

Abstract
Background: Telomerase reverse transcriptase (TERT) promoter mutations play a role in carcinogenesis and are found in both tumors and cancer cell lines. TERT promoter methylation, transcription factor binding, chromatin remodeling, and alternative splicing are also known to play an integral role in TERT regulation.
Methods: Using nanopore Cas9 targeted sequencing, we characterized allele-specific methylation in thyroid cancer cell lines heterozygous for the TERT promoter mutation. Furthermore, using chromatin immunoprecipitation followed by Sanger sequencing, we probed allele-specific binding of the transcription factors GABPA (GA binding protein transcription factor subunit alpha) and MYC, as well as the chromatin marks H3K4me3 and H3K27me3. Finally, using coding single nucleotide polymorphisms and the long-read sequencing, we examined complementary DNA for monoallelic expression (MAE).
Results: We found the mutant TERT promoter allele to be significantly less methylated than wild type, while more methylated in the gene body in heterozygous TERT mutant cell lines. We demonstrated that the transcriptional activators GABPA and MYC bind only to the mutant TERT allele. In addition, the activating and repressive chromatin marks H3K4me3 and H3K27me3, respectively, bind mutant and wild-type alleles exclusively. Finally, in heterozygous mutant cell lines, TERT exhibits MAE from the mutant allele only.
Conclusions: In summary, by employing new long-read sequencing methods, we were able to definitively demonstrate allele-specific DNA methylation, histone modifications, transcription factor binding, and the resulting monoallelic transcription in cell lines with heterozygous TERT mutations.
Keywords: telomerase, DNA methylation, transcriptional regulation, allele specificity
Introduction
The catalytic subunit of telomerase, telomerase reverse transcriptase (TERT), is silenced in somatic cells and transcriptionally active in stem cells and cancer cells (1). Its activation is regulated by multiple factors, including promoter mutations, chromatin status, promoter methylation, transcription factor binding, and alternative splicing of TERT (2–5). It is believed that different types of regulation can work in concert and modulate each other's effects on transcription (6). Previously, we characterized TERT promoter methylation, transcription factor binding, and their relationship to TERT promoter mutations in thyroid cancer cell lines (7). Specifically, we identified a unique methylation pattern suggesting allele-specific methylation in heterozygous TERT mutant thyroid cancer cell lines. Herein, we investigate the allele-specific effects of TERT promoter mutations on promoter methylation, transcription factor binding, chromatin marks, and transcriptional activation.
Activating heterozygous TERT promoter mutations occurs upstream of the translational start site, most commonly at −124 C > T and −146 C > T relative to ATG (8–10). Compared with the −146 C > T mutation, the −124 C > T mutation produces higher levels of TERT promoter transcriptional activation (11). Overall, among differentiated thyroid cancers, 11.7% of papillary thyroid cancers and 11.4% of follicular thyroid cancers harbor the TERT mutation, with incidence associated with advanced stages of disease and aggressive clinicopathologic characteristics, and the TERT mutation is present in the majority of established thyroid cancer cell lines (12–15). Both −124 C > T and −146 C > T mutations create a binding site for the E-twenty-six (ETS) family of transcription factors. Previous work in several cancer types has shown GA binding protein transcription factor subunit alpha (GABPA) to bind at the TERT promoter, including in thyroid cancer (11,16). GABPA binds as a heterotetramer with GABPB to activate transcription of TERT (11). In a recent study, we demonstrated binding of GABPA to the TERT promoter in thyroid cancer cell lines (7). Previous studies in glioblastoma, melanoma, hepatocellular carcinoma, and urothelial carcinoma showed GABPA binding in an allele-specific manner to only the mutant TERT allele (11,17).
Recently, ETS variant 5 (ETV5) was shown to bind at higher levels than GABPA in cell lines derived from highly aggressive anaplastic thyroid cancers, but not shown to predominantly bind in well-differentiated thyroid cancer cell lines (18). Furthermore, allele-specific binding has not been previously demonstrated in TERT mutant well-differentiated thyroid cancer cell lines (16).
TERT promoter methylation may also play a key regulatory role, including inhibition of transcription factor binding and changing of chromatin state (19–21). In other genomic locations, DNA methylation blocks binding of the transcriptional activator MYC (22), which also binds to the TERT minimal promoter to activate TERT transcription (23). Our previous work demonstrated binding of MYC at the minimal TERT promoter and methylation patterns in heterozygous TERT mutant thyroid cancer cell lines in about 50% methylation (7). This suggested that allele-specific methylation in heterozygous mutant cell lines might play a role in regulation.
In support of this, TERT mutant cancers were recently shown to display lower levels of methylation at the TERT promoter than wild-type cancers (24). However, allele-specific methylation at the TERT promoter or allele-specific binding of MYC to TERT has not been shown. This is in part because standard methylation sequencing has mostly been performed in the context of sodium bisulfite pretreatment of DNA, which reverts both TERT promoter mutations back to wild type (C- > T). New nanopore sequencing technology (25) relies on Cas9 targeting and cutting at specific genomic locations, allowing for targeted long-read sequencing. Unlike Illumina sequencing based on florescence, nanopore sequencing detects nucleotide-specific changes in ionic current as a single molecule of DNA moves through a protein pore inserted into a synthetic polymer membrane (26).
Importantly, nanopore sequencing can distinguish cytosine from methyl-cytosine, negating the need for sodium bisulfite pretreatment of DNA. This profiles DNA methylation at the targeted sequence directly, thus enabling one to assess methylation in an allele-specific manner over a much larger genomic region than previously possible.
The TERT mutation is believed to play a role in transcriptional activation, and the TERT mutant allele is associated with RNA polymerase II and activating chromatin marks. In cells heterozygous for the TERT promoter mutation, the mutant allele exhibits activating H3K4me2/3 chromatin marks, while the wild-type allele exhibits silencing H3K27me3 marks (17). Furthermore, a subset of cancer cell lines appear to show monoallelic expression (MAE) of TERT from the TERT mutant promoter (27). Studies have identified heterozygous single nucleotide polymorphisms (SNPs) in the TERT coding region and demonstrated only one allele in the TERT complementary DNA (cDNA) transcript (17,24). However, since the TERT promoter mutation is not represented in the coding sequence, studies have not definitively linked the TERT promoter mutation with MAE from the mutated allele.
In this study, we definitively demonstrate for the first time allele-specific methylation at the TERT promoter and into the gene body, in which the mutant allele is significantly less methylated at the TERT promoter than the wild-type allele, while the mutant allele is more methylated into the gene body than the wild-type allele, consistent with the methylation pattern of actively transcribed genes (28,29). We utilized chromatin immunoprecipitation (ChIP) followed by Sanger sequencing to confirm allele-specific binding of GABPA to the mutant TERT allele in thyroid cancer cell lines and demonstrate for the first time similar allele-specific binding of MYC. Furthermore, we show chromatin marks associated with active transcription (H3K4me3) on only the mutant allele, while repressive chromatin marks (H3K27me3) associate with the wild-type allele.
The allele-specific TERT promoter methylation, differential histone modifications, and activator binding, all result in MAE, demonstrated by phasing a SNP present in exon 2 of TERT with the TERT promoter mutation. In summary, we report the first direct evidence of MAE by the TERT promoter mutation and confirm previous findings in other cancer types of allele-specific binding of GABPA and chromatin marks in thyroid cancer cell lines. Additionally, new long-read sequencing technology allowed the demonstration of allele-specific methylation, allele-specific MYC binding, and MAE of the TERT mutant allele in heterozygous TERT mutant cell lines, deepening our understanding of the mechanism of TERT activation in cancer.
Materials and Methods
Cell lines, culture conditions, and TERT mutation status
Papillary [TPC-1 (30), BCPAP (31)] and follicular thyroid cancer cell lines [FTC-133 (32), FTC-238 (32), and WRO (33)] were kindly provided by Dr. Motoyasu Saji (The Ohio State University Wexner Medical Center, Columbus, OH). Cell lines were grown as previously described (7). The normal thyroid follicular epithelial cell line Nthy-ori-3 (34) was kindly provided by Dr. Thomas Fahey (Weill Cornell Medical Center, New York, NY). Nthy-ori-3 was grown in Roswell Park Memorial Institute-1640 (RPMI-1640; Sigma–Aldrich, St. Louis, MO) medium with 10% heat-inactivated fetal bovine serum (GE Healthcare Life Sciences, Marlborough, MA).
All cell lines were authenticated by short tandem repeat profiling and mycoplasma testing by polymerase chain reaction (PCR)-based MycoDtect Kit within a year of experiments. Cell lines were used below the laboratory's passage 12. Sanger sequencing of the TERT promoter to determine TERT mutation status was conducted as previously described (7).
Nanopore Cas9 targeted sequencing
Nanopore Cas9 targeted sequencing (nCATS) was conducted as previously described (25). Briefly, two crRNAs were designed to target 7.806 kb surrounding the TERT region at chr5:1,288,699–1,296,505: Forward “AAGGCTTAGGGATCACTAAG” and Reverse “AGCGGCAGGTGCCCAGAATA.” crRNAs were mixed at equimolar amounts to a final concentration of 100 μM. After duplex formation, dephosphorylation of the genomic DNA, cleavage and dA tailing, adaptor ligation, and cleanup (25), each sample was sequenced on a nanopore flow cell (version 9.4.1) using the GridION sequencer (Oxford Nanopore Technologies, Oxford, UK).
Data processing for methylation
Analysis of the nanopore data was conducted as previously described (25). Base calling to generate FASTQ reads was performed by the GUPPY algorithm. The resulting reads were aligned to the human genome, hg19, by Minimap2 (35). CpG methylation calling was conducted using nanopolish (36). Reads were phased into wild-type or mutant allele by identifying the promoter motif in FASTQ reads.
ChIP analysis
ChIP analysis was performed as previously described (7). The following antibodies were used (10 μg per ChIP): anti-MYC (No. 9402; Cell Signaling Technology, Danvers, MA), anti-GABPA (No. 27795; Thermo Fisher, Waltham, MA), anti-H3K4me3 (No. 9727; Cell Signaling Technology), anti-H3K27me3 (No. 9733; Cell Signaling Technology), and anti-ETV5 (No. 30023; Thermo Fisher). DNA was purified by MinElute PCR Purification Kit (Qiagen, Hilden, Germany). Quantitative PCR (qPCR) of ChIP and input DNA was carried out as previously described (7), in triplicate, with positive and negative control regions for each antibody described in Supplementary Table S1 and Supplementary Figure S1.
Factor binding was determined by the percentage of input normalization method (37), normalizing the binding at the TERT locus, encompassing the TERT promoter mutation and transcription start site (TSS), relative to DNA input into ChIP. After ChIP, immunoprecipitated DNA and input DNA were amplified by PCR surrounding the TERT −124 C > T mutation and Sanger sequenced (7).
Real-time quantitative telomeric repeat amplification protocol
Cells were lysed in CHAPS lysis buffer (Cell Signaling Technology) and incubated on ice for 20 minutes. Cell debris was removed by centrifugation, and total protein concentration was measured. Negative heat inactivation and no template controls were utilized; positive telomerase quantitation controls were performed using serial dilutions of HEK293T cells (ATCC ACS-4500). The 25 μL reaction contained 1 × SYBR Green (Thermo Fisher), 0.1 μg of TS and ACX primers, and 250 ng of protein extract. qPCR was performed as previously described (38).
TERT expression quantitative reverse transcriptase-PCR
RNA was extracted (7) from cell lines and thyroid tissue using TRIzol isolation and High Pure RNA Kits (Roche, Basel, Switzerland), respectively. SuperScript III Reverse Transcriptase (Thermo Fisher) was utilized to reverse transcribe RNA to cDNA. Full-length TERT transcript levels were detected by qPCR using SYBR Green PCR Master Mix (Thermo Fisher) (39). TERT expression was normalized to GAPDH expression.
Allele-specific transcription characterization
SNPs in the thyroid cancer cell lines were identified by Sanger sequencing of genomic DNA in the TERT 3′ untranslated region (UTR) and by integrative genome viewer examination of the nCATS data in the TERT 5′ UTR and exons 1 and 2. Isolated RNA from the cell lines was DNase I treated and reverse transcribed as above, then amplified at the SNP location (Supplementary Table S2) and Sanger sequenced utilizing the forward primer to determine the genotype.
Results
Allele-specific methylation patterns in thyroid cancer cell lines
DNA isolated from thyroid cancer cell lines underwent enrichment and nanopore sequencing at the TERT promoter by nCATS to determine the methylation status of the TERT promoter. Nanopore methylation calls for all cell lines were compared and found comparable to previously published Illumina bisulfite sequencing of the TERT promoter (7) (Supplementary Fig. S2). Unlike traditional bisulfite treatment, in which the TERT mutation status (C>T) is lost, nanopore sequencing identifies native methylation without bisulfite treatment and thereby maintains the ability to differentiate mutation status between alleles. In the heterozygous TERT mutant cell line TPC-1, a significant drop in methylation surrounding the TERT TSS occurred on the mutant allele, to <5% methylation (Fig. 1).
FIG. 1.

DNA methylation assessed by nCATS shows drop in methylation surrounding TSS and higher levels of methylation in the gene body of mutant TERT allele in heterozygous TPC-1 thyroid cancer cell line. (A) Read-level methylation plots using IGV. Depth of coverage at the TERT gene is shown in the top graph where the coverage at each base is indicated by the gray line plot with the minimum and maximum CpG coverage stated. Each horizontal line in the wild-type (top) and mutant allele (bottom) plots is a single nCATS read. Red denotes a methylated CpG, and blue denotes an unmethylated CpG. The TERT chromosomal coordinates are shown at the bottom to designate position. (B) Average methylation plots. The wild-type allele is depicted in purple, and the mutant TERT allele in orange. Individual dots represent methylation at an individual CpG site. The orange or purple line represents the smoothened level of methylation over the entire area. The black arrow denotes the TSS. TERT exons 1 and 2 are shown by thick black lines. The gene reads from right to left into the gene body are denoted by the dashed arrow. The red line indicates the PCR-amplified region used in subsequent ChIP experiments (Figs. 4 and 5). ChIP, chromatin immunoprecipitation; IGV, Integrative genome viewer; nCATS, nanopore Cas9 targeted sequencing; PCR, polymerase chain reaction; TERT, telomerase reverse transcriptase; TSS, transcription start site.
The mutant allele also showed higher levels of methylation throughout the TERT gene body at or above 75%, while the wild-type allele remained comparatively stable at ∼50–75% methylation over the TERT gene body and promoter. The other heterozygous TERT mutant cell lines BCPAP and FTC-238 followed the same pattern as TPC-1, in which there was a drop in methylation at the TSS with only 20% and 30% of the mutant allele methylated in BCPAP and FTC-238, respectively (Fig. 2; Supplementary Fig. S3). The heterozygous mutant cell lines also exhibited the higher levels of methylation in the gene body. In BCPAP, a drop in methylation in the wild-type allele was observed in the gene body, specifically in the intron between exons 2 and 3.
FIG. 2.

Average methylation plots of the TERT promoter and gene body in homozygous versus heterozygous TERT mutant thyroid cancer cell lines. Methylation levels were in the following thyroid cancer cell lines: wild-type cell line WRO, heterozygous TERT mutant cell lines BCPAP and FTC-238, and homozygous mutant cell line FTC-133. The wild-type allele is depicted in purple, and the mutant TERT allele in orange. Individual dots represent methylation at an individual CpG site. The purple or orange line represents the smoothened methylation over the entire area, showing drops in methylation surrounding the TSS, and higher levels in the gene body, of the mutant allele. The black arrow denotes the TSS. TERT exons 1 and 2 are shown by thick black lines. The gene reads from right to left into the gene body are denoted by the dashed arrow.
The homozygous TERT mutant cell line FTC-133 showed a methylation pattern consistent with the mutant alleles of the heterozygous cell lines, showing a drop in methylation to <15% at the TSS and high gene body methylation. Conversely, the homozygous wild-type cell line WRO showed no significant drop in methylation at the TSS (above 50%) and lower levels of 50–75% methylation in the gene body, reflective of the wild-type alleles in the heterozygous mutant cell lines (Fig. 2). Further examination of TERT promoter methylation revealed low methylation surrounding the TSS in the mutant allele (4.8%, 20%, and 31.8% average methylation for TPC-1, BCPAP, and FTC-238, respectively), while high levels of methylation were seen in the wild-type allele (Fig. 3) (79.9%, 73.1%, and 75.1% average methylation for TPC-1, BCPAP, and FTC-238, respectively).
FIG. 3.

Allele-specific methylation at the TERT promoter in thyroid cancer cell lines by nCATS. Methylation levels at the TERT minimal promoter, including the TSS, GABPA binding site (TERT mutation), and MYC binding site in heterozygous TERT mutant cell lines TPC-1, BCPAP, and FTC-238, wild-type cell line WRO, and homozygous TERT mutant cell line FTC-133. CpG location is denoted by chromosomal location. The wild-type allele is shown in gray with open circles, and the mutant allele is shown in black with solid circles. GABPA, GA binding protein transcription factor subunit alpha.
Furthermore, the regions surrounding the TERT promoter mutation at the GABPA binding site, as well as the MYC binding site, also exhibited significantly less methylation on the mutant allele, with the exception of the GABPA site in FTC-238, which exhibited low methylation of both alleles. Homozygous cell lines followed similar patterns of methylation, with the homozygous mutant cell line exhibiting low levels of methylation at the promoter (<20% methylation), and the homozygous wild-type cell line showing higher levels of methylation (30–65% methylation) (Fig. 3).
Allele-specific transcription factor binding in thyroid cancer cell lines
Since the binding sites for GABPA and MYC were differentially methylated in our heterozygous mutant thyroid cancer cell lines, we investigated whether these factors exhibited allele-specific binding as well. ChIP analysis of the two ETS factors known to bind at the TERT promoter, ETV5 and GABPA, showed GABPA as the predominant factor binding to the TERT promoter in our well-differentiated thyroid cancer cell lines (Supplementary Fig. S4). We therefore performed Sanger sequencing of the TERT promoter on DNA precipitated with either MYC or GABPA antibodies. Sanger sequencing of immunoprecipitated DNA from TPC-1, BCPAP, and FTC-238 cell lines resulted in only the mutant TERT allele, indicating selective binding of GABPA and MYC to the mutant allele (Fig. 4).
FIG. 4.
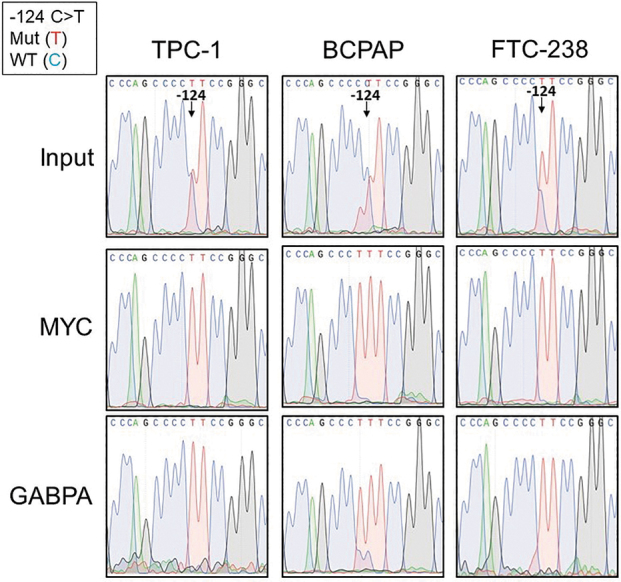
Allele-specific transcription factor binding of MYC and GABPA at the TERT promoter in heterozygous TERT mutant thyroid cancer cell lines. Chromatin immunoprecipitation (ChIP) in the heterozygous TERT mutant thyroid cancer cell lines with either MYC or GABPA antibodies followed by Sanger sequencing of the TERT promoter at the −124 C > T TERT mutation compared with input genomic DNA before ChIP. At the −124 TERT position, the mutant allele is represented by the red curve (T), while the wild-type allele is represented by the blue curve (C). The input genomic DNA shows the presence of both alleles at the −124 position, while the immunoprecipitated DNA shows only the red curve, indicative of the mutant allele.
Allele-specific chromatin marks in thyroid cancer cell lines
To further investigate transcriptional regulation, ChIP followed by Sanger sequencing of the region of the TERT promoter encompassing the TERT mutation and TSS was conducted on DNA immunoprecipitated with antibodies directed against either H3K4me3 (activating) or H3K27me3 (silencing). This revealed the least amount of active chromatin mark, H3K4me3, in the wild-type cell lines Nthy-ori-3 and WRO, an intermediate amount in the heterozygous TERT mutant cell lines (TPC-1, BCPAP, and FTC-238), and the highest amount associated with the homozygous TERT mutant cell line FTC-133 (Fig. 5A). The silent chromatin mark, H3K27me3, displayed the opposite trend, with the highest level in the homozygous wild-type cell lines and the least abundant in the homozygous TERT mutant cell line. To further delineate the effect of the TERT mutation on chromatin marks, we conducted Sanger sequencing of the immunoprecipitated DNA at the TERT locus. In the heterozygous TERT mutant cell lines, we observed an allele-specific association with chromatin marks. The TERT mutant allele preferentially bound to the active H3K4me3, while the wild-type allele bound to the silent H3K27me3 (Fig. 5B).
FIG. 5.

Allele-specific chromatin marks at the TERT minimal promoter in thyroid cancer cell lines. (A) ChIP with either H3K4me3 (activating chromatin mark) or H3K27me3 (silencing chromatin mark) antibodies. Binding was measured relative to 5% input chromatin, and binding at the TERT locus assessed by qPCR. H3K4me3 binding at the TERT promoter was most abundant in the homozygous mutant cell line FTC-133 (black bar), intermediate in the heterozygous mutant cell lines (gray bars), and least abundant in the homozygous wild-type cell lines (white bars with cancer cell line WRO denoted by stipple compared with normal cell line NThy-ori-3). H3K27me3 binding was reciprocal of the H3K4me3 binding. All error bars represent standard error within triplicates. (B) Sanger sequencing of the TERT promoter immunoprecipitated DNA of heterozygous TERT mutant thyroid cancer cell lines at the −124 C > T mutation compared with input genomic DNA before ChIP. At the −124 TERT position, the mutant allele is represented by the red curve (T), while the wild-type allele is represented by the blue curve (C). The input genomic DNA shows the presence of both alleles at the −124 position, while the H3K4me3 immunoprecipitated DNA shows only the red curve, indicative of the mutant allele. H3K27me3 immunoprecipitated DNA shows only the blue curve, indicative of the wild-type allele. qPCR, quantitative PCR.
Monoallelic expression of TERT
To determine if MAE of TERT occurs in our heterozygous TERT mutant thyroid cancer cell lines, we identified SNPs in the TERT coding sequence. As previously described (18), TPC-1 contains a heterozygous G > A SNP in exon 2 (rs2736098). In the heterozygous cell lines BCPAP and FTC-238, a C > T SNP (rs2853690) was identified in the TERT 3′ UTR by Sanger sequencing of the genomic DNA (Fig. 6). Sanger sequencing of cDNA from all three cell lines showed transcripts with only one of the SNP alleles, confirming monoallelic TERT expression (Fig. 6). Furthermore, through long sequencing reads obtained by nCATS, we confirmed the TERT promoter mutation to be on the same allele as the expressed transcript of exon 2 in TPC-1. This expression of only the “A” allele at exon 2 (rs2736098) in TPC-1 directly confirms the expression of TERT only from the mutant TERT allele.
FIG. 6.
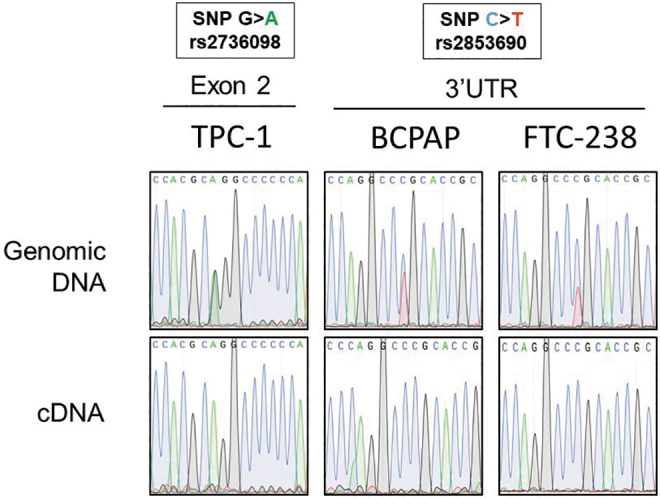
TERT monoallelic expression in heterozygous TERT mutant thyroid cancer cell lines. Sanger sequencing analysis of the exon 2 region and 3′ UTR region of TERT surrounding SNPs rs2736098 and rs2853690, respectively, in amplified genomic DNA, as well as cDNA, from TERT heterozygous mutant cell lines TPC-1, BCPAP, and FTC-238. While the genomic DNA in all cell lines showed both alleles of the SNP, the cDNA showed only one allele in all cell lines. cDNA, complementary DNA; SNP, single nucleotide polymorphism; UTR, untranslated region.
Transcriptional output and telomerase activity
To characterize overall telomerase activation, we measured full-length TERT transcripts, in which the alpha or beta regions were present, in our thyroid cancer cell lines by quantitative reverse transcriptase-PCR. We observed that the wild-type normal thyroid cell line, Nthy-ori-3, had no measurable amount of full-length TERT transcript, and the wild-type WRO showed the least amount of transcript of the cancer cell lines (Fig. 7). Interestingly, FTC-133, the homozygous TERT mutant thyroid cancer cell line, exhibited less TERT transcript than the three heterozygous TERT mutant thyroid cancer cell lines. This pattern also correlated with relative telomerase activity, in which FTC-133 also showed less telomerase activity than the three heterozygous TERT mutant cell lines. The wild-type cell lines showed the least telomerase activity.
FIG. 7.
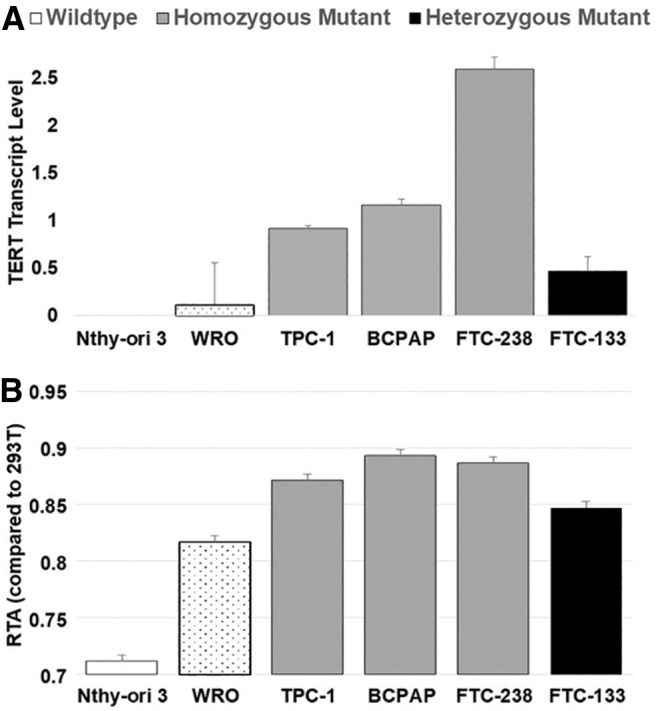
Heterozygous TERT mutant thyroid cancer cell lines exhibit higher levels of TERT transcript and telomerase activity compared with homozygous wild-type or mutant thyroid cancer cell lines. (A) TERT transcript level measured by quantitative reverse transcriptase-PCR in thyroid cancer cell lines, and (B) RTA in thyroid cancer cell lines compared with HEK293T cells. White denotes homozygous wild type (stippling denotes the TERT homozygous wild-type cancer cell line from the normal thyroid cell line), gray denotes heterozygous mutant, and black denotes homozygous mutant thyroid cancer cell lines. Standard deviation is noted. RTA, relative telomerase activity.
Discussion
It is already known that TERT promoter mutations activate TERT transcription and telomerase activity. However, due to limitations in sequencing technology, the allele-specific effects of the heterozygous TERT promoter mutation have not been completely elucidated. Using thyroid cancer cell lines that harbor the TERT mutation (12) and nCATS, we demonstrated allele-specific methylation at the TERT promoter and into the gene body. Specifically, we used ChIP-Sanger sequencing to show both allele-specific GABPA and MYC binding and allele-specific distribution of both active and silent histone modifications (H3K4me3 and H3K27me3). These allele-specific effects correlated directly with MAE of TERT in the heterozygous TERT mutant cell lines as well as overall higher levels of TERT transcription and telomerase activity.
Our characterization of TERT promoter methylation in thyroid cancer cell lines revealed significantly lower methylation levels at the TSS, TERT mutation site, and MYC binding site on the TERT mutant allele compared with the wild-type allele. These findings corroborate previous work that showed overall lower levels of TERT promoter methylation in TERT mutant cancer cell lines (24). Importantly, however, our study extends the work by examining methylation patterns on nonamplified DNA in an allele-specific manner. This demonstrated that only the mutant allele is hypomethylated, with a significant dip (<30% methylated in the mutant allele compared with >70% methylated in the wild-type allele) in methylation restricted to the TSS, thus demonstrating for the first time allele-specific methylation of TERT.
The homozygous wild-type and homozygous TERT mutant cell lines follow the same methylation pattern as the heterozygous alleles. This pattern of promoter hypomethylation and gene body methylation is in line with previously reported methylation patterns for genes with active transcription (28,29,40). Furthermore, our studies of histone modifications support the activation of the TERT mutant allele, as the mutant promoter was associated with activating H3K4me3 marks, while the wild-type allele exhibited silencing H3K27me3 marks, a pattern also seen in other cancer cell types (17).
The TERT promoter mutation (−124 C>T) creates a consensus binding site for the ETS family factors, and we previously demonstrated the factor GABPA bound to the TERT promoter in our cell lines derived from well-differentiated thyroid cancers (7). After a recent study described ETV5 binding at the TERT promoter in thyroid cancer cell lines (18), we investigated ETV5 binding in our thyroid cancer cell lines. We found that ETV5 bound significantly less at the TERT promoter than GABPA in our thyroid cancer cell lines. The recent article functionally characterized ETV5 in three thyroid cancer cell lines, two derived from anaplastic thyroid cancers and one derived from papillary thyroid cancer, finding that ETV5 predominantly bound in the two anaplastic thyroid cancer cell lines, while the papillary thyroid cancer cell line, TPC-1, showed GABPA binding (18). Our GABPA results confirm their findings in TPC-1 and further defines ETS factor involvement in well-differentiated thyroid cancer. ETV5 and a different regulatory mechanism may be present in less-differentiated cancer cells.
We have confirmed, as seen in other cancer types (11,17), that GABPA binds to the TERT mutant allele in an allele-specific manner in thyroid cancer cell lines. Our previous work showed the highest level of GABPA binding in the TERT homozygous mutant, with moderate levels in the heterozygous mutants, and very low levels of binding of GABPA in the wild-type TERT cell line (7). The allele-specific methylation of TERT also affected the MYC binding site at the minimal promoter, which exhibits lower levels of methylation on the mutant allele. As with GABPA, we found MYC binding to the TERT mutant allele in an allele-specific manner.
Our study is the first to show allele-specific binding of MYC. MYC is a known direct activator of TERT transcription (23). Our previous work showed enrichment for MYC at the TERT promoter in the thyroid cancer cell lines (7). As MYC has been shown to be methylation-sensitive in binding to its consensus site at other gene locations (22), the higher level of methylation at the TERT binding site on the wild-type allele may partially prohibit MYC binding, therefore lessening activation. Our results contradict a previous study in cancer cell lines (16) that concluded MYC was a mutation-independent activating factor of TERT. The previous study examined the effects on TERT expression after MYC knockdown in cancer cell lines and therefore could be the result of MYCs role at other loci. Conversely, our study specifically interrogated MYC binding to the TERT promoter, where we did observe mutation-dependent MYC binding.
In addition to the allele-specific binding of activators MYC and GABPA and the activating chromatin mark H3K4me3, we confirmed monoallelic transcription of TERT in the heterozygous TERT mutant thyroid cancer cell lines by utilizing SNPs identified in the coding sequence. Analysis of TPC-1 showed that the TERT mutation and the SNP on the expressed transcript occur on the same allele, providing the first direct evidence of TERT expression solely from the mutant TERT allele. Furthermore, this MAE resulted from monoallelic transcriptional activation of TERT in the thyroid cancer cell lines. As expected, we observed the lowest levels of TERT transcription and telomerase activity in the TERT wild-type cancer cell line, WRO, and the normal thyroid cell line, Nthy-ori-3.
It is important to note that while Nthy-ori-3 is utilized as a normal thyroid cell line, the cell line is immortalized with multiple mutations and chromosomal abnormalities, although none is known to be oncogenic (41). The three heterozygous TERT mutant thyroid cancer cell lines showed the highest levels of TERT expression and telomerase activity. Surprisingly, the TERT homozygous mutant cell line, FTC-133, showed less TERT transcript and telomerase activity than the heterozygous cell lines. Previous study of another TERT homozygous mutant cell line, SW1736, also showed less TERT expression in the cell line than heterozygous mutant cell lines (18). This may suggest additional post-transcriptional and post-translational regulatory processes affecting telomerase activity.
In summary, we have demonstrated and characterized allele-specific methylation at the TERT promoter and into the gene body in heterozygous TERT mutant thyroid cancer cell lines. Furthermore, lower levels of methylation at the GABPA and MYC binding site on the TERT mutant allele correlated with allele-specific binding of these two transcriptional activators. The chromatin mark H3K4me3 further confirmed activation of only the mutant allele, while the H3K27me3 mark also confirmed silencing of the wild-type allele. These regulatory mechanisms result in monoallelic transcription of TERT in heterozygous mutant cell lines, corroborated by unique SNPs that showed TERT transcription originating solely from the mutant TERT allele.
Supplementary Material
Author Disclosure Statement
W.T. has two patents licensed to ONT (US Patent 8,748,091 and US Patent 8,394,584). T.G. and W.T. have received travel funds to speak at symposia organized by Oxford Nanopore Technologies. All other authors declare no potential conflicts of interest.
Funding Information
This work was supported by National Science Foundation grant no. 1746891 (B.A.M.), National Institutes of Health training grant no. T32 GM007445 (B.A.M.), Department of Defense grant no. W81XWH-14-1-0080 (C.B.U.), NIH grant no. R01CA140311 (C.B.U.), and NIH grant no. R01HG009190-NHGRI (W.T.).
Supplementary Material
References
- 1. Yi X, Shay JW, Wright WE. 2001. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res 29:4818–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong MS, Chen L, Foster C, Kainthla R, Shay JW, Wright WE. 2013. Regulation of telomerase alternative splicing: a target for chemotherapy. Cell Rep 3:1028–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, Wasserman JD, Elizabeth C, Alon N, Zhang L, Hovestadt V, Kool M, Jones DTW, Zadeh G, Croul S, Hawkins C, Hitzler J, Wang JCY, Baruchel S, Dirks PB, Malkin D, Pfister S, Taylor MD, Weksberg R, Tabori U. 2013. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol 14:534–542 [DOI] [PubMed] [Google Scholar]
- 4. Kyo S, Takakura M, Fujiwara T, Inoue M. 2008. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci 99:1528–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avin B, Umbricht C, Zeiger M. 2016. Human telomerase reverse transcriptase regulation by DNA methylation, transcription factor binding and alternative splicing (Review). Int J Oncol 49:2199–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conaway JW 2012 Introduction to theme “chromatin, epigenetics, and transcription.” Annu Rev Biochem 81:61–64 [DOI] [PubMed]
- 7. Avin BA, Wang Y, Gilpatrick T, Workman RE, Lee I, Timp W, Umbricht CB, Zeiger MA. 2019. Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosom Cancer 58:530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang FW, Hodis E, Xu MJ, Kryukov G V, Chin L, Garraway LA. 2013. Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. 2013. TERT promoter mutations in familial and sporadic melanoma. Science (80-) 339:959–961 [DOI] [PubMed] [Google Scholar]
- 10. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D, Atkinson S, Saretzki G, Armstrong L, Lako M, Isacson O, Jaenisch R, Mangues R, Batlle E, Lee J, Jaenisch R, Mitalipova M, Mertens F, Netto G, Rasheed B, Riggins G, Rosenquist T, Schiffman M, IeM S, Theodorescu D, Torbenson M, Velculescu V, Wang T, Wentzensen N, Wood L, Zhang M, McLendon R, Bigner D, Kinzler K, Vogelstein B, Papadopoulos N, Yan H, Study AC, Study AOC, (kConFab) KCFC for R into FBC, (GENICA) GEI BC, (SWE-BRCA) SBCS, (HEBON) HB OCRGN, (EMBRACE) E study of B& BMC, (GEMO) GM of CR in BMC, Vergote I, Lambrechts S, Despierre E, Risch H, González-Neira A, Rossing M, Pita G, Doherty J, Alvarez N, Larson M, Fridley B, Schoof N, Chang-Claude J, Cicek M, Peto J, Kalli K, Broeks A, Armasu S, Schmidt M, Braaf L, Winterhoff B, Nevanlinna H, Konecny G, Lambrechts D, Rogmann L, Guénel P, Teoman A, Milne R, Garcia J, Cox A, Shridhar V, Burwinkel B, Marme F, Hein R, Sawyer E, Haiman C, Wang-Gohrke S, Andrulis I, Moysich K, Hopper J, Odunsi K, Lindblom A, Giles G, Brenner H, Simard J, Lurie G, Fasching P, Carney M, Radice P, Wilkens L, Swerdlow A, Goodman M, Brauch H, Garcia-Closas M, Hillemanns P, Winqvist R, Dürst M, Devilee P, Runnebaum I, Jakubowska A, Lubinski J, Mannermaa A, Butzow R, Bogdanova N, Dörk T, Pelttari L, Zheng W, Leminen A, Anton-Culver H, Bunker C, Kristensen V, Ness R, Muir K, Edwards R, Meindl A, Heitz F, Matsuo K, Bois A du, Wu A, Harter P, Teo S, Schwaab I, Shu X, Blot W, Hosono S, Kang D, Nakanishi T, Hartman M, Yatabe Y, Hamann U, Karlan B, Sangrajrang S, Kjaer S, Gaborieau V, Jensen A, Eccles D, Høgdall E, Shen C, Brown J, Woo Y, Shah M, Azmi M, Luben R, Omar S, Czene K, Vierkant R, Nordestgaard B, Flyger H, Vachon C, Olson J, Wang X, Levine D, Rudolph A, Weber R, Flesch-Janys D, Iversen E, Nickels S, Schildkraut J, Idos SS, Cramer D, Gibson L, Terry K, Fletcher O, Vitonis A, Schoot C van der, Poole E, Hogervorst F, Tworoger S, Liu J, Bandera E, Li J, Olson S, Humphreys K, Orlow I, Blomqvist C, Rodriguez-Rodriguez L, Aittomäki K, Salvesen H, Muranen T, Wik E, Brouwers B, Krakstad C, Wauters E, Halle M, Wildiers H, Kiemeney L, Mulot C, Aben K, Laurent-Puig P, Altena A, Truong T, Massuger L, Benitez J, Pejovic T, Perez J, Hoatlin M, Zamora M, Cook L, Balasubramanian S, Kelemen L, Schneeweiss A, Le N, Sohn C, Brooks-Wilson A, Tomlinson I, Kerin M, Miller N, Cybulski C, Henderson B, Menkiszak J, Schumacher F, Wentzensen N, Marchand L Le, Yang H, Mulligan A, Glendon G, Engelholm S, Knight J, Høgdall C, Apicella C, Gore M, Tsimiklis H, Song H, Southey M, Jager A, Ouweland A den, Brown R, Martens J, Flanagan J, Kriege M, Paul J, Margolin S, Siddiqui N, Severi G, Whittemore A, Baglietto L, McGuire V, Stegmaier C, Sieh W, Müller H, Arndt V, Labrèche F, Gao Y, Goldberg M, Yang G, Dumont M, McLaughlin J, Hartmann A, Ekici A, Beckmann M, Phelan C, Lux M, Permuth-Wey J, Peissel B, Sellers T, Ficarazzi F, Barile M, Ziogas A, Ashworth A, Gentry-Maharaj A, Jones M, Ramus S, Orr N, Menon U, Pearce C, Brüning T, Pike M, Ko Y, Lissowska J, Figueroa J, Kupryjanczyk J, Chanock S, Dansonka-Mieszkowska A, Jukkola-Vuorinen A, Rzepecka I, Pylkäs K, Bidzinski M, Kauppila S, Hollestelle A, Seynaeve C, Tollenaar R, Durda K, Jaworska K, Hartikainen J, Kosma V, Kataja V, Antonenkova N, Long J, Shrubsole M, Deming-Halverson S, Lophatananon A, Siriwanarangsan P, Stewart-Brown S, Ditsch N, Lichtner P, Schmutzler R, Ito H, Iwata H, Tajima K, Tseng C, Stram D, Berg D van den, Yip C, Ikram M, Teh Y, Cai H, Lu W, Signorello L, Cai Q, Noh D, Yoo K, Miao H, Iau P, Teo Y, McKay J, Shapiro C, Ademuyiwa F, Fountzilas G, Hsiung C, Yu J, Hou M, Healey C, Luccarini C, Peock S, Stoppa-Lyonnet D, Peterlongo P, Rebbeck T, Piedmonte M, Singer C, Friedman E, Thomassen M, Offit K, Hansen T, Neuhausen S, Szabo C, Blanco I, Garber J, Narod S, Weitzel J, Montagna M, Olah E, Godwin A, Yannoukakos D, Goldgar D, Caldes T, Imyanitov E, Tihomirova L, Arun B, Campbell I, Mensenkamp A, Asperen C van, Roozendaal K van, Meijers-Heijboer H, Collée J, Oosterwijk J, Hooning M, Rookus M, Luijt R van der, Os T, Evans D, Frost D, Fineberg E, Barwell J, Walker L, Kennedy M, Platte R, Davidson R, Ellis S, Cole T, Paillerets BB, Buecher B, Damiola F, Faivre L, Frenay M, Sinilnikova O, Caron O, Giraud S, Mazoyer S, Bonadona V, Caux-Moncoutier V, Toloczko-Grabarek A, Gronwald J, Byrski T, Spurdle A, Bonanni B, Zaffaroni D, Giannini G, Bernard L, Dolcetti R, Manoukian S, Arnold N, Engel C, Deissler H, Rhiem K, Niederacher D, Plendl H, Sutter C, Wappenschmidt B, Borg A, Melin B, Rantala J, Soller M, Nathanson K, Domchek S, Rodriguez G, Salani R, Kaulich D, Tea M, Paluch S, Laitman Y, Skytte A, Kruse T, Jensen U, Robson M, Gerdes A, Ejlertsen B, Foretova L, Savage S, Lester J, Soucy P, Kuchenbaecker K, Olswold C, Cunningham J, Slager S, Pankratz V, Dicks E, Lakhani S, Couch F, Hall P, Monteiro A, Gayther S, Pharoah P, Reddel R, Goode E, Greene M, Easton D, Berchuck A, Antoniou A, Chenevix-Trench G, Dunning A. 2015. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 4:806–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, Choi S, Hong C, He D, Pekmezci M, Wiencke JK, Wrensch MR, Chang SM, Walsh KM, Myong S, Song JS, Costello JF. 2015. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348:1036–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, Sun H, El-Naggar AK, Xing M. 2013. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer 20:603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu R, Xing M. 2016. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer 23:R143–R155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X, Li J, Li X, Liang Z, Gao W, Liang J, Cheng S, Lin Y. 2017. TERT promoter mutation predicts radioiodine-refractory character in distant metastatic differentiated thyroid cancer. J Nucl Med 58:258–265 [DOI] [PubMed] [Google Scholar]
- 15. Yin D, Yu K, Lu R, Li X, Xu J, Lei M, Li H, Wang Y, Liu Z. 2016. Clinicopathological significance of TERT promoter mutation in papillary thyroid carcinomas: a systematic review and meta-analysis. Clin Endocrinol (Oxf) 85:299–305 [DOI] [PubMed] [Google Scholar]
- 16. Liu R, Zhang T, Zhu G, Xing M. 2018. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun 9:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR. 2015. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev 29:2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bullock M, Lim G, Zhu Y, Åberg H, Kurdyukov S, Clifton-Bligh RJ. 2019. The ETS factor ETV5 activates the mutant TERT promoter in thyroid cancer. Thyroid 29:1623–1633 [DOI] [PubMed] [Google Scholar]
- 19. Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL, Qian J. 2015. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics 16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25:1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. 2003. The role of DNA methylation in setting up chromatin structure during development. Nat Genet 34:187–192 [DOI] [PubMed] [Google Scholar]
- 22. Prendergast G, Ziff E. 1991. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science (80-) 251:186–189 [DOI] [PubMed] [Google Scholar]
- 23. Wu K-J, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, Dalla-Favera R. 1999. Direct activation of TERT transcription by c-MYC. Nat Genet 21:220–224 [DOI] [PubMed] [Google Scholar]
- 24. Stern JL, Paucek RD, Huang FW, Ghandi M, Nwumeh R, Costello JC, Cech TR. 2017. Allele-specific DNA methylation and its interplay with repressive histone marks at promoter-mutant TERT genes. Cell Rep 21:3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Sedlazeck FJ, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38:433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng Y, Zhang Y, Ying C, Wang D, Du C. 2015. Nanopore-based fourth-generation DNA sequencing technology. Genom Proteom Bioinf 13:4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, Garraway LA, Kryukov GV. 2015. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis 4:e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ball MP, Li JB, Gao Y, Lee J-H, LeProust EM, Park I-H, Xie B, Daley GQ, Church GM. 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27:361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y-C, Elnitski L. 2019. Aberrant DNA methylation defines isoform usage in cancer, with functional implications. PLoS Comput Biol 15:e1007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanaka J, Ogura T, Sato H, Hatano M. 1987. Establishment and biological characterization of an in vitro human cytomegalovirus latency model. Virology 161:62–72 [DOI] [PubMed] [Google Scholar]
- 31. Fabien N, Fusco A, Santoro M, Barbier Y, Dubois PM, Paulin C. 1994. Description of a human papillary thyroid carcinoma cell line. Morphologic study and expression of tumoral markers. Cancer 73:2206–2212 [DOI] [PubMed] [Google Scholar]
- 32. Rao AS, Goretzki PE, Köhrle J, Brabant G. 2005. Letter re: Id1 gene expression in hyperplastic and neoplastic thyroid tissues. J Clin Endocrinol Metab 90:5906. [DOI] [PubMed] [Google Scholar]
- 33. Estour B, Van Herle AJ, Juillard GJ, Totanes TL, Sparkes RS, Giuliano AE, Klandorf H. 1989. Characterization of a human follicular thyroid carcinoma cell line (UCLA RO 82 W-1). Virchows Arch B Cell Pathol Incl Mol Pathol 57:167–174 [DOI] [PubMed] [Google Scholar]
- 34. Lemoine N, Mayall E, Jones T, Sheer D, McDermid S, Kendall-Taylor P, Wynford-Thomas D. 1989. Characterisation of human thyroid epithelial cells immortalised in vitro by simian virus 40 DNA transfection. Br J Cancer 60:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. 2017. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14:407–410 [DOI] [PubMed] [Google Scholar]
- 37. Haring M, Offermann S, Danker T, Horst I, Peterhansel C, Stam M. 2007. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang S, Tang M, Xin H, Huang J. 2017. Assessing telomerase activities in mammalian cells using the quantitative PCR-based telomeric repeat amplification protocol (qTRAP). Methods Mol Biol 1587:95–101 [DOI] [PubMed] [Google Scholar]
- 39. Wang Y, Meeker AK, Kowalski J, Tsai H-L, Somervell H, Heaphy C, Sangenario LE, Prasad N, Westra WH, Zeiger MA, Umbricht CB. 2011. Telomere length is related to alternative splice patterns of telomerase in thyroid tumors. Am J Pathol 179:1415–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9:465–476 [DOI] [PubMed] [Google Scholar]
- 41. Landa I, Pozdeyev N, Korch C, Marlow LA, Smallridge RC, Copland JA, Henderson YC, Lai SY, Clayman GL, Onoda N, Tan AC, Garcia-Rendueles MER, Knauf JA, Haugen BR, Fagin JA, Schweppe RE. 2019. Comprehensive genetic characterization of human thyroid cancer cell lines: a validated panel for preclinical studies. Clin Cancer Res 25:3141–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.