

Abstract
Background:
Nexilin (NEXN) is a protein of the junctional membrane complex required for development of cardiac T-tubules. Global and cardiomyocyte specific loss of Nexn in mice leads to a rapidly progressive dilated cardiomyopathy and premature death. Therefore, little is known as to the role of NEXN in adult cardiomyocytes. Transverse-axial tubular system (TATS) remodeling are well-known features in heart failure. Although NEXN is required during development for T-tubule formation, its role, if any, in mature T-tubules remains to be addressed.
Methods:
Nexn inducible adult cardiomyocyte-specific KO mice were generated. Comprehensive morphological and functional analyses were performed. Heart samples (n>3) were analyzed by molecular, biochemical and electron microscopy analyses. Isolated single adult cardiomyocytes were analyzed by confocal microscopy and myocyte shortening/re-lengthening and Ca2+ transient studies were conducted.
Results:
Inducible cardiomyocyte specific loss of Nexn in adult mice resulted in a dilated cardiomyopathy with reduced cardiac function (13% reduction in FS%, p<0.05). In vivo and in vitro analyses of adult mouse heart samples revealed that NEXN was essential for optimal contraction and calcium handling, and was required for maintenance of T-tubule network organization (transverse tubular component in icKO reduced by 40% with respect to CTRLs, p<0.05).
Conclusions:
Results here reported revealed NEXN to be a pivotal component of adult junctional membrane complexes required for maintenance of transverse-axial tubular architecture. These results demonstrated that Nexilin plays an essential role in the adult cardiomyocyte and gave further understanding of pathological mechanisms responsible for cardiomyopathy in patients carrying mutations in the Nexilin gene.
Keywords: Cardiomyopathy, T-Tubule, Junctional Membrane Complexes, Heart Failure
Introduction
Junctional membrane complexes (JMCs) are highly specialized protein complexes localized to the dyads of the cardiomyocyte1. JMCs are essential for calcium-induced calcium release, which is fundamental for the efficient rhythmic contraction of the heart, with alterations in JMCs leading to impaired cardiac function and heart failure2–5. In adult cardiomyocytes, dyads are formed by the association of the sarcoplasmic reticulum (SR) terminal cisternae with invaginations of the sarcolemma called transverse (T)-tubules. These structures are essential for fast and efficient excitation-contraction coupling6. In fact, dyads are arranged in an organized network along Z-discs allowing for maximal signal transduction between T-tubules and SR along the whole cardiomyocyte, resulting in optimal contraction7. The cardiomyocyte T-tubule network is structured in a very organized fashion, with transverse components localized in the dyad, and longitudinal branches (axial tubules or A-tubules) connecting the entire tubular system also called the transverse-axial tubular system (TATS)8–16. This arrangement ensures rapid, uniform cell activation, and even subtle changes in the T-tubular system and associated JMC proteins may contribute to heart failure17–21.
Nexilin or NEXN (encoded by NEXN) was isolated as an actin filament-binding protein in rat brain and fibroblasts22 and identified as a Z-disc protein abundant in striated muscles. Multiple mutations in NEXN have been associated with cardiomyopathies, highlighting its importance in maintaining cardiac function23–25. Furthermore, global knockout of Nexn in mice was reported to cause rapidly progressive cardiomyopathy with left ventricular dilation, wall thinning, and decreased cardiac function, resulting in lethality shortly after birth26. Despite these findings, little was known as to the specific role of NEXN in cardiomyocytes, or mechanisms by which global KO of Nexn in mice results in rapidly progressive cardiomyopathy. To address these issues, we created a floxed Nexn mouse line and generated constitutive Nexn early CM-specific knockout (cKO) mice, revealing that NEXN is a pivotal component of JMCs required for T-tubule formation during development27.
Early postnatal lethality of Nexn global and cardiomyocyte-specific KO mice has prevented the study of potential roles for NEXN in adult cardiomyocytes. For this reason, we generated a Nexn inducible adult cardiomyocyte-specific knockout (icKO) mouse model. This allowed us to study the role of NEXN in mature cardiomyocytes, where the T-tubule network is already formed and functional. Our findings demonstrated that loss of NEXN in adult mouse heart leads to cardiac stress, altered cardiac function, and dilated cardiomyopathy. Further analyses also revealed that loss of NEXN in cardiomyocytes led to disrupted T-tubule network organization and calcium handling defects.
Materials & Methods
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal procedures
Floxed Nexn (Nexnf/f) mice generation has been already described in previous work27. Inducible cardiac KO (icKO) mice were generated crossing Nexnf/f mice with mice expressing α-myosin heavy chain (αMHC)-MerCreMer (MCM)28.
The UCSD Animal Care Program maintained all animals and the UCSD Institutional Animal Care and Use Committee approved all experimental procedures. Genotyping was performed using primers reported in Table S1.
Tamoxifen Induction
4-Hydroxytamoxifen (Sigma) was dissolved in peanut oil (Sigma) at a concentration of 5 mg/mL. Adult (2-month-old) Nexnf/f and Nexnf/f-αMHC-MCM mice were treated with 4-hydroxy- tamoxifen by intraperitoneal injection once daily for 4 days at a dose of 40 mg/kg body weight. Experiments were performed 2 weeks after the first tamoxifen injection, a representative scheme of the icKO inducement is reported in Figure 1A.
Figure 1 -. Loss of Nexilin in adult mice results in cardiac stress and left ventricle dilation.
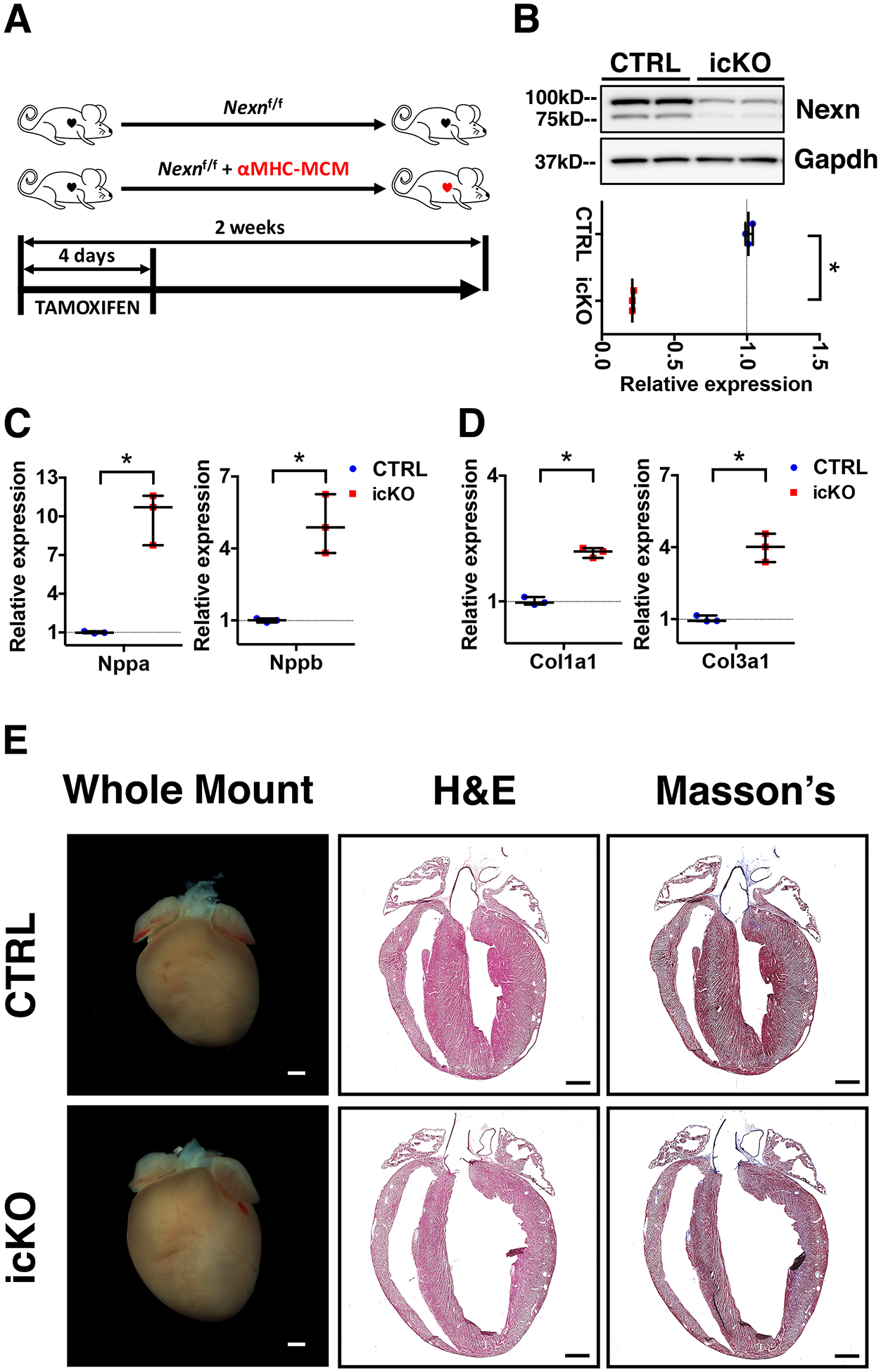
(A) Tamoxifen treatment scheme for icKO of Nexilin; 2 months old mice were injected intraperitoneally. (B) Representative western blot and relative quantification showing effective cardiac KO of Nexilin. (C, D) Quantitative RT-PCR for cardiac stress mRNA markers 2 weeks after tamoxifen treatment. (C) Graphs showing increased expression of Nppa and Nppb in the icKO, cardiac genes encoding respectively for atrial natriuretic factor and brain natriuretic peptide. (D) Graphs showing an increased expression of Col1a1 and Col3a1 in the icKO. (E) Representative whole heart images, 4-chambers view hematoxylin/eosin and Masson’s trichrome stainings images of longitudinal histological sections from icKO and CTRL hearts 2 weeks after tamoxifen treatment. Scale bars 1mm. (*) Statistically significant differences with P value < 0.05
Echocardiography
Mice were anesthetized with 3% isoflurane for 10 seconds and maintained at 0.5% isoflurane during the procedure. Echocardiography was performed by using a VisualSonics, SonoSite FUJIFILM, Vevo 2100 ultrasound system with a linear transducer 32–55MHz. Percentage fractional shortening (%FS) was used as an indicator of systolic cardiac function. Measurements of end-diastolic left ventricular (LV) internal diameter (LVIDd), end-systolic LV internal diameter (LVIDs), end-diastolic interventricular septal thickness (IVSd), and end-diastolic LV posterior wall thickness (LVPWd) were determined from the LV M-mode tracing.
Whole heart imaging, Histology and Immunofluorescence
Hearts from icKO and CTRL mice were processed and staining were performed as previously described27. Antibodies used for Immunofluorescence and their concentrations are listed in Table S2. For whole heart imaging, picture were acquired with an Olympus SZX12 stereomicroscope. Histology images were acquired with a Hamamatsu Nanozoomer 2.0HT Slide Scanner. Immunofluorescence images were obtained using a Zeiss LSM 880 airy scan confocal microscope. Image processing were performed with Fiji software29 and Photoshop CC (Adobe).
Quantitative real-time PCR
Total RNA and cDNA were obtained as previously described27. PCR mixture contained 1 μl diluted cDNA, 5 μl 2× iTaq Universal SYBR Green Supermix (BIO-RAD), and 200 nM of each gene-specific primer in a final volume of 10 μl. Real-time PCRs were performed using a CFX96 Biorad Thermocycler. Primer sequences are reported in Table S1.
Western Blot analysis
Protein lysates were extracted as previously described27 and separated on 4–12% SDS-PAGE gels (Life Technologies), then transferred for 2 hours at 4°C on to a PVDF membrane (BioRad). After blocking, membranes were incubated overnight at 4°C with the primary antibody (listed in Table S2). Blots were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody generated in Rabbit (1:5000) or Mouse (1:5000) (Dako) for 1.5 hours at room temperature. Immunoreactive protein bands were visualized using an enhanced chemiluminescence reagent (Thermo Fisher Scientific) following manufacturer instructions, then analyzed by densitometry and expressed as pixel density normalized to GAPDH. All buffers used have been previously listed30.
Adult cardiomyocyte isolation
Mice were anesthetized with isoflurane hearts were excised and then rinsed in Krebs-Henseleit buffer B (KHB-B) (118 mM NaCl, 4.8 mM KCl, 25 mM HEPES, 1.25 mM K2HPO4, 1.25 mM MgSO4, 11 mM glucose, pH 7.4). Next, every heart was perfused and enzymatically digested as previously described27. Then the hearts were minced in KHB solution with 2% BSA and 1.2mM CaCl2, gently agitated, then filtered through a 100μm polyethylene mesh. Cells were kept in KHB/2%BSA/ CaCl2 solution at room temperature before use.
Cell shortening/re-lengthening and Ca2+ transient studies
Adult cardiomyocytes contractility and calcium transients studies were performed as previously described27, 31, 32. Briefly, the cells were loaded with Fura-2-AM (1.0 μM, 20 mins), simultaneous measurement of intracellular Ca2+([Ca2+]i) and cell contractility was assessed by using a video-based edge-detection system (IonOptix, Milton, MA) at a frequency of 0.5 Hz using a MyoPacer Field Stimulator (IonOptix). A total of 60–80 individual myocytes from 4 mice of each group were recorded and analyzed. The ratio of Fura-2 fluorescence at 340 nm and 380 nm was calculated and the amplitude of intracellular Ca2+transient was determined by the change between the basal and peak ratio (ΔF/F0). The decay portion of the calcium transient (time to 63% decline) was used to measure the calcium decay (Tau). The amplitude of cell contraction was assessed by peak shortening, and the rate of cell relaxation was assessed by the time to 63% re-lengthening (Tau).
T-tubule imaging
Isolated cardiomyocytes were incubated with 50μM Di-8 Anepps (Thermo Fisher Scientific, D3167) for 20 minutes on matrigel (Millipore Sigma, E6909) coated chamber imaging slides (Ibidi). After 2 quick washes, the cells were analyzed using a Zeiss LSM 880 airy scan confocal microscope with an excitation at 458nm and a detection spectrum between 550nm and 740nm. Contracting and healthy cells were selected for imaging and for the entire acquisition cells were kept in KHB/2%BSA/1.2mMCaCl2 with addition of 2,3-Butanedione monoxime 10mM (Sigma-Aldrich) to inhibit excess of contraction. A total of 60 individual cardiomyocytes from 3 mice of each group were analyzed. Images were processed with Fiji software: skeletonization and directionality analyses were performed using plugins previously described14, 29, 33. Extrapolated data from Fourier’s components were analyzed using Prism 6.0 (GraphPad Software, La Jolla, CA) to create average directionality histograms and to perform area under curve measurements.
Electron microscopy
Adult mouse hearts were perfused first with 5ml of 0.1 M sodium cacodylate buffer pH 7.4 and then with 5ml of 2% paraformaldehyde + 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4. Perfusion solutions were oxygenated and ice-cold. Left ventricular tissue was extracted from the hearts and washed in 0.1 M sodium cacodylate buffer pH 7.4, then divided in small samples of a 2mm2 maximum size. After a 1-hour pre-fixation in 2% paraformaldehyde + 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4, the samples were post-fixed in 1% osmium tetroxide. The samples were than stained overnight in 2% uranyl acetate and dehydrated in graded ethanol solutions and embedded in Durcupan ACM (Sigma). Sections were cut between 50 and 70 nm thick using a Leica ultramicrotome and images were recorded on a FEI Technai 12 Spirit electron microscope operated at voltages ranging from 80 to 120 kV.
Statistical Analysis
Data on graphs are expressed as median with IQRs. Statistical analysis was performed using Prism 6.0 (GraphPad Software, La Jolla, CA). Differences between groups were analyzed by 2-tailed Student’s t-test. For data represented in Figure 2A–E two-way ANOVA and Bonferroni’s test for multiple comparison were used. P values <0.05 were considered statistically significant.
Figure 2 -. Nexilin is essential for cardiac function in adult mice.
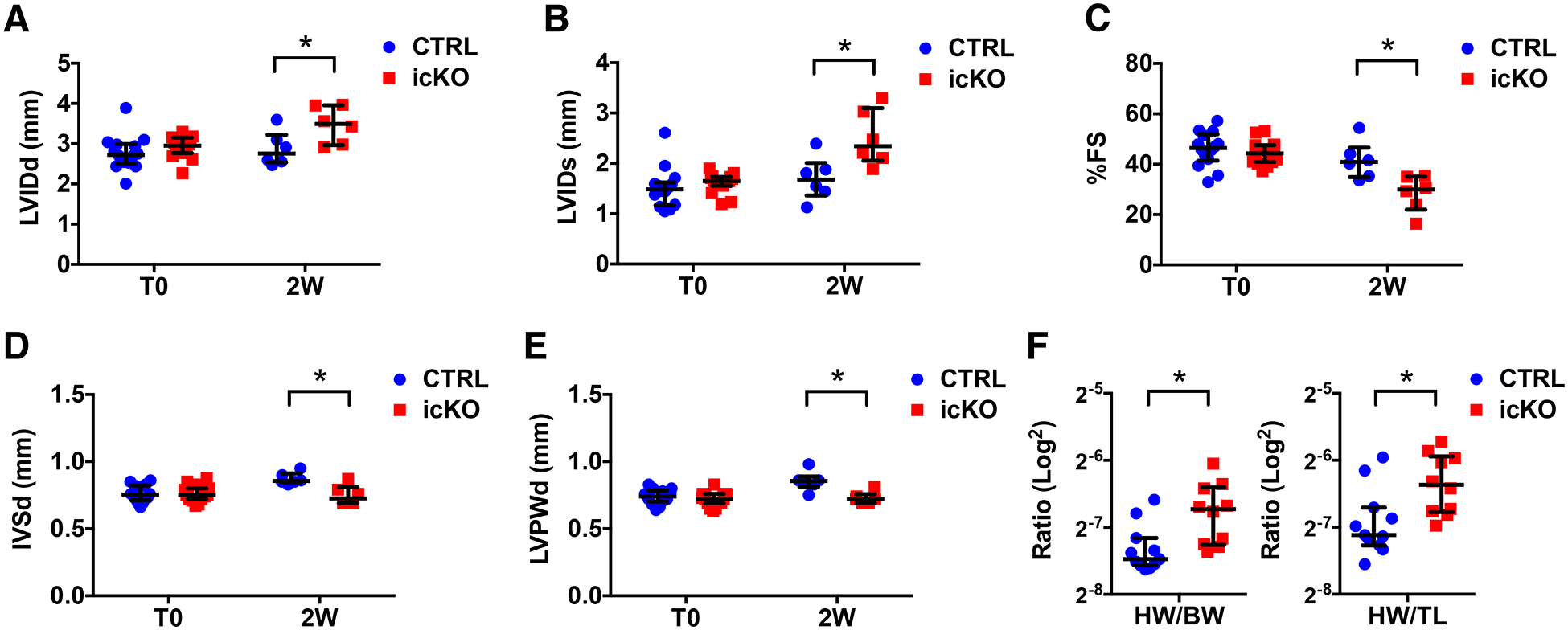
(A-E) Graphs representing transthoracic echocardiographic measurements from CTRL and icKO mice before (T0) and 2 weeks (2W) after tamoxifen treatment (n ≥ 6): (A) left ventricular internal 12 diameter end-diastole (LVIDd), (B) left ventricular internal diameter end-systole (LVIDs), (C) % fractional shortening (FS), (D) Interventricular septal end diastole (IVSd) and (E) Left ventricular posterior wall end diastole (LVPWd). (F) Graphs representing heart weight (HW)/body weight (BW) and HW/tibial length (TL) ratio of CTRL and icKO mice 2 weeks after tamoxifen treatment (n ≥ 10). (*) Statistically significant differences with P value < 0.05.
Results
Loss of Nexilin in adult mouse heart leads to cardiac stress and dilated cardiomyopathy
Early postnatal lethality of constitutive Nexilin (Nexn) cardiomyocyte-specific KOs prevented study of the role of NEXN in adult cardiomyocytes27. To investigate the role of Nexilin (NEXN) in adult heart, we crossed mice harboring the Nexn floxed allele (Nexnf/f) with mice expressing the inducible α-myosin heavy chain (αMHC)-MerCreMer (MCM)28. Mice were induced with tamoxifen (TAM) at two months of age, and analyzed two weeks after the start of TAM to ensure cardiomyocyte-specific KO (icKO) of Nexn. To verify recombination efficiency, we isolated proteins from heart tissue of icKO and littermate control (CTRL) mice and confirmed loss of Nexilin by western blot analysis (Fig. 1A, B).
Quantitative PCR analyses of cardiac stress markers expression levels revealed a significant increase in hearts of icKO when compared to those of CTRLs (Fig. 1C, E). Gross anatomical and histological analyses showed that Nexn-icKO hearts exhibited dilated cardiomyopathy (DCM) and structural alterations (Fig.1E), but further observation via transmission electron and confocal microscopy demonstrated no alterations or disarray in sarcomere structure (Fig. S1).
Nexilin icKO causes abnormal cardiac function in adult mice
We next evaluated cardiac function of icKOs and CTRLs after tamoxifen treatment.
Transthoracic echocardiography confirmed that Nexn-icKOs exhibited increased left ventricular diameter (Fig. 2A, B). Abnormal left ventricular dimensions were accompanied by reduced ejection fraction (Fig. 2C) and ventricular wall thinning (Fig. 2D, E). Cardiac abnormalities in icKO hearts were accompanied by significantly increased heart weight/body weight and heart weight/tibial length ratios (Fig. 2F, G). Altogether, these observations suggested that NEXN had a key role in adult heart function, with its loss leading to cardiac malfunction and DCM.
Nexilin is crucial for Ca2+ homeostasis in adult cardiomyocytes
Data from our previous study showed that constitutive loss of NEXN in embryonic cardiomyocytes resulted in abnormal Ca2+ dynamics and altered expression of junctional and Ca2+ handling proteins27. To investigate effects of NEXN loss in adult cardiomyocytes on calcium homeostasis, we examined adult cardiomyocytes isolated from Nexn icKO and CTRL mice. Calcium imaging with the IonOptix system revealed that the amplitude of Ca2+ transients was significantly reduced, while the decay was slower, in icKO cardiomyocytes compared to CTRL cardiomyocytes (Fig. 3A–C). Consequently, icKO cardiomyocytes showed reduced and altered contractility (Fig. 3D–F) with no significant change in the sarcomere length (Fig. S2). Western blot analyses confirmed abnormal expression levels of junctional and Ca2+ handling proteins in Nexn icKO adult hearts. These results indicated that DCM and abnormal cardiac function observed in icKO mice is most likely caused by altered Ca2+ homeostasis and aberrant expression of dyadic and Ca2+ related proteins.
Figure 3 -. Lack of Nexilin in adult cardiomyocytes alters calcium dynamics.
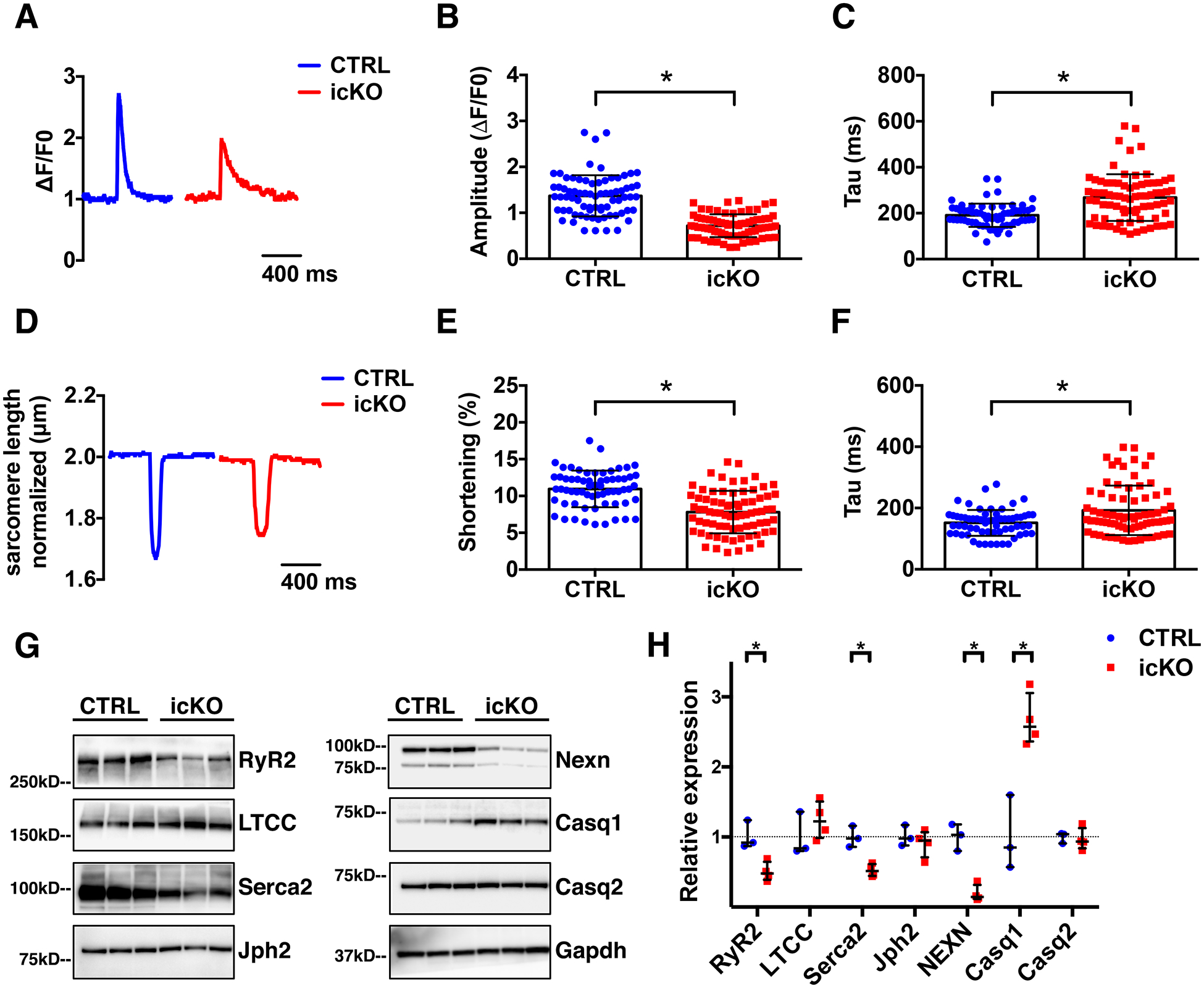
(A) representative Ca2+ transient curve; and (B-C) histograms showing results from quantifications of Ca2+ transient (B) amplitude of the peak (ΔF/F0) and (C) transient decay (Tau) of live cardiomyocytes isolated from CTRL and icKO ventricles, 2 weeks after tamoxifen treatment. (D) Representative graph of cardiomyocyte sarcomere length measurement. (E) Graphs showing measurements of sarcomere length shortening percentage and (F) sarcomere relaxation time (Tau). Cardiomyocytes were labeled with Fura2-AM (n=6 mice). (G) Western blot representative images and (H) relative quantification graphs of Nexilin and Ca2+-handling proteins expression in CTRL and icKO ventricles lysates, 2 weeks after tamoxifen treatment. (*) Statistically significant differences with P value < 0.05.
Nexilin is essential for maintenance of the adult cardiomyocyte TATS
Absence of NEXN in developing cardiomyocytes prevents formation of cardiac T-tubules, ultimately resulting in altered expression of dyadic proteins, and Ca2+ handling defects27. In adult cardiomyocytes, T-tubules are fully formed and organized in a precise network to ensure proper heart function, raising the question as to a potential role for NEXN in mature T-tubules. To investigate T-tubule organization in Nexn icKO hearts, we performed live confocal microscopy imaging of isolated adult cardiomyocytes stained with Di-8 Anepps. This analysis revealed significant disorganization of the T-tubule network, with a significant reduction of the transversal component, in cardiomyocytes isolated from Nexn icKOs relative to those from CTRLs (Fig. 4). Images were acquired under consistent conditions with the background being removed according to a determined threshold. Pictures were skeletonized (Fig.4B, E) and directionality of transverse (T-) and axial (A-) tubular systems analyzed via Fourier transform, using Fiji software to translate images to a directionality histogram. Cardiomyocytes were oriented such that the x-axis of the analyzed image corresponded to the longitudinal axis of the cell (A-tubule components), represented by the 0° bin Gauss fit in the directionality histogram, whereas T-tubule components (y-axis oriented to the transversal tubular system of the cell) were represented by the 90° bin Gauss fit (Fig.4C, F). As shown, the A-tubule components (0°) of the tubular network did not significantly change between icKO and CTRLs, but the transverse component (90°) was significantly decreased in icKO cardiomyocytes relative to CTRLs (Fig.4G, H). Confirming a disorganization of the tubular system, also L-type calcium channel (LTCC) localization appeared disorganized in icKO cardiomyocytes, but the pattern of Ryanodine Receptor type 2 (RyR2) did not change between icKO and CTRL cardiomyocytes (Fig. S3). These data suggest that NEXN not only is necessary for T-tubule formation during development, but is also essential for maintenance and organization of the TATS in adult cardiomyocytes.
Figure 4 -. Nexilin is required for T-tubules organization in adult cardiomyocytes.
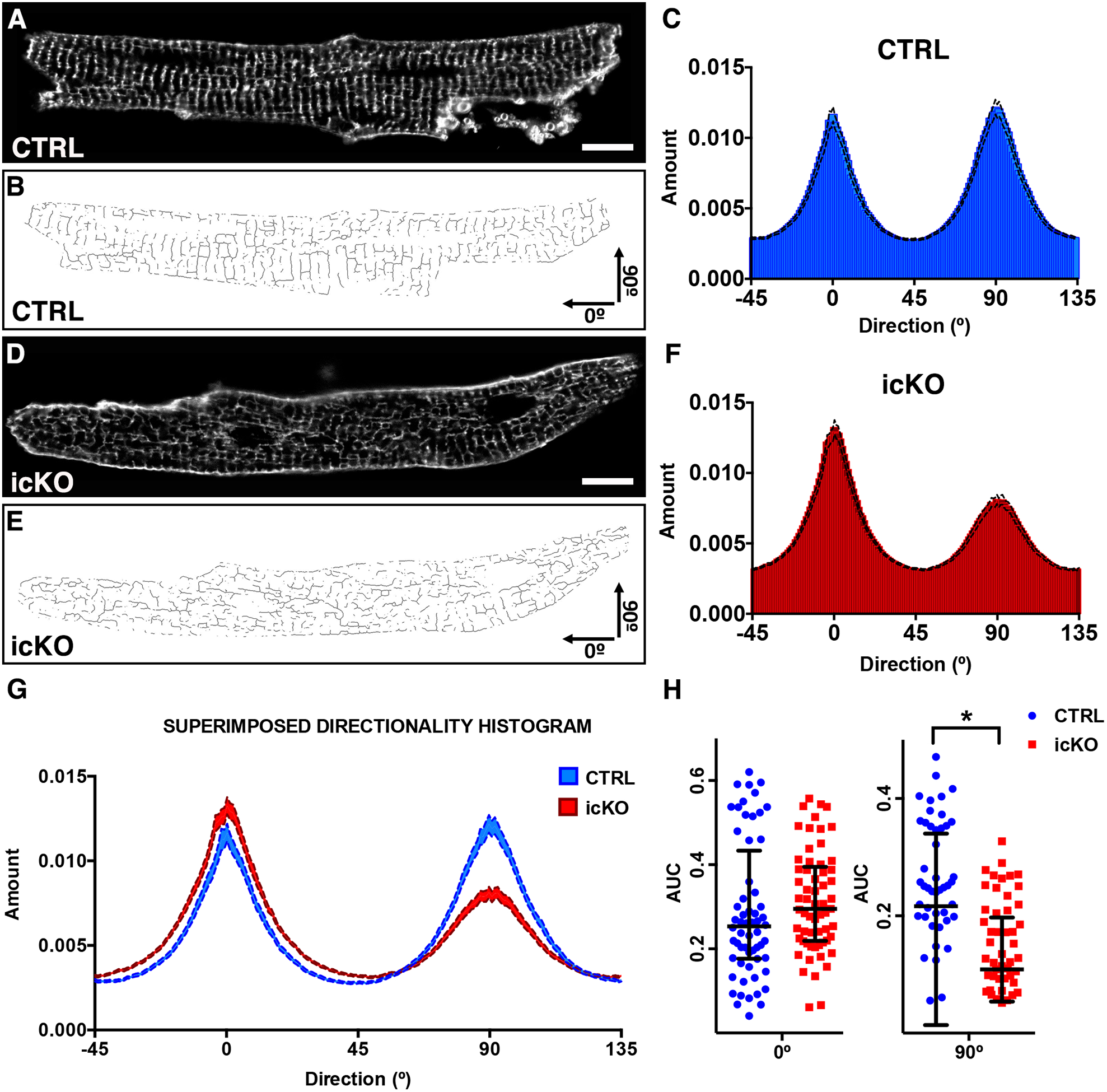
(A, D) representative live confocal images of cardiomyocytes stained with Di-8 Anepps isolated from CTRL and icKO mice 2 weeks after tamoxifen treatment. Scale bars 10μm. (B, E) Relative skeletonized images, oriented in a x/y fashion: transverse tubules represent the 90° component and longitudinal tubules the 0° component. (C, F) Directionality histograms derivated from Fourier transforms of all the analyzed cardiomyocytes images. (G) Graph showing a comparison between directionality histograms from C and F. (H) CTRL and icKO comparison of area under curve (AUC) from 90° and 0° tubular Fourier components gaussian distribution. (*) Statistically significant differences with P value < 0.05.
Discussion
Previous studies highlighted the importance of NEXN in heart and in cardiac development23–25, with global and cardiac KO of Nexn leading to DCM and early postnatal lethality26, 27. The premature death of constitutive Nexn cardiomyocyte knockouts (cKOs) prevented the study of NEXN in adult cardiomyocytes. To overcome this problem, we generated an inducible cardiomyocyte Nexn mouse model (Fig. 1A, B).
Morphological evaluation of the icKO model demonstratsed that NEXN is indispensable for adult heart function. In fact, its loss caused cardiac stress (Fig. 1C, D) and cardiac abnormalities with left ventricular dilation (Fig. 1E). Morphological defects were accompanied by altered cardiac function with significantly reduced ejection fraction (Fig. 2).
Given our previous findings that identified NEXN as a protein of the JMC27, and since morphological abnormalities and reduced cardiac function is a known hallmark for Ca2+ handling and contractility defects34, we performed cardiomyocyte shortening/re-lengthening and Ca2+ transient studies. Results confirmed a crucial role for NEXN in Ca2+ homeostasis and regulation of JMC and Ca2+ handling proteins, with loss of NEXN resulting in reduced contractile ability, alterations in Ca2+ transients and abnormal JMC protein expression (Fig. 3).
Changes in Ca2+ transients are usually a symptom of EC-coupling alterations and since NEXN is required for T-tubule maturation in neonatal cardiomyocytes, we hypothesized that loss of NEXN might cause a disruption in dyad architecture. Indeed, we discovered that absence of NEXN in adult cardiomyocytes significantly reduced the transversal component of the tubular network (Fig. 4), and this disorganization was most likely causative for Ca2+ handling defects that in turn resulted in reduced contractility, altered cardiac function, and DCM.
T-tubules are the transverse component of a much more complicated sarcolemmal structure within the mature cardiomyocyte8. Together with longitudinal (axial) tubules originating from the sarcolemma (A-tubules), T-tubules form an intricate membrane network also named transverse-axial tubule system (TATS) in the cardiomyocyte12, 35. It is increasingly evident that both A-tubules and T-tubules are determinant for optimal signal transduction and functional contraction of the heart36, 37. The correct organization of TATS is prerequisite for normal cardiomyocyte function8–13.
T-tubule network uncoupling and remodeling are well-known features in cardiac diseases and heart failure13, 17–20. It is not yet clear whether this is a consequence or a cause of the related pathology. In DCM, the longitudinal component of the TATS is significantly increased, suggesting a reorganization of the tubular system due to morphological changes induced in diseased cardiomyocytes38. In our model, only the transverse component of TATS was reduced, while the axial component was not significantly altered (Fig. 4), suggesting that the TATS disorganization was not consequent to DCM, but more likely a causative factor due to NEXN absence in the cardiomyocyte. This idea is supported by distinct tubular network phenotypes described in other DCM models, where T-tubules were overall reduced in quantity and intensity rather than being disorganized as observed here39–41. Data from electron and confocal microscopy also confirmed that TATS disorganization was not secondary to alterations in Z-discs or sarcomere disarray (Fig. S1).
Usually, in heart failure models where TATS remodeling has been observed, Junctophilin 2 (Jph2) and LTCC expression levels are significantly reduced21, 42. This was not the case in our NEXN icKO mouse model, where neither LTCC nor Jph2 were significantly decreased. On the other hand, RyR2 and SERCA2 were significantly decreased, while Calsequestrin 1 (CASQ1), the non-cardiac specific form of CASQ, was overexpressed. These alterations might be explained by the TATS disorganization shown in Figure 4, as the observed disruption of T-tubule localization would impair dyad formation, and thus the proximity of RyR2 with LTCC. As proximity of these proteins is determinant for efficient calcium induced calcium release40, 43, 44, perturbations in proximity are likely to account for observed defects in Ca2+ release in Nexn icKO cardiomyocytes, ultimately affecting contractility and resulting in abnormal cardiac function and DCM. Confirming this hypothesis, LTCC localization was altered in icKO cardiomyocytes, while RyR2 localization was not affected by loss of NEXN (Fig. S3), although RyR2 expression was reduced in icKO. This also suggests that NEXN may play a role in the tethering of the sarcolemma to the SR, rather than organization of the SR itself, and future biochemical studies are warranted to better understand the precise molecular interactions between NEXN and other JMC proteins that are necessary for sarcolemma/SR tethering and dyad formation and organization.
Altogether, results reported in this manuscript demonstrated that NEXN is required for maintenance of TATS architecture in adult cardiomyocytes. These results give new insight into molecular mechanisms underlying cardiomyopathy in patients with NEXN mutations, suggesting that therapies involving calcium handling regulation may be beneficial for these patients. However, further studies are warranted to precisely determine how specific alterations in NEXN sequence modify protein function and cause cardiac disease.
Supplementary Material
Clinical Perspective.
What is new?
Adult cardiomyocyte specific inducible KO of Nexn results in dilated cardiomyopathy.
NEXN is required in adult cardiomyocytes for maintaining transversal-axial tubule system organization with loss of NEXN in adult cardiomyocytes leading to impaired calcium handling and altered contraction.
What are the clinical implications?
Identification of NEXN as a new possible target for T-Tubule remodeling in adult heart.
Providing further mechanistic insight into molecular pathways leading to cardiomyopathy in patients with mutations in NEXN.
Acknowledgments
We thank Dr. Jennifer Santini (Microscopy Core of UCSD, La Jolla, CA, USA), Dr. Mark Ellisman and all the members of NCMIR (UCSD, La Jolla, CA, USA), Dr. Kirk Peterson and all Seaweed Lab staff, especially Nancy D. Dalton (UCSD, La Jolla, CA, USA).
Sources of Funding
JC and SEM are funded by grants from the NHLBI and JC holds an American Heart Association Endowed Chair in Cardiovascular Research. OK is supported by the Shenzhen Basic Research Foundation (KCYJ20160428154108239, KQJSCX20170330155020267).
Footnotes
Disclosures
None
References
- 1.London B Defining the Complexity of the Junctional Membrane Complex. Circ Res. 2017;120:11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabiato A Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–14. [DOI] [PubMed] [Google Scholar]
- 3.Protasi F Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front Biosci. 2002;7:d650–8. [DOI] [PubMed] [Google Scholar]
- 4.Venetucci L, Denegri M, Napolitano C and Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–75. [DOI] [PubMed] [Google Scholar]
- 5.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A and Wehrens XH. SPEG (Striated Muscle Preferentially Expressed Protein Kinase) Is Essential for Cardiac Function by Regulating Junctional Membrane Complex Activity. Circ Res. 2017;120:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 7.Scriven DR, Asghari P and Moore ED. Microarchitecture of the dyad. Cardiovasc Res. 2013;98:169–76. [DOI] [PubMed] [Google Scholar]
- 8.Soeller C and Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–75. [DOI] [PubMed] [Google Scholar]
- 9.Simpson FO and Oertelis SJ. The fine structure of sheep myocardial cells; sarcolemmal invaginations and the transverse tubular system. J Cell Biol. 1962;12:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson FO. The Transverse Tubular System in Mammalian Myocardial Cells. Am J Anat. 1965;117:1–17. [DOI] [PubMed] [Google Scholar]
- 11.Forssmann WG and Girardier L. A study of the T system in rat heart. J Cell Biol. 1970;44:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrantini C, Crocini C, Coppini R, Vanzi F, Tesi C, Cerbai E, Poggesi C, Pavone FS and Sacconi L. The transverse-axial tubular system of cardiomyocytes. Cell Mol Life Sci. 2013;70:4695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo A, Zhang C, Wei S, Chen B and Song LS. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. 2013;98:204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner E, Brandenburg S, Kohl T and Lehnart SE. Analysis of tubular membrane networks in cardiac myocytes from atria and ventricles. J Vis Exp. 2014:e51823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong T and Shaw RM. Cardiac T-Tubule Microanatomy and Function. Physiol Rev. 2017;97:227–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo A and Song LS. AutoTT: automated detection and analysis of T-tubule architecture in cardiomyocytes. Biophys J. 2014;106:2729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibrahim M, Al Masri A, Navaratnarajah M, Siedlecka U, Soppa GK, Moshkov A, Al-Saud SA, Gorelik J, Yacoub MH and Terracciano CM. Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 2010;24:3321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE and Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sacconi L, Ferrantini C, Lotti J, Coppini R, Yan P, Loew LM, Tesi C, Cerbai E, Poggesi C and Pavone FS. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc Natl Acad Sci U S A. 2012;109:5815–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibrahim M, Gorelik J, Yacoub MH and Terracciano CM. The structure and function of cardiac t-tubules in health and disease. Proc Biol Sci. 2011;278:2714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H and Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtsuka T, Nakanishi H, Ikeda W, Satoh A, Momose Y, Nishioka H and Takai Y. Nexilin: a novel actin filament-binding protein localized at cell-matrix adherens junction. J Cell Biol. 1998;143:1227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim WK, Zhao X, Fradkin D, Kohler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjaer H, Jorgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Morner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA and Meder B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–35a. [DOI] [PubMed] [Google Scholar]
- 24.Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, Just S, Hess A, Ehlermann P, Weichenhan D, Grimmler M, Liptau H, Hetzer R, Regitz-Zagrosek V, Fischer C, Nurnberg P, Schunkert H, Katus HA and Rottbauer W. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med. 2009;15:1281–8. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Li Z, Wang J, Sun K, Cui Q, Song L, Zou Y, Wang X, Liu X, Hui R and Fan Y. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2010;87:687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aherrahrou Z, Schlossarek S, Stoelting S, Klinger M, Geertz B, Weinberger F, Kessler T, Aherrahrou R, Moreth K, Bekeredjian R, Hrabe de Angelis M, Just S, Rottbauer W, Eschenhagen T, Schunkert H, Carrier L and Erdmann J. Knock-out of nexilin in mice leads to dilated cardiomyopathy and endomyocardial fibroelastosis. Basic Res Cardiol. 2016;111:6. [DOI] [PubMed] [Google Scholar]
- 27.Liu C, Spinozzi S, Chen JY, Fang X, Feng W, Perkins G, Cattaneo P, Guimaraes-Camboa N, Dalton ND, Peterson KL, Wu T, Ouyang K, Fu XD, Evans SM and Chen J. Nexilin Is a New Component of Junctional Membrane Complexes Required for Cardiac T-Tubule Formation. Circulation. 2019;140:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM and Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–5. [DOI] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P and Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Mu Y, Veevers J, Peter AK, Manso AM, Bradford WH, Dalton ND, Peterson KL, Knowlton KU, Ross RS, Zhou X and Chen J. Postnatal Loss of Kindlin-2 Leads to Progressive Heart Failure. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollak AJ, Liu C, Gudlur A, Mayfield JE, Dalton ND, Gu Y, Chen J, Heller Brown J, Hogan PG, Wiley SE, Peterson KL and Dixon JE. A secretory pathway kinase regulates sarcoplasmic reticulum Ca(2+) homeostasis and protects against heart failure. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, Lao DH, Dalton ND, Gu Y, Wang C, Wang M, Liang Y, Lange S, Ouyang K, Peterson KL, Evans SM and Chen J. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu ZQ. Scale space approach to directional analysis of images. Appl Opt. 1991;30:1369–73. [DOI] [PubMed] [Google Scholar]
- 34.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W and Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466:1129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sperelakis N and Rubio R. An orderly lattice of axial tubules which interconnect adjacent transverse tubules in guinea-pig ventricular myocardium. J Mol Cell Cardiol. 1971;2:211–220. [DOI] [PubMed] [Google Scholar]
- 36.Asghari P, Schulson M, Scriven DR, Martens G and Moore ED. Axial tubules of rat ventricular myocytes form multiple junctions with the sarcoplasmic reticulum. Biophys J. 2009;96:4651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukyanenko V, Ziman A, Lukyanenko A, Salnikov V and Lederer WJ. Functional groups of ryanodine receptors in rat ventricular cells. J Physiol. 2007;583:251–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crossman DJ, Ruygrok PN, Soeller C and Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 2011;6:e17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D’Hooge J and Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–46. [DOI] [PubMed] [Google Scholar]
- 40.Seidel T, Navankasattusas S, Ahmad A, Diakos NA, Xu WD, Tristani-Firouzi M, Bonios MJ, Taleb I, Li DY, Selzman CH, Drakos SG and Sachse FB. Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading. Circulation. 2017;135:1632–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I and Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louch WE, Sejersted OM and Swift F. There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol. 2010;2010:503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bers DM and Stiffel VM. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am J Physiol. 1993;264:C1587–93. [DOI] [PubMed] [Google Scholar]
- 44.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW and Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.