

Despite being prevalent in numerous ecologically and clinically relevant bacterial species, the biological role of proteins with a domain of unknown function, DUF1127, is unclear. Experimental models are needed to approach their elusive function. We used the phytopathogen Agrobacterium tumefaciens, a natural genetic engineer that causes crown gall disease, and focused on its three small DUF1127 proteins. They have redundant and pervasive roles in nutrient acquisition, cellular metabolism, and biofilm formation. The study shows that small proteins have important previously missed biological functions. How small basic proteins can have such a broad impact is a fascinating prospect of future research.
KEYWORDS: gene annotation, nutrient transport, phosphate metabolism, small proteins
ABSTRACT
In any given organism, approximately one-third of all proteins have a yet-unknown function. A widely distributed domain of unknown function is DUF1127. Approximately 17,000 proteins with such an arginine-rich domain are found in 4,000 bacteria. Most of them are single-domain proteins, and a large fraction qualifies as small proteins with fewer than 50 amino acids. We systematically identified and characterized the seven DUF1127 members of the plant pathogen Agrobacterium tumefaciens. They all give rise to authentic proteins and are differentially expressed as shown at the RNA and protein levels. The seven proteins fall into two subclasses on the basis of their length, sequence, and reciprocal regulation by the LysR-type transcription factor LsrB. The absence of all three short DUF1127 proteins caused a striking phenotype in later growth phases and increased cell aggregation and biofilm formation. Protein profiling and transcriptome sequencing (RNA-seq) analysis of the wild type and triple mutant revealed a large number of differentially regulated genes in late exponential and stationary growth. The most affected genes are involved in phosphate uptake, glycine/serine homeostasis, and nitrate respiration. The results suggest a redundant function of the small DUF1127 paralogs in nutrient acquisition and central carbon metabolism of A. tumefaciens. They may be required for diauxic switching between carbon sources when sugar from the medium is depleted. We end by discussing how DUF1127 might confer such a global impact on cell physiology and gene expression.
IMPORTANCE Despite being prevalent in numerous ecologically and clinically relevant bacterial species, the biological role of proteins with a domain of unknown function, DUF1127, is unclear. Experimental models are needed to approach their elusive function. We used the phytopathogen Agrobacterium tumefaciens, a natural genetic engineer that causes crown gall disease, and focused on its three small DUF1127 proteins. They have redundant and pervasive roles in nutrient acquisition, cellular metabolism, and biofilm formation. The study shows that small proteins have important previously missed biological functions. How small basic proteins can have such a broad impact is a fascinating prospect of future research.
INTRODUCTION
One of the remaining challenges in the postgenomic age is the large number of annotated domains and proteins of unknown function. Remarkably, almost one-quarter of the close to 18,000 protein families listed in the Pfam database (1) have an unassigned function. More than 3,900 of them either harbor or entirely consist of a domain of unknown function (DUF). More than 2,700 DUFs are annotated in bacteria, and they provide a rich source of novel protein folds and functions. Therefore, it is of prime interest to identify the biological role of DUFs or, in other words, “de-DUF the DUFs” (2). A common problem in the elucidation of DUF functions is that deletion of the corresponding genes often is not associated with obvious phenotypes, suggesting that many DUFs are relevant only under certain conditions or are redundant in the cell. It is important to note, however, that a substantial number of DUFs are essential. In Escherichia coli, 89 of the 359 DUFs (25%) have been reported to be essential and called eDUFs (3).
Identifying the function of short proteins with a DUF poses a particular challenge, since small proteins typically have modulatory functions, for example, on protein function, protein complex formation, or translation control, that are not obvious in standard phenotypic assays (4). In general, the proportion of so-called “small proteins” with a length of 50 amino acids (aa) or fewer is relatively low in any given proteome (5, 6). They have frequently been missed in automated genome annotations with arbitrary cutoffs such as 100 aa to avoid a large number of misannotations (7, 8). In addition to the difficulties in identifying small proteins in silico, they are not amenable to routine experimental procedures, such as standard protein electrophoresis or mass spectrometry (6, 9, 10). Despite these drawbacks, there is an increasing appreciation for small proteins and small DUFs (6, 11–13). Recent proteomics and ribosome profiling studies suggest that bacterial proteomes harbor dozens of small proteins that had been missed in automatic annotations (14, 15), and revealing their hidden functionalities is gaining momentum.
A very prominent but largely unexplored bacterial DUF family is DUF1127, which was first described in 2004 in the Pfam database (release 10.0) (1). Almost all DUF1127 proteins are single-domain proteins (99.8%), and the vast majority (93.4%) have a length of <100 aa. Approximately 15% are shorter than 51 aa and thus qualify as bona fide small proteins. The shortest stand-alone DUF1127 protein has a length of merely 23 aa (UniProt accession no. A0A3S1LPK2, Mesorhizobium). The InterPro database currently lists a DUF1127 in around 17,000 proteins from 4,000 different species (release 77.0) (16); 98.3% of these sequences derive from bacteria, 0.1% from viral genomes, and 1.6% from unclassified sources. Only two DUF1127 proteins are reported in archaea and three in fungi. DUF1127 proteins almost exclusively occur in alpha- (67.2%) and gammaproteobacteria (30.4%). Often, multiple DUF1127 members are predicted in alphaproteobacteria, for example, four each in Agrobacterium tumefaciens, Brucella abortus, and Rhodobacter sphaeroides, whereas typical gammaproteobacteria, such as Escherichia coli and Salmonella enterica, only contain a single member, which is called YjiS (17). The yjiS gene is found in close genomic proximity to the so-called immigration control region, which encodes different sets of restriction enzymes depending on the strain (18). The expression of yjiS in S. enterica is induced under virulence conditions, but the physiological relevance of this response is unknown (19–21).
Information on the expression and regulation of DUF1127 proteins is scarce. The expression of three DUF1127 genes in B. abortus is activated by the LysR-type transcriptional regulator (LTTR) VtlR (22), which is required for survival inside macrophages (23). Deletion of one or all three DUF1127 genes did not affect the ability of the pathogen to infect macrophages, suggesting that other genes are responsible for the virulence phenotype of the vtlR mutant. The search for phenotypic differences between the wild type (WT) and mutants in a broad range of growth media revealed a role of at least one DUF1127 protein in sugar metabolism. The bab2_0512 mutant was unable to utilize l-fucose (24). Like Brucella, many other alphaproteobacteria encode orthologs of the VtlR regulator, which is called LsrB (LysR-type symbiosis regulator B) in the nitrogen-fixing plant symbiont Sinorhizobium meliloti and in the plant pathogen A. tumefaciens (25, 26). Deletion of lsrB in A. tumefaciens reduces the expression of the two DUF1127 genes atu1667 and atu8161 (26), suggesting that regulation of this gene family by LsrB/VtlR regulators is conserved.
In many alphaproteobacteria, DUF1127 genes are found in the genomic context of cuckoo genes. The cuckoo family encompasses small regulatory RNAs (sRNAs) with a typical sequence motif CCUCCUCCC (27) comprising an anti-Shine-Dalgarno sequence (28, 29). The arrangement of a DUF1127 gene directly followed by one or more cuckoo sRNA genes is, for example, found in S. meliloti (30, 31), Rhizobium leguminosarum (32), and B. abortus (33). In R. sphaeroides, the DUF1127 gene RSP_6037 is harbored in an operon with the four cuckoo sRNAs CcsR1 to -4 (34). This operon is under the control of the sigma factors RpoHI and RpoHII, which are both activated by heat and oxidative stress (35, 36). The cuckoo sRNAs indirectly repress several metabolic pathways that consume glutathione and produce reactive oxygen species, thus increasing tolerance against oxidative stress. RSP_6037 negatively impacts expression of CcsR1 to CcsR4 by a yet-unknown mechanism (34).
During our investigations on sRNAs in A. tumefaciens, we noticed that some DUF1127-encoding genes had previously been annotated as sRNAs (37, 38). The model organism A. tumefaciens is a widespread plant pathogen which causes neoplastic crown gall disease in more than 600 plant species from 90 different families (39, 40). Due to its unique ability to transfer a defined piece of DNA, T-DNA, into its host, A. tumefaciens is the most commonly exploited vehicle for genetically engineered plants (41, 42). Despite the widespread use in science and industry, A. tumefaciens still harbors many genes with unknown function. Prior to this study, four DUF1127 proteins had been annotated (Atu1667, Atu1847, Atu8161, and Atu8135). We added three more to this list and systematically studied the expression and biological function of the entire family, which can be divided into two subclasses based on sequence, length, regulation, and functionality. Deletion of the three members of the short DUF1127 class with a length of <50 aa induced striking phenotypes, such as an unusual growth defect and increased biofilm formation. Transcriptome sequencing (RNA-seq) revealed a massively altered transcriptome in the triple mutant, in particular, in late exponential and stationary growth. Cumulatively, our results suggest that the short DUF1127 proteins fulfill a redundant function in the central carbon metabolism of A. tumefaciens.
RESULTS
The genome of A. tumefaciens encodes seven DUF1127 proteins.
The number of annotated DUF1127 proteins in the InterPro database has continuously increased in past years (16). To uncover the full repertoire of this protein family in the model alphaproteobacterium A. tumefaciens C58 (also known as Agrobacterium fabrum), we established a de novo search pipeline. Briefly, all putative open reading frames (ORFs) were extracted from all six reading frames of the four replicons, namely, the circular and linear chromosomes and the At and the Ti plasmids. These ORFs were translated into the corresponding amino acid sequences, and all sequences exceeding the length of 23 aa, which corresponds to the previously shortest annotated DUF1127 protein in InterPro (UniProt accession no. A0A370WQ23) were subjected to further analyses.
Using BLAST (43), the resulting 130,000 sequences were compared with a database assembled from all DUF1127 proteins listed in InterPro. All four previously annotated A. tumefaciens DUF1127 proteins (Atu1667, Atu1847, Atu8135, and Atu8161) were found by this approach (Fig. 1A). In addition, two annotated hypothetical proteins of unknown function (Atu1766 and Atu1865) (44, 45) were assigned to this group (see Fig. S1 in the supplemental material). The seventh candidate from the linear chromosome was previously described as small noncoding RNA L4 (37) and only recently received an ORF designation in the NCBI database (46). According to their size and due to other characteristics (see below), the three candidates shorter than 50 aa are called short DUF1127 proteins (SDPs) further on, and the ones between 72 and 101 aa are long DUF1127 proteins (LDPs) (Fig. 1A). The sequences extending beyond the DUF1127 domain in the LDPs show no similarities among themselves or to other annotated protein domains. A hallmark of all DUF1127 proteins, not only in A. tumefaciens but also in other organisms, is the remarkable overabundance of arginine residues in their sequences. DUF1127 proteins on average contain more than twice as many arginine residues (14.1%) as the standard protein (5.53%; P < 0.0001) according to UniProt (47).
FIG 1.

Seven DUF1127 proteins are predicted and produced in A. tumefaciens. (A) Characteristics of three SDPs and four LDPs. *, protein annotation was incorrect and adjusted at the N terminus (see Fig. S2B in the supplemental material). (B) Western blot analysis of chromosomally integrated C-terminal 3×FLAG fusions of all seven DUF1127 proteins at different growth phases in YEB medium at 30°C. A heat shock was applied for 20 min at 42°C and cold shock for 20 min at 17°C.
Strikingly, five of the seven A. tumefaciens DUF1127 proteins (SDP1 and -2 and LDP2 to -4) are encoded within a 200,000-bp region on the circular chromosome, probably an indication of gene duplication events. LDP1 is located 1 Mbp away on the same replicon, and SDP3 is the only DUF1127 gene located on the linear chromosome. The latter is directly upstream of the cuckoo sRNA gene L5, a genomic arrangement reminiscent of but not identical to the situation in R. sphaeroides (34). Here, four cuckoo sRNAs are in an operon together with the DUF1127 protein RSP_6037. In contrast, SDP3 and the L5 RNA are in separate transcription units, each with its own promoter (see Fig. S2A).
Chromosomally encoded C-terminal 3×FLAG tag fusions of all seven DUF1127 proteins were constructed to examine their expression by Western blotting (Fig. 1B). The corresponding fusion genes replaced the respective wild-type genes at their native locations. The fusion proteins showed different expression patterns over the growth curve. SDP13×FLAG and SDP23×FLAG were present in all growth phases, whereas SDP33×FLAG and LDP33×FLAG peaked in stationary phase. LDP23×FLAG and LDP43×FLAG were predominantly produced during late exponential growth (optical density [OD] at 600 nm of 1.5). LDP13×FLAG was most abundant during early exponential growth and gradually decreased toward stationary phase. Since several DUF1127 genes from other alphaproteobacteria are regulated by the heat shock sigma factor RpoH (35, 48), the expression of Agrobacterium DUF1127 genes was analyzed under heat and cold shock conditions as well. Short-term exposure to heat (20 min at 42°C) induced all three SDP3×FLAG fusions as well as LDP23×FLAG and LDP33×FLAG. The remaining two proteins (LDP13×FLAG and LDP43×FLAG) increased after cold shock (20 min at 17°C).
Overall, the results of the 3×FLAG fusions demonstrate that all seven DUF1127 proteins are authentic proteins in A. tumefaciens. It is noteworthy that immunodetection of the LDPs generally required longer exposure times than for detection of SDPs, in particular, for LDP23×FLAG and LDP43×FLAG. This suggests that the three short proteins are produced at higher levels than the long ones. This observation is also backed up by our RNA-seq data (see below).
The DUF1127 family consists of at least three subclasses.
Approximately two-thirds of all annotated DUF1127 proteins occur in alphaproteobacteria and approximately one-third in gammaproteobacteria. The higher abundance in the first group is in part due to several DUF1127 members within one organism, whereas E. coli, Salmonella, and their close relatives produce only a single DP, which is called YjiS. The seven DUF1127 proteins from A. tumefaciens fall into two groups, well in line with their length. The three SDPs are most similar to each other (44.7% to 66.7% identity) but display only moderate similarity to the four LDPs (23.4% to 40.4% identity), which form a separate subclass. The homology of the SDPs to E. coli YjiS, which is 54 aa long, is equally low (27.1% to 34.0% identity) (Fig. 2A and B).
FIG 2.

DUF1127 proteins can be divided into clusters. (A) SDPs from A. tumefaciens show a high sequence similarity. Identical residues are marked with an asterisk, residues with highly similar properties are marked with a colon, and weakly conserved residues are marked with a period. Arginine residues are shaded in black. (B) The homology tree of all seven DUF1127 proteins from A. tumefaciens and YjiS from E. coli was calculated by Clustal Omega (109). (C) Subdivision of DUF1127 proteins. The stacked bar chart shows the absolute frequency for the lengths of all DUF1127 proteins from InterPro. A kernel density estimation (dashed line) was superimposed. DUF1127 proteins were divided into subclasses according to sequence similarity to either SDPs from A. tumefaciens or to YjiS from E. coli.
To address the question whether the DUF1127 proteins in general can be sorted into distinct subclasses, all DUF1127 proteins from InterPro were categorized into three groups according to sequence similarity: (i) those that display a high similarity to the SDPs from A. tumefaciens, (ii) those with a high similarity to E. coli YjiS (YjiS-like proteins), and (iii) those that could not be assigned to either of these groups (LDPs). As of January 2020, the InterPro database listed 16,537 DUF1127 proteins. The three newly discovered DUF1127 proteins from A. tumefaciens in this study were appended to the list. Sorting these 16,540 proteins in the three categories resulted in 3,168 (19%) proteins in the SDP class, 1,005 (6%) in the YjiS-like class, and 12,367 (74%) without assignment to either of these groups. For complete lists of all entries see Tables S8 to S10. Identical results have been retrieved for a selection of species via a clustering method (see Fig. S3A). The majority of the proteins assigned to the SDPs derive from alphaproteobacteria (72.1%), whereas only 0.2% derive from gammaproteobacteria. In contrast, YjiS-like proteins are found mainly in gammaproteobacteria (88.4%) and rarely in alphaproteobacteria (0.6%). The nonassigned DUF1127 proteins are found partially in alphaproteobacteria (53.3%) and partially in gammaproteobacteria (30.2%). The overall length distribution of all DUF1127 proteins shows four prominent peaks at around 47 aa, 54 aa, and 71 aa and at slightly less than 100 aa (Fig. 2C). SDPs and YjiS-like proteins display a rather narrow distribution within the first two peaks, such that almost all SDPs fall into the peak around 47 aa and YjiS-like proteins are almost exclusively found in the peak at 54 aa. The unassigned DUF1127 proteins are very heterogeneous in sequence and length.
Several other properties support the demarcation of SDPs and YjiS-like proteins from the remaining ones. The size difference already visible in the length distribution (Fig. 2C) was confirmed via Welch’s t test (Fig. S3B). As mentioned above, the DUF1127 is arginine rich. The arginine content of SDPs and YjiS-like proteins is even higher and lies in the range between 16% and 17% (Fig. S3B). The high number of arginine residues is reflected in a positive net charge of DUF1127 proteins, which is most pronounced in SDPs and YjiS-like proteins.
Yet another difference between the subclasses pertains to the number of proteins of the same group in a single species (Fig. S3B). While species producing SDPs on average have 2.3 of them, species with YjiS-like proteins only have an average of 1.5 of them. The relative amount of LDPs per species exceeds those of both other groups, with an average of 3.8. Since this subclass is composed of a rather heterogeneous group of proteins with various extensions, it might fall into several distinct subclasses upon further investigation.
The presence of SDPs coincides with the LysR-type regulator LsrB and cuckoo sRNAs.
DUF1127 proteins have been studied in only a few bacteria, and two findings caught our attention. First, DUF1127 genes in A. tumefaciens and B. abortus are regulated by LysR-type regulators called LsrB and VtlR, respectively (22, 26). Second, a direct genomic neighborhood of DUF1127 genes with cuckoo sRNAs was described in R. sphaeroides (34, 35). To examine whether these reported associations can be generalized, we asked whether DUF1127 proteins, LsrB-type regulators, and cuckoo sRNAs coincide in various species (Fig. 3). Among the selected species were close relatives of A. tumefaciens, such as R. leguminosarum, Neorhizobium galegae, and Pseudorhizobium pelagicum, as well as more distantly related alphaproteobacteria. The analyzed gammaproteobacteria included Pseudomonas aeruginosa, Yersinia enterocolitica, and the Enterobacteriaceae Klebsiella pneumoniae, E. coli, and S. enterica. In this selection, SDPs were exclusively found in alphaproteobacteria except for P. pelagicum. YjiS-like proteins were present only in Enterobacteriaceae, which lacked LDP members.
FIG 3.
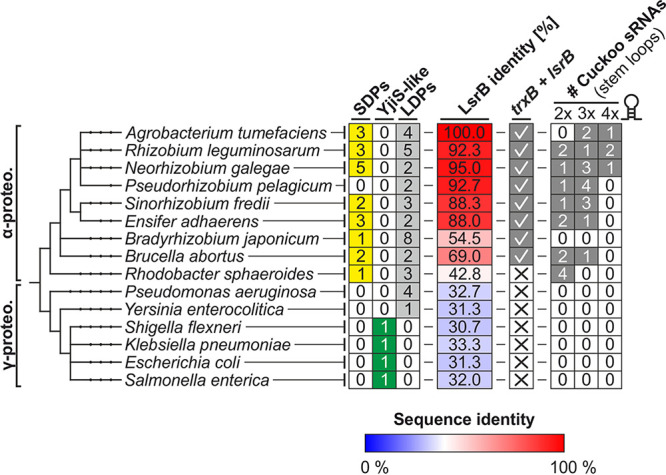
Cooccurrence of DUF1127 proteins with LsrB and cuckoo sRNAs. The tree shows the phylogenetic relations of a selection of alpha- (α-proteo.) and gammaproteobacteria (γ-proteo.). For each strain, the numbers of SDPs, YjiS-like proteins, and LDPs are given. BLAST searches identified the proteins with the highest similarity to LsrB from A. tumefaciens (A9CI74, B5ZXG9, A0A068SS95, A0A081MRV5, C3MD12, W8I232, G7D4X2, Q2YRP4, Q3J274, Q9I1F9, A1JKA5, A0A0H2UWZ6, A0A0H3GNP2, P30864, and Q8ZRM6). Red and blue colors represent proteins with higher and lower sequence identity than 40%, respectively A genomic neighborhood to a trxB homolog is indicated by a check mark. Putative cuckoo sRNA sequences were extracted from each genome by searching for the pattern (CCTCCTCCC-N20–50)≥1-CCTCCTCCC. The numbers of sequences encoding two, three, or four CCUCCUCCC motifs are given.
LysR-type regulators belong to the most abundant class of transcriptional regulators (49). Using A. tumefaciens LsrB as query in a BLAST search, we identified the best hit in each organism and considered it an LsrB-type protein when it had more than 40% sequence identity, which is a recommended cutoff (50). A conserved genomic neighborhood between lsrB and homologs of the thioredoxin reductase gene trxB (Fig. 3), which was reported previously (22), confirmed LsrB homology among the selected Rhizobiales. With the exception of P. pelagicum, there was a strict association between SDPs and LsrB homologs in alphaproteobacteria (Fig. 3).
To determine the prevalence of cuckoo sRNAs, each genome was searched for a pattern coding for at least two CCUCCUCCC motifs: (CCTCCTCCC-N20–50)≥1-CCTCCTCCC. The three known cuckoo sRNAs from A. tumefaciens (28) and the four annotated sRNAs from R. sphaeroides (35) were found in this search. Putative cuckoo sRNAs were identified in all alphaproteobacteria except for Bradyrhizobium japonicum. Gammaproteobacteria seem to lack cuckoo-like sRNAs entirely. Despite a few exceptions, these searches reveal a strong correlation between SDPs, an LsrB-type regulator, and cuckoo RNAs in alphaproteobacteria.
DUF1127-coding genes are reciprocally regulated by LsrB according to their subclasses.
The previously described LsrB/VtlR-dependent expression of SDP1 and SDP2 in A. tumefaciens (26) and of homologous genes in B. abortus (22) motivated us to examine the transcription of all seven DUF1127 genes in A. tumefaciens WT and in an lsrB-deficient mutant (ΔlsrB). Northern blot analyses revealed that the three SDP genes were downregulated in the lsrB mutant, whereas the four LDP genes were upregulated (Fig. 4A). This suggests that the transcription factor LsrB directly or indirectly activates SDP and represses LDP gene expression. This reciprocal regulation supports the biocomputational division into two distinct subclasses described in the previous sections.
FIG 4.

SDP and LDP genes are reciprocally regulated by the transcription factor LsrB. (A) Northern blot analyses of SDP- and LDP-encoding genes in the WT and lsrB mutant. Ethidium bromide (EtBr)-stained 16S or 23S rRNA served as a loading control. (B) Putative LsrB-binding sites marked in red match the TGC-N7-GCA motif, and orange sequences display the shorter TGC-N6-GCA motif. Previously described binding sites of the three DUF1127 genes from B. abortus (bab1_0914, bab2_0514, and bab2_0574) are underlined. The position relative to the transcriptional start site (+1) is indicated.
The binding sites of the B. abortus LsrB homolog VtlR have recently been determined experimentally upstream of three positively regulated DUF1127 genes (22). Based on the common 13-bp-long LTTR-binding motif T-N11-A (51), we searched for LsrB-binding sites upstream of the A. tumefaciens SDP genes. The palindromic motif TGC-N6-7-GCA was found upstream of the transcriptional start sites (TSSs) of the SDP genes from A. tumefaciens as well as in the experimentally determined binding sites from B. abortus (Fig. 4B). Interestingly, the bab1_0914 and bab2_0574 genes contain this motif in tandem, once with a linker of six and once with a linker of seven nucleotides, respectively or vice versa. The location of the putative LsrB-binding site in the A. tumefaciens SDP promoters between −58 and −71 is consistent with activator binding sites. Similar motifs were not found upstream of the four LDP genes (data not shown).
The SDPs affect the abundance of the cuckoo sRNA L5.
The genomic arrangement of a DUF1127 protein gene followed by a gene coding for a cuckoo sRNA is often found in alphaproteobacteria. In A. tumefaciens, the SDP3 gene directly precedes the sRNA gene L5, which is transcribed from its own promoter (37) (Fig. S2A). The L5 RNA is 162 nucleotides (nt) long and forms four stem-loops containing one CCUCCUCCC motif in each loop. The full-length RNA is processed to a shorter product presumably composed of three stem-loops (L5* in Fig. 5). Deletion of the SDP3 gene alone affected neither L5 expression nor processing (data not shown). To test whether the SDPs as a group have an influence on the cuckoo sRNA, we constructed a triple mutant (ΔΔΔ), which lacks all three SDPs, and compared the transcript amounts of L5 and L5* in the WT and mutant at different growth phases by Northern blotting (Fig. 5). In early and late exponential growth (ODs of 0.5 and 1.5, respectively), the amounts of both L5 products were increased in the ΔΔΔ mutant compared to that in the WT, whereas the L5 level was decreased in the mutant in stationary phase. The elevated L5 levels in the triple mutant resemble the situation in R. sphaeroides where the expression of the cuckoo sRNA cluster CcsR1 to -4 is low in the presence of the DUF1127 protein RSP_6037 (34).
FIG 5.
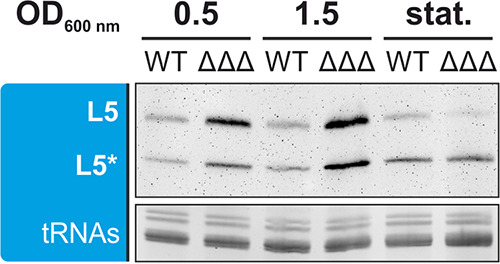
Transcript amounts of L5 and its processed form L5* in the WT and ΔΔΔ mutant at different growth phases, which are indicated as ODs (0.5 and 1.5) and stationary phase (stat.). EtBr-stained tRNAs served as a loading control for Northern blot analysis.
The A. tumefaciens SDP genes are differentially expressed.
To approach the biological function of DUF1127 proteins, we focused on the three SDPs because (i) they contain little more than the DUF1127 domain alone, (ii) they constitute a separate subclass with high similarity among themselves and distinct from the LDPs, (iii) they are positively regulated by the same transcription factor, LsrB, and (iv) they seem to be produced at higher levels than the other DUF1127 proteins. First, we measured the transcript levels of the SDP genes under different growth conditions by Northern blotting. All three transcripts were readily detectable and peaked at different points in the growth curve (Fig. 6A and B). The highest SDP1 expression was observed at the end of the exponential phase (phase II), whereas expression of SDP2 was highest during early exponential growth (phase I). The SDP3 transcript increased throughout the growth curve until it finally reached its maximum in phase III. Overall, the expression patterns show that under any given growth condition, at least one of the three SDPs is produced, and this is consistent with the results of the 3×FLAG tag fusions (Fig. 1B).
FIG 6.
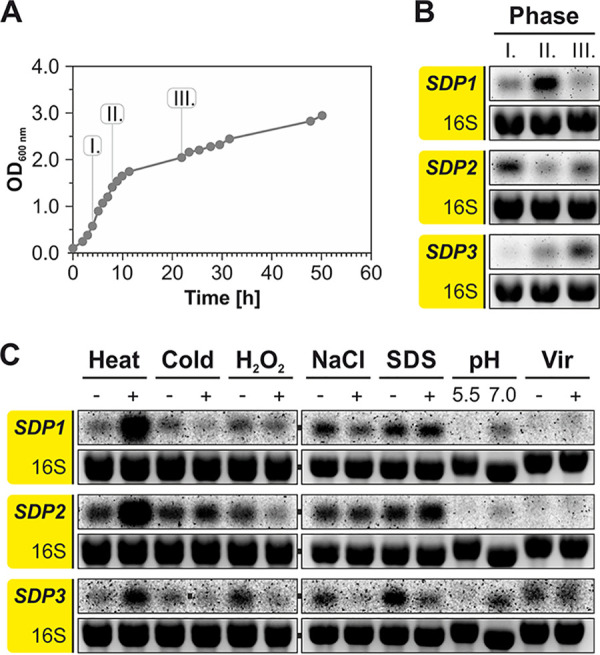
SDP genes are differentially expressed. (A) A. tumefaciens WT was grown in YEB medium at 30°C. (B) Samples for RNA isolation were taken at the indicated time points and subjected to Northern blot analysis. (C) Differential expression of SDP genes under various growth and stress conditions. The following stress conditions were applied for 10 min: heat shock (42°C), cold shock (17°C), oxidative stress (0.01% [wt/vol] H2O2), osmotic stress (1 M NaCl), and cell envelope stress (0.08% [wt/vol] SDS). The pH-dependent expression was tested after continuous growth in minimal AB medium. For virulence induction, cells were grown in AB medium (pH 5.5) supplemented with acetosyringone.
When A. tumefaciens was subjected to various stress conditions, all three SDP genes showed similar expression patterns (Fig. 6C). The most pronounced induction occurred after a heat shock when the temperature was shifted from 30°C to 42°C for 10 min. Cold shock (10 min at 17°C), H2O2-mediated oxidative stress, NaCl-mediated osmotic stress, and SDS-mediated cell envelope stress caused moderate changes of gene expression, typically resulting in downregulation. SDP transcripts were barely detectable in AB minimal medium at low pH, and virulence induction by addition of acetosyringone in AB medium at pH 5.5 did not affect expression relative to that under noninduced conditions.
The lack of all three SDPs causes a distinct growth phenotype.
Several lines of evidence described above suggested a redundant function of the three SDPs in A. tumefaciens. To address their physiological importance, all combinations of single, double, and triple deletion mutants were constructed. Candidates were tested via PCR and Northern blot analysis to verify successful construction of all seven deletion strains (see Fig. S4). Growth experiments with the WT and the seven deletion strains in standard complex medium (yeast extract-beef extract [YEB]) showed no differences in the first 10 h until the end of exponential growth (Fig. 7A). Also later on, the single mutants grew like the WT. The double deletion strains showed slight deviations from the WT. Both strains lacking SDP3 (ΔSDP1 ΔSDP3 and ΔSDP2 ΔSDP3) showed WT-like growth in the first 24 h but reached a lower final OD. Strain ΔSDP1 ΔSDP2 grew slightly better than the WT in late exponential phase but showed a decreased OD thereafter.
FIG 7.
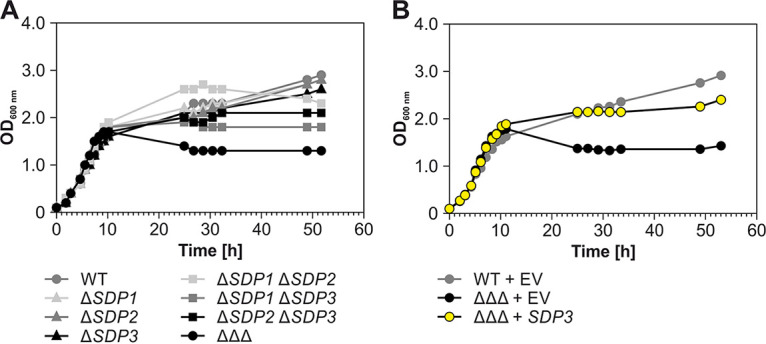
Deletion of the SDP genes causes a growth defect. (A) Growth of all single, double, and triple mutants in YEB medium at 30°C. (B) The growth defect of the ΔΔΔ mutant was complemented by the SDP3 gene, which was reintegrated into the genome via single-crossover homologous recombination. EV, empty vector.
The ΔΔΔ strain reproducibly showed a very peculiar growth phenotype (Fig. 7 and 8A). It grew normally until late exponential phase, where it reached even higher ODs than the WT, between 10 and 12 h of growth. Thereafter, the OD suddenly dropped and remained constant at approximately 1.5 for several days, whereas the WT showed diauxic behavior and continued to grow after a lag phase. A closer look at viable cells and cell morphology showed that the number of viable and intact ΔΔΔ cells started to decrease already when the OD was higher than that for the WT (see Fig. S5). Another striking observation was that the mutant cells were longer than WT cells.
FIG 8.

Sucrose- and osmolarity-dependent growth of the ΔΔΔ mutant. Growth of WT and ΔΔΔ strains in YEB and LB medium with and without 0.5% (wt/vol) sucrose and 1.0% (wt/vol) NaCl. Samples for Northern blot analyses were taken at the indicated time points (see Fig. S6).
Almost-normal growth was restored to the ΔΔΔ mutant by complementation with the SDP3 gene, which was reintroduced into the linear chromosome (Fig. 7B).
Sucrose and salt influence SDP gene expression and growth of the ΔΔΔ mutant.
While testing different growth media, we made the surprising finding that the very robust growth phenotype of the A. tumefaciens ΔΔΔ strain in standard rich medium (yeast extract-beef extract [YEB] medium) described above did not at all occur in Luria-Bertani (LB) rich medium, in which both strains grew equally well to a very high maximal OD of 5.5 (Fig. 8). The two main differences between those media are 0.5% sucrose, which serves as carbon source in YEB, and 1% NaCl present in LB medium. To test if the presence or absence of these two compounds influences the YEB-specific growth defect, we propagated A. tumefaciens WT and the triple mutant in various medium combinations. Growth of both strains was comparable in YEB without sucrose but ceased at an OD of 1.5. Addition of sucrose to LB had no visible impact on growth of both strains, whereas addition of 1% NaCl to YEB diminished the growth differences between both strains such that the triple mutant grew better and the WT grew more poorly than in standard YEB. Addition of 2% NaCl further reduced the differences between both growth curves (data not shown). Conversely, withdrawing the salt from sucrose-supplemented LB induced a growth defect in the ΔΔΔ strain.
Samples were taken from all these cultures to examine the expression of SDP genes by Northern blotting (see Fig. S6). The transcript patterns in standard YEB were the same as described above (Fig. 6B), whereas the peaks of all three genes were shifted to earlier growth phases in the absence of sucrose. Addition of 1% (wt/vol) NaCl to YEB did not affect the expression of SDP1 and SDP3 but shifted the maximum expression of SDP2 toward phase III. In standard LB medium, all three genes showed a parallel early expression peak in phase I. Addition of sucrose to LB restored the expression pattern known from YEB medium independently of the presence of NaCl.
Deletion of all three SDP genes increases cell attachment and biofilm formation.
In search for other phenotypes of the ΔΔΔ mutant, we analyzed various general and A. tumefaciens-specific parameters and observed that the mutant cells aggregated much faster than the WT. This was shown by transferring an aliquot of each strain from a 24-h liquid culture into a microtiter-well plate and incubating for another 24 h under tilting at room temperature. While formation of visible clumps by the WT took at least 6 h, the ΔΔΔ strain started to aggregate already after 5 min (Fig. 9A). Since cell aggregation is often linked to biofilm formation, this process was examined by the crystal violet assay. The mutant produced 5- to 8-fold more biofilm than the WT (Fig. 9B). Another feature typically associated with biofilm formation is the production of certain polysaccharides, such as cellulose and unipolar polysaccharide (UPP), an adhesin at single-cell poles that contact surfaces (52, 53). The WT and triple mutant strains were grown on YEB agar plates supplemented with both a Congo red solution and Fluorescent Brightener 28 (alias calcofluor white) (Fig. 9C). While Congo red is used to visualize biofilm-associated matrix such as cellulose or UPP under regular light (54, 55), Fluorescent Brightener 28 noncovalently binds to β-glycosidically linked polysaccharides such as succinoglycan and is visible under UV light (56, 57). On these plates, the ΔΔΔ strain grew to a smaller diameter than the WT. Moreover, it had a darker color, indicating increased amounts of cellulose and/or UPP. In contrast, it produced less extracellular succinoglycan matrix.
FIG 9.
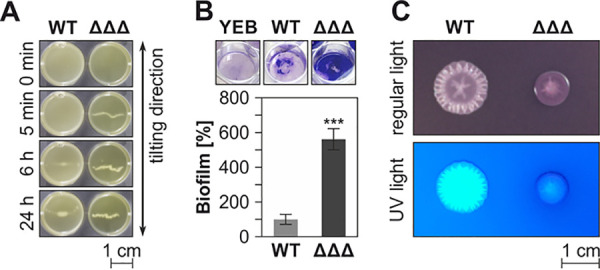
Deletion of the SDP genes results in various phenotypic alterations. (A) Cell aggregation was visualized by prolonged incubation in well plates on a tilting laboratory shaker. (B) Increased biofilm formation in the ΔΔΔ mutant was shown by crystal violet assays. ***, P < 0.001; three biological replicates. Sterile YEB served as a negative control. The top shows the wells with stained biomass prior to resolubilization and photometric quantification. (C) Colony morphology and exopolysaccharide composition were visualized on YEB agar plates supplemented with Congo red dye and Fluorescent Brightener 28.
Since cell aggregation and biofilm formation are critical for attachment of A. tumefaciens to host plants (58), the T-DNA transfer capacities of the WT and triple mutant were compared in a qualitative seedling infection assay with Arabidopsis thaliana (59). In brief, A. tumefaciens WT and mutant were transformed with the pBISN1 plasmid carrying a β-glucuronidase gene under the control of a plant-specific promoter between T-DNA borders. During plant transfection, the reporter gene is integrated into the plant genome of infected tissue, and the produced β-glucuronidase converts a substrate into a blue dye. Both strains were able to genetically engineer the plant, as was visible by the blue staining of leaves due to β-glucuronidase activity (see Fig. S7), and normal T-DNA transfer by the mutant is consistent with the low expression of the SDP genes under virulence conditions (Fig. 6).
An ABC transporter and an alcohol dehydrogenase are overproduced in the ΔΔΔ mutant.
To find reasons for the phenotypic differences between the WT and ΔΔΔ strains, we looked for obvious changes in their proteomes. Samples were taken at the three different growth phases used previously (Fig. 6A), separated by SDS-PAGE, and visualized by Coomassie staining (Fig. 10A). The most striking difference between the WT and mutant was observed in phase II. Here, an additional band in the range between 35 and 40 kDa appeared in the mutant. That area was excised from the WT and ΔΔΔ strain gels and subjected to mass spectrometry. The most abundant protein in the ΔΔΔ sample was PstS (Atu0420; sequence coverage of 52.3% and 18 unique peptides), the substrate-binding protein (SBP) of the ABC transporter complex PstSCAB (60). The second most abundant protein was Adh (Atu2022; coverage was 62.1% and 18 unique peptides), an unclassified NADP-dependent alcohol dehydrogenase. Northern blot analyses confirmed the massive induction of the corresponding pstS and adh genes in the SDP mutant in growth phase II (Fig. 10B).
FIG 10.
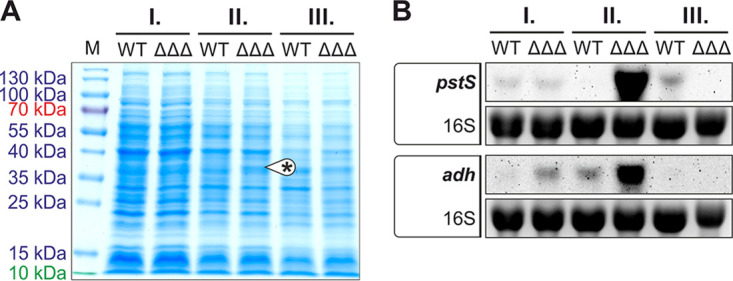
Growth phase-dependent differences in the proteomes of WT and ΔΔΔ strains. (A) Crude extracts from different growth phases were separated by SDS-PAGE and stained with Coomassie brilliant blue. During growth phase II, the ΔΔΔ mutant showed an additional protein band between 35 and 40 kDa. The proteins identified in this band by mass spectrometry were PstS (36.2 kDa) and Adh (37.6 kDa). (B) Growth phase-dependent expression of pstS and adh in WT and ΔΔΔ strains was confirmed by Northern blotting. EtBr-stained 16S rRNA served as a loading control.
Massive changes in the transcriptome of the ΔΔΔ mutant in late growth phases.
To decipher the global changes in gene expression at the transcriptome level, we isolated total RNA from WT and ΔΔΔ cultures at ODs of 0.5 and 1.5 and after 24 h (growth phases I, II, III) (Fig. 6A and 8A) and subjected it to RNA-seq analysis (see Tables S5 to S7). A sequencing depth between 15 and 33 million reads was achieved for each sample. Consistent with the Northern blot experiment (Fig. 10B), the pstS (atu0420) mRNA was among the most highly induced transcripts in the triple mutant at an OD of 1.5. It ranked at position 3 with a 112-fold induction in the mutant compared to that in the WT (Table S6 and Fig. 11) The adh (atu2022) mRNA and the sRNA L5 (C2_1831446F) also were significantly induced (9.7- and 3.1-fold, respectively) (Table S6), again in line with the Northern blots (Fig. 5 and 10B) and thus validating the RNA-seq results.
FIG 11.

Differentially expressed genes in the ΔΔΔ mutant. The volcano plots represent the data retrieved from RNA-seq. Each gene with a significantly altered expression is represented as a dot. Colors indicate genes from different functional groups.
Along with the phenotypical changes of the ΔΔΔ mutant, which occurred only in later growth phases, the differences between the transcriptomes of WT and ΔΔΔ strains increased over time as well. In growth phase I, the expression of only 76 genes was altered in the ΔΔΔ mutant. This number increased dramatically to 896 and 2,575 genes in phases II and III, respectively (Fig. 11). Thirty genes were expressed differently in the triple mutant than in the WT under all three conditions (see Fig. S8). Fifteen of them were upregulated in the ΔΔΔ mutant, 14 were downregulated, and one (atu4442) was upregulated at an OD of 0.5 and then downregulated. Among the upregulated genes were LDP1 and LDP4, suggesting some cross-regulation between the DUF1127 genes. Two sets of genes that were induced in the ΔΔΔ strain in all growth phases are involved in glycine/serine homeostasis, i.e., soxBDAG (atu4310-atu4313) and glyA (atu4314), and in anaerobic nitrate respiration (denitrification), i.e., norDBC (atu4386, atu4388, atu4389).
Several genes from these two sets were found among the most strongly induced genes in the ΔΔΔ mutant at an OD of 1.5 (labeled in yellow and blue in Fig. 11). Since phase II is reached shortly before the growth of both strains begins to differentiate, the number of genes with an altered expression was expected to increase at this point. Among the increased genes were atu4310 to atu4315. They code for the sarcosine oxidase SoxBDAG mentioned above, which converts sarcosine to glycine (61), the serine hydroxymethyltransferase GlyA, which catalyzes the bidirectional conversion between glycine and serine (62), and the formyltetrahydrofolate deformylase PurU, which provides tetrahydrofolate that serves as a cofactor for SoxBDAG (63, 64). The denitrification genes induced in the ΔΔΔ mutant include the nitrite reductase genes nirV (atu4381; 18-fold) and nirK (atu4382; 26-fold). Among the multitude of induced nitric oxide reductase genes, i.e., norD (atu4386), norQ (atu4387), norB (atu4388), norC (atu4389), norE (atu4391), and norF (atu8197), norC showed the strongest induction (36-fold). The activation of denitrification genes might be due to the 5-fold-elevated expression of the denitrification regulator gene nnrU (atu4392).
Fully consistent with the massive induction of the pstS transcript and the PstS protein, the two genes with even higher induction factors in phase II are also known phosphorus starvation genes. The phnD (atu0173) gene codes for the SBP of the phosphonate-ABC transporter PhnDEC and was >230-fold increased. The >220-fold-induced ugpA (atu0305) gene codes for UgpA, the SBP of the ABC transporter UgpBAEC, which imports sn-glycerol 3-phosphate (G3P) under phosphorus-limited conditions (65). Interestingly, all three copies of the ugpBAEC genes (atu0305-atu0308, atu3096-atu3099, and atu5058-atu5063) showed increased expression. The remaining genes of the phosphonate transporter PhnDEC (atu0171-atu0174) and the associated genes phnL (atu0176) and phnK (atu0177) were upregulated as well. This also accounts for the remaining genes of the phosphate transporter PstSCAB (atu0420-atu0423). Moreover, glpQ (atu5061) and ugpQ (atu4212) were induced. Both encode glycerophosphoryl-diester phosphodiesterases that catalyze the same step of degradation of the phosphate-containing lipid phosphatidylcholine (66, 67). In addition, the genes btaAB (atu2119-atu2120) were upregulated. BtaAB is responsible for the formation of the phosphateless betaine lipid diacylglyceryl-N,N,N-trimethylhomoserine (68). Altogether, the RNA-seq experiments revealed a striking upregulation of genes associated with the response to phosphorus limitation and belonging to the pho regulon. The concomitant 25-fold induction of the phoB (atu0425) gene coding for the response regulator PhoB, which is known to be a major activator of phosphorus starvation genes (69), might explain the coordinated upregulation.
Among the genes with decreased expression in the ΔΔΔ mutant, in particular, at an OD of 1.5, were numerous sugar-specific ABC transporter genes (Fig. 11). In addition to the phosphorus uptake systems, only a few ABC transporter genes with unknown substrates were upregulated.
DISCUSSION
The full repertoire of DUF1127 proteins in an alphaproteobacterium.
DUF1127 proteins represent a family of small proteins predominantly found in alpha- and gammaproteobacteria. In general, nature tends to favor short proteins for specialized tasks, which complicates the search for their biological function (70). To provide insights into the functionality of DUF1127 proteins, we chose the plant pathogen A. tumefaciens as a model system, systematically uncovered all family members, and analyzed the phenotypes and gene expression of mutant strains. The first task necessitated the establishment of a stringent biocomputational workflow revisiting the previously annotated family members, since small proteins are notoriously missed in standard genome annotations. Some workflows integrate bioinformatically predicted ribosomal binding sites (RBSs) to improve the identification of nonannotated small ORFs (smORFs) (71). We extracted potential ORFs from the genome of A. tumefaciens without considering possible RBSs and compared them against a database consisting of all DUF1127 proteins listed in InterPro, using the length of the shortest annotated DUF1127 protein (23 aa) as a cutoff. More than 132,000 putative protein sequences were retrieved. Although smORFs are sometimes too short for reliable homology determination (70, 72), our analysis resulted in the successful identification of seven A. tumefaciens proteins with a DUF1127, all of which were found to be expressed at the RNA and protein level. This added three new members to the DUF1127 family and improved the annotation of the previously annotated candidates. Comparison with transcriptome data (37) revealed that Atu1667 (SDP1) is 29 aa shorter at its N terminus than the previously annotated protein (see Fig. S2B in the supplemental material). In the meantime, this inconsistency has been corrected by NCBI (ATU_RS08170). Incorrect N-terminal annotations are frequent in prokaryotic genomes due to the preference for larger ORFs in automated genome annotations (73, 74). A new smORF was discovered in a transcript previously described as an sRNA called L4 (37). Short transcripts often are considered to be too short to contain a protein-coding sequence and thus are annotated as noncoding transcripts. Only recently it was appreciated that transcripts previously believed to be noncoding actually encode small proteins (75, 76). Our analysis revealed two further proteins of unknown function (Atu1766 and Atu1865) as DUF1127 proteins. Interestingly, they have been removed from the DUF1127 list in the current version of InterPro (release 77.0), whereas the UniProtKB database still links them to proteins with a DUF1127. This ambiguity demonstrates the challenges in the reliable identification of short DUFs.
Differential regulation of DUF1127 genes.
Gene expression in bacteria is strictly controlled in response to environmental conditions, and knowing about the differential expression can guide the search for function. Apart from growth phase-dependent variations in gene expression, the production of all three SDPs and two LDPs in A. tumefaciens increased after heat shock, whereas the remaining two LDPs showed higher abundance after cold shock. Expression of RSP_6037 in R. sphaeroides is also induced by heat through the alternative sigma factors RpoHI and RpoHII (35). It is also induced by oxidative stress, which is not the case for the SDP genes in A. tumefaciens. In contrast to R. sphaeroides, A. tumefaciens has only one RpoH homolog that is involved in the heat shock but not oxidative stress response (77).
A major contributor to the regulation of all seven DUF1127 genes in A. tumefaciens is the transcription factor LsrB, and this might be conserved in many alphaproteobacteria, including B. abortus (22, 26). LsrB belongs to the LTTR family, which is ubiquitous in bacteria and constitutes the largest group of DNA-binding proteins (78, 79). A conserved functionality is supported by the finding that the virulence defect of an A. tumefaciens lsrB mutant can be complemented by LsrB orthologs from S. meliloti and B. abortus (26). Despite a fairly loose consensus sequence of LTTRs, we were able to identify a potential LsrB-binding motif in a region 46 to 71 bp upstream of the TSSs of the positively regulated SDP genes. The putative binding motif TGC-N6–7-GCA displays a dyad symmetry with similarity to putative LsrB-binding sites that have been described previously (49). Moreover, it overlaps with the experimentally determined binding site in B. abortus (22). Most LTTRs suppress their own expression until they are dissociated from their own promoter through a specific stimulus (49). In S. meliloti, an LsrB-binding site upstream of lsrB suggests similar autoregulation of LsrB homologs (25). Identification of this stimulus might provide further insights into the function of the LsrB-regulated DUF1127 genes.
In R. sphaeroides, the DUF1127 protein RSP_6037 somehow reduces the transcript amounts of the cuckoo sRNAs CcsR1 to -4, which are encoded in the same operon (34). If this was a universal function of DUF1127 proteins, one would expect a strict cooccurrence of DUF1127 genes with cuckoo sRNA genes. While there are no known cuckoo sRNAs in gammaproteobacteria at all (28), there indeed is a relatively strict cooccurrence of cuckoo sRNAs and SDPs in alphaproteobacteria. Deletion of all three SDP genes in A. tumefaciens increased the amount of the L5 sRNA in early growth phases, similar to the negative correlation between RSP_6037 and CcsR1 to -4 in R. sphaeroides (34). The functional implication of the correlation between LsrB, SDPs, and cuckoo sRNAs deserves further investigation.
Phenotypic and transcriptomic consequences of the lack of short DUF1127 proteins.
A common strategy to unravel the function of proteins of unknown function is the construction and phenotypic characterization of deletion mutants. Such experiments with DUF1127 members have rarely been done. The R. sphaeroides gene RSP_6037 is essential, which might be due to its involvement in C1 metabolism and oxidative stress response (34). Deletion of an SDP gene in B. abortus caused a defect in fucose metabolism (24). While this phenotype was specific to one particular SDP in Brucella, the SDP paralogs of Agrobacterium seem to have a redundant function, because the peculiar growth behavior and other phenotypes required the deletion of all three genes.
Our cumulative results indicate that the Agrobacterium SDPs play an astounding role in various metabolic processes, in particular, phosphate and carbon utilization. Growth curves suggest that the triple mutant fails to switch to alternative carbon sources in the standard growth medium. Among the numerous changes in the transcriptome caused by the absence of the three SDPs, we have selected four particularly interesting classes of genes for further discussion (Fig. 12).
FIG 12.
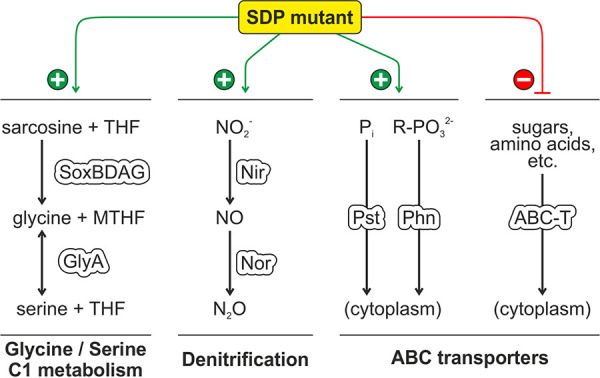
Summary of the most severe effects on gene expression in the A. tumefaciens triple SDP mutant. For details, see the text. MTHF, (5,10-methylene)tetrahydrofolate; R-PO32−, phosphonate.
(i) One of the most striking results is the permanent induction of the SoxBDAG/GlyA pathway (Fig. S8), which is implicated in glycine/serine homeostasis and C1 metabolism (Fig. 12). The tetrahydrofolate (THF)-dependent demethylation of sarcosine (N-methylglycine) generates glycine and 5,10-methylene-THF (MTHF), which can either be used to produce serine by GlyA or serve as methyl donor to other cellular compounds. The reverse reaction from serine to glycine is the major source of C1 units in the cell (80). Shifts in nutrient availability are known to provoke changes in amino acid homeostasis, in particular, of serine, which is the precursor of numerous biomolecules (80). In E. coli, the intracellular level of serine rises upon depletion of glucose as a carbon source (81). If the cell does not counteract, a toxic intracellular level of serine can be reached, because an excess of serine inhibits the synthesis of isoleucine and aromatic amino acids (82, 83). Additionally, serine can be misincorporated into peptidoglycan instead of alanine by MurC (84). This results in a destabilization of the cell wall and causes elongated cells and cell lysis (81). The Agrobacterium SDP mutant showed some morphological irregularities. Typically, cells get shorter when entering stationary growth phase as a result of reductive division and dwarfing (85). The cell size of the ΔΔΔ mutant did not decrease as much as that of the WT, and a fraction of the cells lysed after 24 h of growth in YEB (Fig. S5), which suggests unbalanced cell envelope biogenesis and/or cell division during the transition between growth phases. The induction of GlpQ and UgpQ further points towards an imbalance in glycine/serine homeostasis in the triple SDP mutant. These enzymes participate in phosphatidylcholine degradation (66, 67) and release choline, which is a precursor of glycine and serine through glycine betaine and sarcosine. Inhibition of this conversion by elevated osmolarity (86) might explain the impact of osmolarity on the growth defect of the ΔΔΔ mutant.
(ii) Like the amino acid homeostasis genes described above, denitrification genes were among the permanently overexpressed genes in the SDP mutant, which were further induced in late exponential phase when the ΔΔΔ strain reached a higher OD than the WT (Fig. S8 and Fig. 12). This induction probably reflects the increased need of the mutant for terminal electron acceptors for respiration, which cannot be satisfied by the available oxygen. A similar response was shown in E. coli when growing at high rates (87).
(iii) The most massively induced genes in the ΔΔΔ strain in the transition phase from exponential to stationary phase were genes for phosphorus acquisition systems. It is possible that the mutant exhausted the available phosphorus from the medium earlier than the WT and experienced starvation. Alternatively, changes in cell envelope biosynthesis might have induced the pho regulon. Interestingly, phosphorus limitation is known to enhance surface attachment and biofilm formation in A. tumefaciens (88, 89). This process is driven by the response regulator PhoB. The induction of the PhoB regulon in the SDP mutant might thus explain the increased biofilm formation.
(iv) With the exception of phosphate and phosphonate uptake systems, many ABC transporters were downregulated in the SDP mutant, in particular, those for sugar transporters. Several of them, for example, the substrate binding proteins FrcB (Atu0063) and ChvE (Atu2348), are known to be under control of the sRNA AbcR1 (90). For most others, neither the substrates nor the regulation is known. Cumulatively, the massive rearrangement of the transcription profile in the SDP mutant shows that the DUF1127 domain plays an important role in bacterial metabolism and nutrient acquisition.
How can short arginine-rich proteins have such a profound effect on gene expression?
One of the most pressing open questions is how the absence of the three SDPs in A. tumefaciens can result in the observed phenotypes and dramatic changes in gene expression. The small proteins composed of only the DUF1127 domain with 47 or 48 aa are unlikely to have any enzymatic activity. They probably act through sequestration of intracellular biomolecules. The positive net charge due to the high arginine content suggests anionic interaction partners, which could be lipids, DNA, RNA, or proteins. The B. abortus DUF1127 proteins were found in the membrane fraction (24). The interacting lipid species and the functional relevance of this localization is not known. Other findings such as the influence on cuckoo RNAs suggest some interaction of DUF1127 proteins with RNAs (34). This raises the question of if and how DUF1127 proteins recognize specific RNA sequences such as the cuckoo motif. Our finding that the amount of the sRNA L5 was increased in the ΔΔΔ mutant at least in the early growth phases might suggest that L5 is responsible for the growth defect and the massively altered transcriptome. This, however, does not seem to be the case, since deletion of the L5 gene did not cause any growth defect (data not shown). Other preliminary data also show that the changes in the transcriptome of an L5 mutant barely overlap the changes in the ΔΔΔ mutant, again indicating that the SDPs do not act through the L5 sRNA. This certainly does not exclude the possibility that the positively charged surfaces of DUF1127 proteins sequester certain RNAs and thereby globally change their activity and/or stability.
Yet another option for a global impact on bacterial physiology would be the interaction of DUF1127 proteins with cellular proteins. The SDPs could, for instance, regulate sugar uptake in a manner similar to that of the 43-aa protein SgrT from E. coli, which modulates the activity of the glucose-phosphotransferase system (91) or the 49-aa AcrZ protein, which modulates the multidrug efflux pump AcrB (92, 93). The best-characterized example of a short protein with a global impact on protein function and gene expression is the multipurpose protein PII (94). It is approximately 110 aa long and one of the most widely distributed signal-transducing proteins in nature. The small trimeric protein binds to intracellular metabolites and numerous proteins, among them, metabolic enzymes, transporters, and transcription factors, and thereby controls carbon and nitrogen metabolism. It is conceivable that the DUF1127 domain is an equally versatile multitasking entity.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Detailed lists of all bacterial strains and plasmids that were used in this study are given in Table S1 and Table S2 in the supplemental material. All used oligonucleotides and a description of how plasmids were constructed are listed in Table S3 and Table S4. The composition of YEB medium was adapted from that described by Vervliet et al. (95); 1 mM MgSO4 was used instead of 2 mM. The LB medium used in this study was composed of 0.5% (wt/vol) yeast extract, 1% (wt/vol) tryptone, and 1% (wt/vol) NaCl (96). AB minimal medium was composed of 0.4% (wt/vol) morpholineethanesulfonic acid (MES), 0.2% (wt/vol) NH4Cl, 2.5 mM MgSO4, 2 mM KCl, 1.3 mM KH2PO4, 90 μM CaCl2, and 16.5 μM FeSO4. The pH was adjusted and the medium was autoclaved. Then, 1% (wt/vol) sucrose was added from a sterile 10% (wt/vol) stock solution. If necessary, kanamycin was added to a final concentration of 50 μg/ml. For solid media, 1.8% (wt/vol) agar was added.
Cultivation of bacteria.
For growth curves, 200 ml YEB medium was inoculated with A. tumefaciens in a 1,000-ml baffled flask to an OD of 0.1 and cells were grown at 30°C and at 180 rpm. To test expression of the DUF1127 genes in A. tumefaciens under different stress conditions, cells were grown to an OD of approximately 0.5, and then 25 ml of the subculture was transferred into a 100-ml flask and incubated under the designated conditions. To test gene expression under virulence-induced conditions, cells were grown in AB medium (pH 5.5) for approximately 8 h. Then, 0.1 mM acetosyringone was added and cells were incubated at 23°C and at 130 rpm for another 16 h.
Cell aggregation assay.
A. tumefaciens was inoculated in 200 ml YEB to an OD of 0.1 and grown at 30°C and 180 rpm in baffled flasks for 24 h. Then, 5 ml of the culture was transferred into a six-well plate and incubated at room temperature (RT) on a tilting laboratory shaker at a speed of approximately 30 turns per min for another 24 h. Photographs were taken at different time points.
Congo red and Fluorescent Brightener 28 plates.
One liter of solid YEB was supplemented with 20 ml of a Congo red solution (0.2% [wt/vol] Congo red and 0.1% [wt/vol] Coomassie brilliant blue G) as described in reference 97 and with 1 ml of a 2% Fluorescent Brightener 28 (alias calcofluor white) solution (in dimethyl sulfoxide [DMSO]). Five microliters of bacterial cultures was spotted onto the plates and incubated at 30°C for 4 days. UV light was used for visualization of Fluorescent Brightener 28-stained bacteria.
Biofilm formation assay.
The strains of interest were grown to an OD of approximately 1.5. All wells of a six-well plate were filled with 3 ml bacterial suspension. Another six-well plate filled with sterile YEB served as a blank control. All plates were incubated overnight at 30°C. Cells from one well were resuspended to determine the relative cell number via the OD. The remaining five wells were supplemented with 300 μl of a 1% (wt/vol) crystal violet solution and incubated at RT for 15 min on a tilting laboratory shaker at 30 turns per min. Then, the supernatant was discarded and the wells were washed very carefully with 5 ml distilled water three times. After that, the plates were dried upside down for 10 to 30 min, and pictures were taken. The remaining stained cells then were resuspended in 3 ml of 30% acetic acid. The plate filled with sterile YEB served as a blank for the photometric determination of relative biofilm formation at an OD of 600 nm. Values were normalized according to the corresponding relative cell number. The WT value was defined as 100%.
Seedling infection assay.
The qualitative infection assay was done as described by Wu et al. (59). Briefly, A. tumefaciens was transformed with the pBISN1 plasmid, which carries a β-glucuronidase gene under the control of a plant-specific promoter between T-DNA borders. After coincubation with 3-day-old A. thaliana seedlings, infected sites were stained with X-Gluc (5-bromo-4-chloro-1H-indol-3-yl-β-d-glucopyranosiduronic acid).
Creation of markerless A. tumefaciens deletion mutants.
A detailed scheme of the workflow was described by Wilms et al. (98). Briefly, for the genomic region to be deleted, one DNA fragment upstream and one fragment downstream were amplified via PCR. A length of approximately 300 bp was chosen. The fragments were ligated to each other and cloned into the pK19mobsacB plasmid. A. tumefaciens was transformed with the deletion plasmid via electroporation. Selection was achieved on solid LB supplemented with 50 μg/ml kanamycin. Due to the lack of a compatible oriV, the plasmid could only persist in the strain by integration into the genome via homologous recombination. Retrieved transformants were inoculated in 5 ml liquid LB containing 5% (wt/vol) sucrose and inoculated at 37°C for 6 to 8 h. Cells then were applied on solid LB supplemented with 10% (wt/vol) sucrose to select for plasmid excision mutants. Finally, kanamycin-sensitive clones were tested via PCR for successful deletion.
Isolation of total RNA from A. tumefaciens and Northern blot analysis.
RNA isolation and Northern blot analysis were performed as described by Wilms et al. (98). The primers that were used for the synthesis of digoxigenin-labeled RNA probes by T7 RNA polymerase-based in vitro transcription are listed in Table S4.
RNA-seq.
A. tumefaciens WT and the triple deletion mutant were grown at 30°C in YEB medium. Samples for RNA extraction were taken at ODs of 0.5 and 1.5 and after 24 h. Library preparation, sequencing, and statistical evaluation were done by Novogene Co., Ltd. Sequencing was performed on an Illumina NovaSeq 6000 platform.
Preparation and separation of protein samples via SDS-PAGE.
Depending on the size of the protein to be tested, SDS-PAGE was performed with either 12% (wt/vol) polyacrylamide (PAA) gels according to the protocol by Laemmli (99) or 16.5% (wt/vol) PAA gels according to Schägger (100). The protein-sample buffer (pH 6.8) was composed of 50 mM Tris, 10% (vol/vol) glycerol, 2% (wt/vol) SDS, 1% (vol/vol) β-mercaptoethanol, and 0.1% (wt/vol) bromophenol blue.
Visualization of proteins by SDS-PAGE and Western blot analysis.
For visualization of unspecific proteins, gels were stained with 0.2% (wt/vol) Coomassie R-250 in 50% (vol/vol) methanol and 10% (vol/vol) acetic acid for at least 30 min. Then, gels were washed with destaining solution (45% [vol/vol] methanol, 10% [vol/vol] acetic acid) until the colorization of the protein bands reached the desired intensity. For the detection of specific proteins, SDS-PAGE was followed by Western blotting and immunodetection according to standard protocols (101, 102). Amersham Protran 0.1-μm NC membrane with a pore size of 0.1 μm was used due the small size of some proteins that were blotted. For the immunodetection of 3×FLAG-tagged proteins, an anti-FLAG primary antibody (retrieved from mouse) from Sigma-Aldrich Inc. was used at a dilution of 1:4,000 as well as an anti-mouse antibody–horseradish peroxidase (HRP) conjugate from Biozym Scientific GmbH at a dilution of 1:4,000.
Mass spectrometry sample preparation and measurements.
Sample preparation and analysis by mass spectrometry were performed as described by Cormann et al. with minor changes (103). Protein samples were applied on a polyacrylamide gel and Coomassie stained as described above. Single bands were excised and destained in 25 mM NH4HCO3 and 50% (vol/vol) acetonitrile. The pieces were dried in acetonitrile, followed by an in-gel tryptic digest (0.125 μg trypsin in 25 mM NH4HCO3) overnight. Peptides were eluted from the gel by addition of 1% (vol/vol) formic acid and 50% (vol/vol) acetonitrile. After complete drying of the solution in a vacuum concentrator, samples were resuspended in solution A (0.1% [vol/vol] formic acid, 2% [vol/vol] acetonitrile in water). Peptides were purified with a nanoACQUITY UPLC Symmetry C18 Trap column from Waters GmbH (100-Å pore size, 5-μm particle size, 180 μm in diameter, 20 mm in length). The tryptic peptides were eluted from an ACQUITY UPLC BEH C18 column from Waters GmbH (130-Å pore size, 1.7-μm particle size, 75 μm in diameter, 150 mm in length) at a flow rate of 0.4 μl/min at 45°C in a discontinuous gradient of solution A to solution B (0.1% [vol/vol] formic acid in acetonitrile) over 60 min (2% for 5 min, 5% for 5 min, 30% for 31 min, 85% for 5 min, 95% for 1 min, 2% for 13 min). The ultraperformance liquid chromatography (UPLC) systems were coupled to the mass spectrometer via a SilicaTip emitter (30 μm) from New Objective Inc. Mass spectra were recorded at a range of 300 to 2,000 m/z with a resolution of 240,000. Singly charged ions and ions with an unassigned charge state were rejected from tandem mass spectrometry (MS/MS) spectra. Data analysis was performed as described by Rexroth et al. (104). The corresponding database consisted of all annotated proteins of A. tumefaciens C58 from NCBI to which L4 and common contaminants as human keratin (105) were appended and the C terminus of Atu1667 was corrected. A false-discovery rate of 1% was applied.
Statistical evaluation via t test.
To determine significant differences between two sets of data, an independent two-sample t test was used. Since this kind of test premises a t distribution of the data, an Anderson-Darling test (106) was applied to ensure that a t distribution could not be excluded. Since, for all compared data sets, different variances could be expected, Welch’s t test was applied (107). In all cases, two-tailed tests with an alpha level of 5% were chosen.
In silico prediction of DUF1127 proteins.
To find undiscovered DUF1127 genes in the genome of A. tumefaciens, a de novo search for putative ORFs was performed covering all four replicons. Each DNA sequence between two stop codons whose length could be divided by three was defined as a putative ORF. The applied length cutoff was equal to the shortest annotated DUF1127 protein-encoding ORF (22 codons). Each newly extracted ORF then was translated into a protein sequence and searched via BLAST against the InterPro-DP database. Further criteria were a maximum E value of 10E−20 and a minimum identity of 75% to at least one annotated DP. Sequences with positive matches were checked against annotated genes of A. tumefaciens by comparing the position of their stop codons. Finally, N termini were adjusted to NCBI annotations if available.
Subdividing DUF1127 proteins.
All DUF1127 proteins from the InterPro-DP database were divided into three subclasses according to sequence similarity. The first group was composed of proteins with a high similarity to L4, Atu1667, or Atu8161 from A. tumefaciens. The second group showed a strong homology to YjiS from E. coli MG1655. The final group contained all remaining proteins that could not be assigned to the previous two groups due to the chosen cutoff. Protein homology was determined using the pairwise2.align.globalds function of Biopython (108). Since a close homology could be expected within this protein family, a PAM120 matrix was chosen. Each gap in the alignment received a penalty of −10, and each gap extend was penalized with −0.5. To assign a protein to a specific group, a minimum score of 75 was premised.
Data availability.
RNA-seq data are available at the Gene Expression Omnibus (GEO) database under accession number GSE150941.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to F.N. and M.M.N. (NA 240/13-1 and NO 836/4-1, respectively) in the context of the priority program SPP2002.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD. 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mudgal R, Sandhya S, Chandra N, Srinivasan N. 2015. De-DUFing the DUFs: deciphering distant evolutionary relationships of domains of unknown function using sensitive homology detection methods. Biol Direct 10:38. doi: 10.1186/s13062-015-0069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodacre NF, Gerloff DL, Uetz P. 2013. Protein domains of unknown function are essential in bacteria. mBio 5:e00744-13. doi: 10.1128/mBio.00744-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orr MW, Mao Y, Storz G, Qian SB. 2020. Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res 48:1029–1042. doi: 10.1093/nar/gkz734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiessen A, Pérez-Rodríguez P, Delaye-Arredondo L. 2012. Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes. BMC Res Notes 5:85. doi: 10.1186/1756-0500-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storz G, Wolf YI, Ramamurthi KS. 2014. Small proteins can no longer be ignored. Annu Rev Biochem 83:753–777. doi: 10.1146/annurev-biochem-070611-102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stothard P, Wishart DS. 2006. Automated bacterial genome analysis and annotation. Curr Opin Microbiol 9:505–510. doi: 10.1016/j.mib.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Harrison PM, Kumar A, Lang N, Snyder M, Gerstein M. 2002. A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res 30:1083–1090. doi: 10.1093/nar/30.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garbis S, Lubec G, Fountoulakis M. 2005. Limitations of current proteomics technologies. J Chromatogr A 1077:1–18. doi: 10.1016/j.chroma.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 10.Hemm MR, Paul BJ, Miranda-Ríos J, Zhang A, Soltanzad N, Storz G. 2010. Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J Bacteriol 192:46–58. doi: 10.1128/JB.00872-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews SJ, Rothnagel JA. 2014. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet 15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- 12.Saghatelian A, Couso JP. 2015. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat Chem Biol 11:909–916. doi: 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khitun A, Ness TJ, Slavoff SA. 2019. Small open reading frames and cellular stress responses. Mol Omics 15:108–116. doi: 10.1039/c8mo00283e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanOrsdel CE, Kelly JP, Burke BN, Lein CD, Oufiero CE, Sanchez JF, Wimmers LE, Hearn DJ, Abuikhdair FJ, Barnhart KR, Duley ML, Ernst SEG, Kenerson BA, Serafin AJ, Hemm MR. 2018. Identifying new small proteins in Escherichia coli. Proteomics 18:e1700064. doi: 10.1002/pmic.201700064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver J, Mohammad F, Buskirk AR, Storz G. 2019. Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio 10:e02819-18. doi: 10.1128/mBio.02819-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, Gough J, Haft DR, Huang H, Letunic I, Lopez R, Luciani A, Madeira F, Marchler-Bauer A, Mi H, Natale DA, Necci M, Nuka G, Orengo C, Pandurangan AP, Paysan-Lafosse T, Pesseat S, Potter SC, Qureshi MA, Rawlings ND, Redaschi N, Richardson LJ, Rivoire C, Salazar GA, Sangrador-Vegas A, Sigrist CJA, Sillitoe I, Sutton GG, Thanki N, Thomas PD, Tosatto SCE, Yong SY, Finn RD. 2019. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47:D351–D360. doi: 10.1093/nar/gky1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2013. GenBank. Nucleic Acids Res 41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sibley MH, Raleigh EA. 2004. Cassette-like variation of restriction enzyme genes in Escherichia coli C and relatives. Nucleic Acids Res 32:522–534. doi: 10.1093/nar/gkh194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rollenhagen C, Sorensen M, Rizos K, Hurvitz R, Bumann D. 2004. Antigen selection based on expression levels during infection facilitates vaccine development for an intracellular pathogen. Proc Natl Acad Sci U S A 101:8739–8744. doi: 10.1073/pnas.0401283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kröger C, Colgan A, Srikumar S, Händler K, Sivasankaran SK, Hammarlöf DL, Canals R, Grissom JE, Conway T, Hokamp K, Hinton JCD. 2013. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 14:683–695. doi: 10.1016/j.chom.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Baek J, Lee J, Yoon K, Lee H. 2017. Identification of unannotated small genes in Salmonella. G3 (Bethesda) 7:983–989. doi: 10.1534/g3.116.036939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheehan LM, Budnick JA, Blanchard C, Dunman PM, Caswell CC. 2015. A LysR-family transcriptional regulator required for virulence in Brucella abortus is highly conserved among the α-proteobacteria. Mol Microbiol 98:318–328. doi: 10.1111/mmi.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sternon JF, Godessart P, de Freitas RG, Van der Henst M, Poncin K, Francis N, Willemart K, Christen M, Christen B, Letesson JJ, De Bolle X. 2018. Transposon sequencing of Brucella abortus uncovers essential genes for growth in vitro and inside macrophages. Infect Immun 86:e00312-18. doi: 10.1128/IAI.00312-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budnick JA, Sheehan LM, Kang L, Michalak P, Caswell CC. 2018. Characterization of three small proteins in Brucella abortus linked to fucose utilization. J Bacteriol 200:e00127-18. doi: 10.1128/JB.00127-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo L, Yao SY, Becker A, Rüberg S, Yu GQ, Zhu JB, Cheng HP. 2005. Two new Sinorhizobium meliloti LysR-type transcriptional regulators required for nodulation. J Bacteriol 187:4562–4572. doi: 10.1128/JB.187.13.4562-4572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang G, Li Q, Xing S, Li N, Tang Z, Yu L, Yan J, Li X, Luo L. 2018. The LsrB protein is required for Agrobacterium tumefaciens interaction with host plants. Mol Plant Microbe Interact 31:951–961. doi: 10.1094/MPMI-02-18-0041-R. [DOI] [PubMed] [Google Scholar]
- 27.Reinkensmeier J, Giegerich R. 2015. Thermodynamic matchers for the construction of the cuckoo RNA family. RNA Biol 12:197–207. doi: 10.1080/15476286.2015.1017206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.del Val C, Romero-Zaliz R, Torres-Quesada O, Peregrina A, Toro N, Jiménez-Zurdo JI. 2012. A survey of sRNA families in α-proteobacteria. RNA Biol 9:119–129. doi: 10.4161/rna.18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melançon P, Leclerc D, Destroismaisons N, Brakier-Gingras L. 1990. The anti-Shine-Dalgarno region in Escherichia coli 16S ribosomal RNA is not essential for the correct selection of translational starts. Biochemistry 29:3402–3407. doi: 10.1021/bi00465a037. [DOI] [PubMed] [Google Scholar]
- 30.Capela D, Barloy-Hubler F, Gouzy J, Bothe G, Ampe F, Batut J, Boistard P, Becker A, Boutry M, Cadieu E, Dreano S, Gloux S, Godrie T, Goffeau A, Kahn D, Kiss E, Lelaure V, Masuy D, Pohl T, Portetelle D, Puhler A, Purnelle B, Ramsperger U, Renard C, Thebault P, Vandenbol M, Weidner S, Galibert F. 2001. Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc Natl Acad Sci U S A 98:9877–9882. doi: 10.1073/pnas.161294398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galibert F, Finan TM, Long SR, Pühler A, Abola P, Ampe F, Barloy-Hubler F, Barnet MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dréano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thébault P, Vandenbol M, et al. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- 32.Reeve W, O'Hara G, Chain P, Ardley J, Bräu L, Nandesena K, Tiwari R, Malfatti S, Kiss H, Lapidus A, Copeland A, Nolan M, Land M, Ivanova N, Mavromatis K, Markowitz V, Kyrpides N, Melino V, Denton M, Yates R, Howieson J. 2010. Complete genome sequence of Rhizobium leguminosarum bv trifolii strain WSM2304, an effective microsymbiont of the South American clover Trifolium polymorphum. Stand Genomic Sci 2:66–76. doi: 10.4056/sigs.44642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crasta OR, Folkerts O, Fei Z, Mane SP, Evans C, Martino-Catt S, Bricker B, Yu GX, Du L, Sobral BW. 2008. Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One 3:e2193. doi: 10.1371/journal.pone.0002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Billenkamp F, Peng T, Berghoff BA, Klug G. 2015. A cluster of four homologous small RNAs modulates C1 metabolism and the pyruvate dehydrogenase complex in Rhodobacter sphaeroides under various stress conditions. J Bacteriol 197:1839–1852. doi: 10.1128/JB.02475-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berghoff BA, Glaeser J, Sharma CM, Vogel J, Klug G. 2009. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol Microbiol 74:1497–1512. doi: 10.1111/j.1365-2958.2009.06949.x. [DOI] [PubMed] [Google Scholar]
- 36.Nuss AM, Glaeser J, Berghoff BA, Klug G. 2010. Overlapping alternative sigma factor regulons in the response to singlet oxygen in Rhodobacter sphaeroides. J Bacteriol 192:2613–2623. doi: 10.1128/JB.01605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilms I, Overlöper A, Nowrousian M, Sharma CM, Narberhaus F. 2012. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 9:446–457. doi: 10.4161/rna.17212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee K, Huang X, Yang C, Lee D, Ho V, Nobuta K, Fan JB, Wang K. 2013. A genome-wide survey of highly expressed non-coding RNAs and biological validation of selected candidates in Agrobacterium tumefaciens. PLoS One 8:e70720. doi: 10.1371/journal.pone.0070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Cleene M, De Ley J. 1976. The host range of crown gall. Bot Rev 42:389–466. doi: 10.1007/BF02860827. [DOI] [Google Scholar]
- 40.De Cleene M. 1985. The susceptibility of monocotyledons to Agrobacterium tumefaciens. J Phytopathol 113:81–89. doi: 10.1111/j.1439-0434.1985.tb00829.x. [DOI] [Google Scholar]
- 41.Clough SJ, Bent AF. 1998. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 42.Gelvin SB. 2003. Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 67:16–37. doi: 10.1128/mmbr.67.1.16-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida J, Woo L, Chen Y, Paulsen IT, Eisen JA, Karp PD, Bovee DS, Chapman P, Clendenning J, Deatherage G, Gillet W, Grant C, Kutyavin T, Levy R, Li MJ, McClelland E, Palmieri A, Raymond C, Rouse G, Saenphimmachak C, Wu Z, Romero P, Gordon D, Zhang S, Yoo H, Tao Y, Biddle P, Jung M, Krespan W, Perry M, Gordon-Kamm B, Liao L, Kim S, Hendrick C, Zhao ZY, Dolan M, Chumley F, Tingey SV, Tomb JF, Gordon MP, Olson MV, Nester EW. 2001. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294:2317–2323. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- 45.Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, Mullin L, Houmiel K, Gordon J, Vaudin M, Iartchouk O, Epp A, Liu F, Wollam C, Allinger M, Doughty D, Scott C, Lappas C, Markelz B, Flanagan C, Crowell C, Gurson J, Lomo C, Sear C, Strub G, Cielo C, Slater S. 2001. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science 294:2323–2328. doi: 10.1126/science.1066803. [DOI] [PubMed] [Google Scholar]
- 46.Sayers EW, Beck J, Brister JR, Bolton EE, Canese K, Comeau DC, Funk K, Ketter A, Kim S, Kimchi A, Kitts PA, Kuznetsov A, Lathrop S, Lu Z, McGarvey K, Madden TL, Murphy TD, O'Leary N, Phan L, Schneider VA, Thibaud-Nissen F, Trawick BW, Pruitt KD, Ostell J. 2020. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 48:D9–D16. doi: 10.1093/nar/gkz899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bairoch A. 2013. Release notes for UniProtKB/Swiss-Prot release 2013_04. https://www.uniprot.org/statistics/Swiss-Prot%202013_04.
- 48.Barnett MJ, Bittner AN, Toman CJ, Oke V, Long SR. 2012. Dual RpoH sigma factors and transcriptional plasticity in a symbiotic bacterium. J Bacteriol 194:4983–4994. doi: 10.1128/JB.00449-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maddocks SE, Oyston PCF. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 50.Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng 12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 51.Parsek MR, Ye RW, Pun P, Chakrabarty AM. 1994. Critical nucleotides in the interaction of a LysR-type regulator with its target promoter region. catBC promoter activation by CatR. J Biol Chem 269:11279–11284. [PubMed] [Google Scholar]
- 52.Matthysse AG, Holmes KV, Gurlitz RHG. 1981. Elaboration of cellulose fibrils by Agrobacterium tumefaciens during attachment to carrot cells. J Bacteriol 145:583–595. doi: 10.1128/JB.145.1.583-595.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tomlinson AD, Fuqua C. 2009. Mechanisms and regulation of polar surface attachment in Agrobacterium tumefaciens. Curr Opin Microbiol 12:708–714. doi: 10.1016/j.mib.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Kim J, Koestler BJ, Choi JH, Waters CM, Fuqua C. 2013. Genetic analysis of Agrobacterium tumefaciens unipolar polysaccharide production reveals complex integrated control of the motile-to-sessile switch. Mol Microbiol 89:929–948. doi: 10.1111/mmi.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freeman DJ, Falkiner FR, Keane CT. 1989. New method for detecting slime production by coagulase negative staphylococci. J Clin Pathol 42:872–874. doi: 10.1136/jcp.42.8.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomlinson AD, Ramey-Hartung B, Day TW, Merritt PM, Fuqua C. 2010. Agrobacterium tumefaciens ExoR represses succinoglycan biosynthesis and is required for biofilm formation and motility. Microbiology 156:2670–2681. doi: 10.1099/mic.0.039032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haldane DJM, Robart E. 1990. A comparison of calcofluor white, potassium hydroxide, and culture for the laboratory diagnosis of superficial fungal infection. Diagn Microbiol Infect Dis 13:337–339. doi: 10.1016/0732-8893(90)90027-S. [DOI] [PubMed] [Google Scholar]
- 58.Douglas CJ, Halperin W, Nester EW. 1982. Agrobacterium tumefaciens mutants affected in attachment to plant cells. J Bacteriol 152:1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu HY, Liu KH, Wang YC, Wu JF, Chiu WL, Chen CY, Wu SH, Sheen J, Lai EM. 2014. AGROBEST: an efficient Agrobacterium-mediated transient expression method for versatile gene function analyses in Arabidopsis seedlings. Plant Methods 10:19. doi: 10.1186/1746-4811-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan ZC, Zaheer R, Morton R, Finan TM. 2006. Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res 34:2686–2697. doi: 10.1093/nar/gkl365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chlumsky LJ, Zhang L, Ramsey AJ, Jorns MS. 1993. Preparation and properties of recombinant corynebacterial sarcosine oxidase: evidence for posttranslational modification during turnover with sarcosine. Biochemistry 32:11132–11142. doi: 10.1021/bi00092a024. [DOI] [PubMed] [Google Scholar]
- 62.Plamann MD, Stauffer GV. 1983. Characterization of the Escherichia coli gene for serine hydroxymethyltransferase. Gene 22:9–18. doi: 10.1016/0378-1119(83)90059-8. [DOI] [PubMed] [Google Scholar]
- 63.Nagy PL, Marolewski A, Benkovic SJ, Zalkin H. 1995. Formyltetrahydrofolate hydrolase, a regulatory enzyme that functions to balance pools of tetrahydrofolate and one-carbon tetrahydrofolate adducts in Escherichia coli. J Bacteriol 177:1292–1298. doi: 10.1128/jb.177.5.1292-1298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kvalnes-Krick K, Jorns MS. 1987. Interaction of tetrahydrofolate and other folate derivatives with bacterial sarcosine oxidase. Biochemistry 26:7391–7395. doi: 10.1021/bi00397a029. [DOI] [PubMed] [Google Scholar]
- 65.Schweizer H, Grussenmeyer T, Boos W. 1982. Mapping of two ugp genes coding for the pho regulon-dependent sn-glycerol-3-phosphate transport system of Escherichia coli. J Bacteriol 150:1164–1171. doi: 10.1128/JB.150.3.1164-1171.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Larson TJ, Ehrmann M, Boos W. 1983. Periplasmic glycerophosphodiester phosphodiesterase of Escherichia coli, a new enzyme of the glp regulon. J Biol Chem 258:5428–5432. [PubMed] [Google Scholar]
- 67.Tommassen J, Eiglmeier K, Cole ST, Overduin P, Larson TJ, Boos W. 1991. Characterization of two genes, glpQ and ugpQ, encoding glycerophosphoryl diester phosphodiesterases of Escherichia coli. Mol Gen Genet 226:321–327. doi: 10.1007/BF00273621. [DOI] [PubMed] [Google Scholar]
- 68.Riekhof WR, Andre C, Benning C. 2005. Two enzymes, BtaA and BtaB, are sufficient for betaine lipid biosynthesis in bacteria. Arch Biochem Biophys 441:96–105. doi: 10.1016/j.abb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Makino K, Shinagawa H, Amemura M, Kimura S, Nakata A, Ishihama A. 1988. Regulation of the phosphate regulon of Escherichia coli. Activation of pstS transcription by PhoB protein in vitro. J Mol Biol 203:85–95. doi: 10.1016/0022-2836(88)90093-9. [DOI] [PubMed] [Google Scholar]
- 70.Lipman DJ, Souvorov A, Koonin EV, Panchenko AR, Tatusova TA. 2002. The relationship of protein conservation and sequence length. BMC Evol Biol 2:20. doi: 10.1186/1471-2148-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. 2008. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol 70:1487–1501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rudd KE, Humphery-Smith I, Wasinger VC, Bairoch A. 1998. Low molecular weight proteins: a challenge for post-genomic research. Electrophoresis 19:536–544. doi: 10.1002/elps.1150190413. [DOI] [PubMed] [Google Scholar]
- 73.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nielsen P, Krogh A. 2005. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics 21:4322–4329. doi: 10.1093/bioinformatics/bti701. [DOI] [PubMed] [Google Scholar]
- 75.Yeasmin F, Yada T, Akimitsu N. 2018. Micropeptides encoded in transcripts previously identified as long noncoding RNAs: a new chapter in transcriptomics and proteomics. Front Genet 9:144. doi: 10.3389/fgene.2018.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pang Y, Mao C, Liu S. 2018. Encoding activities of non-coding RNAs. Theranostics 8:2496–2507. doi: 10.7150/thno.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakahigashi K, Ron EZ, Yanagi H, Yura T. 1999. Differential and independent roles of σ32 homolog (RpoH) and an HrcA repressor in the heat shock response of Agrobacterium tumefaciens. J Bacteriol 181:7509–7515. doi: 10.1128/JB.181.24.7509-7515.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perez-Rueda E. 2000. The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res 28:1838–1847. doi: 10.1093/nar/28.8.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schell M. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol 47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 80.Stauffer GV. 2004. Regulation of serine, glycine, and one-carbon biosynthesis. EcoSal Plus 1:3.6.1.2. doi: 10.1128/ecosalplus.3.6.1.2. [DOI] [PubMed] [Google Scholar]
- 81.Kriner MA, Subramaniam AR. 2020. The serine transporter SdaC prevents cell lysis upon glucose depletion in Escherichia coli. Microbiologyopen 9:e960. doi: 10.1002/mbo3.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hama H, Sumita Y, Kakutani Y, Tsuda M, Tsuchiya T. 1990. Target of serine inhibition in Escherichia coli. Biochem Biophys Res Commun 168:1211–1216. doi: 10.1016/0006-291x(90)91157-n. [DOI] [PubMed] [Google Scholar]
- 83.Tazuya-Murayama K, Aramaki H, Mishima M, Saito K, Ishida S, Yamada K. 2006. Effect of l-serine on the biosynthesis of aromatic amino acids in Escherichia coli. J Nutr Sci Vitaminol (Tokyo) 52:256–260. doi: 10.3177/jnsv.52.256. [DOI] [PubMed] [Google Scholar]
- 84.Zhang X, El-Hajj ZW, Newman E. 2010. Deficiency in L-serine deaminase interferes with one-carbon metabolism and cell wall synthesis in Escherichia coli K-12. J Bacteriol 192:5515–5525. doi: 10.1128/JB.00748-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nyström T. 2004. Stationary-phase physiology. Annu Rev Microbiol 58:161–181. doi: 10.1146/annurev.micro.58.030603.123818. [DOI] [PubMed] [Google Scholar]
- 86.Smith LT, Pocard JA, Bernard T, Le Rudulier D. 1988. Osmotic control of glycine betaine biosynthesis and degradation in Rhizobium meliloti. J Bacteriol 170:3142–3149. doi: 10.1128/jb.170.7.3142-3149.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Szenk M, Dill KA, de Graff AMR. 2017. Why do fast-growing bacteria enter overflow metabolism? Testing the membrane real estate hypothesis. Cell Syst 5:95–104. doi: 10.1016/j.cels.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 88.Danhorn T, Hentzer M, Givskov M, Parsek MR, Fuqua C. 2004. Phosphorus limitation enhances biofilm formation of the plant pathogen Agrobacterium tumefaciens through the PhoR-PhoB regulatory system. J Bacteriol 186:4492–4501. doi: 10.1128/JB.186.14.4492-4501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu J, Kim J, Danhorn T, Merritt PM, Fuqua C. 2012. Phosphorus limitation increases attachment in Agrobacterium tumefaciens and reveals a conditional functional redundancy in adhesin biosynthesis. Res Microbiol 163:674–684. doi: 10.1016/j.resmic.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Overlöper A, Kraus A, Gurski R, Wright PR, Georg J, Hess WR, Narberhaus F. 2014. Two separate modules of the conserved regulatory RNA AbcR1 address multiple target mRNAs in and outside of the translation initiation region. RNA Biol 11:624–640. doi: 10.4161/rna.29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lloyd CR, Park S, Fei J, Vanderpool CK. 2017. The small protein SgrT controls transport activity of the glucose-specific phosphotransferase system. J Bacteriol 199:e00869-16. doi: 10.1128/JB.00869-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hobbs EC, Yin X, Paul BJ, Astarita JL, Storz G. 2012. Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proc Natl Acad Sci U S A 109:16696–16701. doi: 10.1073/pnas.1210093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Du D, Neuberger A, Orr MW, Newman CE, Hsu P-C, Samsudin F, Szewczak-Harris A, Ramos LM, Debela M, Khalid S, Storz G, Luisi BF. 2020. Interactions of a bacterial RND transporter with a transmembrane small protein in a lipid environment. Structure 28:625.e6–634.e6. doi: 10.1016/j.str.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Forchhammer K, Selim KA. 2020. Carbon/nitrogen homeostasis control in cyanobacteria. FEMS Microbiol Rev 44:33–53. doi: 10.1093/femsre/fuz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vervliet G, Holsters M, Teuchy H, Van Montagu M, Schell J. 1975. Characterization of different plaque forming and defective temperate phages in Agrobacterium strains. J Gen Virol 26:33–48. doi: 10.1099/0022-1317-26-1-33. [DOI] [PubMed] [Google Scholar]
- 96.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. doi: 10.1128/JB.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Römling U, Sierralta WD, Eriksson K, Normark S. 1998. Multicellular and aggregative behaviour of Salmonella typhimurium strains is controlled by mutations in the agfD promoter. Mol Microbiol 28:249–264. doi: 10.1046/j.1365-2958.1998.00791.x. [DOI] [PubMed] [Google Scholar]
- 98.Wilms I, Möller P, Stock AM, Gurski R, Lai EM, Narberhaus F. 2012. Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens. J Bacteriol 194:5209–5217. doi: 10.1128/JB.00510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 100.Schägger H. 2006. Tricine-SDS-PAGE. Nat Protoc 1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 101.Renart J, Reiser J, Stark GR. 1979. Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: a method for studying antibody specificity and antigen structure. Proc Natl Acad Sci U S A 76:3116–3120. doi: 10.1073/pnas.76.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goldman A, Ursitti JA, Mozdzanowski J, Speicher DW. 2015. Electroblotting from polyacrylamide gels. Curr Protoc Protein Sci 82:10.7.1–10.7.16. doi: 10.1002/0471140864.ps1007s82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cormann KU, Möller M, Nowaczyk MM. 2016. Critical assessment of protein cross-linking and molecular docking: an updated model for the interaction between photosystem II and Psb27. Front Plant Sci 7:157. doi: 10.3389/fpls.2016.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rexroth S, Rexroth D, Veit S, Plohnke N, Cormann KU, Nowaczyk MM, Rögner M. 2014. Functional characterization of the small regulatory subunit PetP from the cytochrome b6f complex in Thermosynechococcus elongatus. Plant Cell 26:3435–3448. doi: 10.1105/tpc.114.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Spät P, Klotz A, Rexroth S, Maček B, Forchhammer K. 2018. Chlorosis as a developmental program in cyanobacteria: the proteomic fundament for survival and awakening. Mol Cell Proteomics 17:1650–1669. doi: 10.1074/mcp.RA118.000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Anderson TW, Darling DA. 1954. A test of goodness of fit. J Am Stat Assoc 49:765–769. doi: 10.1080/01621459.1954.10501232. [DOI] [Google Scholar]
- 107.Welch BL. 1947. The generalisation of student’s problems when several different population variances are involved. Biometrika 34:28–35. doi: 10.2307/2332510. [DOI] [PubMed] [Google Scholar]
- 108.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, De Hoon MJL. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are available at the Gene Expression Omnibus (GEO) database under accession number GSE150941.