

Abstract
Prostate cancer (CaP) is the second leading cause of cancer-related deaths in western men. Because androgens drive CaP by activating the androgen receptor (AR), blocking AR’s ligand-activation, known as androgen deprivation therapy (ADT), is the default treatment for metastatic CaP. Despite an initial remission, CaP eventually develops resistance to ADT and progresses to castration-recurrent CaP (CRPC). CRPC continues to rely on aberrantly activated AR that is no longer inhibited effectively by available therapeutics.
Interference with signaling pathways downstream of activated AR that mediate aggressive CRPC behavior may lead to alternative CaP treatments. Developing such therapeutic strategies requires a thorough mechanistic understanding of the most clinically relevant and druggable AR-dependent signaling events. Recent proteomics analyses of CRPC clinical specimens indicate a shift in the phosphoproteome during CaP progression. Kinases and phosphatases represent druggable entities, for which clinically tested inhibitors are available, some of which are incorporated already in treatment plans for other human malignancies.
Here, we reviewed the AR-associated transcriptome and translational regulon, and AR interactome involved in CaP phosphorylation events. Novel and for the most part mutually exclusive AR-dependent transcriptional and post-transcriptional control over kinase and phosphatase expression was found, with yet other phospho-regulators interacting with AR. The multiple mechanisms by which AR can shape and fine-tune the CaP phosphoproteome were reflected in diverse aspects of CaP biology such as cell cycle progression and cell migration. Furthermore, we examined the potential, limitations and challenges of interfering with AR-mediated phosphorylation events as alternative strategy to block AR function during CaP progression.
Keywords: kinase, phosphatase, castration, hormonal therapy, treatment resistance, coregulator
Introduction
Prostate cancer (CaP) is expected to cause the death of 33,300 American men in 2020, and remains the second leading cause of cancer-related deaths in western men (Siegel et al., 2020). It is well-recognized that the androgen receptor (AR), a ligand-activated transcription factor, drives CaP progression (Huggins and Hodges, 2002, Dai et al., 2017). Therefore, blocking AR activation, known as androgen deprivation therapy (ADT), has been the default treatment for metastatic CaP for almost 80 years. Although ADT is successful in inducing an initial remission, CaP cells eventually adapt to acquire resistance to ADT, and castration-recurrent CaP (CRPC) develops. Adaptations that allow the emergence of CRPC include several mechanisms by which AR activation is restored, such as activating somatic mutations, amplifications and rearrangements in the AR gene, intratumoral metabolism and synthesis of (precursor) androgens, compensatory expression of the AR-related glucocorticoid receptor that is able to take over parts of AR function, all of which have been the subject of excellent reviews before (Narayanan et al., 2016, Watson et al., 2015, Mills, 2014, Dai et al., 2017).
Because AR is still active and continues to drive CaP progression after failure of ADT, alternative means of interfering with its activity are sought. In view of AR’s transcription factor function such efforts have mostly focused on its ligand-independent constitutively active transcription activity, its ability to bind DNA, and to form active transcriptional complexes. Several compounds targeting these aspects of AR function have been developed and/or tested clinically, yet none have moved into clinic (Kumari et al., 2017, Senapati et al., 2019).
Recent genomic and proteomics analyses of CaP models and clinical specimens have provided substantial new evidence for critical roles for AR in various cellular events relevant to aggressive CaP behavior and CaP progression such as cell cycle regulation, epigenetic modifications, DNA repair, and translational regulation (Liu et al., 2019, Schiewer et al., 2012, Xu et al., 2012, Schiewer and Knudsen, 2016, Schiewer and Knudsen, 2019). While delineating the involvement of AR in these cellular processes, it has become clear that AR controls several phosphorylation events in which specific kinases and phosphatases play important roles. In other human malignancies, defining and understanding the phosphorylation-based signaling cascades events that drive disease progression resulted in novel therapeutic strategies, some of which have shown exceptional success rates for therapeutic intervention and overcoming treatment resistance (Lynch et al., 2004, Shaw et al., 2014, Chapman et al., 2011). For instance, in non-small cell lung cancer, the tyrosine kinase inhibitors gefitinib or erlotinib are effective in targeting activating mutations in epidermal growth factor receptor (EGFR), a driver of disease progression (Ciardiello and Tortora, 2008). Upon emergence of somatic mutations, second or even third generation EGFR inhibitors are used to improve outcomes (Mok et al., 2017, Janjigian et al., 2014). The possibility of similar therapeutic successes targeting phosphorylation regulators in CaP is supported by the emerging phospho-proteomic characterizations of clinical CRPC specimens (Faltermeier et al., 2016, Drake et al., 2016) and testing of kinase inhibiting drugs, alone or in combination therapy, in CaP co-clinical trials (Munster et al., 2019, Sweeney et al., 2019).
Here, we review information on AR’s control over kinase and phosphatase expression and its collaboration with such key regulators of phosphorylation events in CaP. We explore the biology that is impacted by AR-associated phosphorylation, its relevance during CaP progression, and the overall potential, limitations and challenges associated with exploiting AR-dependent phosphorylation as alternative strategy to overcome resistance to AR-targeting treatments in CaP (Figure 1).
Figure 1. Differential AR action in CaP.
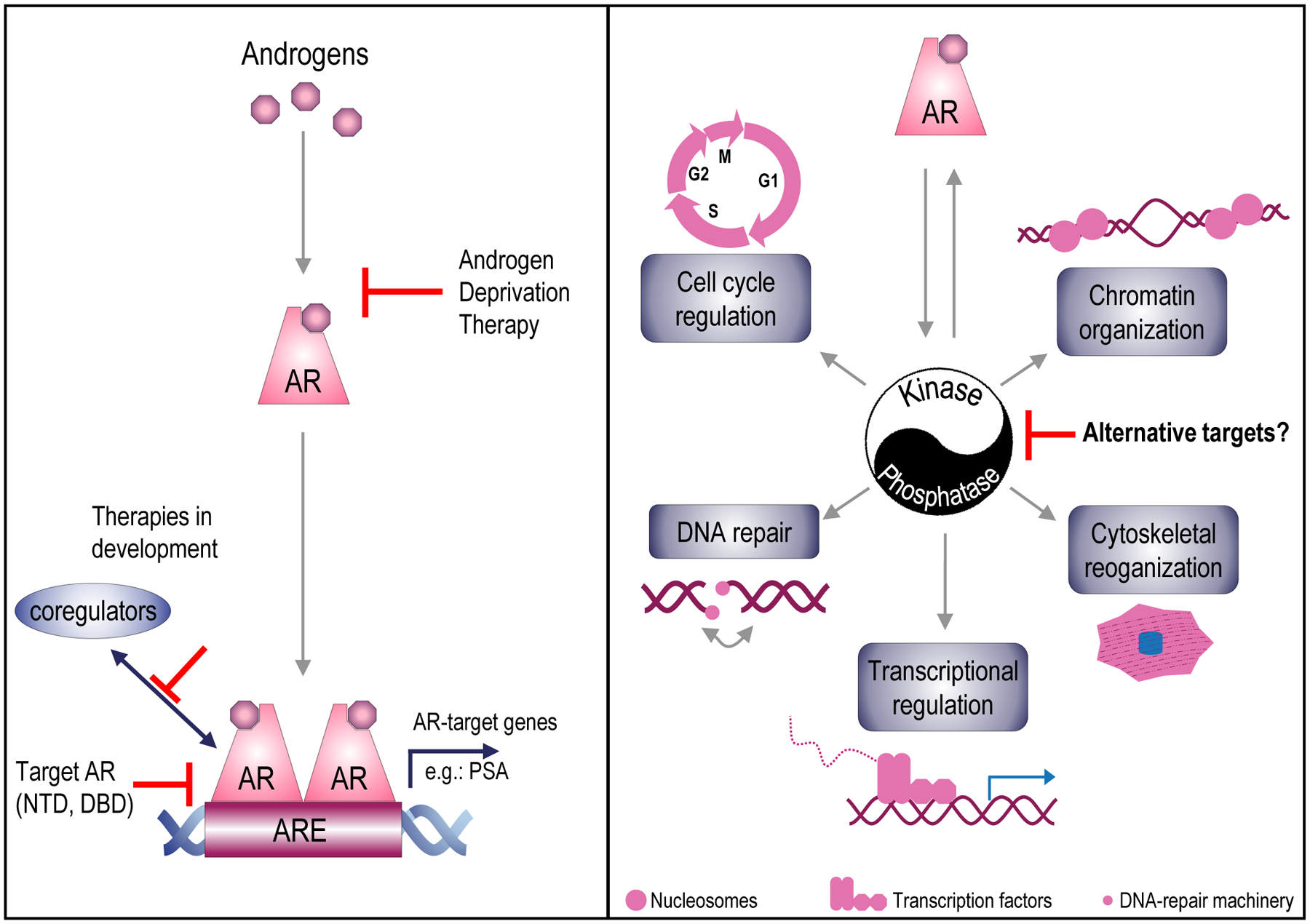
Canonical AR signaling (left panel). Androgens bind to AR. Upon activation, AR homodimerizes, relocates to the nucleus and binds to androgen response elements (AREs) where it interacts with coregulators to transcribe target genes such as the gene encoding Prostate-Specific Antigen (PSA). Androgen deprivation therapy, the first-line standard of care, blocks ligand-activation of AR. Treatment strategies to block AR’s transcription function in an alternative manner include inhibiting AR-coregulator interactions, AR’s constitutively active transcription by the N-terminal domain (NTD), and AR-DNA binding by the DNA-binding domain (DBD)of AR. Schematic representing AR’s regulation of several cellular processes via its control over and interactions with kinases and phosphatases.
1. AR dependence of kinase and phosphatase expression and activity
Control of AR over important phosphorylation events and AR’s collaboration with kinases and phosphatases in CaP has been recognized before. A few well-known examples that immediately come to mind are AR’s regulation of cyclin-dependent kinase (CDK) activity and its interaction with epigenetic modifiers such as TRIM24 (a kinase) and the tumor suppressor PTEN, a phosphatase (Groner et al., 2016, Knudsen et al., 1998, Li et al., 2001). Yet, despite the emerging insights on the important role and the clinical relevance of the phospho-proteome during CRPC progression (Drake et al., 2013, Drake et al., 2012, Drake et al., 2016, Faltermeier et al., 2016), the extent of AR’s contribution to phosphorylation events in CaP is yet to be fully appreciated. Here, we review literature to define more comprehensively the transcriptional and translational control by AR over kinase and phosphatase expression, and to examine alternative mechanisms by which AR impacts phosphorylation-dependent CaP cell biology.
a. Transcriptional control by AR over kinases and phosphatases
First, we explored the possibility that AR regulates the CaP phosphoproteome by directly controlling transcription of genes that encode kinases or phosphatases. To this end, members of a CaP gene signature representing hundreds of bona fide AR target genes (n=452, LNCaP cell dataset) (Liu et al., 2017), i.e. genes that contain at least one Androgen Response Element (ARE) and whose androgen-responsiveness has been experimentally verified, were cross-matched with 536 kinases listed on KinHub (Eid et al., 2017), a resource that maintains and curates knowledge on the human kinome. This approach isolated 28 genes encoding kinases as direct AR target genes. Expression of 13 kinases was androgen-repressed, the remaining 15 were upregulated by AR stimulation. The majority (n=23) of these genes encoded for Ser/Thr kinases while only 5 led to Tyr kinase expression (Table 1). Kinases controlled transcriptionally by AR include for instance CAMKK2, a member of the CAMK family of kinases, which is well-known for its role in lipid metabolism, and has been proposed by several groups as viable therapeutic target in CaP (Massie et al., 2011, Karacosta et al., 2012, White et al., 2018, Penfold et al., 2018). Other more neglected kinases include the discoidin domain receptor family member 1, DDR1, which has been implicated in epithelial-mesenchymal transition (EMT) and metastasis in human cancers (Maeyama et al., 2008, Koh et al., 2015, Yeh et al., 2011), has not yet been extensively studied in CaP, but can be inhibited by nilotinib, a drug that is being tested in clinical trials for leukemia (Jeitany et al., 2018). Comparison with recent phospho-proteome data obtained via mass spectrometry on clinical CRPC cases (Drake et al., 2016) revealed several kinases, for instance MERTK, to be over-activated in CaP compared to benign prostate, suggesting important roles in late stage CaP and supporting their therapeutic potential. For several of these kinases, inhibitors have been developed, some of which have been tested or are being tested in clinical CaP trials, or have been used to treat other malignancies (Table 1).
Table 1. AR-associated kinases.
Kinases that are transcriptionally (top part) or translationally (middle part) regulated by AR and kinases that interact with AR (bottom part). Up/down; kinase expression induced/repressed by AR. Inhibitors; commercially available kinase inhibitors, information obtained from genecards.org. CaP phosphoproteome; site of kinase phosphorylation in CRPC samples obtained from Faltermeier et al., 2016 and Drake et al., 2016. NA, not applicable.
| Gene Symbol | AR-dependence | Type | Family | Inhibitor | CaP phosphoproteome |
|---|---|---|---|---|---|
| AR regulation: | transcriptional | ||||
| BMPR1A | up | Ser/Thr | TKL | DMH-1 | NA |
| BMPR1B | up | Ser/Thr | TKL | LDN-212854 | NA |
| CAMKK2 | up | Ser/Thr | CAMK | A 484954, NH125 | pS495, pS511c |
| CDK8 | down | Ser/Thr | CMGC | Senexin A | NA |
| CDKL5 | down | Ser/Thr | CMGC | None | NA |
| CLK2 | down | Ser/Thr | CMGC | TG003 | NA |
| DDR1 | down | Tyr | TK | DDR1-IN-1, nilotinib | Y520, Y792, Y796 d |
| EPHA3 | up | Tyr | TK | KB004 | Y596 d |
| ERBB2a | down | Tyr | TK | >90 inhibitors | pS1054, pS1078, pS1083 c |
| MAK1a | up | Ser/Thr | CMGC | None | NA |
| MAP2K4 | up | Ser/Thr | STE | MEK inhibitors | pS257 c |
| MAP4K1 | down | Ser/Thr | STE | None | NA |
| MAPK6b | up | Ser/Thr | CMGC | None | pS386, pT389 e |
| MAPK8 | down | Ser/Thr | CMGC | None | pT183 d |
| MAPKAPK3 | down | Ser/Thr | CAMK | None | NA |
| MERTK | up | Tyr | TK | None | NA |
| MYLK | down | Ser/Thr | CAMK | ML 9 Hydrochloride | pS911, pS1768 c |
| PAK2 | down | Ser/Thr | STE | FRAX597 | pS2, pS141 c |
| PRKAA1 | up | Ser/Thr | CAMK | None | pS496 c |
| PRKCA | up | Ser/Thr | AGC | None | pT638, pS657 f |
| PRKD1a | down | Ser/Thr | CAMK | CID 2011756 | pS473 e |
| ROR1 | up | Tyr | TK | None | NA |
| RPS6KA1a | down | Ser/Thr | AGC | BRD 7389 | pS232 e |
| RPS6KA3a | up | Ser/Thr | AGC | AT9283 | pS221 e |
| SGK1a | up | Ser/Thr | AGC | None | NA |
| SNRK | down | Ser/Thr | CAMK | None | NA |
| STK17B | up | Ser/Thr | CAMK | None | NA |
| STK39 | up | Ser/Thr | STE | None | pS385, pS315, pT354 c |
| AR regulation: | translational | ||||
| HIPK3a | NA | Ser/Thr | CMGC | None | NA |
| MAPK6b | NA | Ser/Thr | CMGC | None | pS386, pT389 e |
| MAPKAPK2 | NA | Ser/Thr | CAMK | PF-3644022 | NA |
| PINK1 | NA | Ser/Thr | Other | None | NA |
| TRIM24a | NA | Ser/Thr | Atypical | None | pS1028 c |
| AR regulation: | interactor | ||||
| AKT1 | NA | Ser/Thr | AGC | Nelfinavir, Everolimus, PHT 427 | pT34 c |
| CDK11B | NA | Ser/Thr | CMGC | None | pS265, pS271, pS422 c |
| CDK6 | NA | Ser/Thr | CMGC | LEE011 - CDK4/6 | NA |
| CDK7 | NA | Ser/Thr | CMGC | Alvocidib, seliciclib, BS-181 | pS164 g |
| CDK9 | NA | Ser/Thr | CMGC | Alvocidib, seliciclib, Dinaciclib, AZD 5438 | NA |
| DAPK3 | NA | Ser/Thr | CAMK | None | NA |
| DYRK1A | NA | Ser/Thr | CMGC | Harmine, INDY, ProINDY | pY321 f |
| EGFR | NA | Tyr | TK | >200 inhibitors | pS1039, pS1042 c |
| ERBB2a | NA | Tyr | TK | >90 inhibitors | pS1054, pS1078, pS1083 c |
| GAK | NA | Ser/Thr | Other | None | pS16, pS73, pS826, pS829, pS1185 c |
| GSK3B | NA | Ser/Thr | CMGC | SB 216763, AZD1080 | pY216 d |
| HIPK3a | NA | Ser/Thr | CMGC | None | NA |
| LATS2 | NA | Ser/Thr | AGC | None | NA |
| MAK1a | NA | Ser/Thr | CMGC | None | NA |
| MAPK1 | NA | Ser/Thr | CMGC | FR 180204 | Y187 d |
| MAPK15 | NA | Ser/Thr | CMGC | None | NA |
| NLK | NA | Ser/Thr | CMGC | None | NA |
| PAK6 | NA | Ser/Thr | STE | None | pS560 d |
| PKN1 | NA | Ser/Thr | AGC | lestaurtinib, tofaticinib | pS69, pS916 d |
| PRKD1a | NA | Ser/Thr | CAMK | CID 2011756 | pS473 e |
| PRKDC | NA | Ser/Thr | Atypical | AZD7648, | pT2609, pS2612, pS3205 d |
| RNASEL | NA | Ser/Thr | Other | None | NA |
| RPS6KA1a | NA | Ser/Thr | AGC | BRD 7389 | pS232 e |
| RPS6KA3a | NA | Ser/Thr | AGC | AT9283 | pS221 e |
| SGK1a | NA | Ser/Thr | AGC | None | NA |
| SRC | NA | Tyr | TK | Herbimycin A, MNS, Dasatinib | pS104 e |
| TAF1 | NA | Ser/Thr | Atypical | None | pS1152, pS1155, pS1669, pS1672 e |
| TNK2 | NA | Tyr | TK | AIM-100, Dasatinib | pY827 e |
| TRIM24a | NA | Ser/Thr | Atypical | None | pS1028 c |
, entry occurs in top and bottom part of table;
, entry occurs in top and middle part of table;
phosphorylation site is significantly enriched in CaP but functional consequence of phosphorylation is unknown;
phosphorylation site is significantly enriched in CaP and associated with kinase activation;
phosphorylation site is found but not significantly enriched in CaP and functional consequence of phosphorylation is unknown;
phosphorylation site not significantly enriched in CaP but activates the kinase;
phosphorylation site is significantly enriched in CaP and represses kinase activity. Ser/thr, serine/threonine kinase; Tyr, tyrosine kinase; TKL, Tyrosine Kinase-Like; CAMK, Calcium/Calmodulin dependent-kinase; TK, Tyrosine Kinase; CMGC, members of cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase and CDC-like kinase; AGC, members of PKA, PKG, and PKC; STE, “Sterile” serine/threonine kinases;
These data demonstrate that a significant number of kinases is controlled by AR directly binding to their regulatory gene regions. We performed a similar examination of the AR target gene list for genes encoding any of the 226 phosphatases classified by the Human Gene Nomenclature Committee (HGNC). This analysis returned 13 AR-dependent and transcriptionally regulated phosphatases, consisting of 9 phosphatases that are androgen-stimulated and 4 that are down-regulated upon AR activation, verifying that this class of proteins can be subject to similar AR regulation (Table 2). Among the 13 phosphatases were PTEN and INPP4B, which have been implicated in CaP progression before (Hodgson et al., 2014, Hodgson et al., 2011, Li et al., 1997), whereas others such as SYNJ1 have lesser recognized functions in CaP. Pharmacologic modulation of phosphatase activity is possible, as evidenced by availability of several phosphatase inhibitors, however, only a handful of them are in clinical trials (Bollu et al., 2017).
Table 2. AR-associated phosphatases.
Phosphatases that are transcriptionally (top part) or translationally (middle part) regulated by AR and phosphatase that interact with AR (bottom part). Up/down; phosphatase expression induced/repressed by AR. Inhibitors; commercially available kinase inhibitors, information obtained from genecards.org. CaP phosphoproteome; site of phosphorylation in CRPC samples obtained from Faltermeier et al., 2016 and Drake et al., 2016. NA, not applicable.
| Gene Symbol | AR dependence | Group | Inhibitor | CaP phosphoproteome |
|---|---|---|---|---|
| AR regulation: | transcriptional | |||
| INPP4B | up | Phosphoinositide phosphatases | None | NA |
| INPP5A | down | Phosphoinositide phosphatases | None | NA |
| INPP5D | down | Phosphoinositide phosphatases | None | pS971* |
| PMM2 | up | HAD Asp-based non-protein phosphatases | None | NA |
| PPM1K | up | Protein phosphatases, Mg2+/Mn2+ dependent | None | NA |
| PPP1CB | up | Protein phosphatase catalytic subunits | FK 506 | NA |
| PPP2CB | up | Protein phosphatase catalytic subunits | FK 506 | NA |
| PTEN1 | down | Phosphoinositide phosphatases | SF1670 | NA |
| PTPN11 | up | Protein tyrosine phosphatases non-receptor type | BVT 948, TCS 401 | pY584* |
| PTPN21 | up | Protein tyrosine phosphatases non-receptor type | None | pS658# |
| PTPRM | up | Protein tyrosine phosphatases receptor type | None | NA |
| PTPRR | down | Protein tyrosine phosphatases receptor type | None | NA |
| SYNJ1 | up | Phosphoinositide phosphatases | None | pS830# |
| AR regulation: | translational | |||
| MINPP1 | NA | Phosphoinositide phosphatases | None | NA |
| PHLPP1 | NA | Protein phosphatases, Mg2+/Mn2+ dependent | None | NA |
| PPP4C | NA | Protein phosphatase catalytic subunits | None | NA |
| AR regulation: | interactor | |||
| CDC25A | NA | Class III Cys-based CDC25 phosphatases | NSC 663284, NSC 95397 | NA |
| CDC25B | NA | Class III Cys-based CDC25 phosphatases | NSC 663284, NSC 95397 | NA |
| CTDSP2 | NA | CTD family phosphatases | None | NA |
| PGAM5 | NA | Serine/Threonine protein phosphatases | None | pS80* |
| PPP1CA | NA | Protein phosphatase catalytic subunits | FK 506 | NA |
| PPP1CC | NA | Protein phosphatase catalytic subunits | FK 506 | NA |
| PTEN1 | NA | Phosphoinositide phosphatases | SF1670 | NA |
, entry occurs in top and bottom part of table.
AR regulation of its target genes can be context-dependent and may be influenced by, for instance, the genomic make-up or the AR expression levels of CaP cells. We therefore examined the extent to which the findings of AR-dependence of genes encoding kinases and phosphatases derived from LNCaP cells can be extrapolated to other AR-positive cell lines, such as VCaP. VCaP cells express the TMPRSS2-ERG gene fusion and show AR gene amplification, which can each impact the composition of the AR cistrome (Wasmuth et al., 2020, Yu et al., 2010, Cai et al., 2013, Waltering et al., 2009, Makkonen et al., 2011). Analysis of 2 publically available and independent AR ChIP-Seq data sets derived from VCaP cells (Massie et al., 2011, Asangani et al., 2014) employing the same selection criteria used on LNCaP cell data (i.e. AR binding peaks present with 300 Kb of transcriptional start sites (Liu et al., 2017) revealed androgen-induced AR binding peaks in 27/28 kinase-encoding genes and all, or 13/13, phosphatase-encoding genes (Supplemental Table 1). Moreover, except for the gene encoding CLK2 (two thirds of replicates positive), AR binding sites were present in all biological replicates in androgen-stimulated conditions, indicating that AR regulation of these kinases and phosphatases occurs in multiple AR-positive CaP models.
b. Translational control by AR over kinases and phosphatases
The impact of AR-dependent transcriptional control over its target genes is readily measured via oligoarray, RNA-Seq and similar gene expression assays, and can be validated using AR ChIP-chip, ChIP-Seq or ChIP-exo approaches. The AR-controlled transcriptome has therefore been well-documented and characterized in an array of CaP model systems as well as clinical specimens (Senapati et al., 2019, Kumari et al., 2017, Mills, 2014). The extent to which AR-dependent transcription is translated into CaP proteomes or to which CaP translational events are AR-dependent is poorly understood. This is an important question since recent studies have shown discordance between mRNA levels and protein abundance (Sinha et al., 2019). The lack of our knowledge in this regard, particularly as it regards AR’s control over CaP translation (rates), relates to the well-recognized, and not CaP-specific, limitations in the technical assessment of translational rates. The results from techniques such as polysome profiling, ribo-seq and ribosome profiling can serve as surrogate for translation rates, but until very recently these had not been used on CaP models, let alone in the context of CaP’s AR dependency. A recent study found that 4EBP1, an inhibitor of the translation initiation complex, contains an ARE in its first codon, and upon androgen stimulation of CaP cells, 4EBP1 mRNA levels are induced (Liu et al., 2019). These findings point to an indirect mechanism for AR to shape the CaP proteome. Subsequently, ribosome profiling was done on CaP tissues from a transgenic mouse CaP model driven by prostate-specific PTEN deletion, which mimics aggressive CRPC progression. Tissues from intact versus castrated animals were analyzed, which allowed to verify AR-dependence. The authors report a subset of mRNAs that are differentially regulated at the post-transcriptional level and are subject to AR signaling, which encode 5 kinases and 3 phosphatases (Tables 1 and 2). Remarkable, only one entry (i.e. MAPK6) overlapped between transcriptionally and post-transcriptionally AR-dependent genes, indicating distinct levels of AR control over the expression of phospho-regulators.
c. Alternative AR involvement in kinase and phosphatase action
Direct transcriptional or post-transcriptional control over kinase and phosphatase expression may not capture the full extent of AR’s ability to regulate phosphorylation status of CaP cells.
Mechanisms that involve genomic AR function
It has been long-recognized that the (de)phosphorylation activity of these regulators depends on activating ligands, interactions with adaptors, scaffold proteins and binding patterns that ensure their proper spatial and temporal activation and complex formation within the cell (Pawson and Scott, 1997), or E3 ligases that modulate their action. In CaP, several aspects of such “indirect” modulation of kinase and phosphatase action may involve transcriptional or translational regulation by AR. For instance, AR stimulates the enzymatic pathways that govern synthesis of fatty acids (Heemers et al., 2006), which are critical activators of protein kinase C family members (Bell and Burns, 1991). Similarly, several adaptor proteins (e.g. GRB10) and scaffold proteins (e.g. AKAP12) have been identified as ARE-driven genes (Liu et al., 2017). Kinase and phosphatase-associated E3 ligases such as MID1 and TRIM36 are bona fide AR target genes. MID1 interacts with the kinase PDPK1 and the phosphatase PP2A (Aranda-Orgilles et al., 2011), and thus potentially providing indirect AR control over their respective phospho-proteomes. TRIM36, on the other hand, is an androgen-induced gene whose over-expression in CaP blocks MEK/ERK signaling, while its decreased activity following ADT increases MEK/ERK kinase transduction pathways (Liang et al., 2018).
Literature indicates that AR can employ also yet other mechanisms to influence the CaP proteome. One well-known example is AR-dependent activity of several CDKs, which was not captured by the above analyses, consistent with its regulation by AR-responsive cyclin expression (Knudsen et al., 1998). It is also conceivable that AR controls enzymatic activity of phosphorylation modulators without altering their expression levels. One such mechanism by which AR may steer kinase activity to specific CaP functions is its signaling, via activation of RhoA, to the Ser/Thr kinase PKN1, which then mediates androgen control to the secondary transcription factor Serum Response Factor (SRF) (Venkadakrishnan et al., 2019). SRF controls the immediate early response, cell cycle regulation, and organization of cytoskeleton. AR-RhoA-PKN1 mediated activation of SRF signaling is linked with aggressive CaP behavior and poor outcome (Heemers et al., 2011, Lundon et al., 2017, Prencipe et al., 2018). Interestingly, PKN1 serves also as an AR-associated coregulator, which points towards bidirectional regulation in which AR transactivation is modulated by kinases and phosphatases (Metzger et al., 2008). Review of the RAAR database that contains more than 280 AR-associated coregulators that we compiled before (DePriest et al., 2016) shows that 29 and 6 genes possess kinase and phosphatase moieties, respectively (Tables 1 and 2) (DePriest et al., 2016). Examples include the kinase PRKDC and the phosphatase, CDC25B. Such proteins are part of AR transcriptional complexes at ARE in target genes that are formed in a context-dependent manner, preferentially control subsets of AR target genes and thus aspects of CaP biology (Liu et al., 2017). Differential phosphorylation of transcriptional complex components may underlie some of this specificity (Rasool et al., 2019). Other means of cross talk between AR and phosphorylation machinery are conceivable, such as AR control of non-coding RNAs (miRNA, eRNA, circRNA) that impact kinase or phosphatase expression.
Mechanisms that involve non-genomic AR function
It should be noted that all of the scenarios above consider only AR’s genomic function, i.e. AR exerting its transcription factor function in the cell nucleus. However, non-genomic AR action has been recognized, which involves signal transduction via a membrane-bound or cytoplasmic androgen-activated AR (Leung and Sadar, 2017, Zarif and Miranti, 2016). Non-genomic AR signaling occurs within seconds to minutes, thus much more rapidly than the canonical genomic AR function, and has been shown to activate, for example, Src and MAPK signaling pathways in the cytoplasm (Migliaccio et al., 2000, Migliaccio et al., 2007). These signaling axes have been reported to be constitutively active in CRPC and could thus impact significantly the CaP phosphoproteome during CaP progression.
Delineating the full scope of the mechanisms by which AR intersects with CaP phosphorylation events will be important. Even taking into account this limitation and considering only the information provided in Tables 1 and 2, it is obvious that AR can exert major impact on CaP’s phosphorylation status: a total of 53 kinases and 22 phosphatases are either controlled by AR or intersect with its signaling. The kinome tree for the impacted kinases shows distribution over 7 kinase families, with most enrichment for the CMGC kinase family. AR-associated phosphatases belong to the classes of lipid phosphatases, protein phosphatases and HAD Asp-based phosphatases, while no acid phosphatases, alkaline phosphatases, sugar phosphatases or 5’-nucleotidases are enriched.
2. Clinically relevant CaP biology controlled by AR-dependent kinases and phosphatases
Phosphorylation events control and fine-tune most, if not all, cellular processes. The overview provided in the previous section indicates that AR action can regulate and involve the activity of dozens of kinases and phosphatases, but the impact of subsequent alterations in downstream phosphorylations on CaP cell behavior and clinical CaP progression has not been explored. Even a superficial glance at the proteins listed in Tables 1 and 2, especially when considering the signaling cascades and substrates that are known to be modified by these at the post-translational level, already predicts that a diverse array of cellular functions is affected. For the purpose of this review, we will focus on those aspects of CaP biology that are most relevant to disease progression and/or are considered for therapeutic intervention.
a. Cell proliferation and cell cycle regulation
The ability of cells to proliferate and to progress through the cell cycle is a hallmark of all cancers (Hanahan and Weinberg, 2011). Cell cycle progression is exquisitely regulated by phosphorylation events at critical transition steps, which are tightly controlled by cyclin-CDK interactions. In CaP, one of the best characterized consequences of AR action is control over cell proliferation: lower doses of androgens induce CaP cell proliferation whereas high doses lead to CaP cell differentiation and/or cell death (Damassa et al., 1991, Denmeade and Isaacs, 2010, Sedelaar and Isaacs, 2009, Chuu et al., 2005, Zhau et al., 1996). Although the molecular mechanism(s) by which AR controls CaP cell proliferation remains incompletely understood (Balk and Knudsen, 2008, Schiewer et al., 2012, Wen et al., 2014), biphasic expression patterns and post-translational modifications of regulators of cell cycle checkpoints, particularly at the G1-S phase transition, have long been recognized. Progression from G1 to S phase of the cell cycle following low dose androgen stimulation involves the AR-dependent action of the kinase CDK2, 4, and 6. CDK6 is listed in Table 1 as AR interactor, and activity of CDK6 as well as these 2 other CDKs is indirectly regulated by AR via control over cyclin availability (Knudsen et al., 1998). The substrate for the action of CDK2, 4 and 6 in AR-controlled cells cycle progression is Rb, with Cdk-dependent Rb phosphorylation leading to E2F-controlled cell cycle progression (Markey et al., 2002). In addition to these components of the cell cycle machinery, several other kinases or phosphatases that are either transcriptionally or translationally regulated by AR or are part of the AR interactome at target genes can influence CaP cell proliferation (Table 1). These include multiple kinases that act at different levels of MAP kinase signaling cascades, for which substrates are not as well known, and may mediate resistance to CDK4/6 inhibition (de Leeuw et al., 2018), as well as regulators of MAPK activity, such as RPS6KA1 and RPS6KA3. In addition, ERBB2, AKT/PI3K, SGK3 signaling have all also been described to impact CaP cell proliferation (Gao et al., 2016, Sherk et al., 2008, Shanmugam et al., 2007, Edlind and Hsieh, 2014). Changes in activity and expression of the tumor suppressor and phosphatase PTEN, both transcriptionally regulated by AR and an AR interactor, are well-known to alter CaP cell proliferation and aggressiveness (Wang et al., 2003).
b. Cytoskeleton organization and function
Successful completion of cell division, the final step of cell proliferation, involves spindle formation and reorganization of microtubules, which rely on tightly regulated phosphorylation events. The potential role for AR control over these modifications in CaP cells have yet to be examined. None-the-less, changes in organization and remodeling of the (actin) cytoskeleton are among the most common alterations observed in CaP compared to benign prostate tissues (Haffner et al., 2017, Hemstreet et al., 2000, Purnell et al., 1987). Such alterations also underlie each step of the invasion-metastatic cancer cascade, another key hallmark of cancer (Hanahan and Weinberg, 2011), that starts from local invasion, intravasation and extravasation of cancer cells leading to micrometastases, which develop into macroscopic metastatic cancers. Several of the proteins listed in Table 1 and 2 play role in these key steps. For instance, DDR1, discoidin domain receptor tyrosine kinase 1, is a collagen-induced receptor that activates signal transduction pathways involved in cell adhesion, proliferation, and extracellular matrix remodeling (Lee et al., 2019). Its family member DDR2 was shown to play an essential role in CaP bone metastasis and its silencing decreased CaP cell motility and invasiveness (Yan et al., 2014). Consistent with such a role, inhibition of DDR1 reduced EMT, linked to invasive/metastatic properties, in CaP (Maeyama et al., 2008, Koh et al., 2015, Yeh et al., 2011). Another AR controlled kinase, MERTK is well-known to promotes phagocytosis of apoptotic cells and reduces cytokine response (Graham et al., 2014), and drives invasion and metastasis in melanoma (Schlegel et al., 2013), glioblastoma (Wang et al., 2013) as well as CaP (Faltermeier et al., 2016). Although the kinase MYLK is poorly studied in CaP, because of its involvement in phosphorylating myosin light chains and therefore, cytoskeletal reorganization (Zhi et al., 2005), it is anticipated to similarly impact CaP biology. PAK2 is known to phosphorylate MAPK4, MAPK6 and activate MAPKAPK5 to influence F-actin polymerization, induce cytoskeleton reorganization and cell migration (Li et al., 2011, De la Mota-Peynado et al., 2011, Wang et al., 2018). Other examples include PRKD1 (Luef et al., 2016), which negatively regulates cell migration, and MAPKAPK2, which along with HSP27 increased matrix metaloprotease type 2 (MMP2) activity, leading increased cell invasion (Xu et al., 2006).
c. Transcriptional control
Progression from normal prostate to treatment-naïve localized CaP that is responsive to ADT, and then to CRPC is marked by differential gene expression patterns, which involve also considerable shifts in the transcriptomes and cistromes that are controlled by AR, the major driver of CaP progression (Massie et al., 2011, Stelloo et al., 2015, Sharma et al., 2013) . These alterations in gene expression profiles result from epigenetic and transcriptional events in which several of the AR-associated kinases and phosphatases listed above are known to fulfill critical roles. In addition to roles in cell cycle progression, several CDKs that interact with AR have been implicated also in transcription of AR target genes. CDK6 fulfills such dual roles, and has been reported to coactivate also strongly transcription by the gain-of-function AR T878A mutant that occurs under selective pressure of ADT and is present in CRPC (Lim et al., 2005). Other CDKS, such as CDK7 and 9 have been mainly implicated in AR-dependent transcription (Rasool et al., 2019, Lee et al., 2000, Pawar et al., 2018, Itkonen et al., 2019). Whether kinase activity is relevant for this is not always clear, as it is necessary for some CDKs and dispensable for others. The target of CDK kinase action can differ also. CDK7 interacts with the N-terminal of AR, but has not reported to directly phosphorylate AR (Lee et al., 2000). Instead, CDK7 phosphorylates the MED1 component of Mediator complex, which enhances MED1 interaction with AR. Inhibition of CDK7 using a specific inhibitor (TZH1) blocks MED1 recruitment to chromatin (Rasool et al., 2019). On the other hand, CDK9 phosphorylates AR at serine 81 (Gordon et al., 2010), and a kinase-dead version of CDK9 impairs AR transactivation (Lee et al., 2001). Consistent with roles for several CDKs in regulating AR transactivation, also CDC25A and CDC25B, phosphatases that control CDK activity, can modulate transcription of AR target genes (Chiu et al., 2009, Balk and Knudsen, 2008). Whether this entails CDC’s phosphatase activity or alterations in phosphorylation of AR or associated transcriptional regulators is not known. Other classes of kinases and phosphatases have been implicated in AR transcriptional control. Functionally diverse coregulators such as HIPK3 aka ANPK (androgen-receptor interacting nuclear protein) or DAPK3 interact with AR and activate its transcriptional function but AR has not been shown to be a direct substrate of HIPK3 or DAPK3 (Moilanen et al., 1998, Leister et al., 2008, Felten et al., 2013). AKT1, whose activation and activity is a major issue during endocrine therapy in CaP (Carver et al., 2011), does phosphorylate AR at S210 and S790 (Lin et al., 2001), which leads to suppression of AR-target genes such as p21. PAK6-AR interaction (Lee et al., 2002, Yang et al., 2001) also impacts AR target gene transcription and relies on PAK6’s kinase domain and PAK6-dependent phosphorylation of AR’s DNA binding domain (DBD) domain (Schrantz et al., 2004). Direct phosphorylation and activation of AR by SRC sensitizes AR to intracrine androgen levels, resulting in the engagement of canonical and non-canonical AR-dependent gene signatures (Guo et al., 2006, Chattopadhyay et al., 2017). We identified the histone phosphatase PGAM5 as integral and essential part of AR transcriptional complexes that contain also p53 and the AR-associated coregulator WDR77 and preferentially control CaP cell proliferation and survival (Liu et al., 2017). Whether this involves PGAM5’s phosphatase function was not addressed. Other contributions of the AR-controlled phospho-regulators to the CaP transcriptome are conceivable. As an example, CLK2 is a transcriptionally AR-dependent kinase (Jin et al., 2013) that regulates the spliceosomal complex and alternative splicing in breast cancer and endothelial cells (Eisenreich et al., 2009, Yoshida et al., 2015), suggesting it may influence the spectrum of splice forms expressed.
3. The clinically relevant AR-dependent CaP phosphoproteome
The development of kinase inhibitors and their subsequent therapeutic success in other human malignancies such as non-small cell lung cancer or melanoma (Lynch et al., 2004, Shaw et al., 2014, Chapman et al., 2011) was triggered by observations of activating kinase mutations and/or amplifications. These alterations rendered the kinase function oncogenic and turned it into a driver of cancer progression. In CaP, such somatic alterations in genes encoding kinases or for that matter phosphatases (except PTEN) are rare: only 0.9 to 8 percent (5,226 specimens examined via cBioportal) of the AR-dependent phosphorylation regulators listed in Tables 1 and 2 are affected, with the exception of PTEN deletion, which occurs in 18% of cases/specimens (Figure 4). The absence of comprehensive phospho-proteomics data from clinical CRPC cases further impeded the evaluation of wild-type kinases as valid therapeutic targets and kinase inhibitors as potential novel therapeutics in CaP. Some smaller scale phospho-proteome studies had been conducted using CaP cell lines (Giorgianni et al., 2007) and xenograft models under conditions that represent ADT-naïve CaP and CRPC (Ramroop et al., 2018). Results suggested active signaling by several kinases, including for instance YAP1 and the AR-dependent PAK2, which were put forward as novel viable therapeutic targets (Jiang et al., 2015). Other studies had also recognized that AR can repress major proliferative kinase signaling pathways in CaP, and conversely that ADT can, depending on one’s perspective, either cause unwanted activation of these pathways in CRPC or render these kinases as targets for novel CRPC treatments. The most prominent example is ADT-induced activation of PI3K/AKT signaling, may impact downstream kinase signaling such as that mediated by SGK1 (Carver et al., 2011, Toska et al., 2019). Similar observation have been made for the tyrosine kinase BMX. BMX expression in CaP is suppressed by AR and rapidly increased in response to ADT. BMX contributed to CRPC development in vitro and in vivo by positively regulating the activities of multiple receptor tyrosine kinases, such as MET, FGFR1 (Chen et al., 2013). Inhibition of BMX kinase activity markedly enhanced the response to castration in CaP models (Chen et al., 2018). These studies put forward the concept that modulation of AR activity during CRPC progression may alter the kinase signaling pathways and associated biology that are active in a patient’s CaP.
Figure 4. Somatic alterations in AR-associated kinases and phosphatases.
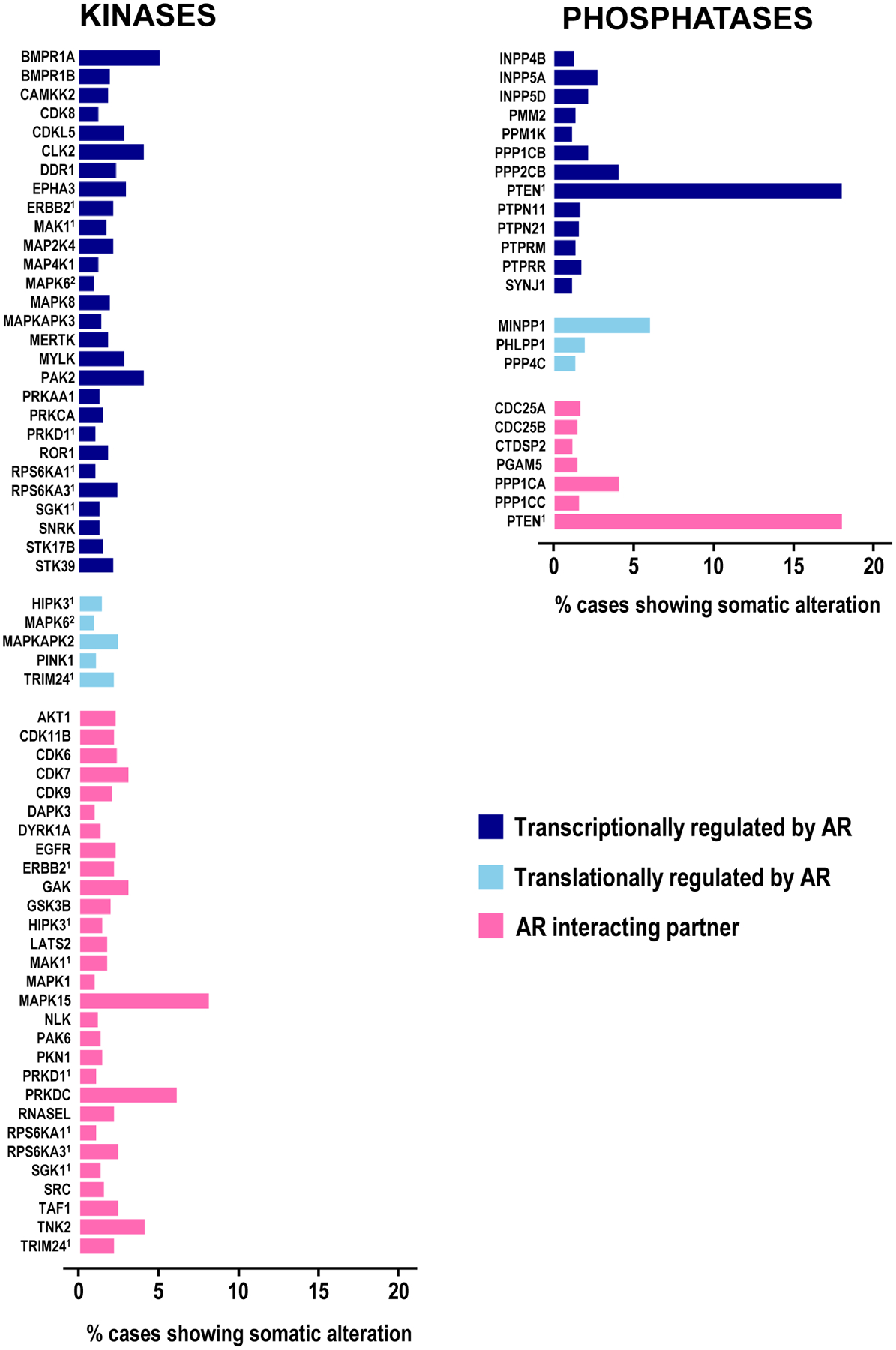
Number of clinical CaP cases with somatic alterations in AR-associated kinases (left panel) and phosphatases (right panel). Data reflect results for cBioportal analyses on 5,226 CaP specimens.
None-the-less, it was not until the concept of personalized medicine gained traction in the CaP field that more in-depth proteomic analysis on clinical CaP specimens were undertaken and activity of some wild-type kinases started to emerge as valid targets for new CaP treatment schemes. A 2012 paper from the Witte group laid the foundation by performing immunohistochemistry for tyrosine phosphorylation marks in patient samples representing progression from benign prostate to CRPC and tissues from a mouse CaP model driven by oncogenes such as AKT, ERG, AR, or K-RASG12V, which showed increased tyrosine phosphorylation marks during both patient and mouse cancer progression (Drake et al., 2012). Subsequent use of phospho-antibodies and mass spectrometry on phospho-tyrosine peptides indicated differential activation of the EGFR, EPHA2, JAK2, SRC and ABL1 tyrosine kinases between these models. These results were the first to suggest that, based on proteomics footprint of an individual patient’s CaP specimen, inhibition of tyrosine kinase, include the AR-dependent EGFR and SRC, could be administered as personalized therapeutics (Drake et al., 2012). Follow-up studies suggesting that kinase activity patterns were similar among the different CaP metastases within the same patient, but differed between patients, increased the attractiveness of this possibility- although the authors indicate some caution since the relatively small number of specimens studied (n=16 may have led this particular study to be somewhat underpowered (Drake et al., 2016). None-the-less, a more comprehensive analysis of clinical samples that involved mass spectrometry on phospho-tyrosine peptides as well as phospho-serine/phospho-threonine-peptides, in combination with genomic and transcriptome analyses and sophisticated bioinformatics tools again confirmed differentially activated kinases in CRPC progression. In the same study, an online portal “pCHIPS” was developed and made publically available as a tool that can define the druggable kinase pathways for drug prioritization in individual patients. Several analyses in the latter study pointed to cell cycle progression, cell migration and AR function as associated with phopsho-proteome alterations and point to wild-type not somatically altered kinases, some of which AR-dependent, being the major determinants of CRPC progression (Drake et al., 2016).
The possibility that wild-type kinase action can induce aggressive CaP behavior was consistent with the results from a functional screen in which the impact of 125 kinases that show either increased mRNA expression, gene amplification or increased protein levels in human CaP on experimental metastasis was examined (Faltermeier et al., 2016). In an in vivo screen, these kinases were overexpressed in murine prostate cells that were tail-vein injected into mice that were subsequently screened for lung metastasis. Twenty of these kinases, namely MAP3K8, NTRK2, ARAF, BRAF, CRAF, MAP2K15, MERTK, FGFR3, FLT3, LYN, MAP3K2, NTRK3, PI3KCa, EGFR, EPHA2, HER2, PDGFRa, FGFR1, SRC, BMX (note again some AR-dependent kinases) (Tables 1 and 2) were able to induce such metastasis. Further validation efforts using human prostate cells validated ARAF, BRAF, CRAF, MERTK and NTRK2 as key drivers of bone and visceral tissue metastasis. Although over-expressing the kinases might not directly correlate to activation of the kinase, and the focus on bone metastasis as endpoint limits implication of kinases involved in treatment-resistance in CaP.
4. Targeting AR-dependent phosphorylation as CaP treatment strategy
The therapeutic successes obtained with kinase inhibition in other human malignancies have led to clinical testing of several kinase inhibitors for CaP treatment. A search of clinicaltrial.gov site, a database of privately and publicly funded clinical studies, using the terms “prostate cancer” and “kinase inhibitor” returned 137 clinical trials which are listed in Supplemental Table 2. The kinase targets for the inhibitors tested fall into 5 broad classes, namely growth factor receptors, tyrosine kinases, cell cycle regulators, Akt/mTOR pathway related kinases and others (Table 3). These include several kinases that are AR-associated, such as EGFR, SRC, CDK6, CDK9, and PRKDC.
Table 3. Overview of clinical trials targeting kinases in metastatic CaP.
Columns indicate class of kinases, number of clinical trials, acronyms of clinical trials (if any), drug targets, and phases of clinical trials. Kinases marked in bold and italics are present in Table 1. Note that 2 clinical trials relating to precision medicine (MATCH and SMMART) were left out; these target several kinases based on the patient’s genotype. An elaborate version of the table can be found in Supplementary Table1.
| Kinase functional class | Number of trials | Trial acronym | Drug targets | Clinical trial phase |
|---|---|---|---|---|
| Growth factor receptors | 99 | INV342, ARS, KHLAD, NRR, GeniPro, QUERGEN, GCP, P1, AZD2171IL/0003, ARCAP, PROSUT, SUN 1120, SMART | VEGFR, PDGFR, FGFR, EGFR, BCR-ABL, KIT, | Phase 1, phase 2 |
| Tyrosine kinases | 14 | BrUOG PR255, READY, PROACT | ALK, ROS1, BCR-ABL, SRC, MEK1, MEK2 | Phase 1, phase 2 |
| Cell cycle regulators | 11 | ATR, AURKA, CDK4, CDK6, CDK9, CHEK1, CHEK2 | Phase 1, phase 2 | |
| Akt/mTOR pathway | 9 | AKT1, FKBP12, MNK1, MNK2, MTOR, CRTC2, PIK3CA, PI3KCB, PRKDC | Phase 1, phase 2 | |
| Other | 2 | FAK, PYK2, PIM1 | Phase 1, phase 2 |
To date, the success of completed trials testing kinase inhibitors as CaP therapeutics has been modest and none of the inhibitors tested have moved into routine clinical practice. Several factors may have contributed to the clinical failure of inhibitors that were successful in preclinical therapeutics studies. Notably, recent studies combining phospho-peptide enrichment with mass spectrometry have shown that CaP cell lines and xenografts poorly model the phospho-proteome present in clinical CaP specimens (Drake et al., 2016). These findings imply that the experimental models used to testing the inhibitor do not fully captured the clinical setting they were used in. In addition, these trials also recruited patients without stratifying them based on the proteomic make-up of their CaP, and thus without knowing the activation status of the kinase being targeted. This approach still leaves the possibility of therapeutic success in a subset of patients with the right target activation (i.e. the outliers, or exceptional responders) that is not reflected in the overall negative trial results obtained for entire patient cohort. Another well-known limitation of most kinase inhibitors is their lack of selectively aka polypharmacology: i.e. the inhibitor is able to inhibit more than one kinase target often at similar IC50s (Klaeger et al., 2017, Davis et al., 2011). This can be especially problematic to the success of treatment when the activity of the target kinase(s) in the patient is not known and when affected kinases have opposite effects on the same CaP pathways or biology. Another important and often underappreciated limitation is that the endpoint used to measure therapeutic success is not adequate for the compound under investigation, leaving investigators essentially unable to judge the efficacy of drugs tested and to misinterpret trial results. An example of this relates to our and others’ testing of the multi-kinase inhibitor, lestaurtinib, as CaP therapeutic (Venkadakrishnan et al., 2019). Lestaurtinib also inhibits the kinase activity of PKN1, which drives CaP progression, and is highly expressed and activated in CRPC. Lestaurtinib had been well-tolerated in phase I clinical trials for advanced carcinomas (Marshall et al., 2005) and was studied clinically for treatment of myelofibrosis and acute myeloid leukemia (Shabbir and Stuart, 2010, Knapper et al., 2006). To date, 2 phase II clinical trials using lestaurtinib have been initiated in CRPC patients. Patients were considered responders if they had >50% decrease in serum PSA, a surrogate marker of CaP progression, compared to baseline. No patients met these response criteria. Instead, serum PSA tended to increase during treatment with lestaurtinib and declined when treatment was stopped. The decision not to move lestaurtinib forward clinically in CRPC was made solely based on the failure to meet the endpoint of PSA response. However, studies in vivo and in vitro CaP models have shown increased PSA production by cells that were growth-arrested by lestaurtinib (Venkadakrishnan et al., 2019, Collins et al., 2007). The implication is that a potentially effective agent to target AR action downstream of activated AR that has a different mechanism of action than traditional ADT and is tolerated by patients, was abandoned mainly because of the use of an inappropriate biomarker to evaluate treatment response.
Appreciating the issues that prevented therapeutic successes using kinase inhibitors may help resolve them and increase the likelihood of future therapeutic successes. Targeting the right kinase in the right patient may be achieved via proteomics analysis on one or more of his CaP tissues. Similar genomic CaP characterizations are the premise for personalized medicine approaches, and the basis for patient stratifications into for instance the NCI MATCH trials (Coyne et al., 2017). Improving the specificity of kinase inhibitors relies in part on a better understanding of the kinase activity. Indeed, for most kinases, the full spectrum of substrates has not yet been described, the possibility of tissue or cancer type substrate specificity is under-appreciated or the extent of substrate overlap among kinases is not known. Technical advances may help resolve these limitations. Several approaches including SILAC, TiO2 enrichment of phophoproteins for a label-free quantification, phospho-specific-immunoprecipitation followed by mass spectrometry, motif analyses using phospho-peptide arrays are extensively used to identify substrates and substrates binding motifs (Wu et al., 2015, Montoya et al., 2011, de Oliveira et al., 2016). Integration of such assays with computational modeling and bioinformatics analyses to better define linear and non-linear substrate motifs, and proper in vitro and in vivo validation of substrate phosphorylation sites using not only kinase domains but full length versions of kinases under investigation, are all likely to expand the bona fide spectrum of kinase substrates. Better knowledge of a kinases substrates or the kinase-substrate interactions will facilitate monitoring kinase-targeting treatment responses. This could be achieved using either relevant phospho-motif specific antibodies or proximity ligation assays on circulating tumor cells obtained from non-invasive liquid biopsies. A second way to improve inhibitor specificity will rely on our enhanced understanding of kinase inhibitor binding pockets or molecular details on substrate-kinase interactions (de Oliveira et al., 2016), which have been proposed as alternative targets to inhibit kinase activity using peptides or peptidomimetics. For many kinases, reliable 3D structural models are not available and crystallization efforts with inhibitors have not yet been undertaken. Ongoing advances in cryoEM are expected to provide this much-needed information.
Conclusions
The majority of the more than 30,000 CaP deaths annually in the US are due to failure of ADT, which prevents ligand-activation of AR. Despite an initial remission, CaP progresses while continuing to rely on AR action, emphasizing the need for novel drugable targets downstream of activated AR. Our literature review uncovered that AR associates with dozens of functionally diverse kinases and phosphatases, and that the ensuing phosphorylation events control CaP biology relevant to aggressive CaP progression. Several of these phosphorylation events were reflected in recently recognized alterations in phospho-proteome in late stage CaP. Kinase inhibitors targeting some of these have been clinically tested and could serve as novel therapies that can overcome CaP’s resistance to ADT, provided some challenges and limitations related to therapeutic efficiency, target specificity and monitoring of treatment responses are addressed adequately.
Supplementary Material
Supplementary Table 1. AR-binding sites in genes encoding kinases and phosphatases in VCaP cells. Table provides an overview of the presence of AR binding sites in transcriptionally-regulated kinases (top part) and phosphatases (bottom part) from Table 1 and Table 2 in 2 independent AR ChIP-seq datasets derived from VCaP cells (Massie et al., 2011, Asangani et al., 2014). AR ChIP-Seq datasets were retrieved via cistrome.org (Massie et. al. 2011: CistromeDB: 2910, 2911; Asangani et. al. 2014: CistromeDB: 44574 – 44577) Data was analyzed and AR binding peaks were mapped using the UCSC genome browser tool. Androgen-induced peaks whose values were at least 3-fold higher than background values were withheld. Yes, presence of an AR peak AR binding site within 300kb of the transcriptional start site of the gene in all (100%) or half (50%) of biological replicates in the respective study; maybe, presence of such peak not certain.
Supplementary Table 2. Overview of clinical trials using kinase inhibitors for CaP treatment.
Figure 2. Kinome tree for AR-associated kinases.
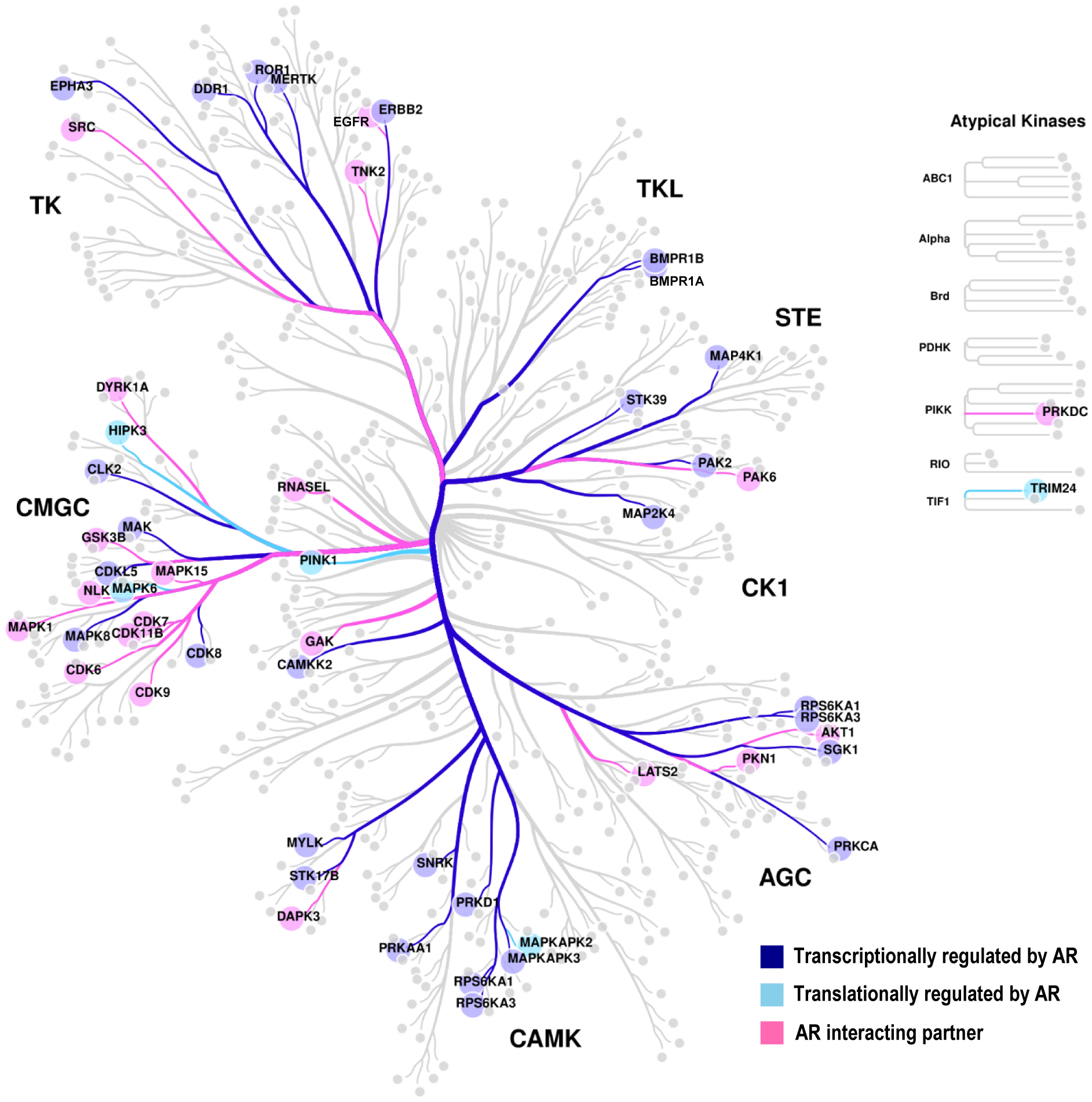
Kinases transcriptionally regulated by AR are marked in dark blue, kinases translationally regulated by AR in sky blue, and AR-interacting partners in pink. Figure generated using online tool, CORAL (Mets et. al. Cell systems, 2018).
Figure 3.
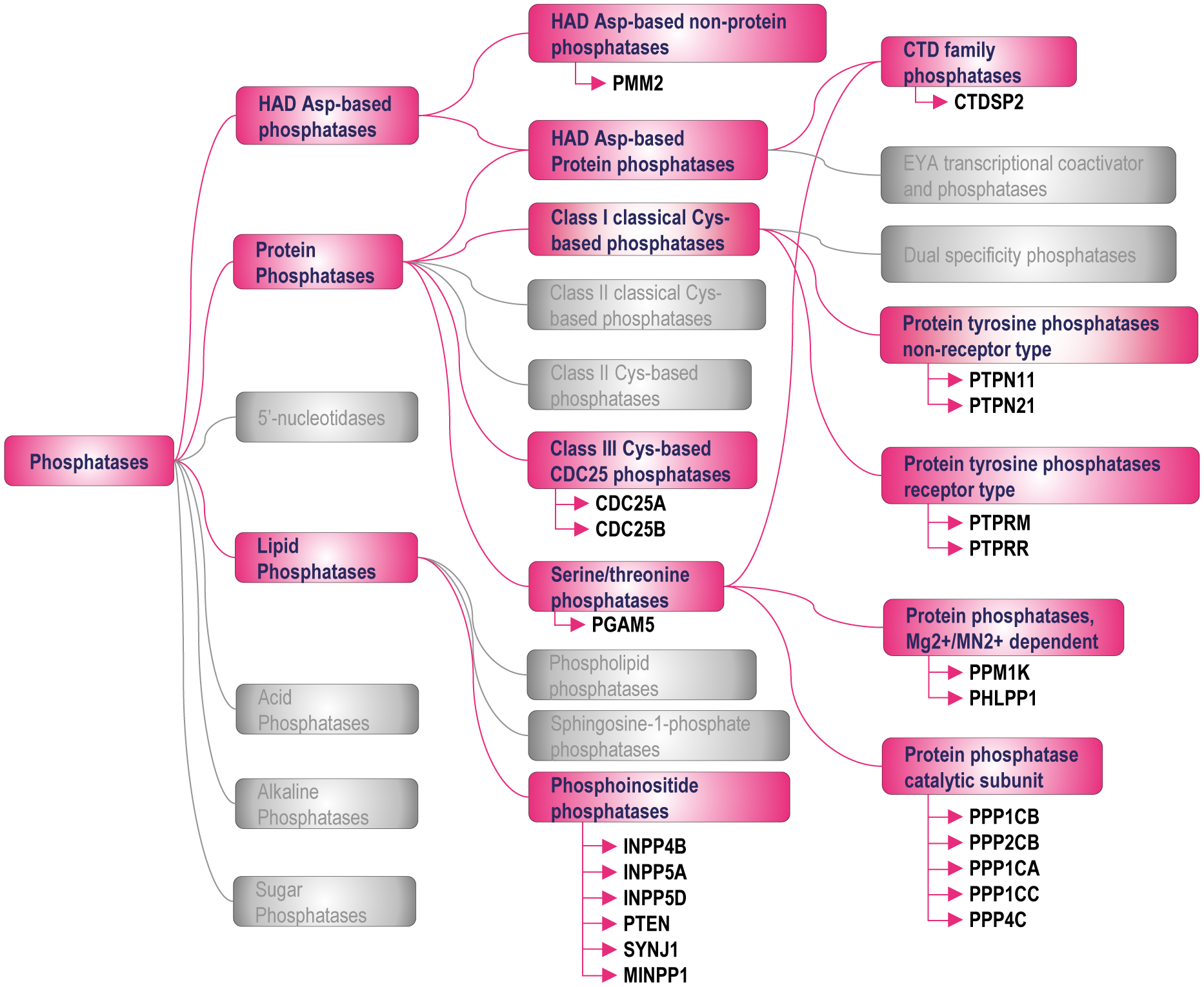
Classification of AR-associated phosphatases.
Funding:
This work was supported the National Cancer Institute (grant number CA166440), the Department of Defense Prostate Cancer Research Program (grant number W81XWH-16-1-0404), and a VeloSano 5 pilot Research Award (to HVH).
Footnotes
Declaration of interest: the authors declare no conflicting interests
References
- ARANDA-ORGILLES B, RUTSCHOW D, ZELLER R, KARAGIANNIDIS AI, KOHLER A, CHEN C, WILSON T, KRAUSE S, ROEPCKE S, LILLEY D, et al. 2011. Protein phosphatase 2A (PP2A)-specific ubiquitin ligase MID1 is a sequence-dependent regulator of translation efficiency controlling 3-phosphoinositide-dependent protein kinase-1 (PDPK-1). J Biol Chem, 286, 39945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASANGANI IA, DOMMETI VL, WANG X, MALIK R, CIESLIK M, YANG R, ESCARA-WILKE J, WILDER-ROMANS K, DHANIREDDY S, ENGELKE C, et al. 2014. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature, 510, 278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALK SP & KNUDSEN KE 2008. AR, the cell cycle, and prostate cancer. Nucl Recept Signal, 6, e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELL RM & BURNS DJ 1991. Lipid activation of protein kinase C. J Biol Chem, 266, 4661–4. [PubMed] [Google Scholar]
- BOLLU LR, MAZUMDAR A, SAVAGE MI & BROWN PH 2017. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin Cancer Res, 23, 2136–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAI C, WANG H, HE HH, CHEN S, HE L, MA F, MUCCI L, WANG Q, FIORE C, SOWALSKY AG, et al. 2013. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest, 123, 1109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARVER BS, CHAPINSKI C, WONGVIPAT J, HIERONYMUS H, CHEN Y, CHANDARLAPATY S, ARORA VK, LE C, KOUTCHER J, SCHER H, et al. 2011. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell, 19, 575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAPMAN PB, HAUSCHILD A, ROBERT C, HAANEN JB, ASCIERTO P, LARKIN J, DUMMER R, GARBE C, TESTORI A, MAIO M, et al. 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med, 364, 2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATTOPADHYAY I, WANG J, QIN M, GAO L, HOLTZ R, VESSELLA RL, LEACH RW & GELMAN IH 2017. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget, 8, 10324–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN S, CAI C, SOWALSKY AG, YE H, MA F, YUAN X, SIMON NI, GRAY NS & BALK SP 2018. BMX-Mediated Regulation of Multiple Tyrosine Kinases Contributes to Castration Resistance in Prostate Cancer. Cancer Res, 78, 5203–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN S, JIANG X, GEWINNER CA, ASARA JM, SIMON NI, CAI C, CANTLEY LC & BALK SP 2013. Tyrosine kinase BMX phosphorylates phosphotyrosine-primed motif mediating the activation of multiple receptor tyrosine kinases. Sci Signal, 6, ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIU YT, HAN HY, LEUNG SC, YUEN HF, CHAU CW, GUO Z, QIU Y, CHAN KW, WANG X, WONG YC, et al. 2009. CDC25A functions as a novel Ar corepressor in prostate cancer cells. J Mol Biol, 385, 446–56. [DOI] [PubMed] [Google Scholar]
- CHUU CP, HIIPAKKA RA, FUKUCHI J, KOKONTIS JM & LIAO S 2005. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res, 65, 2082–4. [DOI] [PubMed] [Google Scholar]
- CIARDIELLO F & TORTORA G 2008. EGFR antagonists in cancer treatment. N Engl J Med, 358, 1160–74. [DOI] [PubMed] [Google Scholar]
- COLLINS C, CARDUCCI MA, EISENBERGER MA, ISAACS JT, PARTIN AW, PILI R, SINIBALDI VJ, WALCZAK JS & DENMEADE SR 2007. Preclinical and clinical studies with the multi-kinase inhibitor CEP-701 as treatment for prostate cancer demonstrate the inadequacy of PSA response as a primary endpoint. Cancer Biol Ther, 6, 1360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COYNE GO, TAKEBE N & CHEN AP 2017. Defining precision: The precision medicine initiative trials NCI-MPACT and NCI-MATCH. Curr Probl Cancer, 41, 182–193. [DOI] [PubMed] [Google Scholar]
- DAI C, HEEMERS H & SHARIFI N 2017. Androgen Signaling in Prostate Cancer. Cold Spring Harb Perspect Med, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAMASSA DA, LIN TM, SONNENSCHEIN C & SOTO AM 1991. Biological effects of sex hormone-binding globulin on androgen-induced proliferation and androgen metabolism in LNCaP prostate cells. Endocrinology, 129, 75–84. [DOI] [PubMed] [Google Scholar]
- DAVIS MI, HUNT JP, HERRGARD S, CICERI P, WODICKA LM, PALLARES G, HOCKER M, TREIBER DK & ZARRINKAR PP 2011. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol, 29, 1046–51. [DOI] [PubMed] [Google Scholar]
- DE LA MOTA-PEYNADO A, CHERNOFF J & BEESER A 2011. Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J Biol Chem, 286, 13603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LEEUW R, MCNAIR C, SCHIEWER MJ, NEUPANE NP, BRAND LJ, AUGELLO MA, LI Z, CHENG LC, YOSHIDA A, COURTNEY SM, et al. 2018. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin Cancer Res, 24, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE OLIVEIRA PS, FERRAZ FA, PENA DA, PRAMIO DT, MORAIS FA & SCHECHTMAN D 2016. Revisiting protein kinase-substrate interactions: Toward therapeutic development. Sci Signal, 9, re3. [DOI] [PubMed] [Google Scholar]
- DENMEADE SR & ISAACS JT 2010. Bipolar androgen therapy: the rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. The Prostate, 70, 1600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEPRIEST AD, FIANDALO MV, SCHLANGER S, HEEMERS F, MOHLER JL, LIU S & HEEMERS HV 2016. Regulators of Androgen Action Resource: a one-stop shop for the comprehensive study of androgen receptor action. Database (Oxford), 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRAKE JM, GRAHAM NA, LEE JK, STOYANOVA T, FALTERMEIER CM, SUD S, TITZ B, HUANG J, PIENTA KJ, GRAEBER TG, et al. 2013. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A, 110, E4762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRAKE JM, GRAHAM NA, STOYANOVA T, SEDGHI A, GOLDSTEIN AS, CAI H, SMITH DA, ZHANG H, KOMISOPOULOU E, HUANG J, et al. 2012. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A, 109, 1643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRAKE JM, PAULL EO, GRAHAM NA, LEE JK, SMITH BA, TITZ B, STOYANOVA T, FALTERMEIER CM, UZUNANGELOV V, CARLIN DE, et al. 2016. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell, 166, 1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDLIND MP & HSIEH AC 2014. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl, 16, 378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EID S, TURK S, VOLKAMER A, RIPPMANN F & FULLE S 2017. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics, 18, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EISENREICH A, BOGDANOV VY, ZAKRZEWICZ A, PRIES A, ANTONIAK S, POLLER W, SCHULTHEISS HP & RAUCH U 2009. Cdc2-like kinases and DNA topoisomerase I regulate alternative splicing of tissue factor in human endothelial cells. Circ Res, 104, 589–99. [DOI] [PubMed] [Google Scholar]
- FALTERMEIER CM, DRAKE JM, CLARK PM, SMITH BA, ZONG Y, VOLPE C, MATHIS C, MORRISSEY C, CASTOR B, HUANG J, et al. 2016. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc Natl Acad Sci U S A, 113, E172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELTEN A, BRINCKMANN D, LANDSBERG G & SCHEIDTMANN KH 2013. Zipper-interacting protein kinase is involved in regulation of ubiquitination of the androgen receptor, thereby contributing to dynamic transcription complex assembly. Oncogene, 32, 4981–8. [DOI] [PubMed] [Google Scholar]
- GAO S, YE H, GERRIN S, WANG H, SHARMA A, CHEN S, PATNAIK A, SOWALSKY AG, VOZNESENSKY O, HAN W, et al. 2016. ErbB2 Signaling Increases Androgen Receptor Expression in Abiraterone-Resistant Prostate Cancer. Clin Cancer Res, 22, 3672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIORGIANNI F, ZHAO Y, DESIDERIO DM & BERANOVA-GIORGIANNI S 2007. Toward a global characterization of the phosphoproteome in prostate cancer cells: identification of phosphoproteins in the LNCaP cell line. Electrophoresis, 28, 2027–34. [DOI] [PubMed] [Google Scholar]
- GORDON V, BHADEL S, WUNDERLICH W, ZHANG J, FICARRO SB, MOLLAH SA, SHABANOWITZ J, HUNT DF, XENARIOS I, HAHN WC, et al. 2010. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol, 24, 2267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAHAM DK, DERYCKERE D, DAVIES KD & EARP HS 2014. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer, 14, 769–85. [DOI] [PubMed] [Google Scholar]
- GRONER AC, CATO L, DE TRIBOLET-HARDY J, BERNASOCCHI T, JANOUSKOVA H, MELCHERS D, HOUTMAN R, CATO ACB, TSCHOPP P, GU L, et al. 2016. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell, 29, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO Z, DAI B, JIANG T, XU K, XIE Y, KIM O, NESHEIWAT I, KONG X, MELAMED J, HANDRATTA VD, et al. 2006. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell, 10, 309–19. [DOI] [PubMed] [Google Scholar]
- HAFFNER MC, ESOPI DM, CHAUX A, GUREL M, GHOSH S, VAGHASIA AM, TSAI H, KIM K, CASTAGNA N, LAM H, et al. 2017. AIM1 is an actin-binding protein that suppresses cell migration and micrometastatic dissemination. Nat Commun, 8, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANAHAN D & WEINBERG RA 2011. Hallmarks of cancer: the next generation. Cell, 144, 646–74. [DOI] [PubMed] [Google Scholar]
- HEEMERS HV, SCHMIDT LJ, SUN Z, REGAN KM, ANDERSON SK, DUNCAN K, WANG D, LIU S, BALLMAN KV & TINDALL DJ 2011. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res, 71, 1978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEEMERS HV, VERHOEVEN G & SWINNEN JV 2006. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol Endocrinol, 20, 2265–77. [DOI] [PubMed] [Google Scholar]
- HEMSTREET GP 3RD, BONNER RB, HURST RE, BELL D & BANE BL 2000. Abnormal G-actin content in single prostate cells as a biomarker of prostate cancer. Cancer Detect Prev, 24, 464–72. [PubMed] [Google Scholar]
- HODGSON MC, DERYUGINA EI, SUAREZ E, LOPEZ SM, LIN D, XUE H, GORLOV IP, WANG Y & AGOULNIK IU 2014. INPP4B suppresses prostate cancer cell invasion. Cell Commun Signal, 12, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HODGSON MC, SHAO LJ, FROLOV A, LI R, PETERSON LE, AYALA G, ITTMANN MM, WEIGEL NL & AGOULNIK IU 2011. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res, 71, 572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGGINS C & HODGES CV 2002. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol, 168, 9–12. [DOI] [PubMed] [Google Scholar]
- ITKONEN HM, POULOSE N, WALKER S & MILLS IG 2019. CDK9 Inhibition Induces a Metabolic Switch that Renders Prostate Cancer Cells Dependent on Fatty Acid Oxidation. Neoplasia, 21, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANJIGIAN YY, SMIT EF, GROEN HJ, HORN L, GETTINGER S, CAMIDGE DR, RIELY GJ, WANG B, FU Y, CHAND VK, et al. 2014. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov, 4, 1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEITANY M, LEROY C, TOSTI P, LAFITTE M, LE GUET J, SIMON V, BONENFANT D, ROBERT B, GRILLET F, MOLLEVI C, et al. 2018. Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol Med, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG N, HJORTH-JENSEN K, HEKMAT O, IGLESIAS-GATO D, KRUSE T, WANG C, WEI W, KE B, YAN B, NIU Y, et al. 2015. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene, 34, 2764–76. [DOI] [PubMed] [Google Scholar]
- JIN HJ, KIM J & YU J 2013. Androgen receptor genomic regulation. Transl Androl Urol, 2, 157–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARACOSTA LG, FOSTER BA, AZABDAFTARI G, FELICIANO DM & EDELMAN AM 2012. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem, 287, 24832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAEGER S, HEINZLMEIR S, WILHELM M, POLZER H, VICK B, KOENIG PA, REINECKE M, RUPRECHT B, PETZOLDT S, MENG C, et al. 2017. The target landscape of clinical kinase drugs. Science, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNAPPER S, BURNETT AK, LITTLEWOOD T, KELL WJ, AGRAWAL S, CHOPRA R, CLARK R, LEVIS MJ & SMALL D 2006. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood, 108, 3262–70. [DOI] [PubMed] [Google Scholar]
- KNUDSEN KE, ARDEN KC & CAVENEE WK 1998. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem, 273, 20213–22. [DOI] [PubMed] [Google Scholar]
- KOH M, WOO Y, VALIATHAN RR, JUNG HY, PARK SY, KIM YN, KIM HR, FRIDMAN R & MOON A 2015. Discoidin domain receptor 1 is a novel transcriptional target of ZEB1 in breast epithelial cells undergoing H-Ras-induced epithelial to mesenchymal transition. Int J Cancer, 136, E508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMARI S, SENAPATI D & HEEMERS HV 2017. Rationale for the development of alternative forms of androgen deprivation therapy. Endocr Relat Cancer, 24, R275–R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE DK, DUAN HO & CHANG C 2000. From androgen receptor to the general transcription factor TFIIH. Identification of cdk activating kinase (CAK) as an androgen receptor NH(2)-terminal associated coactivator. J Biol Chem, 275, 9308–13. [DOI] [PubMed] [Google Scholar]
- LEE DK, DUAN HO & CHANG C 2001. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. J Biol Chem, 276, 9978–84. [DOI] [PubMed] [Google Scholar]
- LEE SR, RAMOS SM, KO A, MASIELLO D, SWANSON KD, LU ML & BALK SP 2002. AR and ER interaction with a p21-activated kinase (PAK6). Mol Endocrinol, 16, 85–99. [DOI] [PubMed] [Google Scholar]
- LEE YC, KURTOVA AV, XIAO J, NIKOLOS F, HAYASHI K, TRAMEL Z, JAIN A, CHEN F, CHOKSHI M, LEE C, et al. 2019. Collagen-rich airway smooth muscle cells are a metastatic niche for tumor colonization in the lung. Nat Commun, 10, 2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEISTER P, FELTEN A, CHASAN AI & SCHEIDTMANN KH 2008. ZIP kinase plays a crucial role in androgen receptor-mediated transcription. Oncogene, 27, 3292–300. [DOI] [PubMed] [Google Scholar]
- LEUNG JK & SADAR MD 2017. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front Endocrinol (Lausanne), 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J, YEN C, LIAW D, PODSYPANINA K, BOSE S, WANG SI, PUC J, MILIARESIS C, RODGERS L, MCCOMBIE R, et al. 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 275, 1943–7. [DOI] [PubMed] [Google Scholar]
- LI P, NICOSIA SV & BAI W 2001. Antagonism between PTEN/MMAC1/TEP-1 and androgen receptor in growth and apoptosis of prostatic cancer cells. J Biol Chem, 276, 20444–50. [DOI] [PubMed] [Google Scholar]
- LI T, ZHANG J, ZHU F, WEN W, ZYKOVA T, LI X, LIU K, PENG C, MA W, SHI G, et al. 2011. P21-activated protein kinase (PAK2)-mediated c-Jun phosphorylation at 5 threonine sites promotes cell transformation. Carcinogenesis, 32, 659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG C, WANG S, QIN C, BAO M, CHENG G, LIU B, SHAO P, LV Q, SONG N, HUA L, et al. 2018. TRIM36, a novel androgen-responsive gene, enhances anti-androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis, 9, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIM JT, MANSUKHANI M & WEINSTEIN IB 2005. Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc Natl Acad Sci U S A, 102, 5156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN HK, YEH S, KANG HY & CHANG C 2001. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci U S A, 98, 7200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU S, KUMARI S, HU Q, SENAPATI D, VENKADAKRISHNAN VB, WANG D, DEPRIEST AD, SCHLANGER SE, BEN-SALEM S, VALENZUELA MM, et al. 2017. A comprehensive analysis of coregulator recruitment, androgen receptor function and gene expression in prostate cancer. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y, HORN JL, BANDA K, GOODMAN AZ, LIM Y, JANA S, ARORA S, GERMANOS AA, WEN L, HARDIN WR, et al. 2019. The androgen receptor regulates a druggable translational regulon in advanced prostate cancer. Sci Transl Med, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUEF B, HANDLE F, KHARAISHVILI G, HAGER M, RAINER J, JANETSCHEK G, HRUBY S, ENGLBERGER C, BOUCHAL J, SANTER FR, et al. 2016. The AR/NCOA1 axis regulates prostate cancer migration by involvement of PRKD1. Endocr Relat Cancer, 23, 495–508. [DOI] [PubMed] [Google Scholar]
- LUNDON DJ, BOLAND A, PRENCIPE M, HURLEY G, O’NEILL A, KAY E, AHERNE ST, DOOLAN P, MADDEN SF, CLYNES M, et al. 2017. The prognostic utility of the transcription factor SRF in docetaxel-resistant prostate cancer: in-vitro discovery and in-vivo validation. BMC Cancer, 17, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYNCH TJ, BELL DW, SORDELLA R, GURUBHAGAVATULA S, OKIMOTO RA, BRANNIGAN BW, HARRIS PL, HASERLAT SM, SUPKO JG, HALUSKA FG, et al. 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med, 350, 2129–39. [DOI] [PubMed] [Google Scholar]
- MAEYAMA M, KOGA H, SELVENDIRAN K, YANAGIMOTO C, HANADA S, TANIGUCHI E, KAWAGUCHI T, HARADA M, UENO T & SATA M 2008. Switching in discoid domain receptor expressions in SLUG-induced epithelial-mesenchymal transition. Cancer, 113, 2823–31. [DOI] [PubMed] [Google Scholar]
- MAKKONEN H, KAUHANEN M, JAASKELAINEN T & PALVIMO JJ 2011. Androgen receptor amplification is reflected in the transcriptional responses of Vertebral-Cancer of the Prostate cells. Mol Cell Endocrinol, 331, 57–65. [DOI] [PubMed] [Google Scholar]
- MARKEY MP, ANGUS SP, STROBECK MW, WILLIAMS SL, GUNAWARDENA RW, ARONOW BJ & KNUDSEN ES 2002. Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res, 62, 6587–97. [PubMed] [Google Scholar]
- MARSHALL JL, KINDLER H, DEEKEN J, BHARGAVA P, VOGELZANG NJ, RIZVI N, LUHTALA T, BOYLAN S, DORDAL M, ROBERTSON P, et al. 2005. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Invest New Drugs, 23, 31–7. [DOI] [PubMed] [Google Scholar]
- MASSIE CE, LYNCH A, RAMOS-MONTOYA A, BOREN J, STARK R, FAZLI L, WARREN A, SCOTT H, MADHU B, SHARMA N, et al. 2011. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J, 30, 2719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- METZGER E, YIN N, WISSMANN M, KUNOWSKA N, FISCHER K, FRIEDRICHS N, PATNAIK D, HIGGINS JM, POTIER N, SCHEIDTMANN KH, et al. 2008. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol, 10, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIGLIACCIO A, CASTORIA G, DI DOMENICO M, DE FALCO A, BILANCIO A, LOMBARDI M, BARONE MV, AMETRANO D, ZANNINI MS, ABBONDANZA C, et al. 2000. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J, 19, 5406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIGLIACCIO A, VARRICCHIO L, DE FALCO A, CASTORIA G, ARRA C, YAMAGUCHI H, CIOCIOLA A, LOMBARDI M, DI STASIO R, BARBIERI A, et al. 2007. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene, 26, 6619–29. [DOI] [PubMed] [Google Scholar]
- MILLS IG 2014. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer, 14, 187–98. [DOI] [PubMed] [Google Scholar]
- MOILANEN AM, KARVONEN U, POUKKA H, JANNE OA & PALVIMO JJ 1998. Activation of androgen receptor function by a novel nuclear protein kinase. Mol Biol Cell, 9, 2527–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOK TS, WU YL, AHN MJ, GARASSINO MC, KIM HR, RAMALINGAM SS, SHEPHERD FA, HE Y, AKAMATSU H, THEELEN WS, et al. 2017. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med, 376, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTOYA A, BELTRAN L, CASADO P, RODRIGUEZ-PRADOS JC & CUTILLAS PR 2011. Characterization of a TiO(2) enrichment method for label-free quantitative phosphoproteomics. Methods, 54, 370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNSTER P, MITA M, MAHIPAL A, NEMUNAITIS J, MASSARD C, MIKKELSEN T, CRUZ C, PAZ-ARES L, HIDALGO M, RATHKOPF D, et al. 2019. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag Res, 11, 10463–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARAYANAN S, SRINIVAS S & FELDMAN D 2016. Androgen-glucocorticoid interactions in the era of novel prostate cancer therapy. Nat Rev Urol, 13, 47–60. [DOI] [PubMed] [Google Scholar]
- PAWAR A, GOLLAVILLI PN, WANG S & ASANGANI IA 2018. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep, 22, 2236–2245. [DOI] [PubMed] [Google Scholar]
- PAWSON T & SCOTT JD 1997. Signaling through scaffold, anchoring, and adaptor proteins. Science, 278, 2075–80. [DOI] [PubMed] [Google Scholar]
- PENFOLD L, WOODS A, MUCKETT P, NIKITIN AY, KENT TR, ZHANG S, GRAHAM R, POLLARD A & CARLING D 2018. CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis. Cancer Res, 78, 6747–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRENCIPE M, FABRE A, MURPHY TB, VARGYAS E, O’NEILL A, BJARTELL A, TASKEN KA, GRYTLI HH, SVINDLAND A, BERGE V, et al. 2018. Role of serum response factor expression in prostate cancer biochemical recurrence. Prostate, 78, 724–730. [DOI] [PubMed] [Google Scholar]
- PURNELL DM, HEATFIELD BM, ANTHONY RL & TRUMP BF 1987. Immunohistochemistry of the cytoskeleton of human prostatic epithelium. Evidence for disturbed organization in neoplasia. Am J Pathol, 126, 384–95. [PMC free article] [PubMed] [Google Scholar]
- RAMROOP JR, STEIN MN & DRAKE JM 2018. Impact of Phosphoproteomics in the Era of Precision Medicine for Prostate Cancer. Front Oncol, 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASOOL RU, NATESAN R, DENG Q, ARAS S, LAL P, SANDER EFFRON S, MITCHELL-VELASQUEZ E, POSIMO JM, CARSKADON S, BACA SC, et al. 2019. CDK7 inhibition suppresses Castration-Resistant Prostate Cancer through MED1 inactivation. Cancer Discov. [DOI] [PMC free article] [PubMed]
- SCHIEWER MJ, AUGELLO MA & KNUDSEN KE 2012. The AR dependent cell cycle: mechanisms and cancer relevance. Mol Cell Endocrinol, 352, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIEWER MJ & KNUDSEN KE 2016. Linking DNA Damage and Hormone Signaling Pathways in Cancer. Trends Endocrinol Metab, 27, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIEWER MJ & KNUDSEN KE 2019. DNA Damage Response in Prostate Cancer. Cold Spring Harb Perspect Med, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLEGEL J, SAMBADE MJ, SATHER S, MOSCHOS SJ, TAN AC, WINGES A, DERYCKERE D, CARSON CC, TREMBATH DG, TENTLER JJ, et al. 2013. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J Clin Invest, 123, 2257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRANTZ N, DA SILVA CORREIA J, FOWLER B, GE Q, SUN Z & BOKOCH GM 2004. Mechanism of p21-activated kinase 6-mediated inhibition of androgen receptor signaling. J Biol Chem, 279, 1922–31. [DOI] [PubMed] [Google Scholar]
- SEDELAAR JP & ISAACS JT 2009. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate, 69, 1724–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENAPATI D, KUMARI S & HEEMERS HV 2019. Androgen receptor co-regulation in prostate cancer. Asian Journal of Urology, In Press. [DOI] [PMC free article] [PubMed]
- SHABBIR M & STUART R 2010. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: from bench to bedside. Expert Opin Investig Drugs, 19, 427–36. [DOI] [PubMed] [Google Scholar]
- SHANMUGAM I, CHENG G, TERRANOVA PF, THRASHER JB, THOMAS CP & LI B 2007. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ, 14, 2085–94. [DOI] [PubMed] [Google Scholar]
- SHARMA NL, MASSIE CE, RAMOS-MONTOYA A, ZECCHINI V, SCOTT HE, LAMB AD, MACARTHUR S, STARK R, WARREN AY, MILLS IG, et al. 2013. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell, 23, 35–47. [DOI] [PubMed] [Google Scholar]
- SHAW AT, KIM DW, MEHRA R, TAN DS, FELIP E, CHOW LQ, CAMIDGE DR, VANSTEENKISTE J, SHARMA S, DE PAS T, et al. 2014. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med, 370, 1189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHERK AB, FRIGO DE, SCHNACKENBERG CG, BRAY JD, LAPING NJ, TRIZNA W, HAMMOND M, PATTERSON JR, THOMPSON SK, KAZMIN D, et al. 2008. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res, 68, 7475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGEL RL, MILLER KD & JEMAL A 2020. Cancer statistics, 2020. CA Cancer J Clin, 70, 7–30. [DOI] [PubMed] [Google Scholar]
- SINHA A, HUANG V, LIVINGSTONE J, WANG J, FOX NS, KURGANOVS N, IGNATCHENKO V, FRITSCH K, DONMEZ N, HEISLER LE, et al. 2019. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell, 35, 414–427 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STELLOO S, NEVEDOMSKAYA E, VAN DER POEL HG, DE JONG J, VAN LEENDERS GJ, JENSTER G, WESSELS LF, BERGMAN AM & ZWART W 2015. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol Med, 7, 1450–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SWEENEY C, PERCENT IJ, BABU S, CULTRERA J, MEHLHAFF BA, GOODMAN OB, MORRIS D, SCHNADIG ID, ALBANY C, SHORE ND, et al. 2019. Phase 1b/2 study of enzalutamide (ENZ) with LY3023414 (LY) or placebo (PL) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) after progression on abiraterone. Journal of Clinical Oncology, 37, 5009–5009. [Google Scholar]
- TOSKA E, CASTEL P, CHHANGAWALA S, ARRUABARRENA-ARISTORENA A, CHAN C, HRISTIDIS VC, COCCO E, SALLAKU M, XU G, PARK J, et al. 2019. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep, 27, 294–306 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKADAKRISHNAN VB, DEPRIEST AD, KUMARI S, SENAPATI D, BEN-SALEM S, SU Y, MUDDULURU G, HU Q, CORTES E, POP E, et al. 2019. Protein Kinase N1 control of androgen-responsive serum response factor action provides rationale for novel prostate cancer treatment strategy. Oncogene. [DOI] [PMC free article] [PubMed]
- WALTERING KK, HELENIUS MA, SAHU B, MANNI V, LINJA MJ, JANNE OA & VISAKORPI T 2009. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res, 69, 8141–9. [DOI] [PubMed] [Google Scholar]
- WANG S, GAO J, LEI Q, ROZENGURT N, PRITCHARD C, JIAO J, THOMAS GV, LI G, ROY-BURMAN P, NELSON PS, et al. 2003. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell, 4, 209–21. [DOI] [PubMed] [Google Scholar]
- WANG Y, MONCAYO G, MORIN P JR., XUE G, GRZMIL M, LINO MM, CLEMENT-SCHATLO V, FRANK S, MERLO A & HEMMINGS BA 2013. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene, 32, 872–82. [DOI] [PubMed] [Google Scholar]
- WANG Y, ZENG C, LI J, ZHOU Z, JU X, XIA S, LI Y, LIU A, TENG H, ZHANG K, et al. 2018. PAK2 Haploinsufficiency Results in Synaptic Cytoskeleton Impairment and Autism-Related Behavior. Cell Rep, 24, 2029–2041. [DOI] [PubMed] [Google Scholar]
- WASMUTH EV, HOOVER EA, ANTAR A, KLINGE S, CHEN Y & SAWYERS CL 2020. Modulation of androgen receptor DNA binding activity through direct interaction with the ETS transcription factor ERG. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed]
- WATSON PA, ARORA VK & SAWYERS CL 2015. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer, 15, 701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEN S, NIU Y, LEE SO & CHANG C 2014. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat Rev, 40, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE MA, TSOUKO E, LIN C, RAJAPAKSHE K, SPENCER JM, WILKENFELD SR, VAKILI SS, PULLIAM TL, AWAD D, NIKOLOS F, et al. 2018. GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr Relat Cancer, 25, 453–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU X, ZAHARI MS, RENUSE S, NIRUJOGI RS, KIM MS, MANDA SS, STEARNS V, GABRIELSON E, SUKUMAR S & PANDEY A 2015. Phosphoproteomic Analysis Identifies Focal Adhesion Kinase 2 (FAK2) as a Potential Therapeutic Target for Tamoxifen Resistance in Breast Cancer. Mol Cell Proteomics, 14, 2887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU K, WU ZJ, GRONER AC, HE HH, CAI C, LIS RT, WU X, STACK EC, LODA M, LIU T, et al. 2012. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science, 338, 1465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU L, CHEN S & BERGAN RC 2006. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene, 25, 2987–98. [DOI] [PubMed] [Google Scholar]
- YAN Z, JIN S, WEI Z, HUILIAN H, ZHANHAI Y, YUE T, JUAN L, JING L, LIBO Y & XU L 2014. Discoidin domain receptor 2 facilitates prostate cancer bone metastasis via regulating parathyroid hormone-related protein. Biochim Biophys Acta, 1842, 1350–63. [DOI] [PubMed] [Google Scholar]
- YANG F, LI X, SHARMA M, ZARNEGAR M, LIM B & SUN Z 2001. Androgen receptor specifically interacts with a novel p21-activated kinase, PAK6. J Biol Chem, 276, 15345–53. [DOI] [PubMed] [Google Scholar]
- YEH YC, WU CC, WANG YK & TANG MJ 2011. DDR1 triggers epithelial cell differentiation by promoting cell adhesion through stabilization of E-cadherin. Mol Biol Cell, 22, 940–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIDA T, KIM JH, CARVER K, SU Y, WEREMOWICZ S, MULVEY L, YAMAMOTO S, BRENNAN C, MEI S, LONG H, et al. 2015. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res, 75, 1516–26. [DOI] [PubMed] [Google Scholar]
- YU J, YU J, MANI RS, CAO Q, BRENNER CJ, CAO X, WANG X, WU L, LI J, HU M, et al. 2010. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell, 17, 443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZARIF JC & MIRANTI CK 2016. The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell Signal, 28, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAU HY, CHANG SM, CHEN BQ, WANG Y, ZHANG H, KAO C, SANG QA, PATHAK SJ & CHUNG LW 1996. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci U S A, 93, 15152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHI G, RYDER JW, HUANG J, DING P, CHEN Y, ZHAO Y, KAMM KE & STULL JT 2005. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc Natl Acad Sci U S A, 102, 17519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. AR-binding sites in genes encoding kinases and phosphatases in VCaP cells. Table provides an overview of the presence of AR binding sites in transcriptionally-regulated kinases (top part) and phosphatases (bottom part) from Table 1 and Table 2 in 2 independent AR ChIP-seq datasets derived from VCaP cells (Massie et al., 2011, Asangani et al., 2014). AR ChIP-Seq datasets were retrieved via cistrome.org (Massie et. al. 2011: CistromeDB: 2910, 2911; Asangani et. al. 2014: CistromeDB: 44574 – 44577) Data was analyzed and AR binding peaks were mapped using the UCSC genome browser tool. Androgen-induced peaks whose values were at least 3-fold higher than background values were withheld. Yes, presence of an AR peak AR binding site within 300kb of the transcriptional start site of the gene in all (100%) or half (50%) of biological replicates in the respective study; maybe, presence of such peak not certain.
Supplementary Table 2. Overview of clinical trials using kinase inhibitors for CaP treatment.