

Abstract
Objectives:
To establish a straightforward single-cell passaging cultivation method that enables high-quality maintenance of human induced pluripotent stem cells without the appearance of karyotypic abnormalities or loss of pluripotency.
Methods:
Cells were kept in culture for over 50 passages, following a structured chronogram of passage and monitoring cell growth by population doubling time calculation and cell confluence. Standard procedures for human induced pluripotent stem cells monitoring as embryonic body formation, karyotyping and pluripotency markers expression were evaluated in order to assess the cellular state in long-term culture. Cells that underwent these tests were then subjected to differentiation into keratinocytes, cardiomyocytes and definitive endoderm to evaluate its differentiation capacity.
Results:
Human induced pluripotent stem cells clones maintained its pluripotent capability as well as chromosomal integrity and were able to generate derivatives from the three germ layers at high passages by embryoid body formation and high-efficient direct differentiation into keratinocytes, cardiomyocytes and definitive endoderm.
Conclusions:
Our findings support the routine of human induced pluripotent stem cells single-cell passaging as a reliable procedure even after long-term cultivation, providing healthy human induced pluripotent stem cells to be used in drug discovery, toxicity, and disease modeling as well as for therapeutic approaches.
Keywords: Human induced pluripotent stem cells, human induced pluripotent stem cells maintenance, human induced pluripotent stem cells differentiation
Introduction
Human induced pluripotent stem cells (hiPSC) unique features bring excitement among scientific and medical communities as an alternative to human embryonic stem cells (hESCs) for research on tissue-specific development, disease modeling, drugs discovery and cell-based therapies. Since iPSCs were reported,1 culture methods have evolved to optimize growth conditions while maintaining pluripotency in order to meet both the special needs of the cells and their rapid demand. However, many laboratories still cultivate iPSCs following out-of-date procedures that remain from the discovery of hESC such as colony passaging,2 which is an approach that usually follows uneven confluency as well as unpredictable growth rates.2,3
To overcome colony passaging–related issues, several groups committed to search for conditions that could support single-cell passaging,4–8 but despite the rapid progress, there is still a lack of standard protocols for iPSC cultivation that take into account differences in genetic background between different cell lines and the effects of long-term culture. Several recent reports present lack of information concerning iPSC genetic integrity,9 especially after long-term cultivation in vitro,10 even though other studies suggest abnormalities to be progressively favored by suboptimal culture conditions such as single-cell passaging11 or high-cell density cultures.12
It is common for laboratories to maintain multiple iPSC cell lineages in culture to avoid the inconsistencies of long-term cultivation; however, it is important to recognize that this approach implies high cost and labor demand and also does not guarantee success for differentiation protocols because working with different clones impairs reproducibility, homogeneity and scalability in differentiations.13 Nevertheless, as long-term maintenance in culture seems likely to promote self-renewal10,14 and limit differentiation through progressive selection of genetic variants,15 the assessment and validation of iPSC cultivation protocols is mandatory to support reproducibility in differentiations.
In this work, we used hiPSC derived from two distinct primary sources to develop a controlled long-term culture methodology that supports single-cell passaging while maintaining pluripotency markers, genomic integrity and ability to generate derivatives by directed differentiation, resulting in high-purity specialized cells.
Methods
Ethics statement
This investigation agrees with the principles outlined in the Declaration of Helsinki and the study protocol was approved by the Ethics Committee for Medical Research on Human Beings of the Institute of Biomedical Sciences from the University of Sao Paulo (#2.009.562). Signed informed consent was obtained from all participants.
hiPSC reprogramming and maintenance
Human erythroblasts and skin fibroblast were reprogrammed for hiPSC generation. Erythroblasts were genetically modified using an episomal vector system previously described in the literature16,17 with slight modifications. Briefly, mononuclear cells (MNCs) were isolated from 10 mL of peripheral blood by Ficoll gradient. Erythroblasts were cultured in a serum-free MNC medium containing the following cytokines diluted in Stem Span Serum Free Expansion Medium (Stemcell Technologies, Canada); 40 ng/mL insulin-like growth factor 1 (IGF1); 100 ng/mL stem cell factor (SCF); 10 ng/mL interleukin 3 (IL-3) and 2 U/mL erythropoietin (EPO). Cells were transfected with plasmids pEB-C5 and pEB-Tg (Addgene, USA) containing reprogramming factors OCT4, SOX2, KLF4, cMYC, LIN28 and SV40T, using the Human CD34 + nucleofector kit and the Nucleofector II device (both from Lonza Group Ltd, Switzerland) following manufacturer’s instructions. Reprogrammed erythroblasts were incubated in mouse embryonic fibroblast (MEF)-coated plates in MEF medium and fetal bovine serum (FBS) ES-Cell Qualified (ESQ; Thermo Fisher, USA) with 20 ng/mL basic fibroblast growth factor (bFGF) overnight. Then, they were transferred into embryonic stem cell (ESC) medium containing Knockout DMEM, Knockout Serum replacement, antibiotic-antimycotic, 200 mM Glutamax, MEM non-essential amino acid solution and 2-mercaptoethanol (all from Thermo Fisher, USA) supplemented with 20 ng/mL bFGF and 0.25 mM sodium butyrate (Sigma Aldrich, EUA). hiPSC colonies were collected from a 6-well MEF-coated plate using Gentle Cell Dissociation Reagent (Stemcell Technologies, Canada) and seeded into Matrigel (BD, USA) coated plates with Essential 8™ medium (E8; Thermo Fisher, USA) supplemented with 10 μM ROCK inhibitor Y-27632 (Stemgent Inc., EUA).
hiPSC lines were derived from fibroblast following Epi5™ Episomal iPSC Reprogramming Kit protocol (Invitrogen) with some modifications. Briefly, fibroblasts were isolated from skin biopsy of a healthy donor by manually processing into smaller pieces using a scalpel. Fibroblasts were cultivated in DMEM High Glucose medium supplemented with 15% bovine fetal serum, 200 mM Glutamax, 100 U/mL penicillin and 100 µg/mL streptomycin (all from Thermo Fisher, USA). Fibroblasts were reprogrammed at third passage and 2.7 × 104 cells were plated in GELTREX™ matrix coated wells from a 12-well plate. The next day, Epi5 Episomal iPSC Reprogramming Kit factors were added in the cell using OptiMEM medium and Lipofectamine 3000 reagents (all from Thermo Fisher, USA). Reprogrammed cells were maintained with Essential 6TM medium (Thermo Fisher, USA) supplemented with 100 ng/mL bFGF (R&D Systems, EUA) and 100 µM of sodium butyrate (Sigma Aldrich, EUA) for 14 days, then the medium was switched to E8 medium and 100 µM of sodium butyrate for more 14 days. From the 14th to 28th day after reprogramming, small colonies emerged and were manually picked and seeded individually into new GELTREX matrix coated wells. After the 28th day, cells were maintained with E8 medium.
Blood and skin samples were collected from four healthy donors ranging in age from 30 to 40 years old: two males (ACP, PC3) and two females (PC2, PC4). From these samples four cell lines were generated, one derived from erythroblasts (ACP) and three derived from skin fibroblasts (PC2, PC3, PC4). After clonal picking and expansion, one clone of ACP (ACP5) and three clones of the lines PC2 (PC2.2, PC2.3, PC2.4), PC3 (PC3.1, PC3.2, PC3.3) and PC4 (PC4.3, PC4.5, PC4.6) were selected. Due to their growth rates and because they were obtained by using different sets of plasmids for reprogramming, thus allowing more interesting comparisons, only ACP5 and PC4 clones (PC4.3, PC4.5, PC4.6) were used for the next steps of long-term cultivation, characterization and differentiation.
From passage 5 and forward, we followed the method described in Figure 1(a). Briefly, hiPSCs were harvested and seeded twice a week after 3 and 4 days of culture. Cell density determination will be explained below. hiPSCs were cultivated with E8 medium and Essential 8 Flex medium™ (E8 Flex) over a GELTREX matrix (all from Thermo Fisher, USA). Passages were performed by incubating the cells with Versene™ supplemented with 10% TrypLE Express (both from Thermo Fisher, USA) for 5–7 min and gentle pipetting for colony dissociation. Cells were centrifuged for 4 min at 300×g and resuspended in E8 medium with 10 nM Y-27632 (Cayman, USA). hESC BR1 (donated by Prof. Dr. Lygia V. Pereira, and previously characterized by Fraga et al.18) was also cultivated as described above and used for comparative analyses when appropriate.
Figure 1.

Single-cell passaging did not impair hiPSC morphology or colony formation capacity. (a) Scheme of the hiPSC maintenance workflow for the week. (b) Cell morphology throughout the days before media changing. Right scale bar represents 1000 µM and the left scale bar represents 100 µM. (c) Cell confluence was monitored by CellCounterAnalyser. (I) Representative scheme of the five recorded photos per 6-well plates, (II) quantification of five different passages recorded 3 days after seeding, data presented as mean ± SD. *p < 0.05; ***p < 0.001 and ****p < 0.0001 by analysis of variance (ANOVA), (III) representative images of the photos recorded for quantification. (d) PDT of hiPSC clones ACP5 (n = 8), PC4.3 (n = 5), PC4.5 (n = 4) and PC4.6 (n = 5) from passage 20 to 60. (e) All PDT values obtained for clones ACP5 (n = 165), PC4.3 (n = 83), PC4.5 (n = 100), PC4.6 (n = 112). Data is presented as mean ± SD. **p < 0.01 versus ACP5 by ANOVA.
Population doubling time calculation
For population doubling time (PDT) calculation, hiPSC were counted first when seeding the cells and then when detaching them for passage. At each time point, cells were counted twice with Trypan blue using a Neubauer’s chamber and mean was used to determine total number of cells. PDT calculation was determined by PDT = t. Log10(2)/(Log10(x) − Log10(x0)), whereby t is expressed in hours. Results were then plotted as mean ± SD for each given day. The significance of the differences among passages was analyzed for each clone through ANOVA and p < 0.05 was considered statistically significant.
Confluence monitoring: CellCountAnalyser software
We used an ImageJ associated software previously published by Busschots et al.19 and upgraded for our team (and so renamed CellCountAnalyser; details can be found in supplemental material Figure S7 and Tables S4 and S5) to monitor and work only with cells in the logarithmic phase of growing. To do so, 24–96 h after cell seeding, five photos were recorded from marked areas that covered the center and borders of cell dishes using EVOS FL Optical microscope (Thermo Fisher, USA). Then using our one-click Python-based platform, cell confluence was individually measured by photo. An excel file containing individual percentage of confluence per photo and mean ± SD calculations was generated. hiPSC dishes were split every time confluence reached 70%–85%.
Karyotype
hiPSCs were grown in a 60-mm plate to reach 70% confluence. At this moment, cells were detached using Versene and collected in a 15-mL tube. Cells were treated with Essential 8 media supplemented with 20 ng/mL Colcemid (Thermo Fisher, USA) for 1 h in 37°C, washed with PBS and treated for 20 min at 37°C with hypotonic solution (1× PBS supplemented with 0.075 M KCl). Cells were washed with 1× PBS and a fixation step was performed using a fixation solution (methanol:acetic acid, 3:1). All centrifugation steps were set at 200×g for 4 min. Conventional chromosome analysis was performed on hiPSC cultures at passages 10, 30 or 50 using GTG banding at 400-band resolution according to standard protocols. A minimal of 10 metaphase cells were analyzed (supplemental material Figure S1). Cell images were captured using the CytoVysion system (Applied Imaging Corporation, USA). hiPSCs were nominated as following: the letters are descriptive of either the donor name (ACP) or the company (PC); as ACP cells are from the same donor, the number is indicative of the clone. For PC cells, the first number indicates the lineage and the following, separated by a dot, indicates the clone.
Integration PCR, RT-PCR and RT-qPCR
To verify if there was any episomal integration into host DNA, we performed an integration polymerase chain reaction (PCR) analysis using three sets of primers (supplemental material Table S1) targeting specific sites of the plasmid’s DNA as described by Chou et al.17 To evaluate gene expression, reverse transcription polymerase chain reaction (RT-PCR) and quantitative RT-PCR (qPCR) were performed using RNA extracted from all clones at specific passages. Detailed information about the primers can be found in supplemental material Table S2. Furthermore, hESCs BR1 cell line18 were used as positive control of pluripotency and human-skin fibroblasts (the somatic cells of origin for PC4 clones) were used as negative control of pluripotency.
Embryoid body differentiation
Embryoid bodies (EBs) were generated as described in previous works20,21 with minor modifications. Briefly, hiPSCs were dissociated, centrifuged and resuspended at 80,000 cells/mL concentration. Drops of 25 µL (with 2000 cells each) were done at a petri dish lid; then the lid was carefully inverted and placed on the top of the dish containing 10 mL of HBSS 1×. After 48 h, cell aggregates were collected and transferred to a low attachment 12-well plate (Sarstedt, Germany) with Essential 6 medium. Half of the medium was replaced every 3 days until Day 13 when RNA was collected using Trizol (Thermo Fisher, USA). End-point RT-PCR to SOX17, MSX1 and PAX6 (endo-, meso- and ectoderm, respectively) and pluripotency marker DNMT3B and NANOG were performed to confirm differentiation efficiency (primers in supplemental material Table S2).
Directed differentiation into keratinocytes
hiPSCs were plated on mitomycin C-inactivated 3T3 cells. After 2 days, defined-KSFM medium (Thermo Fisher, USA) supplemented with 10 ng/mL of BMP4 (R&D Systems, USA) and 1 µM retinoic acid (Sigma Aldrich, USA) were used.21,22From day 4, cells were cultured in a defined between “3T3 cells”.22 Cells were then harvested and seeded in different plates depending on the experiment and characterized by immunostaining and flow cytometry (FC). To induce upper-layer epithelial cells, hiPSC-derived keratinocytes (hiPSC-KCs) were subjected to a 1 µM CaCl treatment for 5–7 days as previously described by Bikle et al.23 Cells were evaluated by immunostaining (K14 and K10; S3 Table) and qRT-PCR using Sybr Green (Thermo Fisher, USA) and K5, K10, IVL primers (S2 Table) and GAPDH was the housekeeping gene.
Pooled primary foreskin human keratinocytes (PHK) were grown in supplemented KGM-GOLD (Lonza, Switzerland) at 37°C and 5% CO2. PHK and iPSC-CKs were used to seed organotypic epithelial cultures as described elsewhere.24,25 Briefly, dermal equivalents were prepared using 80% rat tail collagen type I (Corning, USA), 10% Ham’s F12 10× (Gibco, Life Technologies, USA), 10% of 10× reconstitution buffer (2.2% NaHCO3, NaOH 0.05 M, HEPES 200 mM) and 2 × 105 J2 fibroblasts resuspended in BCS (Cultilab, Brazil). After collagen polymerization of the dermal equivalents 2 × 105 keratinocytes were seeded on top. After 24 h, the cultures were transferred to the medium–air interface and maintained for 9 days. Cultures were harvested by fixation in 10% buffered formalin, embedded in paraffin and then cut into 4 µm sections for hematoxylin–eosin (H&E) staining and K10 immunohistochemistry (supplemental material Table S3).
Directed differentiation into cardiomyocytes
hiPSCs were differentiated using a monolayer directed differentiation method modified from previous reports21 and grown in feeder-free conditions until they reached 60%–70% confluence. Cells were singularized, counted and plated (2.5 × 105 cells/cm²) with E8 with 5 µM of Y-27632 (Cayman Chemical, USA). E8 medium was changed daily until cells reached 100% confluence (Day 0). Medium was then changed to RPMI supplemented with B27 (Thermo Fisher, USA) without insulin (RB-) 4 µM CHIR99021 (Merck Millipore Sigma, USA). After 24 h, medium was changed to RB- supplemented with 10 ng/mL BMP4 (R&D Systems, USA). On Day 2, medium was changed to fresh RB- supplemented with 2.5 µM KY2111 and XAV939 (both from Cayman Chemical, USA). From Day 4 on, medium was changed every 48 h to fresh RPMI supplemented with 213 μg/mL ascorbic acid (Sigma Aldrich, USA), 500 μg/mL DPBS, 35% BSA and 2 µg/mL Plasmocin (InvivoGen, USA). Cells were cultivated for 30 days under the described conditions before passaging as single cells to specific experiments.
Directed differentiation into definitive endoderm
hiPSCs were differentiated toward definitive endoderm (DE) according to Goulart et al.26 with minor modifications. hiPSC cells were seeded at 2.5 × 104 cells/cm2 on GELTREX coated plates and cultured for 3 days until reached 40% confluency. For the next 3 days, the cells were cultured with endoderm differentiation media composed by RPMI 1640 (Thermo Fisher, USA), supplemented with 2% B27 (Thermo Fisher, USA), 100 ng/mL of Activin A (R&D Systems, USA), 25 ng/mL of Wnt3a (R&D Systems, USA), 1% GlutaMAX (Gibco, USA) and 2 µg/mL Plasmocin (InvivoGen, USA), with daily full media changes.
FC and immunofluorescence
Protein expression was analyzed by FC and immunofluorescence (IF). Detailed protocol and information about the antibodies can be found in supplemental material Table S3. For IF, all images were generated in EVOS FL (Thermo Fisher, USA). As for FC, data was acquired using Canto BD equipment and analyzed by FlowJo Software considering 1%–2% of false positive events.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (USA). All descriptive data is presented as the mean ± SD, groups were compared using one-way ANOVA combined with Tukey’s post hoc test and p < 0.05 was considered as statistically significant.
Results
Single-cell passaging does not affect growth rate of hiPSC providing predictable conditions for long-term cultivation
To verify any spontaneous integrations of the vectors used for reprogramming, a set of primers specific to each vector17,27 was used for a PCR analysis (each clone at passages 5 and 11). Only positive control samples (vector DNA) displayed expression of the expected fragments after PCR indicating no DNA integration into hiPSC clones (supplemental material Figure S2).
hiPSCs were cultivated using E8 and E8flex media on GELTREX-coated plates, where cells grew in monolayer maintaining an undifferentiated ESC-like morphology (Figure 1(b)). hiPSC were harvested as single cells by enzymatic dissociation and posterior seeding with ROCK inhibitor (Y-27632), which provided a quite controllable weekly routine (Figure 1(a)) based on predictable growth rates once optimal seeding density was established. To validate optimal seeding density for each one of the clones, we tested different cell densities (data not shown) and tracked the growth rates by monitoring the cellular confluence with CellCountAnalyser (Figure 1(c) and supplemental material Figure S7). hiPSCs were dissociated every time it reached 70%–85% confluency, which is the logarithmic phase of cellular growth. As a result, optimal seeding densities were then established at 40,000 or 20,000 cells per cm2 for ACP5 and at 43,000 or 22,000 cells per cm2 for PC4 clones for passages every 3 or 4 days (Mondays and Thursdays passages, respectively; Figure 1(a) and (b)).
After long-term cultivation, hiPSC PDT was calculated for each clone to assess growth rates. No changes were detected over time, as PDT analysis showed that single-cell passaging had no significant impact in growth rates over 50 passages (Figure 1(d)). In agreement with the experiment of seeding densities, mean PDT for ACP5 (28.3 ± 8.5 h) was indeed statistically different (*p < 0.05) compared to PC4.3 (33.0 ± 11.4 h), PC4.5 (33.9 ± 12.6 h) or PC4.6 (32.8 ± 12.5 h), confirming different growth rates between ACP5 and PC4 clones (Figure 1(e)).
Long-term single-cell passaging does not affect hiPSC pluripotency or elicit chromosomal aberration
After successive single-cell passages, cells were characterized for their pluripotency markers. To assess gene expression, we measured markers NANOG, OCT4, SOX2, LIN28 and DNMT3B by RT-qPCR using hESC lineage BR1 and human-skin fibroblasts cells as positive and negative controls of pluripotency, respectively (Figure 2(a)). As expected, the four hiPSC clones expressed similar amounts of all five pluripotency markers compared to BR1. In addition, these cells also showed significantly higher levels of expression than human-skin fibroblasts (Figure 2(a)). Protein expression was also verified by immunostaining, which confirmed that all clones expressed pluripotency markers OCT4, NANOG and TRA-1-60 (Figure 2(b) and supplemental material Figure S3).
Figure 2.

Immunocytochemistry, FC and RT-qPCR confirmed both protein and genic expression of pluripotency markers in hiPSC clones PC4.3, PC4.5, PC4.6 and ACP5. (a) Expression of pluripotency markers NANOG, OCT4, SOX2, DNMT3B and LIN28 in hiPSC clones at high passages, determined by RT-qPCR. The gene expression of the hiPSC was normalized to that of BR1 (hESC). PC4 clones were obtained by reprogramming fibroblast cells (PC fibro), which were used as negative control for pluripotency. Data is presented as mean ± SD, n = 3. *p < 0.05 by ANOVA with Tukey’s post hoc test. (b) Immunostaining of ACP5 clone at passage 20 showing expression of pluripotency markers OCT4, NANOG (both nuclear) and TRA-1-60 (membrane). Experiments were performed for all hiPSC clones. (c)–(e) Cytometry data of clones at passages 10, 30 and 50 using NANOG, OCT4 and SOX2 markers. In (c), light and dark gray indicate negative control (fibroblasts) and stained cells (iPSC), respectively. Statistical analysis revealed percentage positivity was not different for any marker when comparing clones in (d) (n = 3) or passages (n = 4) in (e).
Furthermore, NANOG, OCT4 and SOX2 were also assessed by FC using clones at passages 10, 30 and 50 to keep track of the clone’s phenotypes over time (Figure 2(c) and supplemental material Figure S4). Results were then analyzed for each marker in two distinctive situations, the first to compare the gene expression between different clones at the same passage, and the second to check gene expression changes over time when comparing the same clone at different passages. We observed no difference between clones (over 95% NANOG + cells and OCT4 + cells, and over 85% SOX2 + cells, Figure 2(d)) or between passages (over 70% NANOG + cells, and over 90% OCT4 + and SOX2 + cells, Figure 2(e)) for any of the markers. This result suggests that not only clones are likely to display a very similar profile under same culture conditions but also that long-term cultivation had not significant impact on cell pluripotency.
Chromosomal abnormalities were checked by performing a G-banding karyotype, which confirmed no aberrations at passages 10, 30 or 50 for clones ACP5, PC4.3, PC4.5 and PC4.6 (Figure 3 and supplemental material Figure S1).
Figure 3.
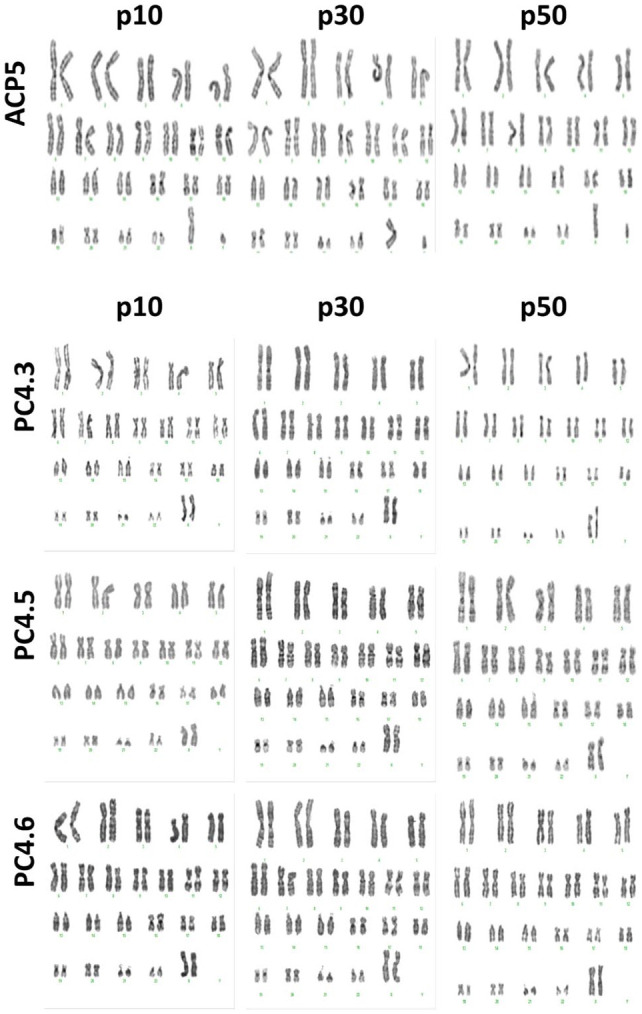
Karyotype confirmed absence of karyotypic abnormalities. G-banding karyotype of ACP5 and PC4 clones after 10, 30 and 50 single-cell passages. No aneuploidies or translocations were detected.
hiPSC clones subjected to single-cell passing preserve their trilineage capacity and display high efficiency for differentiation into keratinocytes, cardiomyocytes and DE
The plasticity of hiPSC was tested in vitro by culturing each clone under conditions to promote EB formation. As expected, after 13 days EBs revealed positive gene expression for markers such as SOX17, MSX1 and PAX6 (endo-, meso- and ectoderm representants, respectively) and decreased expression of DNMT3B and NANOG, two pluripotent markers (supplemental material Figure S5). In contrast, hiPSC clones at passage 20 displayed only high and expected expression of DNMT3B and NANOG when in pluripotency conditions (supplemental material Figure S5).
To further evaluate the differentiation potential of long-term single cell cultivated hiPSCs, we then subjected these cells to directed differentiation protocols to generate keratinocytes, cardiomyocytes and DE using hiPSCs up to passage 70.
Keratinocytes differentiation
hiPSC clones at high passages (25–50) were successfully differentiated into keratinocytes (hiPSC-KCs). After 30 days of differentiation, cells were characterized by immunostaining and FC. FC confirmed a high proportion of hiPSC-KCs positive for epithelial marker K14 (over 85% of keratin 14-positive cells; Figure 4(a)) and no statistical difference of K14 + population percentage was observed between clones (Figure 4(b)). Immunostaining of hiPSC-KCs showed that these cells expressed not only epithelial (CD104, CD49f, K10 (keratin 10) and K14) but also proliferative cell markers (Ki67, P63), indicating a typical profile of proliferative basal-layer epithelial cells (Figure 4(c)).
Figure 4.

Directed differentiation into keratinocytes using hiPSC at high passages yields positive cells for both epithelial and proliferative markers. (a) and (b) FC data showing expression of K14 marker in keratinocytes obtained from hiPSC clones PC4.3, PC4.5, PC4.6 and ACP5. Light and dark gray curves indicate negative control (hiPSC) and stained cells (hiPSC-KC), respectively. Data is presented as mean ± SD, n = 3–6. One-way ANOVA p = 0.114; (c) representative immunostaining of keratinocytes obtained from PC4.3 clone (hiPSC-KC PC4.3). Ki67 and P63 are characteristic nuclear markers of proliferative cells. Surface markers CD104, CD49f and cytoskeleton markers K10, K14 are all epithelial-specific. CD104, CD49f and K14 are expressed especially on proliferative basal layer cells, whereas K10 is mainly expressed on intermediate and outermost layers of epidermis. Nuclei was stained with Hoechst 3342. (d) qRT-PCR of primary keratinocytes (HEK; Thermo Fisher, USA) and iPSC-CK before and after calcium treatment. K5 did not have any significant statistical difference; K10 and IVL expression increased after calcium treatment (n = 3; *p < 0.05; ***p < 0.001; two-way ANOVA). (e) and (f) H&E (e) and K10 (F, brown) staining of epithelial organotypic cultures seeded from primary human keratinocytes (PHK; Lonza, Switzerland) and iPSC-KC. iPSC-KC reproduces epidermal structure and undergoes terminal differentiation.
In addition, we evaluate hiPSC-KC ability to further differentiate into more superficial keratinocytes. In human epithelium, keratinocytes undergo the process of differentiation where cells from inner layers (basal keratinocytes) move up to more superficial levels and switch from producing K14/K5 to produce K1/K10 and further differentiate into keratinized stratum corneum. Several metabolic regulations also occur during keratinocytes differentiation28 being calcium the major regulator of this process.23 Thus, after high concentration of calcium treatment, hiPSC-KC increased K10 and IVL expression similarly to primary keratinocytes (Figure 4(d)); K10-positive cells were also observed by immunostaining (supplemental material Figure S6). Moreover, organotypic cultures from PHKs and hiPSC-KC closely resembled normal epithelium in vivo with morphologically distinct basal, spinous, granular and cornified layers (Figure 4(e)). Upper layer of epithelial cell differentiation was further confirmed by K10 immunohistochemistry; K10-positive cells were restricted to the upper layers of the epithelia and were not detected in the basal and parabasal layers (Figure 4(f)). Our data suggest that long-term single-cell passaged hiPSC clones could generate basal keratinocytes and they are able to further differentiate into upper-layer epithelial cells.
Cardiomyocytes differentiation
We have also used hiPSC up to 70 passages for differentiation into cardiomyocytes. All hiPSC-derived cardiomyocytes (hiPSC-CMs) started to contract approximately at Day 7 of the differentiation protocol, and the beating was sustained through the following days (supplemental material Videos S1 and S2). At Day 30, cells were characterized by immunostaining and FC. Immunostaining showed positive expression of cardiac proteins such as NKX2.5, TNNI1, TNNI3, MYH7, TNNT2 and ACTN2 (Figure 5(a)). FC showed that hiPSC-CMs derived from pluripotent cells from passages 30 to 70 were generated with high efficiency (over 80% ACTN2, TNNT2, TNNI1 and TNNI3 positive cells) and that ACP5 and PC4 lineages generated similar amounts of positive cardiac cells (Figure 5(b) and (c)). Interestingly, we have found that hiPSC-CMs exhibited simultaneous expression of TNNI1 and TNNI3, which are specifically expressed in fetal and mature cardiomyocytes, respectively.
Figure 5.

Directed differentiation into cardiomyocytes using hiPSC at high passages yields positive cells for cardiac markers. (a) Immunostaining of cardiomyocytes (iPSC-CMs ACP5) obtained from ACP5 clone showing protein expression of cardiac specific-markers TNNI1, TNNI3, MYH7, TNNT2, NKX2.5 and ACTN2. Nuclei was stained with Hoechst 3342. (b) and (c) FC data showing protein expression of cardiac-specific markers TNNI1, TNNI3, TNNT2 and ACTN2 in hiPSC-CM. Results were confirmed by performing independent experiments using APC5, PC4.3, PC4.5, PC4.6. Data is presented as mean ± SD, n = 2–5; one-way ANOVA p = 0.9 (ACTN2); 0.2 (TNNI1); 0.9; and 0.1 (TNNT2).
DE differentiation
hiPSC clones at high passages (25–50) were successfully differentiated into DE (hiPSC-DE). Immunostaining shows that the majority of the cell population in all tested cell lines co-express the DE markers FOXA2 and CXCR4 at the end of the DE differentiation protocol (Figure 6(c)), as shown in previous publication.26 FC data indicates that hiPSC-DEs were generated with high efficiency, most of the cells expressing FOXA2 (over 90% were FOX2-positive cells, Figure 6(a)) and no statistical difference were observed among cell lines (ACP5 and PC4) (Figure 6(b)).
Figure 6.

Directed differentiation into DE using hiPSC. (a) and (b) FC data showing expression of FOXA2 marker in DE obtained from hiPSC clones PC4.3, PC4.5, PC4.6 and ACP5. Light and dark gray curves indicate negative control (hiPSC) and positive cells (hiPSC-DE), respectively. Data is presented as mean ± SD, n = 2–4. One-way ANOVA p = 0.7; (c) representative immunostaining of DE markers for all clones double-stained with FOXA2 and CXCR4. Nuclei was stained with Hoechst 3342.
Discussion
In the present study, we have successfully demonstrated that our cultivation method can maintain hiPSC obtained from different somatic cells and different reprogramming systems for over 50 passages under single-cell conditions. To do so, hiPSC were characterized at different timepoints for their expression of pluripotency markers, genetic integrity, growth rates and potential to generate derivatives by both EB formation and direct differentiation into keratinocytes, cardiomyocytes and DE.
To confirm reprogramming success, we first analyzed hiPSC for their pluripotency markers using immunocytochemistry, FC and RT-qPCR. We have found by FC that protein expression of markers OCT4, NANOG and SOX2 showed no alteration nor decreased over time in any of the hiPSC clones, corroborating permanent reprogramming as suggested by previous studies.29,30 Interestingly, even though reprogramming was performed using different sets of plasmids and different somatic cells of origin for lineages ACP and PC4, positivity percentage for each marker showed no difference between hiPSC clones. Consistent with this, RT-qPCR also showed no significant difference among clones for gene expression of pluripotency markers NANOG, OCT4, SOX2, LIN28 or DNMT3B.
Then, we analyzed the quality of the hiPSC concerning genomic alterations. As reprogramming and cultivation processes are recognized as the main causes for the alterations found in genome,9 genomic integrity was assessed by integration PCR and karyotyping. Because of the potential hazardous effects of reprogramming vectors,31 several different integration-free methods to generate iPSC were developed but, in comparison, the use of episomal plasmid vectors for reprogramming has increased efficiency, as shown in previous studies.17,27 Indeed, by using plasmids pEB-C5, pEB-TG and Epi5, we were able to successfully generate hiPSC. Also, PCR confirmed no vector integration into host DNA. It is important to acknowledge that integration of transgenes is not the only mechanism that is capable of altering DNA after reprogramming; however, numerous reports concluded that further changes in the genome related to the vectors are mostly benign and unlikely to be threatening for research or therapy purposes.32,33 Thus, no additional experiments to assess reprogramming-induced genomic alterations were performed.
As for culture-induced alterations, one of the main risks of prolonged cultivation is the progressive selection of genetic variants that are better adapted to in vitro culture environment.14,34 Several studies concerning ESC show that aneuploidies in chromosomes 12, 17 and X are commonly identified after long-term culture.10,14 As recently reviewed by Assou et al.,9 a karyotyping routine is essential for the quality assessment of PSCs, as it can identify many unacceptable genomic abnormalities, which already have been reported to emerge after only five passages.11 However, we have screened ACP5 and PC4 clones at passages 10, 30 and 50 and found no aberrations through G-banding karyotype. Importantly, although we are encouraged by this result, we recognize the need for additional screening to exclude potential infra-karyotypic abnormalities such as 20q11.21 amplification35 or oncogenic mutations,36 which are also unacceptable for studies concerning hiPSC.
We have also assessed hiPSC quality concerning growth rates as PSCs proliferative log phase is required for the best results in many differentiation protocols such as the cardiac ones.37 Thus, tracking cell doubling rates during their maintenance to avoid predictable high confluence and hence cell cycle arrest is strongly desirable, and it can be easily performable by PDT calculation and cell confluence monitoring as a culturing routine. We have assessed PDT and cell confluence from passage 20 to 60, a time range that corresponded to approximately 120 days of cultivation. Statistical analysis of different cultivation timepoints showed no impact in PDT for neither ACP5 nor PC4 clones.
Conversely, other groups found increasing growth rates related to long-term cultivation.11,15 This discrepancy may be explained by the fact that these findings were linked to aneuploidies found within the cells, as a strong correlation between the proportion of cell lines with abnormal karyotypes and population doubling has been previously reported in an extensive study with both ESC and iPSC.38 In addition, our software (CellCountAnalyser) was able to measure cell confluence quicker and more accurately than other software available,19,39–41 giving us the ability to split cells in their log-phase of growing.
A final important finding was the maintenance of the differentiation potential after long-term cultivation, as all hiPSC clones at high passages were able to form EB and to differentiate into ecto-, meso- and endoderm derivatives through directed differentiation. Protein expression analysis of specific markers showed that directed differentiation consistently yielded keratinocytes, cardiomyocytes and DE using hiPSC at high passages. Keratinocyte, cardiomyocytes and DE displayed a high-purity population of cells independently of the passage numbers or hiPSC lineage, as routinely reported from others.13
These facts bring two very important points of discussion concerning the impact of iPSC management on differentiation success. First, low passages should not be used for directed differentiation as early passage hiPSC may retain transient epigenetic memory from adult somatic cell sources impairing PSC plasticity.42 Second, despite being a popular methodology for differentiation in vitro, it is known that EB formation is a process that shows very low reproducibility and often ends up with low purity of the desired differentiated cells,43,44 whereas directed differentiation approaches tend to be more efficient and reliable to obtain specialized cells. This gives great advantage to single-cell passaging methodologies since they facilitate hiPSC directed differentiation by promoting seeding homogeneity and the formation of loosely packed clusters that leads to a more efficient cellular response to signaling molecules.3
Adaptation into single-cell culture was found to be a crucial step for differentiation into lung epithelia,45 and similarly, seeding density has been pointed out as one of the major optimizable factors for cardiomyocyte differentiation.46 Also, the demand of gene editing technologies, such as CRISPR, is a great stimulus to the single-cell iPSC cultivation adoption, once the handling and selection of edited cells are optimized in these cultures.47 Finally, the use of simple tools for cell confluence monitoring, such as our CellCountAnalyser, is essential for increasing reproducibility and predictability during differentiation protocols.
Nonetheless, it is well recognized that single-cell passaging is usually followed by great loss of cellular viability7 and hence several reports suggest it promotes rapid selection of genetically abnormal clones that display increased survival rates. The increasing use of ROCK-inhibitor Y-276328 to promote PSC viability after enzymatic dissociation also concerns scientists because of the lack of reported data on its long-term effects. Currently, the studies addressing this question found no direct effect of Y-27632 on ESC genomic integrity.11 While several studies found chromosomes 17 and 12 to be especially sensitive to adaptation into single-cell passaging,10,11,48 many others did not detect such abnormalities upon long-term culture.49–51 It has been previously suggested that the conflicting data concerning impact of single-cell passaging may be explained by this general designation encompassing different passaging methods, such as EDTA, dispase, TrypLE and trypsin.11,50
A limitation to this study is the lack of direct comparison between the results of single-cell passaging and clump dissociation. However, it does not invalidate the valuable results showing that using long-term single-cell passage with properly addressed quality control verifications and passaging routine can maintain chromosomal integrity and hiPSC differentiation potential. As there are multiple different variables among studies, the impact of single-cell passaging techniques remains subject for further investigation.
Conclusion
Here we presented an easy long-term and single-cell passaging pipeline to cultivate hiPSCs that maintained their characteristics and karyotype under feeder-free conditions that allows a robust hiPSC cultivation routine by regulating cell number in a density- and time-dependent manner. Cells cultivated by this pipeline were able to form EB and successfully directly differentiate into keratinocytes, cardiomyocytes and DE. Together our finding supports the long-term cultivation of hiPSCs in single-cell conditions for further use in cell-modeling and therapy.
Supplemental Material
Supplemental material, SOM-20-0113-R2_SupMaterial_revised_version_FINAL for Long-term single-cell passaging of human iPSC fully supports pluripotency and high-efficient trilineage differentiation capacity by Estela Cruvinel, Isabella Ogusuku, Rosanna Cerioni, Sirlene Rodrigues, Jéssica Gonçalves, Maria Elisa Góes, Juliana Morais Alvim, Anderson Carlos Silva, Vanesca de Souza Lino, Enrique Boccardo, Ernesto Goulart, Alexandre Pereira, Rafael Dariolli, Marcos Valadares and Diogo Biagi in SAGE Open Medicine
Acknowledgments
We thank Prof. Alexander Henning Ulrich for using his FACs equipment. Also, Prof. Monica Matos and Lygia Veiga Pereira for the donation of 3T3 and hESC BR1 cells, respectively. Finally, Amy Gutierrez for reviewing the text.
Footnotes
Declaration of conflicting interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors E.C., I.O., R.C., S.R., J.G., J.M.A., R.D., M.V. and D.B. were employees of PluriCell Biotech during the conduct of the study. E.C., R.D., M.V. and D.B. own shares in PluriCell Biotech (A.C.P., M.V. and D.B. are also co-founders). The other authors report no conflicts.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We acknowledge the financial support of Sao Paulo Research Foundation (FAPESP) (grants #2015/50224-8, #2016/50082-1).
Ethical approval: Ethical approval for this study was obtained from Ethics Committee for Medical Research on Human Beings of the Institute of Biomedical Sciences from the University of São Paulo (2.009.562).
Informed consent: Written informed consent was obtained from all subjects before the study.
ORCID iDs: Juliana Morais Alvim  https://orcid.org/0000-0002-6351-723X
https://orcid.org/0000-0002-6351-723X
Rafael Dariolli
https://orcid.org/0000-0003-0957-1259
Diogo Biagi
https://orcid.org/0000-0002-5872-6133
Supplemental material: Supplemental material for this article is available online.
References
- 1. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131(5): 861–872. [DOI] [PubMed] [Google Scholar]
- 2. Chen KG, Mallon BS, McKay RDG, et al. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014; 14(1): 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen KG, Mallon BS, Hamilton RS, et al. Non-colony type monolayer culture of human embryonic stem cells. Stem Cell Res 2012; 9(3): 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gafni O, Weinberger L, Mansour AA, et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013; 504(7479): 282–286. [DOI] [PubMed] [Google Scholar]
- 5. Krawetz R, Taiani JT, Liu S, et al. Large-scale expansion of pluripotent human embryonic stem cells in stirred-suspension bioreactors. Tissue Eng Part C Methods 2010; 16(4): 573–582. [DOI] [PubMed] [Google Scholar]
- 6. Steiner D, Khaner H, Cohen M, et al. Derivation, propagation and controlled differentiation of human embryonic stem cells in suspension. Nat Biotechnol 2010; 28(4): 361–364. [DOI] [PubMed] [Google Scholar]
- 7. Miñambres R, Guasch RM, Perez-Aragó A, et al. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci 2006; 119(Pt 2): 271–282. [DOI] [PubMed] [Google Scholar]
- 8. Watanabe K, Ueno M, Kamiya D, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 2007; 25(6): 681–686. [DOI] [PubMed] [Google Scholar]
- 9. Assou S, Bouckenheimer J, De Vos J. Concise review: assessing the genome integrity of human induced pluripotent stem cells—what quality control metrics? Stem Cells 2018; 36(6): 814–821. [DOI] [PubMed] [Google Scholar]
- 10. Mayshar Y, Ben-David U, Lavon N, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010; 7(4): 521–531. [DOI] [PubMed] [Google Scholar]
- 11. Bai Q, Ramirez JM, Becker F, et al. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev 2015; 24(5): 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacobs K, Zambelli F, Mertzanidou A, et al. Higher-density culture in human embryonic stem cells results in DNA damage and genome instability. Stem Cell Rep 2016; 6(3): 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujita J, Tohyama S, Kishino Y, et al. Concise review: genetic and epigenetic regulation of cardiac differentiation from human pluripotent stem cells. Stem Cells 2019; 37(8): 992–1002. [DOI] [PubMed] [Google Scholar]
- 14. Harrison NJ, Baker D, Andrews PW. Culture adaptation of embryonic stem cells echoes germ cell malignancy. Int J Androl 2007; 30(4): 275–281; discussion 281. [DOI] [PubMed] [Google Scholar]
- 15. Enver T, Soneji S, Joshi C, et al. Cellular differentiation hierarchies in normal and culture-adapted human embryonic stem cells. Hum Mol Genet 2005; 14(21): 3129–3140. [DOI] [PubMed] [Google Scholar]
- 16. Tofoli FA, Dasso M, Morato-Marques M, et al. Increasing the genetic admixture of available lines of human pluripotent stem cells. Sci Rep 2016; 6: 34699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chou BK, Mali P, Huang X, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res 2011; 21(3): 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fraga AM, Sukoyan M, Rajan P, et al. Establishment of a Brazilian line of human embryonic stem cells in defined medium: implications for cell therapy in an ethnically diverse population. Cell Transplant 2011; 20(3): 431–440. [DOI] [PubMed] [Google Scholar]
- 19. Busschots S, O’Toole S, O’Leary JJ, et al. Non-invasive and non-destructive measurements of confluence in cultured adherent cell lines. MethodsX 2015; 2: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Yang P. In vitro differentiation of mouse embryonic stem (mES) cells using the hanging drop method. J Vis Exp 2008; 17: 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lian X, Zhang J, Azarin SM, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc 2013; 8(1): 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itoh M, Kiuru M, Cairo MS, et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A 2011; 108(21): 8797–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab 2012; 7(4): 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boccardo E, Noya F, Broker TR, et al. HPV-18 confers resistance to TNF-alpha in organotypic cultures of human keratinocytes. Virology 2004; 328(2): 233–243. [DOI] [PubMed] [Google Scholar]
- 25. Boccardo E, Manzini Baldi CV, Carvalho AF, et al. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor-alpha. Carcinogenesis 2010; 31(3): 521–531. [DOI] [PubMed] [Google Scholar]
- 26. Goulart E, De Caires-Junior LC, Telles-Silva KA, et al. Adult and iPS-derived non-parenchymal cells regulate liver organoid development through differential modulation of Wnt and TGF-β. Stem Cell Res Ther 2019; 10(1): 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011; 8(5): 409–412. [DOI] [PubMed] [Google Scholar]
- 28. Eichner R, Sun TT, Aebi U. The role of keratin subfamilies and keratin pairs in the formation of human epidermal intermediate filaments. J Cell Biol 1986; 102(5): 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhattacharya B, Miura T, Brandenberger R, et al. Gene expression in human embryonic stem cell lines: unique molecular signature. Blood 2004; 103(8): 2956–2964. [DOI] [PubMed] [Google Scholar]
- 30. Abeyta MJ, Clark AT, Rodriguez RT, et al. Unique gene expression signatures of independently-derived human embryonic stem cell lines. Hum Mol Genet 2004; 13(6): 601–608. [DOI] [PubMed] [Google Scholar]
- 31. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448(7151): 313–317. [DOI] [PubMed] [Google Scholar]
- 32. Abyzov A, Mariani J, Palejev D, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012; 492(7429): 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhutani K, Nazor KL, Williams R, et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat Commun 2016; 7: 10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews PW. The selfish stem cell. Nat Biotechnol 2006; 24(3): 325–326. [DOI] [PubMed] [Google Scholar]
- 35. Lefort N, Feyeux M, Bas C, et al. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol 2008; 26(12): 1364–1366. [DOI] [PubMed] [Google Scholar]
- 36. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 2011; 39: D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laco F, Woo TL, Zhong Q, et al. Unraveling the inconsistencies of cardiac differentiation efficiency induced by the GSK3β inhibitor CHIR99021 in human pluripotent stem cells. Stem Cell Rep 2018; 10(6): 1851–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amps K, Andrews P, Anyfantis G, et al. Screening a large, ethnically diverse population of human embryonic stem cells identifies a chromosome 20 minimal amplicon that confers a growth advantage. Nat Biotechnol 2012; 29(12): 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaccard N, Griffin LD, Keser A, et al. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnol Bioeng 2014; 111(3): 504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Georg M, Fernández-Cabada T, Bourguignon N, et al. Development of image analysis software for quantification of viable cells in microchips. PLoS ONE 2018; 13(3): e0193605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venter C, Niesler CU. Rapid quantification of cellular proliferation and migration using ImageJ. Biotechniques 2019; 66(2): 99–102. [DOI] [PubMed] [Google Scholar]
- 42. Polo JM, Liu S, Figueroa ME, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 2010; 28(8): 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Später D, Hansson EM, Zangi L, et al. How to make a cardiomyocyte. Dev Camb 2014; 141(23): 4418–4431. [DOI] [PubMed] [Google Scholar]
- 44. James D, Nam HS, Seandel M, et al. Expansion and maintenance of human embryonic stem cell-derived endothelial cells by TGFΒ inhibition is Id1 dependent. Nat Biotechnol 2010; 28(2): 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wong AP, Chin S, Xia S, et al. Efficient generation of functional CFTR-expressing airway epithelial cells from human pluripotent stem cells. Nat Protoc 2015; 10(3): 363–381. [DOI] [PubMed] [Google Scholar]
- 46. Burridge PW, Holmström A, Wu JC. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet 2015; 87: 21.3.1–21.3.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen Y-H, Pruett-Miller SM. Improving single-cell cloning workflow for gene editing in human pluripotent stem cells. Stem Cell Res 2018; 31: 186–192. [DOI] [PubMed] [Google Scholar]
- 48. Maitra A, Arking DE, Shivapurkar N, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet 2005; 37(10): 1099–1103. [DOI] [PubMed] [Google Scholar]
- 49. Bajpai R, Lesperance J, Kim M, et al. Efficient propagation of single cells accutase-dissociated human embryonic stem cells. Mol Reprod Dev 2008; 75(5): 818–827. [DOI] [PubMed] [Google Scholar]
- 50. Kibschull M, Mileikovsky M, Michael IP, et al. Human embryonic fibroblasts support single cell enzymatic expansion of human embryonic stem cells in xeno-free cultures. Stem Cell Res 2011; 6(1): 70–82. [DOI] [PubMed] [Google Scholar]
- 51. Ellerström C, Strehl R, Noaksson K, et al. Facilitated expansion of human embryonic stem cells by single-cell enzymatic dissociation. Stem Cells 2007; 25(7): 1690–1696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, SOM-20-0113-R2_SupMaterial_revised_version_FINAL for Long-term single-cell passaging of human iPSC fully supports pluripotency and high-efficient trilineage differentiation capacity by Estela Cruvinel, Isabella Ogusuku, Rosanna Cerioni, Sirlene Rodrigues, Jéssica Gonçalves, Maria Elisa Góes, Juliana Morais Alvim, Anderson Carlos Silva, Vanesca de Souza Lino, Enrique Boccardo, Ernesto Goulart, Alexandre Pereira, Rafael Dariolli, Marcos Valadares and Diogo Biagi in SAGE Open Medicine