

Abstract
The cytochromes P450 are heme-dependent enzymes that catalyze many vital reaction processes in the human body related to biodegradation and biosynthesis. They typically act as mono-oxygenases; however, the recently discovered P450 subfamily TxtE utilizes O2 and NO to nitrate aromatic substrates such as L-tryptophan. A direct and selective aromatic nitration reaction may be useful in biotechnology for the synthesis of drugs or small molecules. Details of the catalytic mechanism are unknown, and it has been suggested that the reaction should proceed through either an iron(III)-superoxo or an iron(II)-nitrosyl intermediate. To resolve this controversy, we used stopped-flow kinetics to provide evidence for a catalytic cycle where dioxygen binds prior to NO to generate an active iron(III)-peroxynitrite species that is able to nitrate l-Trp efficiently. We show that the rate of binding of O2 is faster than that of NO and also leads to l-Trp nitration, while little evidence of product formation is observed from the iron(II)-nitrosyl complex. To support the experimental studies, we performed density functional theory studies on large active site cluster models. The studies suggest a mechanism involving an iron(III)-peroxynitrite that splits homolytically to form an iron(IV)-oxo heme (Compound II) and a free NO2 radical via a small free energy of activation. The latter activates the substrate on the aromatic ring, while compound II picks up the ipso-hydrogen to form the product. The calculations give small reaction barriers for most steps in the catalytic cycle and, therefore, predict fast product formation from the iron(III)-peroxynitrite complex. These findings provide the first detailed insight into the mechanism of nitration by a member of the TxtE subfamily and highlight how the enzyme facilitates this novel reaction chemistry.
Introduction
The cytochromes P450 (P450s or CYPs) are vital enzymes for human health with, e.g., key functions in the liver related to the metabolism of drugs and xenobiotics; and as such their activity and reactivity is of interest to drug delivery and development.1−5 In addition, they also catalyze key steps in the biosynthesis of natural products and hormones, including estrogen.6−10 Their diverse reactivities and ability to activate inert C–H bonds have resulted in the P450s gaining significant interest due to their potential as biocatalysts in the synthesis of high value chemicals.11−13 However, factors including requirement for expensive redox partners, as well as a lack of knowledge of many facets of their structure and activity, has limited their applications. The P450s contain a heme prosthetic group that is linked to the protein via a proximal coordination to the heme iron through a cysteinate residue.14−16 The P450s typically react as mono-oxygenases, where they transfer one atom of molecular oxygen to a substrate, while the second oxygen atom of O2 is reduced to water.1−4,17−19 In general, P450s, can catalyze diverse reactions, including aliphatic or aromatic hydroxylation, epoxidation and sulfoxidation, in addition to pathways leading to desaturation, C–C bond cleavage and O-dealkylation.20,21 P450 isozymes show substantial differences in catalytic properties and the shape and size of the substrate binding pocket. Consequently, many questions remain related to their catalytic cycle and substrate activation processes.
Recently, two novel P450 subfamilies were discovered that are able to regiospecifically nitrate aromatic amino acids including L-tryptophan and l-tyrosine.22−31 The P450 TxtE isozyme activates L-tryptophan at the 4-position, while the P450 RufO isozyme binds l-tyrosine and adds an NO2 group to the 3-position. In actinobacteria, TxtE catalyzed nitration is part of the biosynthetic pathway of the phytotoxin thaxtomin in Streptomyces scabies, while tyrosine nitration catalyzed by RufO is a key step in the biosynthesis of rufomycin, a natural product peptide antibiotic with activity against Mycobacterium tuberculosis. Furthermore, the l-Phe residue in the N,N-dimethyl-diketopiperazine intermediate in thaxtomin biosynthesis is hydroxylated at the Cα and aromatic ring positions by the bifunctional P450 TxtC.22−24,32−34 The substrate nitration reactions are not restricted to one class of P450 isozymes, as addition of peroxynitrite to the mono-oxygenase P450 3A4 was found to lead to tyrosine nitration efficiently.35 Direct and selective aromatic nitration is difficult to achieve chemically and thus enzyme catalyzed processes have significant potential in biotechnology and therefore we decided to investigate the catalytic mechanism of the P450 TxtE enzyme using a combined experimental and computational approach.
A TxtE crystal structure was reported with the substrate l-Trp bound,24 and an extract of the active site is shown in Figure 1a. The heme iron is bound to the thiolate group of Cys357, the proximal/axial ligand, and the l-Trp substrate is positioned in the active site in a tight orientation with hydrogen bonding interactions to several amino acids, including Arg59 (Figure 1). Unfortunately, due to their transient nature, few catalytic cycle intermediates have been identified and characterized for P450 TxtE. However, it has been established that the enzyme binds L-tryptophan in the active site and utilizes O2 and NO to convert the substrate to L-4-nitrotryptophan selectively, using external redox partners for provision of electrons.22−24 Free superoxide (O2−•) and NO in a water environment react within the diffusion limit to form peroxynitrite efficiently.36 However, on a metal center the order of binding of NO versus O2 determines the reaction products and the formation of nitrate, nitrite, or peroxynitrite, which all react differently with substrates.36 A catalytic cycle of TxtE was hypothesized that proceeds via an iron(III)-peroxynitrite intermediate.22 Previous studies on myoglobin-bound peroxynitrite showed that, with an open substrate-binding pocket that is accessible to protons, the peroxynitrite was quickly converted to nitrate and no aromatic nitration was observed.37
Figure 1.
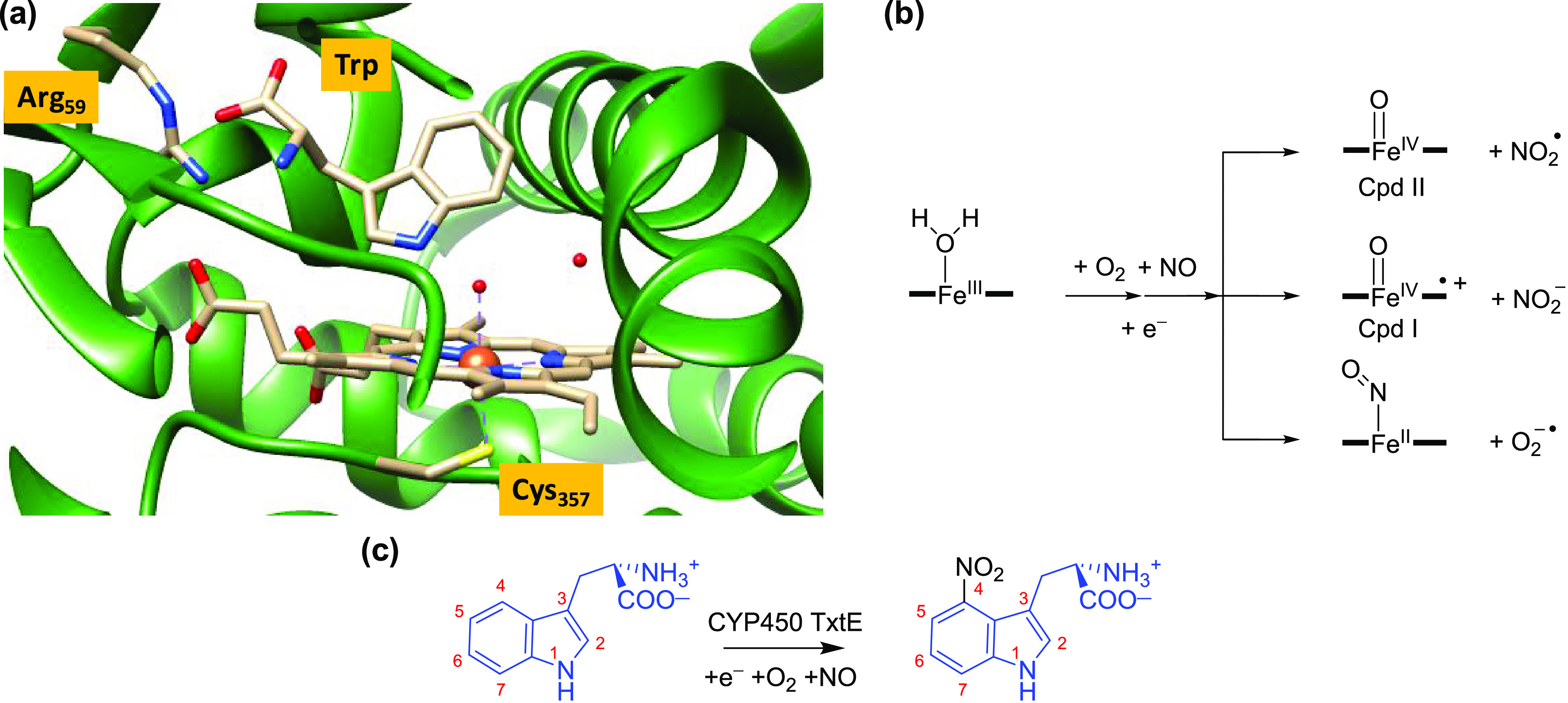
(a) Active site structure of P450 TxtE as taken from the 4TPO PDB file. (b) Proposed reaction mechanism of dioxygen and NO activation of the heme in TxtE and possible reactive intermediates considered here. (c) Overall nitration reaction catalyzed by P450 TxtE.
Little is known about the catalytic cycle of P450 TxtE and only the ferric-H2O resting state (Figure 1a) and the water-displaced ferric states have been characterized.22−24 A P450 TxtE catalytic cycle has been proposed where substrate binding displaces the water molecule from the resting state and, after heme iron reduction, is followed by dioxygen binding to form an iron(II)-dioxygen or an iron(III)-superoxo intermediate, which subsequently reacts with NO to form a ferric-peroxynitrite intermediate.22 The ferric-peroxynitrite complex could react directly with substrate or either homolytically cleave to form an iron(IV)-oxo species called Compound II (CpdII) and NO2•, or heterolytically cleave to give an iron(IV)-oxo heme cation radical called Compound I and NO2–. This would give the P450 nitration isozymes a distinctive catalytic cycle that deviates from the P450 mono-oxygenases, wherein the ferric-superoxo form is reduced and doubly protonated to form CpdI and a water molecule.16−18,38−42 Spectroscopic and kinetic studies characterized CpdI as the active species in the Pseudomonas putida P450 CAM enzyme.43 Although the one-electron reduced form of CpdI, i.e. CpdII, is not part of the mono-oxygenase catalytic cycle, it can be formed upon reduction of CpdI.
Another heme-dependent enzyme, namely nitric oxide synthase (NOS), has also been shown to activate peroxynitrite.44,45 However, in the NOS enzymes, addition of excess peroxynitrite led to the production of the ferrous-nitrosyl species instead. A similar observation was made for P450 BM3 where excess peroxynitrite (ONOO–) yielded iron(II)-NO products rather than the anticipated CpdI or CpdII products.46 This is in contrast to the work of Groves et al.,47 who added NO to oxygenated myoglobin to detect a ferryl-oxo species, i.e., CpdII, and free NO2.47 In particular, the reaction of the iron(III)-superoxo species with NO was found to yield peroxynitrite rapidly. The order by which dioxygen and NO are added to the protein may have an effect on whether ferrous-nitrosyl, CpdI or CpdII is formed, and therefore we decided to study the early stages of the catalytic cycle of TxtE, and particularly focused on the formation of the iron(II)-NO and ferric-peroxynitrite species.
In this study we investigated the order of O2 and NO binding in TxtE and whether free NO2• or NO2– is generated and is involved in the catalytic mechanism of l-Trp nitration by P450 TxtE. In addition, our work focused on understanding the selectivity of substrate nitration and shows that an initial precursor iron(III)-peroxynitrite complex homolytically cleaves into an iron(IV)-oxo heme and NO2•. As substrate, L-tryptophan is located close to the formed NO2• and it is activated rapidly to form nitrated products. Clearly, substrate binding must be a precursor to NO addition to an iron(III)-superoxo intermediate in the catalytic cycle. Our work rationalizes substrate nitration by P450 isozymes and gives insights into how to further engineer P450 enzymes for more efficient biotechnological applications.
Methods
Experiment
Sample Preparation
N-terminal His-tagged TxtE protein was expressed and purified based on a previously described protocol.22 For large-scale protein production 5 × 1 L cultures were grown at 37 °C to an OD600 of ∼0.6 and induced overnight at 15 °C by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside. Cells were harvested by centrifugation and the pellet was resuspended in 100 mL buffer B1 (20 mM Tris-HCl buffer, pH 8.0, 100 mM NaCl, 20 mM imidazole, 1 mM dithiothreitol (DTT) and 10% glycerol). The cells were lysed in the presence of 1 mM phenylmethylsulfonyl fluoride using a French Press (17 000 psi internal cell pressure) followed by sonication for 2 min (Ultrasonic processor, Jencons). After removal of the cellular debris by centrifugation (18 000g for 20 min at 4 °C), the supernatant was applied to a 5 mL HiTrapTM HP affinity column (Nickel Sepharose High Performance, GE Healthcare) equilibrated with buffer B1 (flow rate 2 mL min–1). Unbound proteins were removed by washing with 30 mL buffer B1 and the His6-TxtE fusion protein was eluted with ∼10 mL of buffer B2 (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 200 mM imidazole, 1 mM DTT, 10% glycerol). The protein was further purified by gel filtration using 110 mL Superdex 12 prep grade resin (GE Healthcare) equilibrated with buffer B3 (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM DTT and 10% glycerol). The sample was eluted with ∼64 mL of buffer B3 (flow rate 0.75 mL min–1, column dead volume 40 mL), and fractions containing His6-TxtE were analyzed by electrophoresis (10% SDS-PAGE, Figure S1, Supporting Information), pooled, and concentrated using an Amicon Ultracentrifugal filtration device with a 30 000 molecular weight cutoff membrane (Millipore). Pure His6-TxtE was buffer exchanged into buffer B4 (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, and 30% glycerol), aliquoted and frozen at −80 °C. The purified protein was confirmed to be active using the previously described assay.22 UV–vis absorbance spectra were measured using a Cary 50 UV–vis spectrophotometer (Agilent Technologies).
Stopped-Flow Spectroscopy
Rapid kinetic measurements were carried out using an SX20 rapid mixing stopped-flow spectrophotometer (Applied Photophysics Ltd., Leatherhead) placed inside a Belle Technology anaerobic chamber (oxygen levels <2 ppm) as previously described.48 Proteins and substrates were dissolved in buffer (25 mM Tris, pH 8.0) and degassed, unless otherwise stated. L-Tryptophan (0.5 mM) and sodium dithionite (15 mM) were added to TxtE protein (150 μM) to reduce it to the high-spin iron(II) form. To remove excess dithionite, the protein was passed through a PD10 desalting column equilibrated with 20 mM Tris pH 8 containing 0.15 mM l-Trp (which was kept in the glovebox overnight prior to all measurements and to ensure removal of all oxygen traces) and eluted in a volume of ∼5 mL. To confirm that the protein heme iron was reduced and high-spin (l-Trp was bound), a sample was analyzed by UV–vis spectroscopy in a sealed cuvette. The protein was maintained in a reduced, high-spin state for several hours. All stopped-flow experiments were carried out at 4 °C using a final concentration of 2 μM TxtE and 75 μM l-Trp. For NO experiments with degassed buffer, the sample was bubbled with NO for 10 min to saturate the buffer. To ascertain that the NO did not contain impurities such as small traces of NO2 that could affect the results,36,49−52 some experiments were repeated with purified NO that had first been passed through a 10% (w/v) solution of Ascarite (Sigma-Aldrich) to remove any potential impurities caused by disproportionation of the NO gas from the cylinder. None of these studies gave significantly different results from the experiments with nonpurified NO, see the Supporting Information, Figure S5.
For O2 experiments a sealed tube containing air-saturated buffer (∼200 μM O2) was transferred to the anaerobic chamber. Serial dilutions of each solution were made just prior to use by using degassed buffer as required. Time-resolved spectral changes were recorded using a photodiode array detector, and single wavelength kinetic transients were measured at 435 nm (for O2) or 445 nm (for NO) to determine the rates of gas binding. The rates of NO and O2 binding were measured in single mixing stopped-flow experiments (experimental set up shown schematically in Figure S2, Supporting Information) over a range of concentrations. Double mixing stopped-flow experiments were carried out to determine the order of gas binding to the protein (experimental set up shown schematically in Figure S3, Supporting Information).
The samples from the double mixing stopped-flow experiments were collected after each mix and frozen for further analysis by LC-HRMS. LC-HRMS conditions for analysis of 4-l-nitrotryptophan production were as follows: LC-HRMS was performed on a Sigma Ascentis Express column (C18, 150 × 2.1 mm, 2.7 μm) attached to a Dionex 3000RS UHPLC coupled with a Bruker MaXis ESI-Q-TOF mass spectrometer. The mobile phase A was water containing 0.1% formic acid and mobile phase B was methanol containing 0.1% formic acid. The flow rate was set at 0.2 mL min–1, and the absorbance was measured at 220 nm. The following elution profile was used: 0–5 min −100% A; 5–17.4 min −100% A to 100% B; 17.4- 22.4 min −100% B; then equilibrate back to 100% A.
The mass spectrometer was operated in positive mode with a scan range of 50–2000 m/z. The source conditions were as follows: end plate offset at −500 V; capillary at −4500 V; nebulizer gas (N2) at 1.6 bar; dry gas (N2) at 8 L min–1; dry temperature at 180 °C. Ion transfer conditions were as follows: ion funnel RF at 200 Vpp; multiple RF at 200 Vpp; quadruple low mass at 55 m/z; collision energy at 5.0 eV; collision RF at 600 Vpp; ion cooler RF at 50–350 Vpp; transfer time was set to 121 μs; pre-Pulse storage time was set to 1 μs. Calibration was carried out with sodium formate (10 mM) through a loop injection of 20 μL of standard solution at the beginning of each run.
Theory
Density functional theory calculations were performed on active site model complexes of TxtE based on the crystal structure coordinates of the 4TPO protein data-bank (pdb) file.24 We followed setup procedures as discussed and described previously, but we will summarize the key facts here.53,54 Our initial calculations used a minimal active site model (model A) that included an iron(III)-peroxynitrite-heme (with side chains replaced by hydrogen atoms) and thiolate for the axial cysteinate ligand. A complete tryptophan amino acid was included as substrate, where the carboxylic acid group was deprotonated and the amino group protonated. Hence, the overall charge of model A was −1, and the system was calculated in the individual doublet and quartet spin states. No constraints were put on this model.
A second and more elaborate model (model B) was subsequently studied that included the substrate binding pocket as obtained from the 4TPO pdb file with key hydrogen bonding interactions and salt bridges between substrate and protein (Scheme 1).24 Thus, model B contains model A with one propionate side chain of the heme included and was expanded with part of the substrate binding pocket, namely the Arg59 and Tyr89 side chains, the small peptide chains Asn293-Gln294-Phe295-Thr296-Trp297-Arg298, and Glu394-Phe395, whereby the Gln294 residue was abbreviated to Gly. In the starting structure, the Trp substrate in model B had a zwitterionic state with the amino group protonated and the carboxylate group deprotonated and forming a salt bridge with Arg59. This model has 250 atoms and an overall charge of −1 and is shown in Scheme 1. To make sure that the structure did not deviate too much from the crystal structure coordinates, some geometric constraints were placed on several α-carbon atoms of the protein backbone in the model as identified with a star in Scheme 1. A comparison of starting and final structures of the geometry optimizations showed little changes of the protein part of the model and hence it kept the features of the protein structure intact. In the following section we will identify the model (A or B) after the label of the structure.
Scheme 1. Computational Models Investigated in This Work.

Wavy lines show where a covalent bond was broken.
For Model A, a range of possible reaction pathways for substrate nitration were explored using Gaussian 09,55 see the Supporting Information. Using the obtained insight, we then investigated the low energy pathways with the expanded cluster model B. Based on our previous experience with heme systems, we used well-tested and validated approaches that use density functional theory (DFT) methods.56−58 All geometries were fully optimized without constraints, and transition states were characterized with a single imaginary frequency for the correct mode. For a number of transition state geometries, intrinsic reaction coordinate scans were also performed that connected them to the two local minima adjacent to the transition state (see the Supporting Information).
The hybrid density functional method UB3LYP59,60 was used for all calculations in combination with an LACVP basis set on iron with core potential and 6-31G* on the rest of the atoms (C, N, O, H, and S), basis set BS1.61,62 All optimizations, frequencies, and geometry scans included a conductor-like polarizable continuum model (CPCM) with a dielectric constant mimicking chlorobenzene.63 To obtain more accurate energies, we did single-point calculations with an LACV3P+ basis set on iron with electron core potential and 6-311+G* on the rest of the atoms: basis set BS2. These methods and approaches were used previously and shown to predict the correct regio- and chemoselectivities of reaction mechanisms and to give free energies of activation to within 3 kcal mol–1 of the experiment.64−66
For a selection of small model complexes we tested the effect of an external electric field on the binding energy of NO2 to an indole group using the keyword “Field” as implemented in Gaussian 09.55
Results
Stopped-Flow Analyses
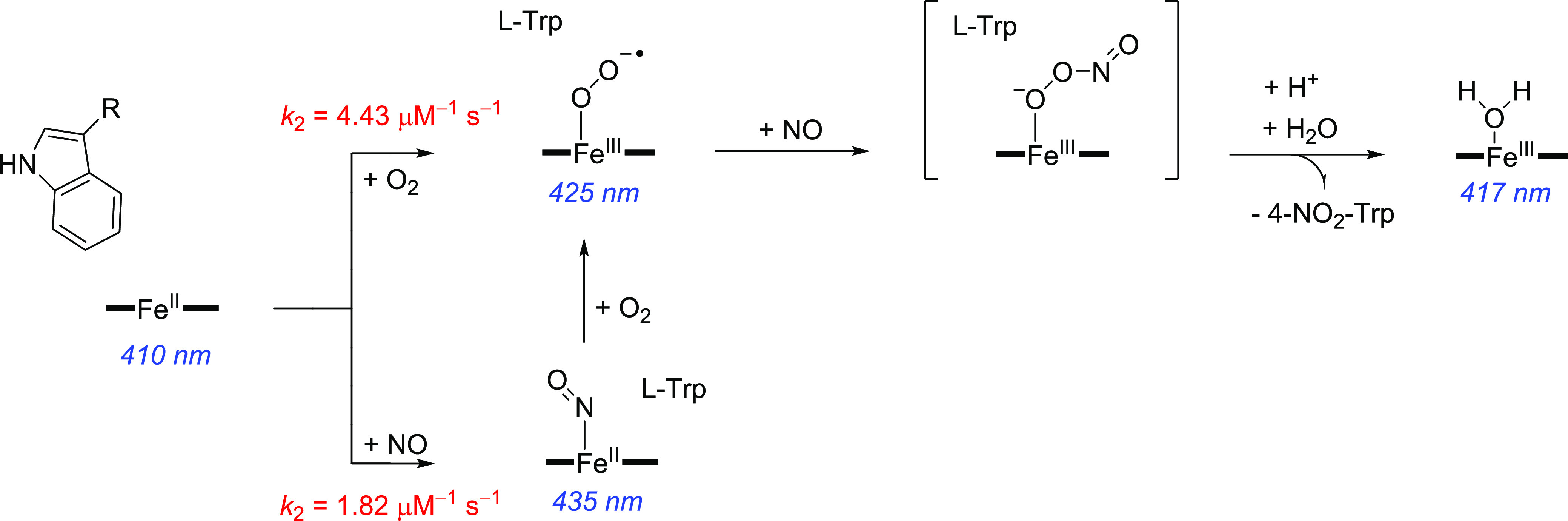
Stopped-flow spectroscopy experiments were performed to determine the order of gas binding. The P450 catalytic cycle for a mono-oxygenation reaction starts with substrate binding in the active site close to the iron(III)-heme in its resting state, followed by release of water from the sixth ligand position of the heme and resulting in conversion of iron from low spin (LS) to high spin (HS). This is followed by a single electron reduction of the heme from HS iron(III) to iron(II) using a redox partner, enabling molecular oxygen binding to form the iron(III)-superoxo complex.1−3,17−19,67,68 A further reduction and two protonation steps result in the formation of Compound I (CpdI), an iron(IV)-oxo heme cation radical species that is the active oxidant in oxygen atom transfer reactions. Trp nitration by P450 TxtE is believed to deviate from this mechanism and upon formation of the iron(III)-superoxo species the enzyme reacts with NO to form an iron(III)-peroxynitrite intermediate or alternatively gives a ferric-nitrosyl complex. Indeed, in all systems studied to date nitrating P450 enzymes are found to be linked to a nitric oxide synthase enzyme that provides NO for the process by reacting l-Arg with O2 on the heme iron.22,24
In order to confirm the mechanism for TxtE and the order of O2 and NO binding, we have used stopped-flow rapid mixing measurements to determine the order of gas binding to the protein. Initial stopped-flow measurements involved mixing the TxtE enzyme against either NO (purified or unpurified) or O2 and showed that characteristic transient iron(II)-NO and iron(III)-superoxo species were formed in each case. Time-resolved spectra, recorded using a photodiode array detector, showed that the absorbance maximum of the heme-iron state transiently shifts from approximately 410 to 425 nm upon binding of O2, and to 435 nm upon binding of NO (Figure 2a), similar to literature reports.69,70 Identical spectral changes were observed when purified NO was used in the measurements, suggesting that any effects from possible impurities caused by disproportionation of the NO source were minimal (see Figure S5, Supporting Information).
Figure 2.

Stopped-flow analysis of the mechanism of gas binding in TxtE. (a) Absorbance spectra measured after 50 ms using stopped-flow photodiode array measurements upon mixing of TxtE iron(II) with either anaerobic buffer, 20 μM O2, or 20 μM NO. (b) Dependence of the rate constant for gas binding to iron(II) heme TxtE on the concentration of either O2 or NO. The data were fitted to a straight line to determine the 2nd order rate constant for O2 or NO binding. (c) Time-resolved spectra at selected time points from double mixing stopped-flow experiments, where iron(II) heme TxtE was first mixed with 20 μM O2, aged for 50 ms, and then mixed with 20 μM NO prior to data collection. The iron(III)-superoxo species at 425 nm is rapidly converted to the water-ligated iron(III) form at 417 nm over 100 ms. (d) Relative amounts of the l-4-nitrotryptophan product formed in samples collected from double mixing stopped-flow experiments, where iron(II) heme TxtE was first mixed with either 20 μM O2 or 20 μM NO, aged for 50 ms, and then mixed with 100 μM NO or O2, respectively. All raw data can be found in the Supporting Information.
Previous studies on dioxygen binding to P450 enzymes also showed a Soret band at 425 nm for the substrate-bound ferric-superoxo species, in agreement with the TxtE data seen here.71 Consequently, the rate of gas binding was measured at the wavelength where the maximum increase in absorbance occurred: in our case we measured the absorbance changes at 435 nm for O2 and at 445 nm for NO over a range of O2 and NO concentrations (Figures S4 and S5 for purified NO, Supporting Information) rather than at the Soret maximum absorbances. In each case, the rate of binding was linearly dependent on the gas concentration, yielding second order rate constants of 4.43 ± 0.03 × 106 M–1 s–1 for O2 binding and 1.82 ± 0.03 × 106 M–1 s–1 for NO binding (Figure 2b). Hence, the rate of binding of O2 is approximately 2.5 times faster than that for NO, implying that O2 is most likely to bind to the enzyme prior to NO. Moreover, this is likely to be particularly evident inside the cell where the concentration of O2 would be expected to be much higher than that of NO.
Next, we attempted to isolate the putative iron(III)-peroxynitrite intermediate and to confirm the order of gas binding in TxtE through a double mixing stopped-flow experiment. To this end, TxtE was first mixed with O2 or NO, allowed to age for 50 ms to form the initial iron(III)-superoxo or iron(II)-NO species, and then mixed with the second gas prior to collection of the time-resolved spectral changes. Measurements where TxtE was first mixed with NO, followed by a second mix with O2, only appeared to show the formation of the iron(II)-NO species at 435 nm on time scales faster than 1 s (Figure S8, Supporting Information). On longer time scales (<100 s) the 435 nm peak slowly disappeared and a new peak at 425 nm was formed, which is likely to represent the iron(III)-superoxo species as the bound NO is gradually displaced by O2 (Figure S8, Supporting Information). Hence, it appears that O2 is unable to react directly with the iron(II)-NO intermediate to form any new species, suggesting that dioxygen must bind prior to NO in order to produce a catalytically active species.
The reverse experiments, involving an initial mix with O2 followed by a second mix with NO, were slightly more complicated as the iron(III)-superoxo species decays over approximately 1 s, even upon mixing with anaerobic buffer. It is converted to a new species with an absorbance maximum at 417 nm, which represents the water-ligated iron(III) form of the enzyme (Figure S9, Supporting Information). Nevertheless, clear differences are observed upon binding of NO to the iron(III)-superoxo complex, implying that binding of dioxygen prior to NO appears to give a more likely catalytic intermediate than the reverse ordering. The iron(III)-superoxo species is converted to the same water-ligated iron(III) species much more rapidly (<100 ms) upon mixing with NO (Figures 2c and S10, Supporting Information), presumably as a result of catalysis to form the l-4-nitrotryptophan product. On longer time scales (>4s) the l-Trp substrate rebinds to this iron(III) to give rise to starting substrate-bound heme spectrum at 390 nm (Figure S8).
Further confirmation of the binding order was provided by additional double mixing experiments, where equal concentrations of O2 and NO were initially mixed and allowed to age for 50 ms, prior to mixing with the enzyme (Figure S9). In this case, there was no observable formation of the iron(II)-NO species at 435 nm. Instead, rapid formation of the iron(III)-superoxo species was observed at 425 nm, followed by the rapid decay to the water-ligated iron(III) species at 417 nm, in a similar manner to the previous binding of NO to the iron(III)-superoxo complex. It was not possible to identify any additional catalytic intermediates in the double mixing photodiode array experiments, which implies that the rate of formation of the proposed peroxynitrite species is likely to be much slower than the subsequent reaction chemistry.
Consequently, in order to provide further evidence that the catalytic iron(III)-peroxynitrite intermediate is formed directly upon NO binding to the iron(III)-superoxo species, we collected samples from the double mixing stopped-flow experiments and measured the amount of l-4-nitrotryptophan product that had been formed (Figures 2d and S10, Supporting Information). Upon mixing of the iron(II)-NO complex with different concentrations of O2, only a small amount of l-4-nitrotryptophan was formed, presumably because the NO is gradually displaced by O2 prior to collecting and freezing the samples from the stopped-flow instrument. However, the amount of the L-4-nitrotryptophan product is significantly higher when the iron(III)-superoxo species is mixed with NO (Figure 2d). Moreover, the amount of product increases with the concentration of NO used in the second mix (Figure S10, Supporting Information). This indicates that efficient reaction chemistry only occurs when O2 binds first to form the iron(III)-superoxo species, followed by NO binding to generate the catalytically relevant iron(III)-peroxynitrite intermediate. The experimentally derived catalytic cycle of P450 TxtE, based on the stopped-flow experiments presented here, is shown in Scheme 2. The work shows that both dioxygen and NO can bind to an iron(II) pentacoordinated heme structure, although the dioxygen binds faster. Subsequently, the iron(III)-superoxo species rapidly reacts with NO and one molecule of substrate to form 4-nitro-L-tryptophan, and returns the enzyme to the resting state. Unfortunately, the peroxynitrite complex was too short-lived to characterize and study in detail, and hence a computational study was performed.
Scheme 2. Reaction Mechanism of TxtE As Derived from Stopped-Flow Measurements.
Computation
To gain further insight into the catalytic cycle of TxtE and related nitrating enzymes, we pursued a computational study using model complexes based on the active site and substrate binding environment of the enzyme. Our cluster model had 250 atoms and is shown above in Scheme 1. Our initial focus was on the potential role of peroxynitrite in the reaction mechanism, but the substrate activation reactions were also investigated. As we propose free NO2 to be formed during the catalytic cycle of TxtE, we decided to explore substrate activation on the C4, C5, C6, and C7-positions of l-Trp. In particular, the reaction energy with zero-point energy (ZPE) correction (ΔE+ZPER) for the formation of iron(III)-peroxynitrite from either iron(III)-superoxo + NO (eq 1) or iron(II)-nitrosyl + O2 (eq 2) was calculated. To this end, the individual molecules in reactions 1 and 2 were calculated with the small model complex A in the absence of substrate and the energy differences for these reactions are shown in Scheme 3.
| 1 |
| 2 |
Scheme 3. UB3LYP/BS2//UB3LYP/BS1+ZPE Calculated Mechanisms for Iron(III)-Peroxynitrite Formation.

At the UB3LYP/BS2//UB3LYP/BS1+ZPE level of theory, both are exothermic, with values of −11.4 kcal mol–1 for reaction 1 and −36.0 kcal mol–1 for reaction 2. Note that, with the large model complex, the energy for reaction 1 was calculated to be ΔE+ZPER = −12.3 kcal mol–1; hence the protein environment has little effect on the iron(III)-peroxynitrite bond formation.
However, the reaction rates for the two processes give the opposite ordering and a barrier height with magnitude ΔE‡+ZPE = 14.0 kcal mol–1 was calculated for reaction 1, while reaction 2 is estimated to be well over 27 kcal mol–1 using gas-phase model complexes. This reaction barrier is in good quantitative agreement with calculations on a biomimetic iron-porphyrin model complex with an axial imidazole group for an O-bound iron(III)-peroxynitrite dissociation into an iron(IV)-oxo and NO2.72 Furthermore, DFT calculations of large cluster models of heme-NO and heme-O2 species showed that O2 binds more strongly than NO.73 It should be mentioned here that the reaction of iron(III)-superoxo with NO gives an O-bound iron(III)-peroxynitrite structure, whereas the reaction of iron(II)-NO with O2 gives the less stable N-bound iron(III)-peroxynitrite that is rapidly released from the iron center and forms a pentacoordinated iron(II)-heme and a free peroxynitrite ion. As such, the DFT calculations on small cluster models support a faster reaction of iron(III)-superoxo with NO than iron(II)-NO with O2. Moreover, they show that iron(II)-NO with dioxygen does not give an iron(III)-peroxynitrite directly but instead a relatively unstable N-bound iron(III)-peroxynitrite that either reacts to form iron(III)-superoxo by NO release or reorients to form the O-bound iron(III)-peroxynitrite complex. The driving force for converting the iron(II)-NO + O2 into an iron(III)-superoxo and a NO molecule was calculated to be exothermic by 24.6 kcal mol–1.
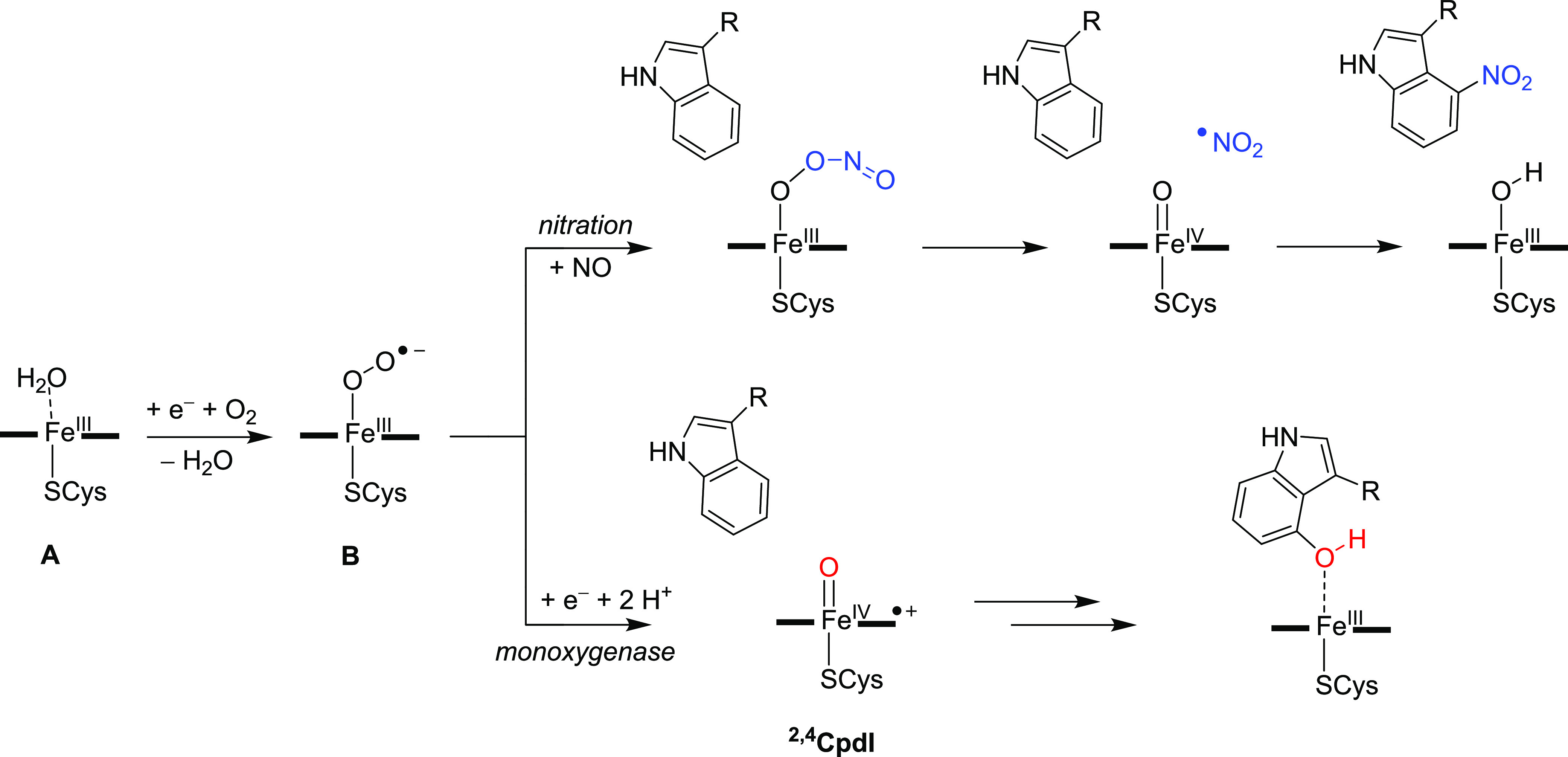
Next, we explored the reaction mechanism of substrate activation in TxtE, leading to substrate nitration and hydroxylation as displayed in Scheme 4. Initially, the work focused on the steps from the iron(III)-peroxynitrite complexes in the doublet and quartet spin states, and we investigated three major reaction pathways. These iron(III)-peroxynitrite complexes (4,2Re) were geometry optimized with a large active site model that includes the substrate and the binding pocket. Pathways 1 and 2 start with a homolytic O–O bond cleavage in the peroxynitrite group to form Compound II, i.e. an iron(IV)-oxo heme and NO2 (complex Int1) via a transition state TS1. Thereafter, pathways 1 and 2 diverge and substrate attack of either NO2 or iron(IV)-oxo were investigated. In pathway 1, the free NO2 radical attacks the substrate directly to form intermediate Int4 via transition state TS4, which after hydrogen atom transfer to the iron(IV)-oxo species via transition state TS5 leads to nitrated Trp and the ferric-hydroxide complex, i.e., protonated Compound II (Int5).
Scheme 4. Mechanistic Pathways Explored for the Reaction of Iron(III)-Peroxynitrite with l-Trp on the C4-Position Using DFT Methods.

In pathway 2, by contrast, an electrophilic addition of the oxo to a carbon atom of the Trp substrate takes place to form a radical intermediate Int2 via a transition state TS2. This is then followed by abstraction of the ipso-proton from the Trp carbon atom by the heme to form the protonated heme structure Int3 via transition state TS3. A proton reshuttle leads to phenol products (Int6) or alternatively the NO2 radical attacks the ipso-position to form nitrated Trp products (Int5).
For the small model complex, a small barrier of ΔE‡+ZPE = 12.2 kcal mol–1 for peroxynitrite dissociation into CpdII and NO2 was found via 2TS1A. Much larger barriers are found for the next steps in the mechanism, namely for the attack of NO2 on the substrate (via 2TS4A) or the oxo attack on the substrate (via 2TS2A). With the small model complex, we find pathway 1 is favored over pathway 2, whereby 2TS2A is calculated to be higher in energy than 2TS4A by at least 4 kcal mol–1 (Tables S4 and S6, Supporting Information). Subsequently, the full mechanism was calculated for nitration at the C4-, C5-, C6-, and C7-positions of l-Trp using the large model B. Furthermore, as substrate is bound in the substrate binding pocket with different residues on both sides, we considered attacks on either side of the aromatic ring, which are designated as the endo- and exo-pathways. In addition, to these two pathways, we also tested a direct attack of heme-bound peroxynitrite onto these carbon-positions of l-Trp to form intermediate Int4 using model A. However, this pathway (pathway 3; Figure S20, Supporting Information) is very high in energy and did not lead to NO2 transfer to substrate, but instead the peroxynitrite structure stayed intact. As such, peroxynitrite will need to be activated first and split into NO2 and an iron(IV)-oxo species in order to be reactive. Consequently, pathway 3 was ruled out for further studies. We also attempted a reaction where NO2 attacks the substrate in structure 2,4Int2A to form either Int4A or Int5A directly. However, no viable low energy pathways were found either.
Subsequently, we focused on the large model structure (model B), and calculated the full reaction landscape starting from a peroxynitrite bound structure. The potential energy landscape for the reaction of the iron(III)-peroxynitrite complex with l-Trp on the lowest energy doublet and quartet spin states for pathway 1 for attack on the C4-exo position are given in Figure 3. The reactant states (ReB) of the heme active site with substrate bound are in a doublet spin ground state as expected for iron(III) complexes, while the quartet spin state is ΔG = 3.6 kcal mol–1 higher in free energy. There is a small barrier of ΔG‡ = 3.3 kcal mol–1 via 2TS1B for homolytic O–O cleavage to form iron(IV)-oxo and an NO2• radical. The quartet spin barrier is much higher in energy, i.e., ΔG‡ = 14.5 kcal mol–1, and therefore the first reaction step will take place on a dominant doublet spin state surface.
Figure 3.

Potential energy landscape (with values in kcal mol–1) of l-Trp nitration at the C4-position via exo-attack in an active site model complex B. All geometries optimized in Gaussian at UB3LYP/BS1. The two energetic values represent ΔE+ZPE (ΔG) data with energies obtained using basis set BS2 and with ZPE and solvent corrections; free energies are given in parentheses at 298 K and contain ZPE, thermal, solvent and entropic corrections. Optimized geometries of the transition states give bond lengths in angstroms and the imaginary frequency in cm–1.
The peroxynitrite-cleaving transition state geometries have substantially elongated O–O distances of 1.902 (1.766) Å for 2TS1B (4TS1B) and visualization of the imaginary frequency shows an O–O bond cleavage mode. The first reaction step is virtually thermoneutral on the doublet spin state and brings the quartet spin state to within a fraction of a kcal mol–1. This is not surprising as, electronically,4,2Int1B can be characterized as an iron(IV)-oxo heme complex, i.e. Compound II (CpdII), with an NO2 radical in close proximity. Indeed the group spin densities (Supporting Information) of 4,2Int1B show a full spin on the NO2 group. The molecular valence orbitals of 4,2Int1B (shown in Figure S29, Supporting Information) confirm that CpdII has an electronic configuration with two unpaired electrons in π* orbitals (π*xz and π*yz) for the antibonding interaction of a 3d iron orbital with a 2p on the oxo group. The π*yz orbital mixes somewhat with a π*NO2 orbital and hence there is a small but weak long-range interaction between CpdII and NO2. The heme-based a2u orbital is doubly occupied in CpdII, in contrast to CpdI where it is singly occupied. Thus, the two unpaired electrons in π*xz and π*yz are ferromagnetically coupled to the NO2• radical (in orbital π*NO2) into an overall quartet spin state or antiferromagnetically coupled into an overall doublet spin state. As such, there are two different spin-configurations for the CpdII structure with overall doublet and quartet spin. These two configurations are close in energy and indeed we find them within 1 kcal mol–1.
In previous work on P450 reactivity by Compound I (CpdI), i.e., the iron(IV)-heme cation radical species which is the one electron oxidized form of CpdII, it was found to also have close lying doublet and quartet spin state structures, but with π*xz1 π*yz1 a2u1 configuration.74−76 Thus, reaction mechanisms with substrates were shown to proceed on competing spin-state surfaces with competing rate-constants. Moreover, examples have been highlighted in the past, where product distributions were different on each of the spin-state surfaces.77,78 Therefore, we calculated the full mechanism of l-Trp nitration on the doublet and quartet spin states for all reactions.
We attempted to optimize the geometry of the product from heterolytic cleavage of peroxynitrite to give either 4,2CpdI + NO2– or an iron(III)-oxo(heme) + NO2+. However, in both cases when we swapped molecular orbitals in 4,2Int1B, the SCF converged back to the original conformation. Therefore, the heterolytic cleavage pathways are high in energy and are unlikely scenarios for the TxtE and can be ruled out. As such we do not expect either NO2+ or NO2– to be involved in the substrate nitration reaction mechanism.
Subsequently, we investigated the addition of NO2 to l-Trp substrate from the 2,4Int1B structures and, as an example, we show the calculated mechanism for activation of the C4-exo-position of l-Trp by CpdII in Figure 3. The C–N bond formation transition states take place via barriers 2,4TS4B,C4-exo. On both the doublet and quartet spin state surfaces, small barriers that are within ΔG‡ = 1 kcal mol–1 of 4,2Int1B are found. Nevertheless, both transition states were characterized as shown in Figure 3 with a single imaginary frequency of i178/i179 cm–1 representing a C–NO2 vibrational stretch. As the transition states represent NO2 transfer to l-Trp and no electron is transferred to CpdII, there are little changes to the geometry of the CpdII part as compared between 2,4Int1B and 2,4TS4B,C4-exo, and geometrically those parts of the structure are the same.
The transition state is a pure NO2• attack on the aromatic ring of l-Trp and no electron transfer to the heme takes place. The shallow intermediate (4,2Int4B,C4-exo) is followed by another low-energy barrier, namely the hydrogen atom transfer from l-Trp to CpdII, which could not be characterized for the C4-exo pathway and hence will proceed very quickly. As such, these reactions will be fast and efficient, and it is unlikely that NO2• will drift out of the substrate binding pocket if substrate is present. Obviously, it is important that NO2• does not drift out of the protein pocket as it is known to activate aromatic amino acids and could react with enzymatic residues if the substrate is not positioned appropriately.35 In particular, the substrate binding pocket of the 4TPO PDB file24 is lined with several aromatic residues, e.g., Phe79, Trp82, Tyr89, Trp297, and Phe395. Clearly, peroxynitrite activation and the production of NO2 should not lead to nitration of any of these active site residues. Therefore, NO2 should be used by the protein as soon as it is formed and, based on the low barriers found here, they clearly implicate a rapid substrate activation.
As our proposed mechanism of l-Trp nitration by P450 enzymes suggests the formation of a free NO2 radical after homolytic cleavage of peroxynitrite, we decided to explore pathways leading to C4-, C5-, C6-, and C7-nitration for model B, and the results are summarized in Table 1. Of course, these mechanisms have two stereochemical isomers, which are designated as the exo- and endo-pathways. Optimized geometries of all 2,4TS4B transition states are given in the Figure S25 (Supporting Information), while the one for C4-exo is given above in Figure 3. As this reaction step proceeds away from the heme center and no electron transfer takes place, it means that the heme CpdII distances, i.e. the Fe–O and Fe–S interactions, are very close for all structures. A little more variation is seen in the C–N bond that is being formed in the transition state. The shortest distances are found for C5-activation whereby the endo-pathway gives the shortest distances of 1.714 Å, while those for the exo-pathway are 1.721 Å. By contrast, much longer C–N distances are found for the C7-activation with values of 1.830/1.831 Å for 2TS4B,C7-endo/4TS4B,C7-endo and 1.827/1.842 Å for 2TS4B,C7-exo/4TS4B,C7-exo.
Table 1. Relative Energies of NO2 Transfer to Various Positions of the l-Trp Substratea.
| pathway | 2TS4B (4TS4B) | 2Int4B (4Int4B) | 2TS5B (4TS5B) | 2Int5B (4Int5B) |
|---|---|---|---|---|
| C4-exo | >0.0 (>0.0) | –3.5 (−3.5) | >-3.0 (>-3.0) | –58.0 (−23.2) |
| C4-endo | 6.6 (>0.0) | 6.4 (−1.4) | 11.0 (15.0) | –49.2 (−14.9) |
| C5-exo | 0.9 (0.9) | –0.6 (0.0) | 16.9 (17.9) | –49.6 (−15.8) |
| C5-endo | >3.0 (>0.0) | 2.7 (−4.2) | 8.3 (10.3) | –57.7 (−23.6) |
| C6-exo | 0.6 (>0.0) | 0.3 (−0.4) | 6.1 (6.6) | –40.8 (−46.9) |
| C6-endo | 3.2 (2.2) | 0.2 (1.1) | 6.4 (6.1) | –45.9 (−40.8) |
| C7-exo | 2.4 (2.4) | 0.2 (0.2) | 2.7 (4.5) | –60.9 (−56.1) |
| C7-endo | >0.0 (>0.0) | –1.6 (−1.5) | 1.8 (3.8) | –56.9 (−27.2) |
Values in kcal mol–1 represent ΔE+ZPE data with energies obtained at UB3LYP/BS2//UB3LYP/BS1 relative to 2ReB.
Next, we looked at the energetics for NO2 transfer to the C4-, C5-, C6-, and C7-positions of the l-Trp substrate through attack from the exo and endo directions. We located C–N bond formation transition states (2,4TS4B) and the subsequent local minima (2,4Int4B), as well as the ipso-hydrogen atom abstraction transition state (2,4TS5B) by CpdII leading to the nitrated product complexes. 2,4Int5B and the energies with respect to 2ReB for all pathways are given in Table 1. Several barriers for C–N bond formation via TS4B are very low in energy and could not be located precisely, but geometry scans find them to be less than 1 kcal mol–1 above the Int1B intermediate. These pathways should lead to addition of NO2 to l-Trp at the C4-exo, C4-endo, C5-endo, C6-exo, and C7-endo positions. As a matter of fact, most TS4B barriers are found within a small window of 4 kcal mol–1 and only doublet spin C4-endo is higher in energy, i.e. its transition state is about 7 kcal mol–1 above 2Int1B.
The NO2 addition local minimum (2,4Int4B) is followed by hydrogen abstraction from the ipso-position by CpdII to form nitrated Trp and ferric-hydroxide. Due to the position of the substrate in the binding pocket, in several cases substantial hydrogen abstraction barriers are found. Thus, the C6- and C7-positions of l-Trp are pointing toward the iron(IV)-oxo group of the heme and hence, when NO2 attacks the substrate at these positions, their hydrogen atoms can easily be abstracted by CpdII. Indeed, TS5B barriers for C6- and C7-nitration of less than 7 kcal mol–1, with respect to isolated reactants, are those mostly found. By contrast, the C5-exo position points away from the heme and consequently hydrogen atom transfer is difficult and high barriers are obtained as a result of major structural rearrangements.
Finally, we investigated the possible aromatic hydroxylation reaction of P450 TxtE (bottom-right pathway in Scheme 4). As shown before, the aromatic hydroxylation by a metal(IV)-oxo species starts with an electrophilic addition of the oxo to the aromatic ring, followed by proton shuttle from the aromatic ipso-position to a nearby proton acceptor group, such as the nitrogen atoms of the porphyrin ring, prior to relay of the proton to the oxygen atom to form hydroxylated product.79−81 This mechanism, i.e., pathway 2 in Scheme 4, was found to be high in energy for the quartet spin state (ΔE‡+ZPE > 30 kcal mol–1 for 4TS2A and 4TS3A) and, although somewhat lower on the doublet spin state surface (>25 kcal mol–1), is still much higher in energy than the NO2 transfer step to the substrate. Therefore, substrate hydroxylation will not be a feasible pathway in the presence of free NO2 radicals. This is in agreement with experimental work on P450 TxtE that failed to identify hydroxylated l-Trp products.
An alternative to the electrophilic aromatic hydroxylation, that started with a hydrogen atom abstraction from the substrate by the iron(IV)-oxo species in Int2, was also investigated. In particular, the hydrogen atom abstraction from the C6 position was explored for the large model system. However, high energy hydrogen atom abstraction free energies of activation of ΔG‡ = 23.5 kcal mol–1 and 22.8 kcal mol–1 for doublet and quartet, respectively, were obtained (Supporting Information, Figure S27). Therefore, a direct hydrogen atom abstraction from the aromatic ring of l-Trp can be ruled out as a viable mechanism as expected given the high C–H bond strength.
Overall, the work shows that the aromatic nitration by P450 TxtE is triggered by a reaction of NO with an iron(III)-superoxo species to form iron(III)-peroxynitrite that releases NO2• rapidly. These NO2• radicals react with the indole ring of l-Trp through addition and the nearby CpdII species abstracts the ipso-hydrogen atom to form the 4-nitrotryptophan products. However, as free NO2• is formed, this means regioselectivity is determined by the tightness of substrate binding and the orientation of Trp in the substrate binding pocket. How this selectivity is determined, we discuss in further detail below.
Discussion
In this work a combined experimental and computational study on the P450 aromatic nitration enzyme TxtE is reported. These studies focus on the catalytic reaction mechanism and the nature of the selectivity of the reaction. Overall, our studies show that the mechanism is distinctly different from conventional P450 mono-oxygenases, where an active species (Compound I, CpdI) is initially formed and subsequently reacts with substrates.1–5 To explain the differences in the catalytic reaction cycle of P450 TxtE and P450 mono-oxygenases in general, we compare the two reaction mechanisms side-by-side in Scheme 5. Both catalytic cycles start from an iron(III)-heme complex that is linked to the protein through a thiolate bridge with a cysteinate residue. The sixth coordination site of the iron(III) is occupied by a water molecule in the resting state (structure A in Scheme 5). Upon substrate binding the water molecule is released and the metal complex undergoes a spin-state change from low-spin to high-spin. DFT calculations showed that the high-spin is preferred over the low-spin in the 5-coordinate iron(III) complex, whereas the spin-state ordering is reversed in 6-coordinate environments.82 Thereafter, the complex is reduced by the redox partner and binds molecular oxygen (complex B in Scheme 5). Beyond complex B, the catalytic cycles of P450 mono-oxygenases and P450 nitration enzymes diverge. Thus, the mono-oxygenases reduce structure B and after two proton transfer steps generate a high-valent iron(IV)-oxo heme cation radical species called CpdI. The P450 TxtE isozymes, by contrast, bind a molecule of NO that reacts with the iron(III)-superoxo intermediate (B) to form an iron(III)-peroxynitrite intermediate.
Scheme 5. Comparative Catalytic Cycles of P450 Nitration and P450 Mono-Oxygenase Enzymes.
As discussed above, several pathways for nitration of l-Trp substrate were calculated here and our lowest energy mechanism involves a reaction that starts with the iron(III)-peroxynitrite splitting into Compound II (CpdII), i.e., iron(IV)-oxo heme and NO2• radical. The latter then attacks the substrate at the C4-position and the ipso-hydrogen atom at C4 moves to CpdII to generate an iron(III)-hydroxo complex. A proton transfer from the protein or solvent then brings the catalytic cycle back into the resting state. At this stage it is not clear whether this proton transfer step happens prior to, or after, product release.
The catalytic cycles of P450 nitration and mono-oxy-genation in Scheme 5 show dramatic differences. Thus, P450 TxtE would require only a single electron from a redox partner and no proton transfer to generate an iron(III)-peroxynitrite as the active species, whereas P450 mono-oxygenases need two electrons and two external protons to form CpdI. However, one proton transfer is needed in P450 TxtE to convert protonated CpdII back to the resting state. To understand these differences in proton relay mechanisms during the catalytic cycle of P450 TxtE and general P450 mono-oxygenases, we analyzed structures of representative nitration and mono-oxygenase enzymes (Figure 4). In particular, Figure 4 displays an extract of the active site structures of P450 TxtE (4TPO PDB as a representative of a nitrating P450)24 and P450 CAM (3WRH PDB as a mono-oxygenase P450 structure).83,84
Figure 4.

Extracts of the crystal structure coordinates of P450 TxtE (left) and P450 CAM (right) as taken from the 4TPO and 3WRH PDB files. Amino acids are labeled as in the PDB files.
P450 CAM hydroxylates camphor regio- and stereoselectively at the C4-position and its mechanism has been extensively studied with experimental and computational methods.85−88 As rationalized and discussed previously,73,87 P450 CAM has a number of active site residues involved in proton-relay during the catalytic cycle. First, a buried Glu366 residue is connected via a chain of water molecules to the alcohol group of Thr252 that is located just above the heme. The Glu366 residue is believed to deliver the first proton in the catalytic cycle of P450 mono-oxygenases,89 while Asp251 delivers the second one. The Asp251 residue points out of the active site, but computational modeling showed that a rotation of the side chain brings the carboxylate group close to the Thr252 alcohol group, and thus Asp251 can deliver a second proton into the substrate binding pocket to complete the formation of CpdI.
An analysis of the TxtE structure of 4TPO shows that the Glu366 residue is missing and is replaced by a Gln residue. Nevertheless, the Gln environment is similar to that of Glu366 in P450 CAM, namely at the end of a cavity surrounded by apolar residues, such as Leu and Ile. Interestingly, Val253 is not seen in P450 TxtE and is replaced by another Thr residue (Thr251). Nevertheless, it is clear from the TxtE structure that one proton source is lacking in the active site. Therefore, proton transfer in P450 TxtE will be considerably slower than in P450 CAM, where one proton may already be stored on the Glu366 residue prior to the start of the catalytic cycle, and which should lead to efficient protonation of the iron(II)-peroxo to form an iron(III)-hydroperoxo complex.
In P450 CAM the second proton in the catalytic cycle is proposed to be delivered through the rotation of Asp251. In the TxtE structure, however, this Asp residue is missing and a proline residue takes its position. Therefore, the proton transfer residues in P450 TxtE have been mutated with respect to typical mono-oxygenase structures, and consequently proton transfer into the active site for the conversion of the iron(III)-superoxo species into CpdI is hampered. This should give the iron(III)-superoxo species a relatively long lifetime until NO is delivered and can form a heme-peroxynitrite complex.
The electron transfer processes that happen in the reaction of the iron(III)-superoxo species with NO to form NO2 via a peroxynitrite intermediate are given in Figure 5 as a valence bond (VB) diagram. VB schemes have been extensively used to explain electron transfer processes in reaction mechanisms and, in particular, were found to be useful to explain regio- and chemoselectivities of reaction processes and make predictions on what bonds are most likely activated in a substrate.90−93 These VB structures give a bond orbital occupied by two electrons as a line separated by two dots, while individual (unpaired) electrons are given as dots. Thus, the isolated iron(III)-superoxo structure has an open-shell singlet ground state with orbital occupation δx2–y22 3dxz2 πyz2 π*yz1 πOO,yz2 π*OO,yz0 πOO,xz2 π*OO,xz1.76 The πOO and π*OO orbitals represent the bonding and antibonding orbitals along the superoxo bond in the xz and yz planes. The metal-type orbitals contain the doubly occupied δx2-y2 orbital, which is a nonbonding orbital in the plane of the heme with lobes pointing toward the meso-carbon atoms. Then there are three electrons along the Fe–O bond in πyz(2) and π*yz(1) for the bonding and antibonding interaction of the metal 3dyz with a 2py on oxygen. This interaction is described as a two-center-three-electron bond between the Fe and O atoms in Figure 5. In the xz plane the 3dxz iron orbital is not involved in interactions with any of the oxygen atoms as the peroxo group is in that plane so, in contrast to CpdII, it is a doubly occupied atomic orbital in the iron(III)-superoxo form: 3dxz2. The approaching NO molecule has one unpaired electron on the nitrogen atom. Upon attack of the NO on the superoxo radical an N–O bond is formed by the pairing up of the π*OO,xz and πN radicals to form the peroxynitrite πNO bond with two electrons. At the same time, the two electrons from the πOO,yz orbital in the iron(III)-superoxo structure become the lone-pair electrons on bridging oxygen of the peroxynitrite group: lpO. This then generates the iron(III)-peroxynitrite intermediate, i.e., the reactant complex Re in Figure 3 and Scheme 4, with orbital configuration δx2–y22 3dxz2 πyz2 π*yz1 πOO,xz2 π*OO,xz0 πNO2 lpO2. Consequently, the bonding and antibonding type electrons involved in the superoxo bond in the iron(III)-superoxo species are redistributed over the chemical system and a single bond remains between the two oxygen atoms, i.e., dioxygen configuration πOO,xz2 π*OO,xz1.
Figure 5.
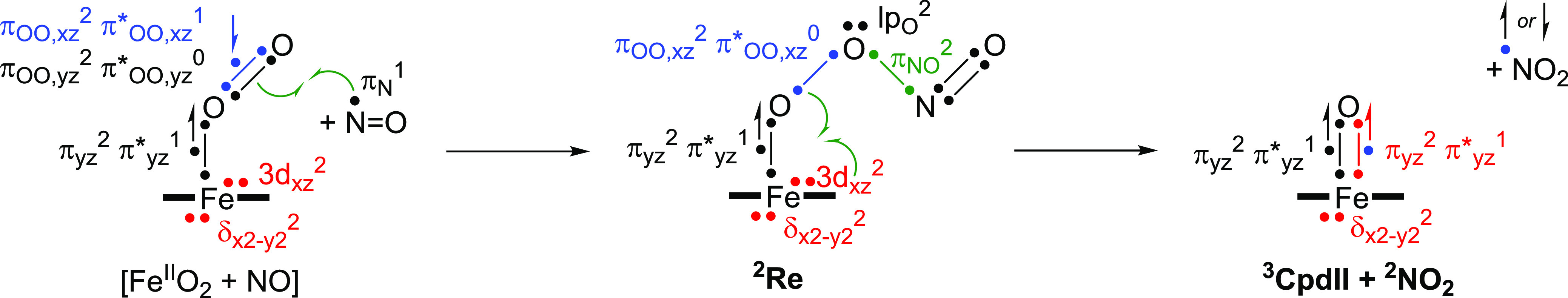
Valence bond scheme for the electron migrations during the conversion of the ferric-superoxo and NO into CpdII and NO2 via a peroxynitrite intermediate.
Next, the O–O bond breaks homolytically to form NO2 and a CpdII fragment by moving one electron from the πOO,xz orbital in 2Re into the Fe–O bond. This electron pairs up with the two electrons of the 3dxz orbital on the iron to form the new two-center three-electron bond with πxz2 π*xz1 configuration. CpdII is in a triplet spin ground state,91 with π*xz1 π*yz1 configuration that is either antiferromagnetically coupled to a radical on NO2 in a doublet spin state or ferromagnetically coupled into a quartet spin state. Indeed, 4,2Int1 are close in energy, while in 4,2Re the doublet and quartet spin states are well separated. The calculations reported in this work show that a reaction of iron(III)-superoxo with NO rapidly forms iron(III)-peroxynitrite complexes. Moreover, the subsequent reaction for homolytic cleavage of peroxynitrite to form NO2 and CpdII has a calculated barrier of only ΔG‡ = 3.3 kcal mol–1 and hence should proceed rapidly. These results are consistent with recent work of the Moënne-Loccoz and Karlin and their co-workers on a biomimetic nonheme Cu(II)-peroxynitrite complex that was found to homolytically cleave into NO2 and a copper(II)-oxo species.72,94
As shown in this work, homolytic cleavage of peroxynitrite gives CpdII and free NO2, which can add to the π-system of the substrate. To find out if a bare indole group in the gas-phase would have a preference for nitration of a specific carbon site in l-Trp, we calculated the C–N bond formation energy by addition of NO2• to methylindole, see Figure 6. As can be seen in all cases, the addition of NO2• to methylindole is endothermic by 10.5–15.3 kcal mol–1 in the gas-phase with a preference for addition at the C4-position. As such, if all carbon atoms of l-Trp are accessible in the protein then a reaction with NO2 should give a dominant C4-nitration. Indeed experimental studies on l-Trp activation by P450 TxtE have confirmed that this is the only detectable product.22−24,32 The results of the calculations reported in Table 1 above show that the lowest NO2 addition barrier is for the C4-exo pathway. However, the table shows several alternative pathways to C5-, C6-, and C7-nitration to be close in energy. Hence, a mixture of products is expected in an open substrate binding pocket. Consequently, a tight binding of substrate and oxidant that brings NO2• and the target position of substrate close is essential for P450 TxtE.
Figure 6.
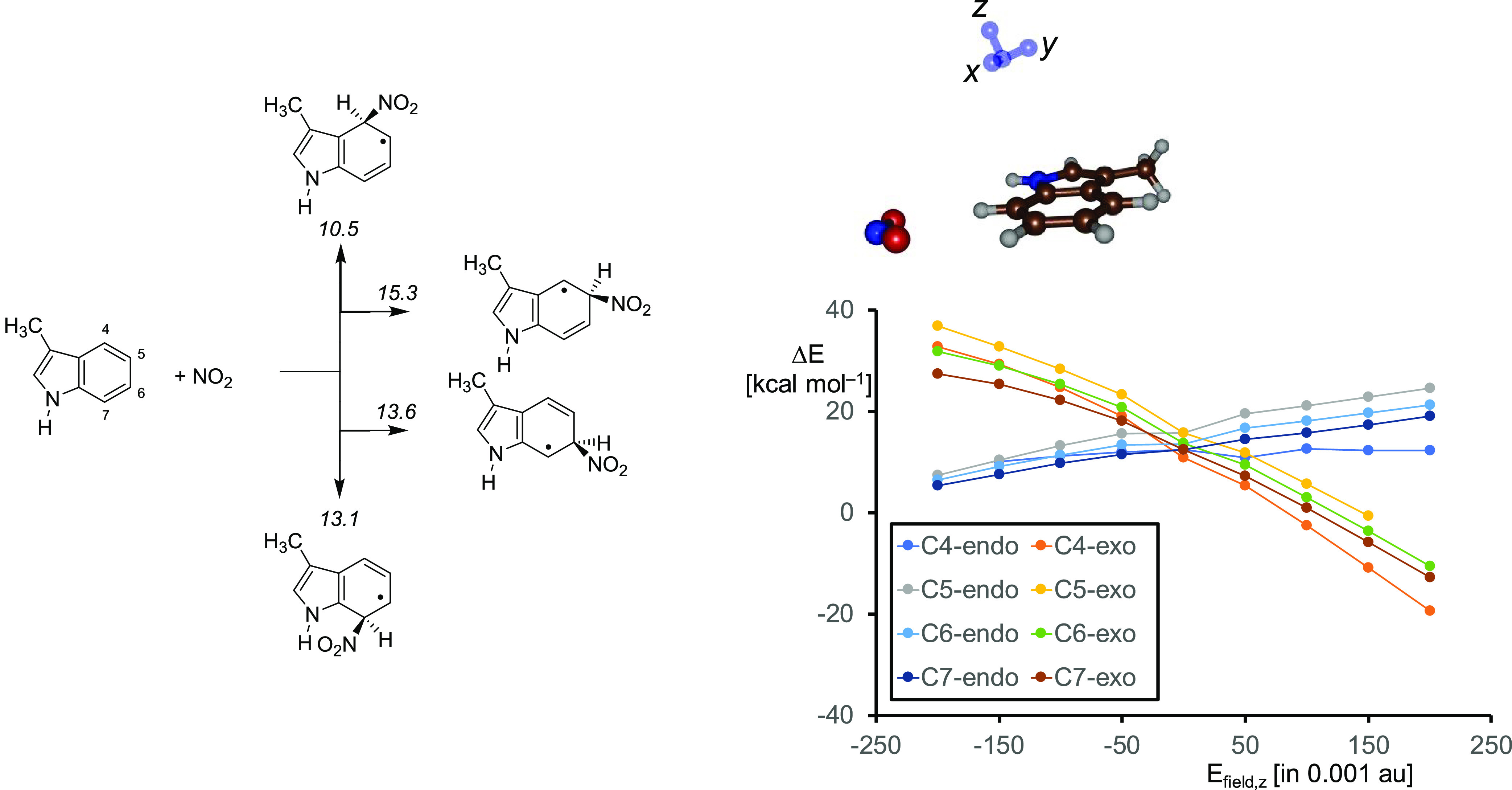
(a) UB3LYP/6-31G* calculated C–N bond formation energies in a bare methylindole molecule. Values are ΔE+ZPE data in kcal mol–1. (b) Electric field effect (with Efield in units of 0.001 au) on the relative energies of NO2 addition to methylimidazole along the z-axis.
Interestingly, the addition of NO2 to the aromatic ring of methylindole is endothermic by well over 10 kcal mol–1, whereas the same step in the large enzymatic structure is almost thermoneutral as seen by the energy differences for model B of Int1 and Int4 in Figure 3. By contrast, for the small model complex without the protein environment (model A) an energy of ΔE+ZPE = 19.4 (doublet) or 18.6 (quartet) kcal mol–1 is obtained for the same reaction step. Therefore, the protein environment stabilizes complex Int4 dramatically and enables a favorable substrate nitration reaction with low barriers.
To find out how the long-range interactions stabilize the NO2 adduct Int4 we took the methylindole structures and calculated their energies through the addition of an external electric field.95,96 Electric fields were applied to the molecular x-, y-, and z-axes, whereby the y-axis is along the C–NO2 bond that is being formed and the z-axis along the C–ipso-H coordinate as shown on the top-right in Figure 6. These applied electric fields are of a magnitude similar to those used in vibrational Stark effect spectroscopy measurements on enzymatic systems and hence mimic realistic perturbations.97 With a field along the x- and y-axis small energy changes are seen, but along the z-axis these changes are major (see Figure 6b). Therefore, the barrier height of the reaction is linked with the movement of the C–H bond from in-plane with respect to the Trp group to make space for the in-coming nitro group. Thus, an applied electrical field along the z-axis stabilizes the NO2 adduct with methylindole dramatically and changes the reaction energy for adduct formation from endothermic by 10 kcal mol–1 to exothermic by 10 kcal mol–1. Particularly, the C4-addition pathway is stabilized with an electric field effect along the z-axis and enhances the selectivity of the reaction. As a consequence, the protein is able to stabilize and drive an otherwise unfavorable reaction through an induced polarized environment. The substrate binding pocket has many polar residues, e.g. Arg59 and Glu394 that bind the carboxylate and ammonium groups of the substrate.24 In addition, the side chains of the polar residues Met88, Tyr89, Met173 and Asn293 contact the indole group of the Trp substrate and through electrostatic and induced dipole moment interactions may affect the selectivity of the reaction, but this will need to be confirmed by future mutation studies.
Conclusions
In this work a combined experimental and computational study is presented for the possible reaction mechanisms of aromatic nitration by a P450 isozyme. Using stopped-flow kinetics, we show that dioxygen binds first to the heme iron followed by NO to generate an iron(III)-peroxynitrite species. We set up several active site model complexes with bound l-Trp and investigated substrate activation pathways leading to nitration as well as to oxidation of the substrate. We found low-energy barriers for the homolytic cleavage of iron(III)-peroxynitrite into NO2 and CpdII. Consequently, the reaction of NO and O2 on an iron(III)-heme center is expected to generate free NO2 radicals efficiently. We then explored NO2 attack on l-Trp and found pathways for activation at the C4, C5, C6 and C7-positions. Although NO2 addition is generally a low-energy process, many pathways encounter relatively high energy hydrogen atom transfer steps to form products. As such, substrate positioning and binding must be essential in P450 TxtE as indeed is observed experimentally,26 where a point mutation in a dynamic loop forming the active site lid caused a complete switch of nitration regiochemistry from C4 to C5.
Acknowledgments
M.Q.E.M. thanks the Malaysian Government and Islamic Science University of Malaysia (USIM) for a studentship. The Punjab Education Endowment Fund (PEEF) in Pakistan is acknowledged for a Ph.D. scholarship to H.A.S. The Biotechnology and Biological Sciences Research Council (BBSRC) is thanked for support to N.S.S., A.W.M., and G.L.C. (grant refs BB/H006265/1 and BB/H006281/1). The assistance of Dr. Lijiang Song with LC-MS measurements is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c05070.
Detailed experimental data, including experimental setup of double mixing experiments, spectroscopic and mass spectrometric data; in addition, full computational data with all optimized geometries on model A and B for the substrate hydroxylation and nitration of l-Trp by an iron(III)-peroxynitrite structure; the data contain absolute and relative energies, group spin densities and charges, and Cartesian coordinates of all optimized geometries; in addition, constraint geometry scan, drawings of optimized geometries, and intrinsic reaction coordinate scans are given (PDF)
Author Present Address
∇ (S.B.) Department of Chemistry, Faculty of Natural & Mathematical Sciences, King’s College London, Britannia House, 7 Trinity Street, London SE1 1DB, United Kingdom.
Author Contributions
○ S.L. and S.M.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R., Ed.; Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, 2005. [Google Scholar]
- Kadish K. M., Smith K. M., Guilard R., Eds.; Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010. [Google Scholar]
- Munro A. W.; Girvan H. M.; McLean K. J. Variations on a (t)heme—novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. 10.1039/B604190F. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauser J. A.; Guengerich F. P. Cytochrome P450 3A4-catalyzed testosterone 6β-hydroxylation stereochemistry, kinetic deuterium isotope effects, and rate-limiting steps. J. Biol. Chem. 2005, 280, 19496–19506. 10.1074/jbc.M501854200. [DOI] [PubMed] [Google Scholar]
- Spinello A.; Pavlin M.; Casalino L.; Magistrato A. A dehydrogenase dual hydrogen abstraction mechanism promotes estrogen biosynthesis: can we expand the functional annotation of the aromatase enzyme?. Chem. - Eur. J. 2018, 24, 10840–1849. 10.1002/chem.201802025. [DOI] [PubMed] [Google Scholar]
- Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; de Visser S. P., Kumar D., Eds.; Royal Society of Chemistry Publishing: Cambridge, U.K., 2011. [Google Scholar]
- Fessner N. D. P450 monooxygenases enable rapid late-stage diversification of natural products via C-H bond activation. ChemCatChem 2019, 11, 2226–2242. 10.1002/cctc.201801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinello A.; Ritacco I.; Magistrato A. The catalytic mechanism of steroidogenic cytochromes P450 from all-atom simulations: Entwinement with membrane environment, redox partners, and post-transcriptional regulation. Catalysts 2019, 9, 81. 10.3390/catal9010081. [DOI] [Google Scholar]
- Grogan G. Cytochromes P450: exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. 10.1016/j.cbpa.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Ang E. L.; Zhao H. Recent developments in the application of P450 based biocatalysts. Curr. Opin. Chem. Biol. 2018, 43, 1–7. 10.1016/j.cbpa.2017.08.006. [DOI] [PubMed] [Google Scholar]
- Li Z.; Jiang Y.; Guengerich F. P.; Ma L.; Li S.; Zhang X. W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 2020, 295, 833–849. 10.1074/jbc.REV119.008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos T. L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova I. F.; Poulos T. L. Understanding the mechanism of cytochrome CYP450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. 10.1039/C2DT31833D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T. C–H bond activation in heme proteins: the role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. 10.1016/j.cbpa.2009.02.028. [DOI] [PubMed] [Google Scholar]
- Meunier B.; de Visser S. P.; Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Huang X.; Groves J. T. Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. 10.1021/acs.chemrev.7b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar K. L.; Scharf D. H.; Litomska A.; Hertweck C. Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem. Rev. 2017, 117, 5521–5577. 10.1021/acs.chemrev.6b00697. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P.; Yoshimoto F. K. Formation and cleavage of C-C bonds by enzymatic oxidation-reduction reactions. Chem. Rev. 2018, 118, 6573–6655. 10.1021/acs.chemrev.8b00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry S. M.; Kers J. A.; Johnson E. G.; Song L.; Aston P. R.; Patel B.; Krasnoff S. B.; Crane B. R.; Gibson D. M.; Loria R.; Challis G. L. Cytochrome P450–catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol. 2012, 8, 814–816. 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Li M.; Xu C.; Wang Z.; Zhou H.; Yang M.; Chen Y.; Tang L.; He J. Structural insights into the mechanism for recognizing substrate of the cytochrome P450 enzyme TxtE. PLoS One 2013, 8, e81526 10.1371/journal.pone.0081526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodani S. C.; Cahn J. K. B.; Heinisch T.; Brinkmann-Chen S.; McIntosh J. A.; Arnold F. H. Structural, functional, and spectroscopic characterization of the substrate scope of the novel nitrating cytochrome P450 TxtE. ChemBioChem 2014, 15, 2259–2267. 10.1002/cbic.201402241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan H. M.; Munro A. W. Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr. Opin. Chem. Biol. 2016, 31, 136–145. 10.1016/j.cbpa.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Dodani S. C.; Kiss G.; Cahn J. K. B.; Su Y.; Pande V. S.; Arnold F. H. Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat. Chem. 2016, 8, 419–425. 10.1038/nchem.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo R.; Zhang Y.; Jiang C.; Hackett J. C.; Loria R.; Bruner S. D.; Ding Y. Engineered P450 biocatalysts show improved activity and regio-promiscuity in aromatic nitration. Sci. Rep. 2017, 7, 842. 10.1038/s41598-017-00897-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H.; Katsuyama Y.; Minami H.; Ohnishi Y. Identification and characterization of a bacterial cytochrome P450 monooxygenase catalyzing the 3-nitration of tyrosine in rufomycin biosynthesis. J. Biol. Chem. 2017, 292, 15859–15869. 10.1074/jbc.M117.791269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G.; Zuo R.; Zhang Y.; Powell M. M.; Zhang P.; Hylton S. M.; Loria R.; Ding Y. One-pot biocombinatorial synthesis of herbicidal thaxtomins. ACS Catal. 2018, 8, 10761–10768. 10.1021/acscatal.8b03317. [DOI] [Google Scholar]
- Zuo R.; Ding Y. Direct aromatic nitration system for synthesis of nitrotryptophans in Escherichia coli. ACS Synth. Biol. 2019, 8, 857–865. 10.1021/acssynbio.8b00534. [DOI] [PubMed] [Google Scholar]
- Buddha M. R.; Tao T.; Parry R. J.; Crane B. R. Regioselective nitration of tryptophan by a complex between bacterial nitric-oxide synthase and tryptophanyl tRNA synthetase. J. Biol. Chem. 2004, 279, 49567–49570. 10.1074/jbc.C400418200. [DOI] [PubMed] [Google Scholar]
- Greule A.; Stok J. E.; De Voss J. J.; Cryle M. J. Unrivalled diversity: the many roles and reactions of bacterial cytochromes P450 in secondary metabolism. Nat. Prod. Rep. 2018, 35, 757–791. 10.1039/C7NP00063D. [DOI] [PubMed] [Google Scholar]
- Alkhalaf L. M.; Barry S. M.; Rea D.; Gallo A.; Griffiths D.; Lewandowski J. R.; Fulop V.; Challis G. L. Binding of distinct substrate conformations enables hydroxylation of remote sites in Thaxtomin D by cytochrome P450 TxtC. J. Am. Chem. Soc. 2019, 141, 216–222. 10.1021/jacs.8b08864. [DOI] [PubMed] [Google Scholar]
- Schopfer F. J.; Baker P. R. S.; Freeman B. A. NO-dependent protein nitration: a cell signaling event or an oxidative inflammatory response?. Trends Biochem. Sci. 2003, 28, 646–654. 10.1016/j.tibs.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Lin H.-L.; Kenaan C.; Zhang H.; Hollenberg P. F. Reaction of human cytochrome P450 3A4 with peroxynitrite: Nitro-tyrosine formation on the proximal side impairs its interaction with NADPH-cytochrome P450 reductase. Chem. Res. Toxicol. 2012, 25, 2642–2653. 10.1021/tx3002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford P. C.; Lorković I. M. Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes. Chem. Rev. 2002, 102, 993–1017. 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- Herold S.; Matsui T.; Watanabe Y. Peroxynitrite isomerization catalyzed by His64 myoglobin mutants. J. Am. Chem. Soc. 2001, 123, 4085–4086. 10.1021/ja010111d. [DOI] [PubMed] [Google Scholar]
- Hersleth H.-P.; Ryde U.; Rydberg P.; Görbitz C. H.; Andersson K. K. Structures of the high-valent metal-ion haem–oxygen intermediates in peroxidases, oxygenases and catalases. J. Inorg. Biochem. 2006, 100, 460–476. 10.1016/j.jinorgbio.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Groenhof A. R.; Ehlers A. W.; Lammertsma K. Proton assisted oxygen-oxygen bond splitting in cytochrome P450. J. Am. Chem. Soc. 2007, 129, 6204–6209. 10.1021/ja0685654. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P.; Munro A. W. Unusual cytochrome P450 enzymes and reactions. J. Biol. Chem. 2013, 288, 17065–17073. 10.1074/jbc.R113.462275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan H. One small step for cytochrome P450 in its catalytic cycle, one giant leap for enzymology. J. Porphyrins Phthalocyanines 2019, 23, 358–366. 10.1142/S1088424619300040. [DOI] [Google Scholar]
- Dubey K. D.; Shaik S. Cytochrome P450: The wonderful nanomachine revealed through dynamic simulations of the catalytic cycle. Acc. Chem. Res. 2019, 52, 389–399. 10.1021/acs.accounts.8b00467. [DOI] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 2010, 330, 933–937. 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- Lang J.; Maréchal A.; Couture M.; Santolini J. Reaction intermediates and molecular mechanism of peroxynitrite activation by NO synthases. Biophys. J. 2016, 111, 2099–2109. 10.1016/j.bpj.2016.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toader V.; Xu X.; Nicolescu A.; Yu L.; Bolton J. L.; Thatcher G. R. J. Nitrosation, nitration, and autoxidation of the selective estrogen receptor modulator raloxifene by nitric oxide, peroxynitrite, and reactive nitrogen/oxygen species. Chem. Res. Toxicol. 2003, 16, 1264–1276. 10.1021/tx025641h. [DOI] [PubMed] [Google Scholar]
- Behan R. K.; Hoffart L. M.; Stone K. L.; Krebs C.; Green M. T. Reaction of cytochrome P450BM3 and peroxynitrite yields nitrosyl complex. J. Am. Chem. Soc. 2007, 129, 5855–5859. 10.1021/ja064590y. [DOI] [PubMed] [Google Scholar]
- Su J.; Groves J. T. Direct detection of the oxygen re-bound intermediates, ferryl Mb and NO2, in the reaction of metmyoglobin with peroxynitrite. J. Am. Chem. Soc. 2009, 131, 12979–12988. 10.1021/ja902473r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ortega A.; Quesne M. G.; Bui S.; Heyes D. J.; Steiner R. A.; Scrutton N. S.; de Visser S. P. Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. 10.1021/jacs.5b03836. [DOI] [PubMed] [Google Scholar]
- Lim M. D.; Lorković I. M.; Ford P. C. The preparation of anaerobic nitric oxide solutions for the study of heme model systems in aqueous and nonaqueous media: some consequences of NOx impurities. Methods Enzymol. 2005, 396, 3–17. 10.1016/S0076-6879(05)96001-1. [DOI] [PubMed] [Google Scholar]
- Aga R. G.; Hughes M. N. The preparation and purification of NO gas and the use of NO releasers: the application of NO donors and other agents of nitrosative stress in biological systems. Methods Enzymol. 2008, 436, 35–48. 10.1016/S0076-6879(08)36003-0. [DOI] [PubMed] [Google Scholar]
- Cole L. J.; Huston W. M.; Moir J. W. B. Delivery of nitric oxide for analysis of the function of cytochrome c’. Methods Enzymol. 2008, 436, 21–33. 10.1016/S0076-6879(08)36002-9. [DOI] [PubMed] [Google Scholar]
- Hartung J. Organic radical reactions associated with nitrogen monoxide. Chem. Rev. 2009, 109, 4500–4517. 10.1021/cr900085j. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Borowski T.; de Visser S. P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. - Eur. J. 2016, 22, 2562–2581. 10.1002/chem.201503802. [DOI] [PubMed] [Google Scholar]
- Ghafoor S.; Mansha A.; de Visser S. P. Selective hydrogen atom abstraction from dihydroflavonol by a nonheme iron center is the key step in the enzymatic flavonol synthesis and avoids byproducts. J. Am. Chem. Soc. 2019, 141, 20278–20292. 10.1021/jacs.9b10526. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Cantú Reinhard F. G.; Sainna M. A.; Upadhyay P.; Balan G. A.; Kumar D.; Fornarini S.; Crestoni M. E.; de Visser S. P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. - Eur. J. 2016, 22, 18608–18619. 10.1002/chem.201604361. [DOI] [PubMed] [Google Scholar]
- Yang T.; Quesne M. G.; Neu H. M.; Cantú Reinhard F. G.; Goldberg D. P.; de Visser S. P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. 10.1021/jacs.6b05027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantú Reinhard F. G.; de Visser S. P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound I?. Chem. - Eur. J. 2017, 23, 2935–2944. 10.1002/chem.201605505. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3658. 10.1063/1.444267. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Cantú Reinhard F. G.; Faponle A. S.; de Visser S. P. Substrate sulfoxidation by an iron(IV)-oxo complex: benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. 10.1021/acs.jpca.6b09765. [DOI] [PubMed] [Google Scholar]
- Timmins A.; Saint-André M.; de Visser S. P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C–H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. 10.1021/jacs.7b02839. [DOI] [PubMed] [Google Scholar]
- Cantú Reinhard F. G.; Barman P.; Mukherjee G.; Kumar J.; Kumar D.; Kumar D.; Sastri C. V.; de Visser S. P. Keto-enol tautomerization triggers an electrophilic aldehyde deformylation reaction by a nonheme manganese(III)-peroxo complex. J. Am. Chem. Soc. 2017, 139, 18328–18338. 10.1021/jacs.7b10033. [DOI] [PubMed] [Google Scholar]
- Quaroni L. G.; Seward H. E.; McLean K. J.; Girvan H. M.; Ost T. W.; Noble M. A.; Kelly S. M.; Price N. C.; Cheesman M. R.; Smith W. E.; Munro A. W. Interaction of nitric oxide with cytochrome P450 BM3. Biochemistry 2004, 43, 16416–16431. 10.1021/bi049163g. [DOI] [PubMed] [Google Scholar]
- Munro A. W.; Girvan H. M.; Mason A. E.; Dunford A. J.; McLean K. J. What makes a P450 tick?. Trends Biochem. Sci. 2013, 38, 140–150. 10.1016/j.tibs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Ouellet H.; Lang J.; Couture M.; Ortiz de Montellano P. R. Reaction of Mycobacterium tuberculosis cytochrome P450 enzymes with nitric oxide. Biochemistry 2009, 48, 863–872. 10.1021/bi801595t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra A.; Denisov I. G.; Sligar S. G. Spectroscopic features of cytochrome P450 reaction intermediates. Arch. Biochem. Biophys. 2011, 507, 26–35. 10.1016/j.abb.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ost T. W. B.; Clark J.; Mowat C. G.; Miles C. S.; Walkinshaw M. D.; Reid G. A.; Chapman S. K.; Daff S. Oxygen activation and electron transfer in flavocytochrome P450 BM3. J. Am. Chem. Soc. 2003, 125, 15010–15020. 10.1021/ja035731o. [DOI] [PubMed] [Google Scholar]
- Sharma S. K.; Schaefer A. W.; Lim H.; Matsumura H.; Moënne-Loccoz P.; Hedman B.; Hodgson K. O.; Solomon E. I.; Karlin K. D. Six-coordinate peroxynitrite low-spin iron(III) porphyrinate complex-the product of the reaction of nitrogen monoxide (·NO(g)) with a ferric-superoxide species. J. Am. Chem. Soc. 2017, 139, 17421–17430. 10.1021/jacs.7b08468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M.-S.; Huang M.-J.; Watts J. D. Binding of O2 and NO to heme in heme-nitric oxide/oxygen-binding (H-NOX) proteins. A theoretical study. J. Phys. Chem. B 2013, 117, 10103–10114. 10.1021/jp403998u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T. Evidence for sulfur-based radicals in thiolate compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. 10.1021/ja991541v. [DOI] [Google Scholar]
- Shaik S.; de Visser S. P.; Ogliaro F.; Schwarz H.; Schröder D. Two-state reactivity (TSR) mechanisms of hydroxylation and epoxidation by cytochrome P450 revealed by theory. Curr. Opin. Chem. Biol. 2002, 6, 556–567. 10.1016/S1367-5931(02)00363-0. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Kumar D.; de Visser S. P.; Altun A.; Thiel W. Theoretical perspective on the structure and mechanism of cyto-chrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- Ji L.; Faponle A. S.; Quesne M. G.; Sainna M. A.; Zhang J.; Franke A.; Kumar D.; van Eldik R.; Liu W.; de Visser S. P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation?. Chem. - Eur. J. 2015, 21, 9083–9092. 10.1002/chem.201500329. [DOI] [PubMed] [Google Scholar]
- Pickl M.; Kurakin S.; Cantú Reinhard F. G.; Schmid P.; Pöcheim A.; Winkler C. K.; Kroutil W.; de Visser S. P.; Faber K. Mechanistic studies of fatty acid activation by CYP152 peroxygenases reveal unexpected desaturase activity. ACS Catal. 2019, 9, 565–577. 10.1021/acscatal.8b03733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser S. P.; Shaik S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 2003, 125, 7413–7424. 10.1021/ja034142f. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Milko P.; Schyman P.; Usharani D.; Chen H. Trends in aromatic oxidation reactions catalyzed by cytochrome P450 enzymes: A valence bond modeling. J. Chem. Theory Comput. 2011, 7, 327–339. 10.1021/ct100554g. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. - Eur. J. 2006, 12, 8168–8177. 10.1002/chem.200600376. [DOI] [PubMed] [Google Scholar]
- Balding P. R.; Porro C. S.; McLean K. J.; Sutcliffe M. J.; Marechal J.-D.; Munro A. W.; de Visser S. P. How do azoles inhibit cytochrome P450 enzymes? A density functional study. J. Phys. Chem. A 2008, 112, 12911–12918. 10.1021/jp802087w. [DOI] [PubMed] [Google Scholar]
- Kishimoto A.; Takagi K.; Amano A.; Sakurai K.; Mi-Zushima T.; Shimada H. Structure of a P450cam intermediate. Protein databank entry 3WRH.
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov R.; Makris T. M.; Kofman V.; Werst D. E.; Sligar S. G.; Hoffman B. M. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 2001, 123, 1403–1415. 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- Kamachi T.; Yoshizawa K. A Theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- Schöneboom S.; Cohen S.; Lin H.; Shaik S.; Thiel W. Quantum mechanical/molecular mechanical investigation of the mechanism of C-H hydroxylation of camphor by cytochrome P450cam: theory supports a two-state rebound mechanism. J. Am. Chem. Soc. 2004, 126, 4017–4034. 10.1021/ja039847w. [DOI] [PubMed] [Google Scholar]
- Lai R.; Li H. Hydrogen abstraction of camphor catalyzed by cytochrome P450cam: A QM/MM Study. J. Phys. Chem. B 2016, 120, 12312–12320. 10.1021/acs.jpcb.6b09923. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Hirao H.; de Visser S. P.; Zheng J.; Wang D.; Thiel W.; Shaik S. New features in the catalytic cycle of cytochrome P450 during the formation of Compound I from Compound 0. J. Phys. Chem. B 2005, 109, 19946–19951. 10.1021/jp054754h. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]
- Faponle A. S.; Quesne M. G.; de Visser S. P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production?. Chem. - Eur. J. 2016, 22, 5478–5483. 10.1002/chem.201600739. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Senthilnathan D.; Singh D.; Kumar D.; Maldivi P.; Sorokin A. B.; de Visser S. P. Origin of the enhanced reactivity of h-nitrido-bridged diiron(IV)-oxo porphyrinoid com-plexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. 10.1021/acscatal.5b02720. [DOI] [Google Scholar]
- Li X.-X.; Postils V.; Sun W.; Faponle A. S.; Solà M.; Wang Y.; Nam W.; de Visser S. P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant?. Chem. - Eur. J. 2017, 23, 6406–6418. 10.1002/chem.201700363. [DOI] [PubMed] [Google Scholar]
- Liu J. L.; Siegler M. A.; Karlin K. D.; Moënne-Loccoz P. Direct resonance Raman characterization of a peroxynitrito copper complex generated from O2 and NO and mechanistic insights into metal-mediated peroxynitrite decomposition. Angew. Chem., Int. Ed. 2019, 58, 10936–10940. 10.1002/anie.201904672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik S.; de Visser S. P.; Kumar D. External electric field will control the selectivity of enzymatic-like bond activations. J. Am. Chem. Soc. 2004, 126, 11746–11749. 10.1021/ja047432k. [DOI] [PubMed] [Google Scholar]
- Timmins A.; Fowler N. J.; Warwicker J.; Straganz G. D.; de Visser S. P. Does substrate positioning affect the selectivity and reactivity in the hectochlorin biosynthesis halogenase?. Front. Chem. 2018, 6, 513. 10.3389/fchem.2018.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Fried S. D.; Boxer S. G. A Preorganized electric field leads to minimal geometrical reorientation in the catalytic reaction of ketosteroid isomerase. J. Am. Chem. Soc. 2020, 142, 9993–9998. 10.1021/jacs.0c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.