153298 and R21 AI137709 (to KDJ), F32 DA044692 (to MEO), R01 AI149444 (to SMC), and T32GM008541 (to MAT through Biomolecular Pharmacology Program); the Natural Sciences and Engineering Research Council of Canada PGSD3–502274 (to LL); the Fulbright Scholar Program (to ALN); and the Skaggs Institute for Chemical Biology (to LL, KDJ).
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393). Additionally, this research used resources beamline 17-ID-1 AMX of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.
Purified BoNT/A was produced by William H. Tepp (UW-Madison). We would like to thank Mark S. Hixon for helpful discussions regarding kinetics for covalent inhibitors. This is Scripps Research Institute Manuscript #29999.
Abbreviations Used
- ACC
automatic column chromatoraphy
- BoNT/A
botulinum neurotoxin serotype A
- CAM
chloramphenicol
- CB
carbenicillin
- CDC
Centers for Disease Control
- DIPEA
N,N-diisopropylethylamine
- FBS
fetal bovine serum
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HC
heavy chain
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- hiPSC
human induced pluripotent stem cell
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LB
lysogeny broth
- LC
light chain
- LDS
lithium dodecyl sulfate
- MTS
methanethiosulfonate
- MTSEA
(2-aminoethyl)methanethiosulfonate
- MTSPA
(3-aminopropoyl)methanethiosulfonate
- NaBH3
sodium borohydride
- NaMTS
sodium methanethiosulfonate
- OTX
O-(Tetrahydro-2H-pyran-2-yl)hydroxylamine
- PAINS
pan-assay interference compounds
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SNAP-25
synaptosomal-associated protein of 25 kDa
- SNARE
soluble N-ethylmaleimide sensitive factor attachment receptor
- TPEN
N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- ZBG
zinc-binding group
Footnotes
Supporting Information.
The following files are available free of charge.
Additional discussion and experimental details for crystallography, enzyme assays, and cell assay (PDF)
1H NMR and 13C NMR spectra of intermediate and final compounds (PDF)
Molecular formula strings of, 20–23, 31–33, 37, 38, 53–57, and 59 (CSV)
Covalent docking of compound 32 in BoNT/A LC (PDB)
Accession Codes
X-ray diffraction data, coordinates, and structure factors for the X-ray crystal structures are deposited with the PDB (www.wwpdb.org) under the accession numbers 6XCB (BoNT/A LC/20), 6XCC (BoNT/A LC/21), 6XCD (BoNT/A LC/22), 6XCE (BoNT/A LC/53), 6XCF (BoNT/A LC/59). Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- 1.Schantz EJ; Johnson EA, Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol. Rev 1992, 56, 80–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnon SS; Schechter R; Inglesby TV; et al. , Botulinum toxin as a biological weapon: Medical and public health management. J. Am. Med. Assoc 2001, 285, 1059–1070. [DOI] [PubMed] [Google Scholar]
- 3.Cocco A; Albanese A, Recent developments in clinical trials of botulinum neurotoxins. Toxicon 2018, 147, 77–83. [DOI] [PubMed] [Google Scholar]
- 4.Jankovic J, Botulinum toxin in clinical practice. J. Neurol. Neurosurg. Psychiatry 2004, 75, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yiannakopoulou E, Serious and Long-Term Adverse Events Associated with the Therapeutic and Cosmetic Use of Botulinum Toxin. Pharmacology 2015, 95, 65–69. [DOI] [PubMed] [Google Scholar]
- 6.Willis B; Eubanks LM; Dickerson TJ; Janda KD, The Strange Case of the Botulinum Neurotoxin: Using Chemistry and Biology to Modulate the Most Deadly Poison. Angew. Chem. Int. Ed 2008, 47, 8360–8379. [DOI] [PubMed] [Google Scholar]
- 7.Osborne SL; Latham CF; Wen PJ; Cavaignac S; Fanning J; Foran PG; Meunier FA, The janus faces of botulinum neurotoxin: Sensational medicine and deadly biological weapon. J. Neurosci. Res 2007, 85, 1149–1158. [DOI] [PubMed] [Google Scholar]
- 8.Wein LM; Liu Y, Analyzing a bioterror attack on the food supply: The case of botulinum toxin in milk. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(CDC), C. f. D. C. a. P. Botulism Annual Summary, 2016; Center for Disease Control and Prevention (CDC): Atlanta, Georgia, 2017. [Google Scholar]
- 10.(CDC), C. f. D. C. a. P. Botulism Annual Summary, 2017; Center for Disease Control and Prevention (CDC): Atlanta, Georgia, 2019. [Google Scholar]
- 11.Passaro DJ; Werner SB; McGee J; Mac Kenzie WR; Vugia DJ, Wound Botulism Associated With Black Tar Heroin Among Injecting Drug Users. JAMA 1998, 279, 859–863. [DOI] [PubMed] [Google Scholar]
- 12.Qureshi IA; Qureshi MA; Vellipuram A-R; Kassar D, Is black tar heroin use associated with wound botulism? A report of two Hispanic patients. Clin. Case. Rep 2018, 6, 1391–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peak CM; Rosen HR; Kamali A; Poe A; Shahkarami M; Kimura AC; Jain S; McDonald E Wound Botulism Outbreak Among Persons Who Use Black Tar Heroin — San Diego County, California, 2017–2018; 2019; pp 1415–1418. [DOI] [PMC free article] [PubMed]
- 14.Koriazova LK; Montal M, Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol 2002, 10, 13–18. [DOI] [PubMed] [Google Scholar]
- 15.Simpson LI, The binary toxin produced by Clostridium botulinum enters cells by receptor-mediated endocytosis to exert its pharmacologic effects. J. Pharmacol. Exp. Ther 1989, 251, 1223–1228. [PubMed] [Google Scholar]
- 16.Blasi J; Chapman ER; Link E; Binz T; Yamasaki S; De Camilli P; Sudhof TC; Niemann H; Jahn R, Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [DOI] [PubMed] [Google Scholar]
- 17.Pellizzari R; Rossetto O; Schiavo G; Montecucco C, Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos. Trans. R. Soc. B 1999, 354, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tacket CO; Shandera WX; Mann JM; Hargrett NT; Blake PA, Equine antitoxin use and other factors that predict outcome in type A foodborne botulism. Am. J. Med 1984, 76, 794–798. [DOI] [PubMed] [Google Scholar]
- 19.Simpson L, The life history of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [DOI] [PubMed] [Google Scholar]
- 20.Pellett S; Bradshaw M; Tepp WH; Pier CL; Whitemarsh RCM; Chen C; Barbieri JT; Johnson EA, The Light Chain Defines the Duration of Action of Botulinum Toxin Serotype A Subtypes. mBio 2018, 9, e00089–18 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitemarsh RCM; Tepp WH; Johnson EA; Pellett S, Persistence of Botulinum Neurotoxin A Subtypes 1–5 in Primary Rat Spinal Cord Cells. PLoS One 2014, 9, e90252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moritz MS; Tepp WH; Bradshaw M; Johnson EA; Pellett S, Isolation and Characterization of the Novel Botulinum Neurotoxin A Subtype 6. mSphere 2018, 3, e00466–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin L; Olson ME; Eubanks LM; Janda KD, Strategies to Counteract Botulinum Neurotoxin A: Nature’s Deadliest Biomolecule. Acc. Chem. Res 2019, 52, 2322–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen AY; Adamek RN; Dick BL; Credille CV; Morrison CN; Cohen SM, Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev 2019, 119, 1323–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boldt GE; Kennedy JP; Janda KD, Identification of a Potent Botulinum Neurotoxin A Protease Inhibitor Using in Situ Lead Identification Chemistry. Org. Lett 2006, 8, 1729–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Čapková K; Yoneda Y; Dickerson TJ; Janda KD, Synthesis and structure–activity relationships of second-generation hydroxamate botulinum neurotoxin A protease inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 6463–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stowe GN; Šilhár P; Hixon MS; Silvaggi NR; Allen KN; Moe ST; Jacobson AR; Barbieri JT; Janda KD, Chirality Holds the Key for Potent Inhibition of the Botulinum Neurotoxin Serotype A Protease. Org. Lett 2010, 12, 756–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seki H; Pellett S; Šilhár P; Stowe GN; Blanco B; Lardy MA; Johnson EA; Janda KD, Synthesis/biological evaluation of hydroxamic acids and their prodrugs as inhibitors for Botulinum neurotoxin A light chain. Biorg. Med. Chem 2014, 22, 1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silvaggi NR; Boldt GE; Hixon MS; Kennedy JP; Tzipori S; Janda KD; Allen KN, Structures of Clostridium botulinum Neurotoxin Serotype A Light Chain Complexed with Small-Molecule Inhibitors Highlight Active-Site Flexibility. Chem. Biol 2007, 14, 533–542. [DOI] [PubMed] [Google Scholar]
- 30.Šilhár P; Silvaggi NR; Pellett S; Čapková K; Johnson EA; Allen KN; Janda KD, Evaluation of adamantane hydroxamates as botulinum neurotoxin inhibitors: Synthesis, crystallography, modeling, kinetic and cellular based studies. Biorg. Med. Chem 2013, 21, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boldt GE; Kennedy JP; Hixon MS; McAllister LA; Barbieri JT; Tzipori S; Janda KD, Synthesis, Characterization and Development of a High-Throughput Methodology for the Discovery of Botulinum Neurotoxin A Inhibitors. J. Comb. Chem 2006, 8, 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thyagarajan B; Potian JG; Garcia CC; Hognason K; Čapková K; Moe ST; Jacobson AR; Janda KD; McArdle JJ, Effects of hydroxamate metalloendoprotease inhibitors on botulinum neurotoxin A poisoned mouse neuromuscular junctions. Neuropharmacology 2010, 58, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith GR; Caglič D; Čapek P; Zhang Y; Godbole S; Reitz AB; Dickerson TJ, Reexamining hydroxamate inhibitors of botulinum neurotoxin serotype A: Extending towards the β-exosite. Bioorg. Med. Chem. Lett 2012, 22, 3754–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D; Mobashery S, Mechanism-based Inhibition of Metalloproteinases. Frontiers in Medicinal Chemistry - Online 2004, 1, 77–83. [Google Scholar]
- 35.Stura EA; Le Roux L; Guitot K; Garcia S; Bregant S; Beau F; Vera L; Collet G; Ptchelkine D; Bakirci H; Dive V, Structural Framework for Covalent Inhibition of Clostridium botulinum Neurotoxin A by Targeting Cys(165). J. Biol. Chem 2012, 287, 33607–33614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Čapková K; Hixon MS; Pellett S; Barbieri JT; Johnson EA; Janda KD, Benzylidene cyclopentenediones: First irreversible inhibitors against botulinum neurotoxin A’s zinc endopeptidase. Bioorg. Med. Chem 2010, 20, 206–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardinale SC; Butler MM; Ruthel G; Nuss JE; Wanner LM; Li B; Pai R; Peet NP; Bavari S; Bowlin TL, Novel Benzimidazole Inhibitors of Botulinum Neurotoxin/A Display Enzyme and Cell-Based Potency. Botulinum J 2011, 2, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B; Cardinale SC; Butler MM; Pai R; Nuss JE; Peet NP; Bavari S; Bowlin TL, Time-dependent botulinum neurotoxin serotype A metalloprotease inhibitors. Biorg. Med. Chem 2011, 19, 7338–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bremer PT; Hixon MS; Janda KD, Benzoquinones as inhibitors of botulinum neurotoxin serotype A. Biorg. Med. Chem 2014, 22, 3971–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garland M; Babin BM; Miyashita S-I; Loscher S; Shen Y; Dong M; Bogyo M, Covalent Modifiers of Botulinum Neurotoxin Counteract Toxin Persistence. ACS Chem. Biol 2019, 14, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saunders MJ; Graves SW; Sklar LA; Oprea TI; Edwards BS, High-throughput multiplex flow cytometry screening for botulinum neurotoxin type a light chain protease inhibitors. Assay Drug Dev. Technol 2010, 8, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noguchi N, Ebselen, a useful tool for understanding cellular redox biology and a promising drug candidate for use in human diseases. Arch. Biochem. Biophys 2016, 595, 109–112. [DOI] [PubMed] [Google Scholar]
- 43.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H, Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [DOI] [PubMed] [Google Scholar]
- 44.Hanson MA; Stevens RC, Cocrystal structure of synaptobrevin-II bound to botulinum neurotoxin type B at 2.0 Å resolution. Nat. Struct. Biol 2000, 7, 687–692. [DOI] [PubMed] [Google Scholar]; *The authors would like to note that this publication has been retracted due to inconsistencies in an X-ray crystal structure that is unrelated to the discussion in this manuscript. As such, PDB: 1F82 remains a valid X-ray crystal structure of BoNT/B LC.
- 45.Agarwal R; Eswaramoorthy S; Kumaran D; Binz T; Swaminathan S, Structural Analysis of Botulinum Neurotoxin Type E Catalytic Domain and Its Mutant Glu212→Gln Reveals the Pivotal Role of the Glu212 Carboxylate in the Catalytic Pathway. Biochemistry 2004, 43, 6637–6644. [DOI] [PubMed] [Google Scholar]
- 46.Agarwal R; Binz T; Swaminathan S, Structural Analysis of Botulinum Neurotoxin Serotype F Light Chain: Implications on Substrate Binding and Inhibitor Design. Biochemistry 2005, 44, 11758–11765. [DOI] [PubMed] [Google Scholar]
- 47.Jin R; Sikorra S; Stegmann CM; Pich A; Binz T; Brunger AT, Structural and Biochemical Studies of Botulinum Neurotoxin Serotype C1 Light Chain Protease: Implications for Dual Substrate Specificity. Biochemistry 2007, 46, 10685–10693. [DOI] [PubMed] [Google Scholar]
- 48.Arndt JW; Yu W; Bi F; Stevens RC, Crystal Structure of Botulinum Neurotoxin Type G Light Chain: Serotype Divergence in Substrate Recognition. Biochemistry 2005, 44, 9574–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masuyer G; Zhang S; Barkho S; Shen Y; Henriksson L; Košenina S; Dong M; Stenmark P, Structural characterisation of the catalytic domain of botulinum neurotoxin X - high activity and unique substrate specificity. Sci. Rep 2018, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arndt JW; Chai Q; Christian T; Stevens RC, Structure of Botulinum Neurotoxin Type D Light Chain at 1.65 Å Resolution: Repercussions for VAMP-2 Substrate Specificity. Biochemistry 2006, 45, 3255–3262. [DOI] [PubMed] [Google Scholar]
- 51.Silvaggi NR; Boldt GE; Hixon MS; Kennedy JP; Tzipori S; Janda KD; Allen KN, Structures of Clostridium botulinum Neurotoxin Serotype A Light Chain complexed with small-molecule inhibitors highlight active-site flexibility. Chem. Biol 2007, 14, 533–42. [DOI] [PubMed] [Google Scholar]
- 52.Caglič D; Krutein MC; Bompiani KM; Barlow DJ; Benoni G; Pelletier JC; Reitz AB; Lairson LL; Houseknecht KL; Smith GR; Dickerson TJ, Identification of Clinically Viable Quinolinol Inhibitors of Botulinum Neurotoxin A Light Chain. J. Med. Chem 2014, 57, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai H; Feng M; Roxas-Duncan V; Dakshanamurthy S; Smith LA; Yang DCH, Quinolinol and peptide inhibitors of zinc protease in botulinum neurotoxin A: Effects of zinc ion and peptides on inhibition. Arch. Biochem. Biophys 2009, 491, 75–84. [DOI] [PubMed] [Google Scholar]
- 54.Deshpande SS; Sheridan RE; Adler M, Efficacy of certain quinolines as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1997, 35, 433–445. [DOI] [PubMed] [Google Scholar]
- 55.Bremer PT; Adler M; Phung CH; Singh AK; Janda KD, Newly Designed Quinolinol Inhibitors Mitigate the Effects of Botulinum Neurotoxin A in Enzymatic, Cell-Based, and ex Vivo Assays. J. Med. Chem 2017, 60, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Konstantinović J; Kiris E; Kota KP; Kugelman-Tonos J; Videnović M; Cazares LH; Terzić Jovanović N; Verbić TŽ; Andjelković B; Duplantier AJ; Bavari S; Šolaja BA, New Steroidal 4-Aminoquinolines Antagonize Botulinum Neurotoxin Serotype A in Mouse Embryonic Stem Cell Derived Motor Neurons in Postintoxication Model. J. Med. Chem 2018, 61, 1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shine N; Suryadi K, New FRET Substrate for Botulinum Neurotoxin Type A. In 7th International Conference on Basic and Therapeutic Aspects of Botulinum and Tetanus Toxins, Santa Fe, NM, USA, 2011. [Google Scholar]
- 58.Capková K; Hixon MS; McAllister LA; Janda KD, Toward the discovery of potent inhibitors of botulinum neurotoxin A: development of a robust LC MS based assay operational from low to subnanomolar enzyme concentrations. Chem. Comm 2008, 3525–3527. [DOI] [PubMed] [Google Scholar]
- 59.Liu S; Zhou B; Yang H; He Y; Jiang Z-X; Kumar S; Wu L; Zhang Z-Y, Aryl Vinyl Sulfonates and Sulfones as Active Site-Directed and Mechanism-Based Probes for Protein Tyrosine Phosphatases. J. Am. Chem. Soc 2008, 130, 8251–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maurer T; Fung HL, Comparison of methods for analyzing kinetic data from mechanism-based enzyme inactivation: application to nitric oxide synthase. AAPS PharmSci 2000, 2, E8–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mons E; Jansen IDC; Loboda J; van Doodewaerd BR; Hermans J; Verdoes M; van Boeckel CAA; van Veelen PA; Turk B; Turk D; Ovaa H, The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc 2019, 141, 3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silva DG; Ribeiro JFR; De Vita D; Cianni L; Franco CH; Freitas-Junior LH; Moraes CB; Rocha JR; Burtoloso ACB; Kenny PW; Leitão A; Montanari CA, A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett 2017, 27, 5031–5035. [DOI] [PubMed] [Google Scholar]
- 63.Feltrup TM; Singh BR, Development of a Fluorescence Internal Quenching Correction Factor to Correct Botulinum Neurotoxin Type A Endopeptidase Kinetics Using SNAPtide. Anal. Chem 2012, 84, 10549–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eubanks LM; Hixon MS; Jin W; Hong S; Clancy CM; Tepp WH; Baldwin MR; Malizio CJ; Goodnough MC; Barbieri JT; Johnson EA; Boger DL; Dickerson TJ; Janda KD, An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatheway CH; Snyder JD; Seals JE; Edell TA; Lewis GE Jr., Antitoxin levels in botulism patients treated with trivalent equine botulism antitoxin to toxin types A, B, and E. J. Infect. Dis 1984, 150, 407–412. [DOI] [PubMed] [Google Scholar]
- 66.Zhu K; Borrelli KW; Greenwood JR; Day T; Abel R; Farid RS; Harder E, Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model 2014, 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- 67.Hu J; Van den Steen PE; Sang Q-XA; Opdenakker G, Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov 2007, 6, 480–498. [DOI] [PubMed] [Google Scholar]
- 68.Jacobsen JA; Major Jourden JL; Miller MT; Cohen SM, To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta Mol. Cell. Res 2010, 1803, 72–94. [DOI] [PubMed] [Google Scholar]
- 69.Agrawal A; Romero-Perez D; Jacobsen JA; Villarreal FJ; Cohen SM, Zinc-Binding Groups Modulate Selective Inhibition of MMPs. ChemMedChem 2008, 3, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bremer PT; Pellett S; Carolan JP; Tepp WH; Eubanks LM; Allen KN; Johnson EA; Janda KD, Metal Ions Effectively Ablate the Action of Botulinum Neurotoxin A. J. Am. Chem. Soc 2017, 139, 7264–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baldwin MR; Bradshaw M; Johnson EA; Barbieri JT, The C-terminus of botulinum neurotoxin type A light chain contributes to solubility, catalysis, and stability. Protein Expression Purif 2004, 37, 187–195. [DOI] [PubMed] [Google Scholar]
- 72.Silvaggi NR; Wilson D; Tzipori S; Allen KN, Catalytic Features of the Botulinum Neurotoxin A Light Chain Revealed by High Resolution Structure of an Inhibitory Peptide Complex. Biochemistry 2008, 47, 5736–5745. [DOI] [PubMed] [Google Scholar]
- 73.Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 74.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Terwilliger TC; Grosse-Kunstleve RW; Afonine PV; Moriarty NW; Zwart PH; Hung L-W; Read RJ; Adams PD, Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D. Biol. Crystallogr 2008, 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 77.Liebschner D; Afonine PV; Moriarty NW; Poon BK; Sobolev OV; Terwilliger TC; Adams PD, Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. Sect. D. Biol. Crystallogr 2017, 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schrodinger LLC, The PyMOL Molecular Graphics System, Version 1.8 2015.
- 79.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA, OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 80.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Madhavi Sastry G; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 82.Malizio CJ; Goodnough MC; Johnson EA, Purification of Clostridium botulinum Type A Neurotoxin In Bacterial Toxins: Methods and Protocols, Holst O, Ed. Humana Press: Totowa, NJ, 2000; pp 27–39. [DOI] [PubMed] [Google Scholar]
- 83.Pellett S; Tepp WH; Clancy CM; Borodic GE; Johnson EA, A neuronal cell-based botulinum neurotoxin assay for highly sensitive and specific detection of neutralizing serum antibodies. FEBS Lett 2007, 581, 4803–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mosmann T, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- 85.Irwin JJ; Duan D; Torosyan H; Doak AK; Ziebart KT; Sterling T; Tumanian G; Shoichet BK, An Aggregation Advisor for Ligand Discovery. J. Med. Chem 2015, 58, 7076–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.